

Abstract
Cellular senescence is a well-established driver of aging and age-related diseases. There are many challenges to mapping senescent cells in tissues such as the absence of specific markers and their relatively low abundance and vast heterogeneity. Single-cell technologies have allowed unprecedented characterization of senescence; however, many methodologies fail to provide spatial insights. The spatial component is essential, as senescent cells communicate with neighboring cells, impacting their function and the composition of extracellular space. The Cellular Senescence Network (SenNet), a National Institutes of Health (NIH) Common Fund initiative, aims to map senescent cells across the lifespan of humans and mice. Here, we provide a comprehensive review of the existing and emerging methodologies for spatial imaging and their application toward mapping senescent cells. Moreover, we discuss the limitations and challenges inherent to each technology. We argue that the development of spatially resolved methods is essential toward the goal of attaining an atlas of senescent cells.
Cellular senescence refers to the irreversible growth arrest that occurs when cells become exposed to a variety of stressors. Induction of senescence alters almost every aspect of cell biology, from marked changes in transcriptome and proteome, epigenetic remodeling of chromatin and changes in quantity and functionality of organelles to enhanced secretion of pro-inflammatory molecules commonly known as the senescence-associated secretory phenotype (SASP)1 (Fig. 1).
Fig. 1 |. Senescence is a complex cell fate that alters almost every aspect of cell biology.
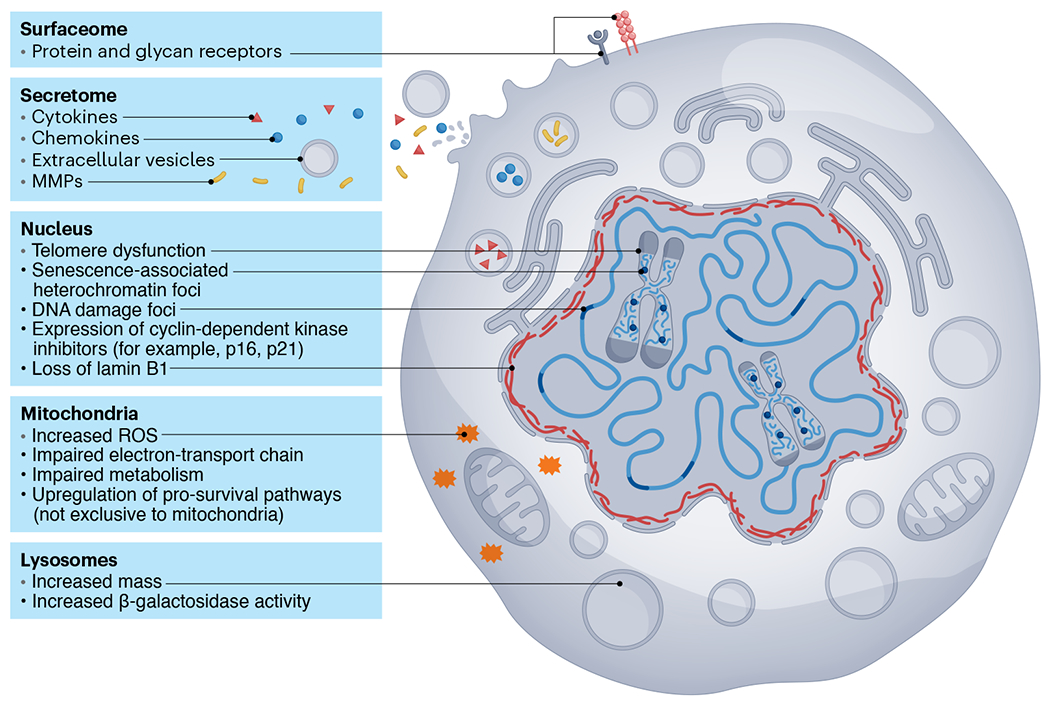
Some changes observed during senescence involve (1) alterations in protein and glycan receptors, (2) a pro-inflammatory SASP, (3) multiple nuclear abnormalities, such as DNA damage, telomere dysfunction, chromatin alterations and modifications to the nuclear envelope, (4) mitochondrial dysfunction and (5) changes in lysosomal mass and functionality. MMP, matrix metalloproteinase; ROS, reactive oxygen species.
Cells bearing senescence-associated markers have been shown to accumulate in most tissues during aging and age-related diseases. Importantly, several studies have shown that elimination of senescent cells in mice either genetically or by using drugs that can kill senescent cells (senolytics) alleviates several pathologies during aging and age-related disorders2. Therefore, these cells are thought to represent promising therapeutic targets to delay or even reverse functional deficits in aging. It should be noted, however, that senescent cells also have important physiological roles and that their clearance may be detrimental in certain conditions3–5.
Despite multiple molecular changes being described in senescent cells, the field is increasingly recognizing that these cells are incredibly difficult to detect in tissues. One major challenge is that there is no single, stand-alone marker to identify a senescent cell. None of the markers used to detect senescent cells are individually specific. For example, the commonly used marker senescence-associated β-galactosidase (SA-β-Gal) at pH 6 can be detected in vitro in confluent and immortalized cells6 and in activated macrophages7, while cell cycle kinase inhibitors p16INK4a and p21CIP1 can be expressed irrespectively of senescence in certain contexts7,8. For these reasons, the field collectively suggested that a multi-marker approach should be applied to detect senescent cells in vivo, and the idea of ever identifying a single universal senescence marker is regarded with widespread skepticism1.
Another major challenge in detecting senescent cells is that these cells are thought to be relatively rare, with some estimates indicating that they can be present below 5% in aged tissues9. This low abundance makes it paramount that single-cell methodologies have sufficient resolution and throughput to be able to detect senescence-associated markers and cover a sufficient area of tissue. Adding to this challenge, some of the senescence-associated markers require imaging platforms capable of visualizing subcellular structures, as we will discuss later.
The advance of all omics technologies in conjunction with increasingly affordable new achievements in microfluidic and microprinting fields has powered the revolution of single-cell big data. In addition, the convergence of these technological innovations with a new wave of bioinformatic methodologies is altering the way in which we approach science and discovery. This technological revolution has already provided the community with extraordinary resources such as Tabula Muris10 and Tabula Sapiens11. Several studies have used single-cell technologies to successfully identify senescence-associated signatures in the context of aging12,13. There are, however, several technical factors that limit the information that one can extract from such analyses, particularly as they rely on dissociated cell suspensions. Preparation and preservation of the cell suspension can affect the final proportion of cells that are sampled. Some cell types are too large and incompatible with microfluidic-based single-cell isolation. In fact, size is a major issue in the preservation and detection of senescent cells, as they are characterized by increased cell volume and therefore can be irretrievably lost in these analyses. Adding to these limitations, inferring spatial relationships from single-cell suspensions is almost impossible.
The spatial aspect is of major importance in understanding senescence, as senescent cells communicate with neighboring cells, impacting their function as well as the composition of extracellular space. Senescent cells can induce senescence in neighboring cells14,15 and, through the SASP, recruit immune cells to their vicinity. The SASP can have a major impact on various biological processes, including cell proliferation, angiogenesis, inflammation16, epithelial-to-mesenchymal transition17, tissue repair18 and wound healing3 (Fig. 2). The nature of this response is dependent on cell type and physiological context. Therefore, spatially resolved methods capable of quantitatively identifying senescent cells by a complex, multi-factorial molecular signature and mapping their location and interactions within the microenvironment are urgently needed.
Fig. 2 |. Senescence communicates with neighboring cells and alters their function via the SASP.
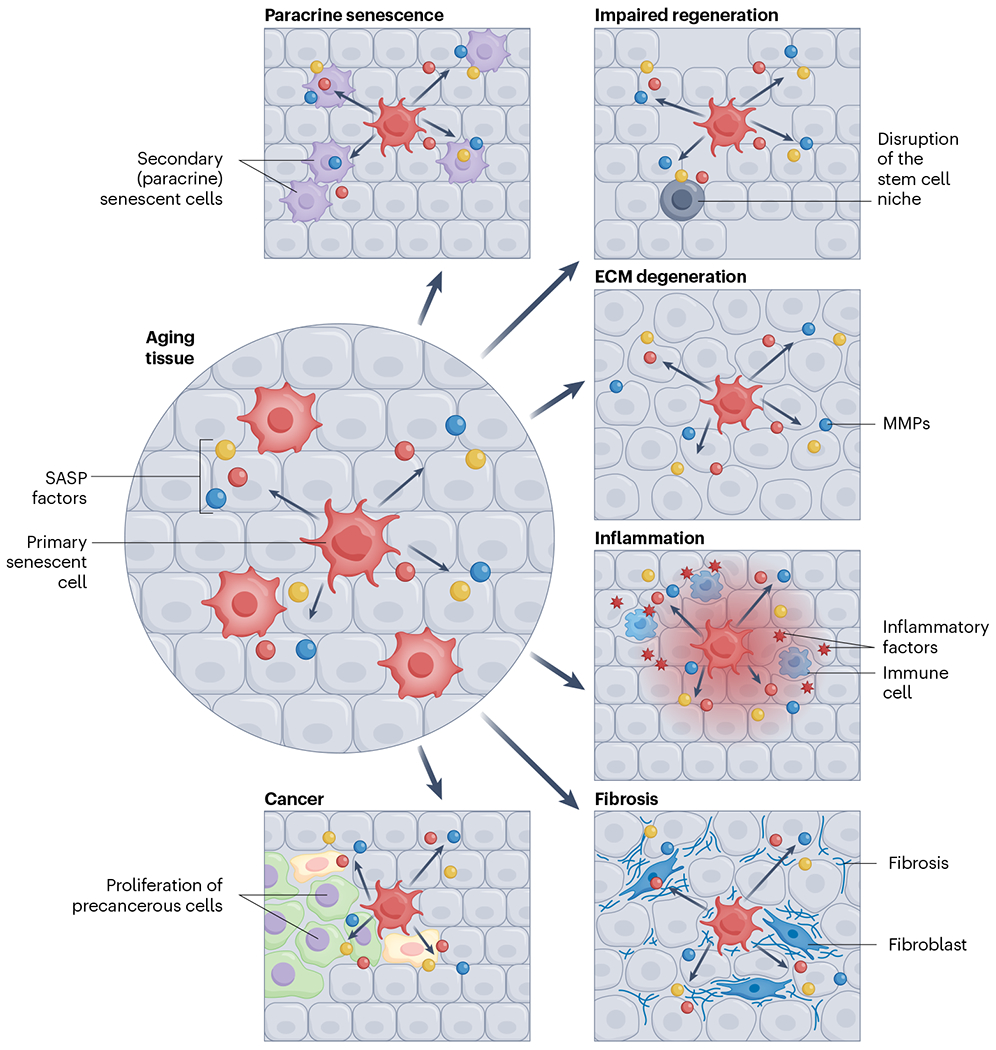
Senescent cells can (1) spread senescence to surrounding cells, (2) disrupt stem cell niches and thereby impair tissue regeneration, (3) lead to extracellular matrix (ECM) degeneration, resulting in aberrant tissue architecture, (4) drive the recruitment of immune cells and exacerbate tissue inflammation, (5) affect tissue fibrosis and (6) stimulate the proliferation of precancerous cells.
