

Abstract
The conserved protein HMCES crosslinks to abasic (AP) sites in ssDNA to prevent strand scission and the formation of toxic dsDNA breaks during replication. Here, we report a non‐proteolytic release mechanism for HMCES‐DNA‐protein crosslinks (DPCs), which is regulated by DNA context. In ssDNA and at ssDNA‐dsDNA junctions, HMCES‐DPCs are stable, which efficiently protects AP sites against spontaneous incisions or cleavage by APE1 endonuclease. In contrast, HMCES‐DPCs are released in dsDNA, allowing APE1 to initiate downstream repair. Mechanistically, we show that release is governed by two components. First, a conserved glutamate residue, within HMCES' active site, catalyses reversal of the crosslink. Second, affinity to the underlying DNA structure determines whether HMCES re‐crosslinks or dissociates. Our study reveals that the protective role of HMCES‐DPCs involves their controlled release upon bypass by replication forks, which restricts DPC formation to a necessary minimum.
Keywords: abasic (AP) sites, DNA repair, DNA replication, DNA‐protein crosslinks, HMCES
Subject Categories: DNA Replication, Recombination & Repair; Post-translational Modifications & Proteolysis
DNA context controls dissociation of the abasic‐site protector HMCES upon bypass of replication forks to restrict DPC formation to the necessary minimum.
Introduction
Covalent crosslinks between proteins and DNA (DNA‐protein crosslinks, DPCs) are dangerous lesions caused by a variety of endogenous and exogenous sources, including widely used chemotherapeutic agents (Tretyakova et al, 2015; Stingele et al, 2017). DPCs are toxic because they interfere with DNA replication (Duxin et al, 2014). Therefore, cells possess conserved repair mechanisms that target DPCs in replication‐dependent and ‐independent manners (Weickert & Stingele, 2022). DPC repair involves the proteolytic destruction of the protein adduct by DPC‐specific proteases of the SPRTN/Wss1 family or by proteasomal degradation (Stingele et al, 2014, 2016; Vaz et al, 2016; Larsen et al, 2019). Failure to degrade DPCs has drastic consequences; complete loss of SPRTN is lethal in mammalian cells, while partial loss‐of‐function results in premature aging and predisposition to liver cancer (Lessel et al, 2014; Maskey et al, 2014, 2017). Despite the severe phenotypes associated with the absence of SPRTN alone, several additional proteases appear to target DPCs (Borgermann et al, 2019; Bhargava et al, 2020; Dokshin et al, 2020; Kojima et al, 2020; Serbyn et al, 2020). The diversity of repair mechanisms underlines the threat posed by DPCs. However, some DPCs have important physiological roles. The human protein HMCES forms crosslinks with abasic (AP) sites to protect genome integrity (Mohni et al, 2019).
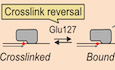
AP sites are frequent endogenous DNA lesions, which arise spontaneously or enzymatically during base excision repair and active DNA demethylation (Thompson & Cortez, 2020). AP sites exist in equilibrium between a closed‐ring furanose and an open‐ring aldehyde form. The latter is prone to undergo spontaneous β‐elimination, resulting in strand scission and DNA single‐strand break (SSB) formation, which can also arise enzymatically upon AP site cleavage by AP endonucleases and lyases (Krokan & Bjoras, 2013; Amidon & Eichman, 2020). If such SSBs form in double‐stranded DNA (dsDNA), they are swiftly repaired by the cellular SSB repair machinery (Abbotts & Wilson, 2017). In contrast, incision of AP sites in ssDNA, for example, at the replication fork, will result in the formation of toxic DSBs (Mehta et al, 2020; Semlow et al, 2022). To prevent such a catastrophic scenario, the conserved catalytic SOS response‐associated peptidase (SRAP) domain of HMCES (Fig 1A) associates with replication forks to crosslink to AP sites in ssDNA (Mohni et al, 2019). Crosslinking occurs between the N‐terminal cysteine residue of the SRAP domain (methionine is proteolytically removed) and an AP site, resulting in the formation of a thiazolidine ring, which prohibits strand scission (Fig 1B and C; Halabelian et al, 2019; Thompson et al, 2019; Wang et al, 2019). DPC formation has been suggested to be initiated by the N‐terminal amino group attacking the AP sites' open‐ring aldehyde form (Halabelian et al, 2019; Thompson et al, 2019; Wang et al, 2019). The resulting Schiff‐base intermediate is then converted into a thiazolidine ring upon reaction with the sulfhydryl group of Cys2 (Fig 1B).
Figure 1. HMCES‐DNA‐protein crosslinks are reversible.
- SRAP domain sequence alignment highlighting key active site residues in H. sapiens, X. laevis, S. cerevisiae and E. coli HMCES homologues (Cys2 = orange, Glu127 = red and His210 = green).
- Proposed reaction mechanism of SRAP domain crosslinking to an AP site.
- Crystal structure of HMCES' active site crosslinked to an AP site. PDB: 6OE7 (Halabelian et al, 2019). DNA is shown in grey. Active site residues are coloured as in (A). Interatomic distances (Å) are labelled.
- Schematic of the assay shown in (E) and (G). HMCESFL and HMCESSRAP (WT or active site variants) were incubated for 1 h at 37°C with a Cy5‐labelled 30mer oligonucleotide containing either a dT or an AP site at position 15. Afterwards, non‐crosslinked HMCES was inactivated by heat denaturation at 60°C for 5 min. A second 6‐FAM‐labelled 30mer oligonucleotide containing an AP site was added and formation of 6‐FAM DPCs was assessed after an additional incubation for 120 min.
- HMCESFL‐ and HMCESSRAP‐WT and HMCESSRAP‐C2S‐DPC formation with Cy5‐ and 6‐FAM‐oligonucleotides was analysed using denaturing SDS–PAGE. Incised DNA is caused by spontaneous hydrolysis of the AP site.
