

Abstract
Our knowledge of the diversity of eukaryotic viruses has recently undergone a massive expansion. This diversity could influence host physiology through yet unknown phenomena of potential interest to the fields of health and food production. However, the assembly processes of this diversity remain elusive in the eukaryotic viromes of terrestrial animals. This situation hinders hypothesis-driven tests of virome influence on host physiology. Here, we compare taxonomic diversity between different spatial scales in the eukaryotic virome of the mosquito Culex pipiens. This mosquito is a vector of human pathogens worldwide. The experimental design involved sampling in five countries in Africa and Europe around the Mediterranean Sea and large mosquito numbers to ensure a thorough exploration of virus diversity. A group of viruses was found in all countries. This core group represented a relatively large and diverse fraction of the virome. However, certain core viruses were not shared by all host individuals in a given country, and their infection rates fluctuated between countries and years. Moreover, the distribution of coinfections in individual mosquitoes suggested random co-occurrence of those core viruses. Our results also suggested differences in viromes depending on geography, with viromes tending to cluster depending on the continent. Thus, our results unveil that the overlap in taxonomic diversity can decrease with spatial scale in the eukaryotic virome of C. pipiens. Furthermore, our results show that integrating contrasted spatial scales allows us to identify assembly patterns in the mosquito virome. Such patterns can guide future studies of virome influence on mosquito physiology.
Keywords: virome, mosquito, Culex pipiens, metagenomics
Introduction
An impressive expansion in the known diversity of viruses has recently taken place, thanks to metagenomics (Kuhn et al. 2019). This expansion has been followed by major improvements in the emerging field of community ecology of viruses. For example, viral communities or viromes have been shown to play a major role in the regulation of the carbon cycle in the oceans (Brum et al. 2016) or in bacteriome dynamics (Manrique, Dills, and Young 2017; Huang et al. 2021) (here virome is defined as the community of viruses infecting a host or a community of hosts as in Harvey and Holmes 2022). However, current knowledge is largely biased toward the viromes of unicellular hosts. Our understanding of the ecology of the eukaryotic virome in terrestrial animals largely lags behind (Anthony et al. 2015; Stulberg et al. 2016). For example, we still know little about the potential differences in virome assembly due to environmental filtering (abiotic factors like climate), biotic filtering (ecological interactions among species like competition), and stochastic processes (e.g. random events during the spread of viruses in a host population) (Anthony et al. 2015; Shi et al. 2017; Bergner et al. 2019; Pettersson et al. 2019; Arze et al. 2021) (the term assembly here defines both the taxonomical diversity and its abundance in a virome as used in community ecology (Kraft and Ackerly 2014)). This situation severely hampers, among others, studies on the influence of the eukaryotic virome on the physiology of those hosts.
The eukaryotic virome of mosquitoes has received considerable attention due to its potential influence on the transmission of human pathogens with a huge impact on health worldwide (Almeida et al. 2021). Similarly to the situation in other eukaryotic viromes, most studies on the mosquito virome have focused on describing new viruses (e.g. Fauver et al. 2016; Shi et al. 2016, 2017; Atoni et al. 2018; Sadeghi et al. 2018). These viruses are mostly RNA viruses, the dominant group in terrestrial eukaryotes (Wolf et al. 2018), and are thought to be mosquito commensals that do not infect vertebrates (Elrefaey et al. 2020). Moreover, they are often supposed to be vertically transmitted although other transmission modes have been observed (Hall et al. 2016; Gaye et al. 2020; Ye et al. 2020; Altinli, Schnettler, and Sicard 2021). Interestingly, a few mosquito-specific viruses have been shown to influence the infection rate of mosquitoes by human pathogens under laboratory conditions (Bolling et al. 2012; Goenaga et al. 2015; Hall-Mendelin et al. 2016; Baidaliuk et al. 2019; Ye et al. 2020; White et al. 2021) and, very recently, in the field (Olmo et al. 2023).
The improvement in the known viral diversity in mosquitoes contrasts with the paucity of studies on its assembly patterns. For example, little is known about the potential influence of spatial scale on virome assembly (Shi et al. 2019, 2020), a main question in community ecology (Agrawal et al. 2007; Logue et al. 2011). The study of different spatial scales is particularly important to determine the distribution of viral diversity among individuals or populations of a host species. Two main scenarios can be envisaged concerning diversity distribution. In the first scenario, deterministic processes lead to assemblies with viruses that are shared by all host individuals or populations, which is a large overlap in taxonomic diversity between viromes. For example, the specificity of the interactions between host and viral proteins can allow high transmissibility of certain viruses and favor their spread to all individuals of a host species. The group of viruses found in all individuals or populations is usually called the core virome (Shi et al. 2019; Coatsworth et al. 2022; Konstantinidis et al. 2022). The second scenario involves the dominance of random processes in virome assembly and, thus, a limited overlap in taxonomic diversity among the viromes of hosts. For example, random events can influence virus spread in a host population, leading to differences in virome diversity among individuals in the population (Dolan, Whitfield, and Andino 2018). Disentangling which scenario takes place is important to fully understand virome assembly in mosquitoes and its potential impact on pathogen transmission. For example, the identification of the core taxa is often a key step in the study of microbial communities since core taxa are likely to influence host physiology (Shade and Handelsman 2012; Berg et al. 2020). However, estimating the overlap in taxonomic diversity, and thus which viruses belong to the core, is not trivial due to potential differences at different scales. For example, all hosts in a given region may share certain viruses, thus suggesting a core virome. Nevertheless, enlarging the analysis to other regions may find low infection rates, even absence, of the same viruses. To date, several studies have explored the potential existence of a core virome in different mosquitoes (Shi et al. 2017, 2019; Pettersson et al. 2019; He et al. 2021; Parry, James, and Asgari 2021; Thongsripong et al. 2021; Coatsworth et al. 2022; Feng et al. 2022; Konstantinidis et al. 2022). Nevertheless, most current studies have analyzed a single spatial scale (Shi et al. 2017; Pettersson et al. 2019; He et al. 2021; Thongsripong et al. 2021; Coatsworth et al. 2022; Feng et al. 2022; Konstantinidis et al. 2022). Moreover, mosquito numbers in those studies were usually low (i.e. below fifty individuals) (Pettersson et al. 2019; Shi et al. 2019; Thongsripong et al. 2021; Coatsworth et al. 2022), a situation that can lead to a limited exploration of the viral diversity. Overall, these limits in experimental designs hamper to determine whether a core virome takes place at different spatial scales (e.g. within and between host populations) and which viruses belong to the core.
Here, we analyze the influence of spatial scale on the diversity of eukaryotic viruses associated with the common house mosquito Culex pipiens Linnaeus, 1758. This mosquito is the main vector of important human pathogens, like West Nile virus. Moreover, C. pipiens has nearly a worldwide distribution, probably through introductions from Africa and Europe (Fonseca et al. 2004). We have analyzed the virome of C. pipiens both between and within five countries situated around the Mediterranean Sea and in Africa and Europe (Fig. 1). To ensure an in-depth exploration of viral diversity, we analyzed between hundreds and thousands of individuals per country and from the main habitats of C. pipiens in the study area. Using a blend of meta-transcriptomics (Shi et al. 2016, 2017; Shi, Zhang, and Holmes 2018) and polymerase chain reaction (PCR) analyses, we revealed that the overlap in taxonomic diversity decreased with decreasing spatial scale. That is, a group of viruses was found in the five countries. However, a limited overlap in taxonomic diversity took place between mosquitoes within a country, along with random coinfection patterns in individual mosquitoes. Moreover, our results suggest differences in virome assembly between European and African countries. Phylogenetic and beta-diversity analyses suggested that the observed geographic structuration was probably not due to strict isolation of virus populations but rather to differences in virus relative abundancy.
Figure 1.
An overview of the mosquito sampling sites around the Mediterranean Basin. (A–E) Magnifications of the sampling regions in each country. The panels are provided here to allow an overview of the sampling sites. A larger version of the panel, with scale bars, is provided in Figure S6. The detailed site information can be found in Table S4. (A) Egypt; (B) Spain; (C) Tunisia; (D) France; and (E) Italy.
Materials and Methods
Mosquito sampling
Mosquitoes were captured in five countries (Egypt, France, Italy, Spain, and Tunisia; Fig. 1). The genetics of the C. pipiens complex has been shown to be relatively homogeneous in the region (Aardema et al. 2020). In each country, mosquitoes were sampled in several sites (Fig. S1 and Table S1). Sites were representative of habitats of C. pipiens in the study region, including marshlands, riverine woodlands, crop fields, or urbanized areas. Sites were grouped into two categories based on habitat anthropization (Table S1). The two categories were broadly defined to take into account differences in the diversity and availability of habitats between countries. A ‘high’ anthropization level was broadly defined as a peri-urban site, located less than 500 m from the perimeter of a densely populated area. Similarly, a ‘low’ level was largely defined as any agricultural or natural site with a low population density.