This Review represents the collective knowledge from the imaging mapping working group from the SenNet, an NIH Common Fund initiative. The SenNet Consortium is invested in the development and application of spatial omics technologies not only to characterize and map senescence cells in tissues but also to establish potential relationships between senescent cells and their environment19. Here, we describe some current methodologies used by the SenNet Consortium to map senescent cells in tissues in a spatially resolved manner, ranging from low to high throughput. We also describe current developments in image-analysis approaches and the emergent use of deep learning methods to detect senescent cells. Importantly, we discuss limitations of available technologies and challenges of applying them to the detection of senescent cells. We also speculate about the opportunities and challenges that the ongoing revolution in single-cell spatially resolved methodologies will bring to the field of cellular senescence.
Imaging-based technologies to detect senescent cells in tissue sections
Imaging-based technologies are essential for advancing senescence research. Over the years, various methods have been developed to achieve spatially resolved measurements of messenger RNA (mRNA) or proteins in biological samples. Here, we describe some of the methods, ranging from low plex to multiplex, that are currently being used within the SenNet Consortium (Fig. 3). Our purpose is not to compile a comprehensive list of markers or to make any considerations about their suitability and specificity. We intend to briefly describe some of the most frequently used techniques and how they have been used in the context of senescent cell detection in tissue sections as well as their advantages and limitations.
Fig. 3 |. Spatially resolved methods for mapping senescent cells and studying senescence-associated pathology.
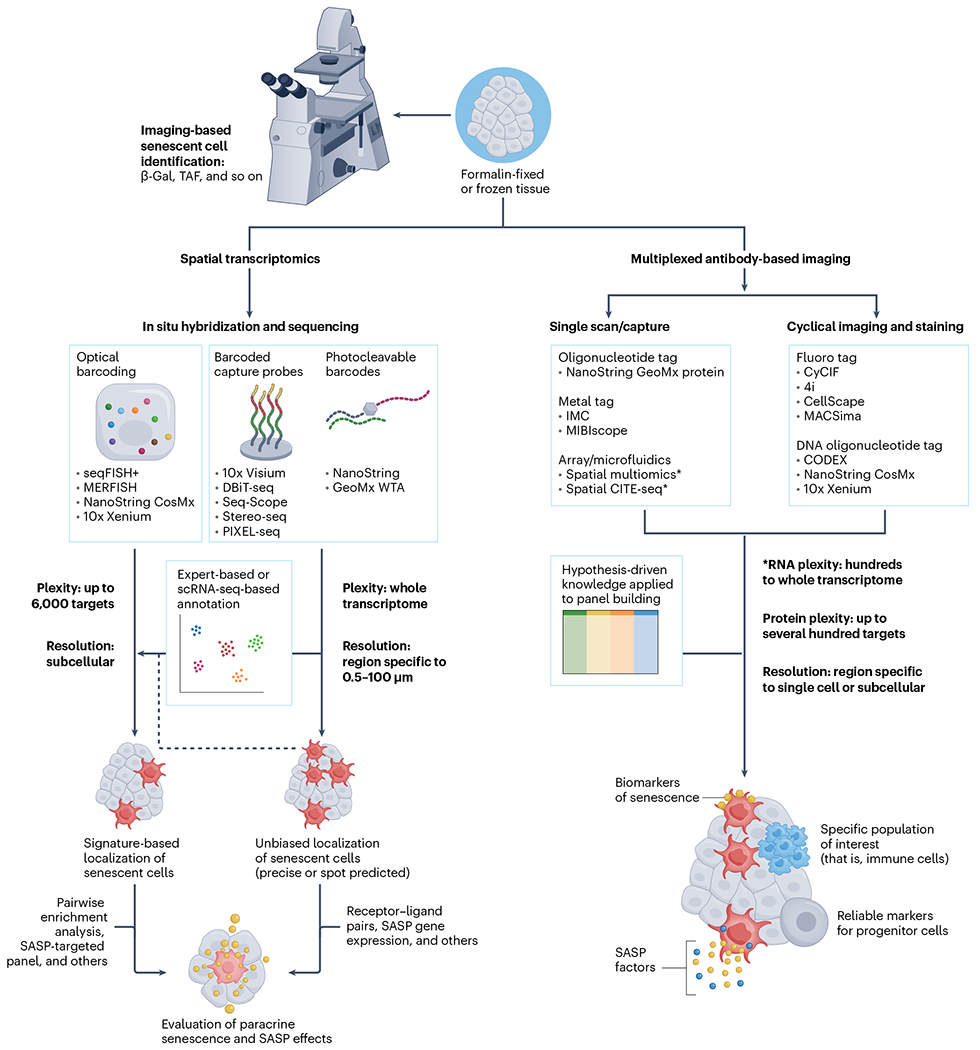
Three major approaches that have been proposed or are already being used for detection of senescent cells in freshly frozen or FFPE tissue samples. Imaging-based methods require light or a fluorescent microscope and include detection of established features of senescent cells, such as SA-β-Gal or TAF using commercially available assay kits. Various spatial transcriptomic methods can be generally assigned to those using in situ or ex situ sequencing technologies, depending on whether complementary DNA (cDNA) amplification and signal detection are performed at the physical transcript location or whether the transcript location is barcoded using a probe that is hybridized to target RNA, providing spatial information during standard sequencing procedures. Spatial transcriptomic methods can be used for both precise localization of cells expressing signature genes with subcellular resolution or for unbiased identification and investigation of senescence hotspots in larger tissue areas, covering the full transcriptome but trading plexity for spatial resolution. A plethora of downstream analysis techniques can help with evaluation of cellular neighborhoods and the impact of senescent cells on surrounding tissues. Multiplexed antibody-based methods are serving as high-plex, high-throughput approaches to characterize the protein milieu of senescent cells and their neighbors. A key feature of these methods is the ability to focus on specific cell populations or areas with abundance of particular biomolecules of interest (including glycans and lipids) during the data-acquisition step. Depending on the hypotheses or research questions, panels can be built to not only localize and distinguish senescent cells from other cells but also to evaluate (patho-)physiological effects on surrounding tissues by investigating SASP factor distribution and markers of major cellular biology processes, such as the DNA damage, endoplasmic reticulum stress, mitochondrial dysfunction, and so on. WTA, whole transcriptome analysis.
Low-plex imaging methods
Thus far, most studies aiming to identify senescent cells at the single-cell level in a spatially resolved manner have relied on the detection of one or few types of biomolecules using methods such as histochemistry, immunohistochemistry, immunofluorescence in situ hybridization (ImmunoFISH), fluorescence in situ hybridization (FISH) and RNA in situ hybridization (RNA-ISH). Here, we will provide some examples.
Histochemical methods.
Detection of the activity of the lysosomal enzyme SA-β-Gal at pH 6.0 is a common method used to detect senescent cells20. Limitations of this methodology for identification of senescent cells are that it requires frozen tissue sections (not applicable to formalin-fixed paraffin-embedded (FFPE) samples) and its diffused cytoplasmic staining can be challenging to evaluate. Another histochemical assay to detect senescent cells in tissues is the detection of lipofuscin using dyes such as Sudan Black B or analogs21. Advantages of this method are that it can be applied to FFPE tissues and combined with other immunohistochemical techniques.
Immunohistochemistry and immunofluorescence.
Expression of p21CIP1, p16INK4A, p53, components of the DNA damage response (DDR; for example, γH2A.X and 53BP1)22, decreased lamin B1 (ref. 23), loss of nuclear high-mobility group box 1 (HMGB1)24 and expression of GLB1 (ref. 25) (which encodes lysosomal β-d-galactosidase) are common senescence-associated markers assessed in tissues by immunohistochemistry and immunofluorescence. In immunohistochemical techniques, common challenges in visualizing senescence-associated markers are low levels of antigen expression, lack of specificity of the primary antibody, variability between antibodies even from the same manufacturer, cross-reactivity between primary and/or secondary antibodies, autofluorescence, quality of tissue fixation and others. Notably, many of these issues also affect highly multiplexed, antibody-based spatial imaging platforms. In addition, it is well established within the senescence community that many commercially available antibodies that target mouse p16INK4A are unspecific, making its detection in tissues unreliable. As such, we advise careful antibody validation to ensure that quality reagents are used, as, regrettably, often the information in commercial antibody datasheets or in academic publications does not necessarily withstand further experimental scrutiny.
ImmunoFISH, FISH and RNA-ISH.
ImmunoFISH has been widely used in the senescence field to detect telomere-associated DNA damage foci (TAF)26,27. This method involves evaluating colocalization between DDR foci (frequently using antibodies targeting γH2A.X and 53BP1) and FISH signals from telomere-specific peptide nucleic acid probes by fluorescence microscopy. Centromere FISH has also been used to detect senescence-associated distension of satellites (SADS), also a senescence-associated marker, in multiple tissues28–30. Both methods require high-resolution fluorescence imaging, generation of z stacks (to acquire three-dimensional (3D) images) and have high analytical burden and therefore are not amenable to large-scale studies. RNA-ISH has also been used to visualize single RNA molecules encoding senescence-associated markers (for example, p16INK4A, p21CIP1 and specific SASP components) in different tissues13,16,31. These methods can be applied to both frozen and FFPE tissue sections. Factors such as tissue processing and autofluorescence greatly affect image quality and readout accuracy.
