

Abstract
CHARMM is rich in methodology and functionality as one of the first programs addressing problems of molecular dynamics and modeling of biological macromolecules and their partners, e.g., small molecule ligands. When combined with the highly developed CHARMM parameters for proteins, nucleic acids, small molecules, lipids, sugars, and other biologically relevant building blocks, and the versatile CHARMM scripting language, CHARMM has been a trendsetting platform for modeling studies of biological macromolecules. To further enhance the utility of accessing and using CHARMM functionality in increasingly complex workflows associated with modeling biological systems, we introduce pyCHARMM, Python bindings, functions, and modules to complement and extend the extensive set of modeling tools and methods already available in CHARMM. These include access to CHARMM function-generated variables associated with the system (psf), coordinates, velocities and forces, atom selection variables and force field related parameters. The ability to augment CHARMM forces and energies with energy terms or methods derived from machine learning or other sources, written in Python, CUDA or OpenCL and expressed as Python callable routines is introduced together with analogous functions callable during dynamics calculations. Integration of Python-based graphical engines for visualization of simulation models and results is also accessible. Loosely coupled parallelism is available for workflows such as free energy calculations, using MBAR/TI approaches or high-throughput multisite λ-dynamics (MSλD) free energy methods, string path optimization calculations, replica exchange and molecular docking with a new Python-based CDOCKER module. CHARMM accelerated platform kernels through the CHARMM/OpenMM API, CHARMM/DOMDEC and CHARMM/BLaDE API are also readily integrated into this Python framework. We anticipate that pyCHARMM will be a robust platform for the development of comprehensive and complex workflows utilizing Python and its extensive functionality as well as an optimal platform for users to learn molecular modeling methods and practices within a Python-friendly environment such as Jupyter Notebooks.
Keywords: molecular modeling, molecular dynamics, free energy, docking, machine learning
Graphical abstract

Introduction
Understanding how biological macromolecular systems (proteins, nucleic acids, lipid membranes, carbohydrates, and their complexes) function is a major objective of current research by computational chemists and biophysicists. The utility of atomic models with realistic microscopic interactions in the investigation of biomolecules, as well as other chemical systems, has been established through what now represents nearly five decades of computational studies to address biological questions.1 These methods and applications have been described in a plethora of books and reviews.2–11 Moreover, such studies have now reached a point where computational models play an important role in the design and interpretation of experiments.12 This is of particular importance where molecular simulations are used to obtain information that is difficult to determine experimentally.13,14
With the continued evolution of the field of biomolecular simulation and the complexity of questions explored and phenomena investigated, maintaining maximum flexibility and availability of a wide range of computational methods for the exploration and integration of novel ideas in research and its applications is essential. Access to software platforms for the development and application of computational biophysical methods has spurred the introduction of several general-purpose programs distributed in academic and commercial environments. Many of these were described in two special issues of the Journal of Computational Chemistry (JCC)15–20 and elsewhere.21–23 Additionally, resources that enable complex simulation models of biological macromolecules to be set-up, run and analyzed have emerged, with CHARMM-GUI seeing the broadest range of functionality.24–47
CHARMM (Chemistry at HARvard Molecular Mechanics) is a general and flexible molecular simulation and modeling program that uses classical (empirical, fixed charge and polarizable, and semi-empirical) and quantum mechanical (QM) (semi-empirical or ab initio) energy functions for molecular systems of many different classes, sizes, and levels of heterogeneity and complexity. This functionality is integrated into a single executable (approximately 1,000,000 lines of modular Fortran, C/C++, CUDA and OpenCL code) and calculations are accessed using CHARMM through the interpreted CHARMM scripting language. This feature in CHARMM is unique relative to the other major packages noted above, and has been provided since the introduction of CHARMM in 1983.48 The script-level programmability of CHARMM has enabled many algorithmic ideas and complex simulation schemes to be tested and applied without the need to develop software routines to be compiled and integrated with the executable version of the program on a particular platform. This ability to prototype methodological ideas prior to committing them to code has been a key driver in the impressive range of methods available in CHARMM.
Within the CHARMM scripting language a set of command structures, including GOTO, STREam, and IF-ELSE-ENDIf structures, corresponding to the respective control-flow statements in most programming languages, provide the basis for the powerful high-level scripting function that permits the general and flexible control of complicated simulation protocols and facilitates the prototyping of new methods as just noted. The various functionalities of CHARMM can be combined in myriad ways using these command structures. The order of accessible CHARMM commands at any point in the script is controlled by the data required by the command. One example would be calculation of the energy: the energy cannot be calculated until i) the topology and parameter files describing the fragment (or residue) libraries needed to generate the system of interest are read; ii) the sequence of said residues is specified; iii) the data structures associated with that sequence are generated, i.e., the psf is read and any patching is performed; iv) and the coordinates are either built or read from a file. While other programs used in the field enable some of this flexibility through shell scripting or related interpreted language scripting that is exercised during the running of the program, these programs largely rely on fixed input files to describe a single computational workflow or rely on external programs and pre-processing to achieve related objectives. The programmability of CHARMM through its scripting language is unique and was instrumental in establishing both complex workflows in structure refinement as well as simplified interfaces for specific modeling tasks, both realized in the MMTSB ToolSet developed in 2004.49
pyCHARMM Architecture, Organization and Functionality
Given the architecture of CHARMM and its extensive programmability using the CHARMM scripting language, it is a natural evolution to augment the CHARMM scripting language to leverage the popularity, familiarity and utility of the Python language.50 This task was accomplished by implementing a thin API for calling CHARMM routines from Python, called pyCHARMM.
Installation of pyCHARMM starts with obtaining the latest version of the CHARMM source code and compiling CHARMM as a shared, callable library (as opposed to a binary executable). Separately, using the pip package manager, pyCHARMM – expressed as a Python package – is installed. To successfully complete the process, an environment variable called CHARMM_LIB_DIR is defined to inform the Python module of the location of the CHARMM shared library. It is this library that the modules of pyCHARMM use to access CHARMM functionalities. The detailed steps of installation are provided in the SI.
A successful installation of the pyCHARMM package in a location appropriate to Python creates a thin Python API - where each module consists of Python bindings to Fortran functions that collectively constitute a thin Fortran API, a.k.a. the pyCHARMM API. Through these functions, access is provided to the core CHARMM functionalities and users only interact directly with the Python API. This architecture, through careful marshalling, ensures that many of the key CHARMM data structures can be bi-directionally passed between the two APIs. Figure 1 illustrates the architecture of the installation. From the user’s end, calls to functions are made which eventually access Fortran/C++ routines in CHARMM via Python bindings to these routines. A Python binding in a module calls a Fortran function defined in a Fortran module file and they typically have a one-to-one relation. As Fortran routines are successfully executed, the returned data travel back to the end-user.
