

Abstract
Monoclonal antibody (mAb) therapies have rapidly become a powerful class of therapeutics with applications covering a diverse range of clinical indications. Though most widely used for the treatment of cancer, mAbs are also playing an increasing role in the defense of viral infections, most recently with palivizumab for prevention and treatment of severe RSV infections in neonatal and pediatric populations. In addition, during the COVID-19 pandemic, mAbs provided a bridge to the rollout of vaccines; however, their continued role as a therapeutic option for those at greatest risk of severe disease has become limited due to the emergence of neutralization resistant Omicron variants. Although there are many techniques for the identification of mAbs, including single B cell cloning and immunization of genetically engineered mice, the low cost, rapid throughput and technological simplicity of antibody phage display has led to its widespread adoption in mAb discovery efforts. Here we used our 27-billion-member naïve single-chain antibody (scFv) phage library to identify a panel of neutralizing anti-SARS-CoV-2 scFvs targeting diverse epitopes on the receptor binding domain (RBD). Although typically a routine process, we found that upon conversion to IgG, a number of our most potent clones failed to maintain their neutralization potency. Kinetic measurements confirmed similar affinity to the RBD; however, mechanistic studies provide evidence that the loss of neutralization is a result of structural limitations likely arising from initial choice of panning antigen. Thus this work highlights a risk of scFv-phage panning to mAb conversion and the importance of initial antigen selection.
Keywords: antibody discovery, neutralizing antibodies, phage display, phage panning, SARS-CoV-2
Introduction
At the start of the COVID-19 pandemic, there was an urgent need for the development of novel therapeutics to bridge the gap until the rollout of vaccines and development of herd immunity. Antibodies served to fill this void with the rapid development and Emergency Use Authorization of bamlanivimab/etesevimab (Lilly) and casirivimab/imdevimab (Regeneron). To generate these monoclonal antibodies (mAb), Lilly utilized B cells isolated from an early SARS-CoV-2 patient, and Regeneron leveraged their VelociSuite technology to rapidly identify antibodies by immunization of transgenic mice genetically modified to possess a human immune system (Baum et al., 2020; Jones et al., 2021). Though highly effective due to the ability to isolate antibodies that have undergone affinity maturation in vivo, both of these methods are time consuming and technically challenging, requiring specialized equipment and resources. In addition, regulatory constraints related to blood sample availability, Institutional Review Board (IRB) approvals and biocontainment issues at institutional, local and regional levels further hampered timely accessibility of convalescent blood samples from SARS-CoV-2 infected patients.
Phage display is a powerful and technologically simple platform that allows for the rapid identification of high affinity antibodies through panning and enrichment of massive naïve libraries (Alfaleh et al., 2020). Filamentous phage are comprised of a circular, single stranded DNA encapsulated by ~2700 copies of the major coat protein (pVIII) and 3–5 copies of the minor coat proteins (pIII, pVI, pVII and pIX) (Ebrahimizadeh and Rajabibazl, 2014). With thousands of copies of pVIII and structural constraints only allowing fusion of small peptides, antibody fragments are typically fused to the N-terminus of the pIII protein which naturally assembles at the head of the virus and is required for bacterial infection (Noren and Noren, 2001; Silacci et al., 2005; Sellmann et al., 2020; Moreno et al., 2022). To simplify the downstream molecular biology, phagemids are commonly used in library repertoire construction where the pIII-single chain variable fragment (scFv) fusion is expressed off a plasmid containing the f1 origin of replication (Peterson et al., 1999). When bacteria containing the phagemid are coinfected with helper phage, both wild type (WT) pIII and pIII-scFv fusions are expressed and simultaneously integrated into new phage molecules. As such, each phage now contains a mixture of WT and mutant pIII molecules, maintaining infectivity while allowing display of the scFv of interest (O’connell et al., 2002). Low integration rate of the pIII-scFv fusion also helps to reduce avidity effects that would be observed if multiple scFvs were expressed on each phage, allowing for the selection of high affinity antibodies.
The Marasco Lab previously built a 27-billion-member naïve phagemid library which has been used by our lab over the past two decades to isolate potent antibodies against influenza, flaviviruses, Middle Eastern Respiratory Syndrome (MERS), SARS-CoV and countless oncology targets (Sui et al., 2004, 2009; Xu et al., 2010; Tang et al., 2014; Chang et al., 2022). Through this platform, we rapidly identified 39 SARS-CoV-2 specific clones against recombinant S1 and receptor binding domain (RBD), which were then cloned into scFv-Fc expression vectors for detailed characterization. Epitope binning revealed that we had a diverse panel of antibodies targeting different regions of the RBD, whereas neutralization assays showed that they had a range of neutralization abilities, ranging from half-maximal inhibitory concentration (IC50) values of <1 nM to >500 nM. However upon conversion to immunoglobulin G (IgG), certain clones displayed a complete loss of neutralization activity while maintaining strong kinetic measurements toward recombinant RBD, suggesting we have identified multiple structurally constrained epitopes that are sensitive to neutralization but incompatible with larger and more rigid IgGs. This challenge highlights the inherent risk of antibody discovery using the single-chain format, as some of the IgGs that lost neutralization were among our most potent neutralizing scFv-Fcs.
Methods
Phage panning
Peripheral B cells from 57 healthy donors were used to create two, non-immunized scFv-phage libraries totaling 2.7 × 1010 members (Sui et al., 2004). 1.66 × 1012 pfu of scFv-phage from each library was combined and used to perform three rounds of panning against SARS-CoV-2 S1 protein (Sino Biologicals, Wayne, PA) or SARS-CoV-2 RBD protein expressed in our lab. Briefly, SARS-CoV-2 RBD or S1 proteins were passively absorbed onto Nunc MaxiSorp Immuno tubes (Thermo Fisher Scientific) overnight in PBS. S1 coated tubes were incubated with our phage library, followed by extensive PBS/PBS-T (PBS + 0.05% Tween-20) washes to remove nonspecific phage. Bound phage were eluted with 100 mM triethylamine and neutralized with 1 M Tris–HCl, pH 7.5. The neutralized phage solution was used to infect exponentially growing TG1 E.coli cells, and the infected bacterial cells were plated out on 2xYT + 1% glucose plates. The next day, the TG1 colonies were scraped from the plates and VCS-M13 helper phage was added (MOI 20) to rescue the enriched phage library. The purified library was then used for subsequent rounds of panning, and the SARS-CoV-2 S1 and RBD coating concentration was decreased in each round to increase the affinity of the enriched antibodies.
