

Abstract
Musa acuminata is a main wild contributor to banana cultivars. Here, we reported a haplotype-resolved and telomere-to-telomere reference genome of M. acuminata by incorporating PacBio HiFi reads, Nanopore ultra-long reads, and Hi-C data. The genome size of the two haploid assemblies was estimated to be 469.83 Mb and 470.21 Mb, respectively. Multiple assessments confirmed the contiguity (contig N50: 16.53 Mb and 18.58 Mb; LAI: 20.18 and 19.48), completeness (BUSCOs: 98.57% and 98.57%), and correctness (QV: 45.97 and 46.12) of the genome. The repetitive sequences accounted for about half of the genome size. In total, 40,889 and 38,269 protein-coding genes were annotated in the two haploid assemblies, respectively, of which 9.56% and 3.37% were newly predicted. Genome comparison identified a large reciprocal translocation involving 3 Mb and 10 Mb from chromosomes 01 and 04 within M. acuminata. This reference genome of M. acuminata provides a valuable resource for further understanding of subgenome evolution of Musa species, and precise genetic improvement of banana.
Subject terms: Structural variation, Comparative genomics, DNA sequencing, Natural variation in plants
Background & Summary
The wild relatives of domesticated crops, i.e. crop wild relatives (CWRs), generally possess genetic diversity helpful in developing more productive and resilient crop varieties, thereby providing a wide practical gene pool for genetic improvement of crops1. In order to address the challenges and threats posed by emerging diseases and climate change, CWRs appear to be a source for solutions to manage both biotic and abiotic stresses2,3. At present, combining huge sequence information and precise gene-editing tools provides a route to transform CWRs into ideal crops2. Therefore, a high-quality reference genome of CWR germplasm is an important prerequisite for efficiently introducing potential useful genes into breeding programmes. Thanks to the advances in sequencing technologies and analytical tools, many high-quality reference genomes for crops as well as their important wild relatives have been generated. These genetic resources will thus facilitate the identification of structural variants and incorporation of the variants from CWRs into crop gene pools.
Banana domestication started at least 7000 years ago in Southeast Asia4. Hybridization between various species and subspecies of the Musa genus led to the development of modern bananas with high production5. To date, most banana cultivars were derived from Musa acuminata (A genome), a complex of subspecies geographically segregated in distinct Southeast Asian continental regions and islands6. Four particular M. acuminata subspecies have been raised as the main contributors of edible banana cultivars, which are banksii, burmannica, malaccensis, and zebrina4. Several large structural variants in these subspecies were identified and suggested to be associated with the domestication of banana7–11. Genome research first started in the subspecies malaccensis. The first draft genome of M. acuminata ssp. malaccensis was assembled by incoporating Sanger and Roche/454 reads, with sequence errors corrected by Illumina data12. This assembly was anchored along the Musa linkage groups of the genetic map built with SSR and DArT markers. The double-haploid genotype (DH-Pahang) was used in this study for reducing genome complexity and facilitating assembly process. Recently the telomere-to-telomere (T2T) reference genome of DH-Pahang has been constructed using Nanopore data and polished with Nanopore and Illumina reads, with continuity improved significantly13. Although DH genotype could miss some important genetic information, these genome resources have significantly facilitated the studies of banana domestication and genome evolution. With advances in the sequencing technologies and biosoftwares, heterozygosity would not be the consistent hurdle. Currently, more and more haplotype-resolved and T2T genomes have been published, such as lychee14 and apple15, providing unprecedented insights into subgenome evolution and domesticated history.
In this study, we assembled a haplotype-resolved and telomere-to-telomere reference genome of M. acuminata ssp. malaccensis by incorporating PacBio HiFi reads, Nanopore ultra-long reads, and high throughput chromatin conformation capture (Hi-C) paired reads. An unphased reference genome was first constructed and used for guiding haplotype-resolved scaffolding (Fig. 1). Multiple assessment methods were applied to evaluate the quality of the haplotype-resolved assembly. A comprehensive genome comparison between this assembly and the previous reference of the DH genotype identified a large reciprocal translocation involving 3 Mb and 10 Mb from chromosomes 01 and 04. Furthermore, the 3-Mb segment (34,734,628 to 37,810,715 bp in chromosome 04) was suggested to be associated with flower development pathway, such as anther/stamen development. The haplotype-resolved genome of M. acuminata will help to obtain a better understanding of potential structural variants, allele specific expression and subgenome evolution of Musa species, and serve as reliable reference for banana breeding programmes.
Fig. 1.

The workflow of generating haplotype-resolved and telomere-to-telomere reference genome for M. acuminata. The unphased reference genome was constructed following the workflow on the left yellow panel. Then a haplotype-resolved and telomere-to-telomere reference genome was produced according to the scheme on the right blue panel. Green boxes represent raw sequencing data; white boxes represent the tools used in this pipeline; pink boxes represent intermediate data; blue boxes represent post-analysis process.
Methods
Sample collection and sequencing
The M. acuminata sample used for DNA and RNA extraction was obtained from South China Botanical Garden, Chinese Academy of Sciences, Guangzhou, China. Tissues were immediately frozen in liquid nitrogen and preserved at −80 °C for DNA/RNA extraction. The CTAB method was used to extract high quality genomic DNA from leaf tissue samples.
A standard SMRTbell library was constructed using SMRTbell Express Template Prep Kit 2.0 according to the manufacturer’s recommendations (Pacific Biosciences, CA, USA) and sequenced on a PacBio Sequel II platform. This yielded 32.39 Gb HiFi data, covering ~65 × coverage of the haploid genome size. The N50 length of the HiFi reads was 17.32 kb. A nanopore library was constructed with the Oxford Nanopore SQK-LSK109 kit following the manufacturers’ instructions and sequenced on a PromethION platform. Totally 20.80 Gb ONT data were obtained, covering ~42 × coverage of the haploid genome size. The N50 length was 86.86 kb. A Hi-C library was constructed based on cross-linked genomic DNA and sequenced on an Illumina NovaSeq platform (Illumina, San Diego, CA, USA). In total, 134 Gb Hi-C data were obtained, covering ~268 × coverage of the haploid genome size. The 15.58 Gb NGS data were obtained using the Illumina NovaSeq platform, covering ~31 × coverage of the haploid genome size (Table 1).
