

Summary
The mechanistic tie between genome-wide association study (GWAS)-implicated risk variants and disease-relevant cellular phenotypes remains largely unknown. Here, using human induced pluripotent stem cell (hiPSC)-derived neurons as a neurodevelopmental model, we identify multiple schizophrenia (SZ) risk variants that display allele-specific open chromatin (ASoC) and are likely to be functional. Editing the strongest ASoC SNP, rs2027349, near vacuolar protein sorting 45 homolog (VPS45) alters the expression of VPS45, lncRNA AC244033.2, and a distal gene, C1orf54. Notably, the transcriptomic changes in neurons are associated with SZ and other neuropsychiatric disorders. Neurons carrying the risk allele exhibit increased dendritic complexity and hyperactivity. Interestingly, individual/combinatorial gene knockdown shows that these genes alter cellular phenotypes in a non-additive synergistic manner. Our study reveals that multiple genes at a single GWAS risk locus mediate a compound effect on neural function, providing a mechanistic link between a non-coding risk variant and disease-related cellular phenotypes.
Keywords: GWAS, chromatin accessibility, synergistic, neuropsychiatric disorders, human iPS cells, schizophrenia, CROP-seq, noncoding variants, Micro-C, neuron, scRNA-seq, isogenic, CRISPR-Cas9 gene editing, common risk variants, allele-specific open chromatin
Graphical abstract
Highlights
-
•
Many neuropsychiatric GWAS risk SNPs alter chromatin accessibility in NGN2-neurons
-
•
A schizophrenia-risk SNP cis-regulates three genes via allele-specific chromatin looping
-
•
The schizophrenia-risk SNP enhances dendritic complexity and neuronal activity
-
•
Multiple genes in a single GWAS risk locus act in a non-additive synergistic fashion
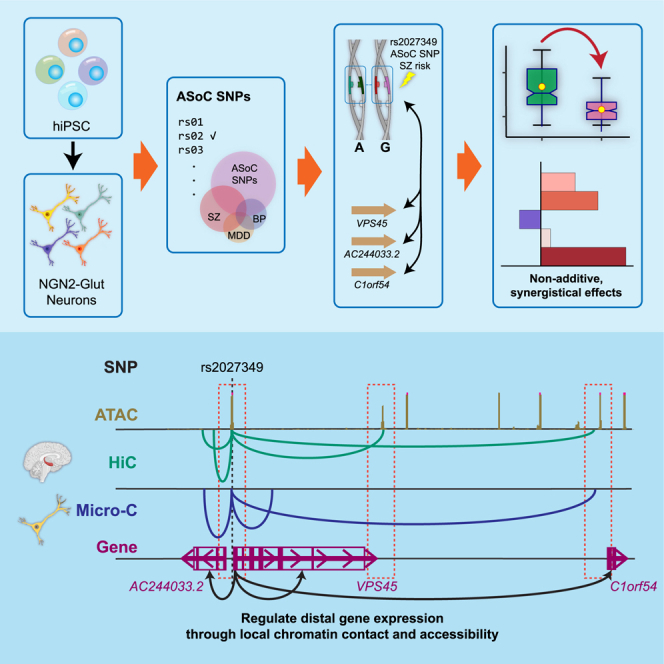
Zhang et al. identified thousands of potentially functional non-coding SNPs showing allele-specific open chromatin. They prioritized schizophrenia GWAS risk variants for functional studies in NGN2 neurons. Multiple genes in a single schizophrenia GWAS risk locus were found to act in a non-additive synergistic fashion, contributing to disease-relevant neuronal changes.
Introduction
Genome-wide association studies (GWASs) of schizophrenia (SZ) and other neuropsychiatric disorders have identified hundreds of common risk loci.1,2,3,4,5 Most neuropsychiatric risk variants reside in non-coding sequences that likely alter gene expression. However, despite the progress made from studying human post-mortem brains (e.g., PsychENCODE),6,7,8,9 animal models,10 and, more recently, human induced pluripotent stem cell (hiPSC) models,11,12,13,14 causal disease genes and the underlying molecular mechanisms remain elusive for most neuropsychiatric disease risk loci.
A challenge for understanding the causal mechanisms underlying these associations is that each GWAS locus often encompasses multiple genes and many common risk variants that are equally associated with disease due to linkage disequilibrium (LD). We have recently shown that allele-specific open chromatin (ASoC) is an effective functional readout of regulatory single-nucleotide polymorphisms (SNPs), displaying differential allelic chromatin accessibility in heterozygous individuals,13 i.e., two alleles of a SNP show read imbalance in assay for transposase-accessible chromatin using sequencing (ATAC-seq).13,15 Compared with expression quantitative trait locus (eQTL) mapping, e.g., PsychENCODE,16 GTEx,17 and brain eQTL,18 ASoC mapping has the advantage of directly identifying putatively functional variants rather than those merely in LD.13 Interestingly, our integrative analysis of neuronal ASoC variants with brain eQTL and Hi-C data suggests that many ASoC variants may cis-regulate their adjacent and distal genes through chromatin contacts.13 However, such postulated long-range cis-regulatory effects involving multiple genes in the same GWAS locus and their relevance to cellular phenotypic change have not been empirically established.
Here, by using hiPSC-derived and NEUROG2 (NGN2)-induced excitatory neurons (NGN2-Glut)19 as a neuro developmental model,11,20,21,22,23 we identified many putatively functional SZ GWAS risk variants showing ASoC, and thus likely to be functional, and demonstrated that a single SZ-risk SNP cis-regulates two adjacent genes, VPS45 and a long non-coding RNA (lncRNA, AC244033.2), as well as a distal gene that encodes chromosome 1 open reading frame 54 (C1orf54). More importantly, we found that all three genes synergistically contribute to the cellular and molecular alterations relevant to neuropsychiatric disorders.
Results
ASoC mapping in NGN2-Glut identifies functional SZ-risk SNPs
To identify functional SZ GWAS risk variants that may alter chromatin accessibility and gene expression in NGN2-Glut, we carried out ATAC-seq15 and mapped ASoC variants in neurons of 20 hiPSC lines (Figures 1A, S1A, and S1B and STAR Methods). We noted that the differentiated neurons were overall pure excitatory and comparable between lines (Figures S1C–S1E). We obtained 48–66 million paired-end ATAC-seq reads per sample (Table S1 and Figures S1F–S1I) and identified 183,692 open chromatin peaks using MACS2.24 These NGN2-Glut and our previously reported hiPSC-derived glutamatergic neurons (iN-Glut) were overall similar to each other (Figure 1A) despite some observed different patterns (Figure S1J).13
Figure 1.

Allele-specific open chromatin (ASoC) mapping in NGN2-Glut identifies strong ASoC at the SZ-associated VPS45 locus
(A) The strategy of generating NGN2-glutamatergic excitatory neurons from hiPSCs for bulk ATAC-seq and RNA-seq analysis.
(B) Circus plot showing SZ-credible-risk SNPs that exhibited ASoC (FDR <0.05) in NGN2-Glut, iN-Glut, or both.
(C) Allelic ratio correlation of SZ-associated ASoC SNPs (FDR <0.05) in iN-Glut and NGN2-Glut, n = 26.
(D) Cell-type-specific and allele-specific ATAC-seq read pile-ups flanking ASoC SNP rs2027349 at the SZ-associated VPS45 locus. The top track shows the PGC3 SZ GWAS probabilities of all the SNPs in this region. Aquamarine, total reads that contain rs2027349; dark blue, reads containing reference allele (G); dark red, reads containing alternative allele (A).
Following our recently developed approach to mapping putatively functional non-coding variants that show differential allelic chromatin accessibility (i.e., ASoC),13 we identified 8,205 ASoC SNPs (false discovery rate [FDR] <0.05) out of 163,066 heterozygous SNPs in open chromatin regions (OCRs) (Figure S2A and Table S2). We focused on identifying putatively functional SZ GWAS risk SNPs25 showing ASoC in NGN2-Glut over iN-Glut (Table S3).13 For the list of SZ credible SNPs at the 108 GWAS risk loci (multiple SNPs per locus),25 we found 31 ASoC SNPs at 26 SZ-risk loci in NGN2-Glut, of which 14 were heterozygous, and 7 of them also showed ASoC in iN-Glut (Figure 1B and Table S3). Of the 17 SZ loci with genome-wide significant (GWS) risk SNPs showing ASoC in iN-Glut,13 9 had a nominally significant difference of ASoC (p < 0.05) in NGN2-Glut. Overall, there was a significant overlap of SZ-risk SNPs showing ASoC (FDR <0.05) between the two datasets (24-fold enrichment, p < 2.5 × 10−6; two-tailed Fisher’s exact test) (Figure S2B and Table S3). The allelic ratio of the chromatin accessibility of all SZ credible SNPs also showed a modest correlation between NGN2-Glut and iN-Glut (R = 0.41; Figure S2C), which was much stronger for ASoC SNPs (R = 0.81; Figure 1C). Thus, our ASoC mapping in NGN2-Glut reproducibly identified putative functional SZ-risk SNPs that altered chromatin accessibility in neurons generated by two different methods.
We then evaluated to what extent the NGN2-neuron ASoC SNPs overlapped with GWAS risk variants (including LD proxies with R2 > 0.8) of other neuropsychiatric disorders and with brain/neuron eQTL. For bipolar disorder (BP) (n = 64)5 and major depressive disorder (MDD) (n = 102),4 we found that five GWS SNPs of five BP risk loci and six GWS SNPs of six MDD risk loci also showed ASoC (Table S2), representing an overlap to a smaller extent than SZ (26/108 risk loci; Table S3). Further systematic GWAS risk enrichment testing for major neuropsychiatric disorders and brain traits (see STAR Methods) identified the strongest enrichment of ASoC SNPs in SZ (>16-fold) but not in BP, MDD, or autism (Figure S2D). Moreover, we found that about 16% of ASoC SNPs were also eQTLs in human fetal brains,26 adult brains,1,16 or hiPSC-derived dopaminergic neurons,27 representing 1.3- to 1.8-fold enrichment (vs. non-ASoC SNPs; p = 2.3 × 10−9 to 2.5 × 10−18, two-tailed Fisher’s exact test) of brain/neuron eQTLs (Table S2 and Figure S2E). Thus, NGN2-Glut ASoC SNPs were most enriched for SZ-risk variants relative to other neuropsychiatric or neurodegenerative disorders and likely affected gene expression.
To nominate SZ-risk SNPs for further functional study, we examined loci with GWS SNPs showing ASoC in both datasets. Consistent with the iN-Glut data,13 rs2027349 at the VPS45 locus remained the most significant ASoC in NGN2-Glut (Figure 1D), followed by the ASoC SNP rs12895055 at the BCL11B locus (Table S3 and Figure S2F). rs2027349 at the VPS45 locus is in high LD with the SZ GWAS index SNP rs12138231 (R2 = 0.94) and was fine-mapped as the only likely causal SNP at this locus in our recent study.13 The other two replicated ASoC SNPs also showing GWS association with SZ were at the PBRM1/GNL3 (rs10933) and BAG5 (rs7148456) loci (Table S3 and Figure S2F). Therefore, these four SZ-associated ASoC SNPs were prioritized for functional testing for regulatory potential.
Multiplex CRISPRi in NGN2-Glut confirmed the regulatory function of the ASoC sequence flanking rs2027349
Applying a previously validated CRISPR droplet sequencing (CROP-seq) approach13,28 to NGN2-Glut, we examined whether the OCR sequences that flank the four prioritized SZ-associated ASoC SNPs at the VPS45 (rs2027349), BCL11B (rs12985055), PBRM1/GNL3 (rs10933), and BAG5 (rs7148456) loci have regulatory potential, using the same set of guide RNAs (gRNAs).13 The gRNA pool targeted 20 SNP loci, each with three gRNAs.13 Lenti-gRNAs were used to transduce NGN2-Glut on day 20, and the perturbed neurons were collected on day 29 for single-cell RNA sequencing (scRNA-seq) (Figure 2A and STAR Methods). NGN2-Glut appeared to be heterogeneous (Figures 2B–2E and S3A); to maximize the study power, we included all the MAP2+/SOX2− neurons of the Glut+ clusters (n = 8,921) in differential expression (DE) analysis, of which 4,057 neurons were infected by a single gRNA (Figures S3B and S3C).
Figure 2.

Multiplex CRISPRi combined with scRNA-seq in NGN2-Glut identifies regulatory sequences flanking SZ-associated ASoC SNPs
(A) Modified CROP-seq approach to screen cis-targets of the 20 SNP sites in NGN2-Glut. Two iPSC lines (CD0000009, CD0000011) were used.
(B) Uniform Manifold Approximation and Projection (UMAP) plot showing the 11 clusters of the 10,247 cells used in the scRNA-seq analysis.
(C) UMAP plot showing the normalized expression of MAP2.
(D) UMAP plot showing the expression pattern of glutamatergic markers SLC17A7 (vGlut1) and SLC17A6 (vGlut2).
(E) Violin plots showing expression of neuron-specific markers.
(F) Gene track-expression plot showing the cis effects of CROP-seq gRNAs targeting rs2027349 (VPS45 site). ∗FDR <0.05.
(G) Same as (F), but for three CROP-seq gRNAs targeting rs7148456 (BAG5 site).
(H) UMAP plot showing cells assigned to one of the three VPS45 gRNAs (red) or control gRNAs (targeting GFP; blue).
(I) The effects of VPS45 gRNAs on the expression level of VPS45. Kruskal-Wallis test (non-parametric) was used (shown are Dunn’s multiple comparisons adjusted p; ∗∗∗p < 0.001).
(J) Top 10 enriched GO terms of genes up- or downregulated by ASoC-targeting gRNA-2 at the rs2027349 locus (VPS45). Red line, FDR 0.05.
We focused on genes within ±500 kb of a targeting SNP/gRNA to identify the regulatory SNP site and its cis target by testing the DE of cis genes between gRNA-targeted and non-targeted neurons (Student’s t test; see STAR Methods and Table S4). We found that sequences flanking three (rs2027349 near VPS45, rs10933 near PBRM1/GNL3, and rs7148456 near BAG5) of the four prioritized ASoC SNPs had cis-target genes with adjusted p < 0.05 (Figures 2F, 2G, and S3D–S3F and Table S4). All cis targets (adjusted p < 0.05) showed reduced expression by respective gRNAs, consistent with the expected transcriptional repression by KRAB in CRISPR interference (CRISPRi).29,30 VPS45 near rs2027349 showed the most significant reduction (Figures 2H and 2I), which was consistent with rs2027349 exhibiting the strongest ASoC in NGN2-Glut among all the SZ-risk SNPs (Figure 1B and Table S3).
To assess the biological relevance of CRISPRi at the three loci with cis targets (Table S4), we analyzed the transcriptomic effects of the CRISPRi. For rs2027349, the two gRNAs, VPS45-2 and VPS45-3, yielded 167 and 162 differentially expressed genes (DEGs), respectively (Table S5). The gRNA BAG5-2 targeting rs7148456 gave 69 DEGs, while gRNA PBRM1-3 targeting rs10933 gave only 18 DEGs (Figure S3G). Our further gene ontology (GO) term analysis for each set of DEGs showed that DEGs from CRISPRi at the VPS45 locus, but not at the PBRM1/GNL3 or BAG5 locus, were enriched for neural GO terms (Figure 2J and Table S6). This suggests that CRISPRi perturbation at rs2027349 near VPS45 elicited the most biologically relevant transcriptomic changes.