Screening of the enriched library was performed by picking single bacterial colonies from the third round of panning and culturing them in 96 well plates. Small scale rescue was performed via VCS-M13 helper phage, and the phage supernatant was used to screen via SARS-CoV-2 RBD or S1 coated enzyme-linked immunoassay (ELISA) plates. Positive wells were selected for colony PCR and subsequent Sanger sequencing (Quintara Biosciences).
Protein Expression and Purification
Recombinant SARS-CoV-2 protein production
hACE2 (transOMIC) and SARS-CoV-2 RBD/S1 (SinoBiologics) cDNA were purchased and cloned into our mammalian expression vector. Stabilized SARS-CoV-2 spike trimer expressing plasmid was obtained through BEI and HexaPro was a kind gift from Dr Jason McLellan’s Lab (UT Austin) (Stabilized spike produced under HHSN272201400008C and obtained through BEI Resources, NIAID, NIH: Vector pCAGGS Containing the SARS-Related Coronavirus 2, Wuhan-Hu-1 Spike Glycoprotein Gene (soluble, stabilized), NR-52394). All proteins were expressed in the Expi293F system and cells were transiently transfected by Expifectamine 293 following the standard protocol. 4-5 days post-transfection, supernatants were clarified and incubated overnight with Ni-NTA resin (Qiagen). They were subsequently purified via gravity flow column and buffer exchanged by centrifugation in Amicon centrifugal filters. Avi tagged proteins were biotinylated by Avidity’s BirA biotinylation kit following standard protocols. Protein concentration was measured on a Nanodrop 100 using the MW and extinction coefficients calculated on ExPASy’s ProtParam.
Antibody production
Recombinant scFv-Fc, IgG and Fab antibodies were produced in Expi293F cells (ThermoFisher Scientific). Mammalian expression vectors encoding the antibodies were transiently transfected using Expifectamine 293 following the standard protocol and cultivated for 4 days. The harvested supernatants were incubated with Protein A-Sepharose 4B resin (Invitrogen) or Ni-NTA Superflow (Qiagen) overnight at 4°C followed by purification via gravity flow columns (BioRad) and buffer exchanged by centrifugation in Amicon centrifugal filters. Protein concentration was measured on a Nanodrop 100 using the MW and extinction coefficients calculated on ExPASy’s ProtParam.
SDS PAGE
Samples were mixed with 4× NuPAGE LDS Sample Buffer and ran on a Bolt 4–12% Bis-Tris gel (Invitrogen) following standard protocols. The gel was then removed from the casing and stained with Coomassie Brilliant Blue R250 staining solution (BioRad). Destaining was performed overnight using a solution of 50% methanol, 40% ddH2O, 10% acetic acid. Prior to imaging, gel was rehydrated in ddH2O.
Biolayer interferometry binding assays
All assays were performed in 96-well black plates on an OctetRed96 instrument with shaking at 1000 RPM. Curve fitting was performed using a 1:1 binding model in the data analysis software. Mean KD, Kon, Kdis values were determined with a global fit applied to all data.
SARS-CoV-2 RBD kinetic binding assay
Streptavidin biosensors were hydrated for >10 min in 1X PBS supplemented with 0.05% v/v Tween-20. Biotinylated SARS-CoV-2 RBD or S1 at 1–5 ug/ml and antibodies in 2-fold serial dilutions from 50 nM were prepared in PBS-T. The experiment included the following steps at 25°C: (i) baseline in PBS-T (60 s); (ii) loading of RBD onto sensors (90 s); (iii) baseline PBS-T (60 s); (iv) association of antibodies for Kon measurement (90 s); (v) dissociation of antibodies for Kdis measurement in PBST (300 s); (vi) sensor regeneration and neutralization in 0.1 M glycine (pH 2.5) and PBS-T (30 s).
SARS-CoV-2 RBD competitive binding assay
Streptavidin biosensors were pre-equilibrated for a minimum of 10 min in 1X PBS-T. Biotinylated RBD or S1 at 1–5 ug/ml and antibodies/proteins (saturating at 250 nM and competing at 125 nM) were prepared in PBS-T. The experiment included the following steps at 25°C: (i) baseline in PBS-T (60 s); (ii) loading of RBD onto biosensors (30 s); (iii) baseline in PBS-T (60 s); (iv) association of saturating antibodies (or buffer as a control) (600 s); (v) baseline in PBS-T (60 s); (vi) association of competing antibody/proteins (300 s); (vii) sensor regeneration and neutralization in 0.1 M glycine (pH 2.0) and PBS-T (30 s).
Pseudovirus neutralization assay
SARS-CoV-2 pseudovirus was produced by transfecting Lenti-X cells (Takara Bio) with Lipofectamine 3000. Three days post transfection the supernatant was harvested and filtered through a 0.45 um filter to remove cellular debris. A total of 10,000 293T-angiotensin converting enzyme-2 (ACE2) cells/well were seeded in 96 well white bottom plates the day before the assay. Clarified pseudoviral supernatant was mixed 1:1 with the appropriate antibody dilution and incubated at RT for 1 h. Media was carefully removed from the culture plate and then replaced with 60 ul of the pseudovirus/Ab mixture. The plate was incubated for 48 h (37°C, 5% CO2) before supernatant was removed and cells were lysed via passive lysis buffer (Promega) following standard protocols. Luciferase expression was detected by the addition of Promega Bio-Glo and plates were read on a PolarStar Omega.