Several mosquito collections were carried out in each site between April and October 2015. After overnight trapping, mainly using CO2-baited CDC-type traps, mosquitoes were identified to the species level on a chill table using morphological determination keys. Non-engorged females of C. pipiens sensu lato were distributed in pools of around thirty individuals (mean pool size = 24 individuals, Table S2) and stored at −80 °C until further processing. An additional sampling was carried out in France in 2018. The latter sampling followed the same protocol as in 2015 but for pool size. In the former collection, mosquitoes were either dispatched in pools of six individuals or individualized (Table S2).
Library preparation and sequencing
Mosquito viromes were analyzed using meta-transcriptomics (Shi et al. 2016). Ten libraries (one library per country and anthropization level) were prepared for Illumina sequencing as previously described (Cheval et al. 2011). Briefly, mosquito pools were homogenized in ice-cold 1X phosphate buffered saline (Tissue Lyser; Qiagen). An aliquot of 150 µl was used for total RNA isolation with the Nucleospin RNA virus kit (Macherey Nagel). Ten RNA samples were generated by pooling aliquots of RNA extractions from all the pools from the same country and anthropization level. The ten samples were retro-transcribed to cDNA (SuperScript IV reverse transcriptase; Invitrogen) with random hexamers. Then, cDNA was amplified using multiple displacement amplification with phi29 polymerase and random hexamers. Libraries were prepared with the TruSeq DNA Nano Library Prep kit (Illumina). Libraries were sequenced at an expected depth of 80 million reads on a HiSeq2000 (Illumina) in a 150-bp single-read format. Library preparations and sequencing were outsourced to DNAVision (Charleroi, Belgium).
Generation of viral operational taxonomic units
We used an updated version of the SnakeVir bioinformatics pipeline (version 1.2.0) to identify potential viral sequences from reads (Gil et al. 2021). Briefly, library adapters and low-quality sequences in reads were removed with Cutadapt 1.6 (quality cutoff = 30, minimum read length = 60) (Martin 2011). Reads from rRNA sequences from Diptera and bacteria were (Buchfink et al. 2015) then removed through mapping with BWA 0.7.15 (default options) (Li and Durbin 2009) against SILVA rRNA bases (Gil et al. 2021). The remaining reads were de-novo assembled with Megahit version 1.1.2 (default options) (Li et al. 2015). The resulting contigs were used in a second de-novo assembly with CAP3 (Huang and Madan 1999). A contig was considered a virus-like sequence if a viral sequence was found as the first best hit by Diamond after comparison with the NCBI nr database (release 234, 10/2019; e-value cutoff = 10–3). Then, virus-like contigs were screened for sequences potentially derived from endogenous viral elements (EVEs) using BLASTn against the nt database of the National Center for Biotechnology Information (NCBI) (release 249, 6/2022; e-value cutoff = 10–3). Contigs with a non-viral best hit with a coverage above 25 per cent were considered as potential EVEs and removed.
The number of reads per virus-like contig and library was quantified through mapping with BWA 0.7.15 (Li and Durbin 2009). Duplicate reads had been previously removed with the markdup tool in Samtools (Li et al. 2009). Contigs sharing the same best hit were grouped into a viral operational taxonomic unit (vOTU) as previously described (Shi et al. 2019; Gil et al. 2021). Names of vOTUs were generated by combining the best-hit name and the mean percent identity at the amino-acid level of the contigs with the best hit. The percent identity was placed at the end of the best-hit name after a double underscore. The vOTUs were considered to include the virus species of their best hit if their contigs had mean percent identities above 90 per cent against their best hit. Spaces and hyphens in vOTU names were replaced by dots and underscores, respectively, to facilitate analysis in R software.
The taxonomy of vOTUs was generated with SnakeVir v1.2.0 (Gil et al. 2021). Since many best hits had not yet been assigned to a family, the vOTUs were grouped into an arbitrary taxonomic level, here called ‘cluster’, similar to the family rank (Gil et al. 2021). The cluster was defined as the family of the best hit whenever available in GenBank. For those best hits without a full taxonomic classification, the most likely family was defined from the associated literature and BLAST searches. Clusters were named using the family name without the ‘viridae’ suffix. If analyses failed to indicate a likely family for a given vOTU, the cluster was generated as the name of the most probable order of the vOTU and replacing the ‘virales’ suffix with ‘_unclass’.
Validation of vOTUs
Two arbitrary detection thresholds were applied to the list of vOTUs as previously described (Gil et al. 2021). The first threshold was applied to each library and involved removing all vOTUs with less than ten reads in each library. After this step, the resulting dataset was processed to match the second threshold. In the second threshold, the sum of reads of a given vOTU in all libraries had to be at least 100 reads to consider the vOTU as detected. Any vOTU with less than 100 reads in total was removed from the dataset. We next validated that taxon conservation was not due to contamination during sample processing and sequencing. To do so, the vOTUs in the ten libraries of this study were compared to those found in six libraries sequenced in parallel and processed in the same laboratory for another project. These libraries were derived from adult females of the mosquito Aedes vexans from Senegal and have been previously described (Gil et al. 2021). This comparison found twenty-five vOTUs in common. Twelve vOTUs were not considered potential contaminants because they had a mean read abundance at least two logs higher in the C. pipiens libraries. None of the remaining vOTUs was part of the most prevalent vOTUs in the C. pipiens libraries or was present in all libraries.
Analysis of published viromes of C. pipiens
A search for articles with robust characterizations of the virome of C. pipiens was done in PubMed (October 2021). The criteria defined to select datasets included (1) the minimum number of mosquito individuals was set to 100, (2) the minimal sequencing depth was set to 1 million reads per library, and (3) the virome diversity should be dominated by viruses from arthropods. Four studies were selected for the analysis (Bennouna et al. 2019; Pettersson et al. 2019; Stanojevic et al. 2020; He et al. 2021). All studies analyzed populations in a single country (China, Morocco, Serbia, or Sweden; Table S3). There were important methodological differences among studies (Table S3). For example, the studies widely differed in mosquito number, library preparation, or sequencing depth. Moreover, the studies had mosquito numbers, sequencing depths, or a yield in virus-like reads largely below the ones in this study. Hence, we limited the comparison to the presence of the core taxons found in our dataset.
PCR detection of selected virus taxa
We developed quantitative reverse transcription polymerase chain reactions using SYBR green chemistry for the detection of several vOTUs. First, we selected vOTUs in four core clusters—the Mesoni, Ifla, Nege, and Reo clusters—for the analysis. We also selected vOTUs in two additional clusters present in nine libraries, the Chu and Bunya_unclass clusters. At least three primer pairs were designed and tested per vOTU. Primer design was done with LightCycler Probe design Software 2.0 (Roche) and based on the contigs of each vOTU. Primer pairs were used in RT-qPCR reactions using mosquito pools that had been shown to be either positive or negative toward the targeted vOTU (three pools of each type). For each vOTU, we selected the primer pair with the lowest cycle threshold (Ct). Moreover, primer pairs were selected only if (1) they provided Ct values below 30 when tested on positive pools, (2) the resulting amplicon derived from the targeted vOTU as shown with amplicon sequencing, and (3) no amplification was observed in negative pools. The PCR output was not satisfactory in terms of specificity and sensibility for vOTUs in the Reo (not shown), and vOTUs of this cluster were not included in the analysis. Moreover, the PCR design for the most prevalent vOTU in the Nege was not satisfactory (Dezidougou.virus__98; not shown) and was discarded from the analysis. A satisfactory PCR design was obtained for Culex.negev_like.virus.1__99, and the second most prevalent vOTU in the Nege. Culex.negev_like.virus.1__99 was thus included in the analysis. The final set of five vOTUs and their prevalence among libraries are presented in Fig. S2. The primer pairs can be found in Table S4.
RT-qPCR conditions were as follows: first, total RNA was reverse transcribed using the RevertAid First Strain cDNA kit (ThermoFisher) and random primers following the manufacturer’s instructions. Then, quantitaive PCRs (qPCR) were carried out with the Brilliant III-Sybrgreen QPCR Master Mix (Agilent) in a total volume of 20 µl consisting of 2 µl of cDNA template, 0.3 µM of the forward and reverse primers, 10 µl of Master Mix 2X, and RNase-free H2O. PCR parameters were 95°C for 10 min, 40 cycles of 95°C for 20 s, 60°C for 20 s (55° for Culex.Bunyavirus.2__96), and a melting curve analysis involving 95°C for 10 s, 65°C for 60 s, and 97°C for 1 s. Amplicons were Sanger sequenced (Genewiz, Germany) whenever an unclear melting curve was observed.