Precise allelic editing of the ASoC SNP at the VPS45 locus reveals a complex gene regulation relevant to neurodevelopmental disorders
We next examined the functional effects of the precise allele editing of rs2027349 (Figure 3A). Using the isogenic pairs of hiPSC lines (two donors) of three different genotypes (AA, AG, and GG) of rs2027349 that we have previously generated by CRISPR-Cas9 editing,13 we derived NGN2-Glut and performed transcriptomic DE analysis between different genotypes (Figure 3A). For the DE of cis genes within a 500 kb interval of rs2027349 (Figure 3B and Table S5), we found that cis-gene expression changes between AA vs. GG neurons were correlated (Spearman’s ρ = 0.602) with those between AG vs. GG (Figures 3B and S4A), confirming the consistency of the allelic effect in samples with different genotypes. Allele A of rs2027349 was associated with an increased expression of a VPS45 transcript isoform (ENST00000369130.3) but not with total mRNAs of VPS45 in NGN2-Glut (Figures 3B, 3C, and S4B). Interestingly, we found that allele A also significantly increased the expression of an lncRNA (AC244033.2) (FDR <0.005) proximal to VPS45 but transcribed in the opposite direction, as well as a distal gene (C1orf54) that is ∼200 kb downstream of the edited SNP (FDR <0.023) (Figure 3B and Table S7). The increased expression of these cis-target genes of rs2027349 in AA neurons was independently verified by quantitative polymerase chain reaction (qPCR) and was consistently significant in both donor lines (Figures S4C–S4G). The allelic effects of rs2027349 on the three cis-target genes were consistent across different maturation stages (20–50 days) of NGN2-Glut neurons (Figures S4H–S4J). These results suggest a complex cis-regulation of multiple genes (VPS45, AC244033.2, and C1orf54) by a single SZ-risk SNP (rs2027349) at the VPS45 locus.
Figure 3.

CRISPR-Cas9 editing of rs2027349 in NGN2-Glut cis-regulates multiple genes and leads to disease-relevant transcriptomic changes
(A) CRISPR-Cas9 editing of rs2027349, neuron differentiation, and experiment details.
(B) cis effects of rs2027349 genotypes on local gene expression within a 500 kb window of rs2027349. The genotype (GG) was used as the baseline. Two or three clones per genotype of two iPSC lines were used.
(C) Changing G to A (rs2027349) significantly increases the expression of VPS45 transcript ENST00000369130.3. Kruskal-Wallis (non-parametric) test; ∗∗∗p < 0.001.
(D) Volcano plot showing differential expression of genes (n = 14,999) in NGN2-Glut after rs2027349 editing. Some DEGs (FDR <0.05) highlighted are also SZ-risk genes. The p values were derived from a combinational analysis of all three genotypes (AA, AG, and GG). −log2FC was between AA and GG neurons.
(E) Sunburst plot showing strong enrichment of synapse-related ontology terms among the downregulated DEGs in AA neurons (SynGO31).
(F) MAGMA gene-set GWAS enrichment analysis of DEGs for neuropsychiatric disorders/traits shows high SZ and neuroticism enrichment.
(G) Upset plot showing the overlap of the DEGs from rs2027349 editing (AA vs. GG neurons) against a list of DEGs associated with five neuropsychiatric disorders in post-mortem brains. Red connections emphasize genes related to SZ.
(H) An xy scatterplot showing the correlation of the log2FC of DEG sets in (G). Shown are corresponding ρ and p values.
VPS45 has been suggested to play a role in vesicle-mediated protein trafficking and neurotransmitter release.32 While AC244033.2 and C1orf54 have no known function, cis- and trans-acting lncRNAs are known to be pervasive and may have profound roles in neurodevelopment.33,34 We thus reasoned that the transcriptomic changes associated with the SZ-risk allele A of rs2027349 might be relevant to neurodevelopment and synaptic function. Our DE analysis identified 687 upregulated genes and 731 downregulated genes (FDR <0.05) in AA neurons (vs. GG) (Figure 3D). Gene set enrichment analysis (GSEA) using WebGestalt35 showed that the upregulated genes were strongly enriched for GO terms related to neuron differentiation, neurogenesis, and nervous system development. In contrast, the downregulated genes were strongly enriched for GO terms such as synaptic signaling and synaptic membrane (Table S8 and Figures S5A and S5B). Since synaptic abnormalities have been implicated by SZ GWAS,36 we further analyzed the DEGs by synaptic GO (SynGO)31 to confirm the relevance of transcriptomic changes to synaptic function. Consistent with the result from WebGestalt analysis, the downregulated genes in AA neurons were significantly enriched for synaptic genes (pre- and post-synapse; Figure 3E and Table S9). While the upregulated genes were not (Figure S5C and Table S10), some noteworthy upregulated genes were found in SynGO terms such as synaptic vesicle (SLC17A7 and SYN3) and post-synaptic ribosome (Figure S5D). These results show that the SZ-risk allele of rs2027349 affects global gene expression related to neurodevelopment and synaptic function.
Subsequently, we assessed whether the transcriptional changes associated with the A allele of rs2027349 are relevant to neuropsychiatric disorders. We first examined the enrichment of DEGs for those associated with SZ. Of the 55 PGC3-prioritized single SZ-risk genes36 that were also expressed in NGN2-Glut, 10 were downregulated and 4 were upregulated in AA neurons (Figure 3D and Table S7), representing a 4.6-fold enrichment of SZ-risk genes among downregulated genes (Fisher’s exact test, p = 0.0002, two-tailed) and a 1.9-fold non-significant enrichment in upregulated genes (Figure S5E). Analysis of all the dysregulated genes within SZ loci in AA neurons showed significant enrichment of different SZ GWAS gene sets and human post-synaptic density (PSD) gene sets often associated with SZ risk (Figure S5F).25,36,37,38 We also performed MAGMA analysis using GWAS summary statistics39,40 and found that the downregulated genes in AA neurons, but not the upregulated ones, showed strong enrichment of GWAS risk of SZ and, to a less extent, of neuroticism, BP, and MDD (Figure 3F).
We further examined whether the transcriptional changes in AA neurons (vs. GG) are enriched for or resemble the transcriptional differences in post-mortem brain tissues of major psychiatric disorders. For overlapping DEGs (FDR <0.05) between datasets (Figure 3G), we found significant positive correlations between their expression changes in AA neurons and individuals with SZ (ρ = 0.34, p = 2 × 10−11), BP (ρ = 0.37, p = 5.7 × 10−5), and ASD (ρ = 0.30, p = 1.9 × 10−5) using the PsychENCODE datasets (Figures 3G, 3H, and S5G).16 The transcriptome-wide expression changes in AA neurons and individuals with SZ, BP, and ASD also exhibited significant correlations (ρ = 0.1–0.15, p = 2.0 × 10−37 to 6.9 × 10−68; n = 13,816 genes) (Figure S5H). The expression changes in AA neurons did not correlate significantly with transcriptional changes associated with MDD or AAD (Figures S5G and S5H). These results suggest that the ASoC SNP rs2027349 mediates a complex cis-gene regulation involving multiple genes (VPS45, AC244033.2, and C1orf54) at the VPS45 locus in NGN2-Glut that may lead to transcriptional changes relevant to neurodevelopment, synaptic function, and major neuropsychiatric disorders.
Editing rs2027349 at the VPS45 locus alters neurodevelopmental phenotypes in NGN2-Glut neurons
We first examined day 32 NGN2-Glut of rs2027349-edited isogenic lines for their neuronal morphology and did a Sholl analysis for dendritic complexity (Figures 3A and 4A). Two-way ANOVA (genotype × distance) analysis showed that genotype had a significant effect on the number of intersections (dendritic branching) (p < 0.0001) (Figure 4B). The number of intersections significantly differed at 80–160 μm from the soma, with GG showing a reduced dendritic branching (Figures 4A and 4B). AA and GG neurons differed most at 100 μm from the soma (p < 0.0001, Šídák post hoc test).
Figure 4.

CRISPR-Cas9 editing of rs2027349 at VPS45 locus alters neuron development in NGN2-Glut
(A) Representative traces of dendrites from rs2027349 AA, AG, and GG neurons. Scale bar: 100 μm.
(B) Sholl analysis of rs2027349 AA (n = 97), AG (n = 47), and GG (n = 101) neurons. Marked data points indicate significant distance differences between the AA and the GG genotypes (two-way repeated-measures ANOVA with Šídák multiple test correction). Data were from three independent experiments.
(C) Representative images of GFP-labeled dendrites of NGN2-Glut neurons and synaptic puncta labeled with post-synaptic density 95 (PSD-95, DLG4) and Synapsin I (SYN1); scale bar: 5 μm.
(D–F) Punctum density of PSD-95, SYN1, and PSD-95+/SYN1+, respectively. Each dot represents synaptic density from one neuron (AA, n = 77; GG, n = 75; AG, n = 39). Data were from two independent experiments (Kruskal-Wallis test, non-parametric); ∗p < 0.05; ∗∗p < 0.01; ∗∗∗∗p < 0.0001. Data were from two clones per genotype of donor line CD0000011. Consistent results for the second donor (CD0000012) are shown in Figure S6.
We next assessed the effect of rs2027349 editing on dendritic protrusions (Figures S6A and S6B). Although we did not observe a significant difference in protrusion density between genotypes (Figure S6B), we found that the density and area of both pre-synaptic (Synapsin 1; SYN1) and post-synaptic (PSD-95, DLG4) puncta in AA neurons were significantly higher than in GG neurons (Figures 4C–4E, S6C, and S6D). The differences were most robust for the colocalized SYN1+/PSD-95+ punctum density, labeling bone fide synapses. AG neurons displayed an intermediate phenotype significantly different from AA and GG (Figures 4F and S6E). We also evaluated the neuronal morphometric alterations in the isogenic SNP-edited lines (AA and GG) from an independent donor, which replicated the reduced dendritic branching and synaptic punctum density (SYN1) in GG neurons (Figures S6F and S6G). Thus, consistent with its transcriptional effects, rs2027349 influences dendritic complexity and synapse development in NGN2-Glut.
Editing rs2027349 at the VPS45 locus affects synaptic function in NGN2 neurons
We then used multielectrode arrays (MEAs) to evaluate neuronal population dynamics in NGN2-Glut of rs2027349-edited isogenic lines (Figures 3A, 5A, and 5B). We found that isogenic neurons exhibited differences in mean firing rate and the number of bursts between days 41 and 74 (post-neural induction), when neurons showed the most active firing (Figures 5C, 5D, and S6H). On days 50, 53, and 56, the mean firing rates were about 50% higher (p = 0.004–0.006, Student’s t test, two-tailed), and the average number of bursts per 10 min was about 65% more (p = 7.8 × 10−5 to 4.0 × 10−6, Student’s t test, two-tailed) in AA compared with GG neurons, with AG neurons displaying an intermediate phenotype (Figures 5C and 5D). Network burst frequency was found to be different only at day 50, suggesting the impact may be restricted to specific developmental stages.
Figure 5.

CRISPR-Cas9 editing of rs2027349 alters neural network and electrophysiological activity in NGN2-Glut
(A and B) Example raster plots of neuronal firing events in MEA.
(C and D) Mean firing rate and burst numbers in day 50, 53, and 56 NGN2-Glut with different rs2027349 genotypes from two independent experiments. Each point represents one replicate. Kruskal-Wallis (non-parametric) test; ∗∗∗p < 0.001.
(E) Representative two-photon pseudo-color Ca2+ imaging time-series images showing a neuron and its firing activities in a 275 s span; scale bar: 20 μm.
(F and G) Representative neuron firing signals from five cells of genotype AA (F) or GG (G).
(H–J) Ca2+ transient frequency and amplitudes in AA (n = 124) and GG (n = 166) neurons from two independent experiments. (H) Cumulative probability plot of Ca2+ transient interevent intervals (IEIs) in AA and GG neurons, two-sample KS test, two-tailed; p < 0.001. (I) Violin and boxplot showing the distribution of Ca2+ transient frequency in AA and GG neurons. Yellow point indicates the mean. Student’s t test (non-parametric, two-tailed); ∗∗∗p < 0.001.
(J) Violin and boxplot showing the amplitude (dF/F0) distribution in AA and GG neurons. Yellow point indicates the mean. Student’s t test (non-parametric, two-tailed); ∗p < 0.05. Data were obtained from two clones per genotype of donor line CD0000011. Consistent results for the second donor (CD0000012) are shown in Figure S6.
We next assessed the effects of SNP editing by Ca2+ imaging (see STAR Methods) in AA and GG neurons at day 35 after neuron induction (Figures 3A and 5E–5G). We found that the neuronal Ca2+ transient (firing) frequency in AA neurons was significantly higher than in GG neurons (p < 0.001, Student’s t test, two-tailed) with shorter interevent intervals in AA neurons (p < 0.001, two-sample KS test) (Figures 5H and 5I). There was also a significant (p = 0.042) reduction in the amplitude of Ca2+ transients in AA neurons (Figure 5J). We found that Ca2+ transients were strongly correlated (synchronization index range from 0.7 to 0.9) between different neuronal cultures of each genotype; however, no significant difference in synchrony index was found between AA and GG neurons (Figures 5F, 5G, S6L, and S6M). Furthermore, with an independent donor line, we confirmed the observed higher frequency of Ca2+ transients (p < 0.001, Student’s t test, two-tailed), shorter interevent intervals (p < 0.001, two-sample KS test), and a non-significant trend toward a reduction in the amplitude of Ca2+ transients in AA neurons (Figures S6I–S6K). The increased frequency of Ca2+ transients in AA neurons was consistent with the results from the MEA experiments (Figures 5A–5D). Together, these results suggest that the SZ-risk allele of rs2027349 is associated with hyperactivity of NGN2 neurons, consistent with previously reported neuronal hyperactivity associated with SZ-risk alleles.12,14,41,42
All three genes, VPS45, AC244033.2, and C1orf54, at the VPS45 locus altered neural phenotypes in a non-additive and synergistic style
Given that the ASoC SNP rs2027349 can cis-regulate multiple genes (VPS45, AC244033.2, and C1orf54) (Figures 3B and 3C) and alter neuronal phenotypes (Figures 4 and 5), we attempted to determine which gene(s) at this locus likely contributed to the SZ-risk allele-associated neuronal phenotypes. Therefore, we made CRISPR-edited AA NGN2-Glut with individual short hairpin RNA (shRNA)-mediated gene knockdown (KD) of VPS45, AC244033.2, or C1orf54 (EGFP as a control, see STAR Methods) and attempted to reverse risk-allele-associated cellular phenotypes. We first confirmed the expected reduction of expression in NGN2-Glut for each KD gene (Figures 6A–6C and S7A and Table S11). We then assessed the effect of KD in AA neurons to mimic the phenotypes observed in GG neurons. We found that shRNA of C1orf54 (shRNA_C1orf54) and, to a lesser extent, shRNA of AC244033.2 (shRNA_lncRNA) reversed dendritic complexity in AA neurons (Figures 6D and S7B). While we found that VPS45 KD did not alter dendritic complexity (Figures 6D and S7B), our Ca2+ imaging showed that KD of VPS45, as well as lncRNA and C1orf54, all partially reversed the higher Ca2+ transient frequency of AA neurons (Figure 6E). Together, these results suggest that VPS45, AC244033.2, and C1orf54 all contribute to the phenotypic changes in rs2027349-edited AA neurons.