Plaque reduction neutralization test
A series of 10 half-log dilutions was prepared in triplicate for each antibody or antibody mixture in Dulbecco’s Phosphate Buffered Saline (DPBS) (Gibco). Each dilution was incubated at 37°C and 5% CO2 for 1 h with an equal volume of 1000 plaque forming units/ml (PFU/ml) of SARS-CoV-2 (isolate USA-WA1/2020), diluted in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco) containing 2% fetal bovine serum (Gibco) and antibiotic–antimycotic (Gibco). Controls included DMEM containing 2% fetal bovine serum and antibiotic–antimycotic only as a negative control, 1000 PFU/ml SARS-CoV-2 incubated with DPBS, and 1000 PFU/ml SARS-CoV-2 incubated with DMEM. Two hundred microliters of each dilution or control were added to confluent monolayers of Vero E6 cells in triplicate and incubated for 1 h at 37°C and 5% CO2. The plates were gently rocked every 5–10 min to prevent monolayer drying. The monolayers were then overlaid with a 1:1 mixture of 2.5% Avicel® RC-591 microcrystalline cellulose and carboxymethylcellulose sodium (DuPont Nutrition & Biosciences, Wilmington, DE) and 2X Modified Eagle Medium (Temin’s modification, Gibco) supplemented with 2X antibiotic–antimycotic (Gibco), 2X GlutaMAX (Gibco) and 10% fetal bovine serum (Gibco). Plates were incubated at 37°C and 5% CO2 for 2 days. The monolayers were fixed with 10% neutral buffered formalin and stained with 0.2% aqueous Gentian Violet (RICCA Chemicals, Arlington, TX) in 10% neutral buffered formalin for 30 min, followed by rinsing and plaque counting. The IC50 were calculated using GraphPad Prism 8.
FACS binding
293T cells were transduced to stably express membrane bound SARS-CoV-2 spike protein. 2E5 cells were washed and resuspended in cold MACS rinsing buffer + BSA (Miltenyi) before adding to antibodies diluted in cold MACS buffer. Cells were incubated at 4°C for 1 h to allow for antibody binding, after which they were washed 2× with MACS buffer before incubation with fluorescently labeled anti-hFc (BioLegend) for 20 min at 4°C. Cells were washed 3× with cold MACS buffer before being fixed with 1% paraformaldehyde (Boston BioProducts). Cells were analyzed on a BD Canto II with HTS reader. Samples were run in triplicate.
FACS S1 disassociation
293T-Spike cells were washed, resuspended at 4E6 cells/ml in MACS buffer+ and aliquoted at 50 ul per well in a V bottom 96 well plate. Antibodies were diluted to 200 nM in MACS buffer+. MACS buffer+ contains 20 uM cycloheximide to inhibit protein synthesis. Ab dilution and cell plates were first incubated at 37°C for 15 min to equilibrate the plates. At the desired time points, 50 ul of Ab dilution was transferred to the corresponding well in the 96 well plate and mixed via pipetting. The plate was maintained at 37°C during the entire time course. After the last time point, the cell plates were rapidly transferred to ice and quenched with ice cold MACS buffer. The plate was washed 2× with MACS buffer, followed by resuspension in anti-hFc-APC (Biolegend) for 20 min at 4°C. Cells were washed 3× with MACS buffer before fixation by 1% PFA. Cells were analyzed immediately on a BD Canto II with HTS reader. Samples were run in duplicate.
Results
Phage panning
Discovery of novel SARS-CoV-2 antibodies began with phage panning campaigns for recombinantly expressed RBD and S1 domains of the spike. Phage panning is a stochastic process and highly dependent on the orientation and order in which the target protein is presented. Three unique panning campaigns were performed; 3xS1, 3xRBD, and to take advantage of panning flexibility, rescued phage from the second round of the S1 panning was also applied to RBD protein for a parallel third round, resulting 2xS1-1xRBD (Fig. S1). Circa 1300 random colonies were selected from these three pannings and cultured for screening by phage ELISA followed by sequencing of positive wells. Sequence alignment revealed the three panning campaigns resulted in 65 unique clones, 39 of which were able to be cloned and expressed as scFv-Fcs (Fig. S2).
Kinetic screening of anti-spike antibodies
ScFv-Fcs were transiently transfected into Expi293F cells and purified via protein A resin. Kinetics were measured via biolayer interferometry (BLI) to determine the apparent Kon, Kdis and KD values for each antibody (Fig. 1A). As antigen was first loaded onto each sensor followed by the antibody, the bivalent nature of the antibody introduces the possibility of avidity effects. However low antigen coating concentrations were used, which allows for spatial separation of the loaded antigens, decreasing the ability of an antibody to simultaneously interact with multiple ligands. Of the 39 antibodies, 30 showed strong binding to the S1 domain of the spike protein and displayed a range of kinetic characteristics. The majority of antibodies bound to S1 with nanomolar affinity, with 25% of them having off-rates below the limit of detection (LOD) of the OctetRed instrument (<1.0E-7 s−1). Antibodies with fast off-rates tended to be partially balanced out by a fast on-rate, as the four highest off-rate antibodies (Abs 37, 16, 21, 20) did not have the lowest KDs. The highest KD values of our anti-SARS-CoV-2 antibodies were ~1E-8 M, held by Abs 25 and 2-10, both of which have substantially lower on-rates compared with the rest of the antibody pool.
Fig. 1.