The five RT-qPCR designs were used to screen a collection of mosquitoes from six countries (Table S2). In total, 359 pools (7,906 mosquitoes) were screened with each RT-qPCR to estimate the infection rate per country. Moreover, 184 mosquitoes were individually screened with each RT-qPCR to estimate coinfection rates in individuals. The statistical analyses used to estimate both the infection and coinfection rates are detailed later.
Phylogenetic analysis
First, vOTUs were selected for phylogenetic analysis only if detected in at least two European countries and the two African countries. Moreover, for a given vOTU, contigs from the different countries had to encompass the same genomic region with at least 10X read coverage over 500 bp. This analysis selected three vOTUs: Alphamesonivirus.1__97, Culex.Iflavi_like.virus.4__96, and Culex.Bunyavirus.2__96. The contigs of those vOTUs had mean identities at the nucleotide level above 90 per cent with their best hits, and the same synteny of open reading frames was found in the genome of their best hit. The contigs in the vOTUs were thus considered as belonging to the same species as their best hits.
The contigs selected for Alphamesonivirus.1__97 and Culex.Iflavi_like.virus.4__96 represented nearly full genomes. The contigs of Culex.Bunyavirus.2__96 represented a nearly complete L segment. In addition to those contigs, homologous sequences from different countries found in GenBank were also included. The search for homologous sequences unveiled that sequences from Culex picorna-like virus 1 and Aedes Ifla-like virus belong to the same species as Culex Iflavi-like virus 4. Similarly, sequences from Culex Bunya-like virus and Bunyanwera environmental sample belong to the same species as Culex Bunyavirus 2.
Alignments were generated with Mega X using the ClustalW algorithm for each group of sequences (Kumar et al. 2018). All ambiguously aligned regions were removed using TrimAL (Capella-Gutiérrez, Silla-Martínez, and Gabaldón 2009). The alignments had 16,000, 8,800, and 7,000 nucleotide positions for the sequences from Alphamesonivirus 1, Culex Iflavi-like virus 4, and Culex Bunyavirus 2, respectively. For each alignment, IQ-Tree (v.1.6.1) (Minh et al. 2020) and its module modelfinder were used to determine the best-fit model of nucleotide substitution. The TN+F+R3 model (Tamura and Nei substitution model with empirical base frequency and gamma distributions for three categories) was selected for each alignment. Maximum-likelihood phylogenetic trees were then produced with 1,000 bootstrapping replications. Trees were visualized and annotated with the R package ggtree (Yu et al. 2017).
Statistical analyses
Most statistical analyses are described along with the results and were conducted using R version 3.5.1. The associated figures were generated with the ggplot2 package. Analyses of alpha and beta diversities were carried out, unless specifically stated, at the cluster level to avoid potential biases in vOTU identification. The Jaccard and Bray-Curtis dissimilarities were estimated using the vegan package (Oksanen et al. 2019) from count data normalized using Total Sum Normalization (Mcknight et al. 2019). Rarefaction curves were generated with the vegan package (Oksanen et al. 2019). Maximum-likelihood estimates of infection rates per virus, country, and year were obtained with the binGroup package (Biggerstaff 2008). A probabilistic analysis of virus coinfection in individual mosquitoes was done with the coocur package (Griffith, Veech, and Marsh 2016).
Results
Overlap in taxonomic diversity in the virome of C. pipiens around the Mediterranean Basin
We used meta-transcriptomics (Shi et al. 2016) to characterize the eukaryotic virome of C. pipiens in five countries in Africa and Europe (Fig. 1). Two libraries were generated per country, each comprising 711 females on average (min/max = 105/2,475; Table S5). To maximize the exploration of viral diversity in each country, the two libraries were derived from sites with either a high or a low anthropization level (Table S5). Meta-transcriptomic analysis of the ten libraries provided 715 million reads (Table S5). Among them, 41 million reads (5.7 per cent) mapped to virus-like sequences associated with 324 vOTUs (Shi et al. 2019). The number of virus-like reads significantly differed between libraries (Wilcoxon signed-rank exact test, P = 0.001). We did not detect a significant effect of raw reads or mosquito number on the yield of virus-like reads (Spearman’s rank correlation, P > 0.076). A rarefaction curve analysis further supported that sampling effort, in terms of viral reads, was adequate even in libraries with lower mosquito numbers (Fig. S3).
After quality filtering, the dataset comprised 201 vOTUs, distributed over 44 clusters (an arbitrary taxon similar to a family, see Materials and Methods), and 99.98 per cent of the total virus-like reads. The dataset was dominated by vOTUs related to viruses detected in arthropods (167 vOTUs, 99.90 per cent of the total virus-like reads). We were only interested in vOTUs potentially infecting C. pipiens. We thus discarded the vOTUs related to non-arthropod hosts. None of the non-arthropod vOTUs was found in more than six libraries. The final dataset consisted of 167 vOTUs and 36 clusters, mainly RNA viruses.
Seventy-seven percent of the vOTUs had identities at the amino-acid level below 90 per cent with their best hits (i.e. with the accession with the lowest e-value found by Diamond in GenBank) (Fig. S4). This situation suggests the presence of numerous new viruses, as usually observed in studies on the mosquito virome (Pettersson et al. 2019; Shi et al. 2019, 2020). Selected sequences were submitted to GenBank (BioProject PRJNA806751). Sequence submission was done with the Gsub tool, a new tool allowing easy annotation and submission of virus contigs (Supplementary Annex 1). Most names of new virus species were provided by children participating in the 2021 Science Days in Montpellier (France). A detailed description of this diversity is beyond the scope of this study and was not further explored here.
Cluster overlap between libraries and countries is presented in Fig. 2A. Ten clusters were present in all the libraries (27 per cent of the clusters; hereafter referred to as ‘core clusters’; Fig. 2A). Moreover, half of the clusters were present in all countries (eighteen clusters, Fig. 2A). Core clusters tended to rank higher in terms of read abundance, but some of them had low frequencies in most libraries (i.e. <0.1 per cent, see clusters Mononega_unclass and Picorna_unclass in Fig. 2B). More generally, readdistribution among clusters was rather heterogeneous, with a few clusters providing most of the reads both globally and in each library (Fig. 2B).
Figure 2.
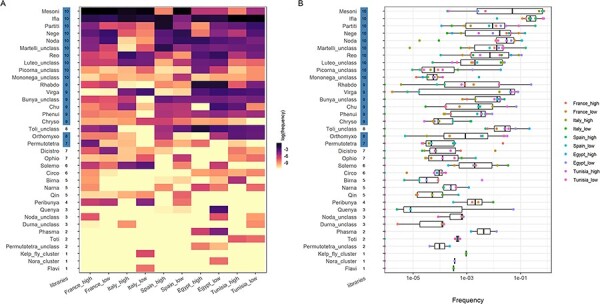
The cluster diversity and abundance in the eukaryotic virome of Culex pipiens in the Mediterranean Basin. (A) A heatmap showing the relative abundance of clusters among libraries. Libraries are shown on the x-axis with the European countries on the left and the African ones on the right to facilitate the comparison. The terms ‘high’ and ‘low’ in the library names stand for the level of habitat anthropization. Clusters are ranked on the y-axis in a descending order, first by the number of libraries with the presence and, then, by the total read number. The number of libraries in which a given cluster was found is shown on the right of the cluster name. This number appears highlighted if the cluster was detected in all countries. Tile color stands for relative abundancy in reads in a given library: the darker the tile color, the higher the relative frequency of the cluster. (B) Boxplots with relative abundance of clusters. Clusters are ranked as in (A). Dots indicate the frequency in a given library and the dot color stands for the library.
We also observed an overlap of vOTUs among libraries (Fig. 3, Table S6). Seven core clusters included a vOTU that was present in all libraries (hereafter referred to as core vOTUs, Fig. 3, Table S6). The other core clusters (Partiti, Mononega_unclass and Nege) had a vOTU present in at least eight libraries (Fig. 3, Table S6).
Figure 3.