Figure 1:
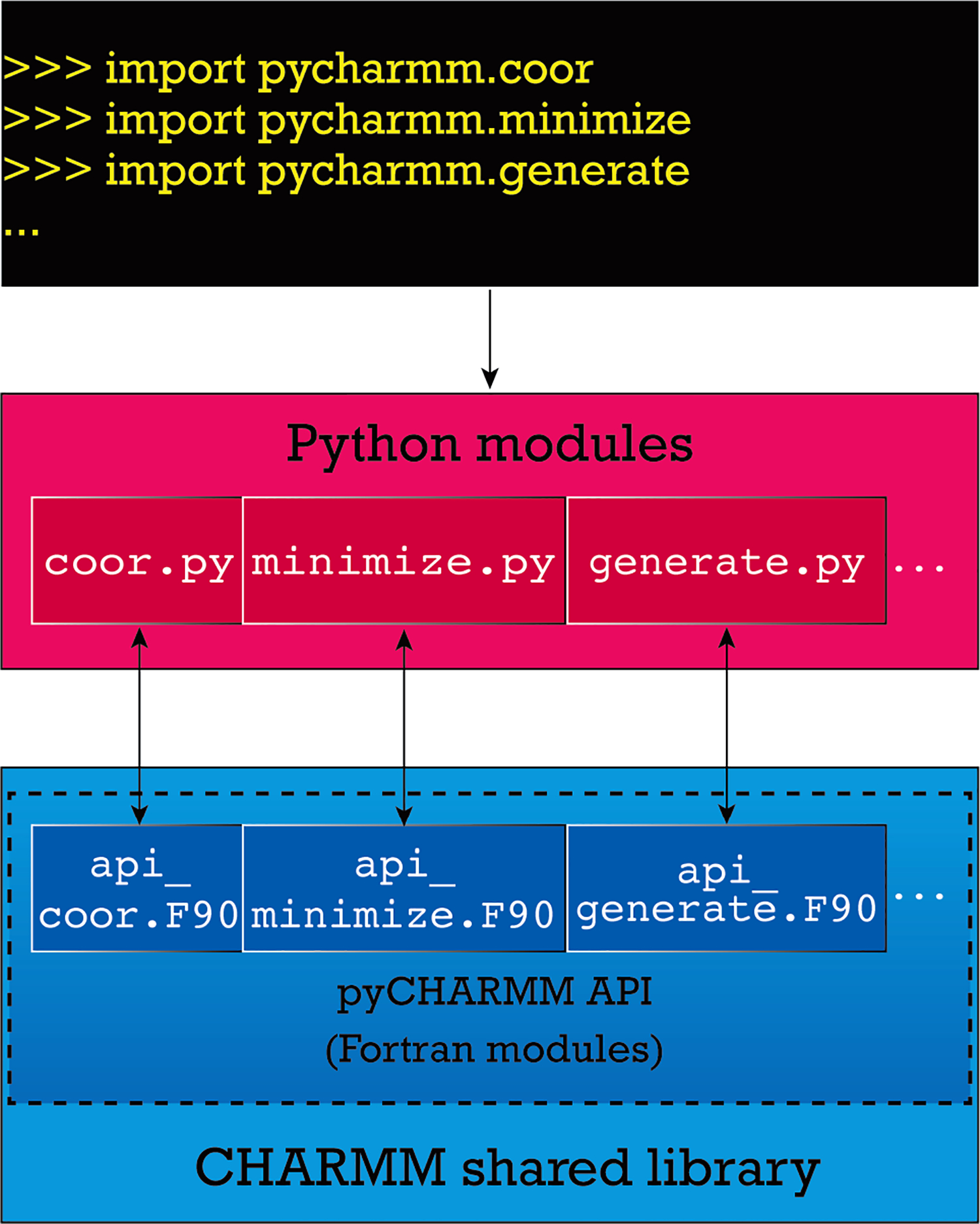
pyCHARMM architecture. (Top) The end user writes a Python script to execute CHARMM commands by their Python expressions after importing the necessary Python modules in pyCHARMM. (Middle) The functions available to the user are the Python modules in the Python API, which in many cases represent bindings to the Fortran routines in the pyCHARMM API (or the Fortran module). The pyCHARMM API is a collection of Fortran modules that is included with the CHARMM source code.
The current pyCHARMM APIs consist of the functional components of CHARMM and provide the interfaces between CHARMM functionality and its expression in Python. For example, as illustrated in Figure 1, the API file api_coor.F90 provides an interface between CHARMM coordinate functionality as part of the coordinate manipulation commands (doc/corman.info) and the pyCHARMM commands, api_minimize.F90 interfaces with the CHARMM ABNR, SD and OpenMM (OMM) minimizers, api_generate.F90 contains the interface to Python instantiations of psf generation and manipulation commands (from doc/struct.info) and api_ic.F90 enables the internal coordinate manipulation commands from CHARMM (doc/ic.info). A list of current pyCHARMM APIs is presented in Table S1.
The CHARMM commands and functionality are then expressed through Python modules. These modules provide bindings to the CHARMM commands denoted by the module name and they must be imported into the python script before they can be used. For example import pycharmm.shake provides the Python bindings to set-up and execute the SHAKE command and import pycharmm.nbonds allows the manipulation of the non-bonded data structure (e.g., see doc/nbonds.info). Likewise, the module psf.py provides an interface to the psf-based data structures and access to the variables that reside within them. Table S2 lists the Python modules provided as part of the CHARMM distribution for use in pyCHARMM.
In the Python expression of CHARMM, the basic CHARMM scripting keywords have been largely maintained to provide continuity in the structure of CHARMM commands through pyCHARMM. This was deliberate to conveniently enable an experienced CHARMM user to transfer their current CHARMM scripts to pyCHARMM scripts as well as allow new CHARMM users to utilize the extensive libraries of CHARMM scripts that are available on the world-wide-web. The pyCHARMM modules can also be conveniently integrated with other functionalities from external Python modules and libraries for the creation of complicated frameworks for simulations and/or analyses. For instance, the module energy_func.py represents an abstraction of the CHARMM user energy term and enables one to call any user-supplied energy function at every energy evaluation during pyCHARMM execution. This function can also utilize Python interfaces to machine learned energy functions such as the TorchANI Neural Network Potential in PyTorch,51 as well as implementations of CUDA or OpenCL utilizing established Python APIs, e.g, NVIDIA CUDA Python 11.7.1, Numba, PyCUDA, and PyOpenCL.
A Python module in pyCHARMM typically represents a single command in CHARMM and is documented with `Docstrings`, which allow the user to access documentation at the command line or at the Python session prompt. Alternatively, a user can also visit pyCHARMM’s documentation located at https://github.com/clbrooksiii/pyCHARMM-Workshop that provides details about the usage of each Python module, the CHARMM commands they provide bindings to (with examples and links to the command’s documentation webpage), and their member functions along with their syntactic usage, input parameters and return values.
While the core of CHARMM functionality is directly represented by Python modules, in the present implementation not every CHARMM command is expressed by an explicit Python binding. Nevertheless, all CHARMM commands are available for use in Python scripting through the lingo.py module, which is an interface to the CHARMM script command interpreter. This module can also be combined with script.py to create callable custom python functions for repeated tasks.
Python Mapping of “Standard” Molecular Modeling Tasks in pyCHARMM
It is instructive to illustrate the correspondence between a simple task in CHARMM scripting language and pyCHARMM. In Figure 2, we illustrate this for the case of building a blocked alanine residue, minimizing its conformation in vacuum, and computing its energy. The emphasis is on the correspondence between the CHARMM script commands and the structure of the Python bindings to those commands. The close correspondence was a design feature instituted in this manner to allow users to take advantage of the wide range and distribution of CHARMM scripts as they begin learning to program their molecular modeling applications in pyCHARMM.
Figure 2:

Building an alanine dipeptide. An illustration of the correspondence between CHARMM scripting language commands and their expression as Python functions. Shown are the specific tasks of building the structure of Ala-dipeptide, assigning coordinates to it, and setting up a list of non-bonded interaction parameters for energy minimization calculations.