Kinetic and competition results with corresponding epitope binning of anti-SARS-CoV-2 antibodies. (A) Kinetic measurements were performed using biotinylated S1 protein and the panel of anti-SARS-CoV-2 antibodies was competed against reference antibodies CR3022 and Ab 12 and recombinant ACE2 in a BLI-based competition assay. Competition and percent blockade is determined by comparison to an unblocked sensor. Competition is indicated by a black block, and percent blockade is shown on a sliding scale, with red blocks displaying near complete inhibition and green blocks showing minimal inhibition. (B) Competition matrices and SARS-CoV cross reactivity were used to create antibody sub-bins, each with a different competition pattern or binding properties. The majority of antibodies bound to the RBD, with one-third targeting the CR3022 epitope and two-third targeting the ACE2 interface. N.d., not determined
Epitope mapping of anti-spike antibodies
First pass antibody clustering was done by BLI-based competition matrices of sequentially numbered antibodies, allowing for the selection of a representative clone for each cluster based on kinetic properties. General antibody bins were then defined in BLI studies by competition with two RBD targeted antibodies, ACE2 blocking antibody Ab 12 or non-ACE2 blocking CR3022. These results lead to the identification of three broad bins: Bin 1 targeted S1 outside of the RBD, Bin 2 targeted the CR3022 epitope of the RBD and Bin 3 targeted the ACE2 binding region (Yuan et al., 2020; Chang et al., 2022). Sub-bins were identified by detailed competition matrices and as shown in Figure 1B, allowed for further subdivision of the Ab 12 group into six sub-bins. One Ab 12 sub-bin (Bin 3F) was unique in that all antibodies tested (4 of 6 in Bin 3F) competed with Ab 12 but surprisingly did not inhibit ACE2 binding (>75% blockade). The remaining Ab 12 competing sub-bins all inhibited ACE2 binding and were defined by varying competition patterns with other antibodies within the group. For example, Ab 27 competes with Ab 12 and ACE2, whereas Ab 26 blocks Ab 12, partially blocks ACE2, and does not block Ab 27. Ab 12 is the most potent ACE2 binding inhibitor, however Abs 14, 27, 29 and 38 all block >85% of ACE2-RBD binding and represent three separate sub-bins in the Ab 12 group, suggesting they bind related but discrete epitopes from each other (Fig. 1B).
CR3022 is known to bind to the RBD opposite of the ACE2 binding interface, and it does not block ACE2 binding. However, all of the antibodies in the CR3022 group significantly inhibited ACE2 binding to RBD, suggesting that they do not bind an identical epitope to CR3022. Cross binding assays of our antibodies with SARS-CoV spike revealed that only Ab 2-7 cross-reacts and cross neutralizes and thus binds to a different epitope compared with the rest of the bin and CR3022 (Chang et al., 2022).
Authentic virus neutralization of representative scFv-Fcs
Based on the epitope binning results, representative antibodies were chosen from each sub-bin for authentic virus neutralization assays using plaque reduction neutralization tests (PRNT). Figure 2A shows a panel of scFv-Fcs with varying virus neutralization potency. Potent antibodies with IC50 < 10 nM came from Bins 3A (Abs 7, 12), 3C (Abs 14, 18, 27) and 3E (Ab 38) with Ab 12 and Ab 27 showing the most potent activity with IC50s of 0.87 nM and 0.43 nM respectively. Even though antibodies in Bins 3B and 3F do not compete extensively with ACE2, those that were tested displayed moderate neutralization, suggesting a different mechanism of neutralization. Representative antibodies from Bin 2 were tested against authentic virus and Ab 2-7 (Bin 2B) displayed moderate neutralization with an IC50 of ~47 nM (Fig. 2B), whereas Ab 28 (Bin 2C) did not neutralize and since this was the highest affinity antibody in the group, additional members of the group were not tested. Interestingly, non-RBD targeting Ab 5 was also able to neutralize authentic virus, albeit with a much higher IC50 of 588.7 nM.
Fig. 2.

Neutralization activity of anti-SARS-CoV-2 scFv-Fcs. (A) Selected antibodies were tested against authentic SARS-CoV-2 virus in full neutralization curves. (B) Anti-SARS-CoV-2 scFv-Fcs demonstrate a wide range of neutralization activity, with Abs 12 and 27 on the potent end and Abs 28 and 23 on the weak/non-neutralizing end. Bin 3 antibodies typically have the most potent activity and Bins 1-2 display considerably weaker activity.
IgG conversion and testing
Abs 5, 12, 14, 27, 29, 38 and 2-7 were selected for conversion to IgG per convention in our antibody discovery pipeline. This allowed us to cover the most potent neutralizing scFv-Fcs (Abs 12, 14, 27) as well as antibodies from Bin 1 (Ab 5) and Bin 2 (Ab 2-7). Kinetic experiments with recombinant RBD comparing converted IgGs with the original scFv-Fcs demonstrated minor differences in affinity, however this variation was likely due to instrument noise/sensitivity suggesting that the IgGs would neutralize authentic virus at comparable levels (Fig. 3A). To test this, side-by-side authentic virus neutralization assays were performed. Abs 12 and 38 maintained or showed minor decreases in neutralization efficiency, however unexpectedly, a number of the IgGs exhibited a marked loss in neutralization efficiency, with IC50s decreasing by >100-fold if not disappearing completely (Fig. 3B).
Fig. 3.

Characterization of converted IgGs. (A) Kinetic measurements via BLI of selected antibodies show that conversion from scFv-Fc to IgG does not have a significant effect on the KD values for these antibodies when binding the RBD. (B) scFv-Fcs and IgGs were tested in parallel SARS-CoV-2 neutralization assays (PRNT). Abs 12 and 38 showed minimal loss in neutralization efficacy, however Abs 14, 27, 29 and 2-7 displayed substantial loss. Of the three most potent scFv-Fcs, Abs 12 and 38 maintain their ability to neutralize as IgGs, whereas the IC50 for Abs 14 and 27 shifts to the right by 40- and 950-fold respectively. In this assay, Ab 29 and 2-7 IgGs appear to lose all neutralization ability; however, due to the high IC50 for the scFv-Fcs, this may be due to the curve shifting further to the right than our assay tests.