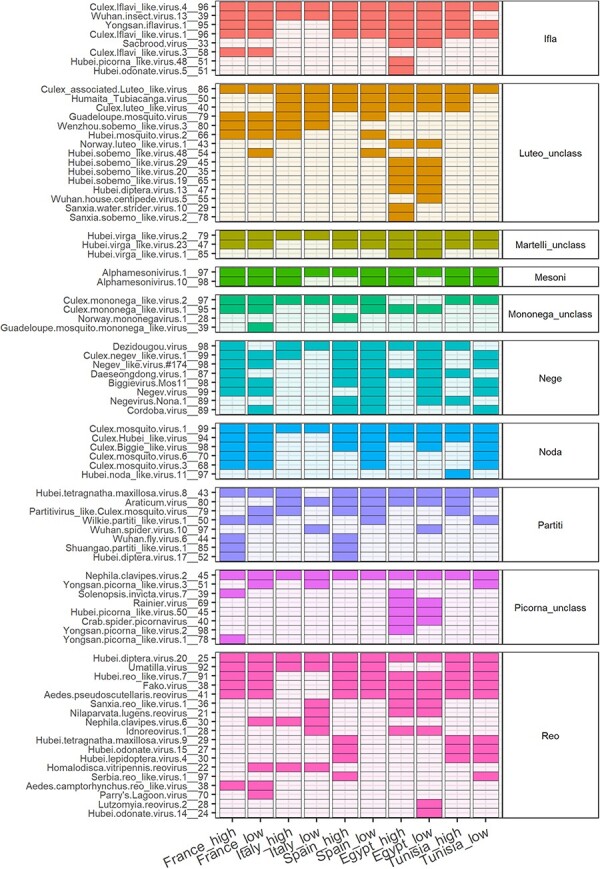
The prevalence of vOTUs from core clusters in the eukaryotic virome of Culex pipiens among libraries. The names of vOTUs and the associated clusters appear on the left and right of the heatmap, respectively. Tiles with darker color indicate the detection in a given library. Tile color stands for the cluster to facilitate visualization.
Presence of core taxa of the virome of C. pipiens in other regions and mosquito species
We performed a literature analysis to investigate whether the core taxa identified in our study were associated with C. pipiens in other regions and other core viromes of mosquitoes. First, our dataset was compared to four studies on the virome of C. pipiens from China, Morocco, Serbia, and Sweden (Bennouna et al. 2019; Pettersson et al. 2019; Stanojevic et al. 2020; He et al. 2021). Despite important methodological differences between studies (Table S3), we observed numerous core clusters shared among countries (Fig. S5). All core clusters were detected in Morocco and Sweden. Seven and six out of ten clusters were found in the studies from China and Serbia, respectively. An overlap of core vOTUs was also observed among studies (Fig. S5). All core clusters, except for the Partiti cluster, had vOTUs shared in other countries (Fig. S5).
Next, we searched the literature for studies on the core virome in mosquitoes over a large geographical scale (i.e. several continents). We found only one study (Shi et al. 2020). In that study, Shi et al. compared the viromes from field populations of Aedes aegypti from North America, Africa, Asia, and Oceania. The core virome in A. aegypti did not fully overlap with that observed in C. pipiens here. Only seven out of twenty-three core clusters were shared between the two mosquitoes (Fig. 4).
Figure 4.
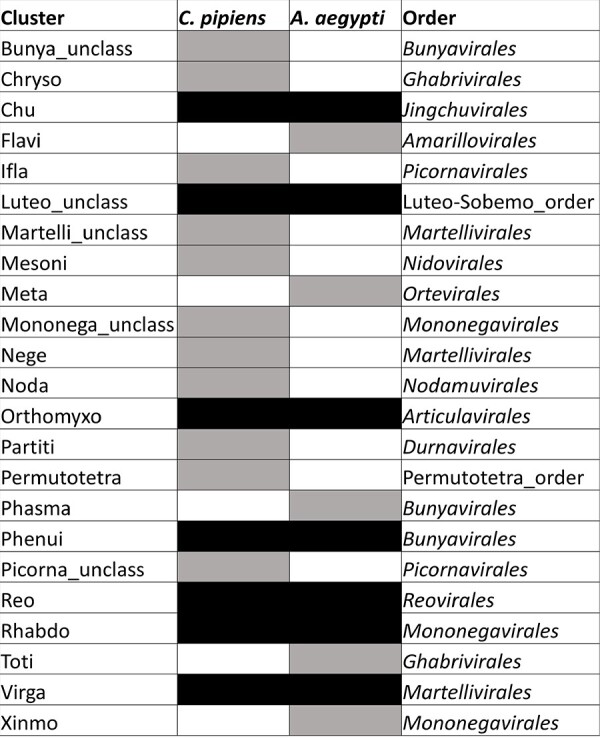
Core clusters in the viromes of Culex pipiens and Aedes aegypti. Clusters present in all countries in our dataset (n = 18) were compared to those found associated with A. aegypti in at least two continents (n = 12; data from Shi et al. 2020; microorganisms). Gray tiles indicate the detection in a single mosquito species and black tiles indicate the detection in the two species. Since the cluster does not always match a family, the order of each cluster is provided in the last column to facilitate the taxonomic placement of clusters.
Limited overlap in taxonomic diversity within countries
We explored the distribution of a subset of the virome in C. pipiens at a different spatial scale, that of the country. We estimated the infection rates of five vOTUs, including two core vOTUs, in a collection of mosquito pools from each country (359 pools, 7,906 mosquitoes in total; Table S2). None of the vOTUs reached an infection rate above 10 per cent in any of the countries (Fig. 5A). We had expected high infection rates of core viruses as previously observed in other mosquito species with a similar approach (80–100 per cent infection rates) (Shi et al. 2019). Thus, we extended the analysis to another country to further explore differences in infection rates. We estimated the infection rate of a core virus, Alphamesonivirus.1__97, in forty-four pools (1,300 females; Table S2) collected in Morocco in 2016 (Bennouna et al. 2019). The infection rate lied below 10 per cent in the Moroccan samples, like in the other five countries in 2015 (Fig. 5B).
Figure 5.
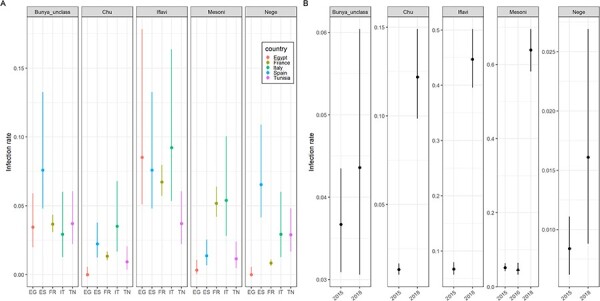
Infection rates of five vOTUs in Culex pipiens females. The five vOTUs were Culex.Bunyavirus.2__98 (Bunya_unclass), Culex.mosquito.virus.4__97 (Chu), Culex.Iflavi_like.virus.4__96 (Iflavi), Alphamesonivirus.1__97 (Mesoni), and Culex.negev_like.virus.1__99 (Nege). Infection rates are grouped in panels depending on the cluster of the vOTU. Dots indicate the maximum-likelihood estimates of the infection rates (infected females per 100 females) and vertical segments stand for the 95 per cent confidence intervals. Estimates were obtained using pools of mosquitoes. (A) Infection rates of the five vOTUs in the five countries sampled in 2015. EG: Egypt, ES: Spain, FR: France, IT: Italy, and TN: Tunisia. (B) Infection rates of the five virus taxa in France in 2015 and 2018. The infection rate of the vOTU in the Mesoni cluster in Morocco in 2016 is also included to facilitate the comparison (marked with a triangle).
We observed significant differences in infection rates between vOTUs (Kruskal–Wallis, P = 0.014). More precisely, the infection rates of Culex.Iflavi_like.virus.4__96 were significantly higher than those of Culex.Bunyavirus.2__98 and Culex.mosquito.virus.4__97 (Conover–Iman test, P = 0.010 and 0.002, respectively). Moreover, significant differences in infection rates were observed between countries for a given virus (Fig. 5A). We did not detect a significant effect of the country on the infection rates of all viruses (Kruskal–Wallis, P = 0.37).
We then explored whether infection rates could change among years. To do so, 94 pools of 6 females and 184 individualized females collected in France in 2018 (748 females in total) were screened for the five vOTUs. Infection rates in 2018 were often significantly higher than that in 2015 (Fig. 5B). For example, infection rates of Alphamesonivirus.1__97 and Culex.Iflavi_like.virus.4__96 increased from less than 10 per cent in 2015 to around 50 per cent in 2018. Overall, our results showed that the infection rates of core vOTUs could be relatively low and differed between countries and years.