- Quantification of DPC formation assays shown in (E), left panel: DPC formation to Cy5 oligonucleotide, right panel: DPC formation to 6‐FAM oligonucleotide.
- DPC formation of HMCESSRAP‐WT and variants (E127A or H210A) with Cy5‐ and 6‐FAM‐oligonucleotides was analysed using denaturing SDS–PAGE. Incised DNA is caused by spontaneous hydrolysis of the AP site.
- Quantification of DPC formation assays shown in (G), left panel: DPC formation to Cy5 oligonucleotide, right panel: DPC formation to 6‐FAM oligonucleotide.
Data information: Bar graphs in (F) and (H) show the mean of three independent experiments ± SD. Two WT data points are common between (F) and (H).
Source data are available online for this figure.
The protective function of HMCES‐DPCs is particularly important, when cells face substantial amounts of AP sites, for example, upon exposure to genotoxic agents (Srivastava et al, 2020), overexpression of the cytosine deaminase APOBEC3A (Mehta et al, 2020; Biayna et al, 2021), or AID‐induced somatic hypermutation (Wu et al, 2022). In addition, HMCES‐DPCs were shown in Xenopus egg extracts to arise as intermediates of replication‐coupled DNA inter‐strand crosslink (ICL) repair (Semlow et al, 2022). Unhooking of an AP site‐induced ICL (AP‐ICL) by a DNA glycosylase yields an AP site, to which HMCES crosslinks. In egg extracts, HMCES‐DPCs appear to be mainly degraded by the SPRTN protease (Semlow et al, 2022), which requires unfolding of the protein adduct by the FANCJ helicase (Yaneva et al, 2023). While human SPRTN cleaves HMCES‐DPCs in vitro if FANCJ is present (Yaneva et al, 2023), it is unclear to what extent SPRTN is required for repair in mammalian cells, where proteasomal HMCES‐DPC degradation has been reported (Mohni et al, 2019). Notably, HMCES‐DPCs are slowly lost over time in egg extracts when SPRTN is depleted and proteasomal activity inhibited (Semlow et al, 2022), suggesting that additional mechanisms may resolve HMCES‐DPCs. Recent work indicated that SRAP‐DPCs can undergo reversal in principle (Paulin et al, 2022), but it remained unclear how reversal can be reconciled with the need to protect AP sites in ssDNA.
Here, we use in vitro reconstitution to dissect the principles of a non‐proteolytic release mechanism for HMCES‐DPCs. We demonstrate that DPC release is determined by DNA context and occurs in two steps. First, a conserved glutamate residue located in HMCES' active site catalyses the reversal of the thiazolidine crosslink. Second, HMCES either re‐crosslinks, if affinity to the underlying DNA structure is high, or releases the AP site, if affinity is low. As a consequence, HMCES efficiently protects AP sites in ssDNA and at ssDNA‐dsDNA junctions but releases them once the DPC is bypassed by the replication machinery and transferred into dsDNA.
Results
HMCES‐DNA‐protein crosslinks are reversible
Once HMCES‐DPCs form, they appear stable over several days at room temperature in vitro (Thompson et al, 2019). To test whether HMCES remains irreversibly attached during incubation or constantly cycles between a crosslinked and a non‐crosslinked state, we designed an assay to assess the reversibility of HMCES‐DPCs (Fig 1D, schematic). First, we generated AP sites by incubating a Cy5‐labelled 30mer DNA oligonucleotide containing a deoxyuridine (dU) at position 15 with uracil‐DNA glycosylase (UDG). DNA containing dT instead of dU served as a control. DPCs were then generated by addition of recombinant full‐length HMCES (HMCESFL) or the catalytic SRAP domain (HMCESSRAP). Next, reactions were exposed to a short heat treatment (5 min, 60°C), which inactivates free HMCES while not affecting crosslinked HMCES (Yaneva et al, 2023). Finally, a 6‐FAM‐labelled AP site‐containing DNA oligonucleotide was added to all reactions to test whether HMCES can be released from the Cy5‐oligonucleotide and re‐crosslink to the 6‐FAM‐oligonucleotide. Indeed, we observed formation of DPCs between 6‐FAM‐labelled DNA and HMCESFL and HMCESSRAP (Figs 1E, lanes 6 and 7, F, and EV1A), suggesting that some DPCs between HMCES and the Cy5‐oligonucleotide reverted which in turn allowed re‐crosslinking to the 6‐FAM‐oligonucleotide. 6‐FAM‐DPCs did not form if a Cy5‐dT‐oligonucleotide was used (Fig 1E, lanes 2 and 3), indicating that inactivation of free HMCES was efficient, or if HMCES' catalytic cysteine was replaced by serine (HMCESSRAP‐C2S) (Fig 1E, lane 8).