Antibody binding potential and characteristics
Fluorescence-activated cell sorting (FACS) staining was performed on selected IgG and scFv-Fc pairs to determine if RBD and full spike binding differed. Similar to the neutralization experiments, the IgG for Abs 14, 27 and 2-7 experienced significant decreases in binding to cell surface expressed spike compared with the original scFv-Fcs (Fig. 4). As previous data demonstrated that these IgGs bind to the RBD with comparable affinity compared with the scFv-Fcs (Fig. 3A), the loss of binding to intact trimeric spike on the cell surface suggests that a structural and/or energetic constraint may be responsible.
Fig. 4.
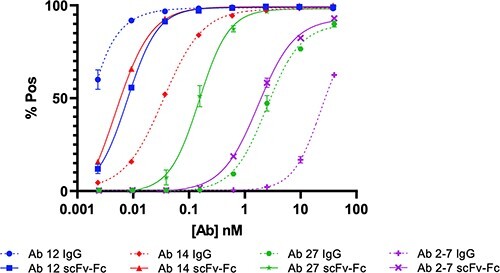
FACS binding curves comparing selected IgG and scFv-Fc pairs. (A) FACS binding curves with 293T-Spike cells show a pronounced decrease in binding for Abs 14, 27 and 2-7, whereas Ab 12 shows an increase in binding.
Mechanism of neutralization for scFv-Fcs and selected IgGs
The majority of our antibodies have similar affinity and as shown in Fig. S3, there is not a strong correlation between KD, Kon, Kdis and neutralization (IC50), rather the greatest correlation is seen between ACE2 blockade and IC50, supporting the idea that epitope plays a more important role in neutralization than affinity. This is further supported by the fact that there are some antibodies with strong binding kinetics that do not neutralize as expected. For example, Ab 29 has a fast on-rate and an off-rate below the LOD of the octet (KD > 1E-12 M) whereas Ab 14 has an equivalent on-rate but a >103 fold faster off-rate, leading to a KD of 5.94E-10 M. Both block the ACE2/RBD binding in competition assays, thus it would be expected that Ab 29 would be a more potent neutralizer. However, based on scFv-Fc virus neutralization curves, Ab 14 is nearly 10-fold more potent than Ab 29 (IC50 of 1.86 versus 17.6 nM, respectively). To parse out these differences between binding affinity and neutralization, we looked at a potential mechanism of action for a representative group of scFv-Fcs.
When the spike protein binds to ACE2, a drastic conformational shift occurs in which the S1 domain dissociates from the spike and S2 enters a post-fusion state enabling membrane fusion (Cai et al., 2020). Premature shedding of the S1 domain is an effective and irreversible method of viral neutralization as the post fusion spike is not able to fuse with the target cell membrane (Jin et al., 2021). To see if our antibodies were able to mimic ACE2 and initiate the transformation into the post-fusion conformation, we performed a spike shedding FACS experiment as previously described by Wec et al. (2020) (Huang et al., 2022). Briefly, trimeric spike expressing cells are incubated with saturating levels of antibodies, and the level of bound antibody was measured via FACS at different time points. Figure 5A shows that over time, the fluorescence of Ab 14 incubated cells decreases to levels comparable to that of cells incubated with ACE2. Ab 29, on the other hand, also decreases slightly but not to levels equivalent to ACE2 or Ab 14, suggesting that the epitope that Ab 14 targets is a stronger initializer of S1 shedding compared with the Ab 29 epitope.
Fig. 5.

Spike shedding induced by anti-SARS-CoV-2 antibody binding. (A) Selected scFv-Fcs were tested in a spike shedding experiment using 293T cells stably expressing the SARS-CoV-2 spike. Percent change is relative to the median fluorescence signal for each sample at the 5 min time point, and antibody traces are colored based on their epitope sub-bins. Bin 2 antibodies Ab 2-7 and Ab 28 do not lead to shedding of the spike as seen by the increase in fluorescent signal over time. Antibodies in Bin 3E (Abs 17, 19, 35) trigger minimal to low shedding (green traces), whereas Abs in Bin 3C (Abs 14, 18, 27, 38) all lead to significant levels of spike shedding. Ab 12 (Bin 3A) leads to the most significant amount of shedding, whereas sub bin member Ab 7 induces significantly less shedding. (B) Selected scFv-Fcs were tested against their respective IgGs. Abs 12 and 38 display similar levels of spike shedding as an IgG and scFv-Fc, correlating to a minimal change in IC50 in in vitro virus neutralization assays. Abs 14, 27 and 29 show pronounced decreases in the amount of spike shedding in the IgG compared with the original scFv-Fc, providing a rational to their drastic loss of neutralization efficacy seen with in vitro neutralization assays.
Levels of spike shedding generally followed the mAb bin groupings. For example, antibodies in Bins 3A and 3C lead to significant amounts of spike shedding, compared with antibodies in Bin 3F that displayed a flatter profile. Bin 3F antibodies are surprising as they have modest IC50 values (~20 nM), but do not substantially block ACE2 or lead to spike shedding, suggesting a different mechanism of action. Looking at antibodies in other bins, it is clearly seen that Bin 2 antibodies (Abs 28 and 2-7) do not lead to spike shedding in this assay as the fluorescence increases over time, suggesting that Ab 2-7 neutralizes by sterically blocking ACE2.
The spike shedding assay was next utilized to examine the mechanistic difference between scFv-Fcs and their converted IgGs. We first looked at Abs 12 and 38, which remain potent neutralizing Abs following conversion, and as shown in Figure 5, incubation with either format leads to similar levels of spike shedding. Conversely, incubation with Abs 14, 27 or 29 IgG leads to a substantial reduction in spike shedding compared with the scFv-Fc, correlating to their reduced neutralization efficacy. This suggests that while the scFv-Fcs are able to utilize spike shedding as a second mechanism of action to neutralize viral infection, the IgGs are unable to do so and similar to Bin 2 antibodies rely solely on their ability to block ACE2 binding leading to decreased neutralization.