All methods mentioned here have the limitation of detecting one or relatively few biomolecules simultaneously and are therefore not adequate to characterize the molecular heterogeneity of senescent cells in a spatially resolved manner. There are, however, several advantages: these techniques are established, widely used and inexpensive; as such, protocols are relatively easy to apply and are generally reproducible.
Super-resolution microscopy.
Super-resolution microscopy has emerged as having great potential to investigate biological samples at unprecedented levels of detail, as new and powerful fluorescence-based methods now permit imaging cellular compartments at a resolution beyond that of the diffraction limit of light (<200 nm). Super-resolution microscopy includes techniques such as structured illumination microscopy, photo-activated localization microscopy, stimulated emission depletion and stochastic optical reconstruction microscopy (STORM), which have been reviewed extensively elsewhere32. Several features associated with cellular senescence can benefit from the increased resolution afforded by these techniques. For example, structured illumination microscopy was used to estimate the density of nuclear pores33. Features that require detection of colocalized signals can also benefit from the increased resolution afforded by super-resolution methods. A prominent example is detection of DNA damage, which is achieved, for example, by detection of colocalized foci of 53BP1 and γH2A.X within the nucleus as evidence of DNA segments with chromatin alterations reinforcing senescence34 or TAF35. The increased resolution can make it possible to better identify these foci and quantify their number in individual cells. Senescent cells are known to display substantial changes in the organization of chromatin and DNA within the nucleus36. The ability of STORM to provide a detailed rendering of the structure of chromatin fibers37 makes it ideal to investigate changes in heterochromatin distribution within senescent nuclei, such as the loss of lamin-associated domains38, which has been primarily investigated via electron microscopy techniques39. Recent advances in 3D DNA FISH such as OligoSTORM can also be used to study, in more detail, local changes in the 3D structure of senescent chromosomes in individual cells that have been identified by genomic techniques such as high-throughput chromosome conformation capture (Hi-C)40,41.
Super-resolution imaging is still facing limitations that challenge its widespread adoption. For example, there are only a few effective photoswitchable dyes that can be used in multi-color STORM. To overcome this limitation, many laboratories have taken advantage of microfluidic systems to image multiple cycles of STORM using the same dye. This method, however, is technically limiting and expensive. In addition, the recording of millions to billions of single-molecule localizations of fluorescent probes in techniques such as STORM comes at the cost of a substantially more demanding analytical and storage infrastructure. This is particularly relevant when applying these techniques to image senescent cells in tissues, given their very low abundance. A solution to overcome this problem is to use conventional widefield microscopy to identify putative senescent cells, which are then targeted for super-resolution imaging.
High-plex imaging methods
Spatial proteomics.
Spatially resolved proteomic approaches have the potential to characterize the density and microanatomical location of senescent cells, to define the impact of senescent cells on their microenvironment and the immune cell types interacting with them and to identify the distribution of key protein senescence effectors within human tissues. To date, there has been a disproportionate use of changes in mRNA levels to detect senescent cells. However, for many senescence-associated markers, it is not yet clear whether changes in mRNA correspond to a change in protein expression. Furthermore, high-plex proteomics can potentially aid in concurrently identifying senescence-associated features otherwise undetectable by transcriptomics such as morphologic changes to organelles, protein mislocalization and post-translational modifications. Hence, the application of proteomics will be critical for defining senescent cell biology.
Fundamental considerations that define the value of spatial proteomic platforms include (1) the dynamic range of expressed protein detection, (2) the minimum distance between measured protein features (spatial resolution) and (3) the total area of spatially resolved tissue analyzed (spatial throughput). The majority of current high-plex spatial proteomic platforms use panels of tagged antibodies to detect and quantify between ten and 60 proteins, although some have reported higher numbers. The benefits, limitations and validation of these tagged antibody approaches have been recently reviewed in depth42. Antibody-based approaches have an advantage over spatial transcriptomics in that multiple functional biomarkers can be simultaneously detected and localized to specific subcellular compartments. As an alternative to antibody-based methods, innovations for unbiased mass spectrometry-based studies are evolving to query a larger swath of the proteome and metabolome with spatial orientation in tissues, although these methods are still fairly limited in throughput and resolution43,44, restricting their application to mapping rare senescent cells.
However, there are challenges. For instance, expression of p16INK4A protein ranges from undetectable to qualitatively low in aged tissues and is an example of a senescence-associated marker that requires a consistent and robust limit of detection sensitivity for accurate studies.
A high degree of subcellular resolution is also critical in the evaluation of biomarker distribution, as certain senescence patterns are linked to specific cellular compartments. For example, HMGB1, a ubiquitous and predominantly nuclear protein, localizes to the cytoplasm and is secreted during senescence24. Similarly, levels of lamin B1, which is a component of the nuclear lamina, decrease during senescence23, and its detection requires sufficient resolution to visualize a 30–100-nm-thick structure. Because both these senescence-associated markers rely on evaluating their absence or lower expression in a heterogeneous tissue, their detection may be particularly challenging and require imaging platforms with higher resolution.
Thorough in situ characterization of DDR foci also requires high-resolution imaging to detect and quantify distinct intranuclear puncta, as DDR foci are often under 1 μm34,35. Probes targeting SA-β-Gal will stain the lysosomes specifically, which vary in size from 100 nm to over 1 μm20. This highlights the importance of selecting the optimal platform based on its resolution (Table 1).
Table 1 |.
Comparison of spatial proteomic methods
| Platform | Supplier | Compatible tissue preparation | Max plexity | Resolution limit | Tissue area limit | Standard FOV | Stitch capability | Cyclic/all in one | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|
| Stain | Detection | |||||||||
| PhenoCycler (fmr. CODEX) | Akoya Biosciences | FFPE, FF | >100 | 0.25 μm | 612 mm2 | 0.5 × 0.7 mm | Yes | All in one | Cyclic | 101 |
| CosMx SMI | NanoString | FFPE, FF | 64–100 | <100 nm | 375 mm2 | 0.7×0.9 mm | Yes | All in one | Cyclic | 50 |
| MIBI-TOF | Ionpath | FFPE, FF | >40 | 350 nm | >800 mm2 | 0.8×0.8 mm | Yes | All in one | All in one | 102 |
| IMC/Hyperion Imaging System | Standard BioTools (fmr. Fluidigm) | FFPE, FF | >40 | 1 μm | >675 mm2 | 0.25×0.25 mm | Yes | All in one | All in one | 103 |
| MACSima | Miltenyi | FFPE, FF cells | >100 | 106 nm | >800 mm2 | 0.3 × 0.5 mm | Yes | Cyclic | Cyclic | 104 |
| CellScape | Canopy | FFPE, FF cells | 75 | 182 nm | 320 mm2 | 0.796–2.714 mm2 | Yes | Cycle | Cyclic | 105 |
FF, fresh frozen; fmr., formerly; FOV, field of view; max, maximum.
Selection of an appropriate spatial proteomic platform to study senescence requires not only consideration of image-capture capabilities but also technical advantages and limitations in characterizing senescence. Iterative immunofluorescence methods, such as MACSima and CellScape, are capable of imaging with enough resolution to detect subtle changes in organelle structure and morphology (100–200 nm per pixel) and do not require specialized antibody conjugations, which are time consuming and expensive and have the potential to negatively affect their ability to recognize targets. Methods such as cyclic immunofluorescence (CyCIF)45 and iterative indirect immunofluorescence imaging (4i)46, which do not require proprietary instrumentation, allow the elution of antibodies in relatively mild conditions that preserve the sample structure up to and possibly over 20 rounds of elution and imaging, allowing the detection of over 40 protein targets. Iterative indirect immunofluorescence imaging was already successfully used to identify senescence-associated markers and molecular signatures from a set of 48 cell cycle proteins47, hence achieving a similar plex level as that of commercial platforms without requiring a dedicated instrument, apart from a standard microfluidic system.
However, all iterative immunofluorescence methods share several caveats in their application. Repeated rounds of staining and bleaching may alter epitope stability and are subject to incomplete fluorophore inactivation between cycles. The added length of time for each subsequent staining incubation also hinders analytical throughput relative to other spatial proteomic innovations, making it more challenging and time consuming to identify rare cell types.
Platforms using barcoded antibodies (PhenoCycler, CosMx) implement a single antibody staining step followed by transient hybridization detection cycles with fluorescently tagged oligonucleotides. In these platforms, the diversity of barcode conjugates makes them unparalleled in their plex limit, potentially exceeding 100 targets on a single tissue section. Image resolution using these systems is similar to that of the aforementioned cyclical staining methods, although fluorescence-based detection may still suffer from high noise-to-signal ratios for low-expression targets such as p16INK4A, particularly in tissues that have undergone harsh fixation. Mass spectrometry-based platforms, which use up to 40 different metal-conjugated antibodies in a single staining and detection step, permit highly sensitive protein detection. Metal tagging can circumvent other issues common to fluorescence-based approaches such as tissue autofluorescence; however, protocols for mass tagging antibodies can negatively impact their sensitivity. Furthermore, mass imaging obliterates the tissue sample, rendering multiplexed ion beam imaging (MIBI) and imaging mass cytometry (IMC) incompatible with sequential analyses. Irrespective of staining and detection modalities, platforms that limit the imageable tissue area (CosMx, MACSima, CellScape, PhenoCycler, IMC) may altogether miss populations of senescent cells, which may be unequally distributed, in subsections of larger tissues.
Attempts to characterize senescence spatially at the protein level must balance the breadth of tissue coverage with the depth of the data generated (Table 1). Protocols and reagents must be carefully considered and thoroughly validated in appropriate tissues. High-plex imaging requires substantial time investment in the generation and optimization of conjugated antibody panels, in addition to the time required to acquire images. Moreover, even established workflows may require several days to weeks to capture, process and analyze enough raw imaging data to effectively characterize senescent cell features within a single tissue slide. The validation of high-quality antibodies for senescence and SASP biomarkers, optimized conjugation of these reagents to necessary reporter systems, continued improvement in both spatial resolution and throughput and determination of the data sufficiency for spatial identification of the SASP using each marker are key milestones to drive forward human senescence research with spatial proteomic techniques.