The tasks illustrated in Figure 2 also include reading the CHARMM topology and parameter files, reading a protein sequence, and in the end writing a CHARMM psf file and coordinates in CHARMM CARD and PDB formats. The Python bindings for such commands in pyCHARMM are contained in the read.py and write.py modules as noted in Table S2. Also included are psf generation and terminal patching, non-bonded specification, energy calculation and minimization.
A key point to note from this illustrative example is the order in which pyCHARMM functions can be called. When using pyCHARMM, Pythonic functions – Python expressions of the CHARMM commands – must be called in the same order in which the corresponding CHARMM commands would typically be called. For instance, functions to read the topology and parameter files must precede functions to build or read the structures (using one of the many ‘struct’ module commands like READ SEQUence/GENErate or READ PSF). Likewise, coordinates for all the atoms in the system must be defined before the functions to build the simulation box (CRYSTal) and setup images are called when setting up the periodic boundary conditions.
Running Dynamics using pyCHARMM
In CHARMM, the DYNAmic command is used to run molecular dynamics. In pyCHARMM, to do the same, an object of class `DynamicsScript` must be invoked and then the object’s member function `run()` must be called. An illustrative example in Figure 3 shows how the CHARMM DYNAmic command corresponds to the use of an instance of `DynamicsScript`.
Figure 3:

Configuring and performing a molecular dynamics run using the ‘DynamicsScript’ class in pyCHARMM. The correspondence with the DYNAmic command in CHARMM in terms of the use of command keywords as function arguments to instantiate an object of the class is shown and the presence of different categories of run parameters is illustrated. In Figure S1, the use of Python dictionaries for the same purpose is shown.
The example shows the use of function arguments used to instantiate the object. These arguments correspond exactly to the keywords that accompany the DYNAmic command in CHARMM. For example, the keyword “NSTEP”, which is used for the number of steps of integration, is supplied as a function argument by the same name (lower cased). Likewise, these function arguments can be used to specify the frequency-spec parameters (e.g., frequency of updating non-bonded lists), temperature-spec parameters (e.g., temperature and pressure conditions), unit-spec parameters (e.g., I/O channels) and options-spec parameters (e.g., method of velocity assignment) for the run. A complete list of keywords accompanying the DYNAmic command, with detailed documentation for each of the keywords, can be found at https://academiccharmm.org/documentation/version/c47b1/dynamc. One should also note that there are certain keywords as components of the DYNAmic command which do not have an associated value but whose presence in the command dictates their use. For example, the keyword “START” in the command instructs CHARMM to initiate a new dynamics run as opposed to extending a previous run (done using the RESTART keyword instead). To use them in pyCHARMM, the function argument (of the same name as the keyword) should be supplied with a ‘True’ or ‘False’ value to achieve the equivalent effect, e.g., ‘start’ = True. Conveniently, pyCHARMM also accommodates the use a Python dictionary with the aforementioned parameters instead of/in conjunction with the function arguments (Figure S1).
Of added note is the fact that the ‘DynamicsScript’ class is derived from the ‘CommandScript’ base class in pyCHARMM. Other derivatives are the ‘NonBondedScript’ and ‘UpdateNonBondedScript’. Likewise, they can be instantiated in the same way where the command keywords can be provided as function arguments or Python dictionaries or both. Then a member function ‘run()’ executes the associated action. In Figure 2, the use of ‘NonBondedScript’ class is shown.
pyCHARMM Script Factory
The ‘script_factory’ function in the pyCHARMM module gives users a convenient way to run any CHARMM command like they would when running CHARMM. This feature allows them to execute CHARMM commands that, at present, do not have a corresponding pyCHARMM function. The function takes a string as the first argument, which must correspond to a CHARMM command, and returns an object of the ‘CommandScript’ class. Subsequently, an object of the newly minted class can be instantiated while simultaneously providing arguments for the CHARMM command as function arguments or as keys in a Python dictionary. These arguments, akin to the instantiation of the DynamicsScript class object mentioned above, must correspond to the keywords that accompany the command for which the class was created. Note that keywords, whose presence only directs CHARMM to invoke or revoke specific operations of that command, must be passed with a True or False value as opposed to keywords expected to be accompanied by a value, which must be passed as an argument along with that value. As a child of the CommandScript base class, the returned class has a ‘run()’ method which can be called to execute the command. Table 1 presents an example usage of the script_factory function to run the ‘SKIPenergy’ command in CHARMM, which directs the ‘ENERgy’ command to selectively exclude or include terms when calculating and printing out energy values.
Table 1:
Example usage of the script_factory function to execute a CHARMM command without a corresponding pyCHARMM function.
| SKIPenergy – EXCL BOND ANGL DIHE | import pycharmm NewSkipClass = pycharmm.script_factory(‘SKIPenergy’) my_skip = NewSkipClass(excl = “bond angl dihe”) my_skip.run() |
The ‘script_factory’ function and the ‘charmm_script’ function in the lingo.py module are essentially the same. But whereas the latter would have to be fully written every time a certain command is called (for example in a loop), the class returned by ‘script_factory’ associated with that command can be used repeatedly and more conveniently to do the same.
Preserving and Extending Parallelization
CHARMM has multiple accelerated platform kernels for molecular dynamics simulations and related modeling tasks, such as the CHARMM/OpenMM interface, CHARMM/DOMDEC52 and CHARMM/BLaDE53. They can all be accessed in pyCHARMM to run individual simulations on CPU and/or GPU hardware. In addition, the Python package mpi4py54, 55 can be used to run multiple simulations simultaneously, with flexible distribution of work across parallel processors enabling loosely-coupled parallelism via MPI. These two levels of parallelization can lead to significant speed-ups at par with CHARMM. However, tightly coupled MPI parallelism, as used in large parallel DOMDEC simulations, is not currently accessible in pyCHARMM.
Python Integrated Workflows Facilitate CHARMM-based Simulations in pyCHARMM
A key advantage of pyCHARMM is the ready integration with a broad range of Python functionality. This may include existing Python-based toolkits for preparing initial structures for molecular modeling and simulations (e.g., RDKit,56 PROPKA,57 Pybel,58 and PDB2PQR59, 60), analysis of molecular dynamics simulations, such as MDAnalysis,61, 62 and MDTraj,63 and visualization of molecular systems and trajectories, including VMD-Python,64 PyMOL, py3Dmol and NGLView.65 On one hand, users could prepare a system using a single Python script and closely monitor the changes made during the process, for example, in a Python-friendly environment like Jupyter Notebooks.66 This makes pyCHARMM a powerful tool for beginners to learn molecular modeling and simulation. On the other hand, it allows for easy implementation of various pipelines using pyCHARMM and even enables end-to-end application in some cases. For instance, by combining pyCHARMM with free energy calculation tools like FastMBAR67 or pymbar,68 one can implement a pipeline to rapidly compute absolute solvation free energies of small molecules using the MBAR/TI approach (see Figure 4 and AbsoluteSolvation on the GitHub site pyCHARMM-Workshop). Here, multiple molecular dynamics simulations need to be performed at various λ values for a given molecule. Then, potential energies are computed for each trajectory under perturbed thermodynamic states (i.e., with different λvalues), which are used in FastMBAR,67 a Python solver for large scale MBAR equations, for free energy calculations. Due to the ready integration of pyCHARMM with FastMBAR and other Python modules such as numpy69 and pandas,70 simulation and analysis can be performed without exiting the Python environment, and this pipeline allows for end-to-end prediction of absolute solvation free energy of a given small molecule. This is illustrated in a simple example Jupyter Notebook in the GitHub repository noted above. The results of running a set of such calculations on a subset of the FreeSolv database71 using the CGENFF force field and program are shown in Figure 4B, where we illustrate application of the integrated workflow of such calculations using pyCHARMM. Similarly, we can perform high-throughput multisite λ-dynamics (MSλD) simulations72 using pyCHARMM to calculate relative binding free energies for multiple small molecules to a target protein. In this case, the Python tool msld_py_prep73 may be utilized to identify the maximum common substructure of the set of small molecules, followed by charge renormalization. The adaptive landscape flattening (ALF) algorithm,74 which is available as a series of Python scripts, may be used to flatten the free energy landscapes in the alchemical space, thus enhancing sampling. By integrating msld_py_prep and ALF with pyCHARMM using Python scripts it becomes much easier to perform such MSλD simulations, allowing one to compute the relative free energies of many molecules in a single simulation.