Bivalent binding is required for spike shedding
One possible explanation for the decreased cell surface binding seen with the IgGs is that they are unable to simultaneously bind both arms, leading to a decrease in avidity. Therefore we next looked to examine how this would affect spike shedding by producing a monovalent Fab format of our most characterized antibody (Ab 12). Figure 6 shows the effect of simultaneous binding as both bivalent (IgG, scFv-Fc) molecules demonstrate similar levels of S1 shedding, whereas the monovalent Fab is unable to induce shedding. Pseudovirus experiments further confirm this result as the decreased levels of spike shedding correspond to an appreciable difference in neutralization efficacy (Fig. S4). This demonstrates that for antibodies targeting the RBD saddle-ridge domains and possibly Bin 3C and 3D epitopes, simultaneous binding of both arms is required to trigger spike shedding and provides a potential rationale for the loss of neutralization observed following conversion of some scFv-Fcs to IgGs (Greaney et al., 2021; Chang et al., 2022).
Fig. 6.

Spike shedding assay comparing mono- versus bivalent binding. scFv-Fc, Fab and IgG formats of Ab 12 were tested in a spike shedding experiment. Only the bivalent scFv-Fc and IgG lead to spike shedding, whereas the monovalent Fab signal remains constant throughout the experiment.
Discussion
Since the sequence of the SARS-CoV-2 spike protein was first published in early January 2020, a large number of neutralizing antibodies have been identified from convalescent patient B-cells, animal immunization and phage display (Hansen et al., 2020; Li et al., 2020; Jones et al., 2021; Zhao et al., 2021; Yuan et al., 2022). In this work, we utilized our 27-billion-member naïve phage-Ab library to identify a large panel of high affinity, anti-SARS-CoV-2 antibodies targeting the S1 domain of the spike. Many of these antibodies block the ACE2-RBD interaction and neutralize SARS-CoV-2 virus in vitro, whereas one (Ab 5) moderately neutralizing antibody targeted S1 outside of the RBD. Selected scFv-Fcs were then converted into IgG format without an appreciable decrease in affinity, however in vitro neutralization assays surprisingly resulted in substantially decreased potency. Using a panel of representative scFv-Fcs, we delved into the mechanism of neutralization and further observed that many of our most potent scFv-Fcs benefited from a secondary mode of action, where binding initiated the switch to the post fusion conformation, permanently disabling the spike protein and preventing future membrane fusion. Examination of different antibody formats also revealed the importance of avidity and simultaneous engagement of antibody arms as monovalent Fab fragments were not able to induce shedding.
At the most basic level, for an antibody to neutralize SARS-CoV-2 infection, it needs to block the ACE2-RBD binding, thereby preventing infection of host cells. Though we identified a number of antibodies that fall into that category, we also identified a neutralizing sub-bin (3F) that targets the RBD but does not substantially block ACE2-RBD binding. Though these were all high affinity antibodies, there appeared to be a theoretical limit to their neutralization potential as none of the tested Abs were able to achieve subnanomolar IC50 values. By delving further into our most potent neutralizing scFv-Fcs, we were able to demonstrate that they not only blocked ACE2-RBD binding, but also benefited from a second mode of action where they triggered irreversible spike shedding, permanently disabling the viral particle from infecting cells and leading to potent neutralization (Figs 2 and 5).
The next step in a typical antibody discovery campaign would be converting the scFv-Fcs into IgGs for further development. Though it is possible that conversion results in an antibody with decreased or increased affinity due to conformational changes resulting from removal of the single-chain linker, this is generally a routine step leading to an IgG without appreciable changes in affinity (Quintero-Hernández et al., 2007; Steinwand et al., 2014; Rangnoi et al., 2021). As expected, initial screening of the new IgGs using BLI indicated minor differences in affinity to soluble RBD, likely a result of instrument noise/sensitivity. To our surprise, the majority of these IgGs exhibited severely reduced neutralization capabilities (Fig. 3B). To first explore the difference in these formats, FACS binding curves were generated for selected scFv-Fc/IgG pairs. These curves revealed that while the affinity for soluble RBD was unchanged, the ability to bind cell surface displayed full length spike was severely diminished for IgGs with decreased neutralization (Fig. 4). One potential reason for this decrease in binding has been previously demonstrated using cryo-EM data and modeling of Ab 2-7, as the smaller scFvs are afforded easy access to the epitope while structural constraints only observed with the trimeric spike protein block binding of the Fab/IgG formats due to steric clashes between the CH1 and N-terminal domain (NTD). A second possible explanation for this difference in binding is that the epitope is oriented in a way that both arms of the IgG are unable to simultaneously bind, leading to a loss of avidity and thus resulting in the rightward shift in the binding curve.
Utilizing an established spike shedding assay, we explored the effect of IgG conversion on spike shedding and if simultaneous binding of both IgG arms is required. Comparing levels of scFv-Fc and IgG induced shedding clearly indicated that the loss of spike shedding is correlated to a loss of neutralization efficacy. This is best embodied by Ab 29, which as an scFv-Fc triggers moderate spike shedding that is completely abrogated upon conversion to IgG, a pattern mirrored in PRNT assays (Figs 3 and 5). To examine if the loss of a binding arm could be responsible for this difference, we compared the levels of spike shedding for bi- and monovalent fragments of Ab 12 (Yan et al., 2021). While both scFv-Fc and IgG formats were able to induce significant levels of spike shedding, the monovalent Fab was unable to replicate this phenomenon. Though only one antibody was tested in these formats, this result provides evidence suggesting the importance of simultaneous binding and supports the idea that these IgGs are only able to bind with one Fab arm at a time, further supporting the premise that ACE2 blockade alone is not sufficient to produce high potency neutralizing antibodies, rather a secondary mechanism of action is required.