Spatial transcriptomics
Current spatial gene expression profiling methods can identify transcripts’ locations at a subcellular level. A targeted gene panel (multiplexed error-robust FISH (MERFISH), sequential FISH (seqFISH)+, CoxMx, spatially resolved transcript amplicon readout mapping (STAR-Map)) could enhance detection of senescence-associated markers, while unbiased methodologies (Visium, deterministic barcoding in tissue for spatial omics sequencing (DBiT-seq), Seq-Scope) could be more appropriate for systematic and unbiased discovery-based approaches. While markers can help to identify specific cell types, senescence is a cell state with temporal and phenotypic heterogeneous characteristics that will depend on cell type, organ, stimulus and physiologic and temporal conditions. Unbiased spatial transcriptomics may be an invaluable tool to map and identify senescent cells to define where they originate or accumulate in human tissues across lifespan. Although there are multiple spatial transcriptomic methods, here we describe some of the strategies and methodologies currently in use by the SenNet Consortium as well as some of their advantages and pitfalls (Table 2 and Fig. 3).
Table 2 |.
Comparison of array-based spatial transcriptomic methods
| ST method | DNA features | Barcode sequencing on each array | Feature diameter(s) (μm) | Feature center distance(s) (μm) | Percentage of array area covered by featuresa | Feature density (features per mm2) | Array substrate | Tissue used for RNA capturingb | cDNA synthesis and amplification | Mean UMIsc | Dataset |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Visium | Spotted DNA | No | 55 | 100 | 27% | ~110 | Glass | Mouse OB, 10 μm thick, tissue fixation and permeabilization | TSO and PCR | 15,377/55 μm | GSE153859 (ref. 106) |
| DBiT-seq | Microfluidic wells | No | (1) 10 (2) 25 (3) 50 |
(1) 30 (2) 53 (3) 100 |
(1) 11% (2) 22% (3) 25% |
(1) 1,100 (2) 360 (3) 100 |
PDMS | Mouse embryo, 7 μm thick, tissue fixation and permeabilization | TSO and PCR | ~5,000/10 μm | GSE137986 (ref. 57) |
| Slide-seqV2 | Assembled beads | Yes | 10 | 10 | 78% | 1×104 | Glass | Mouse OB, 10 μm thick, freshly frozen | TSO and PCR | 497/10 μm | (ref. 107) |
| HDST | Assembled beads | Yes | 2 | 3 | 34% | 1.07×105 | Silicon | Mouse OB, 10 μm thick, tissue fixation and permeabilization | TSO and PCR | 12/10 μm or 2/2 μm | GSE130682 (ref. 108) |
| Seq-Scope | Illumina DNA clusters | Yes | ≥0.5 | ≤0.8 | ≤70% | ≤1.5×106 | Linear PAA coating | Mouse colon, 10 μm thick, tissue fixation and permeabilization | RPE and PCR | ~2,743/10 μm | GSE169706 (ref. 61) |
| Stereo-seq | DNA nanoballs | Yes | 0.22 | (1) 0.5 (2) 0.715 | (1) 15% (2) 7% | (1) 4×106 (2) 1.96×106 | Silicon | Mouse OB, 10 μm thick, tissue fixation and permeabilization | TSO and PCR | ~1,450/10 μm or 59/2 μm | GSE153164 (ref. 62) |
| PIXEL-seq | Polonies | No | ~1 | ~1 | >90% | ≤1×106 | Cross-linked PAA gel | Mouse OB, 10 μm thick, freshly frozen | TSO and PCR | 977/10 μm or 47/2 μm | GSE186097 (ref. 64) |
HDST, high-definition spatial transcriptomics; OB, olfactory bulb; PAA, polyacrylamide; PDMS, polydimethylsiloxane; RPE, random priming and extension; ST, spatial transcriptomic; TSO, template-switching oligonucleotide.
The percentages for Visium, DBiT-seq, Slide-seqV2, HDST and Stereo-seq were calculated based on reported feature sizes, densities and spatial patterns. The percentage for Seq-Scope was measured by analyzing a reported cluster image of the flow cell hybridized with the highest template concentration, 100 pM.
Different RNA-capturing conditions were used, and some probably captured RNA from multiple cell layers from a whole tissue section. Different amplification methods have different yields. Multiple-displacement amplification amplifies cDNA fragments and typically gives a higher yield than PCR amplification of full-length cDNA.
Unique molecular identifier (UMI) counts were obtained from the original publications. Feature gaps typically were not considered in the calculations.
Targeted methods.
FISH has been used to image RNA transcripts and allows the accurate localization and quantification of RNA molecules in single cells. However, there are technical challenges to the application of FISH to the identification of multiple mRNA molecules simultaneously, related to the limited number of distinct color channels. A solution to this challenge was the development of advanced highly multiplexed RNA-detection methods that use RNA-ISH probes coupled with combinatorial barcodes. Cyclic hybridization and imaging of fluorescent reporter probes (complementary to barcodes) enables highly multiplexed in situ imaging and spatial localization of RNA probes at high resolution. Platforms using these techniques include seqFISH48, MERFISH49, NanoString CosMx50 and 10x Genomics Xenium.
The advantages of FISH-based methodologies are (1) high-plex number above hundreds, (2) high detection efficiency and high number of transcripts per cell in comparison with sequencing-based technologies, (3) single-cellular to subcellular resolution owing to advancement in machine learning-based segmentation algorithms, for example, Cellpose51, (4) standardized commercial platforms and reagents (MER-SCOPE, CosMx), (5) compatibility with FFPE and freshly frozen tissue sections and mouse and human tissues and (6) a highly advanced computation algorithm to allow for discovery of cell-to-cell interactions, cell community identification and ligand–receptor delineation. This feature is ideal for discovering rare senescent cells and their spatial niche as well as intercellular secretome signaling pathways. However, current limitations to the targeted methods include (1) size of the imaging area and therefore low throughput for imaging large tissue areas, (2) long duration of imaging acquisition, (3) large dataset size, (4) limited and pre-selected panels of markers and (5) the need for expensive specialized equipment and custom-designed probe panels.
Unbiased methods.
Visium.
The company 10x Genomics currently offers spatial barcoding-based transcript detection (Visium) in flash-frozen and FFPE tissues, providing robust spatially resolved whole-transcriptome evaluation at the cost of relatively low resolution and discontinuous detection area (barcoded spot diameter, ~50 μm; 100 μm between spots; 6.5 × 6.5-mm tissue-capture area). Already there are reports of Visium being used to investigate the spatial characteristics of cellular senescence in mouse52 and human brains53 as well as to decipher age-dependent tissue-regeneration capabilities54. An important advantage of this methodology is the possibility for retrospective analysis of stored FFPE blocks55, which allows designing experiments around a desired age range.
Visium version 2 slides have a tissue-capture area almost three times larger (11 × 11 mm) than the original, while Visium HD will offer continuous mRNA capture across the whole tissue section. The 10x Xenium platform claims to provide subcellular-level spatial mRNA and protein detection for several hundred targets at a time, using prebuilt tissue-specific probe panels (up to 300–400 genes at the moment), with up to 100 customer-specific probes added on top. Importantly, owing to the non-destructive nature of the assay, the same slides can be transferred for downstream unbiased Visium gene expression profiling. This provides a great opportunity for targeted and whole-transcriptome gene expression data integration.
Although Visium is an unbiased spatial transcriptomic solution, its low resolution (100 μm center to center; by comparison, the naked human eye has a resolution of ~40 μm) makes it unsuitable for profiling single cells. Therefore, isolating the senescent cell transcriptome and characterizing their spatial location will not be precise. Despite this limitation, Visium may still be useful for certain applications in which a coarser-resolution analysis is sufficient.
NanoString GeoMx is a spatial omics platform for analysis of RNA (whole transcriptome) or proteins (over 100-plex) that uses photocleavable oligonucleotide barcodes that are coupled to RNA probes or antibodies. UV light directed onto selected geometric regions of interest (or cell types based on fluorescence profiles) releases the probes, which are then collected to determine transcript or protein abundance56. Limitations include specific area or cell number requirements within region-of-interest (ROI) selection; thus, while specific cell types may be selected using morphology markers, this method cannot resolve down to the single-cell level.
An example of the application of Visium in combination with the NanoString GeoMx platform to identify senescence in the liver is illustrated in Fig. 4.
Fig. 4 |. Examples of exploratory strategies used by the University of Minnesota Tissue Mapping Center to identify senescence in liver samples using both the 10x Genomics Visium and the NanoString GeoMx platforms.