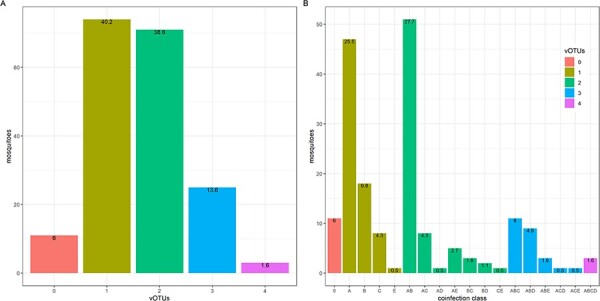
Frequent random coinfection in individual mosquitoes
We explored the potential non-random co-occurrence of different viruses within individual mosquitoes. We used the screen of the five vOTUs in 184 individualized females presented above. Those females had been collected in France in 2018, the year with the highest infection rates for most vOTUs (Fig. 5). This analysis showed that 54 per cent of the females were infected by at least two different vOTUs (Fig. 6A). Different combinations of vOTUs were observed among individuals (Fig. 6B). A probabilistic approach was used to infer co-occurrence patterns in the ten vOTU pairs (Veech 2013). One pair could not be included in the final analysis because its expected co-occurrence was below 1 per cent (the pair Culex.negev.like_virus.1__99/Culex.mosquito.virus.1__97). Only one pair out of the remaining nine pairs showed an association deviating from random (positive association between Culex.Iflavi_like.virus.4__96 and Culex.Bunyavirus.2__96, P = 0.004; other pairs: P > 0.061).
Figure 6.
The distribution of vOTUs in individual Culex pipiens females. The five vOTUs were Culex.Bunyavirus.2__98 (Bunya_unclass cluster), Culex.mosquito.virus.4__97 (Chu cluster), Culex.Iflavi_like.virus.4__96 (Iflavi cluster), Alphamesonivirus.1__97 (Mesoni cluster), and Culex.negev_like.virus.1__99 (Nege cluster). (A) The distribution of the number of vOTUs in individual mosquitoes. Bar color stands for the number of vOTUs per mosquito. The relative frequencies of each coinfection group are shown on top of the bars (in percentages). (B) The distribution of combinations of specific vOTUs (i.e. coinfection classes) found in individual C. pipiens females. Relative frequencies of each coinfection class are shown in percentages on top of the bars. Bars are colored following the number of vOTUs per mosquito as in (A). Each capital letter stands for a given vOTU as follows. A: Alphamesonivirus.1__97, B: Culex.Iflavi_like.virus.4__96, C: Culex.Bunyavirus.2__98, D: Culex.negev.like_virus.1__99, and E: Culex.mosquito.virus.4__97.
Influence of geography on virome assembly
We explored the virome heterogeneity observed between libraries in Fig. 2A. Analyses of alpha and beta diversities were restrained to the influence of the geographical region (i.e. the continent), the only factor for which the experimental design (e.g. number of replicate libraries) allowed a robust analysis. Thus, other variables, like the habitat anthropization, were included in the analyses only to test their potential confounding effect. Analyses were carried out with the cluster dataset instead of the vOTU dataset to avoid potential problems in vOTU identification due to the lack of precision of the best-hit-based clustering. Similar results were found with the vOTU dataset (not shown).
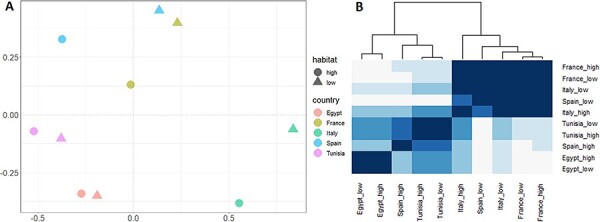
The continent, the habitat anthropization, and the number of mosquitoes or sampling sites per library did not show a significant influence on cluster richness, Simpson and Shannon indexes (Kruskal–Wallis test, P > 0.33; Fig. S6). Jaccard and Bray-Curtis dissimilarities were significantly lower and higher than 0.5, respectively (Wilcoxon test, P < 0.005; Fig. S7A). Moreover, Bray-Curtis dissimilarities, but not Jaccard dissimilarities, were significantly higher between viromes from different continents than between viromes from the same continent (Wilcoxon test, P = 4 × 10−5 and 0.26, respectively; Fig. S7B). No significant difference was found in the two dissimilarities when analyzed depending on habitat anthropization (Wilcoxon test, P > 0.63; Fig. S7C). These results suggested that the viromes tended to share taxa although with different relative frequencies, especially between viromes from different continents.
Virome assemblies in the ten libraries were compared using non-parametric multidimensional scaling and Bray-Curtis dissimilarities, as well as hierarchical clustering (Fig. 7). Viromes tended to aggregate according to continent and country although not perfectly (Fig. 7). A significant influence of continent on virome assembly, but not of habitat anthropization, was found with a PERMANOVA (continent: R2 = 0.445, P = 0.01; habitat anthropization: R2 = 0.062, P = 0.35).
Figure 7.
(A) A non-metric multidimensional scaling plot on Bray-Curtis dissimilarities between the viromes in the ten libraries. (B) A heatmap showing Pearson’s correlation coefficients between the viromes in the ten libraries. A dendrogram based on hierarchical clustering is shown at the top. Tile color represents Pearson’s coefficients, with color becoming darker along with the Pearson’s coefficient. The terms ‘high’ and ‘low’ in library names stand for anthropization level in collection sites.
Virus phylogenies do not suggest strict isolation of virus populations in the Mediterranean Basin
The potential influence of geography on virome assembly could be explained, among others, by differences in virus genetics around the Mediterranean Basin. The existence of the Mediterranean Sea for over 6 million years should have allowed for the isolation, and thus divergent evolution, of virus populations located in Africa and Europe. If the viromes contain different genotypes of the same virus species, the interactions between virus species may differ, leading to specific virome assemblies. We thus explored this question using a phylogenetic approach. Phylogenetic trees were generated with the sequences of three taxonomically distant viruses (Culex Bunyavirus 2, Alphamesonivirus 1, and Culex Iflavi-like virus 4) from each country, together with sequences from other countries (Fig. 8). Sequence clustering based on geography was only clear for Culex Bunyavirus 2.
Figure 8.
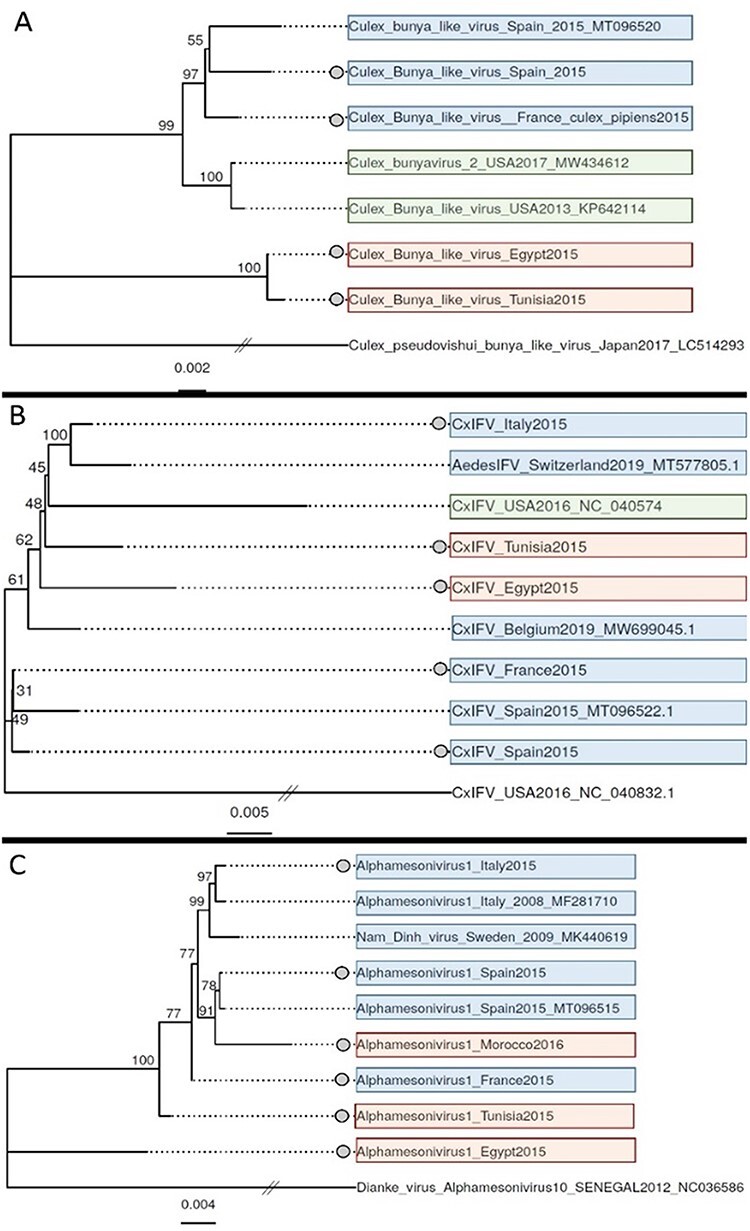
Maximum-likelihood phylogenetic trees of three vOTUs: Culex.Bunyavirus.2__98 (A), Culex.Iflavi_like.virus.4__96 (CxIFV) (B), and Alphamesonivirus.1__97 (C). Sequences from this study are marked with a dot on the left of the sequence name. A divergent species from the same family was used as an outgroup to root each tree (the lowest branch in all the trees). The scale bar represents the number of nucleotide substitutions per site. Background color stands for geographical origin (Europe: blue, Africa: red, and America: green). GenBank accession numbers of sequences from other studies are provided on the branch names.