Table 1.
Summary of sequencing data of Musa acuminata ssp. malaccensis for haplotype-resolved and telomere-to-telomere assembly and genome annotation.
| Sequencing | Clean base (Gb) | Clean reads | N50 length (bp) | Depth (X) | Sample | Application |
|---|---|---|---|---|---|---|
| HiFi | 32.39 | 1,793,624 | 17,320 | 64.78 | Leaf | Assembly |
| HiC | 134.00 | 894,989,890 | 2 × 150 | 268 | Leaf | Chromosome construction |
| ONT | 20.80 | 439,578 | 86,861 | 41.6 | Leaf | Gap filling |
| Illumina | 15.58 | \ | 2 × 150 | 31.16 | Leaf | Genome evaluation |
| RNA-seq | 6.6 | \ | 2 × 150 | \ | Flower | Genome annotation |
| RNA-seq | 7.1 | \ | 2 × 150 | \ | Fruit | Genome annotation |
| RNA-seq | 6.1 | \ | 2 × 150 | \ | Leaf | Genome annotation |
| RNA-seq | 7.1 | \ | 2 × 150 | \ | Root | Genome annotation |
Additionally, total RNA was extracted from four tissues, including root, leaf, flower, and fruit, using the NEBNext® Ultra™ II Directional RNA Library Prep Kit for Illumina® (New England Biolabs, MA, USA). Paired-end 150-bp reads were also generated by the Illumina NovaSeq platform. These yielded a total of 26.90 Gb raw RNAseq data (Table 1). All sequencing were carried out at Anhui Double Helix Gene Technology Co., Ltd. (Anhui, China).
Genome size and heterozygosity estimation
CCS software (https://github.com/PacificBiosciences/ccs) with default parameters was used to generate the consensus reads (HiFi reads). Based on the obtained high-accurate HiFi reads, the K-mer distribution was analysed with jellyfish16 with jellyfish count -C -m 21 -s 100000000 and jellyfish histo -h 1000000. The results were subsequently imported to GenomeScope v2.017 with K-mer length = 21 and Ploidy = 2. The genome size of M. acuminata was estimated to be 450.43 Mb with the 21 K-mer, about 14% shorter than DH-Pahang genome size (523.00 Mb) estimated by flow cytometry12. The heterozygosity rate was estimated to be 0.59% (Fig. 2).
Fig. 2.
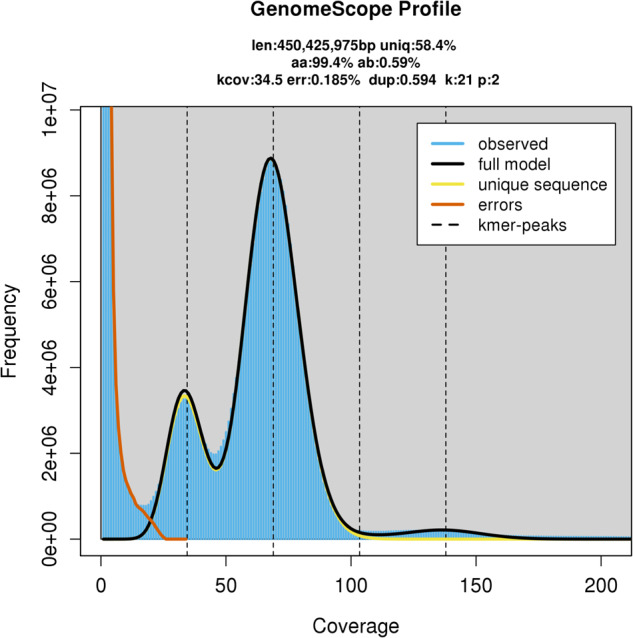
The GenomeScope profile of M. acuminata based on 21 K-mer.
De novo haplotype-resolved genome assembly
Fastp v0.23.218 was performed to filter Hi-C reads with default parameters. Subsequently, hifiasm v0.16.1-r37519 was carried out to generate the primary unphased draft genome based on HiFi and Hi-C reads. This generated a 491.54 Mb draft genome with an N50 of 26.62 Mb, and only 20 contigs consisted of 90% length of the genome (Table 2). Then, ragtag v2.1.020 with default parameters was first used to sort, orientate, and cluster the primary contigs guided by the T2T version of M. acuminata ssp. malaccensis DH-Pahang genome13 (Hereafter MAv4). Meanwhile, the primary contigs were anchored into 11 pseudo-chromosomes using Juicer v1.621 and 3D-DNA v18092222 in turn. Then, based on the assembly file obtained from ragtag and the hic file from Juicer and 3D-DNA, Juicebox v2.20.0023 was introduced for visualizing Hi-C data and manual correction in order to obtain a high-quality reference genome. Finally, there were only 17 gaps in the high-quality reference genome. For gap filling, ONT assembly was constructed by NextDenovo (https://github.com/Nextomics/NextDenovo) with read-cutoff = 1k and genome_size = 500 M. Then this draft ONT assembly was polished by Nextpolish24 based on the HiFi reads and the Illumina reads with default parameters. Subsequently minimap2 v2.24-r112225 with default parameters was used to map the polished ONT assembly to the primary reference genome. We examined the breakpoint with the Integrative Genomics Viewer (IGV) tool26 and manually filled the gaps based on the alignment results. After using ONT assembly to fill all remaining gaps, a high-quality reference genome named MA was generated. The genome size of this unphased assembly is 471.04 Mb with an anchored rate of 95.83%. The Hi-C heatmap confirmed the contiguity of the assembly (Supplementary Figure S1).