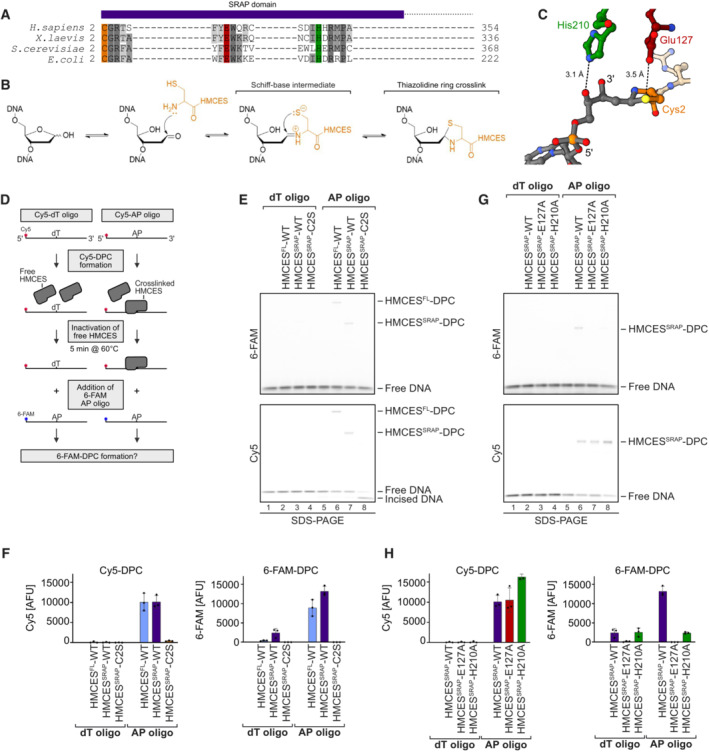
Figure EV1. HMCES‐DNA‐protein crosslinks are reversible. Related to Fig 1 .
- Coomassie‐stained SDS–PAGE gel showing recombinant purified human HMCESFL‐WT, HMCESSRAP‐WT, HMCESSRAP‐C2S, HMCESSRAP‐R98E, HMCESSRAP‐E127A, HMCESSRAP‐H210A and HMCESSRAP‐R98E/E127A proteins used in this study.
- DPC formation of HMCESFL and HMCESSRAP (WT or indicated variants). dU‐containing DNA (0.1 μM) was incubated alone or with UDG and increasing concentrations of HMCES (0.025, 0.05, 0.1, 0.2, 0.4, 0.8, 1.6 and 3.2 μM), as indicated for 1 h at 37°C prior to analysis by denaturing SDS–PAGE.
- Quantification of HMCES‐DPC formation assays shown in (B).
Data information: Data in (C) represent the mean of three individual experiments ± SD.
Next, we asked whether HMCES‐DPC reversal occurs spontaneously or whether it is an enzymatic process. The active site of HMCES features, in addition to the catalytic cysteine at position 2, two highly conserved amino acid residues, Glu127 and His210 (Fig 1A and C). Structural data suggest that both residues stabilize the transient Schiff‐base intermediate during DPC formation (Halabelian et al, 2019; Thompson et al, 2019; Wang et al, 2019). Nonetheless, substitution of the corresponding glutamate residue in the prokaryotic HMCES orthologue YedK results in only reduced DPC formation (Thompson et al, 2019; Wang et al, 2019), while the effect of substituting the histidine remains controversial with reports of decreased and increased DPC formation (Thompson et al, 2019; Wang et al, 2019). Consistently, we observed that human HMCESSRAP with substitution of Glu127 (E127A) or His210 (H210A) were able to form DPCs with Cy5‐labelled AP site‐containing DNA in our assay (Figs 1G, lanes 7 and 8, Cy5 scan, H, EV1B and C). However, HMCESSRAP‐E127A and ‐H210A variants did not form DPCs with the subsequently added 6‐FAM‐oligonucleotide (Fig 1G, lanes 7 and 8, 6‐FAM scan, and H). These results suggest that stabilization of the Schiff‐base intermediate by Glu127 and His210 is not essential for DPC formation per se but may rather be important to reverse thiazolidine ring formation, perhaps explaining the strict conservation of both residues during evolution. In agreement with a recent study (Paulin et al, 2022), we conclude that HMCES‐DPCs are reversible, that released HMCES retains the ability to re‐crosslink, and that release is an enzymatic process requiring conserved active‐site residues.
Release of HMCES‐DPCs is determined by DNA context
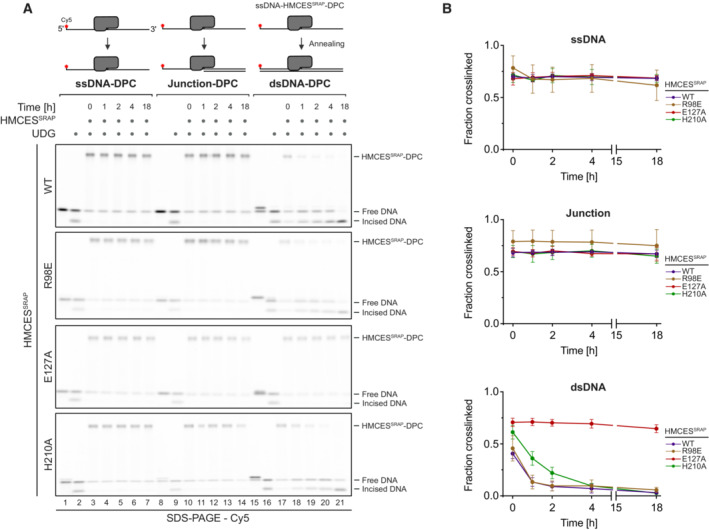
The fact that HMCES‐DPCs are reversible raises the question of whether the release is regulated. AP sites must be protected in ssDNA to prohibit strand breakage, but HMCES‐DPC formation may be less favourable in dsDNA, where it would prohibit initiation of AP site repair by AP endonucleases. In line with the need to stabilize AP sites in ssDNA, DPC formation by HMCESSRAP‐WT occurs efficiently in ssDNA and at ssDNA‐dsDNA junctions with a 5′‐flap (Figs 2A and B) (Mohni et al, 2019; Thompson et al, 2019). In contrast, DPC formation does not occur in dsDNA (Fig 2A and B) (Mohni et al, 2019). It has been speculated that this is due to HMCES not being able to accommodate AP sites in its active site if dsDNA is present on the 3′‐site of the lesion (Thompson et al, 2019). Interestingly, however, HMCES‐DPC formation occurred efficiently, when the DNA strand across the AP site contained a nick or a 4‐nucleotide gap (Fig 2C and D). This indicates that HMCES does not necessarily require long stretches of ssDNA to form a DPC, but rather relies on the bendability of the substrate DNA induced by a nick or gap.