Discussion
A relatively high overlap in taxonomic diversity was found in the eukaryotic virome of C. pipiens in five countries around the Mediterranean Basin. Core viruses were phylogenetically distant and represented a large fraction of the virome in terms of read abundancy. Moreover, many core viruses of C. pipiens in this study have also been found in populations sampled in other regions. The large geographical spread of core viruses could be explained by the dispersal of virus-carrying mosquitoes, either naturally (e.g. windblown mosquitoes) or by humans (Shi et al. 2017). The observed degree of overlap in taxonomic diversity could not be fully expected from the experimental design. Here, sampling involved mosquito populations separated by an important physical barrier, the Mediterranean Sea. Furthermore, the two samples from each country were obtained each from a different level of habitat anthropization, a factor that impacts viral diversity (Junglen et al. 2009). Differences in continent and anthropization level were thus expected to lead to differences in taxonomic diversity between libraries.
To our knowledge, only one study has performed an in-depth comparison of the virome of a mosquito species over a large geographical scale (Shi et al. 2020). Similarly to our results, the overlap in taxonomic diversity in the virome of A. aegypti between continents included numerous and diverse viral taxa (Shi et al. 2020). Interestingly, the core viromes in A. aegypti and C. pipiens differed in taxonomic diversity. Hence, our results suggest that core viromes can differ among mosquito species. Given the important differences in methodology between the two studies, further analyses using the same methodology are required to compare the core viromes of A. aegypti and C. pipiens over a large geographical range.
Contrary to the observations at the between-country scale, the overlap in taxonomic diversity was limited between mosquitoes from the same country. Our results thus suggest a new phenomenon: the level of overlap in taxonomic diversity in the C. pipiens virome can depend on the spatial scale, with overlap decreasing along with scale. Whether this phenomenon takes place in the eukaryotic virome of other mosquitoes or terrestrial animals is yet unclear due to the limited number of studies exploring different scales simultaneously (Anthony et al. 2015; Bergner et al. 2020; Arze et al. 2021). A previous study on mosquito viromes, geographically restricted to a Caribbean island, suggests that other situations are possible (Shi et al. 2019). The latter study showed infection rates above 90 per cent for several viruses in A. aegypti and Culex quinquefasciatus adults, a situation suggesting a large overlap in taxonomic diversity between individuals.
The coinfection rates in individual mosquitoes were analyzed to explore the potential non-random coinfection of certain viruses. Contrary to previous studies (Shi et al. 2019; Batson et al. 2021; Parry, James, and Asgari 2021), the large number of individuals allowed a probabilistic analysis of co-occurrence. Mosquitoes were often infected by more than one virus, as previously observed in C. pipiens and other mosquito species (Shi et al. 2019; Batson et al. 2021; Parry, James, and Asgari 2021). However, coinfection by most virus couples did not deviate from random co-occurrence. Thus, the core viruses included in our analysis do not seem to be always transmitted together. Overall, our prevalence and co-occurrence data provide insights into an open question in virome ecology, that of the potential similarity in assembly patterns between the virome and the bacteriome (Brum Jennifer et al. 2015; Thongsripong et al. 2021). The low prevalence of certain core viruses contrasts the high prevalence and co-occurrence of core taxa in the bacteriome of mosquitoes, including C. pipiens (Muturi et al. 2016; Guégan et al. 2018).
Our results suggested an influence of the geographical region on virome assembly in C. pipiens, with viromes cluster depending on their African or European origin. Jaccard and Bray-Curtis dissimilarities suggested that clustering was mainly due to differences in relative abundancy between viruses although specificities in taxonomic diversity were also observed. Moreover, the virus phylogenies obtained did not suggest a strict isolation of virus populations in the study area (Fig. 8). Thus, viruses would be shared and exchanged in the study area, but the resulting viromes would tend to differ in relative abundancy among virus taxa.
The observed patterns in virome assembly require further support due to limits in our experimental design. First, our list of core taxa may not be exhaustive. Our meta-transcriptomic approach is considered to be among the most sensitive for virus detection (Zhang, Shi, and Holmes 2018), and the number of mosquitoes was higher than in other studies (Pettersson et al. 2019; Shi et al. 2019). Nevertheless, we cannot exclude that other core viruses have not been detected due to, among others, low viral loads or biases in the library preparation (Gil et al. 2021). Especially, the detection of core viruses with low loads represents a challenge with metagenomics due to (1) its relatively low sensitivity and (2) a high rate of false positives favored by random amplification. Experimental designs with, among others, larger sample numbers and negative controls at different steps are required to overcome this challenge. Second, certain taxa considered here as part of the core may not be detected in all host populations if the spatial or temporal extent of the sampling is increased. Furthermore, our study only analyzed adult females. Whether assembly patterns in the mosquito virome are conserved along the life cycle or between sexes are questions that remain poorly studied (Shi et al. 2020; Coatsworth et al. 2022). Third, the analyses of infection rates involved a limited set of viruses. We chose to include the two core viruses with read counts and contig lengths warranting a robust detection. However, other core viruses may always have high infection rates in C. pipiens populations. Future work on a larger panel of core viruses could thus unveil the coexistence of contrasted infection rates. Another point that requires further study is the potential influence of geography on the mosquito virome. Here, the number of replicate libraries and genomic sequences could not allow a robust analysis of this question. Moreover, meta-transcriptomics is not a fully quantitative method and, thus, the differences in abundance between viruses can only be interpreted in relative terms. Thus, studies with both a larger number of replicates and geographical ranges, as well as truly quantitative analyses of species abundance within viromes, are needed.
Uncovering deterministic patterns in the eukaryotic viromes of terrestrial animals is considered to be challenging (Anthony et al. 2015; Bergner et al. 2020). This study shows that integrating different spatial scales allows to identify deterministic patterns in the C. pipiens virome. Our data can represent a base for hypothesis-driven studies on, among others, the potential role of the C. pipiens virome in pathogen transmission. For example, the core viruses identified here represent candidates for studies on their influence on pathogen transmission. Moreover, the virome differences observed among individuals or regions could participate to the complex epidemiological patterns associated with mosquito-borne arboviruses (Olmo et al. 2023).
Supplementary Material
Acknowledgements
We thank the personnel of the EID Méditerranée (Agencies of Arles, Canet-en-Roussillon, Frejorgues and Montcalm) and Lou Carlier for their involvement in mosquito collection. We also thank Emmanuel Albina for his contribution in the initial phases of the project.
Contributor Information
Patricia Gil, ASTRE, CIRAD, INRAE, University of Montpellier, Montpellier, Languedoc-Roussillon 34398, France.
Antoni Exbrayat, ASTRE, CIRAD, INRAE, University of Montpellier, Montpellier, Languedoc-Roussillon 34398, France.
Etienne Loire, ASTRE, CIRAD, INRAE, University of Montpellier, Montpellier, Languedoc-Roussillon 34398, France.
Ignace Rakotoarivony, ASTRE, CIRAD, INRAE, University of Montpellier, Montpellier, Languedoc-Roussillon 34398, France.
Florian Charriat, ASTRE, CIRAD, INRAE, University of Montpellier, Montpellier, Languedoc-Roussillon 34398, France.
Côme Morel, ASTRE, CIRAD, INRAE, University of Montpellier, Montpellier, Languedoc-Roussillon 34398, France.
Thierry Baldet, ASTRE, CIRAD, INRAE, University of Montpellier, Montpellier, Languedoc-Roussillon 34398, France.
Michel Boisseau, ASTRE, CIRAD, INRAE, University of Montpellier, Montpellier, Languedoc-Roussillon 34398, France.
Albane Marie, EID Mediterranée, Montpellier 34000, France.
Benoît Frances, EID Mediterranée, Montpellier 34000, France.
Gregory L’Ambert, EID Mediterranée, Montpellier 34000, France.
Mohamed Bessat, Department of Parasitology, Faculty of Veterinary Medicine, Alexandria University, Alexandria 5410012, Egypt.
Yehia Otify, Department of Parasitology, Faculty of Veterinary Medicine, Alexandria University, Alexandria 5410012, Egypt.
Maria Goffredo, Istituto Zooprofilattico Sperimentale dell’Abruzzo e del Molise ‘G. Caporale’, Teramo 64100, Italy.