Table 2.
Summary of genome assembly of Musa acuminata ssp. malaccensis genome.
| Assembly | MA | MAH1 | MAH2 |
|---|---|---|---|
| contigs (> = 0 bp) | 141 | 275 | 206 |
| contigs (> = 1000 bp) | 141 | 275 | 206 |
| contigs (> = 5000 bp) | 141 | 275 | 206 |
| contigs (> = 10000 bp) | 141 | 275 | 206 |
| contigs (> = 25000 bp) | 132 | 235 | 194 |
| contigs (> = 50000 bp) | 112 | 180 | 139 |
| Total length (> = 0 bp) | 491,526,655 | 500,781,154 | 484,357,301 |
| Total length (> = 1000 bp) | 491,526,655 | 500,781,154 | 484,357,301 |
| Total length (> = 5000 bp) | 491,526,655 | 500,781,154 | 484,357,301 |
| Total length (> = 10000 bp) | 491,526,655 | 500,781,154 | 484,357,301 |
| Total length (> = 25000 bp) | 491,334,973 | 499,946,784 | 484,102,230 |
| Total length (> = 50000 bp) | 490,674,857 | 498,063,881 | 482,072,416 |
| contigs | 141 | 275 | 206 |
| Largest contig | 50,229,097 | 50,630,355 | 50,002,820 |
| Total length | 491,526,655 | 500,781,154 | 484,357,301 |
| GC (%) | 39.58 | 39.89 | 39.69 |
| N50 | 26,620,819 | 16,527,116 | 18,582,139 |
| N90 | 7,537,575 | 4,161,334 | 6,090,445 |
| auN | 26,629,771 | 20,028,603 | 19,951,612 |
| L50 | 7 | 9 | 10 |
| L90 | 20 | 33 | 28 |
Note: MA represents the primary contig sets, while MAH1 and MAH2 represent contigs in haplotype1 and contigs in haplotype2.
To obtain a haplotype-resolved genome, a similar pipeline was applied (Fig. 1). Two primary haploid assemblies were first generated by hifiasm. Further genome assembly statistics were performed with QUAST27 with default parameters. Accumulative lengths of the two haploid assemblies were 500.78 Mb and 484.36 Mb with N50 of 16.53 Mb and 18.58 Mb, respectively (Table 2). After Hi-C scaffolding processes, 469.83 Mb and 470.21 Mb were anchored to 11 chromosomes respectively, with an anchored rate of 93.82% and 97.08% (Table 3). The genome sizes of the two haploid assemblies were slightly longer than that of MAv4 (468.82 Mb)13, and represented approximately 90% of DH-Pahang genome size (523.00 Mb) estimated by flow cytometry12. All 66 gaps in the two haploid assemblies were filled. Finally, the haplotype-resolved and telomere-to-telomere reference genome for M. acuminata was obtained; and the two haploid assemblies were named MAH1 and MAH2. The circos28 software was introduced to draw the genome features shown in Fig. 3. The Hi-C heatmap confirmed this assembly as a complete and reliable haplotype-resolved reference genome (Fig. 4).
Table 3.
The lengths of the pseudo-chromosomes of Musa acuminata ssp. malaccensis genomes.
| CHR | MAH1 | MAH2 | MAv4 | |||
|---|---|---|---|---|---|---|
| Length(bp) | Contigs | Length(bp) | Contigs | Length(bp) | Contigs | |
| CHR01 | 50,630,355 | 1 | 50,002,820 | 1 | 41,765,374 | 7 |
| CHR02 | 36,099,580 | 1 | 36,147,835 | 2 | 34,826,099 | 1 |
| CHR03 | 43,044,357 | 4 | 42,983,428 | 4 | 43,931,233 | 4 |
| CHR04 | 37,851,086 | 6 | 37,817,432 | 5 | 45,086,258 | 1 |
| CHR05 | 46,163,207 | 7 | 45,655,733 | 9 | 46,513,039 | 4 |
| CHR06 | 43,119,214 | 3 | 41,967,602 | 3 | 43,117,521 | 1 |
| CHR07 | 37,893,627 | 6 | 37,753,859 | 3 | 39,373,400 | 5 |
| CHR08 | 51,441,117 | 4 | 51,151,883 | 6 | 51,314,288 | 4 |
| CHR09 | 46,958,856 | 3 | 47,015,202 | 3 | 47,719,527 | 1 |
| CHR10 | 42,015,131 | 6 | 45,690,726 | 5 | 40,511,255 | 8 |
| CHR11 | 34,610,164 | 3 | 34,028,409 | 3 | 34,663,808 | 1 |
| Total | 469,826,694 | 44 | 470,214,929 | 44 | 468,821,802 | 37 |
Fig. 3.

The Overview of M. acuminata genome assembly and features. The tracks represent the following elements (from outer to inner): (a) Karyotypes of the 22 chromosome sequences, (b) TRF-183bp centromeric repeat density, (c) Copia density, (d) Transposable element (TE) density, (e) GC contents, (f) Gene density. The innermost is syntenic relationships.
Fig. 4.