Figure 6.

VPS45, AC244033.2 (lncRNA), and C1orf54 interactively altered neural phenotypes in NGN2-Glut at the rs2027349 locus
(A–C) Box-whisker plots showing that shRNA-mediated single and triple KD in AA neurons reversed the gene expression patterns to GG neurons. GAPDH was the endogenous control for normalization in qPCR assay. AA_EGFP_KD was used as the control for all. Saltire mark indicates the mean. Kruskal-Wallis test (non-parametric). For individual KD, two clones per donor line (CD0000011) and three biological replicates per clone were used. For triple KD, three biological replicates from one clone were used.
(D) Sholl analysis of individual KDs; refer to Figure S7B for statistics. Two clones per genotype from two independent experiments were used.
(E) Calcium imaging analysis of neuron firing frequency for gene KD. Kruskal-Wallis (non-parametric) test with Dunn’s multiple comparisons and adjusted p values. n = 181, 245, 229, 276, and 152 neurons. One or two clones per genotype from two independent experiments were used. ∗∗∗∗p < 0.0001.
(F) Hierarchical clustering of log2FC for neurodevelopmental genes downregulated in rs2027349-edited GG neurons and their expression changes in each shRNA KD in AA neurons.
(G) Enrichment of the up-/downregulated genes in each shRNA KD in AA neurons among the neurodevelopmental (i.e., neuron differentiation related) genes downregulated in rs2027349-edited GG neurons.
(H) Same as (F), using synaptic genes upregulated in GG neurons.
(I) Same as (G), examining the enrichment among synaptic genes upregulated in GG neurons. In (G) and (I), Fisher’s exact test (two-tailed) was used to estimate the enrichment; OR, odds ratio, ∗∗∗p < 0.001.
(J) Summary of the correlations from linear regression model fitting. Whiskers, ±95% CI; fill, −log10p.
(K) Pie chart showing the number and proportions of genes in each synergistic category in the combinatorial vs. the additive model.
(L) Competitive GSEA of DEGs in “more upregulated” (more.up) and “more downregulated” (more.down) categories based on 698 curated neural gene sets and stratified by eight neural categories.
(M and N) Bar plots of overrepresentation analysis in (L) using a hypergeometric test of the gene sets at FDR <0.05 and ranked by FDR value. Red lines, FDR 0.05.
To further understand how each gene (VPS45, AC244033.2, or C1orf54) contributes to the neural phenotypic effects, we examined their impacts at the molecular level using RNA-seq (Figures S7C and S7D and STAR Methods) and compared the list of DEGs of each KD condition with DEGs in rs2027349-edited GG (vs. AA) neurons (Tables S11 and S12). Since the number of the DEGs (FDR <0.05) for each KD condition was large, with 28%, 49%, and 44% of the expressed genes for KD in VPS45, AC244033.2, and C1orf54, respectively (Figures S7F–S7H), we focused our analyses on a subset of DEGs most relevant to our observed neuronal phenotypic changes by rs2027349 editing. These selected genes belong to the enriched GO terms (from rs2027349 editing) related to neuron development/differentiation (downregulated in GG) or those related to synaptic signaling (upregulated in GG; Figures 6F–6I and Tables S8 and S12). We then asked whether each subset of those DEGs was enriched for any shRNA KD-associated DEGs with a directional expression change consistent with the rs2027349-editing effect (Figures 6F–6I). We found that neuronal differentiation genes with reduced expression in SNP-edited GG neurons were significantly enriched for genes downregulated by shRNA KD of C1orf54 (OR = 2.1, p = 0.009; Fisher’s exact test) but not AC244033.2 or VPS45 (Figures 6F and 6G and Table S12). For synaptic signaling genes with increased expression in SNP-edited GG neurons, we observed an excess of genes upregulated by shRNA KD of VPS45 (OR = 2.6, p = 0.004; Fisher’s exact test) and AC244033.2 (OR = 2.6, p = 0.001; Fisher’s exact test), but not C1orf54 (Figures 6H and 6I and Table S12). These results support that all three genes contribute to the phenotypic changes in rs2027349-edited neurons, likely by influencing the expression of different sets of biologically relevant genes.
We next tested if the transcriptional effects of rs2027349 editing could be explained by the effects of individual KD (VPS45, AC244033.2, or C1orf54) in NGN2-Glut. We first fit a linear regression model where we regressed expression fold changes from the SNP editing, as a response variable, against expression changes from KD of three individual genes. For all the DEGs from the SNP editing (GG vs. AA) (Table S12), our model fitting explained 3% of the SNP editing effects across genes (adjusted R2 = 0.03, p < 4.3 × 10−9), with an unexpected negative correlation (Figures 6J and S7E). However, when we introduced the interaction terms between the expression effects of individual KDs, we observed a marked improvement in model performance, explaining 7% of the variation of the SNP editing effects (adjusted R2 = 0.07, p < 2.2 × 10−16). The gene × gene interaction terms were highly significant, with VPS45_KD × C1orf54_KD (β = 0.26, p = 1.4 × 10−5) and VPS45_KD × lncRNA_KD (β = 0.16, p = 6.7 × 10−5) positively correlated with the SNP editing effect (Figure 6J). The highly significant interaction terms of different shRNA KDs were consistent with our further GO-term enrichment analysis of DEGs from each shRNA KD, where we found that overlapping genes between different KD conditions showed stronger enrichment for biologically relevant GO terms, i.e., relating to neurodevelopment and synaptic signaling (Figures S7F–S7H and Table S13). These results suggest that VPS45, AC244033.2, and C1orf54 contribute to the phenotypic changes in rs2027349-edited neurons in a non-additive fashion.
To further explore whether VPS45, AC244033.2, and C1orf54 at the same SZ-risk locus have any synergistic effect, as recently demonstrated for SZ-risk genes of different loci,12,43 we performed a combinatorial triple KD of all three genes in AA neurons (Figures 6A–6C and S8A and Table S11) as described above for single-gene KD (also see STAR Methods). The triple KD achieved similar magnitudes of gene expression changes in individual genes (Figure 6A). Using the same linear model above, we found an improvement in model fitting using the empirical data from the triple KD, which explained 9% (vs. 7% with individual gene KD plus interaction terms) of the variation of the SNP editing effects (adjusted R2 = 0.09, p < 3.7 × 10−15) (Figure 6J). We then followed an established method for testing gene synergistic effect at the transcriptomic level12,43 using RNA-seq data from individual gene KDs and the triple KD from the same donor line (Figure S8A). We found that about 15% of genes were either more upregulated (more.up; n = 1,227) or more downregulated (more.down; N = 1,506) than expected under the additive model (synergy FDR <0.1) (Figures 6K and S8B–S8G). Further GSEA using the gene sets curated as part of the synergistic analytic pipeline12,43,44 showed that booth more.up and more.down genes were strongly enriched for genes associated with SZ and BP, while to a lesser extent with autism and antipsychotic usage (Figures 6L–6N). In contrast, the DEGs with additive effects or those in the combinatorial KD did not show enrichment for genes associated with neuropsychiatric disorders (Figures S8H–S8I). Consistent with the non-additive transcriptional effects of the three cis-target genes, the combinatorial KD exhibited a stronger reduction of neural dendritic complexity and calcium signaling than individual gene KDs in AA neurons (Figures 6D, 6E, and S7B). These results suggest that VPS45, AC244033.2, and C1orf54 contribute to the neural phenotypic changes by synergistically enhancing transcriptional effects relevant to neuropsychiatric disorders.
rs2027349 affects distal gene expression through local chromatin looping and accessibility
Finally, to mechanistically understand the cis-SNP editing effect on all three genes, we examined chromatin looping in a publicly available brain capture Hi-C dataset45 and in our own NGN2-Glut Micro-C (i.e., a promoter Capture Hi-C) dataset (Figure 7A and Table S14). We found that the rs2027349 site had long-range chromatin contacts with promoter regions of many genes, including VPS45/AC244033.2 and C1orf54 in both hippocampal neurons45,46 and in our NGN2-Glut neurons (Figure 7A), which supports the cis-regulatory effect of rs2027349-editing on the distal C1orf54 in NGN2-Glut.
Figure 7.

Long-range chromatin interactions and changes in local chromatin accessibility contribute to the cis effect of rs2027349 on multiple genes
(A) ATAC-seq and Capture Hi-C data from human brain tissue46 and Micro-C data from NGN2-Glut neurons. Orange arcs show the multiple long-range interactions between the rs2027349 locus and several high-accessibility sites (ATAC-seq peaks) in proximity. Bold arcs (red in hippocampal DG neurons and blue in NGN2-Glut neurons) indicate the site-specific interaction between rs2027349 and the promoter region of C1orf54.
(B) CRISPR-Cas9 editing of rs2027349 altered both local and remote chromatin accessibility. Note the higher regional OCR peaks (normalized pile-up intensity) on the rs2027349 locus and regions proximal to the C1orf54 gene in AA neurons. ATAC-seq data were from two clones per genotype of line CD0000011.
To further test whether the differential allelic transcriptional effect of rs2027349 may be mediated by differential allelic chromatin looping or chromatin accessibility, we directly compared the Micro-C sequencing reads of the two different alleles (A and G) of rs2027349 in heterozygous NGN2-Glut between two donor lines. We found an allelic imbalance of chromatin looping, with allele A having more Micro-C reads (Figure 7A, inset), suggesting enhanced chromatin looping associated with allele A of rs2027349. We then examined the differences in local chromatin between the isogenic CRISPR-edited neurons around the edited rs2027349 site (AA vs. GG) in NGN2-Glut, and we found that rs2027349 editing altered the enhancer/promoter OCR peaks of both VPS45/AC244033.2 and C1orf54 loci, with AA neurons showing higher regional OCR peaks than in GG neurons (Figure 7B). The higher regional OCR peaks at the VPS45/AC244033.2 and C1orf54 loci were consistent with the increased expression of all three genes by SNP editing in AA neurons (Figures 3B and 3C). Both Micro-C and ATAC-seq data in NGN2-Glut thus supported a distal regulatory effect of rs2027349, likely by affecting chromatin looping and local chromatin accessibility.
Discussion
Functional interpretation of non-coding GWAS risk variants is a major challenge in genetics. Combining analyses of allelic chromatin accessibility, CRISPRi screening, and precise SNP editing in NGN2-Glut, we prioritized regulatory SZ-risk variants affecting neural chromatin accessibility and gene expression. We tied the SNP function to neural phenotypic changes for a functional SZ-risk variant at the VPS45 locus. More importantly, we demonstrated a complex gene regulation paradigm where a single SZ-risk locus mediates the expression of both proximal and distal genes, which synergistically confer non-additive effects on disease-relevant cellular phenotypes.
Long-range gene regulation for neuropsychiatric genetic risk variants has been supported by brain promoter Capture Hi-C study.45 However, such postulated long-range cis-regulatory effects involving multiple genes and their relevance to cellular phenotypes have not been well established. Here, we showed that a single GWAS risk SNP (rs2027349) at the VPS45 locus exhibited the strongest ASoC and affected the expression of both its adjacent genes (VPS45 and AC244033.2) and a distal gene C1orf54 (∼200 kb away) (Figures 3B and 3C). Interestingly, the transcriptional effect of rs2027349 editing can be better explained by gene × gene interactions (VPS45_KD × C1orf54_KD and VPS45_KD × lncRNA_KD) than by individual genes (Figure 6J), i.e., non-additivity. The observed non-additive effect of multiple genes at a single GWAS risk locus was further corroborated by the demonstrated synergistic effects of all three cis-target genes (VPS45, C1orf54, and the lncRNA AC244033.2) (Figures 6K–6N), which is analogous to the recently reported synergistic transcriptomic effect of SZ-risk SNPs from different loci.12 Although polygenic risk variants/genes for complex disorders have been commonly considered to be additive for liability, the additivity of liability of disease risk variants/genes and the biological non-additivity or synergistic effects of multiple risk genes illustrated here and by others12 in the hiPSC model are not mutually exclusive; it is well recognized that good fit to an additive model does not imply that the underlying genes do not interact at a mechanistic level, i.e., epistasis.47 Our results provide empirical support for complex gene regulation with compound effects from multiple genes at a single SZ GWAS risk locus.
Evaluating the pathophysiological relevance of these neural phenotypic outcomes for SZ is challenging largely due to the need for defined disease-relevant cellular phenotypes. For SZ, studies of post-mortem brains mostly point to reduced dendritic spine density and dendritic arborization,48 while at the level of neuronal function, both pharmacology and genetic animal models of SZ converge on hypofunction of glutamatergic synapses despite the reasonable skepticism as to how accurately animal models can be reflective of SZ.49 However, in general, these cellular phenotypes of SZ postulated from studies of human post-mortem brain and animal models were not fully recapitulated by the hiPSC modeling of common or rare risk factors.50 Our observed increase in neuronal function by the risk allele of rs2027349, although seemingly inconsistent with the documented reduction in dendritic complexities and synaptic function for SZ,48,49,51,52 is similar to the effect of an SZ GWAS risk variant at the MIR137 locus,14 as well as the effects of rare SZ-risk variants with high penetrance (e.g., 16p11 duplication, loss-of-function of SHANK3).41,42 Such seemingly inconsistent cellular phenotypes may be the result of genetic pleiotropy across major psychiatric disorders.53 Alternatively, given that both neural hyperfunction12,13,14,41,42 and hypofunction51,52 were reported for SZ, our results provide further support for a neuronal homeostatic model of neuropsychiatric disorders where either excess or inadequate synaptic signaling output may contribute to pathophysiology.54,55 The relevance of our observed neurobiological phenotypes to SZ was further corroborated at the molecular level by the fact that the transcriptional changes associated with the SZ risk allele, rs2027349, were strongly enriched for SZ GWAS (Figure 3F) and correlated with transcriptomic changes in SZ post-mortem brains (Figures 3G, 3H, S5G, and S5H). Nonetheless, future studies of animal models carrying the knockout allele of the implicated risk genes (VPS45, AC244033.2, and C1orf54) at the rs2027349 locus may help establish the causal link between the observed neurobiological phenotypes and SZ pathophysiology.