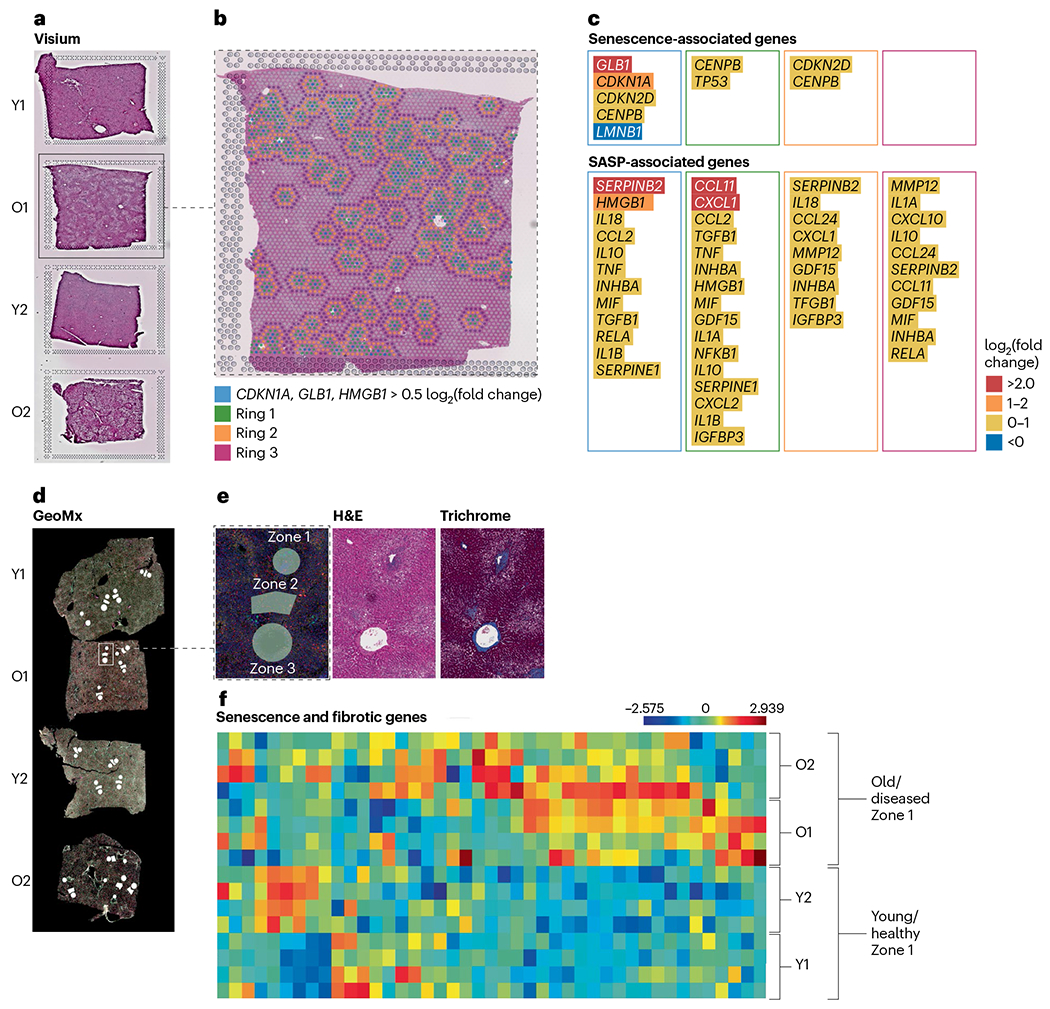
Tissues are from old, diseased liver (O1 and O2) or from young, healthy liver (Y1 and Y2). a, A spatial discovery approach was used with the Visium platform. H&E staining of tissues on the Visium slide is shown. b, Identification of 166 senescent spots in tissue O1 with differential expression of three senescence-associated genes (CDKN1A, GLB1 and HMGB1). To investigate paracrine effects of senescence spots, clusters were created for the senescent spots (blue) and the surrounding area using three target rings (green, yellow and purple). c, Senescence and SASP-associated gene expression in each cluster demonstrates that senescence-associated gene expression is highest in the center blue spots, but SASP gene expression is also elevated in the surrounding area. d, A hypothesis-driven approach was used with the GeoMx platform focused on liver anatomical structures to compare senescence gene expression across tissues. ROIs were selected based on liver zone 1 (periportal), zone 2 (mid-lobular) and zone 3 (pericentral) and marked in white on the GeoMx immunofluorescent image. e, ROI selections in O1 using serial sections of H&E and Masson’s trichrome staining for ROI determination. f, Differential gene expression of senescence- and fibrosis-associated genes in zone 1 of all four tissues with increased expression in old, diseased liver (O1 and O2).
DBiT-seq.
DBiT-seq is an unbiased multiomic approach that allows for spatial barcoding of the transcriptome in tissues57. The flexibility of this microfluidic-based approach means that it can be extended to other omics analyses such as the epigenome and the proteome. In fact, co-profiling of the transcriptome and a panel of antibodies in a methodology known as spatial co-indexing of transcriptomes and epitopes for multiomic mapping by next-generation sequencing (CITE-seq) has already been applied58. Deng and colleagues also demonstrated spatial cleavage under targets and tagmentation (CUT&Tag) to study chromatin modifications at the cellular level59. Recently, DBiT-seq has also been applied to co-mapping the epigenome–transcriptome at the cellular level, allowing for this analysis to be performed in one single tissue section58. DBiT-seq is also compatible with FFPE tissue blocks, allowing the use of archived material60. Although DBiT-seq may not be able to resolve senescent cells at single-cell resolution, the workflow is being optimized to allow for the combination of DBiT-seq and other high-resolution multiplex immunofluorescence imaging techniques on the same tissue slide. This would allow for integration of transcriptomic, proteomic and epigenomic data for the positive identification of senescent cells, which are typically rare.
High-resolution methods: Seq-Scope, Stereo-seq and PIXEL-seq.
The low frequency of senescent cells in tissues and their heterogeneity requires methods capable of precisely locating senescent cells and distinguishing them from surrounding cells. Furthermore, many of the senescence-associated transcripts, such as those encoding cell cycle-dependent kinase inhibitors, are not very abundantly expressed; therefore, high-resolution and high-capture output are essential for spatial analysis of cell senescence.
Recently, several ultra-high-resolution technologies such as Seq-Scope61, Stereo-seq62 and polony-indexed library sequencing (PIXEL-seq)63 have been developed, which could prove ideal for identification and characterization of senescent cells and their interactions with the environment. In addition to providing submicrometer resolution, which is comparable to that of optical microscopy, these methods show highly efficient transcriptome-capture efficiency, up to ~23 unique transcripts per μm2 (Table 2). Considering that single cells can occupy up to 100–500 μm2, this output is comparable to that of conventional single-cell RNA sequencing (scRNA-seq), enabling sensitive detection of senescent transcripts. Importantly, conventional microscopy is compatible with these transcriptomic procedures, as demonstrated by Seq-Scope (hematoxylin and eosin (H&E) histology61) and Stereo-Seq (4,6-diamidino-2-phenylindole (DAPI) fluorescence62). Furthermore, PIXEL-seq uses DNA arrays called ‘polony gels’, which capture tissue RNA from a single-cell layer touching the gel, similar to confocal microscopy, and restrain template diffusion, thus improving single-cell resolution63. Therefore, microscopy-based identification of senescence-associated markers using other methods can be combined with transcriptomic data in a straightforward manner. Among these technologies, Seq-Scope and PIXEL-seq are currently being optimized for sensitive detection of cell senescence in tissues, as part of the SenNet technology development and adaptation program. Potentially, these methods could be also expanded to capture other spatial omics features, such as proteomic signatures, chromatin accessibility and epigenome structure.
Another emerging challenge is the application of image-analysis methods to the detection of senescent cells, which requires the development of a different set of complex tools. In the next section, we describe some of the methodologies currently in use and being developed in this area.
Image data analysis of senescent cells
Nuclear and cell segmentation
Identification of cell objects is an essential first component for spatial mapping. Segmentation can be used to define cell shape and other topological properties that are important features for cell type assignment64,65. In addition, errors in identifying cell or nucleus boundaries can directly impact the assignment of expression levels for genes or proteins in cells, making it harder to uniquely identify cells expressing known senescence-associated markers. Current cell-identification methods (Fig. 5) can largely be classified into three types: (1) methods that rely on nucleus or membrane staining (for example, DAPI and H&E), (2) methods that use the profiled proteins and RNA directly for segmentation and (3) segmentation-free methods. Most methods developed to date belong to group 1 and focus on segmentation based on stained cells or nuclei51,66–70. Although generalizable to a variety of spatial profiling methods, such methods have several drawbacks. First, molecular and auxiliary stains can be misaligned. In addition, not all cells in the same tissue express the same membrane marker, making it hard to select a single stain for cell segmentation. Methods in group 2 solve this by using spatial distribution of RNA or protein molecules to improve segmentation. Examples of such approaches include ranking markers for cell segmentation (RAMCES)71, membrane pattern-based cell segmentation (MPCS)72 and Cytokit73, which use proteomic data, and Baysor74, probabilistic cell typing by in situ sequencing (pciSeq)75 and joint cell segmentation and cell type annotation (JSTA)76, which use RNA for the same purpose. While addressing the image-analysis issue, such methods usually require additional information, including knowledge of the expected cell types and their markers72,76 or cell shape74,75. Finally, methods in group 3 are segmentation free. For example, spot-based spatial cell type analysis by multidimensional mRNA density estimation (SSAM)77 can directly assign cell cluster labels to pixels without grouping them to cells. However, these methods cannot be easily extended to cell-based analyses, including cell trajectory inference78–80 and cell–cell communication81,82, which are both important for studying senescent cell networks and modeling senescence development. Although current methods demonstrate various levels of success for the general purpose of cell-based analysis, further evaluations are needed in the context of senescent cell detection and modeling.
Fig. 5 |. Image data analysis of senescent cells.