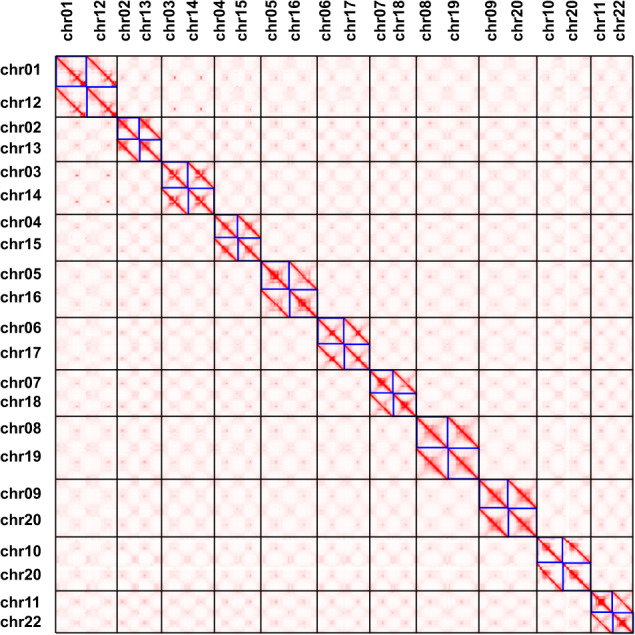
The Hi-C heatmap of haplotype-resolved genome of M. acuminata. 11 chromosome pairs were defined.
Genome quality assessment
Multiple methods were combined to evaluate the quality of genome assembly. First, the HiFi, Illumina, and RNAseq reads were aligned to the phased genome using minimap2 v2.24-r1122, BWA v0.7.17-r118829, and HiSAT2 v2.2.130 with default parameters, respectively. BamTools v2.5.131 was used to calculate the read mapping rates. The results showed a HiFi coverage rate of 99.86% and 99.87% on MAH1 and MAH2 assemblies, respectively. The mapping rate of Illumina reads reached up to 99.98% in both haploid assemblies. The mapping rate of RNAseq reads ranged from 92.44% to 97.34% (Table 4). Second, the LTR Assembly Index (LAI) calculated from LTR_retriever v2.9.032 was used to assess the genome assembly quality. The LAI of MAH1 and MAH2 reached up to 20.18 and 19.48, respectively, indicating that our phased assembly reached the standard of a golden reference. Third, the completeness of the haplotype-resolved genome was evaluated by BUSCO v5.4.333 against the ‘embryophyta_odb 10’ database. In total, 98.57% (1,591 of 1,614) of the complete BUSCO genes were identified (Table 5). Finally, the consensus quality value (QV) of the genome was assessed by Merqury v1.334 with meryl k = 19 count, showing 45.97 and 46.12 of QV (Genome accuracy >99.99%) for MAH1 and MAH2, respectively (Table 6, Supplementary Figure S2).
Table 4.
Assessment of genome quality based on mapping with RNAseq reads.
| Tissues | Data size (Gb) | reads count | Mapping rate (%) | |
|---|---|---|---|---|
| MAH1 | MAH2 | |||
| Flower | 6.6 | 59145701 | 97.34 | 97.29 |
| Fruit | 7.1 | 52110912 | 97.19 | 97.17 |
| Leaf | 6.1 | 45400245 | 96.50 | 96.67 |
| Root | 7.1 | 58396498 | 92.47 | 92.44 |
Table 5.
BUSCO results of MAH1 (C: 98.57%) and MAH2 (C: 98.57%).
| MAH1 | MAH2 | |
|---|---|---|
| Complete BUSCOs (C) | 1591 | 1591 |
| Complete and single-copy BUSCOs (S) | 1515 | 1515 |
| Complete and duplicated BUSCOs (D) | 76 | 76 |
| Fragmented BUSCOs (F) | 8 | 7 |
| Missing BUSCOs (M) | 15 | 16 |
| Total BUSCO groups searched | 1614 | 1614 |
Note: The lineage dataset is embryophyta_odb10.
Table 6.
The consensus quality values of MAH1 and MAH2.
| CHR | k_asm | k_total | Error rate | QV |
|---|---|---|---|---|
| CHR01 | 22610 | 50630337 | 2.35E-05 | 46.2877 |
| CHR02 | 12507 | 36099562 | 1.82E-05 | 47.3903 |
| CHR03 | 18092 | 43043340 | 2.21E-05 | 46.5509 |
| CHR04 | 20933 | 37848568 | 2.91E-05 | 45.3586 |
| CHR05 | 27201 | 46174067 | 3.10E-05 | 45.0845 |
| CHR06 | 22142 | 43118196 | 2.70E-05 | 45.6809 |
| CHR07 | 20602 | 37896126 | 2.86E-05 | 45.4333 |
| CHR08 | 21345 | 51440721 | 2.18E-05 | 46.6068 |
| CHR09 | 21992 | 46957838 | 2.47E-05 | 46.081 |
| CHR10 | 20417 | 42012613 | 2.56E-05 | 45.9204 |
| CHR11 | 17834 | 34622309 | 2.71E-05 | 45.6675 |
| CHR12 | 21732 | 50002802 | 2.29E-05 | 46.4056 |
| CHR13 | 12313 | 36147317 | 1.79E-05 | 47.464 |
| CHR14 | 19174 | 42981910 | 2.35E-05 | 46.2923 |
| CHR15 | 19132 | 37815414 | 2.66E-05 | 45.7456 |
| CHR16 | 26457 | 45652741 | 3.05E-05 | 45.1556 |
| CHR17 | 19902 | 41966584 | 2.50E-05 | 46.0266 |
| CHR18 | 18447 | 37752841 | 2.57E-05 | 45.8968 |
| CHR19 | 20139 | 51149365 | 2.07E-05 | 46.8347 |
| CHR20 | 22081 | 47014184 | 2.47E-05 | 46.0687 |
| CHR21 | 23517 | 45688708 | 2.71E-05 | 45.6707 |
| CHR22 | 15233 | 34027391 | 2.36E-05 | 46.277 |
| MAH1 | 225675 | 469843677 | 2.53E-05 | 45.9712 |
| MAH2 | 218127 | 470199257 | 2.44E-05 | 46.1223 |
Repeat and gene annotation
The extensive de novo TE annotator (EDTA)35 was used to fully screen and group repeat elements. Briefly, a de novo repeat library constructed by RepeatModeler v2.0.136 was imported to RepeatMasker v4.1.1 (http://repeatmasker.org/) to predict repeats. Then, Repbase37 was introduced to predict homology repeats in RepeatMasker. In total we identified 235.46 Mb (50.11%) and 234.61 Mb (49.90%) repetitive sequences in MAH1 and MAH2, respectively. Among these, long terminal repeats (LTR) that accounted for 36.61% in MAH1 and 34.19% in MAH2 were the most abundant repeat elements (Supplementary Table S1). These results were comparable with the findings in the previous T2T DH genome version (Repeat elements: 52.62%; LTR: 34.85%)13.