Figure 4:

(a) Illustration of the workflow in the absolute solvation free energy calculation pipeline. Based on the thermodynamic cycle, the absolute solvation free energy is obtained from the difference between ΔGvaccum and ΔGwater, i.e., the free energy change of molecule annihilation in vacuum and water, respectively. To compute ΔGvaccum and ΔGwater, multiple molecular dynamics simulations are performed at different λ values and potential energies are computed for each trajectory under perturbed thermodynamic states. These simulations and energy calculations can be executed in parallel to reduce the overall wall-time. Finally, FastMBAR is utilized to calculate the free energy difference between different alchemical states. (b) The absolute solvation free energies of 206 representative molecules from the FreeSolv database71 obtained using a protocol written in pyCHARMM based on the examples noted above is compared with their experimental values. The dashed line denotes the y=x line. Representative molecules were obtained by sampling from 50 functional fingerprint clusters into which all 642 molecules of the FreeSolv database was partitioned. Individual molecules were parameterized with CGenFF force field and program.75, 76 A representative example of this workflow is included in the GitHub repository as noted above. A detailed spreadsheet containing the molecules SMILES strings, the computed free energies and the experimental free energies taken from the FreeSolv database71 are included in the SI.
Although the original CHARMM scripting could also be used to develop complex workflows, preparing such scripts can be daunting for researchers who have little experience in molecular simulation and modeling. For example, to perform rigid docking with the CDOCKER module in CHARMM,77, 78 the CHARMM script contains more than 400 lines for grid generation, ligand initial placement, i.e., random translation and rotation from the pocket center, hundreds of docking trials with the simulated annealing algorithm, clustering and sorting of docked poses, and energy minimization with explicit all-atom representations. For flexible CDOCKER,79 additional scripting is needed to select flexible side chains of the receptor, and to use the genetic algorithm to search for optimal docking poses. With pyCHARMM, these complex workflows have been compiled into a single Python module (pycharmm.cdocker) and standard docking calculation can be performed through a single pyCHARMM command (see Figure 5 and CDOCKER_WYu on the GitHub site pyCHARMM-Workshop). This greatly simplifies the usage of CDOCKER in CHARMM and is anticipated to enhance its accessibility and utility for a larger community. Also, pyCHARMM CDOCKER reduces I/O, which leads to reduced wall-time for a given docking trial. Therefore, more extensive searching can be performed to further improve the docking accuracy.80
Figure 5:

Illustration of pyCHARMM CDOCKER. These complex workflows have been compiled into a single Python module (pycharmm.cdocker) and standard docking calculation can be performed through one-line of Python code. In this example, a ligand has been docked at a binding site after CDOCKER searched a cubic volume of edge length of 25.762 Å, centered at (12.33 Å, 33.48 Å, 19.70 Å). See the GitHub site pyCHARMM-Workshop for complete examples of rigid receptor - flexible ligand CDOCKER and flexible CDOCKER.
Another powerful feature in CHARMM is the ability of augmenting CHARMM forces and energies from user-defined energy terms in molecular modeling applications, e.g., molecular dynamics simulations, minimization, etc. With pyCHARMM, we believe that it becomes exceptionally easy to use this feature, for instance, in enhanced sampling techniques where novel, custom biasing potentials need to be incorporated, or in simulations with machine learning based force fields. In the SI (see NeuralNetExample on the GitHub site pyCHARMM-Workshop), we illustrate a simple example of using the TorchANI force field51 in pyCHARMM to examine the potential energy as a function of the dihedral angle of butane. TorchANI is a Python implementation of the ANI neural network potentials based on PyTorch, which provides accurate energies and forces for organic molecules at the QM level. With a concise pyCHARMM script, one can set up the TorchANI model and incorporate it into CHARMM calculations through the user-defined energy term. Such extensibility, customization and user-friendliness of pyCHARMM may dramatically simplify the setup of simulations with novel potential energy functions. For example, mixed machine-learning/empirical energy functions have been used in reactive molecular dynamics simulations to study ligand binding to proteins, which can carry out the necessary sampling to reach relevant time scales that were impossible with QM/MM approaches.81, 82 Also, hybrid machine learning/molecular mechanics potentials have shown to be highly accurate in relative binding free energy calculations.83 With pyCHARMM, such simulations may be more accessible to the scientific community.
As discussed in the previous section, pyCHARMM preserves and even extends the parallel capabilities of CHARMM. A particularly useful feature is in the case of loosely coupled parallelism through the Python module mpi4py.54, 55 For instance, in the pipeline for absolute solvation free energy calculations as mentioned above (see Figure 4 and AbsoluteSolvation on the GitHub site pyCHARMM-Workshop for more details), mpi4py is utilized to run multiple simulations at different λ windows simultaneously, with flexible distribution of work across parallel processors (GPUs in this instance). This greatly reduces the wall time of end-to-end prediction of free energies. Similar parallelization schemes can be realized in other complex workflows as well, such as the string method for path optimization,84–87 replica exchange,88, 89 umbrella sampling,90 weighted ensemble,91–93 and even machine-learning based enhanced sampling methods like diffusion-map-directed molecular dynamics simulations,94–96 For example, here we provide an implementation of the string method using harmonic Fourier beads (HFB)85 to find the optimal path between two metastable states of an alanine dipeptide (see Figure 6 and Aladipeptide_HFBString_MPI on the GitHub site pyCHARMM-Workshop). In the space of a set of collective variables, which are usually Cartesian coordinates of selected atoms, multiple beads (i.e., conformations of the system) are equally distributed along the string. During each cycle of path optimization, all beads evolve independently, using either energy minimization (to find the minimum energy path) or molecular dynamics simulation (to find the minimum free energy path). Such a path evolution procedure can be easily parallelized through mpi4py, by distributing the beads across multiple processors. After this, information from all beads is gathered and the path is updated through the HFB interpolator. Similarly, for replica exchange simulations, where multiple copies of the same system (i.e., replicas) are simulated under different thermodynamic states and they are exchanged periodically based on the Metropolis criterion, these simulations can run in parallel using mpi4py to allocate resources for each replica and to gather information from all replicas to determine how exchange is attempted and whether an exchange should be accepted (see ReplicaExchangeExample on the GitHub site pyCHARMM-Workshop for details). Besides speeding up the overall calculations in these complex workflows, another great advantage of such loosely coupled parallelization through mpi4py is that it allows researchers to modify the parallel aspects of the workflow without having to change the source code of CHARMM. Thus, pyCHARMM enables prototyping new methodological ideas in a facile manner.
Figure 6:

Demonstration of the string method applied to determine the minimum energy path between two metastable states of an alanine dipeptide in vacuum using the harmonic Fourier beads method.85 The initial path is shown in red and the path after 2000 iterations is shown in black. See the GitHub site pyCHARMM-Workshop for a complete example.