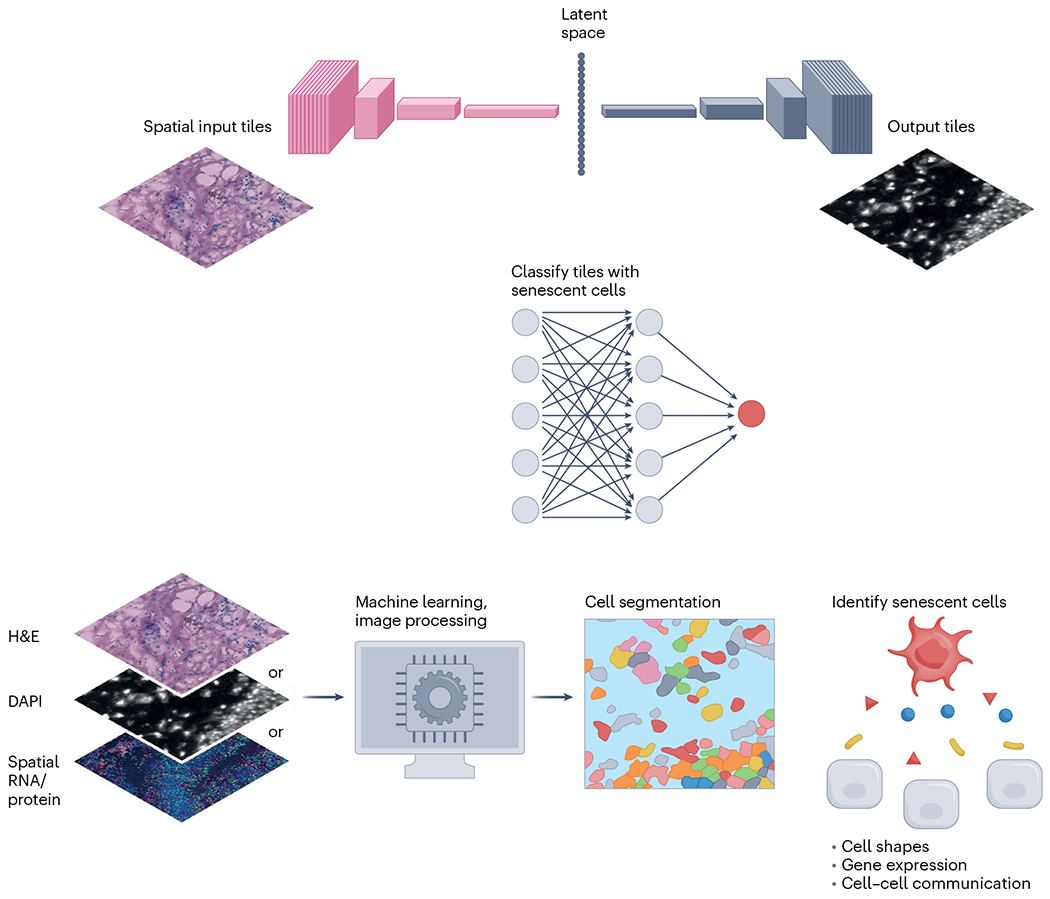
Top, platform comparisons can be performed by registering image tiles across different technology platforms, such as protein, RNA or H&E, and then identifying key features in a shared latent space through encoder–decoder approaches. Latent space features may be used to more robustly identify tiles containing senescent cells. Bottom, many machine learning or image-processing methods have been developed for segmenting cells from nucleus- or membrane-staining images as well as spatial RNA or protein data. Cell segmentation can be used to define cell shapes or determine gene expression of cells, which can further be used to model cell–cell communication in tissues. Such information is important for identifying senescent cells.
Integration with scRNA-seq data for cell type deconvolution
Spatial transcriptomic platforms such as Visium and Slide-seq83 measure gene expression profiles of spatial spots that may contain multiple cells from distinct cell types. When scRNA-seq data from the same tissue are available, computational methods such as SPOTlight, robust cell type decomposition (RCTD), Tangram and conditional autoregressive deconvolution (CARD)84–87 deconvolute the spatial transcriptomic data and infer the proportion of different cell types within each spatial spot. These methods can potentially infer the spatial locations of senescent cells if the senescent cells are annotated in the scRNA-seq data, but there could be important challenges. First, deconvolution methods may struggle to reliably detect differences between senescent and non-senescent cells within each cell type, which could be much smaller than the differences across cell types. Second, senescent cell density in some tissues could be overwhelmed by more prevalent cell types, causing a failure in detection of senescent cells. Third, reference scRNA-seq datasets with annotated senescent cells are lacking in many types of tissues. Finally, although there are studies benchmarking the performance of deconvolution methods in a more general setting88, the performance of deconvoluting senescent cells is largely unknown. Such a benchmark is necessary to determine whether methods specifically designed for deconvoluting senescent cells are needed.
Tensor analysis of senescent cell populations
Current scRNA-seq data for senescent cell analysis often include measurements from multiple individuals, multiple time points, dozens of cell types and thousands of genes. The resulting data can be longitudinal, high dimensional and high order. Such data are naturally organized as tensors aligned over multiple directions representing individuals, time, cell types, genes and so on, but are less suited to traditional statistical modeling and methods. Instead, tensor-based statistical models and methods represent a better toolbox for senescent cell scRNA-seq population analysis. For example, tensor clustering will probably be important to reveal subpopulations among individuals and occurrence patterns among single-cell gene expression profiles89. Tensor regression may also yield interpretable models for phenotype prediction based on sequencing data90.
Tile-based image analysis
An alternative to identification of senescent cells individually is to computationally classify larger image tiles (also known as patches, typically 50–100 μm wide) by whether they contain a senescent cell (Fig. 5). This multiple-instance learning approach sacrifices cellular resolution, but it circumvents the uncertainties of cell segmentation within a tissue slice, caused by, for example, partial cells or regions of high cell density. Secondly, the patch scale subsumes cell–cell spatial relationships and extracellular microenvironmental features, providing a potentially richer and more phenotypically relevant predictive feature space. Lastly, patch-based approaches provide a common scale for cross-platform data comparisons, enabling evaluation of senescence classification methods across technologies with different resolutions. Such comparisons are possible through machine learning methods to convert data from different platforms (for example, CODEX, Visium, H&E), using spatially aware encoder–decoder deep learning approaches91, potentially reducing the need for markers to be measured simultaneously across platforms. These tile-based approaches will be valuable for analyzing SenNet tissues in two dimensions and potentially 3D92, complementing cell community and graph methods that analyze the spatial relationships of segmented cells93.
Three-dimensional and multi-modal analysis
Alternative approaches may provide 3D information to further delineate spatial relationships in larger volumes. For example, optical clearing renders whole tissue more transparent and less refractive and therefore more amenable to deeper confocal, multiphoton or light sheet microscopic detection of low-plex fluorescence markers94. While these approaches can profile volumes at high resolution, they are limited by the number and types of probes, for example, owing to poor penetration of antibodies into deeper tissue. Another approach is serial section reconstruction, which has been used to recreate 3D structure from histological slide scans95 and spatial transcriptomics92. Multi-modal image registration holds the promise of combination of 3D structure obtained by low-plex methods and spatial profiling data obtained only in select planes of the volume. Serial and multi-modal image registration is trivial owing to warping of tissue sections, and rigid and deforming image-registration methods are available to address this such as probabilistic alignment of spatial transcriptomic experiments (PASTE)92, CODA95, Autograd Image Registration Laboratory (AirLab)96 and elastix97 and its ImageJ–Fiji–QuPath6 implementation Warpy98. While cell-level reconstruction across tissue sections has been attained92,98, this may be challenging in tissues with smaller cells or nuclei. Combining serial section approaches with deep learning-based image segmentation has allowed volumetric resolution of anatomical structures95. These approaches can improve our ability to predict where senescent cells occur as well as how and when they interact with the microenvironment.
Use of deep learning methods to identify senescent cells
Simple imaging-based methods coupled with deep learning predict cellular senescence
Deep learning methods have been recently developed to detect senescent cells in culture with phase-contrast imaging99 and tissues with H&E and DAPI staining65. Interestingly, p21CIP1-positive nuclei exhibited higher predicted levels of senescence in multiple tissues, while proliferating cells (indicated by 5-ethynyl-2′-deoxyuridine (EdU) incorporation) showed lower levels65. Further analysis of skin biopsies revealed that predicted senescence in the dermis of more than 160 individuals increased with age and showed expected associations with disease outcomes. This approach using deep learning for the identification of senescent cells was originally developed with human fibroblasts in cell culture, but, using image transformation to focus on nuclear morphology, these deep learning models were applied to diverse cell types in culture and tissues (Fig. 6a).
Fig. 6 |. Nuclear morphology predicts senescence in tissues.
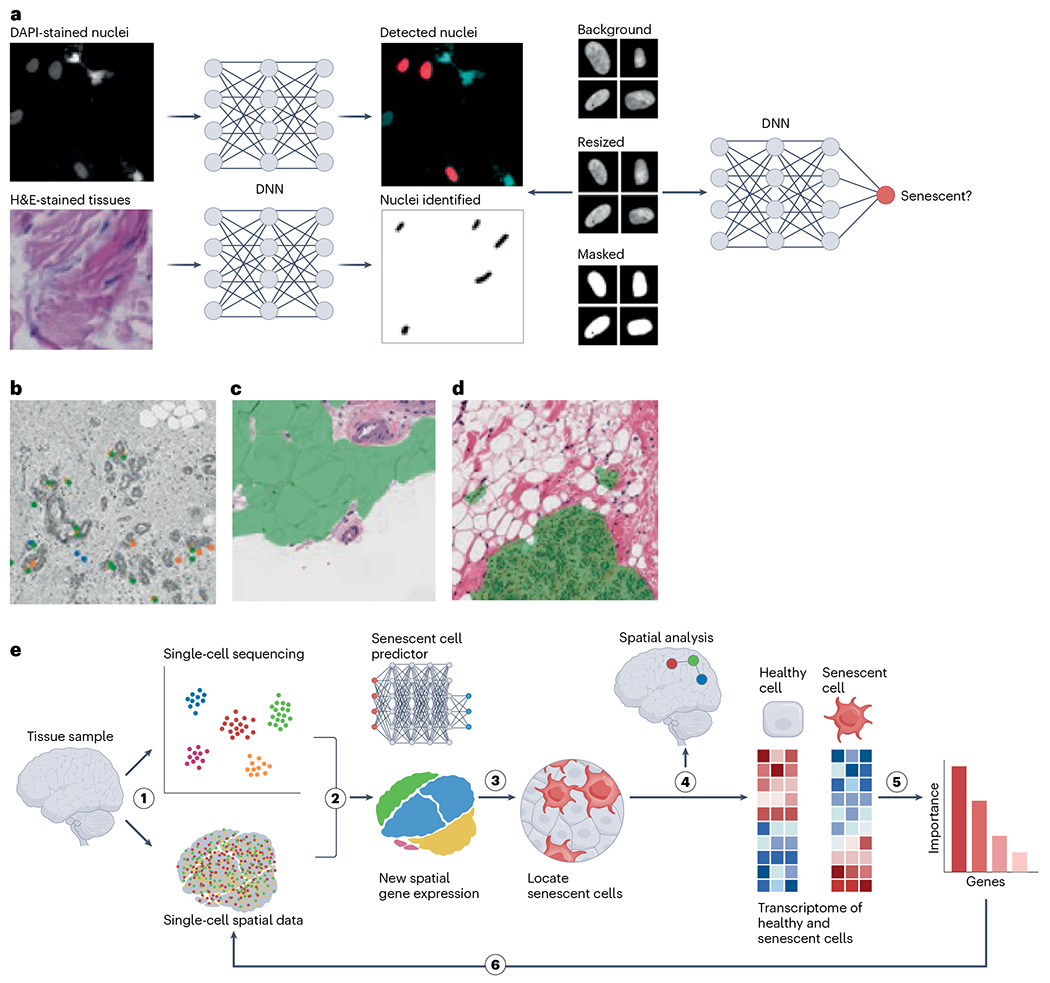
a, Workflow for detecting nuclei in culture or tissue, normalizing images and predicting senescence (tissue and cell image samples are reproduced with permission from ref. 65, Springer Nature America, Inc.). DNN, deep neural network. b, Nuclei with prediction scores above the 95th percentile for several models; orange, replicative senescence model; green, irradiation-induced senescence model; blue, AAD model, which was trained on multiple drug treatments including antimycin A, atazanavir–ritonavir and doxorubicin. c, Segmentation of adipose regions of breast tissue. d, Segmentation of epithelial regions of breast tissue. e, Workflow to identify markers, perform spatial analysis and refine morphology-based predictors by integrating single-cell and spatial transcriptomics.