Giuseppe Mancini, Istituto Zooprofilattico Sperimentale dell’Abruzzo e del Molise ‘G. Caporale’, Teramo 64100, Italy.
Núria Busquets, IRTA. Programa de Sanitat Animal. Centre de Recerca en Sanitat Animal (CReSA), Campus de la Universitat Autònoma de Barcelona (UAB), Bellaterra 08193, Spain.
Lotty Birnberg, IRTA. Programa de Sanitat Animal. Centre de Recerca en Sanitat Animal (CReSA), Campus de la Universitat Autònoma de Barcelona (UAB), Bellaterra 08193, Spain.
Sandra Talavera, IRTA. Programa de Sanitat Animal. Centre de Recerca en Sanitat Animal (CReSA), Campus de la Universitat Autònoma de Barcelona (UAB), Bellaterra 08193, Spain.
Carles Aranda, IRTA. Programa de Sanitat Animal. Centre de Recerca en Sanitat Animal (CReSA), Campus de la Universitat Autònoma de Barcelona (UAB), Bellaterra 08193, Spain; Servei de Control de Mosquits del Consell Comarcal del Baix Llobregat, Barcelona 08980, Spain.
Emna Ayari, Institut de la Recherche Vétérinaire de Tunisie - Université Tunis El Manar, Tunis 1068, Tunisia.
Selma Mejri, Institut de la Recherche Vétérinaire de Tunisie - Université Tunis El Manar, Tunis 1068, Tunisia.
Soufien Sghaier, Institut de la Recherche Vétérinaire de Tunisie - Université Tunis El Manar, Tunis 1068, Tunisia.
Amal Bennouna, Department of Animal Pathology and Public Health, Hassan II Agronomy & Veterinary Institute, Rabat BP 6202, Morocco.
Hicham El Rhaffouli, Veterinary Division, FAR Military Health Service, Meknes 11080, Morocco.
Thomas Balenghien, ASTRE, CIRAD, INRAE, University of Montpellier, Montpellier, Languedoc-Roussillon 34398, France; Department of Animal Pathology and Public Health, Hassan II Agronomy & Veterinary Institute, Rabat BP 6202, Morocco.
Ghita Chlyeh, Département de Production, Protection et Biotechnologies Végétales, Unité de Zoologie, Institute of Agronomy and Veterinary Medicine Hassan II, Rabat BP 6202, Morocco.
Ouafaa Fassi Fihri, Department of Animal Pathology and Public Health, Hassan II Agronomy & Veterinary Institute, Rabat BP 6202, Morocco.
Julie Reveillaud, ASTRE, CIRAD, INRAE, University of Montpellier, Montpellier, Languedoc-Roussillon 34398, France.
Yannick Simonin, ASTRE, CIRAD, INRAE, University of Montpellier, Montpellier, Languedoc-Roussillon 34398, France; Pathogenesis and Control of Chronic Infections, University of Montpellier, INSERM, EFS, Montpellier 34394, France.
Marc Eloit, Institut Pasteur, Université Paris Cité, Pathogen Discovery Laboratory, Paris 75015, France; Institut Pasteur, The OIE Collaborating Centre for Detection and Identification in Humans of Emerging Animal Pathogens, Paris 75724, France; École nationale vétérinaire d’Alfort, Maisons-Alfort 94700, France.
Serafin Gutierrez, ASTRE, CIRAD, INRAE, University of Montpellier, Montpellier, Languedoc-Roussillon 34398, France.
Data availability
Raw reads and selected contigs were deposited in GenBank associated with BioProject PRJNA806751.
Supplementary data
Supplementary data are available at Virus Evolution Journal online.
Funding
This work was supported by the 7th Framework Programme of the European Commission (grant FP7-613996 Vmerge), the Montpellier University of Excellence Program (MUSE, ArboSud project), and the Direction Générale de l’Alimentation from the French Ministry of Agriculture and Food (DGAl grant agreement: SPA17 n0079-E). The contents of this publication are the sole responsibility of the authors and do not necessarily reflect the views of the European Commission.
Conflict of interest:
None declared.
References
- Aardema M. L. et al. (2020) ‘Global Evaluation of Taxonomic Relationships and Admixture within the Culex pipiens Complex of Mosquitoes’, Parasites & Vectors, 13: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal A. A. et al. (2007) ‘Filling Key Gaps in Population and Community Ecology’, Frontiers in Ecology and the Environment, 5: 145–52. [Google Scholar]
- Almeida D. E. et al. (2021) ‘The Virome of Vector Mosquitoes’, Current Opinion in Virology, 49: 7–12. [DOI] [PubMed] [Google Scholar]
- Altinli M., Schnettler E., and Sicard M. (2021) ‘Symbiotic Interactions between Mosquitoes and Mosquito Viruses’, Frontiers in Cellular and Infection Microbiology, 11: 694020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony S. J. et al. (2015) ‘Non-random Patterns in Viral Diversity’, Nature Communications, 6: 8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arze C. A. et al. (2021) ‘Global Genome Analysis Reveals a Vast and Dynamic Anellovirus Landscape within the Human Virome’, Cell Host & Microbe, 29: 1305–1315.e6. [DOI] [PubMed] [Google Scholar]
- Atoni E. et al. (2018) ‘Metagenomic Virome Analysis of Culex Mosquitoes from Kenya and China’, Viruses, 10: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baidaliuk A. et al. (2019) ‘Cell-Fusing Agent Virus Reduces Arbovirus Dissemination in Aedes Aegypti Mosquitoes In Vivo’, Journal of Virology, 93: e00705–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batson J. et al. (2021) ‘Single Mosquito Metatranscriptomics Identifies Vectors, Emerging Pathogens and Reservoirs in One Assay’, Elife, 10: e68353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennouna A. et al. (2019) ‘Identification of Eilat Virus and Prevalence of Infection among Culex pipiens L. Populations, Morocco, 2016’, Virology, 530: 85–8. [DOI] [PubMed] [Google Scholar]
- Berg G. et al. (2020) ‘Microbiome Definition Re-visited: Old Concepts and New Challenges’, Microbiome, 8: 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergner L. M. et al. (2019) ‘Using Noninvasive Metagenomics to Characterize Viral Communities from Wildlife’, Molecular Ecology Resources, 19: 128–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2020) ‘Demographic and Environmental Drivers of Metagenomic Viral Diversity in Vampire Bats’, Molecular Ecology, 29: 26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggerstaff B. J. (2008) ‘Confidence Intervals for the Difference of Two Proportions Estimated from Pooled Samples’, Journal of Agricultural, Biological, and Environmental Statistics, 13: 478–96. [Google Scholar]
- Bolling B. G. et al. (2012) ‘Transmission Dynamics of an Insect-specific Flavivirus in a Naturally Infected Culex pipiens Laboratory Colony and Effects of Co-infection on Vector Competence for West Nile Virus’, Virology, 427: 90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brum J. R. et al. (2015) ‘Patterns and Ecological Drivers of Ocean Viral Communities’, Science 348; 1261498. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2016) ‘Seasonal Time Bombs: Dominant Temperate Viruses Affect Southern Ocean Microbial Dynamics’, The ISME Journal, 10: 437–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B., Xie C., and Huson D. H. (2015) ‘Fast and Sensitive Protein Alignment using DIAMOND’, Nat Methods, 12: 59–60. doi: 10.1038/nmeth.3176 [DOI] [PubMed] [Google Scholar]
- Capella-Gutiérrez S., Silla-Martínez J. M., and Gabaldón T. (2009) ‘Trimal: A Tool for Automated Alignment Trimming in Large-scale Phylogenetic Analyses’, Bioinformatics, 25: 1972–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheval J. et al. (2011) ‘Evaluation of High-throughput Sequencing for Identifying Known and Unknown Viruses in Biological Samples’, Journal of Clinical Microbiology, 49: 3268–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coatsworth H. et al. (2022) ‘Intrinsic Variation in the Vertically Transmitted Core Virome of the Mosquito Aedes Aegypti’, Molecular Ecology, 31: 2545–61. [DOI] [PubMed] [Google Scholar]
- Dolan P. T., Whitfield Z. J., and Andino R. (2018) ‘Mechanisms and Concepts in RNA Virus Population Dynamics and Evolution’, Annual Review of Virology, 5: 69–92. [DOI] [PubMed] [Google Scholar]
- Elrefaey A. M. et al. (2020) ‘Understanding the Mechanisms Underlying Host Restriction of Insect-Specific Viruses’, Viruses, 12: 964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauver J. R. et al. (2016) ‘West African Anopheles gambiae Mosquitoes Harbor a Taxonomically Diverse Virome Including New Insect-specific Flaviviruses, Mononegaviruses, and Totiviruses’, Virology, 498: 288–99. [DOI] [PubMed] [Google Scholar]
- Feng Y. et al. (2022) ‘A Time-series Meta-transcriptomic Analysis Reveals the Seasonal, Host, and Gender Structure of Mosquito Viromes’, Virus Evol, 8: veac006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca D. M. et al. (2004) ‘Emerging Vectors in the Culex pipiens Complex’, Science, 303: 1535–8. [DOI] [PubMed] [Google Scholar]