Figure 2. Formation of HMCES‐DPCs is determined by DNA context.
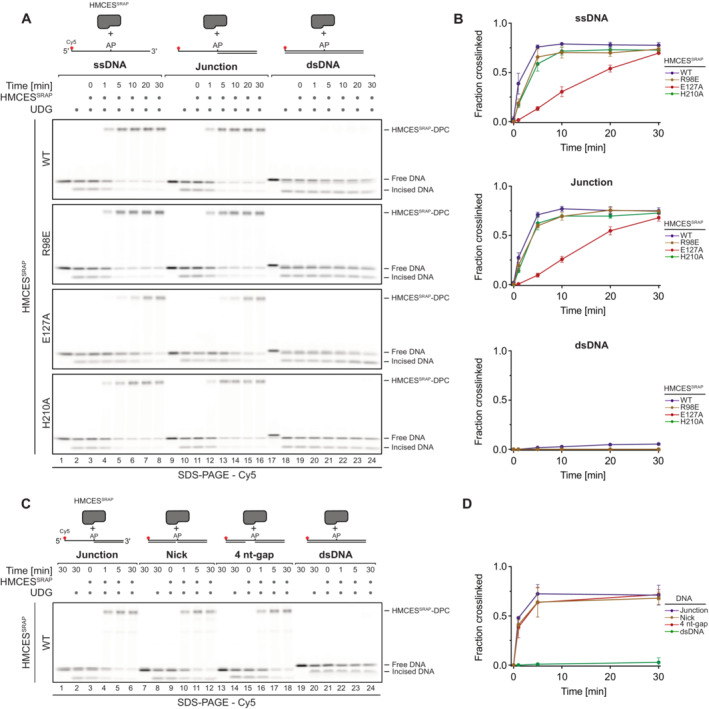
- Kinetics of DPC formation by HMCESSRAP (WT, R98E, E127A or H210A variants) to ssDNA, junction DNA and dsDNA. Corresponding reverse oligonucleotides were annealed to ssDNA to create DNA junction and dsDNA prior to adding HMCESSRAP. To ssDNA, a non‐complementary oligonucleotide was added as control. HMCESSRAP‐WT and variants were incubated with different DNA structures for the indicated amount of time at 37°C prior to separation by denaturing SDS–PAGE.
- Quantification of DPC formation assays shown in (A)
- Kinetics of DPC formation by HMCESSRAP‐WT to junction DNA and dsDNA containing a nick or a 4 nt‐gap. Corresponding reverse oligonucleotides were annealed to create junction DNA and dsDNA containing a nick or a 4 nt‐gap prior to adding HMCESSRAP‐WT. HMCESSRAP‐WT was incubated with the indicated DNA structures for the indicated amount of time at 37°C prior to separation by denaturing SDS–PAGE.
- Quantification of DPC formation assays shown in (C).
Data information: Data in (B) and (D) represent the mean of three independent experiments ± SD.
Source data are available online for this figure.
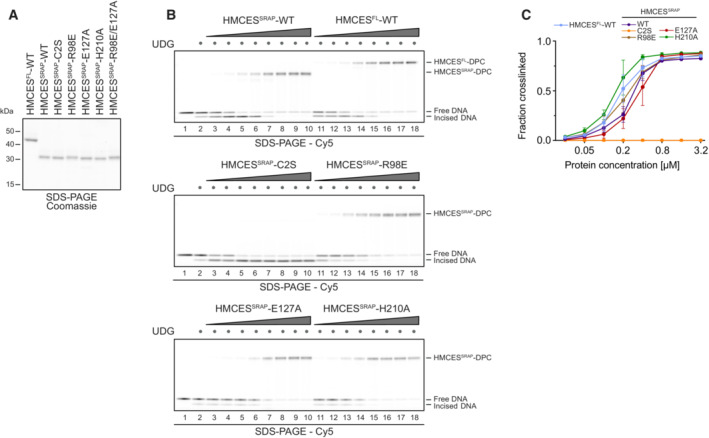
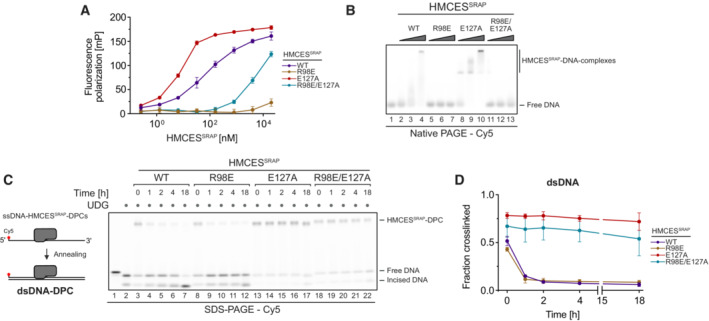
Substitution of Glu127 and His210 had no effect on the specificity of DPC formation, but HMCESSRAP‐E127A crosslinked slower (Fig 2A and B), which may be related to a recently proposed role for Glu127 in AP site ring opening (Paulin et al, 2022). To understand whether DNA context also influences DPC release, we first generated DPCs between HMCESSRAP and an AP site in ssDNA before annealing complementary reverse oligonucleotides to generate either a ssDNA‐dsDNA junction or fully dsDNA (Fig 3A, schematic). Strikingly, HMCESSRAP‐DPCs were stable in ssDNA or ssDNA‐dsDNA junctions but reversed in dsDNA (Fig 3A and B). In contrast, HMCESSRAP variants H210A and E127A were partially (H210A) or entirely (E127A) defective for reversal in dsDNA (Fig 3A and B). HMCESSRAP‐E127A was previously reported to display increased DNA binding (Wang et al, 2019), which we confirmed using fluorescence polarization (Fig EV2A) and electrophoretic mobility shift assays (Fig EV2B). To test whether the inability of this variant to reverse is related to increased DNA binding, we combined E127A with a substitution of Arg98 (R98E), which is located within the HMCESSRAP‐ssDNA interface (Halabelian et al, 2019). DPC formation and release were not affected by the R98E substitution (Figs 2A and B, 3A and B, and EV1B and C), despite severely reduced DNA‐binding activity (Fig EV2A and B) (Mohni et al, 2019). In combination with E127A, substitution of Arg98 decreased DNA binding below WT levels (Fig EV2A and B) but did not restore the ability to revert the crosslink (Fig EV2C and D). Thus, the reversal defect of HMCESSRAP‐E127A‐DPCs is unrelated to increased DNA‐binding affinity. We conclude that DPC release is not only an active process requiring Glu127 (and partially His210) but is also determined by DNA context. DPC release displays opposite specificity to DPC formation, which correlates with the biological need to protect AP sites in ssDNA but not in dsDNA.