Image-based senescence assessment provides a unique and new approach to map and analyze cellular senescence in situ in human tissues. However, understanding the role of senescence in tissue is particularly challenging owing to its heterogeneity, with diverse cell types organizing to form complex structures. While the accuracy for individual nuclei is difficult to establish in tissues, deep learning predictors can offer overall assessment of senescence in tissues along with differential analysis across ages, medical conditions or other factors. For example, the SenNet Tissue Mapping Center at the Buck Institute applied the deep learning predictor to breast tissue biopsies from 67 healthy individuals, obtained from the Komen Tissue Bank, scoring 460,000 nuclei (Fig. 6b). By tracking the location of senescent cells within the breast tissue architecture, this project is exploring the spatial relationship between senescent and non-senescent cells. Additionally, deep learning methods can be used to automatically segment tissue types to study regional differences (Fig. 6c,d). High-precision prediction senescence scores per tissue type can then be correlated with transcriptional, histological and proteomic data, among other factors. Image-based senescence prediction can potentially provide a unique method for SenNet to better characterize the role of senescence in human aging and disease.
Integrative approaches using deep learning for fine-resolution tissue mapping of spatial transcriptomics
Incorporation of the imaging-based deep learning method described above into single-cell transcriptomic technologies offers an unprecedented solution for training models that can map senescent cells in tissues in health and disease. A proposed example of such a pipeline is delineated in this section. This starts with a time-series scRNA-seq and/or single-nucleus RNA-sequencing (snRNA-seq) experiment to acquire transcriptome changes during induction of cellular senescence for a certain cell type. Next, these data can be used to train a deep learning model to predict the senescent cell burden and extract top predictive features in an unsupervised manner, which would represent the markers of cell senescence for this specific cell type in vitro. These preliminary senescence-associated markers will be used in designing specific probes for in situ single-cell techniques such as CosMx or STARmap100 and performing spatial alignment of scRNA-seq and/or snRNA-seq data acquired from the same tissue sample86. This is particularly important as it can overcome the gene-throughput limitations of CosMx and STARmap. Further extending the existing method65, which maps nucleus morphology to cell senescence, the senescent cells will also be precisely located based on their transcriptome signatures in the tissue. This will be achieved by obtaining a transcriptome of senescent cells and training the deep learning model to extract the top features as markers of cellular senescence in vivo. Consequently, these markers can be used to map senescent cells in the same tissue type, and the list of markers will be refined after multiple iterations of the proposed experiments (Fig. 6h).
Conclusions and future perspectives
While there is growing excitement about the therapeutic potential of targeting senescence in the context of aging and age-related disease, our understanding of this phenotype and its complexity in vivo is very much in its infancy. Since Hayflick’s seminal discovery more than 60 years ago, our field has made great strides in describing senescence and its underlying mechanisms, particularly in cultured cells; however, limitations in the available technology have hampered the characterization of senescence in tissues. It is our prediction that the current developments in single-cell spatially resolved platforms, image analyses and deep learning technologies will likely revolutionize the way in which we understand senescence in vivo.
We foresee that, in the future, we will be able to perform multimodal analyses of the proteome, the transcriptome, the epigenome and the metabolome of senescent cells at subcellular and single-molecule resolution. Quantum leaps in imaging throughput including imaging speed and area will enable spatial mapping of senescent cells and their microenvironment in 3D. Advancements in computational biology will allow us to easily integrate dissociative and non-dissociative methods, processing mega 3D spatial datasets and ultimately constructing comprehensive 3D cellular senescence networks in organs.
Acknowledgements
This research was supported by the NIH Common Fund, through the Office of Strategic Coordination and the Office of the NIH Director under awards UG3CA268103 (J.F.P., A.B.L., S.V., D.J.), U54AG075931 (A.U.G., A.L.M., D.B., H.C., J.K.A., M.B., M. Königshoff., O.E., Q.H., T.K.), UG3CA268202 (N.N.), U54AG075941 (J.H.C.), UG3CA268091 (J.H.L.), UG3CA268096 (R.D., X.F.), U54AG075931 (Z.B.-J.), U24CA268108 (Z.B.-J.), U54AG076041 (L.J.N., A.C.N., E.L.T., S.P., F.R., G.B.), U54AG075936 (A.R.Z., Z.J.), U54AG079758 (P.D.A., Q.Z., R.A.P.), U54AG076040 (H.P., J.P.), U54 AG075932 (D.F., A.A.G., C.B., F.W., I.H.), UG3CA268112 (V.S., M. Kumar.). We thank A. Impagliazzo for her work on the illustrations.
Footnotes
Competing interests
J.H.L. is an inventor on pending patent applications related to Seq-Scope. All other authors declare no competing interests.
References
- 1.Gorgoulis V. et al. Cellular senescence: defining a path forward. Cell 179, 813–827 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Robbins PD et al. Senolytic drugs: reducing senescent cell viability to extend health span. Annu. Rev. Pharmacol. Toxicol 61, 779–803 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Demaria M. et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 31, 722–733 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Born E. et al. Eliminating senescent cells can promote pulmonary hypertension development and progression. Circulation 147, 650–666 (2023). [DOI] [PubMed] [Google Scholar]
- 5.Reyes NS et al. Sentinel p16INK4a+ cells in the basement membrane form a reparative niche in the lung. Science 378, 192–201 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Severino J, Allen RG, Balin S, Balin A & Cristofalo VJ Is β-galactosidase staining a marker of senescence in vitro and in vivo? Exp. Cell Res 257, 162–171 (2000). [DOI] [PubMed] [Google Scholar]
- 7.Hall BM et al. Aging of mice is associated with p16Ink4a- and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging 8, 1294–1315 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ogrodnik M, Salmonowicz H, Jurk D & Passos JF Expansion and cell-cycle arrest: common denominators of cellular senescence. Trends Biochem. Sci 44, 996–1008 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Biran A. et al. Quantitative identification of senescent cells in aging and disease. Aging Cell 16, 661–671 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tabula Muris C. et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tabula Sapiens C. et al. The Tabula Sapiens: a multiple-organ, single-cell transcriptomic atlas of humans. Science 376, eabl4896 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang X. et al. Characterization of cellular senescence in aging skeletal muscle. Nat. Aging 2, 601–615 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ogrodnik M. et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 20, e13296 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Acosta JC et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol 15, 978–990 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu M et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med 24, 1246–1256 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lagnado A et al. Neutrophils induce paracrine telomere dysfunction and senescence in ROS-dependent manner. EMBO J. 40, e106048 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laberge RM, Awad P, Campisi J & Desprez PY Epithelial–mesenchymal transition induced by senescent fibroblasts. Cancer Microenviron. 5, 39–44 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krizhanovsky V et al. Senescence of activated stellate cells limits liver fibrosis. Cell 134, 657–667 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee PJ et al. NIH SenNet Consortium to map senescent cells throughout the human lifespan to understand physiological health. Nat. Aging 2, 1090–1100 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dimri GP et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl Acad. Sci. USA 92, 9363–9367 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Georgakopoulou EA et al. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging 5, 37–50 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang C et al. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 8, 311–323 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Freund A, Laberge RM, Demaria M & Campisi J Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 23, 2066–2075 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davalos AR et al. p53-dependent release of alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell Biol 201, 613–629 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun J et al. A Glb1-2A-mCherry reporter monitors systemic aging and predicts lifespan in middle-aged mice. Nat. Commun 13, 7028 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hewitt G et al. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun 3, 708 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herbig U, Ferreira M, Condel L, Carey D & Sedivy JM Cellular senescence in aging primates. Science 311, 1257 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Ogrodnik M et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun 8, 15691 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Farr JN et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med 23, 1072–1079 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swanson EC, Manning B, Zhang H & Lawrence JB Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J. Cell Biol 203, 929–942 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogrodnik M et al. Obesity-induced cellular senescence drives anxiety and impairs neurogenesis. Cell Metab. 29, 1061–1077 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valli J et al. Seeing beyond the limit: a guide to choosing the right super-resolution microscopy technique. J. Biol. Chem 297, 100791 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boumendil C, Hari P, Olsen KCF, Acosta JC & Bickmore WA Nuclear pore density controls heterochromatin reorganization during senescence. Genes Dev. 33, 144–149 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodier F et al. DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J. Cell Sci 124, 68–81 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson R et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 38, e100492 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rocha A, Dalgarno A & Neretti N The functional impact of nuclear reorganization in cellular senescence. Brief. Funct. Genomics 21, 24–34 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ricci MA, Manzo C, García-Parajo MF, Lakadamyali M & Cosma MP Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell 160, 1145–1158 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Shah PP et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 27, 1787–1799 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Cecco M et al. Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell 12, 247–256 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chandra T et al. Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 10, 471–483 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sati S et al. 4D genome rewiring during oncogene-induced and replicative senescence. Mol. Cell 78, 522–538 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hickey JW et al. Spatial mapping of protein composition and tissue organization: a primer for multiplexed antibody-based imaging. Nat. Methods 19, 284–295 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taylor MJ, Liyu A, Vertes A & Anderton CR Ambient single-cell analysis and native tissue imaging using laser-ablation electrospray ionization mass spectrometry with increased spatial resolution. J. Am. Soc. Mass Spectrom 32, 2490–2494 (2021). [DOI] [PubMed] [Google Scholar]