Conclusion
In this paper, we have presented a novel Python-based instantiation of CHARMM, pyCHARMM, as well as the Python bindings, functions and modules that complement and extend the extensive set of modeling tools and methods already available in CHARMM. All of the CHARMM commands are available in pyCHARMM, either as direct Python bindings or through the CHARMM command interpreter module lingo.py. This allows users to take advantage of utilizing the wide range and distribution of CHARMM scripts as they begin learning to program their molecular modeling applications in pyCHARMM. Moreover, due to its ready integration with other Python-based visualization toolkits such as NGLView or py3Dmol, users could closely monitor the changes made at each step, especially within a Python-friendly environment like Jupyter Notebooks. Therefore, we believe that pyCHARMM will be an optimal platform for users to learn molecular modeling and simulation. With pyCHARMM new methods can become immediately available to the larger community through repositories of pyCHARMM scripts and Jupyter Notebooks. For instance, the complex workflow of rigid and flexible docking through CDOCKER has been compiled into a single Python module (pycharmm.cdocker) and standard docking calculations can be performed with a one line Python call. This makes pyCHARMM CDOCKER more user-friendly and will enhance its accessibility and utility for a larger community.
pyCHARMM should also serve as a robust platform for the development of comprehensive and complex workflows for researchers with more experience in molecular simulation and modeling. As a complete and programmable package in the Python framework, pyCHARMM can be readily integrated with other Python functions and toolkits. This allows for easy implementation of various pipelines, such as alchemical free energy calculations using MBAR/TI approaches or high-throughput free energy calculations with MSλD. Another powerful feature in pyCHARMM is the ability to augment CHARMM forces and energies with user-defined energy terms, especially machine learning based force fields. This may be particularly useful in developing novel enhanced sampling methods as well as in simulations with hybrid machine learning/molecular mechanics force fields. pyCHARMM also provides a straightforward means of implementing parallel calculations. Moreover, multiple accelerated platform kernels, such as the CHARMM/OpenMM interface, CHARMM/DOMDEC and CHARMM/BLaDE, are available in pyCHARMM. Loosely coupled parallelization achieved through the Python module mpi4py, for instance, in alchemical free energy calculations, string path optimization calculations, and replica exchange significantly enhance the configurability and facile implementation of such calculations. This not only speeds up the overall calculations in these complex workflows, but also makes modification of existing workflows exceptionally easy. Taken all together, pyCHARMM permits the general and flexible control of complicated simulation protocols and facilitates the prototyping of new methodological ideas.
We finally summarize by noting that CHARMM and subsequently pyCHARMM are available free of charge to non-profit laboratories through the established CHARMM licensed distribution at www.academiccharmm.org. This distribution provides full access to all sources needed to install pyCHARMM. In addition, we have developed an evolving GitHub site pyCHARMM-Workshop (https://github.com/clbrooksiii/pyCHARMM-Workshop) that includes many of the examples discussed in this paper as Jupyter Notebooks or python scripts. With the work we present here it is our intention to provide a Python-based platform for molecular modeling and simulation that brings the extensive development of methods and techniques from CHARMM to the broad community of novice and expert molecular modelers.
Supplementary Material
Acknowledgements
We would like to acknowledge the contributions from all the Brooks group members for their assistance in testing and trying the evolving pyCHARMM development and for participating in the first pyCHARMM workshop carried out in the summer 2022. Furthermore, we acknowledge contributions and participation from members of the groups of Carol Post, Alex MacKerell and Markus Meuwly in this same workshop. We are grateful for support from the National Institute of Health through grant GM130587.
Footnotes
Notes
The authors declare no competing financial interest.
Data Availability
All data, examples, documentation files and tutorial files that are described and discussed here are available in the GitHub repository (https://github.com/clbrooksiii/pyCHARMM-Workshop). The full source and documentation for CHARMM and pyCHARMM are licensed and available free of charge to non-profit and academic laboratories at https://academiccharmm.org/program. The release version of CHARMM/pyCHARMM described here corresponds to c47b2.
References:
- (1).Macuglia D; Roux B; Ciccotti G The emergence of protein dynamics simulations: how computational statistical mechanics met biochemistry. Eur. Phys. J. H 2022, 47, 13. DOI: 10.1140/epjh/s13129-022-00043-y. [DOI] [Google Scholar]
- (2).Hockney RW; Eastwood JW Computer Simulation Using Particles; McGraw-Hill, 1981. [Google Scholar]
- (3).McCammon JA; Harvey S Dynamics of Proteins and Nucleic Acids; Cambridge University Press, 1987. [Google Scholar]
- (4).Brooks CL III; Karplus M; Pettitt BM Proteins: A Theoretical Perspective of Dynamics, Structure, and Thermodynamics; John Wiley & Sons, 1988. [Google Scholar]
- (5).Allen MP; Tildesley DJ Computer Simulation of Liquids; Oxford University Press, 1989. [Google Scholar]
- (6).van Gunsteren WF; Weiner PK; Wilkinson AJ Computer Simulation of Biomolecular Systems. Theoretical and Experimental Applications. van Gunsteren WF, Weiner PK, Wilkinson AJ, Eds.; ESCOM: Leiden, 1993. [Google Scholar]
- (7).Becker OM; MacKerell AD Jr.; Roux B; Watanabe M Computational Biochemistry and Biophysics. Marcel Dekker: New York, 2001. [Google Scholar]
- (8).Roux B Computational Modeling and Simulations of Biomolecular Systems; World Scientific, 2022. [Google Scholar]
- (9).Karplus M; McCammon JA Molecular dynamics simulations of biomolecules. Nat. Struct. Biol 2002, 9 (9), 646–652. DOI: 10.1038/nsb0902-646 From NLM Medline. [DOI] [PubMed] [Google Scholar]
- (10).Karplus M; Barbara P Molecular Dynamics Simulations of Biomolecules. In Accts. Chem. Res, 2002. [DOI] [PubMed] [Google Scholar]