- Gaye A. et al. (2020) ‘Vector Competence of Anthropophilic Mosquitoes for a New Mesonivirus in Senegal’, Emerg Microbes Infect, 9: 496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil P. et al. (2021) ‘A Library Preparation Optimized for Metagenomics of RNA Viruses’, Molecular Ecology Resources, 21: 1788–807. [DOI] [PubMed] [Google Scholar]
- Goenaga S. et al. (2015) ‘Potential for Co-Infection of a Mosquito-Specific Flavivirus, Nhumirim Virus, to Block West Nile Virus Transmission in Mosquitoes’, Viruses, 7: 5801–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith D. M., Veech J. A., and Marsh C. J. (2016) ‘Cooccur: Probabilistic Species Co-Occurrence Analysis in R’, Journal of Statistical Software, Code Snippets, 69: 1–17. [Google Scholar]
- Guégan M. et al. (2018) ‘The Mosquito Holobiont: Fresh Insight into Mosquito-microbiota Interactions’, Microbiome, 6: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall R. A. et al. (2016) ‘Commensal Viruses of Mosquitoes: Host Restriction, Transmission, and Interaction with Arboviral Pathogens’, Evolutionary Bioinformatics Online, 12: 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall-Mendelin S. et al. (2016) ‘The Insect-specific Palm Creek Virus Modulates West Nile Virus Infection in and Transmission by Australian Mosquitoes’, Parasites & Vectors, 9: 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey E., and Holmes E. C. (2022) ‘Diversity and Evolution of the Animal Virome’, Nature Reviews. Microbiology, 20: 321–34. [DOI] [PubMed] [Google Scholar]
- He X. et al. (2021) ‘Metagenomic Sequencing Reveals Viral Abundance and Diversity in Mosquitoes from the Shaanxi-Gansu-Ningxia Region, China’, PLoS Neglected Tropical Diseases, 15: e0009381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D. et al. (2021) ‘Enhanced Mutualistic Symbiosis between Soil Phages and Bacteria with Elevated Chromium-induced Environmental Stress’, Microbiome, 9: 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, and Madan A. (1999) ‘CAP3: A DNA Sequence Assembly Program’, Genome Res, 9: 868–77. doi: 10.1101/gr.9.9.868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junglen S. et al. (2009) ‘Examining Landscape Factors Influencing Relative Distribution of Mosquito Genera and Frequency of Virus Infection’, Ecohealth, 6: 239–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidis K. et al. (2022) ‘Defining Virus-carrier Networks that Shape the Composition of the Mosquito Core Virome of a Local Ecosystem’, Virus Evol, 8: veac036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft N. J. B., and Ackerly D. D. (2014) ‘Assembly of Plant Communities’, in Monson, R. K. (ed.) Ecology and the Environment, pp. 67–88. New York, NY: Springer. [Google Scholar]
- Kuhn J. H. et al. (2019) ‘Classify Viruses – The Gain Is Worth the Pain’, Nature, 566: 318–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S. et al. (2018) ‘MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms’, Molecular Biology and Evolution, 35: 1547–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. (2009) ‘The Sequence Alignment/Map Format and SAMtools’, Bioinformatics, 25: 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D. et al. (2015) ‘MEGAHIT: An Ultra-fast Single-node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph’, Bioinformatics, 31: 1674–6. [DOI] [PubMed] [Google Scholar]
- Li H., and Durbin R. (2009) ‘Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform’, Bioinformatics, 25: 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue J. B. et al. (2011) ‘Empirical Approaches to Metacommunities: A Review and Comparison with Theory’, Trends in Ecology & Evolution, 26: 482–91. [DOI] [PubMed] [Google Scholar]
- Manrique P., Dills M., and Young M. J. (2017) ‘The Human Gut Phage Community and Its Implications for Health and Disease’, Viruses, 9: 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. (2011) ‘Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads’, EMBnet j, 17: 10. doi: 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- Mcknight D. T. et al. (2019) ‘Methods for Normalizing Microbiome Data: An Ecological Perspective’, Methods in Ecology and Evolution, 10: 389–400. [Google Scholar]
- Minh B. Q. et al. (2020) ‘IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era’, Molecular Biology and Evolution, 37: 1530–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muturi E. J. et al. (2016) ‘Culex pipiens and Culex Restuans Mosquitoes Harbor Distinct Microbiota Dominated by Few Bacterial Taxa’, Parasites & Vectors, 9: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen J. et al. 2019. Vegan: Community Ecology Package. R package version 2.5-6. <https://cran.r-project.org/web/packages/vegan/index.html>.
- Olmo R. P. et al. (2023) ‘Mosquito Vector Competence for Dengue Is Modulated by Insect-specific Viruses’, Nat Microbiol, 8: 135–49. [DOI] [PubMed] [Google Scholar]
- Parry R., James M. E., and Asgari S. (2021) ‘Uncovering the Worldwide Diversity and Evolution of the Virome of the Mosquitoes Aedes aegypti and Aedes albopictus’, Microorganisms, 9: 1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson J. H. et al. (2019) ‘Meta-Transcriptomic Comparison of the RNA Viromes of the Mosquito Vectors Culex pipiens and Culex torrentium in Northern Europe’, Viruses, 11: 1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadeghi M. et al. (2018) ‘Virome of >12 Thousand Culex Mosquitoes from Throughout California.’, Virology, 523: 74–88. [DOI] [PubMed] [Google Scholar]
- Shade A., and Handelsman J. (2012) ‘Beyond the Venn Diagram: The Hunt for a Core Microbiome’, Environmental Microbiology, 14: 4–12. [DOI] [PubMed] [Google Scholar]
- Shi M. et al. (2016) ‘Redefining the Invertebrate RNA Virosphere’, Nature, 540: 539–43. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2017) ‘High-Resolution Metatranscriptomics Reveals the Ecological Dynamics of Mosquito-Associated RNA Viruses in Western Australia’, Journal of Virology, 91: e00680–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C. et al. (2019) ‘Stable Distinct Core Eukaryotic Viromes in Different Mosquito Species from Guadeloupe, Using Single Mosquito Viral Metagenomics’, Microbiome, 7: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2020) ‘Stability of the Virome in Lab- and Field-Collected Aedes albopictus Mosquitoes across Different Developmental Stages and Possible Core Viruses in the Publicly Available Virome Data of Aedes Mosquitoes’, mSystems, 5: e00640–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M., Zhang Y.-Z., and Holmes E. C. (2018) ‘Meta-transcriptomics and the Evolutionary Biology of RNA Viruses’, Virus Research, 243: 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanojevic M. et al. (2020) ‘Depicting the RNA Virome of Hematophagous Arthropods from Belgrade, Serbia’, Viruses, 12: 975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stulberg E. et al. (2016) ‘An Assessment of US Microbiome Research’, Nat Microbiol, 1: 15015. [DOI] [PubMed] [Google Scholar]
- Thongsripong P. et al. (2021) ‘Metagenomic Shotgun Sequencing Reveals Host Species as an Important Driver of Virome Composition in Mosquitoes’, Scientific Reports, 11: 8448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veech J. A. (2013) ‘A Probabilistic Model for Analysing Species Co-occurrence’, Global Ecology and Biogeography, 22: 252–60. [Google Scholar]
- White A. V. et al. (2021) ‘Mosquito-infecting Virus Espirito Santo Virus Inhibits Replication and Spread of Dengue Virus’, Journal of Medical Virology, 93: 3362–73. [DOI] [PubMed] [Google Scholar]
- Wolf Y. I. et al. (2018) ‘Origins and Evolution of the Global RNA Virome’, MBio, 9: e02329–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye G. et al. (2020) ‘Transmission Competence of a New Mesonivirus, Yichang Virus, in Mosquitoes and Its Interference with Representative Flaviviruses’, PLoS Neglected Tropical Diseases, 14: e0008920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G. et al. (2017) ‘Ggtree: An R Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data’, Methods in Ecology and Evolution, 8: 28–36. [Google Scholar]
- Zhang Y. Z., Shi M., and Holmes E. C. (2018) ‘Using Metagenomics to Characterize an Expanding Virosphere’, Cell, 172: 1168–72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw reads and selected contigs were deposited in GenBank associated with BioProject PRJNA806751.