Figure 3. Release of HMCES‐DPCs is determined by DNA context.
- DPC reversal kinetics of indicated variants in ssDNA, DNA junction and dsDNA. DPCs were pre‐formed in ssDNA before corresponding reverse oligonucleotides were annealed (for ssDNA reactions, a non‐complementary oligonucleotide was added). DPC reversal was then monitored after incubation for the indicated amount of time at 37°C using denaturing SDS–PAGE.
- Quantification of DPC reversal assays using HMCESSRAP‐WT and variants shown in (A).
Data information: Data in (B) represent the mean of three independent experiments ± SD.
Source data are available online for this figure.
Figure EV2. Release of HMCES‐DPCs is determined by DNA context. Related to Fig 3 .
- Fluorescence polarization measurements of Cy5‐labelled ssDNA (25 nM) incubated with increasing concentrations of HMCESSRAP‐WT, HMCESSRAP‐R98E, HMCESSRAP‐E127A or HMCESSRAP‐R98E/E127A for 20 min on ice prior to measuring fluorescence polarization.
- Non‐covalent DNA binding of indicated HMCESSRAP variants was assessed by electrophoretic mobility shift assay. A Cy5‐labelled 30‐mer ssDNA (0.1 μM) was incubated with HMCESSRAP (0, 0.125, 0.5 or 2 μM) for 20 min at 4°C prior to analysis by native PAGE.
- DPC reversal kinetics of indicated HMCESSRAP variants in dsDNA. A corresponding reverse oligonucleotide was annealed to HMCESSRAP‐DPCs, before incubation for the indicated amount of time at 37°C prior to separation by denaturing SDS–PAGE.
- Quantification of DPC reversal assays shown in (C).
Data information: Data in (A) and (D) represent the mean of three independent experiments ± SD.
Release of HMCES‐DPCs is determined by binding affinity to the underlying DNA
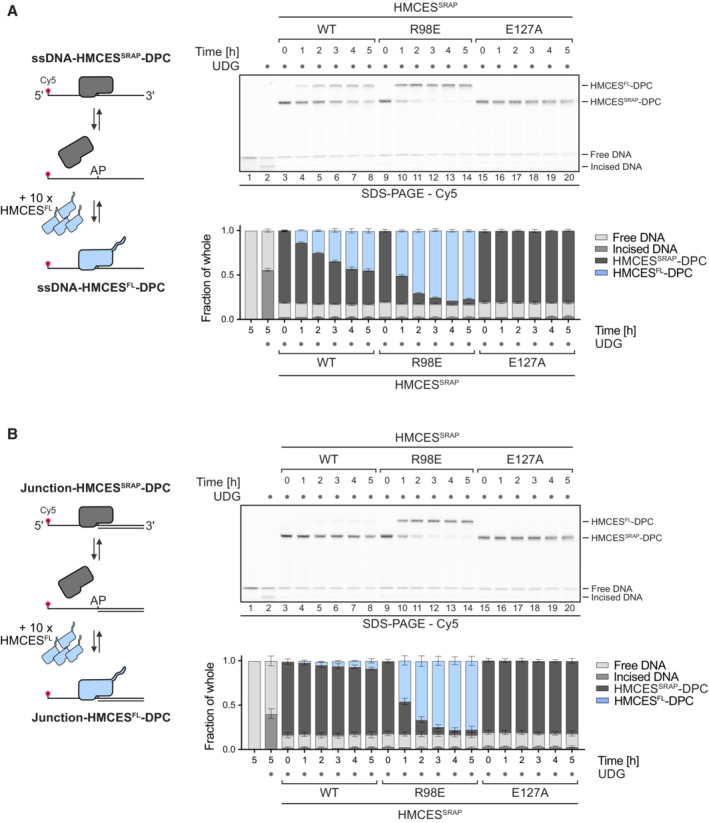
Next, we wanted to understand how DNA context controls the release of HMCESSRAP‐DPCs. Our results so far could be explained by a model in which all HMCES‐DPCs constantly revert independent of DNA context and that specificity is only determined by HMCES' ability to reform the crosslink after release, which does not occur in dsDNA. However, it remained unclear how HMCES could efficiently protect AP sites, if it would constantly dissociate from the lesion. To gain more detailed insights into DPC reversal in different DNA structures, we first generated HMCESSRAP‐DPCs in ssDNA and at ssDNA‐dsDNA junctions (DPCs in dsDNA released too quickly to be assessed by this assay). We then added HMCESFL in 10‐fold excess to outcompete HMCESSRAP upon release of the AP site (Fig 4A and B, schematic). This set‐up allowed us to evaluate release of HMCESSRAP‐DPCs by monitoring the appearance of HMCESFL‐DPCs over time. Notably, HMCESSRAP‐DPCs were released over time in ssDNA (Fig 4A, lanes 3–8) but were much more stable at ssDNA‐dsDNA junctions (Fig 4B, lanes 3–8). DPCs formed by the E127A variant were not released in either setting (Fig 4A, lanes 15–20 and B, lanes 15–20). We wondered whether the enhanced release of WT‐DPCs from ssDNA was related to the previously reported preferential binding of the SRAP domain to ssDNA‐dsDNA junctions compared to ssDNA (Thompson et al, 2019). Accordingly, we hypothesized that the active site of HMCES may constantly cycle between a crosslinked and a non‐crosslinked state independent of DNA context, but that actual dissociation from the underlying DNA substrate would in addition be determined by binding affinity. To test this idea, we asked whether the reduced DNA‐binding affinity of HMCESSRAP‐R98E would affect reversal. Indeed, HMCESSRAP‐R98E‐DPCs reversed much more rapidly in ssDNA and at ssDNA‐dsDNA junctions than WT‐DPCs (Fig 4A, lanes 9–14 and B, lanes 9–14). Taken together, these results suggest that HMCES‐DPC release is governed by two major components. First, the principal capacity of Glu127 to catalyse reversal ensures cycling of the active site between a crosslinked and a non‐crosslinked state. Second, the binding strength to the underlying DNA structure then determines whether HMCES re‐crosslinks or dissociates while in the non‐crosslinked state, which occurs if affinity is low (e.g., within dsDNA or in the context of R98E‐DPCs).