Standard MAKER3 v3.01.0338 pipeline was used to annotate genes. All high-confidence protein sequences in swiss-prot39 database were imported for homology prediction. Transcripts from the 4 tissues, including root, leaf, flower and fruit, were used for gene prediction. Then AUGUSTUS v3.3.2 and SNAP v20131129 were used to train the ab-initio gene models. Finally, the MAKER3 pipeline was run again to obtain high-quality gene annotations. Functional characterization of the predicted coding genes was performed using eggNOG-mapper v240 based on the eggNOG v5.0 database41. A total of 40,889 and 38,269 protein-coding genes were annotated in MAH1 and MAH2, respectively. The total lengths of protein-coding genes were 148.54 Mb and 144.95 Mb, respectively. Average lengths of genes were 3.63 kb and 3.79 kb. Based on the eggNOG-Mapper results, 59,143 (74.72%) genes were functionally annotated (Table 7). Besides, BUSCO scores of protein-coding genes in MAH1 and MAH2 were up to 89.41% and 90.27% (Table 8).
Table 7.
Statistics of protein-coding genes in MAH1 and MAH2.
| MAH1 | MAH2 | |
|---|---|---|
| Number of protein coding genes | 40,889 | 38,269 |
| Total length of protein coding gene (bp) | 148,543,347 | 144,954,069 |
| Average length of protein coding gene (bp) | 3,632 | 3,787 |
| Total exon length (bp) | 48,236,300 | 49,463,483 |
| Average length of exon (bp) | 264 | 272 |
| Genes with one more exon | 28,286 | 25,849 |
| Genes with GO terms | 59,143 | |
Table 8.
Summary of BUSCO analysis of protein-coding genes in MAH1 (C: 89.41%) and MAH2 (C: 90.27%).
| MAH1 | MAH2 | |
|---|---|---|
| Complete BUSCOs (C) | 1443 | 1457 |
| Complete and single-copy BUSCOs (S) | 1374 | 1385 |
| Complete and duplicated BUSCOs (D) | 69 | 72 |
| Fragmented BUSCOs (F) | 75 | 74 |
| Missing BUSCOs (M) | 96 | 83 |
| Total BUSCO groups searched | 1614 | 1614 |
Note: The lineage dataset is embryophyte_odb10.
Identification of telomeres and centromeres
TIDK v0.2.1 (https://github.com/tolkit/telomeric-identifier) was used to find telomeres. In total 36 telomeres were found (Table 9). Plant centromeric regions are generally characterized by the presence of short tandem repeats that are highly enriched in these regions42, accompanied by a collapse in the density of LTR elements such as Copia. By identifying these distinctive features, centromeric regions can be located. We predicted centromeric regions according to the workflow in Shi et al.43, which employed the above approach. Using Tandem Repeats Finder v4.0944 with the parameters: trf genomes.fa 2 7 7 80 10 50 500 -f -d -m, we screened 183 bp, 148 bp, 124 bp, 125 bp, and 191 bp tandem repeat units as candidates based on sorted results and IGV results (Supplementary Table S2, Supplementary Figure S3). The centromeric regions were defined according to the density of 183 bp tandem repeat unit, which was the highest enriched centromeric repeat unit. Finally, all centromeric regions have been captured successfully (Table 10, Supplementary Figure S3).
Table 9.
Summary of telomere information of Musa acuminata ssp. malaccensis genome.
| CHR | Left Start | Left End | Left Length | Right Start | Right End | Right Length |
|---|---|---|---|---|---|---|
| CHR1 | 1 | 13,811 | 13,811 | 50,618,246 | 50,630,355 | 12,109 |
| CHR2 | NA | NA | NA | 36,081,437 | 36,099,580 | 18,143 |
| CHR3 | 1 | 11,011 | 11,011 | 43,033,809 | 43,043,358 | 9,549 |
| CHR4 | 1 | 11,620 | 11,620 | NA | NA | NA |
| CHR5 | NA | NA | NA | NA | NA | NA |
| CHR6 | 1 | 16,814 | 16,814 | NA | NA | NA |
| CHR7 | 1 | 16,051 | 16,051 | 37,869,370 | 37,896,144 | 26,774 |
| CHR8 | 1 | 18,389 | 18,389 | 51,435,804 | 51,440,739 | 4,935 |
| CHR9 | 1 | 10,157 | 10,157 | 46,918,312 | 46,957,856 | 39,544 |
| CHR10 | 1 | 5,537 | 5,537 | 41,999,867 | 42,012,631 | 12,764 |
| CHR11 | 1 | 11,515 | 11,515 | 34,613,922 | 34,622,327 | 8,405 |
| CHR12 | 1 | 12,705 | 12,705 | 49,988,939 | 50,002,820 | 13,881 |
| CHR13 | 1 | 10,262 | 10,262 | 36,111,649 | 36,147,335 | 35,686 |
| CHR14 | 1 | 17,885 | 17,885 | 42,973,945 | 42,981,928 | 7,983 |
| CHR15 | 1 | 9,436 | 9,436 | NA | NA | NA |
| CHR16 | NA | NA | NA | NA | NA | NA |
| CHR17 | 1 | 16,814 | 16,814 | 41,892,879 | 41,966,602 | 73,723 |
| CHR18 | 1 | 5,264 | 5,264 | 37,711,352 | 37,752,859 | 41,507 |
| CHR19 | 1 | 12,873 | 12,873 | 51,144,443 | 51,149,383 | 4,940 |
| CHR20 | 1 | 7,546 | 7,546 | 46,984,301 | 47,014,202 | 29,901 |
| CHR21 | 1 | 14,084 | 14,084 | 45,671,080 | 45,688,726 | 17,646 |
| CHR22 | 1 | 11,515 | 11,515 | 33,998,426 | 34,027,409 | 28,983 |
Table 10.