- (11).Karplus M; Kuriyan J Molecular dynamics and protein function. Proc. Natl. Acad. Sci. USA 2005, 102 (19), 6679–6685. DOI: 10.1073/pnas.0408930102 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Peiffer AL; Garlick JM; Wu Y; Soellner MB; Brooks CL III; Mapp AK TMPRSS2 inhibitor discovery facilitated through an in silico and biochemical screening platform. bioRxiv 2021. DOI: 10.1101/2021.03.22.436465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Yang W; Gao YQ; Cui Q; Ma J; Karplus M The missing link between thermodynamics and structure in F1-ATPase. Proc. Natl. Acad. Sci. USA 2003, 100 (3), 874–879. DOI: 10.1073/pnas.0337432100 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Mao HZ; Weber J Identification of the betaTP site in the x-ray structure of F1-ATPase as the high-affinity catalytic site. Proc. Natl. Acad. Sci. USA 2007, 104 (47), 18478–18483. DOI: 10.1073/pnas.0709322104 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Banks JL; Beard HS; Cao Y; Cho AE; Damm W; Farid R; Felts AK; Halgren TA; Mainz DT; Maple JR; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem 2005, 26 (16), 1752–1780. DOI: 10.1002/jcc.20292 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kale L; Schulten K Scalable molecular dynamics with NAMD. J. Comput. Chem 2005, 26 (16), 1781–1802. DOI: 10.1002/jcc.20289 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Christen M; Hunenberger PH; Bakowies D; Baron R; Burgi R; Geerke DP; Heinz TN; Kastenholz MA; Krautler V; Oostenbrink C; et al. The GROMOS software for biomolecular simulation: GROMOS05. J. Comput. Chem 2005, 26 (16), 1719–1751. DOI: 10.1002/jcc.20303 From NLM Medline. [DOI] [PubMed] [Google Scholar]
- (18).Case DA; Cheatham TE 3rd; Darden T; Gohlke H; Luo R; Merz KM Jr.; Onufriev A; Simmerling C; Wang B; Woods RJ The Amber biomolecular simulation programs. J. Comput. Chem 2005, 26 (16), 1668–1688. DOI: 10.1002/jcc.20290 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Van Der Spoel D; Lindahl E; Hess B; Groenhof G; Mark AE; Berendsen HJ GROMACS: fast, flexible, and free. J. Comput. Chem 2005, 26 (16), 1701–1718. DOI: 10.1002/jcc.20291 From NLM Medline. [DOI] [PubMed] [Google Scholar]
- (20).Brooks BR; Brooks CL III; Mackerell AD Jr.; Nilsson L; Petrella RJ; Roux B; Won Y; Archontis G; Bartels C; Boresch S; et al. CHARMM: the biomolecular simulation program. J. Comput. Chem 2009, 30 (10), 1545–1614. DOI: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Eastman P; Friedrichs MS; Chodera JD; Radmer RJ; Bruns CM; Ku JP; Beauchamp KA; Lane TJ; Wang LP; Shukla D; et al. OpenMM 4: A Reusable, Extensible, Hardware Independent Library for High Performance Molecular Simulation. J. Chem. Theory Comput 2013, 9 (1), 461–469. DOI: 10.1021/ct300857j From NLM PubMed-not-MEDLINE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Eastman P; Pande VS OpenMM: A Hardware Independent Framework for Molecular Simulations. Comput. Sci. Eng 2015, 12 (4), 34–39. DOI: 10.1109/MCSE.2010.27 From NLM PubMed-not-MEDLINE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Eastman P; Swails J; Chodera JD; McGibbon RT; Zhao Y; Beauchamp KA; Wang LP; Simmonett AC; Harrigan MP; Stern CD; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol 2017, 13 (7), e1005659. DOI: 10.1371/journal.pcbi.1005659 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Choi YK; Kern NR; Kim S; Kanhaiya K; Afshar Y; Jeon SH; Jo S; Brooks BR; Lee J; Tadmor EB; et al. CHARMM-GUI Nanomaterial Modeler for Modeling and Simulation of Nanomaterial Systems. J. Chem. Theory Comput 2022, 18 (1), 479–493. DOI: 10.1021/acs.jctc.1c00996 From NLM PubMed-not-MEDLINE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Guterres H; Park SJ; Cao Y; Im W CHARMM-GUI Ligand Designer for Template-Based Virtual Ligand Design in a Binding Site. J. Chem. Inf. Model 2021, 61 (11), 5336–5342. DOI: 10.1021/acs.jcim.1c01156 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Park S; Choi YK; Kim S; Lee J; Im W CHARMM-GUI Membrane Builder for Lipid Nanoparticles with Ionizable Cationic Lipids and PEGylated Lipids. J. Chem. Inf. Model 2021, 61 (10), 5192–5202. DOI: 10.1021/acs.jcim.1c00770 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhang H; Kim S; Giese TJ; Lee TS; Lee J; York DM; Im W CHARMM-GUI Free Energy Calculator for Practical Ligand Binding Free Energy Simulations with AMBER. J. Chem. Inf. Model 2021, 61 (9), 4145–4151. DOI: 10.1021/acs.jcim.1c00747 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Guterres H; Park SJ; Zhang H; Im W CHARMM-GUI LBS Finder & Refiner for Ligand Binding Site Prediction and Refinement. J. Chem. Inf. Model 2021, 61 (8), 3744–3751. DOI: 10.1021/acs.jcim.1c00561 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Choi YK; Park SJ; Park S; Kim S; Kern NR; Lee J; Im W CHARMM-GUI Polymer Builder for Modeling and Simulation of Synthetic Polymers. J. Chem. Theory Comput 2021, 17 (4), 2431–2443. DOI: 10.1021/acs.jctc.1c00169 From NLM PubMed-not-MEDLINE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Gao Y; Lee J; Smith IPS; Lee H; Kim S; Qi Y; Klauda JB; Widmalm G; Khalid S; Im W CHARMM-GUI Supports Hydrogen Mass Repartitioning and Different Protonation States of Phosphates in Lipopolysaccharides. J. Chem. Inf. Model 2021, 61 (2), 831–839. DOI: 10.1021/acs.jcim.0c01360 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kim S; Oshima H; Zhang H; Kern NR; Re S; Lee J; Roux B; Sugita Y; Jiang W; Im W CHARMM-GUI Free Energy Calculator for Absolute and Relative Ligand Solvation and Binding Free Energy Simulations. J. Chem. Theory Comput 2020, 16 (11), 7207–7218. DOI: 10.1021/acs.jctc.0c00884 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lee J; Hitzenberger M; Rieger M; Kern NR; Zacharias M; Im W CHARMM-GUI supports the Amber force fields. J. Chem. Phys 2020, 153 (3), 035103. DOI: 10.1063/5.0012280 From NLM Medline. [DOI] [PubMed] [Google Scholar]
- (33).Qi Y; Lee J; Cheng X; Shen R; Islam SM; Roux B; Im W CHARMM-GUI DEER facilitator for spin-pair distance distribution calculations and preparation of restrained-ensemble molecular dynamics simulations. J. Comput. Chem 2020, 41 (5), 415–420. DOI: 10.1002/jcc.26032 From NLM Medline. [DOI] [PubMed] [Google Scholar]
- (34).Park SJ; Lee J; Qi Y; Kern NR; Lee HS; Jo S; Joung I; Joo K; Lee J; Im W CHARMM-GUI Glycan Modeler for modeling and simulation of carbohydrates and glycoconjugates. Glycobiology 2019, 29 (4), 320–331. DOI: 10.1093/glycob/cwz003 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Qi Y; Lee J; Klauda JB; Im W CHARMM-GUI Nanodisc Builder for modeling and simulation of various nanodisc systems. J. Comput. Chem 2019, 40 (7), 893–899. DOI: 10.1002/jcc.25773 From NLM Medline. [DOI] [PubMed] [Google Scholar]
- (36).Lee J; Patel DS; Stahle J; Park SJ; Kern NR; Kim S; Lee J; Cheng X; Valvano MA; Holst O; et al. CHARMM-GUI Membrane Builder for Complex Biological Membrane Simulations with Glycolipids and Lipoglycans. J. Chem. Theory Comput 2019, 15 (1), 775–786. DOI: 10.1021/acs.jctc.8b01066 From NLM Medline. [DOI] [PubMed] [Google Scholar]