The exact molecular mechanisms by which the SZ-risk variant rs2027349 functions remain to be determined. Our results suggest that these genes were not regulated by each other (Figure 6A). Furthermore, the rs2027349-flanking sequence has direct chromatin contact with the promoter sequence of distal C1orf54 in brain tissues and in our assayed NGN2-Glut neurons, and allele A of rs2027349 seemed to be associated with stronger chromatin looping (Figure 7A). Furthermore, local chromatin accessibility at the promoter/enhancer regions of VPS45, AC244033.2, and C1orf54 was more robust in rs2027349 AA lines (Figure 7B). These results suggest a possible cis-coregulation between these genes that may be mediated through promoter/enhancer-promoter chromatin interactions,45,56 as observed for HLA-DQB1 and HLADRB1.57
Limitations of the study
Although our observed effect of rs2027349 editing on neuronal phenotypes was supported by the transcriptomic (i.e., DE) analyses, these results still need to be interpreted cautiously, since DE analysis, even for isogenic hiPSC lines, can be confounded by clonal variations,58 a general challenge for the field. However, such limitations have been mitigated using multiple lines/clones of all three rs2027349 genotypes (AA, AG, and GG). Moreover, despite the broadly consistent effects of the ASoC loci on cis-target gene expression in the two different donor lines used in CRISPRi and in rs2027349 editing, we did observe differences in the effect size between the donor lines, which may be partially attributed to different genetic backgrounds and highlights the future need for scaling up functional assays with more donor lines. Finally, it is noteworthy that, although the reduced systems’ “buffering” to genetic or environmental perturbations in a simple cellular model like hiPSC-derived neurons may enable the detection of the biological function of a single genetic risk variant/gene,50,59,60 our observed solid biological effect of the SZ-risk variant rs2027349 does not imply disease causality. At the individual level, polygenic disease occurs only when perturbations from many genetic risks (or protective) loci and environmental factors break down the systems’ robustness or buffering capacity.61 Therefore, studying individual disease-risk variants/genes in hiPSC models does not represent a paradigm that can recapitulate the in vivo effects of polygenic risk factors, rather enabling us to gain a mechanistic understanding of disease-risk variants/genes. Collectively, our study uncovers a complex gene regulation at a single SZ GWAS risk locus where an ASoC SNP alters chromatin accessibility of multiple putative risk genes that have non-additive cellular transcriptional and phenotypic effects in excitatory neurons, providing a novel mechanistic link between a non-coding SZ GWAS risk variant and disease-related phenotypes.
Some figures in the article were created with BioRender.com under an institute subscription plan with publishing rights for journals and other academic purposes to the Chicago Immunoengineering Innovation Center, BSD, at the University of Chicago.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-MAP2 | Synaptic Systems | Cat. #: HS-188 011, RRID: AB_2661868 |
| Anti-vGlut1 | Synaptic Systems | Cat. #: 135 011BT, RRID: AB_2884913 |
| Anti-GFP | Abcam | Cat. #: ab13970, RRID: AB_300798 |
| anti-Synapsin I D12G5 XP | Cell Signaling | Cat. #: 5297, RRID: AB_2616578 |
| Anti- PSD-95 MAGUK scaffolding protein antibody | UC Davis/NIH NeuroMab Facility | Cat. #: K28/43, RRID: AB_2877189 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat. #: A-21202, RRID: AB_141607 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat. #: A-21203, RRID: AB_141633 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody | Thermo Fisher Scientific | Cat. #: A-21206, RRID: AB_2535792 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat. #: A-21207, RRID: AB_141637 |
| Chemicals, peptides, and recombinant proteins | ||
| mTeSR1 Plus | STEMCELL Technologies | Cat. #: 100-0276 |
| ReLeSR | STEMCELL Technologies | Cat. #: 05872 |
| Matrigel matrix | Corning | Cat. #: 354234 |
| Accutase | STEMCELL Technologies | Cat. #: 07920 |
| Neuralbasal Medium | Thermo Fisher Scientific | Cat. #: 21103049 |
| 50x B27 Supplement | Thermo Fisher Scientific | Cat. #: 17504044 |
| 100x Glutamax | Thermo Fisher Scientific | Cat. #: 35050061 |
| Doxycycline | Sigma-Aldrich | Cat. #: D3072-1ML |
| Puromycin | Thermo Fisher Scientific | Cat. #: J67236.8EQ |
| Hygromycin | Millipore | Cat. #: 400053 |
| BDNF | PeproTech | Cat. #: 450-02 |
| GDNF | PeproTech | Cat. #: 450-03 |
| NT-3 | PeproTech | Cat. #: 450-10 |
| Digitonin | Promega | Cat. #: G9441 |
| Blasticidin | Thermo Fisher Scientific | Cat. #: A1113903 |
| Fluor-4 AM | Thermo Fisher Scientific | Cat. #: F14217 |
| ProLong Diamond Antifade Mountant | Thermo Fisher Scientific | Cat. #: P36961 |
| Fugene HD | Promega | Cat. #: E2311 |
| Lipofectamine 3000 | Thermo Fisher Scientific | Cat. #: L3000001 |
| Critical commercial assays | ||
| TaqMan Universal PCR Master Mix | Thermo Fisher Scientific | Cat. #: 4324018 |
| High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor | Thermo Fisher Scientific | Cat. #: 4374967 |
| RNeasy Plus Mini Kit | Qiagen | Cat. #: 74134 |
| Chromium Next Gem Cell 3’ Kit 3.1 | 10x Genomics | Cat. #: 100269 |
| 24-well MEA plate | Axion BioSystems | Cat. #: M384-tMEA-24W |
| Deposited data | ||
| Code used in the manuscript | This manuscript | DOI: 10.5281/zenodo.8180188 |
| ATAC-seq and RNA-seq data | GSE188491 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE188491 |
| scRNA-seq data | SRR16919429 | https://dataview.ncbi.nlm.nih.gov/object/SRR16919429 |
| Experimental models: Cell lines | ||
| Human HEK293T cells | ATCC | Cat. #: CRL-3216 RRID: CVCL_0063 |
| Human: iPS cell from sample NG-47827 (F) | Rutgers University Cell and DNA Repository (RUCDR) | Cell ID: 01C08162 |
| Human: iPS cell from sample 92712 (M) | Rutgers University Cell and DNA Repository (RUCDR) | Cell ID: 04C27190 |
| Human: iPS cell from sample 79588 (M) | Rutgers University Cell and DNA Repository (RUCDR) | Cell ID: 04C28905 |
| Human: iPS cell from sample 69153 (M) | Rutgers University Cell and DNA Repository (RUCDR) | Cell ID: 04C37433 |
| Human: iPS cell from sample 18526 (F) | Rutgers University Cell and DNA Repository (RUCDR) | Cell ID: 05C38571 |
| Human: iPS cell from sample 29934 (M) | Rutgers University Cell and DNA Repository (RUCDR) | Cell ID: 05C39664 |
| Human: iPS cell from sample NG-73088 (M) | Rutgers University Cell and DNA Repository (RUCDR) | Cell ID: 05C43356 |
| Human: iPS cell from sample 44414 (F) | Rutgers University Cell and DNA Repository (RUCDR) | Cell ID: 05C43758 |
| Human: iPS cell from sample NG-81739 (M) | Rutgers University Cell and DNA Repository (RUCDR) | Cell ID: 05C45915 |
| Human: iPS cell from sample 81566 (F) | Rutgers University Cell and DNA Repository (RUCDR) | Cell ID: 05C46807 |
| Human: iPS cell from sample 75610 (M) | Rutgers University Cell and DNA Repository (RUCDR) | Cell ID: 05C46837 |
| Human: iPS cell from sample 48181 (M) | Rutgers University Cell and DNA Repository (RUCDR) | Cell ID: 05C48054 |
| Human: iPS cell from sample NG-55047 (F) | Rutgers University Cell and DNA Repository (RUCDR) | Cell ID: 05C49221 |
| Human: iPS cell from sample NG-11293 (M) | Rutgers University Cell and DNA Repository (RUCDR) | Cell ID: 06C52191 |
| Human: iPS cell from sample NG-40965 (M) | Rutgers University Cell and DNA Repository (RUCDR) | Cell ID: 06C52565 |
| Human: iPS cell from sample 16125 (F) | Rutgers University Cell and DNA Repository (RUCDR) | Cell ID: 06C52573 |
| Human: iPS cell from sample NG-77642 (F) | Rutgers University Cell and DNA Repository (RUCDR) | Cell ID: 06C53368 |
| Human: iPS cell from sampleNG-23159 (M) | Rutgers University Cell and DNA Repository (RUCDR) | Cell ID: 06C54426 |
| Human: iPS cell from sample NG-42857 (M) | Rutgers University Cell and DNA Repository (RUCDR) | Cell ID: 07C71166 |
| Human: iPS cell from sample NG-80080 (M) | Rutgers University Cell and DNA Repository (RUCDR) | Cell ID: 07C65853 |
| Oligonucleotides | ||
| See Table S15 | ||
| Recombinant DNA | ||
| Lenti-dCas9-KRAB-blast | Xie et al.29 | RRID: Addgene_89567 |
| pMD2.G | https://www.addgene.org/12259/ | RRID: Addgene_12259 |
| psPAX2 | https://www.addgene.org/12260/ | RRID: Addgene_12260 |
| FUW-M2rtTA | Hockemeyer et al.62 | RRID: Addgene_20342 |
| pTet-O-Ngn2-puro | Zhang et al.19 | RRID: Addgene_52047 |
| CROPseq-Guide-Puro | Datlinger et al.28 | RRID: Addgene_86708 |
| pMDLg/pRRE | Dull et al.63 | RRID: Addgene_12251 |
| pRSV-Rev | Dull et al.63 | RRID: Addgene_12253 |
| Software and algorithms | ||
| MAGMA software and population data | de Leeuw et al.39 | https://ctg.cncr.nl/software/magma |
| H-MAGMA codes and annotation files | Sey et al.39 | https://github.com/thewonlab/H-MAGMA |
| Miniconda | Anaconda Inc. | https://docs.conda.io/en/latest/miniconda.html |
| Pluritest | N/A | https://www.pluritest.com |
| Trimmomatic v 0.32 | Bloger et al.64 | https://github.com/timflutre/trimmomatic |
| Bowtie2 2.4.0 | Langmead et al.65 | https://github.com/BenLangmead/bowtie2 |
| Samtools 1.11.0 | Li et al.66 | https://htslib.org |
| GATK 4.1.8.1 | McKenna et al.67 | https://gatk.broadinstitute.org/ |
| MACS2 2.2.7.1 | Zhang et al.24 | https://github.com/macs3-project/MACS |
| R 4.1.1 | R Core Team | https://cran.r-project.org/ |
| SNPsplit | Kruger et al.68 | https://www.bioinformatics.babraham.ac.uk/projects/SNPsplit/ |
| deepTools 3.5.1 | Ramirez et al.69 | https://deeptools.readthedocs.io/en/develop/ |
| STAR 2.7.0 | Dobin et al.70 | https://github.com/alexdobin/STAR |
| Cell Ranger 4.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| Seurat 3.2 | Stuart et al.71 | https://satijalab.org/seurat/index.html |
| WebGestalt 2019 | Wang et al.35 | http://www.webgestalt.org/ |
| Synaptic Gene Ontologies (SynGO) | Koopmans et al.31 | https://syngoportal.org/ |
| ImageJ 2 | Schneider et al.72 | https://imagej.nih.gov/ij/download.html |
| NIS-Elements Advanced | Nikon Inc. | N/A |
| GraphPad Prism 9 | GraphPad Inc. | https://www.graphpad.com/ |
| Neural Metrics Analysis Tool | Axion Biosystems | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Dr. Jubao Duan (jduan@uchicago.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and study participant details
The 20 hiPSC lines used for generating NGN2-Glut neurons for ATAC-seq were the same lines as used in our previously study13. Briefly, hiPSCs were derived from cryopreserved lymphocytes (CPLs) using the genome-integration-free Sendai virus method (Cytotune Sendai Virus 2.0; Invitrogen). The purity of hiPSC culture was confirmed by immunofluorescence (IF) staining of pluripotency markers (OCT4, SSEA4, NANOG, and TRA-1-60). The pluripotency of the hiPSCs was further confirmed by Pluritest with RNA-seq data, as described in our previous study13. Lines CD0000011 and CD0000012, which are heterozygous (A/G) at SNP site rs2027349, were used for CRISPR/Cas9 editing into all three genotypes, 2-3 clones per genotype13. The isogenic CRISPR-edited clones were all confirmed to be free of major off-target editing by Sanger sequencing of the top 5 predicted off-target sites13. Lines CD000009 and CD000011 that stably express dCas9-KRAB-BSD13 were used for CRISPRi (CROP-seq). None of the three lines used for CRISPR/Cas9 editing or CRISPRi have an extreme SZ polygenic risk score73. The NorthShore University HealthSystem Institutional Review Board (IRB) approved the study.
Method details
hiPSC culture
The hiPSCs were maintained in mTeSR Plus media (STEMCELL) on a matrigel (Corning) coated 6-well plate with media refreshment every other day. Cells were passaged at 1:10-1:20 every 4-6 days using ReLeSR (STEMCELL) following the vendor’s protocol.
Generation of NGN2-Glut neurons
NGN2-Glut neurons were prepared as described19 with modifications. Briefly, on Day 0, 5 × 105 hiPSCs were dissociated using Accutase (STEMCELL) and replated in 1.2 ml mTeSR Plus media containing 5 μM ROCK inhibitor, rtTA virus and Ngn2-puro or Ngn2-Hygro virus on 6-well plates. On Day 1, media was refreshed with mTeSRPlus containing 2 μg/ml doxycycline. From days 2 to 4, media was refreshed daily with Neurobasal Medium supplemented with 1× Glutamax/B27, 2 μg/ml doxycycline, and 2 μg/ml puromycin or 200 μg/ml hygromycin. On day 5, cells were dissociated with Accutase and resuspended in Neurobasal Medium supplemented with 1× Glutamax/B27, 2 μg/ml doxycycline, and 10 ng/ml BDNF/GDNF/NT-3. Cells were replated at 2 × 105/cm2 on 0.1% PEI/Matrigel coated 12-well plates. For morphological immunostaining, 5 × 104 iNs were replated onto 12 mm coverslips (with 5 × 104 rat astrocytes pre-cultured) in Neurobasal Medium supplemented with 1× Glutamax/B27, 2 μg/ml doxycycline, 10 ng/ml BDNF/GDNF/NT-3, and 1% FBS. From Day 6, media was refreshed every three days with a half volume change. Doxycycline was withdrawn from Day 14 onwards. ATAC-seq samples were harvested at Day 20; RNA-seq/qPCR samples were harvested at Day 30. For cells cultured on coverslips, GFP transfection was performed at Day 30 using Lipofectamine 3000 (Thermofisher) at 1:2 DNA:reagent ratio with 1 μg plasmid per coverslip. On Day 32, coverslips were fixed and stained for morphological analysis.
Immunocytochemistry
Characterization of hiPSCs and neurons were performed as described13 with minor modifications. Briefly, fresh cells were fixed in 4% PFA for 15 min at room temperature. After three PBS washes, cells were permeabilized by 1% Triton X-100 in PBS for 15 min at room temperature. Subsequently, samples were blocked with 3% BSA in 0.1% PBST (0.1% Triton X-100 in PBS) for one hour at room temperature. After blocking, samples were incubated with primary antibodies diluted in blocking buffer at 4°C overnight. After three PBS washes, samples were incubated with secondary antibodies diluted in blocking buffer for one hour at room temperature. After another three PBS washes, samples were incubated with 1 μg/ml DAPI for 10 min at room temperature. Samples were then mounted on glass slides using ProLong™ Diamond Antifade Mountant (Thermofisher) overnight before taking images. Primary antibodies used were anti-MAP2 (Synaptic System, 1:700), anti-vGlut1 (Synaptic Systems, 1:100), anti-GFP (chicken, Abcam ab13970, 1:10,000), anti-Synapsin I (rabbit, Cell Signaling #5297, 1:200), and anti-PSD-95 (mouse, NeuroMab clone K28/43 1:1000). Secondary antibodies used were Alexa 488 donkey anti-mouse (1:1000), Alexa 594 donkey anti-mouse (1:1000), Alexa 488 donkey anti-rabbit (1:1000), and Alexa 594 donkey anti-rabbit (1:1000). Images were taken using a Nikon C2 confocal microscope with 20× or 40× lenses as needed.