Figure 4. Release of HMCES‐DPCs is determined by binding affinity to the underlying DNA.
-
A, BCompetition assay between HMCESFL and indicated HMCESSRAP variants. HMCESSRAP‐DPCs in ssDNA (A) or at ssDNA‐dsDNA junctions (B) were pre‐formed and then incubated with 10‐fold excess of HMCESFL for the indicated amount of time at 37°C prior to separation by denaturing SDS gel (upper panels).
Data information: Quantification of competition assay: Bar graphs show the mean of three independent experiments ± SD (lower panels).
Source data are available online for this figure.
Release of HMCES‐DPCs restricts crosslink formation to physiologically relevant situations
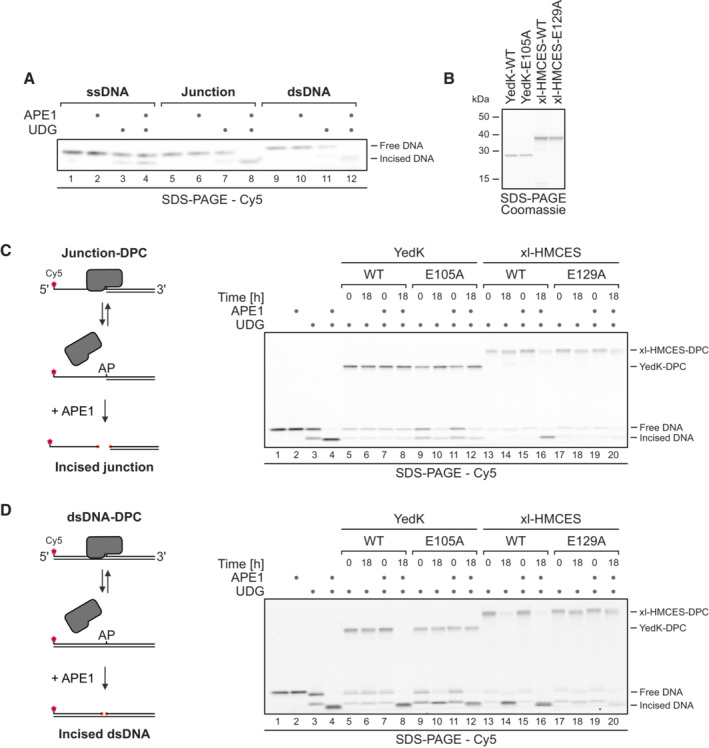
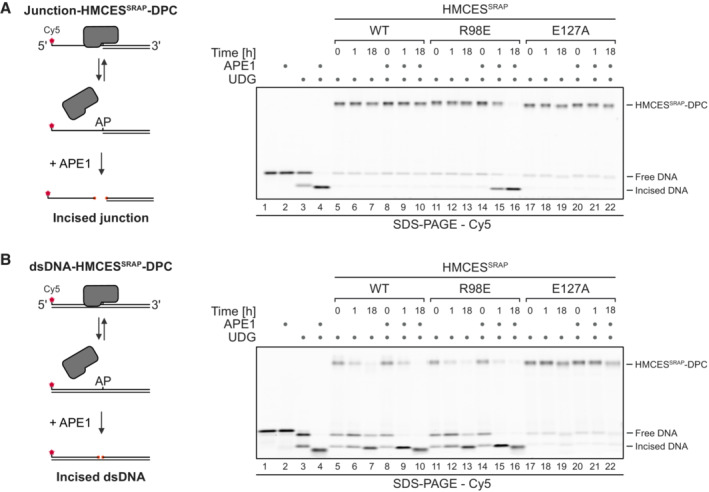
Next, we wanted to understand how DPC release relates to HMCES' ability to block APE1 endonuclease from incising AP sites. APE1 efficiently cleaves AP sites at ssDNA‐dsDNA junctions and in dsDNA but shows little activity in ssDNA (Fig EV3A; Wilson et al, 1995). Therefore, we generated HMCESSRAP‐WT, ‐R98E and ‐E127A‐DPCs at ssDNA‐dsDNA junctions and in dsDNA and incubated them with APE1. As reported previously (Mohni et al, 2019), WT‐DPCs shielded AP sites from APE1 incision at ssDNA‐dsDNA junctions (Fig 5A, lanes 8–10). In contrast, R98E‐DPCs failed to protect against APE1 (Fig 5A, lanes 14–16), suggesting that the increased release of this variant compromises its ability to protect AP sites against APE1 incision. In dsDNA, both WT‐ and R98E‐DPCs did not prevent AP site cleavage (Fig 5B, lanes 8–10 and 14–16, respectively), while E127A‐DPCs fully blocked incision (Fig 5A and B, lanes 20–22). Therefore, failure to undergo auto‐release would lead to the inhibition of AP site repair in dsDNA.