Antibodies discovered in our lab start as phage tethered scFvs, which are rapidly converted into scFv-Fcs for further characterization and screening. Part of the challenge begins with the target protein that is used for our phage display campaigns. By starting with recombinant RBD, we ensure that our antibodies target this sensitive part of the spike protein, however the entire RBD is available for phage-Ab binding as the structural constraints enforced by the remainder of the spike protein are missing. A similar spike RBD panning strategy with our scFv-phage library was used during the SARS-CoV and MERS outbreaks to rapidly isolate IgGs with potent neutralization activity (Sui et al., 2004; Tang et al., 2014). However, in our anti-SARS-CoV-2 RBD/S1 antibody discovery campaign, FACS binding and spike shedding assays provided evidence that although the affinity of the larger and more rigid IgGs remain largely unchanged, a number of potent scFv-Fcs were nevertheless unable to successfully convert to IgG1, presumable due to the inability to engage both binding arms of the IgGs. As a result, these IgGs lose their ability to induce spike shedding and display a corresponding reduction in neutralization. Though starting this antibody discovery campaign by panning with trimeric full-length spike may have mitigated these constraints, this and our previous anti-CoV discovery efforts were initiated immediately following release of the spike sequence when only recombinant RBD and S1 were readily available as development of stabilized trimeric spike proteins had yet to occur (Hsieh et al., 2020). Following development of the HexaPro spike, panning against the recombinant spike protein would have been possible, however using the full-length spike would require additional epitope screening to identify S2 targeted Abs that are potentially cross reactive against spike epitopes conserved between endemic coronaviruses but also generally exhibit less potent neutralization activity (Kreer et al., 2020; Amanat et al., 2021; Shiakolas et al., 2021; Liu and Wilson, 2022). In addition, a cross-panning strategy similar to the S1/RBD panning used in this work could have been applied using HexaPro spike/RBD, which would lead to the selection of RBD-specific antibodies while taking into account the steric constraints of a trimeric spike. Thus the work presented in this manuscript draws attention to the importance of antigen selection in antibody discovery campaigns, as how the ‘bait’ is presented can have significant influence on the IgG convertibility of the resulting neutralizing antibodies.
Abbreviations
- ACE2
angiotensin converting enzyme-2
- BLI
biolayer interferometry
- ELISA
enzyme-linked immunoassay
- FACS
fluorescence-activated cell sorting
- IC50
half-maximal inhibitory concentration
- IgG
immunoglobulin G
- IRB
Institutional Review Board
- LOD
limit of detection
- mAb
monoclonal antibody
- MERS
Middle Eastern Respiratory Syndrome
- NTD
N-terminal domain
- PRNT
plaque reduction neutralization test
- RBD
receptor binding domain
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- scFv
single-chain variable fragment
- WT
wildtype
Author contributions
Matthew Chang (Conceptualization [equal], Formal analysis [equal], Investigation [equal], Methodology [equal], Supervision [equal], Writing—Original draft [equal], Writing—Review & editing [equal]), Hanzhong Ke (Conceptualization [supporting], Supervision [supporting]), Laura Losada Miguéns (Investigation [supporting]), Christian Coherd (Investigation [supporting]), Katrina Nguyen (Investigation [supporting]), Maliwan Kamkaew (Investigation [supporting]), Rebecca Johnson (Formal analysis [supporting], Investigation [supporting]), Nadia Storm (Formal analysis [supporting], Investigation [supporting]), Anna Honko (Formal analysis [supporting], Investigation [supporting], Methodology [equal], Supervision [equal], Writing—Review & editing [supporting]), Quan Zhu (Conceptualization [equal], Funding acquisition [equal], Investigation [equal], Methodology [equal], Supervision [equal]), Anthony Griffiths (Formal analysis [equal], Investigation [equal], Methodology [equal], Supervision [equal], Writing—Review & editing [supporting]), and Wayne Marasco (Conceptualization [equal], Formal analysis [equal], Funding acquisition [lead], Methodology [equal], Supervision [lead], Writing—Original draft [equal], Writing—Review & editing [equal]).
Conflict of interest
The authors report there are no competing interests to declare.
Funding
This work was supported by the Massachusetts Consortium on Pathogen Readiness (MassCPR) under Grant [280870.5116709.0016 to W.A.M.]; National Institute of Allergy and Infectious Diseases under Grant [1R01AI161152-01A1 to W.A.M.].
Data availability
The data that support the findings of this study are available from the corresponding author, W.A.M., upon reasonable request.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The following reagent was obtained through BEI Resources, NIAID, NIH: Modified pαH Vector Containing the SARS-Related Coronavirus 2, Wuhan-Hu-1 HexaPro Spike Glycoprotein Ectodomain, NR-53587.
Edited by: Dr Timothy Whitehead and Dr Roberto Chica
Contributor Information
Matthew R Chang, Department of Cancer Immunology & Virology, Dana-Farber Cancer Institute, Boston, MA 02215, USA.
Hanzhong Ke, Department of Cancer Immunology & Virology, Dana-Farber Cancer Institute, Boston, MA 02215, USA; Department of Medicine, Harvard Medical School, Boston, MA 02115, USA.
Laura Losada Miguéns, Department of Cancer Immunology & Virology, Dana-Farber Cancer Institute, Boston, MA 02215, USA.
Christian Coherd, Department of Cancer Immunology & Virology, Dana-Farber Cancer Institute, Boston, MA 02215, USA.
Katrina Nguyen, Department of Cancer Immunology & Virology, Dana-Farber Cancer Institute, Boston, MA 02215, USA.
Maliwan Kamkaew, Department of Cancer Immunology & Virology, Dana-Farber Cancer Institute, Boston, MA 02215, USA.
Rebecca Johnson, Department of Virology, Immunology, and Microbiology and National Emerging Infectious Diseases Laboratories, Boston University School of Medicine, Boston, MA 02118, USA.
Nadia Storm, Department of Virology, Immunology, and Microbiology and National Emerging Infectious Diseases Laboratories, Boston University School of Medicine, Boston, MA 02118, USA.
Anna Honko, Department of Virology, Immunology, and Microbiology and National Emerging Infectious Diseases Laboratories, Boston University School of Medicine, Boston, MA 02118, USA.