Sholl analysis
GFP-transfected neurons were imaged on a Nikon C2+ confocal microscope with a 10× objective lens and NIS-Elements software. Neurons were only imaged if a visible axon and at least one defined primary dendrite could be identified. Following the acquisition, dendrites were traced and images binarized in ImageJ (National Institutes of Health, Bethesda, MD). For each image, the axon was identified and eliminated from quantification. Sholl analysis was performed using the ‘Sholl analysis’ plugin for ImageJ. The center of the soma was manually defined, and concentric circles spaced 10 μm apart were used to quantify dendritic complexity in all cells.
For Sholl analysis, a two-way repeated measures ANOVA was performed to detect an effect of genotype-distance interaction. A post hoc correction was then applied to the analysis to correct for multiple testing and determine the significance of each data point at each distance interval. For synaptic puncta and protrusion analyses, data sets were tested for normality using D’Agostino and Pearson omnibus normality test. A one-way ANOVA (for parametric data) or a Kruskal Wallis test (for non-parametric data) with post hoc correction was used to determine significance between groups. All data were analyzed in GraphPad Prism version 9. Results were considered as significant if p < 0.05. All data are reported as mean ± SEM. None of the data was removed as outliers.
Puncta and protrusion density analysis
GFP-transfected and immunostained neurons were imaged using a 63× oil immersion objective lens on a Nikon C2+ confocal microscope with NIS-Elements software. Each channel was acquired sequentially to prevent fluorescence bleed-through with 2× line averaging for each channel and 1.5× digital zoom. Image parameters were optimized for each batch (i.e., all neurons from the same differentiation) to limit the over- or undersaturation of signal. All images within a batch were acquired using identical settings for each channel to allow comparison between groups. Dendritic branches were imaged on multiple z-planes to capture full dendritic branches and maximum intensity projections of z-stacks created in Image J were used for downstream puncta analysis. Regions of interest (ROI) on the dendrite (∼100 μm) were selected using the GFP-filled cell as an outline. To identify puncta, a set threshold method was used for each experiment and applied identically to all conditions. The area and intensity of each punctum above the set threshold were recorded, and mean values were calculated for each cell. For puncta density analysis, the number of puncta was recorded and divided by the total area of the ROI calculated in ImageJ. For protrusion density analysis, a region of the dendrite (∼100 μm) was selected, and all protrusion <15 μm were manually counted and recorded.
MEA analysis
NGN2-Glut were prepared as described in a previous section. On Day 28, neurons were dissociated using Accutase and replated at 1 × 105 cells/well with 1.5 × 104 cells/well rat astrocytes onto a 0.1% PEI coated 24-well MEA plate (Axion BioSystems# M384-tMEA-24W). Media was refreshed every three days using Neurobasal Medium supplemented with 1× Glutamax/B27, 10 ng/ml BDNF/GDNF/NT-3, and 1% FBS. MEA data was recorded 24 hrs post media refreshment at 37°C with 5% CO2. On the day of recording, the plate was loaded into Axion Maestro MEA reader (Axion Biosystems) and allowed to rest for 3 min, then recorded for 10 min. Data files were batch-processed using Neural Metrics Tool (Axion Biosystems) with four biological replicates for each condition. For analysis, bursts were identified using an inter-spike interval (ISI) threshold requiring a minimum number of 5 spikes with a maximum ISI of 100 msec. Network bursts were analyzed using the envelope mode with threshold factor 1.25, minimum inter-burst interval (IBI) 100 msec, minimum of 25% active electrodes, and 75% burst inclusion. The synchrony index was calculated using a cross-correlogram synchrony window of 20 msec.
Calcium imaging
NGN2-Glut were replated on rat astrocyte-coated coverslips on D5 DIV. On D35 DIV, neurons were labeled with 5 μM Fluo-4-AM at 37°C for 20 min. Coverslips were then transferred to CO2 saturated 1× ACSF (126 mM NaCl, 2.5mM KCl, 1 mM NaH2PO4, 26.2 mM NaHCO3, 2.5 mM CaCl2, 1.3 mM MgSO4, 11 mM D-Glucose) perfusion system for imaging. After 10-15 min perfusion to wash out extra dyes, time-lapse images were acquired at 1.8 Hz for 10 min using a Nikon A1R multiphoton microscope at 820 nm. Quantified regional intensity values were tabulated as one-dimensional numerical vectors indexed by time, and a Savitzky-Golay filter (locally estimated scatterplot smoothing) was applied for data smoothing. Baseline intensity level (for both peak detection and dF/F0 calculation) was performed using the R baseline package74. The maximum peak intensities were subsequently captured by detecting the maximum values on the rolling margins of each peak window (see the Extended Data code), and frequencies (the inversion of firing rate) were calculated using their corresponding peak positions. Amplitude (dF/F0) was calculated using the standard method by dividing the calibrated peak value at any specific position over the baseline intensity value at the same position as determined by the baseline package, and for each cell, one dF/F0 was calculated by taking the means of the dF/F0 values from all peaks from the same cell. The Synchrony index was calculated using the original spike train distance model75 by spike distance and temporal distance normalized as mean resultant length for inter-sample measuring and visualization. Images analysis and quantification were performed in ImageJ and R.
ATAC-seq samples preparation
ATAC-seq samples were prepared as previously described13,76. Briefly, 75,000 viable cells were lysed in ATAC-Resuspension Buffer (RSB) containing 0.1% NP-40, 0.1% Tween-20, and 0.01% Digitonin for 3 min on ice. Nuclei were washed and resuspended in a transposition mixture. Reactions were incubated at 37°C for 30 min on a thermomixer at 1,000 rpm. Transposed DNA was purified by DNA Clean and Concentrator-5 Kit (Zymo). Eluted DNA was shipped to the University of Minnesota Genomic Center for library preparation and ATAC-seq.
Lentivirus preparation
Lentivirus particles were packaged in fresh HEK 293T culture (passaged ≤ 15 times) as previously described13. Briefly, 2 × 106 cells were replated in T25 flasks one day before transfection. At the day of transfection, media was refreshed without antibiotics. pLenti-dCas9-KRAB-BSD (Addgene # 89567) was co-transfected with pMD2.G (Addgene #12259) and psPAX2 (Addgene #12260) at a 4:2:3 molar ratio; other viral vectors including FUW-M2rtTA (Addgene # 20342), pTet-O-Ngn2-puro (Addgene # 52047), CROPseq-Guide-Puro gRNA (Addgene # 86708), and pTet-O-Ngn2-hygro were co-transfected with pMDLg/pRRE (Addgene #12251), pMD2.G (Addgene #12259), and pRSV-Rev (Addgene #12253) at 1:1:1:1 molar ratio. Transfection was performed using FuGENE HD (Promega) following the vendor’s instructions. 24 hrs post transfection, media was refreshed with Neurobasal Medium supplemented with 1× Glutamax/B27. 48 hrs post transfection, the supernatant was collected and centrifuged at 500 × g for 5 min to remove debris. The virus-containing media was aliquoted into 1.5 ml low protein binding tubes and stored at -80°C until use. Viral RNA was extracted and the titer (MOI) was measured using the Lenti-X qRT-PCR titration kit from Takara (Cat. #: 631235) following vendor’s instructions.
CROP-seq in NGN2-Glut neurons
hiPSC lines (Line CD000009 and CD0000011) that stably express dCas9-KRAB-BSD were established as previously described13. Briefly, on day 0, hiPSCs were replated as small clumps using ReLeSR (STEMCELL) at 10-20% confluence. On Day 1, refresh media with mTeSR Plus containing dCas9-KRAB-BSD virus. 24 hrs post transduction, media was refreshed with mTeSR Plus containing 5 μg/ml Blasticidin. Antibiotics selection was refreshed daily and maintained for 10 days.
NGN2-Glut were prepared from hiPSCs stably expressing dCas9-KRAB-BSD using NGN2-hygro virus as described in a previous section (Generation of NGN2-Glut neurons). From Days 2-4, cells were selected using 150 μg/ml hygromycin (Sigma). On Day 20, 5x106 NGN2-Glut were dissociated using Accutase and replated in Neurobasal Medium supplemented with 1× Glutamax/B27, 10 ng/ml BDNF/GDNF/NT-3, 5 μM ROCK inhibitor, and CROPseq-Guide-Puro virus (vector carrying gRNA targeting genes of interest, MOI=0.2) as described in the original CROP-seq article28. 48 hrs post-transduction, cells were selected using 2 μg/ml puromycin. 7 days after selection, cells were harvested using Accutase for single cell RNA-seq preparation using Chromium Next GEM Single Cell 3' Reagent Kits v3.1 (10× Genomics).
Gene knockdown (KD) in NGN2-Glut neurons
Short hairpin RNA (shRNA) sequences were designed using Thermofisher online designer (rnaidesigner.thermofisher.com/rnaiexpress/insert.do) following antisense-loop-sense pattern. Refer to Table S15 for shRNA sequences. Single-strand shRNA oligos were cloned into BsmBI digested CROPseq-Guide-Puro (Addgene# 86708) vector using NEBuilder® HiFi DNA Assembly Master Mix (New England Biolabs) following vendor’s protocols. Oligo sequence targeting GFP was served as control. Lentivirus particles were prepared as described in previous sections (Lentivirus preparation). Two independently generated, SNP-edited AA (rs2027349) hiPSC lines (A11, H12) derived from donor CD11 were used for shRNA KD. Stable knockdown (KD) hiPSC lines were established as follows. On Day 0, hiPSCs were replated as small clumps using ReLeSR (STEMCELL) at 10-20% confluency. On Day 1, media was refreshed with mTeSRPlus containing CROPseq-KDgRNA-Puro lenti-shRNA virus at MOI of 10. After 48 hrs of lenti-shRNA transduction, media was refreshed with mTeSRPlus containing 0.8 μg/ml puromycin to select for hiPSC stably expressing a targeting shRNA. Puromycin selection was maintained for 10 days with a daily change of new media. For the triple shRNA KD (VPS45, AC244033.2, and C1orf54), stable triple KD hiPSCs were generated by pooling three lenti-shRNA viruses at MOI=10 of each, and positive cells were subsequently selected under 0.8 μg/ml puromycin for 7 d. NGN2-Glut neurons were further generated from the hiPSC lines stable expressing shRNA by using Ngn2-hygro lentivirus transduction as described in a previous section (Generation of NGN2-Glut). Total RNA was isolated from DIV30 cultures using RNeasy Plus kit (QIAGEN) for qPCR quantification and RNA-seq.
ATAC-seq read aligning
All raw sequence reads generated by Illumina HiSeq 2000 had been demultiplexed at the University of Minnesota Genomics Center and provided as 2×75 bp paired-end FASTQ files (targeting 60 M reads per sample). Adapter remnants, low-quality reads, and low QSEQ short sequences near either end of reads were processed by Trimmomatic (ILLUMINACLIP:NexteraPE-PE.fa:2:30:7, SLIDINGWINDOW:3:18, MINLENGTH:26). The processed sequences were separated into paired-end and single-end FASTQ files per sample and only paired-end reads were retained for subsequent mapping. The FASTQ files were individually mapped against the human genome reference file including decoy sequences (GRCh38p7.13/hg38, 1000 Genome Project) using bowtie2 (-x 2000, -mm --qc-filter –met 1 –sensitive –no-mixed -t) and subsequently merged and sorted as BAM-formatted files using samtools, with only uniquely mapped reads (MAPQ > 30) retained. Picard tools MarkDuplicate was then used to remove all PCR and optical duplicated reads from the BAM file.
To further eliminate allelic bias towards reference alleles during the aligning step, we performed WASP calibration on the generated raw BAM files77. Briefly, we first called the VCF file profiles on all SNP sites that were not reference alleles of all 20 samples individually using GATK HaplotypeCaller, and subsequently collapsed the individual VCF files into one summary VCF file containing all non-reference sites of all 20 individuals. Subsequently, this SNP list was used as the basis of WASP calibration and re-alignment, and a new WASP-calibrated new BAM file set was collected as the final output for the following peak calling and ASoC SNP call77.
The insert size distribution histograms of each sample were individually generated using Picard CollectInsertSizeMetrics. All analyzed ATAC-seq samples passed standard QC based on the characteristic nucleosomal periodicity of the insert fragment size distribution and high signal-to-noise ratio around transcription start sites (TSS).
ATAC-seq peak calling and ASoC SNP calling
To increase sample size and sensitivity for peak detection, the BAM files of the processed reads of each sample were first sub-sampled to 30 M pair-end reads per sample (the smallest sample size) and the 20 sub-sampled BAM files were merged as the source file for peak calling. MACS224 was used to generate peak files (narrowPeak format) with recommended settings at FDR = 0.05 (-f BAMPE, --nomodel, --call-summits --keep-dup-all -B). Peaks that fell within the ENCODE blacklisted regions were removed. Also, we removed peaks falling within chromosomes X and Y, and the mitochondrial genome regions.
GATK (version 4.1.8.1) was used for ASoC SNP calling, as recommended by the GATK Best Practices (https://software.broadinstitute.org/gatk/best-practices/)67. As noted above, WASP-calibrated BAM files (without sub-sampling) generated from ATAC-seq pipeline were used as input. Variants were called using the discovery mode of HaplotypeCaller with human GRCh38 (hg38) genome and the corresponding dbSNP version 153, and only reads with MAPQ score ≥30 were used (-stand_call_conf 30). Subsequently, recalibration of SNPs and Indels were performed in tandem using the VariantRecalibrator function (-an DP -an QD -an FS -an SOR -an MQ -an ReadPosRankSum -mode SNP -tranche 100.0 -tranche 99.9 -tranche 99.5 -tranche 90.0 -mG 4) and applied using ApplyRecalibration. Databases used for VariantRecalibrator including the hg38 versions of HapMap v3.3 (priority = 15), 1000G_omni v2.5 (priority = 12), Broad Institute 1000G high confidence SNP list phase 1 (priority = 10), Mills 1000G golden standard INDEL list (priority = 12), and dbSNP v153 (priority = 2). Each sample within the cell type was processed individually, and heterozygous SNP sites with tranche level >99.5% were extracted. To reduce bias introduced by any acquired (or “de novo”) mutations during cell growth, only SNPs with corresponding rs# records found in dbSNP v153 were retained. All heterozygous sites that passed the filter above (20 samples) were merged by CombineVariants to produce the master VCF file of the cell type.