Figure EV3. Auto‐release of HMCES‐DPCs restricts crosslink formation to physiologically relevant situations. Related to Fig 5 .
-
AAPE1 incision of an AP site in ssDNA, DNA junction and dsDNA. A Cy5‐labelled 30‐mer ssDNA was incubated alone or with UDG for 1 h at 37°C. Corresponding reverse oligonucleotides for DNA junction or dsDNA were annealed (for ssDNA, a non‐complementary oligonucleotide was added). Next, samples were incubated with APE1 for 18 h at 37°C before separation by denaturing SDS–PAGE.
-
BCoomassie‐stained SDS–PAGE gel showing recombinant purified bacterial YedK‐WT, YedK‐E105A, xl‐HMCES‐WT and xl‐HMCES‐E129A proteins used in this study.
-
C, DAPE1 incision of an AP site protected by the indicated YedK‐WT‐DPC or YedK‐E105A‐DPC and xl‐HMCES‐WT‐DPC or xl‐HMCES‐E129A‐DPC at ssDNA‐dsDNA junctions (C) or within dsDNA (D). Free dU‐containing DNA was incubated alone or in the presence of UDG and YedK/xl‐HMCES for 1 h at 37°C. Next, corresponding reverse oligonucleotides were annealed to generate an ssDNA‐dsDNA junction (C) or dsDNA (D), and reactions were incubated alone or with APE1 for the indicated amount of time at 37°C prior to separation by denaturing SDS–PAGE.
Figure 5. Auto‐release of HMCES‐DPCs restricts crosslink formation to physiologically relevant situations.
-
A, BAPE1 incision of an AP site protected by the indicated HMCESSRAP‐DPC variants at ssDNA‐dsDNA junctions (A) or within dsDNA (B). Free dU‐containing DNA was incubated alone or in the presence of UDG and HMCESSRAP for 1 h at 37°C. Next, corresponding reverse oligonucleotides were annealed to generate an ssDNA‐dsDNA junction (A) or dsDNA (B), and reactions were incubated alone or with APE1 for the indicated amount of time at 37°C prior to separation by denaturing SDS–PAGE.
Source data are available online for this figure.
Importantly, we observed comparable results (with slight variations) when using the prokaryotic HMCES‐orthologue YedK or Xenopus laevis HMCES (xl‐HMCES) (Fig EV3B), indicating that the capacity of SRAP domain DPCs to auto‐release is conserved across different species. YedK‐DPCs were stable at ssDNA‐dsDNA junctions, prohibiting cleavage of the AP site by APE1 (Fig EV3C, lanes 7–8). In dsDNA, APE1 was able to cleave the AP site, indicating release of the DPC (Fig EV3D, lanes 7–8). AP site cleavage in dsDNA was not observed upon replacing Glu105 (corresponding to Glu127 in HMCES, Fig 1A) with alanine (Fig EV3D, lanes 11–12). Of note, in contrast to human HMCESSRAP, release of the DPC was barely detectable in the absence of APE1 (Fig EV3D, lanes 5–6). While the protection of AP sites at ssDNA‐dsDNA junctions against APE1 cleavage by xl‐HMCES‐DPCs was less strong than observed for the human or prokaryotic protein (Fig EV3C, lanes 15–16), substitution of Glu129 (corresponding to Glu127 in HMCES, Fig 1A) entirely blocked reversal at ssDNA‐dsDNA junctions and in dsDNA (Fig EV3C, lanes 19–20, and D, lanes 19–20, respectively).
Collectively, these data show that HMCES‐DPC release must be finely balanced to (i) ensure protection of AP sites at ssDNA‐dsDNA junctions against potentially catastrophic APE1 incisions (which is compromised upon hyper‐release in the HMCESSRAP‐R98E variant) and to (ii) allow deprotection of AP site in dsDNA so that APE1 can initiate repair (which is compromised upon hypo‐reversal in the HMCESSRAP‐E127A variant).