- (37).Hsu PC; Bruininks BMH; Jefferies D; Cesar Telles de Souza P; Lee J; Patel DS; Marrink SJ; Qi Y; Khalid S; Im W CHARMM-GUI Martini Maker for modeling and simulation of complex bacterial membranes with lipopolysaccharides. J. Comput. Chem 2017, 38 (27), 2354–2363. DOI: 10.1002/jcc.24895 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kim S; Lee J; Jo S; Brooks CL III; Lee HS; Im W CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. J. Comput. Chem 2017, 38 (21), 1879–1886. DOI: 10.1002/jcc.24829 From NLM PubMed-not-MEDLINE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Qi Y; Lee J; Singharoy A; McGreevy R; Schulten K; Im W CHARMM-GUI MDFF/xMDFF Utilizer for Molecular Dynamics Flexible Fitting Simulations in Various Environments. J. Phys. Chem. B 2017, 121 (15), 3718–3723. DOI: 10.1021/acs.jpcb.6b10568 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Jo S; Cheng X; Lee J; Kim S; Park SJ; Patel DS; Beaven AH; Lee KI; Rui H; Park S; et al. CHARMM-GUI 10 years for biomolecular modeling and simulation. J. Comput. Chem 2017, 38 (15), 1114–1124. DOI: 10.1002/jcc.24660 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Lee J; Cheng X; Swails JM; Yeom MS; Eastman PK; Lemkul JA; Wei S; Buckner J; Jeong JC; Qi Y; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput 2016, 12 (1), 405–413. DOI: 10.1021/acs.jctc.5b00935 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Qi Y; Cheng X; Lee J; Vermaas JV; Pogorelov TV; Tajkhorshid E; Park S; Klauda JB; Im W CHARMM-GUI HMMM Builder for Membrane Simulations with the Highly Mobile Membrane-Mimetic Model. Biophys. J 2015, 109 (10), 2012–2022. DOI: 10.1016/j.bpj.2015.10.008 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Qi Y; Ingolfsson HI; Cheng X; Lee J; Marrink SJ; Im W CHARMM-GUI Martini Maker for Coarse-Grained Simulations with the Martini Force Field. J. Chem. Theory Comput 2015, 11 (9), 4486–4494. DOI: 10.1021/acs.jctc.5b00513 From NLM Medline. [DOI] [PubMed] [Google Scholar]
- (44).Jo S; Cheng X; Islam SM; Huang L; Rui H; Zhu A; Lee HS; Qi Y; Han W; Vanommeslaeghe K; et al. CHARMM-GUI PDB manipulator for advanced modeling and simulations of proteins containing nonstandard residues. Adv. Protein Chem. Struct. Biol 2014, 96, 235–265. DOI: 10.1016/bs.apcsb.2014.06.002 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Wu EL; Cheng X; Jo S; Rui H; Song KC; Davila-Contreras EM; Qi Y; Lee J; Monje-Galvan V; Venable RM; et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem 2014, 35 (27), 1997–2004. DOI: 10.1002/jcc.23702 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Qi Y; Cheng X; Han W; Jo S; Schulten K; Im W CHARMM-GUI PACE CG Builder for solution, micelle, and bilayer coarse-grained simulations. J. Chem. Inf. Model 2014, 54 (3), 1003–1009. DOI: 10.1021/ci500007n From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Kognole AA; Lee J; Park SJ; Jo S; Chatterjee P; Lemkul JA; Huang J; MacKerell AD Jr.; Im W CHARMM-GUI Drude prepper for molecular dynamics simulation using the classical Drude polarizable force field. J. Comput. Chem 2022, 43 (5), 359–375. DOI: 10.1002/jcc.26795 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Brooks BR; Bruccoleri RE; Olafson BD; States DJ; Swaminathan S; Karplus M CHARMM - A Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J. Comput. Chem 1983, 4 (2), 187–217. DOI: DOI 10.1002/jcc.540040211. [DOI] [Google Scholar]
- (49).Feig M; Karanicolas J; Brooks CL III. MMTSB Tool Set: enhanced sampling and multiscale modeling methods for applications in structural biology. J. Mol. Graph 2004, 22 (5), 377–395. [DOI] [PubMed] [Google Scholar]
- (50).van Rossum G Python Programming Language. In USENIX Annual Technical Conference, 2007; p 36. [Google Scholar]
- (51).Gao X; Ramezanghorbani F; Isayev O; Smith JS; Roitberg AE TorchANI: A Free and Open Source PyTorch-Based Deep Learning Implementation of the ANI Neural Network Potentials. J. Chem. Inf. Model 2020, 60 (7), 3408–3415. DOI: 10.1021/acs.jcim.0c00451 From NLM Medline. [DOI] [PubMed] [Google Scholar]
- (52).Hynninen AP; Crowley MF New faster CHARMM molecular dynamics engine. J. Comput. Chem 2014, 35 (5), 406–413. DOI: 10.1002/jcc.23501 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Hayes RL; Buckner J; Brooks CL III. BLaDE: A Basic Lambda Dynamics Engine for GPU-Accelerated Molecular Dynamics Free Energy Calculations. J. Chem. Theory Comput 2021, 17 (11), 6799–6807. DOI: 10.1021/acs.jctc.1c00833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Dalcín L; Paz R; Storti M MPI for Python. J. Parall. Distrib. Comput 2005, 65 (9), 1108–1115. [Google Scholar]
- (55).Dalcin L; Fang Y-LL mpi4py: Status update after 12 years of development. Comput. Sci. Eng 2021, 23 (4), 47–54.33967632 [Google Scholar]
- (56).RDKit: Open-source cheminformatics. https://www.rdkit.org.
- (57).Olsson MH; Sondergaard CR; Rostkowski M; Jensen JH PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput 2011, 7 (2), 525–537. DOI: 10.1021/ct100578z From NLM PubMed-not-MEDLINE. [DOI] [PubMed] [Google Scholar]
- (58).O’Boyle NM; Morley C; Hutchison GR Pybel: a Python wrapper for the OpenBabel cheminformatics toolkit. Chem. Cent. J 2008, 2, 5. DOI: 10.1186/1752-153X-2-5 From NLM PubMed-not-MEDLINE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Dolinsky TJ; Czodrowski P; Li H; Nielsen JE; Jensen JH; Klebe G; Baker NA PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nuc. Acids Res 2007, 35 (Web Server issue), W522–525. DOI: 10.1093/nar/gkm276 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Dolinsky TJ; Nielsen JE; McCammon JA; Baker NA PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nuc. Acids Res 2004, 32 (Web Server issue), W665–667. DOI: 10.1093/nar/gkh381 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Michaud-Agrawal N; Denning EJ; Woolf TB; Beckstein O MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem 2011, 32 (10), 2319–2327. DOI: 10.1002/jcc.21787 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Gowers RJ; Linke M; Barnoud J; Reddy TJE; Melo MN; Seyler SL; Dotson DL; Domanski J; Buchoux S; Kenney IM; et al. DAnalysis: A Python package for the rapid analysis of molecular dynamics simulations. In Proceedings of the 15th Python in Science Conference, Austin, TX, 2016; Rostrup, S. B. a. S., Ed.; SciPy: pp 102–109. [Google Scholar]
- (63).McGibbon RT; Beauchamp KA; Harrigan MP; Klein C; Swails JM; Hernandez CX; Schwantes CR; Wang LP; Lane TJ; Pande VS MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J 2015, 109 (8), 1528–1532. DOI: 10.1016/j.bpj.2015.08.015 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Humphrey W; Dalke A; Schulten K VMD: visual molecular dynamics. J. Mol. Graph 1996, 14 (1), 33–38, 27–38. DOI: 10.1016/0263-7855(96)00018-5 From NLM Medline. [DOI] [PubMed] [Google Scholar]
- (65).Nguyen H; Case DA; Rose AS NGLview-interactive molecular graphics for Jupyter notebooks. Bioinformatics 2018, 34 (7), 1241–1242. DOI: 10.1093/bioinformatics/btx789 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Kluyver T; Ragan-Kelley B; Perez F; Granger B; Bussonnier M; Frederic J; Kelley K; Hamrick J; Grout J; Corlay S; et al. Jupyter Notebooks-a publishing format for reproducible computational workflows. Positioning and Power in Academic Publishing: Players, Agents and Agendas 2016, 87–90. DOI: 10.3233/978-1-61499-649-1-87. [DOI] [Google Scholar]