Summary of centromere information of Musa acuminata ssp. malaccensis genome.
| CHR | start | end | length | start_trf_id | end_trf_id |
|---|---|---|---|---|---|
| CHR01 | 34,370,592 | 38,397,126 | 4,026,534 | TRF_12565 | TRF_14496 |
| CHR02 | 7,524,984 | 11,726,285 | 4,201,301 | TRF_23444 | TRF_26360 |
| CHR03 | 21,803,558 | 22,962,260 | 1,158,702 | TRF_44924 | TRF_45373 |
| CHR04 | 20,301,989 | 21,676,450 | 1,374,461 | TRF_61392 | TRF_62137 |
| CHR05 | 24,142,990 | 25,898,645 | 1,755,655 | TRF_78752 | TRF_79866 |
| CHR06 | 23,280,519 | 23,708,701 | 428,182 | TRF_96837 | TRF_97118 |
| CHR07 | 20,753,904 | 22,245,710 | 1,491,806 | TRF_113137 | TRF_114095 |
| CHR08 | 21,057,435 | 22,701,427 | 1,643,992 | TRF_128874 | TRF_129777 |
| CHR09 | 24,616,864 | 28,481,467 | 3,864,603 | TRF_152152 | TRF_154884 |
| CHR10 | 17,354,345 | 18,650,231 | 1,295,886 | TRF_178427 | TRF_179172 |
| CHR11 | 15,992,185 | 17,570,998 | 1,578,813 | TRF_193271 | TRF_194294 |
| CHR12 | 34,320,878 | 36,937,480 | 2,616,602 | TRF_214097 | TRF_215243 |
| CHR13 | 7,460,959 | 11,858,861 | 4,397,902 | TRF_224519 | TRF_227526 |
| CHR14 | 21,657,257 | 22,734,720 | 1,077,463 | TRF_246003 | TRF_246434 |
| CHR15 | 20,157,001 | 21,828,979 | 1,671,978 | TRF_262341 | TRF_263283 |
| CHR16 | 22,536,071 | 24,398,251 | 1,862,180 | TRF_278747 | TRF_280033 |
| CHR17 | 21,596,438 | 23,729,965 | 2,133,527 | TRF_296609 | TRF_297937 |
| CHR18 | 20,698,855 | 21,941,522 | 1,242,667 | TRF_313322 | TRF_314065 |
| CHR19 | 20,950,265 | 23,593,891 | 2,643,626 | TRF_328834 | TRF_330287 |
| CHR20 | 24,652,418 | 27,307,919 | 2,655,501 | TRF_352211 | TRF_354226 |
| CHR21 | 18,468,410 | 18,926,218 | 457,808 | TRF_383621 | TRF_383906 |
| CHR22 | 16,235,391 | 17,087,235 | 851,844 | TRF_400050 | TRF_400648 |
Characterization of a reciprocal translocation in Musa acuminata
Nucmer v4.0.0rc145 was used to obtain the syntenic relationship between MAH1 and MAH2 with default parameters. Then the delta-filter was launched with parameters ‘-i 90 -l 15000’. In the same way, our haplotype-resolved assembly was aligned against MAv4 using nucmer. Mummerplot command was used to generate the dot plots (Supplementary Figure S4). Syri v1.6.346 with default parameters was used for identifying structural variants between MAH1 and MAH2 (Fig. 5). Overall, 47 translocations with a cumulative size of 2.70 Mb (~0.57%), 23 inversions with a cumulative size of 11.30 Mb (~2.40%), and 53 duplications with a cumulative size of 1.33 Mb (~0.28%) were defined. These structural variants were generally heterozygous, representing more complete genetic information compared with the double-haploid MAv4 genome.
Fig. 5.

The sequence collinearity and structural variants between MAH1 and MAH2.
MCscan tools47 were used to search for the syntenic relationships between the two haploid assemblies and MAv4 at the gene level. Briefly, ‘jcvi.compara.catalog’ module with ‘--cscore = 0.99’ and ‘jcvi.compara.synteny’ module with ‘--minspan = 30’ were used to build the syntenic regions; then, syntenic relationships were visualized by ‘jcvi.graphics.karyotype’ module. Besides, potential structural variants and heterozygous regions were shown in Supplementary Figure S5. A reciprocal translocation involving 3 Mb and 10 Mb from chromosome 01 and 04 was identified (Fig. 6a). These reciprocal translocation gene blocks were located in the translocated regions identified in whole genome alignment results (Supplementary Figure S4C,D). The 10-Mb segment from 261,650 to 10,745,936 bp in chromosome 01 of MAH1 was linked to 44,882,868 to 34,419,170 bp in chromosome 04 of MAv4 (Supplementary Figure S5). The 3-Mb segment from 34,734,628 to 37,810,715 bp in chromosome 04 of MAH1 was linked to 122,362 to 3,101,126 bp in chromosome 01 of MAv4. The reciprocal translocation between MAH2 and MAv4 was located in the similar genomic regions. The huge difference in chromosome length in chromosome 01 and chromosome 04 between MAH1/2 and MAv4 was also derived from this reciprocal translocation, while other chromosome lengths and genomic total lengths were comparable (Table 3).
Fig. 6.