To maximize the power to detect ASoC, we pooled SNPs for all the called heterozygous SNPs locations from the 20 samples as justified by the high concordance of allele-specific effects across individuals13. Finally, the VCF files were filtered and only biallelic SNP sites (GT: 0/1) with minimum read depth count (DP) ≥ 20 and minimum reference or alternative allele count ≥ 2 were retained. The binomial p-values (non-hyperbolic) were calculated using the binom.test(x, n, P = 0.5, alternative = “two.sided”, conf.level = 0.95) from the R package, and Benjamini & Hochberg correction was applied to all qualified SNPs as the correcting factor of R function p.adjust(x, method = "FDR"). We set the threshold of ASoC SNP at FDR value = 0.05.
The read pileup statistics proximal to SNP sites were generated using samtools mpileup function, and differential of allele-specific reads was performed using the SNPsplit Perl package (www.bioinformatics.babraham.ac.uk/projects/SNPsplit/)68. The final readouts from both read pileup and SNP-specific reads were visualized using the R package Gviz. In addition, when comparing the changes of chromatin accessibility caused by genotypes across samples, read counts were scaled and normalized using the deepTools package bamCoverage function and re-scaled to reads per genomic content (RPGC) as the base unit69. We confirmed no obvious mapping bias to reference alleles by visualizing the volcano plots that graph the allelic read-depth ratios against -log2p-values in scatter plots (Figure S2A).
Principal component analysis (PCA) for ATAC-seq data
To compare the cellular property between our NGN2-Glut neurons and different hiPSC-derived neuronal cell lines generated in our lab, as well as with previously assayed fetal brain cells, we used chromatin accessibility data from our previously published studies13 as well as DNase I hypersensitive assay from the fetal brain (NIH Roadmap)78. Subsequently, we applied our above described cross-cell-type non-overlapping OCR peak interval (n = 666,614) (GTF file, genome lift-over was performed when necessary) to the 20 lines of NGN2-Glut data using featureCounts79 to get per-interval read count.
Disease GWAS risk enrichment test for NGN2-derived ASoC SNPs by TORUS
The Bayesian hierarchical model (TORUS) was applied to perform an SNP-based enrichment analysis to explore whether ASoC SNPs are enriched in any of the interested diseases in our investigation80. Briefly, TORUS assumes that every variant is a risk variant or not, represented by a binary indicator variable (1 or 0). The prior probability of the indicator of a SNP being 1 depends on its annotations. Subsequently, TORUS links GWAS effect sizes of SNPs and their annotations using the following formula:
where GWAS effect size βj follows a spike-and-slab distribution a priori, and πj is the prior probability for the j-th SNP in a certain locus, modelled by a logistic link with annotation djk (for the k-th annotation) for SNP j. Usually a normal prior distribution is used for g(.). For binary annotations (1 if a SNP has that annotation, 0 otherwise), the parameter αk, is the log odds ratio of k-th annotation, and measures enrichment of risk variants in the k-th annotation, relative to all SNPs in the genome that do not have that annotation. TORUS uses the summary statistics of the entire genome to estimate the enrichment parameters.
For GWAS enrichment test (Figure S2D), we used ASoC SNPs derived from NGN2-Glut. All the annotations are encoded as binary (1 if a SNP has an annotation, and 0 otherwise). The GWAS datasets used for enrichment/TORUS analysis were from multiple sources, including both neuropsychiatric disorders and control disorders/traits, as listed below. Univariate analysis was performed to assess the enrichment of ASoC SNPs in each of the GWAS datasets. The GWAS summary datasets used in the analysis are:
MDD, ukb-b-12064 https://gwas.mrcieu.ac.uk/datasets/ukb-b-12064/ https://gwas.mrcieu.ac.uk/files/ukb-b-12064/ukb-b-12064.vcf.gz
ADHD, ieu-a-1183, https://gwas.mrcieu.ac.uk/datasets/ieu-a-1183/, https://gwas.mrcieu.ac.uk/files/ieu-a-1183/ieu-a-1183.vcf.gz
ASD, ieu-a-1185, https://gwas.mrcieu.ac.uk/datasets/ieu-a-1185/, https://gwas.mrcieu.ac.uk/files/ieu-a-1185/ieu-a-1185.vcf.gz
BMI, ukb-b-19953, https://gwas.mrcieu.ac.uk/datasets/ukb-b-19953/, https://gwas.mrcieu.ac.uk/files/ukb-b-19953/ukb-b-19953.vcf.gz
Insomnia, ukb-b-3957, https://gwas.mrcieu.ac.uk/datasets/ukb-b-3957/, https://gwas.mrcieu.ac.uk/files/ukb-b-3957/ukb-b-3957.vcf.gz
T2D, ukb-b-13806, https://gwas.mrcieu.ac.uk/datasets/ukb-b-13806/, https://gwas.mrcieu.ac.uk/files/ukb-b-13806/ukb-b-13806.vcf.gz
Intelligence, ebi-a-GCST006250, https://gwas.mrcieu.ac.uk/datasets/ebi-a-GCST006250/, https://gwas.mrcieu.ac.uk/files/ebi-a-GCST006250/ebi-a-GCST006250.vcf.gz
Bipolar (BP), ukb-b-6906, https://gwas.mrcieu.ac.uk/datasets/ukb-b-6906/, https://gwas.mrcieu.ac.uk/files/ukb-b-6906/ukb-b-6906.vcf.gz
Schizophrenia (SZ), ieu-b-42, https://gwas.mrcieu.ac.uk/datasets/ieu-b-42/, https://gwas.mrcieu.ac.uk/files/ieu-b-42/ieu-b-42.vcf.gz
Alzheimer’s (AD), ieu-b-5067, https://gwas.mrcieu.ac.uk/datasets/ieu-b-5067/, https://gwas.mrcieu.ac.uk/files/ieu-b-5067/ieu-b-5067.vcf.gz
Neuroticism, ukb-b-4630, https://gwas.mrcieu.ac.uk/datasets/ukb-b-4630/, https://gwas.mrcieu.ac.uk/files/ukb-b-4630/ukb-b-4630.vcf.gz
Bulk RNA-seq and differential expression (DE) analysis
Total RNA was isolated using RNeasy plus kit (QIAGEN) following the vendor’s instructions and quantified by a NanoDrop-8000 spectrometer. The isolated RNA was subsequently sent to the sequencing provider (GENEWIZ) for further processing and sequencing analysis. RNA-seq files were provided in the format of 2×150 bp paired-end FASTQ files. Briefly, libraries were prepared using the NEB Nextera kit with customized adapters. Since the sequencing facility had performed pre-cleaning on raw reads, no Trimmomatic adapter-trimming was performed. 20∼30 M reads were recovered from each sample. Raw files were subsequently mapped to human hg38 genome (GRCh38p7.13) using STAR v2.7.070 with the following parameters:
(--outSAMtype BAM SortedByCoordinate --quantMode GeneCounts --outSAMattrIHstart 0 --outSAMstrandField intronMotif --outSAMmultNmax 1 --outFilterIntronMotifs RemoveNoncanonical --outBAMcompression 10 --outBAMsortingThreadN 20 --outBAMsortingBinsN 20 --outFilterMultimapNmax 1 --outFilterMismatchNmax 1 --outSJfilterReads Unique --limitBAMsortRAM 10000000000 --alignSoftClipAtReferenceEnds No --quantTranscriptomeBAMcompression 10 10).
PCR and optical duplicates were further removed using Picard MarkDuplicates, and unique mappers (MAPQ = 255) were retained for downstream analysis. GENCODE v35-based gene annotations, transcript length, and GC percentage were used81. For gene-based quantifications, the number of fragments at the meta-feature (gene) level was directly collected from the output of STAR, and only genes expressed in at least one cell type with counts per million (CPM) >1 in at least half of the sample within the group were retained for subsequent analysis. CPM numbers were used to plot the correlation heatmap and PCA analysis. For transcript-based quantifications (such as in the case of quantifying the expression level of different VPS45 isoforms), the pseudo-alignment probabilistic model of Kallisto was applied82. The PCA map was generated using R packages, gplots and ggplot2, respectively. DE gene analysis was performed using EdgeR83 by fitting the dataset into a gene-wise negative binomial generalized linear model with quasi-likelihood as glmQLFit() and evaluated by glmQLFTest(). Cell line and batch numbers were introduced when establishing the design matrix and served as blocking factors, whenever applicable.
CPM values generated by EdgeR was used as the basis for making the PCA plot to visualize the clustering effect of different gene knockdown groups. Essentially, only the top 15,000 DE genes were used for calculating the PCs using R prcomp() function and the plot was subsequently generated using the fviz_pca() function from R package factoextra (Figure S8B).
Total RNA extraction and qPCR analysis
For bulk RNA-seq and qPCR, RNA was isolated using RNeasy plus kit (QIAGEN) following the vendor’s instructions and quantified using NanoDrop-8000 spectrometer. For qPCR, RT was performed using High-Capacity cDNA Reverse Transcription Kit (Thermofisher). RT conditions were as follows: 25°C for 10 min, 37°C for 120 min, 85°C for 5 min. qPCR was performed on a Roche 480 II Lightcycler, using gene-specific FAM-labelled TaqMan probes, GAPDH, ABL1 and CASC3 as internal controls. qPCR conditions were as follows: 95°C for 10 min, 45 cycles of (95°C for 20 s, 60°C for 1 min). Biological replicates (3 to 6) were included for all conditions and three technical replicates were used during qPCR.
Gene expression correlation between DE genes from SNP editing and for various disorders
The correlation of expression profiles of DE genes between VPS45 CRISPR/Cas9-edited lines (AA vs.GG) and post-mortem brain tissues of various psychiatric disorders (MDD, BP, ASD, AAD, SZ) were compared by integrating GTEx/PsychENCODE and several other data sources16,84,85. For each DE gene set, genes with FDR < 0.05 were used in the overlapped gene counts and correlation analysis. The R package upsetR was used to make the Upset plot. For correlation analysis, Spearman’s ρ was calculated using R function cor.test(method = “spearman”, alternative = “t”).
Single-cell RNA-seq data processing and analysis
10× Genomics Cell Ranger v4 was used to process raw sequencing data. Briefly, sequencing data were aligned to the human GRCh38/hg38 genome with spike-in gRNA sequences as artificial chromosomes (20 bp gRNA sequence and 250 bp downstream plasmid backbone per each gRNA, together with one additional full sequence of CROP-Seq-Guide-Puro construct also included as a decoy to capture vector-borne sequences). The alignment was performed using STAR 2.7.0 included in Cell Ranger v4 and a customized spiked-in version of GENCODE v28 GTF file containing the aforementioned gRNA sequences and vector backbone as annotations. The filtered gene count matrix output from Cell Ranger was used in subsequent R analysis. In all, we achieved a total mapping rate of 88.7%, with 58.1% mapping rate to the human transcriptome.
The R package Seurat v 3.2 was used to extract information and assemble data tables from the output files to generate the digital gene expression matrix, which was constructed based on per-gene UMI count. Since the Cell Ranger package had already performed preliminary QC filtering and removed low-counting barcodes (i.e., cells) from its filtered output data set (filtered_bc_matrix), we directly imported the output of these processed barcode matrices as raw data. Outputs from one capture (using a mix of CD000009 + CD000011) were imported, which resulted in a total of 10,247 cells used in the initial analysis. Cell line identity de-multiplexing was performed with demuxlet on the 10,247 barcodes using previously published array-derived genotyping information identified 2,280 singletons from CD000009 and 6,979 singletons from CD000011. The Seurat matrix was re-normalized using SCTransform() and analyzed on the generated SCT object. Cells with > 20% mitochondria content were removed from analysis after SCTransform. A cell assigned with a unique RNA was defined as the UMI count of its dominant gRNA in the cell being at least three times more than the sum of UMI counts from all other gRNAs. Only MAP2+ cells that express glutamatergic neuron marker genes (SLC17A6 or SLC17A7) falling within clusters 2, 3, 4, 5, 6, and 7 were used (Figure 2B). Eventually, 4,057 cells survived the filter and were designated as cells with uniquely assigned gRNA for subsequent study. Dimension reduction, UMAP, clustering, and pseudo-bulk DE analysis were performed using the package Seurat and EdgeR. Graphs were made in Seurat and ggplot2.
MAGMA analysis
We performed MAGMA analysis using MAGMA version 1.08b39 to evaluate the enrichment for the GWAS risk of several psychiatric disorders (SZ, Neuroticism, ASD, BD, AAD, MDD)3,4,36,86,87,88, as well as the GWAS setting of Crohn’s disease that served as a control set89. Specifically, we started by compiling the MAGMA-required gene annotation data files using a GRCh37/hg19 vcf file. With the gene-SNP annotation file, we then performed gene-level analysis on SNP p-values using the reference SNP data of 1,000 Genomes European panel (g1000_eur, --bfile) and the pre-computed SNP p-values from each disorder’s GWAS data set. The sample size (ncol=) was also directly taken from the column of sample sizes per SNP column of the datasets or extracted from the affiliated README data. Subsequently, the result files (--gene-results) from the gene-level analysis were read in for competitive gene-set analysis (--set-annot), where we used default setting (‘correct=all’) to control for gene sizes in the number of SNPs and the gene density (a measure of within-gene LD). The gene-set analysis produced the output files (.gsa.out) with competitive gene-set analysis results that contained the effect size (BETA) and the statistical significance of the enrichment of each gene set (All DE genes/upregulated/downregulated) for each disorder’s GWAS data set. The heatmap graph was plotted using R package gplots.
Gene set over-representation analysis (ORA) with WebGestalt
We performed ORA using WebGestalt35. Briefly, lists of DE genes from RNA-seq analysis were input into the web-based interface of WebGestalt to output the enriched (or over-represented) GO-terms. All the expressed genes in NGN2-Glut were used as a reference gene list for the enrichment analysis. The GO-terms included three different categories: biological processes, cellular components, and molecular function. The fold of enrichment and the -log10 p-value for the enriched GO-terms were plotted as volcano plots, which were outputted directly from the browser and then polished and re-arranged for clarity purposes in Adobe Illustrator.
SynGO GE and ontology analysis
SynGo GE and ontology analysis were performed as documented31 using the “location” domain and colorized by either “enrichment analysis” or “gene count”, as indicated on the generated Sunburst plots. Briefly, the list of DE genes was used as the SynGO input list, and all the expressed genes in NGN2-Glut neurons were used as the background gene list. The exact gene list per term was further extracted using the select term function and exported for annotation.
Gene set enrichment analysis
Lists of DE genes were divided into three subsets: all dysregulated genes, upregulated and downregulated (all at FDR < 0.05). DE genes were overlapped with gene sets relevant to SZ, including genes implicated by GWAS and the postsynaptic density proteome25,90,91. The GWAS list included two subsets: “Single genes” subset included all GWS loci implicating single genes, and a second, larger subset combining all genes within single-gene or multigenic GWAS loci. A hypergeometric test was used to calculate the extent of over- or under-enrichment of disorder-relevant genes compared to a chance finding. All genes expressed in NGN2-Glut neurons were used as a background set.
Comparison of shRNA KD results between the two cell lines
In order to compare the concordance of shRNA KD behavior between the two independently generated, SNP-edited AA (rs2027349) cell lines (A11, H12), we performed RNA-seq using shRNA targeting EGFP, VPS45, AC244033.2, and C1orf54 on the two cell lines. Three biological replicates were used for each line, and the DE gene list generated within each line was paired with the corresponding DE gene list from the other line to form a union set. The union set of genes of each KD condition was used as the index for the corresponding log2FC values in either line for regression analysis. Pearson’s R was used to show the correlation between the log2FC values from different lines.