- (67).Ding X; Vilseck JZ; Brooks CL III. Fast Solver for Large Scale Multistate Bennett Acceptance Ratio Equations. J. Chem. Theory Comput 2019, 15 (2), 799–802. DOI: 10.1021/acs.jctc.8b01010 From NLM PubMed-not-MEDLINE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Shirts MR; Chodera JD Statistically optimal analysis of samples from multiple equilibrium states. J. Chem. Phys 2008, 129 (12), 124105. DOI: 10.1063/1.2978177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Harris CR; Millman KJ; van der Walt SJ; Gommers R; Virtanen P; Cournapeau D; Wieser E; Taylor J; Berg S; Smith NJ; et al. Array programming with NumPy. Nature 2020, 585 (7825), 357–362. DOI: 10.1038/s41586-020-2649-2 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).McKinney W Data Structures for Statistical Computing in Python; 2010. DOI: 10.25080/Majora-92bf1922-00a. [DOI]
- (71).Mobley DL; Guthrie JP FreeSolv: a database of experimental and calculated hydration free energies, with input files. J. Comput.-Aided Mol. Des 2014, 28 (7), 711–720. DOI: 10.1007/s10822-014-9747-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Knight JL; Brooks CL III. Multisite lambda Dynamics for Simulated Structure-Activity Relationship Studies. J. Chem. Theory Comput 2011, 7 (9), 2728–2739. DOI: 10.1021/ct200444f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Vilseck JZ; Cervantes LF; Hayes RL; Brooks CL III. Optimizing Multisite lambda-Dynamics Throughput with Charge Renormalization. J. Chem. Inf. Model 2022, 62 (6), 1479–1488. DOI: 10.1021/acs.jcim.2c00047 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Hayes RL; Armacost KA; Vilseck JZ; Brooks CL III. Adaptive Landscape Flattening Accelerates Sampling of Alchemical Space in Multisite lambda Dynamics. J. Phys. Chem. B 2017, 121 (15), 3626–3635. DOI: 10.1021/acs.jpcb.6b09656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Vanommeslaeghe K; MacKerell AD Jr. Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model 2012, 52 (12), 3144–3154. DOI: 10.1021/ci300363c From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Vanommeslaeghe K; Raman EP; MacKerell AD Jr. Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model 2012, 52 (12), 3155–3168. DOI: 10.1021/ci3003649 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Wu GS; Robertson DH; Brooks CL III; Vieth M Detailed analysis of grid-based molecular docking: A case study of CDOCKER - A CHARMm-based MD docking algorithm. J. Comput. Chem 2003, 24 (13), 1549–1562. [DOI] [PubMed] [Google Scholar]
- (78).Ding X; Wu Y; Wang Y; Vilseck JZ; Brooks CL III. Accelerated CDOCKER with GPUs, Parallel Simulated Annealing, and Fast Fourier Transforms. J. Chem. Theory Comput 2020, 16 (6), 3910–3919. DOI: 10.1021/acs.jctc.0c00145 From NLM Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Wu YJ; Brooks CL III. Flexible CDOCKER: Hybrid Searching Algorithm and Scoring Function with Side Chain Conformational Entropy. J. Chem. Inf. Model 2021, 61 (11), 5535–5549. DOI: 10.1021/acs.jcim.1c01078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Wu Y Development and Application of CDOCKER Docking Methodology. 2022.
- (81).Meuwly M Machine Learning for Chemical Reactions. Chem. Rev 2021, 121 (16), 10218–10239. DOI: 10.1021/acs.chemrev.1c00033. [DOI] [PubMed] [Google Scholar]
- (82).Soloviov M; Das AK; Meuwly M Structural Interpretation of Metastable States in Myoglobin–NO. Angew. Chem., Int. Ed 2016, 55 (34), 10126–10130. [DOI] [PubMed] [Google Scholar]
- (83).Rufa DA; Macdonald HEB; Fass J; Wieder M; Grinaway PB; Roitberg AE; Isayev O; Chodera JD Towards chemical accuracy for alchemical free energy calculations with hybrid physics-based machine learning/molecular mechanics potentials. BioRxiv 2020. [Google Scholar]
- (84).Weinan E; Ren W; Vanden-Eijnden E String method for the study of rare events. Phys. Rev. B 2002, 66 (5), 052301. [DOI] [PubMed] [Google Scholar]
- (85).Khavrutskii IV; Arora K; Brooks CL III. Harmonic Fourier beads method for studying rare events on rugged energy surfaces. J. Chem. Phys 2006, 125 (17), 174108. DOI: 10.1063/1.2363379. [DOI] [PubMed] [Google Scholar]
- (86).Maragliano L; Fischer A; Vanden-Eijnden E; Ciccotti G String method in collective variables: Minimum free energy paths and isocommittor surfaces. J. Chem. Phys 2006, 125 (2), 024106. [DOI] [PubMed] [Google Scholar]
- (87).Ren W; Vanden-Eijnden E Finite temperature string method for the study of rare events. J. Phys. Chem. B 2005, 109 (14), 6688–6693. [DOI] [PubMed] [Google Scholar]
- (88).Sugita Y; Okamoto Y Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett 1999, 314 (1–2), 141–151. [Google Scholar]
- (89).Liu P; Kim B; Friesner RA; Berne BJ Replica exchange with solute tempering: A method for sampling biological systems in explicit water. Proc. Natl. Acad. Sci. USA 2005, 102 (39), 13749–13754. DOI: 10.1073/pnas.0506346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Torrie GM; Valleau JP Non-Physical Sampling Distributions in Monte-Carlo Free-Energy Estimation - Umbrella Sampling. J. Comput. Phys 1977, 23 (2), 187–199. DOI: Doi 10.1016/0021-9991(77)90121-8. [DOI] [Google Scholar]
- (91).Dickson A; Brooks CL III. WExplore: hierarchical exploration of high-dimensional spaces using the weighted ensemble algorithm. J. Phys. Chem. B 2014, 118 (13), 3532–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Suarez E; Lettieri S; Zwier MC; Stringer CA; Subramanian SR; Chong LT; Zuckerman DM Simultaneous computation of dynamical and equilibrium information using a weighted ensemble of trajectories. J. Chem. Theory Comput 2014, 10 (7), 2658–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Huber GA; Kim S Weighted-ensemble Brownian dynamics simulations for protein association reactions. Biophys. J 1996, 70 (1), 97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Coifman RR; Lafon S; Lee AB; Maggioni M; Nadler B; Warner F; Zucker SW Geometric diffusions as a tool for harmonic analysis and structure definition of data: Diffusion maps. Proc. Natl. Acad. Sci. USA 2005, 102 (21), 7426–7431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Preto J; Clementi C Fast recovery of free energy landscapes via diffusion-map-directed molecular dynamics. Phys. Chem. Chem. Phys 2014, 16 (36), 19181–19191. [DOI] [PubMed] [Google Scholar]
- (96).Zheng W; Rohrdanz MA; Clementi C Rapid exploration of configuration space with diffusion-map-directed molecular dynamics. J. Phys. Chem. B 2013, 117 (42), 12769–12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data, examples, documentation files and tutorial files that are described and discussed here are available in the GitHub repository (https://github.com/clbrooksiii/pyCHARMM-Workshop). The full source and documentation for CHARMM and pyCHARMM are licensed and available free of charge to non-profit and academic laboratories at https://academiccharmm.org/program. The release version of CHARMM/pyCHARMM described here corresponds to c47b2.