The large reciprocal translocation between chromosome 01 and chromosome 04. (a) Genomic synteny between DH-Pahang (MAv4) and wild M. acuminata. (b) Hi-C heatmap of chromosome 01-04. (c) The possible chromosome structures in wild M. acuminata. The targeted PCR result suggests that homozygous reciprocal translocations (only 1T4) exist in our sample, consistent with our whole genome alignment results in sequence and gene levels.
We further performed GO enrichments based on the extracted genes located in the translocated regions using TBtools v1.10848. The genes in the 10-Mb segment of MAH1 were not significantly enriched in any biological process, while those on the 3-Mb segment were enriched in several pathways associated with flower development (Supplementary Table S3), including anther development (GO:0048653), stamen development (GO:0048443), regulation of flower development (GO:0009909), and floral whorl development (GO:0048438). For further validation, we used nucleotide BLAST tools in National Center for Biotechnology Information (NCBI) with default parameters to align identified genes to non-redundant database, and checked gene functions manually.
Data Records
All raw sequencing reads have been deposited in the National Center for Biotechnology Information (NCBI) under BioProject ID PRJNA962682, and the National Genomics Data Center (NGDC) under BioProject ID PRJCA018611. The PacBio HiFi, Nanopore, Hi-C, Illumina sequencing reads have been deposited in the NCBI Sequence Read Archive database with accession group numbers SRP43512749. Genome assembly is available from GenBank with accession number GCA_030219345.150. The genome annotation files have been submitted to the online open access repository Figshare database51, including a high-quality reference genome that we constructed for guidance.
Technical Validation
Manual correction for chromosome scaffolding
For constructing a high-quality reference genome, we used Juicebox to manually correct the reference genome based on Hi-C alignments (Supplementary Figure S1). Finally, 471.04 Mb (95.83%) contigs were anchored to 11 pseudo-chromosomes. Then, we started to orient, sort and group our haplotype-resolved genome based on this high-quality reference genome. We also used Juicebox to manually correct the haplotype-resolved genome based on Hi-C alignments. In total, 469.83 Mb (93.82%) and 470.21 Mb (97.08%) contigs were anchored to 11 chromosome pairs, respectively. We further examined the Hi-C alignments in chromosome 01 and chromosome 04 in Juicebox (Fig. 6b), and confirmed the accurate assemblies of chromosome 01 and chromosome 04. Besides, chromosome 01 consists of only one contig (Table 3), further confirming its high continuity.
Targeted PCR confirmed the reciprocal translocation between Chr01 and Chr04
Based on the genomic syntenic analysis between our assembly and MAv4, we identified a large reciprocal translocation from chromosomes 01 and 04, corresponding to the translocation found in a previous study9. In that study, three pairs of primers were designed to amplify the breakpoints located along the reference and hypothesized chromosome structures, thereby showing the presence of chromosomes 01, 04, and 1T4 resulting from the translocation. Here we used the same primer pairs to perform targeted PCR to validate the chromosome structures found in our sample (Fig. 6c). DNA was extracted from leaf tissue of M. acuminata ssp. malaccensis. PCR was performed in 50-μL volumes containing 2.5 ng of gDNA, 1 μL of specific primers, 32 μL of distilled, deionized water, and 0.5 μL of TaKaRa LA Taq® (Vazyme) using an Eastwin Life Science EDC810 PCR amplification system. The reaction conditions for thermal cycling were 94 °C for 5 min, followed by 35 cycles of 94 °C for 45 s, 56 °C for 45 s, and 72 °C for 60 s. Thereafter, PCR products were visualized by 2% agarose gel-electrophoresis with a 100 bp DNA ladder. Only the breakpoint of chromosome 1T4 was amplified in our studied sample, suggesting that the reciprocal translocation involving 3 and 10 Mb segments from chromosomes 01 and 04 existed in both haploid genomes of the M. acuminata sample (Fig. 6c). This finding was consistent with our whole genome alignment results in sequence and gene levels.
Supplementary information
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 32070237, 31261140366), and the Strategic Priority Research Program of Chinese Academy of Sciences (Grant No. XDB31000000).
Author contributions
H.R.H., X.J.G. and Y.Z. designed and supervised the research; X.L., R.A. and H.R.H. wrote the manuscript; X.L., R.A. and X.W. analysed the data; X.L. and W.M.L. collected the experimental materials. All authors contributed to manuscript revision, read and approved the submitted version.
Code availability
No special code was used for this study. All software mentioned in methods could be found in the community. If no detail parameters were mentioned for the software, default parameters were used as suggested by the developer.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Xin Liu, Rida Arshad.
Supplementary information
The online version contains supplementary material available at 10.1038/s41597-023-02546-9.
References
- 1.Brozynska M, Furtado A, Henry RJ. Genomics of crop wild relatives: expanding the gene pool for crop improvement. Plant Biotechnol. J. 2016;14:1070–1085. doi: 10.1111/pbi.12454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bohra A, et al. Reap the crop wild relatives for breeding future crops. Trends Biotechnol. 2022;40:412–431. doi: 10.1016/j.tibtech.2021.08.009. [DOI] [PubMed] [Google Scholar]
- 3.Castaneda-Alvarez NP, et al. Global conservation priorities for crop wild relatives. Nat. Plants. 2016;2:16022. doi: 10.1038/nplants.2016.22. [DOI] [PubMed] [Google Scholar]
- 4.Perrier X, et al. Multidisciplinary perspectives on banana (Musa spp.) domestication. Proc. Natl. Acad. Sci. USA. 2011;108:11311–11318. doi: 10.1073/pnas.1102001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davey MW, et al. A draft Musa balbisiana genome sequence for molecular genetics in polyploid, inter- and intra-specific Musa hybrids. BMC Genom. 2013;14:683. doi: 10.1186/1471-2164-14-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perrier X, et al. Combining biological approaches to shed light on the evolution of edible bananas. Ethnobot. Res. App. 2009;7:199–216. [Google Scholar]
- 7.Shepherd K. Cytogenetics Of The Genus Musa (International Network for the Improvement of Banana and Plantain, 1999).