Linear regression analysis of gene x gene interaction
To evaluate which individual gene KD and/or their interaction terms could better explain the transcriptional effects of rs2027349 (VPS45) editing in NGN2-Glut, we employed a general linear regression model to correlate the log2FC of DE genes in GG neurons (vs AA) and their log2FC under different shRNA KD conditions in AA neurons. Because AA neurons showed higher expression of the three local genes of interest (VPS45, AC244033.2, and C1orf54) at the rs2027349 locus as a result of rs2027349 editing, if a gene or gene × gene interaction could explain or mediate the transcriptional effect of the SNP editing, we would expect a significant correlation between log2FC of DE genes in GG neurons (vs AA) and their log2FC in a gene KD, or a combination of KD of two genes in AA neurons. Because of the noisy nature of the gene KD experiment that showed an excessive number of DE genes, we only analyzed the list of 1,267 significant DE genes in GG neurons (vs AA). We included individual gene KD and their interaction terms in the following model:
weighted_fit_df <-
- lm(logFC.groupGG ∼
- logFC_VPS45_KD + logFC_lncRNA_KD + logFC_C1orf54_KD +
- (logFC_lncRNA_KD ∗ logFC_C1orf54_KD) +
- (logFC_VPS45_KD ∗ logFC_lncRNA_KD) +
- (logFC_VPS45_KD ∗ logFC_C1orf54_KD),
data = raw_df)
in which raw_df is the data frame that contained all DE expression data. The derived beta coefficient and p-value were used to infer the association of SNP editing effects and individual gene KD and their interaction terms.
Analysis of gene synergistic effects
The experimental design and evaluation of synergy-driving gene expression were generally modelled following the established protocols12,43. Briefly, raw reads from the bulk RNA-seq data (refer to the section [Bulk RNA-seq and differential expression analysis] above) of individual KD from line H12 and the triple KD (i.e., combined KD) in the same line H12 were used and further batch-corrected with the ComBat_seq() function from the R sva package92. A CPM cut-off of 0.4 was applied based on the counts-over-CPM plot to remove lowly-expressed genes, leaving approximately 18,000 genes for subsequent linear model building. The expression data in CPM were log-transformed using the voom() function from the limma package, and weights were computed for heteroscedasticity adjustments. The lmFit() function from limma package was applied to build the linear model. Comparison groups for individual experiments, as well as the additive and synergistic models, were defined using the makeContrasts() function as described below:
contrast_matrix <-
makeContrasts(additive = (C1orf54_KD + lncRNA_KD + VPS45_KD - 3 ∗ single_KD_ctrl),
combinatorial = (combined_KD – combined_KD_control),
synergistic = (combined_KD -
C1orf54_KD - lncRNA_KD - VPS45_KD -
combined_KD_control +
3 ∗ single_KD_ctrl),
levels = voom_design_matrix)
The DEG results were determined by empirical Bayes moderation with eBayes() and decideTests() function from the limma package.
An FDR value of 0.1 was used to calculate the synergy coefficient and percentage of synergistic DEGs43. When performing enrichment analyses (competitive GSEA and gene ontology over-representation analysis), we employed the same curated gene sets that have been successfully applied for analysing the synergistic effects of common SZ risk variants in hiPSC-derived cortical neurons due to the extensive similarity of the experimental model used12. An FDR value of 0.05 was applied to the synergistic effect in gene ontology over-representation analysis.
Micro-C and 3D genome analysis
To assay the 3D chromatin looping in NGN2-Glut at the VPS45 locus, we performed a Micro-C using Dovetail® Targeted Enrichment Pan Promoter Panel (i.e., a promoter Capture Hi-C). About 500 K NGN2-Glut of two iPSC lines (CD0000011 and CD0000012; see sample description) heterozygous for rs2027349 were generated as described previously. DIV/30 NGN2-Glut cells were dissociated with accutase at 37ºC for 45min. After counting, 500K-1M cells were collected as pellet at 3,000 × g for 5 min and snap-frozen in liquid nitrogen. The samples were sent to Dovetail Genomics (Now Cantata Bio; Scotts Valley, CA) for library preparation and sequencing for data analyses. The Micro-C library was prepared using the Dovetail® Micro-C Kit according to the manufacturer’s protocol. Briefly, the chromatin was fixed with disuccinimidyl glutarate (DSG) and formaldehyde in the nucleus. The cross-linked chromatin was then digested in situ with micrococcal nuclease (MNase). Following digestion, the cells were lysed with SDS to extract the chromatin fragments and the chromatin fragments were bound to Chromatin Capture Beads. Next, the chromatin ends were repaired and ligated to a biotinylated bridge adapter followed by proximity ligation of adapter-containing ends. After proximity ligation, the crosslinks were reversed, the associated proteins were degraded, and the DNA was purified and then converted into a sequencing library using Illumina-compatible adaptors. The target enrichment of the Micro-C library was performed using a Dovetail® Target Enrichment Kit (human pan promoter panel) following the user guide. Briefly, a hybridization of biotin-labelled promoter probe panel with Micro-C library pool was performed, followed by binding the hybridized targets to streptavidin beads to capture and enrich the library. The post-capture PCR was performed to ascertain the library quality (both quantity and size distribution between 150 bp to 1 kb with an average fragment length of 375 – 425 bp. Each enriched library was then sequenced to 150 million reads (2×75 bp pair-end) on the Illumina NovaSeq Platform.
For Micro-C data analyses, the sequencing reads in fastq files was mapped to a refence genome using bwa to use the BWA-MEM algorithm at Cantata Bio (Scotts Valley, CA). The mapping pipeline identified ligation junctions and recorded valid pairs of reads in the final bam files after removing PCR duplicates. Overall, we had 48-49% uniquely mapped non-dup reads for both libraries (CD0000011 and CD0000012). CHiCAGO (Capture Hi-C Analysis of Genomic Organisation) was then used to detect chromatin looping interactions. All probes of the same promoter were pooled together as bait and the interactions were called at the bait level. Local chromatin loops were extracted in the form of a contact matrix and visualized using WashU EpiGenome Browser (https://www.epigenomegateway.wustl.edu). For testing the allelic difference of chromatin looping at the rs2027349 site, we compared the number of Micro-C sequence reads of each allele for both lines and a binomial test was used to calculate the statistical significance of the differential allelic distribution.
Quantification and statistical analysis
Unless otherwise specified, Student’s t-test (between two groups) or the Kruskal Wallis test with Dunn’s multiple comparisons and p-value adjustment (more than two groups) was used to determining significance between groups. Samples were assumed to be unpaired and have non-parametric distribution unless otherwise specified. Data were analyzed using R 4.1.1 and GraphPad Prism 9. Results were considered as significant if p < 0.05 (∗: P < 0.05; ∗∗: P < 0.01; ∗∗∗: P < 0.001; ∗∗∗∗: P < 0.0001). All data are reported as mean ± SEM.
Acknowledgments
We thank Molecular Genetics of Schizophrenia (MGS) investigators for collecting samples that were used for deriving hiPSCs. Funding was provided by NIH grants R01AA023797 and R01MH125528 (to Z.P.P.); R01MH110531 (to X.H.); R01MH097216 (to P.P); and R01MH106575, R01MH116281, and R01AG063175 (to J.D.).
Author contributions
S.Z. analyzed the ATAC-seq, bulk, and scRNA-seq data and performed genetic/genomic analyses. S.Z. and H.Z. performed the experiments, analyzed data, and wrote the manuscript. M.P.F. performed immunostaining, neural imaging and analyses, and wrote the manuscript. A.K. helped with neuron differentiation. V.A.B. and L.E.D. performed neural imaging and analyses. M.D.S. and N.H.P. assisted with immunostaining and analysis. A.R.S. helped with clinical phenotypes, data interpretation, and manuscript writing. Z.P.P. guided the neuron differentiation. Y.Z. helped with the CROP-seq data analysis and X.S. helped with the GWAS SNP enrichment analysis. X.H. supervised the data analyses of Y.Z. and X.S. and wrote the manuscript. P.P. supervised the neuronal phenotype analyses and wrote the manuscript. J.D. conceived the study, supervised the experiments and analyses, and wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: August 28, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2023.100399.
Supplemental information
ASoC SNPs were cross-referenced to a different disease GWAS index and their proxy SNPs (r2 ≥ 0.8) and different brain eQTL SNPS, related to Figures 1 and S2E
Genes within 500 kb of the targeting gRNAs were tested for differential expression between ASoC-specific gRNA-targeting cells and non-targeting cells, related to Figures 2F, 2G, and S3D–S3F
GO terms with enrichment FDR <0.05 are listed, related to Figure 2J
PGC3_SZ GWAS-prioritized single genes are also indicated, related to Figures 3B and 3D
Differentially expressed genes (FDR <0.05) were used in WebGestalt for GO-term enrichment analysis and the top 10 most enriched terms were listed, related to Figures 3D, S5A, and S5B
Neurons infected with shRNAs targeting GFP were used as control cells, related to Figures 6A–6C and S7A
Genes that belong to the enriched neural GO terms are indicated, related to Figures 6F–6I
Top 10 enriched GO terms are listed for each gene set, related to Figures 6J and S7F–S7H
Data were extracted from the NGN2-Glut neuron Micro-C experiment, related to Figure 7A
Data and code availability
The ATAC-seq, and RNA-seq data have been deposited to GEO under GSE188491. scRNA CROP-seq data is accessible under SRA SRR16919429. All codes used in the analysis are accessible at https://zenodo.org/record/8180188 (https://doi.org/10.5281/zenodo.8180188).
References
- 1.GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Demontis D., Walters R.K., Martin J., Mattheisen M., Als T.D., Agerbo E., Baldursson G., Belliveau R., Bybjerg-Grauholm J., Bækvad-Hansen M., et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet. 2019;51:63–75. doi: 10.1038/s41588-018-0269-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grove J., Ripke S., Als T.D., Mattheisen M., Walters R.K., Won H., Pallesen J., Agerbo E., Andreassen O.A., Anney R., et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019;51:431–444. doi: 10.1038/s41588-019-0344-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howard D.M., Adams M.J., Clarke T.K., Hafferty J.D., Gibson J., Shirali M., Coleman J.R.I., Hagenaars S.P., Ward J., Wigmore E.M., et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 2019;22:343–352. doi: 10.1038/s41593-018-0326-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mullins N., Forstner A.J., O'Connell K.S., Coombes B., Coleman J.R.I., Qiao Z., Als T.D., Bigdeli T.B., Børte S., Bryois J., et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat. Genet. 2021;53:817–829. doi: 10.1038/s41588-021-00857-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gandal M.J., Zhang P., Hadjimichael E., Walker R.L., Chen C., Liu S., Won H., van Bakel H., Varghese M., Wang Y., et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science. 2018;362:eaat8127. doi: 10.1126/science.aat8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rajarajan P., Borrman T., Liao W., Schrode N., Flaherty E., Casiño C., Powell S., Yashaswini C., LaMarca E.A., Kassim B., et al. Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science. 2018;362:eaat4311. doi: 10.1126/science.aat4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amiri A., Coppola G., Scuderi S., Wu F., Roychowdhury T., Liu F., Pochareddy S., Shin Y., Safi A., Song L., et al. Transcriptome and epigenome landscape of human cortical development modeled in organoids. Science. 2018;362:eaat6720. doi: 10.1126/science.aat6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li M., Santpere G., Imamura Kawasawa Y., Evgrafov O.V., Gulden F.O., Pochareddy S., Sunkin S.M., Li Z., Shin Y., Zhu Y., et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science. 2018;362:eaat7615. doi: 10.1126/science.aat7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nestler E.J., Hyman S.E. Animal models of neuropsychiatric disorders. Nat. Neurosci. 2010;13:1161–1169. doi: 10.1038/nn.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wen Z., Nguyen H.N., Guo Z., Lalli M.A., Wang X., Su Y., Kim N.S., Yoon K.J., Shin J., Zhang C., et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature. 2014;515:414–418. doi: 10.1038/nature13716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schrode N., Ho S.M., Yamamuro K., Dobbyn A., Huckins L., Matos M.R., Cheng E., Deans P.J.M., Flaherty E., Barretto N., et al. Synergistic effects of common schizophrenia risk variants. Nat. Genet. 2019;51:1475–1485. doi: 10.1038/s41588-019-0497-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang S., Zhang H., Zhou Y., Qiao M., Zhao S., Kozlova A., Shi J., Sanders A.R., Wang G., Luo K., et al. Allele-specific open chromatin in human iPSC neurons elucidates functional disease variants. Science. 2020;369:561–565. doi: 10.1126/science.aay3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forrest M.P., Zhang H., Moy W., McGowan H., Leites C., Dionisio L.E., Xu Z., Shi J., Sanders A.R., Greenleaf W.J., et al. Open chromatin profiling in hiPSC-derived neurons prioritizes functional noncoding psychiatric risk variants and highlights neurodevelopmental loci. Cell Stem Cell. 2017;21:305–318.e8. doi: 10.1016/j.stem.2017.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang D., Liu S., Warrell J., Won H., Shi X., Navarro F.C.P., Clarke D., Gu M., Emani P., Yang Y.T., et al. Comprehensive functional genomic resource and integrative model for the human brain. Science. 2018;362:eaat8464. doi: 10.1126/science.aat8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de la Torre-Ubieta L., Stein J.L., Won H., Opland C.K., Liang D., Lu D., Geschwind D.H. The dynamic landscape of open chromatin during human cortical neurogenesis. Cell. 2018;172:289–304.e18. doi: 10.1016/j.cell.2017.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.G.TEx Consortium Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y., Pak C., Han Y., Ahlenius H., Zhang Z., Chanda S., Marro S., Patzke C., Acuna C., Covy J., et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013;78:785–798. doi: 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Christian K.M., Song H., Ming G.L. Using two- and three-dimensional human iPSC culture systems to model psychiatric disorders. Adv. Neurobiol. 2020;25:237–257. doi: 10.1007/978-3-030-45493-7_9. [DOI] [PubMed] [Google Scholar]