- 44.Buczak K et al. Spatially resolved analysis of FFPE tissue proteomes by quantitative mass spectrometry. Nat. Protoc 15, 2956–2979 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Lin JR, Fallahi-Sichani M, Chen JY & Sorger PK Cyclic immunofluorescence (CycIF), a highly multiplexed method for single-cell imaging. Curr. Protoc. Chem. Biol 8, 251–264 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gut G, Herrmann MD & Pelkmans L Multiplexed protein maps link subcellular organization to cellular states. Science 361, eaar7042 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Stallaert W et al. The structure of the human cell cycle. Cell Syst. 13, 230–240 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eng C-HL et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 568, 235–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xia C, Fan J, Emanuel G, Hao J & Zhuang X Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc. Natl Acad. Sci. USA 116, 19490–19499 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He S et al. High-plex imaging of RNA and proteins at subcellular resolution in fixed tissue by spatial molecular imaging. Nat. Biotechnol 40, 1794–1806 (2022). [DOI] [PubMed] [Google Scholar]
- 51.Stringer C, Wang T, Michaelos M & Pachitariu M Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods 18, 100–106 (2021). [DOI] [PubMed] [Google Scholar]
- 52.Kiss T et al. Spatial transcriptomic analysis reveals inflammatory foci defined by senescent cells in the white matter, hippocampi and cortical grey matter in the aged mouse brain. GeroScience 44, 661–681 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu P et al. The landscape of human tissue and cell type specific expression and co-regulation of senescence genes. Mol. Neurodegener 17, 5 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tower RJ et al. Spatial transcriptomics reveals metabolic changes underly age-dependent declines in digit regeneration. eLife 11, e71542 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gracia Villacampa E et al. Genome-wide spatial expression profiling in formalin-fixed tissues. Cell Genom. 1, 100065 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Merritt CR et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol 38, 586–599 (2020). [DOI] [PubMed] [Google Scholar]
- 57.Liu Y et al. High-spatial-resolution multi-omics sequencing via deterministic barcoding in tissue. Cell 183, 1665–1681 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu Y et al. High-plex protein and whole transcriptome co-mapping at cellular resolution with spatial CITE-seq. Nat. Biotechnol, 10.1038/s41587-023-01676-0 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Deng Y et al. Spatial-CUT&Tag: spatially resolved chromatin modification profiling at the cellular level. Science 375, 681–686 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu Y, Enninful A, Deng Y & Fan R Spatial transcriptome sequencing of FFPE tissues at cellular level. Preprint at bioRxiv 10.1101/2020.10.13.338475 (2020). [DOI] [Google Scholar]
- 61.Cho C-S et al. Microscopic examination of spatial transcriptome using Seq-Scope. Cell 184, 3559–3572 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen A et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 185, 1777–1792 (2022). [DOI] [PubMed] [Google Scholar]
- 63.Fu X et al. Polony gels enable amplifiable DNA stamping and spatial transcriptomics of chronic pain. Cell 185, 4621–4633 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang W et al. Identification of cell types in multiplexed in situ images by combining protein expression and spatial information using CELESTA. Nat. Methods 19, 759–769 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heckenbach I et al. Nuclear morphology is a deep learning biomarker of cellular senescence. Nat. Aging 2, 742–755 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sage D & Unser MA Teaching image-processing programming in Java. IEEE Signal Process. Mag 20, 43–52 (2003). [Google Scholar]
- 67.Bannon D et al. DeepCell Kiosk: scaling deep learning-enabled cellular image analysis with Kubernetes. Nat. Methods 18, 43–45 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schmidt U, Weigert M, Broaddus C & Myers G Cell detection with star-convex polygons. In Medical Image Computing and Computer Assisted Intervention – MICCAI 2018 (MICCAI 2018. Lecture Notes in Computer Science), vol 11071) (eds Frangi A et al. ) 265–273 (Springer, 2018). [Google Scholar]
- 69.Ronneberger O, Fischer P & Brox T U-Net: convolutional networks for biomedical image segmentation. In Medical Image Computing and Computer-Assisted Intervention—MICCAI 2015 (MICCAI 2015. Lecture Notes in Computer Science, vol 9351) (eds. Navab N et al. ) 234–241 (Springer, 2015). [Google Scholar]
- 70.Hollandi R et al. nucleAIzer: a parameter-free deep learning framework for nucleus segmentation using image style transfer. Cell Syst. 10, 453–458 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dayao MT, Brusko M, Wasserfall C & Bar-Joseph Z Membrane marker selection for segmenting single cell spatial proteomics data. Nat. Commun 13, 1999 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dimopoulos S, Mayer CE, Rudolf F & Stelling J Accurate cell segmentation in microscopy images using membrane patterns. Bioinformatics 30, 2644–2651 (2014). [DOI] [PubMed] [Google Scholar]
- 73.Czech E, Aksoy BA, Aksoy P & Hammerbacher J Cytokit: a single-cell analysis toolkit for high dimensional fluorescent microscopy imaging. BMC Bioinformatics 20, 448 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Petukhov V et al. Cell segmentation in imaging-based spatial transcriptomics. Nat. Biotechnol 40, 345–354 (2022). [DOI] [PubMed] [Google Scholar]
- 75.Qian X et al. Probabilistic cell typing enables fine mapping of closely related cell types in situ. Nat. Methods 17, 101–106 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Littman R et al. Joint cell segmentation and cell type annotation for spatial transcriptomics. Mol. Syst. Biol 17, e10108 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Park J et al. Cell segmentation-free inference of cell types from in situ transcriptomics data. Nat. Commun 12, 3545 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li D et al. TraSig: inferring cell–cell interactions from pseudotime ordering of scRNA-seq data. Genome Biol. 23, 73 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ding J, Sharon N & Bar-Joseph Z Temporal modelling using single-cell transcriptomics. Nat. Rev. Genet 23, 355–368 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Song Q, Wang J & Bar-Joseph Z scSTEM: clustering pseudotime ordered single-cell data. Genome Biol. 23, 150 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li D, Ding J & Bar-Joseph Z Identifying signaling genes in spatial single-cell expression data. Bioinformatics 37, 968–975 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yuan Y & Bar-Joseph Z GCNG: graph convolutional networks for inferring gene interaction from spatial transcriptomics data. Genome Biol. 21, 300 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rodriques SG et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Elosua-Bayes M, Nieto P, Mereu E, Gut I & Heyn H SPOTlight: seeded NMF regression to deconvolute spatial transcriptomics spots with single-cell transcriptomes. Nucleic Acids Res. 49, e50 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cable DM et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol 40, 517–526 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Biancalani T et al. Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram. Nat. Methods 18, 1352–1362 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ma Y & Zhou X Spatially informed cell-type deconvolution for spatial transcriptomics. Nat. Biotechnol 40, 1349–1359 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chu T, Wang Z, Pe’er D & Danko CG Cell type and gene expression deconvolution with BayesPrism enables Bayesian integrative analysis across bulk and single-cell RNA sequencing in oncology. Nat. Cancer 3, 505–517 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Han R, Luo Y, Wang M & Zhang AR Exact clustering in tensor block model: statistical optimality and computational limit. J. R. Stat. Soc. B Stat. Methodol 84, 1666–1698 (2022). [Google Scholar]
- 90.Wu M, Huang J & Ma S Identifying gene–gene interactions using penalized tensor regression. Stat. Med 37, 598–610 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Burlingame EA et al. SHIFT: speedy histological-to-immunofluorescent translation of a tumor signature enabled by deep learning. Sci. Rep 10, 17507 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zeira R, Land M, Strzalkowski A & Raphael BJ Alignment and integration of spatial transcriptomics data. Nat. Methods 19, 567–575 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Palla G et al. Squidpy: a scalable framework for spatial omics analysis. Nat. Methods 19, 171–178 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tian T, Yang Z & Li X Tissue clearing technique: recent progress and biomedical applications. J. Anat 238, 489–507 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kiemen AL et al. CODA: quantitative 3D reconstruction of large tissues at cellular resolution. Nat. Methods 19, 1490–1499 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sandkühler R, Jud C, Andermatt S & Cattin PC AirLab: Autograd Image Registration Laboratory. Preprint at arXiv 10.48550/arXiv.1806.09907 (2018). [DOI] [Google Scholar]
- 97.Klein S, Staring M, Murphy K, Viergever MA & Pluim JP elastix: a toolbox for intensity-based medical image registration. IEEE Trans. Med. Imaging 29, 196–205 (2010). [DOI] [PubMed] [Google Scholar]
- 98.Chiaruttini N et al. An open-source whole slide image registration workflow at cellular precision using Fiji, QuPath and elastix. Front. Comput. Sci 3, 10.3389/fcomp.2021.780026 (2022). [DOI] [Google Scholar]
- 99.Kusumoto D et al. Anti-senescent drug screening by deep learning-based morphology senescence scoring. Nat. Commun 12, 257 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang X et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 361, eaat5691 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Black S et al. CODEX multiplexed tissue imaging with DNA-conjugated antibodies. Nat. Protoc 16, 3802–3835 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Keren L et al. MIBI-TOF: a multiplexed imaging platform relates cellular phenotypes and tissue structure. Sci. Adv 5, eaax5851 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Giesen C et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 11, 417–422 (2014). [DOI] [PubMed] [Google Scholar]
- 104.Kinkhabwala A et al. MACSima imaging cyclic staining (MICS) technology reveals combinatorial target pairs for CAR T cell treatment of solid tumors. Sci. Rep 12, 1911 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hennig C, Adams N & Hansen G A versatile platform for comprehensive chip-based explorative cytometry. Cytometry A 75, 362–370 (2009). [DOI] [PubMed] [Google Scholar]
- 106.Lebrigand K et al. The spatial landscape of gene expression isoforms in tissue sections. Nucleic Acids Res. 51, e47 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stickels RR et al. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat. Biotechnol 39, 313–319 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vickovic S et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 16, 987–990 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]