- 8.Hippolyte I, et al. A saturated SSR/DarT linkage map of Musa acuminata addressing genome rearrangements among bananas. BMC Plant Biol. 2010;10:65. doi: 10.1186/1471-2229-10-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martin G, et al. Evolution of the banana genome (Musa acuminata) is impacted by large chromosomal translocations. Mol. Biol. Evol. 2017;34:2140–2152. doi: 10.1093/molbev/msx164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dupouy M, et al. Two large reciprocal translocations characterized in the disease resistance-rich burmannica genetic group of Musa acuminata. Ann. Bot. 2019;124:319–329. doi: 10.1093/aob/mcz078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin G, et al. Chromosome reciprocal translocations have accompanied subspecies evolution in bananas. Plant J. 2020;104:1698–1711. doi: 10.1111/tpj.15031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D’Hont A, et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature. 2012;488:213–217. doi: 10.1038/nature11241. [DOI] [PubMed] [Google Scholar]
- 13.Belser C, et al. Telomere-to-telomere gapless chromosomes of banana using nanopore sequencing. Commun. Biol. 2021;4:1047. doi: 10.1038/s42003-021-02559-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu G, et al. Two divergent haplotypes from a highly heterozygous lychee genome suggest independent domestication events for early and late-maturing cultivars. Nat. Genet. 2022;54:73–83. doi: 10.1038/s41588-021-00971-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun X, et al. Phased diploid genome assemblies and pan-genomes provide insights into the genetic history of apple domestication. Nat. Genet. 2020;52:1423–1432. doi: 10.1038/s41588-020-00723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marcais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ranallo-Benavidez TR, Jaron KS, Schatz MC. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020;11:1432. doi: 10.1038/s41467-020-14998-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng H, Concepcion GT, Feng X, Zhang H, Li H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods. 2021;18:170–175. doi: 10.1038/s41592-020-01056-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alonge M, et al. RaGOO: fast and accurate reference-guided scaffolding of draft genomes. Genome Biol. 2019;20:224. doi: 10.1186/s13059-019-1829-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Durand NC, et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016;3:95–98. doi: 10.1016/j.cels.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dudchenko O, et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 2017;356:92–95. doi: 10.1126/science.aal3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Durand NC, et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 2016;3:99–101. doi: 10.1016/j.cels.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu J, Fan J, Sun Z, Liu S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics. 2020;36:2253–2255. doi: 10.1093/bioinformatics/btz891. [DOI] [PubMed] [Google Scholar]
- 25.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krzywinski M, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barnett DW, Garrison EK, Quinlan AR, Stromberg MP, Marth GT. BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics. 2011;27:1691–1692. doi: 10.1093/bioinformatics/btr174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ou S, Jiang N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 2018;176:1410–1422. doi: 10.1104/pp.17.01310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manni M, Berkeley MR, Seppey M, Simao FA, Zdobnov EM. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021;38:4647–4654. doi: 10.1093/molbev/msab199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rhie A, Walenz BP, Koren S, Phillippy AM. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome biol. 2020;21:245. doi: 10.1186/s13059-020-02134-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ou S, et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 2019;20:275. doi: 10.1186/s13059-019-1905-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flynn JM, et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA. 2020;117:9451–9457. doi: 10.1073/pnas.1921046117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bao W, Kojima KK, Kohany O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA. 2015;6:11. doi: 10.1186/s13100-015-0041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Campbell MS, Holt C, Moore B, Yandell M. Genome annotation and curation using MAKER and MAKER-P. Curr Protoc Bioinformatics. 2014;48:4.11.11–14.11.39. doi: 10.1002/0471250953.bi0411s48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cantalapiedra CP, Hernandez-Plaza A, Letunic I, Bork P, Huerta-Cepas J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 2021;38:5825–5829. doi: 10.1093/molbev/msab293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huerta-Cepas J, et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47:D309–D314. doi: 10.1093/nar/gky1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Melters DP, et al. Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol. 2013;14:R10. doi: 10.1186/gb-2013-14-1-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi X, et al. The complete reference genome for grapevine (Vitis vinifera L.) genetics and breeding. Hortic. Res. 2023;10:uhad061. doi: 10.1093/hr/uhad061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marcais G, et al. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018;14:e1005944. doi: 10.1371/journal.pcbi.1005944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goel M, Sun H, Jiao WB, Schneeberger K. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol. 2019;20:277. doi: 10.1186/s13059-019-1911-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tang H, et al. Synteny and collinearity in plant genomes. Science. 2008;320:486–488. doi: 10.1126/science.1153917. [DOI] [PubMed] [Google Scholar]
- 48.Chen C, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant. 2020;13:1194–1202. doi: 10.1016/j.molp.2020.06.009. [DOI] [PubMed] [Google Scholar]
- 49.2023. NCBI Sequence Read Archive. SRP435127
- 50.Liu X, 2023. Musa acuminata subsp. malaccensis genome assembly. GenBank. GCA_030219345.1
- 51.Liu X, 2023. The phased telomere-to-telomere reference genome of Musa acuminata, a main contributor to banana cultivars. Figshare. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- 2023. NCBI Sequence Read Archive. SRP435127
- Liu X, 2023. Musa acuminata subsp. malaccensis genome assembly. GenBank. GCA_030219345.1
- Liu X, 2023. The phased telomere-to-telomere reference genome of Musa acuminata, a main contributor to banana cultivars. Figshare. [DOI] [PMC free article] [PubMed]
Supplementary Materials
Data Availability Statement
No special code was used for this study. All software mentioned in methods could be found in the community. If no detail parameters were mentioned for the software, default parameters were used as suggested by the developer.