- 21.De Los Angeles A., Fernando M.B., Hall N.A.L., Brennand K.J., Harrison P.J., Maher B.J., Weinberger D.R., Tunbridge E.M. Induced pluripotent stem cells in psychiatry: an overview and critical perspective. Biol. Psychiatry. 2021;90:362–372. doi: 10.1016/j.biopsych.2021.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Townsley K.G., Brennand K.J., Huckins L.M. Massively parallel techniques for cataloguing the regulome of the human brain. Nat. Neurosci. 2020;23:1509–1521. doi: 10.1038/s41593-020-00740-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernando M.B., Ahfeldt T., Brennand K.J. Modeling the complex genetic architectures of brain disease. Nat. Genet. 2020;52:363–369. doi: 10.1038/s41588-020-0596-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schizophrenia Working Group of the Psychiatric Genomics Consortium Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Brien H.E., Hannon E., Hill M.J., Toste C.C., Robertson M.J., Morgan J.E., McLaughlin G., Lewis C.M., Schalkwyk L.C., Hall L.S., et al. Expression quantitative trait loci in the developing human brain and their enrichment in neuropsychiatric disorders. Genome Biol. 2018;19:194. doi: 10.1186/s13059-018-1567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jerber J., Seaton D.D., Cuomo A.S.E., Kumasaka N., Haldane J., Steer J., Patel M., Pearce D., Andersson M., Bonder M.J., et al. Population-scale single-cell RNA-seq profiling across dopaminergic neuron differentiation. Nat. Genet. 2021;53:304–312. doi: 10.1038/s41588-021-00801-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Datlinger P., Rendeiro A.F., Schmidl C., Krausgruber T., Traxler P., Klughammer J., Schuster L.C., Kuchler A., Alpar D., Bock C. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods. 2017;14:297–301. doi: 10.1038/nmeth.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie S., Duan J., Li B., Zhou P., Hon G.C. Multiplexed engineering and analysis of combinatorial enhancer activity in single cells. Mol. Cell. 2017;66:285–299.e5. doi: 10.1016/j.molcel.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 30.Yeo N.C., Chavez A., Lance-Byrne A., Chan Y., Menn D., Milanova D., Kuo C.C., Guo X., Sharma S., Tung A., et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat. Methods. 2018;15:611–616. doi: 10.1038/s41592-018-0048-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koopmans F., van Nierop P., Andres-Alonso M., Byrnes A., Cijsouw T., Coba M.P., Cornelisse L.N., Farrell R.J., Goldschmidt H.L., Howrigan D.P., et al. SynGO: an evidence-based, expert-curated knowledge base for the synapse. Neuron. 2019;103:217–234.e4. doi: 10.1016/j.neuron.2019.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dulubova I., Yamaguchi T., Gao Y., Min S.W., Huryeva I., Südhof T.C., Rizo J. How Tlg2p/syntaxin 16 'snares' Vps45. EMBO J. 2002;21:3620–3631. doi: 10.1093/emboj/cdf381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGuire A.L., Gabriel S., Tishkoff S.A., Wonkam A., Chakravarti A., Furlong E.E.M., Treutlein B., Meissner A., Chang H.Y., López-Bigas N., et al. The road ahead in genetics and genomics. Nat. Rev. Genet. 2020;21:581–596. doi: 10.1038/s41576-020-0272-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ang C.E., Ma Q., Wapinski O.L., Fan S., Flynn R.A., Lee Q.Y., Coe B., Onoguchi M., Olmos V.H., Do B.T., et al. The novel lncRNA lnc-NR2F1 is pro-neurogenic and mutated in human neurodevelopmental disorders. Elife. 2019;8:e41770. doi: 10.7554/eLife.41770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J., Vasaikar S., Shi Z., Greer M., Zhang B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017;45:W130–W137. doi: 10.1093/nar/gkx356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trubetskoy V., Pardiñas A.F., Qi T., Panagiotaropoulou G., Awasthi S., Bigdeli T.B., Bryois J., Chen C.Y., Dennison C.A., Hall L.S., et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature. 2022;604:502–508. doi: 10.1038/s41586-022-04434-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.International Schizophrenia Consortium. Purcell S.M., Wray N.R., Stone J.L., Visscher P.M., O'Donovan M.C., Sullivan P.F., Sklar P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirov G., Pocklington A.J., Holmans P., Ivanov D., Ikeda M., Ruderfer D., Moran J., Chambert K., Toncheva D., Georgieva L., et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry. 2012;17:142–153. doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sey N.Y.A., Hu B., Mah W., Fauni H., McAfee J.C., Rajarajan P., Brennand K.J., Akbarian S., Won H. A computational tool (H-MAGMA) for improved prediction of brain-disorder risk genes by incorporating brain chromatin interaction profiles. Nat. Neurosci. 2020;23:583–593. doi: 10.1038/s41593-020-0603-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Leeuw C.A., Mooij J.M., Heskes T., Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015;11:e1004219. doi: 10.1371/journal.pcbi.1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blizinsky K.D., Diaz-Castro B., Forrest M.P., Schürmann B., Bach A.P., Martin-de-Saavedra M.D., Wang L., Csernansky J.G., Duan J., Penzes P. Reversal of dendritic phenotypes in 16p11.2 microduplication mouse model neurons by pharmacological targeting of a network hub. Proc. Natl. Acad. Sci. USA. 2016;113:8520–8525. doi: 10.1073/pnas.1607014113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yi F., Danko T., Botelho S.C., Patzke C., Pak C., Wernig M., Südhof T.C. Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science. 2016;352:aaf2669. doi: 10.1126/science.aaf2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schrode N., Seah C., Deans P.J.M., Hoffman G., Brennand K.J. Analysis framework and experimental design for evaluating synergy-driving gene expression. Nat. Protoc. 2021;16:812–840. doi: 10.1038/s41596-020-00436-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fromer M., Roussos P., Sieberts S.K., Johnson J.S., Kavanagh D.H., Perumal T.M., Ruderfer D.M., Oh E.C., Topol A., Shah H.R., et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 2016;19:1442–1453. doi: 10.1038/nn.4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song M., Yang X., Ren X., Maliskova L., Li B., Jones I.R., Wang C., Jacob F., Wu K., Traglia M., et al. Mapping cis-regulatory chromatin contacts in neural cells links neuropsychiatric disorder risk variants to target genes. Nat. Genet. 2019;51:1252–1262. doi: 10.1038/s41588-019-0472-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y., Song F., Zhang B., Zhang L., Xu J., Kuang D., Li D., Choudhary M.N.K., Li Y., Hu M., et al. The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol. 2018;19:151. doi: 10.1186/s13059-018-1519-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barton N.H., Keightley P.D. Understanding quantitative genetic variation. Nat. Rev. Genet. 2002;3:11–21. doi: 10.1038/nrg700. [DOI] [PubMed] [Google Scholar]
- 48.Moyer C.E., Shelton M.A., Sweet R.A. Dendritic spine alterations in schizophrenia. Neurosci. Lett. 2015;601:46–53. doi: 10.1016/j.neulet.2014.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coyle J.T., Ruzicka W.B., Balu D.T. Fifty Years of Research on Schizophrenia: The Ascendance of the Glutamatergic Synapse. Am. J. Psychiatry. 2020;177:1119–1128. doi: 10.1176/appi.ajp.2020.20101481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Muhtaseb A.W., Duan J. Modeling common and rare genetic risk factors of neuropsychiatric disorders in human induced pluripotent stem cells. Schizophr. Res. 2022 doi: 10.1016/j.schres.2022.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Penzes P., Cahill M.E., Jones K.A., VanLeeuwen J.E., Woolfrey K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011;14:285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duan J., Sanders A.R., Gejman P.V. From Schizophrenia Genetics to Disease Biology: Harnessing New Concepts and Technologies. J. Psychiatr. Brain Sci. 2019;4:e190014. doi: 10.20900/jpbs.20190014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ruderfer D.M., Ripke S., McQuillin A., Boocock J., Stahl E.A., Pavlides J.M.W., Mullins N., Charney A.W., Ori A.P., Loohuis L.M.O., et al. Genomic dissection of bipolar disorder and schizophrenia, including 28 subphenotypes. Cell. 2018;173:1705–1715.e16. doi: 10.1016/j.cell.2018.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramocki M.B., Zoghbi H.Y. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature. 2008;455:912–918. doi: 10.1038/nature07457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Landek-Salgado M.A., Faust T.E., Sawa A. Molecular substrates of schizophrenia: homeostatic signaling to connectivity. Mol. Psychiatry. 2016;21:10–28. doi: 10.1038/mp.2015.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Javierre B.M., Burren O.S., Wilder S.P., Kreuzhuber R., Hill S.M., Sewitz S., Cairns J., Wingett S.W., Várnai C., Thiecke M.J., et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell. 2016;167:1369–1384.e19. doi: 10.1016/j.cell.2016.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gutierrez-Arcelus M., Baglaenko Y., Arora J., Hannes S., Luo Y., Amariuta T., Teslovich N., Rao D.A., Ermann J., Jonsson A.H., et al. Allele-specific expression changes dynamically during T cell activation in HLA and other autoimmune loci. Nat. Genet. 2020;52:247–253. doi: 10.1038/s41588-020-0579-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pak C., Danko T., Mirabella V.R., Wang J., Liu Y., Vangipuram M., Grieder S., Zhang X., Ward T., Huang Y.W.A., et al. Cross-platform validation of neurotransmitter release impairments in schizophrenia patient-derived NRXN1-mutant neurons. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2025598118. e2025598118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Merkle F.T., Eggan K. Modeling human disease with pluripotent stem cells: from genome association to function. Cell Stem Cell. 2013;12:656–668. doi: 10.1016/j.stem.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 60.Zhang S., Sanders A.R., Duan J. Modeling PTSD neuronal stress responses in a dish. Nat. Neurosci. 2022;25:1402–1404. doi: 10.1038/s41593-022-01172-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Masel J., Siegal M.L. Robustness: mechanisms and consequences. Trends Genet. 2009;25:395–403. doi: 10.1016/j.tig.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hockemeyer D., Soldner F., Cook E.G., Gao Q., Mitalipova M., Jaenisch R. A drug-inducible system for direct reprogramming of human somatic cells to pluripotency. Cell Stem Cell. 2008;3:346–353. doi: 10.1016/j.stem.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dull T., Zufferey R., Kelly M., Mandel R.J., Nguyen M., Trono D., Naldini L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998;72:8463–8471. doi: 10.1128/JVI.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Krueger F., Andrews S.R. SNPsplit: Allele-specific splitting of alignments between genomes with known SNP genotypes. F1000Res. 2016;5:1479. doi: 10.12688/f1000research.9037.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yao Y., Guo W., Zhang S., Yu H., Yan H., Zhang H., Sanders A.R., Yue W., Duan J. Cell type-specific and cross-population polygenic risk score analyses of MIR137 gene pathway in schizophrenia. iScience. 2021;24:102785. doi: 10.1016/j.isci.2021.102785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liland K.H., Almøy T., Mevik B.H. Optimal choice of baseline correction for multivariate calibration of spectra. Appl. Spectrosc. 2010;64:1007–1016. doi: 10.1366/000370210792434350. [DOI] [PubMed] [Google Scholar]
- 75.Victor J.D., Purpura K.P. Nature and precision of temporal coding in visual cortex: a metric-space analysis. J. Neurophysiol. 1996;76:1310–1326. doi: 10.1152/jn.1996.76.2.1310. [DOI] [PubMed] [Google Scholar]
- 76.Xu J., Carter A.C., Gendrel A.V., Attia M., Loftus J., Greenleaf W.J., Tibshirani R., Heard E., Chang H.Y. Landscape of monoallelic DNA accessibility in mouse embryonic stem cells and neural progenitor cells. Nat. Genet. 2017;49:377–386. doi: 10.1038/ng.3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.van de Geijn B., McVicker G., Gilad Y., Pritchard J.K. WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat. Methods. 2015;12:1061–1063. doi: 10.1038/nmeth.3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bernstein B.E., Stamatoyannopoulos J.A., Costello J.F., Ren B., Milosavljevic A., Meissner A., Kellis M., Marra M.A., Beaudet A.L., Ecker J.R., et al. The NIH roadmap epigenomics mapping consortium. Nat. Biotechnol. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liao Y., Smyth G.K., Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019;47:e47. doi: 10.1093/nar/gkz114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wen X. Molecular QTL discovery incorporating genomic annotations using Bayesian false discovery rate control. Ann. Appl. Stat. 2016;10 [Google Scholar]
- 81.Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S., et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bray N.L., Pimentel H., Melsted P., Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 83.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mayfield R.D., Harris R.A., Schuckit M.A. Genetic factors influencing alcohol dependence. Br. J. Pharmacol. 2008;154:275–287. doi: 10.1038/bjp.2008.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gaiteri C., Sibille E. Differentially expressed genes in major depression reside on the periphery of resilient gene coexpression networks. Front. Neurosci. 2011;5:95. doi: 10.3389/fnins.2011.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nagel M., Jansen P.R., Stringer S., Watanabe K., de Leeuw C.A., Bryois J., Savage J.E., Hammerschlag A.R., Skene N.G., Muñoz-Manchado A.B., et al. Meta-analysis of genome-wide association studies for neuroticism in 449,484 individuals identifies novel genetic loci and pathways. Nat. Genet. 2018;50:920–927. doi: 10.1038/s41588-018-0151-7. [DOI] [PubMed] [Google Scholar]
- 87.Stahl E.A., Breen G., Forstner A.J., McQuillin A., Ripke S., Trubetskoy V., Mattheisen M., Wang Y., Coleman J.R.I., Gaspar H.A., et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet. 2019;51:793–803. doi: 10.1038/s41588-019-0397-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhou Z., Yuan Q., Mash D.C., Goldman D. Substance-specific and shared transcription and epigenetic changes in the human hippocampus chronically exposed to cocaine and alcohol. Proc. Natl. Acad. Sci. USA. 2011;108:6626–6631. doi: 10.1073/pnas.1018514108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu J.Z., van Sommeren S., Huang H., Ng S.C., Alberts R., Takahashi A., Ripke S., Lee J.C., Jostins L., Shah T., et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015;47:979–986. doi: 10.1038/ng.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pardiñas A.F., Holmans P., Pocklington A.J., Escott-Price V., Ripke S., Carrera N., Legge S.E., Bishop S., Cameron D., Hamshere M.L., et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 2018;50:381–389. doi: 10.1038/s41588-018-0059-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bayés A., van de Lagemaat L.N., Collins M.O., Croning M.D.R., Whittle I.R., Choudhary J.S., Grant S.G.N. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat. Neurosci. 2011;14:19–21. doi: 10.1038/nn.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang Y., Parmigiani G., Johnson W.E. ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2020;2:lqaa078. doi: 10.1093/nargab/lqaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
ASoC SNPs were cross-referenced to a different disease GWAS index and their proxy SNPs (r2 ≥ 0.8) and different brain eQTL SNPS, related to Figures 1 and S2E
Genes within 500 kb of the targeting gRNAs were tested for differential expression between ASoC-specific gRNA-targeting cells and non-targeting cells, related to Figures 2F, 2G, and S3D–S3F
GO terms with enrichment FDR <0.05 are listed, related to Figure 2J
PGC3_SZ GWAS-prioritized single genes are also indicated, related to Figures 3B and 3D
Differentially expressed genes (FDR <0.05) were used in WebGestalt for GO-term enrichment analysis and the top 10 most enriched terms were listed, related to Figures 3D, S5A, and S5B
Neurons infected with shRNAs targeting GFP were used as control cells, related to Figures 6A–6C and S7A
Genes that belong to the enriched neural GO terms are indicated, related to Figures 6F–6I
Top 10 enriched GO terms are listed for each gene set, related to Figures 6J and S7F–S7H
Data were extracted from the NGN2-Glut neuron Micro-C experiment, related to Figure 7A
Data Availability Statement
The ATAC-seq, and RNA-seq data have been deposited to GEO under GSE188491. scRNA CROP-seq data is accessible under SRA SRR16919429. All codes used in the analysis are accessible at https://zenodo.org/record/8180188 (https://doi.org/10.5281/zenodo.8180188).