Quan Zhu, Department of Cancer Immunology & Virology, Dana-Farber Cancer Institute, Boston, MA 02215, USA; Department of Medicine, Harvard Medical School, Boston, MA 02115, USA.
Anthony Griffiths, Department of Virology, Immunology, and Microbiology and National Emerging Infectious Diseases Laboratories, Boston University School of Medicine, Boston, MA 02118, USA.
Wayne A Marasco, Department of Cancer Immunology & Virology, Dana-Farber Cancer Institute, Boston, MA 02215, USA; Department of Medicine, Harvard Medical School, Boston, MA 02115, USA.
References
- Alfaleh, M.A., Alsaab, H.O., Mahmoud, A.B. et al. (2020) Front. Immunol., 11. 10.3389/fimmu.2020.01986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amanat, F., Thapa, M., Lei, T. et al. (2021) Cell, 184, 3936–3948.e10. 10.1016/j.cell.2021.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum, A., Fulton, B.O., Wloga, E. et al. (2020) Science, 369, 1014. 10.1126/science.abd0831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, Y., Zhang, J., Xiao, T. et al. (2020) Science, 369, 1586–1592. 10.1126/science.abd4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, M.R., Tomasovic, L., Kuzmina, N.A. et al. (2022) Nat. Commun., 13, 5814. 10.1038/s41467-022-33030-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimizadeh, W. and Rajabibazl, M. (2014) Curr. Microbiol., 69, 109–120. 10.1007/s00284-014-0557-0. [DOI] [PubMed] [Google Scholar]
- Greaney, A.J., Starr, T.N., Gilchuk, P. et al. (2021) Cell Host Microbe, 29, 44–57.e9. 10.1016/j.chom.2020.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, J., Baum, A., Pascal, K.E. et al. (2020) Science, 369, 1010–1014. 10.1126/science.abd0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh, C.L., Goldsmith, J.A., Schaub, J.M., et al. (2020) 1505, 1501–1505. 10.1126/science.abd082. [DOI] [Google Scholar]
- Huang, Q., Han, X. and Yan, J. (2022) Emerg. Microbes Infect., 11, 2412–2422. 10.1080/22221751.2022.2125348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, D., Wei, J. and Sun, J. (2021) Biochem. Biophys. Res. Commun., 566, 45–52. 10.1016/j.bbrc.2021.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, B.E., Brown-Augsburger, P.L., Corbett, K.S. et al. (2021) Sci. Transl. Med., 13, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreer, C., Zehner, M., Weber, T. et al. (2020) Cell, 182, 843–854.e12. 10.1016/j.cell.2020.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W., Chen, C., Drelich, A. et al. (2020) Proc. Natl. Acad. Sci. U. S. A., 117, 29832–29838. 10.1073/pnas.2010197117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H. and Wilson, I.A. (2022) Immunol. Rev., 310, 76–92. 10.1111/imr.13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno, E., Valdés-Tresanco, M.S., Molina-Zapata, A. et al. (2022) BMC. Res. Notes, 15, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren, K.A. and Noren, C.J. (2001) Methods, 23, 169–178. 10.1006/meth.2000.1118. [DOI] [PubMed] [Google Scholar]
- O’connell, D., Becerril, B., Roy-Burman, A. et al. (2002) J. Mol. Biol., 321, 49–56. 10.1016/S0022-2836(02)00561-2. [DOI] [PubMed] [Google Scholar]
- Peterson, F.C., Anderson, P.J., Berliner, L.J. et al. (1999) Protein Expr. Purif., 15, 16–23. 10.1006/prep.1998.0984. [DOI] [PubMed] [Google Scholar]
- Quintero-Hernández, V., Juárez-González, V.R., Ortíz-León, M. et al. (2007) Mol. Immunol., 44, 1307–1315. 10.1016/j.molimm.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Rangnoi, K., Rüker, F., Wozniak-Knopp, G. et al. (2021) ACS Omega, 6, 25258–25268. 10.1021/acsomega.1c03044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellmann, C., Pekar, L., Bauer, C. et al. (2020) Mol. Biotechnol., 62, 228–239. 10.1007/s12033-020-00236-0. [DOI] [PubMed] [Google Scholar]
- Shiakolas, A.R., Kramer, K.J., Wrapp, D. et al. (2021) Cell Rep. Med., 2, 100313. 10.1016/j.xcrm.2021.100313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silacci, M., Brack, S., Schirru, G. et al. (2005) Proteomics, 5, 2340–2350. 10.1002/pmic.200401273. [DOI] [PubMed] [Google Scholar]
- Steinwand, M., Droste, P., Frenzel, A. et al. (2014) MAbs, 6, 204–218. 10.4161/mabs.27227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui, J., Hwang, W.C., Perez, S. et al. (2009) Nat. Struct. Mol. Biol., 16, 265–273. 10.1038/nsmb.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui, J., Li, W., Murakami, A. et al. (2004) Proc. Natl. Acad. Sci., 101, 2536–2541. 10.1073/pnas.0307140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, X.C., Agnihothram, S.S., Jiao, Y. et al. (2014) Proc. Natl. Acad. Sci., 111, PMID: 24778221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wec, A.Z., Wrapp, D., Herbert, A.S. et al. (2020) Science, 369, 731–736. 10.1126/science.abc7424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, C., Lo, A., Yammanuru, A. et al. (2010) PloS One, 5. 10.1371/journal.pone.0009625. [DOI] [Google Scholar]
- Yan, R., Wang, R., Ju, B. et al. (2021) Cell Res., 31, 517–525. 10.1038/s41422-021-00487-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, M., Wu, N.C., Zhu, X. et al. (2020) Science, 368, 630–633. 10.1126/science.abb7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, T.Z., Garg, P., Wang, L. et al. (2022) MAbs, 14, 1–17. 10.1080/19420862.2021.2002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, S., Zhang, H., Yang, X. et al. (2021) Nat. Commun., 12, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, W.A.M., upon reasonable request.