SPRTN‐dependent proteolysis is the dominant mechanism for HMCES‐DPC removal in Xenopus egg extracts
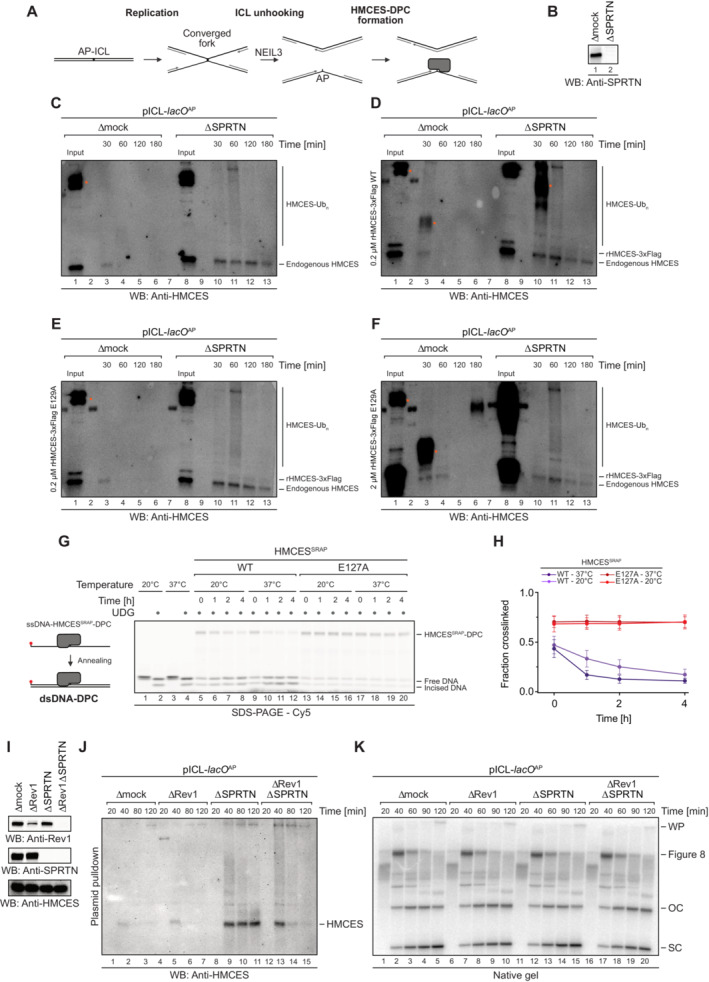
Next, we sought to test whether crosslink reversal contributes to HMCES removal in a more physiological system. We monitored HMCES‐DPC stability during replication‐coupled repair of a plasmid containing a site‐specific AP‐ICL (pICL‐lacO AP) in Xenopus egg extracts supplemented with WT recombinant 3xFlag‐tagged Xenopus laevis HMCES protein (rHMCES‐3xFlag) or E129A‐mutated rHMCES‐3xFlag. As shown in Fig EV3C and D, the WT protein undergoes efficient DPC reversal from dsDNA in vitro while the E129A‐mutated protein does not. Similar to endogenous HMCES present in egg extract, both the WT and the E129A‐mutated protein were barely detectable on chromatin isolated from extract containing SPRTN but accumulated on chromatin isolated from SPRTN‐depleted extract (Fig EV4A–F). We therefore conclude that, relative to reversal, SPRTN‐dependent proteolysis represents the primary mechanism for HMCES‐DPC removal during ICL repair in egg extracts. However, technical challenges prevented us from determining whether reversal contributes to HMCES‐DPC resolution when proteolysis is blocked (Fig EV4A–K, see figure legend for discussion).
Figure EV4. Resolution of HMCES‐DPCs during replication‐coupled ICL repair. Related to Fig 5 .
-
ASchematic depiction of the NEIL3‐dependent repair of an AP‐ICL, a lesion that forms when an AP site reacts with a nucleobase of the opposing DNA strand forming a covalent crosslink (Price et al, 2014). In Xenopus egg extracts, such crosslinks are primarily unhooked by the NEIL3 glycosylase (Semlow et al, 2016), which yields an AP site leading to formation of an HMCES‐DPC.
-
B–FIn the absence of SPRTN, the intact HMCES‐DPC is presumably bypassed by TLS and transferred into dsDNA. To test whether this triggers autorelease, we analysed the stability of DPCs formed by wild‐type and E129A‐mutated Xenopus laevis rHMCES‐3xFlag proteins during ICL repair in egg extract. pICL‐lacO AP was replicated in mock‐ or SPRTN‐depleted extracts (B) supplemented with WT or E129A rHMCES‐3xFlag. At the time points indicated, plasmid was recovered under stringent conditions, the DNA was digested and released proteins were separated by SDS–PAGE. HMCES‐DPCs were detected using an antibody raised against the SRAP domain that permits simultaneous monitoring of endogenous HMCES protein and the recombinant 3xFlag‐tagged HMCES (which migrates slower during SDS–PAGE due to the 3xFlag). In this experimental setup, the endogenous protein serves as a control for the effects of SPRTN depletion and autorelease. Like the endogenous HMCES (C), both WT (D) and E129A‐mutated rHMCES‐3xFlag (E) were stabilized by SPRTN depletion, implying that proteolysis is the dominant mechanism for removing HMCES‐DPC under these conditions. However, it was challenging to assess the relative behaviour of tagged WT and mutant protein because DPCs formed by the wild‐type recombinant protein (like those formed by the endogenous protein) are resolved slowly in SPRTN‐depleted extract (on the timescale of hours, somewhat slower than the timescale for observed for reversal in vitro). Additionally, the E129A‐mutated recombinant flag‐tagged protein crosslinked less efficiently than endogenous HMCES, making it difficult to detect even when present in large excess (F). We are, therefore, unable to determine from these data whether HMCES‐DPC reversal occurs during ICL repair in egg extract under the conditions tested. Orange dots denote non‐specific bands or bands corresponding to contaminating IgG.
-
GOne explanation for the discrepancy in the degree of reversal observed between the in vitro reconstitution and egg extract systems could be that the in vitro reactions were all performed at 37°C, while replication in egg extracts must be performed at 20°C. Therefore, we assessed reversal of HMCESSRAP‐WT or ‐E127A‐DPCs in dsDNA at the indicated temperatures for the indicated amount of time before analysis by denaturing SDS–PAGE. Indeed, autocatalytic reversal was significantly delayed at 20°C.
-
HQuantification of DPC reversal assays using HMCESSRAP‐WT and ‐E127A shown in (G).
-
IThe extracts used in the replication reactions shown in (J and K) were immunoblotted for SPRTN, Rev1 and HMCES.
-
JAs an alternative additional strategy to determine whether reversal contributes to HMCES‐DPC resolution, we tested whether REV1 depletion results in stabilization of HMCES‐DPCs, reasoning that blocking TLS (and transfer of the DPC into dsDNA) may inhibit reversal. pICL‐lacO AP was replicated in mock‐, REV1‐, SPRTN‐ or REV1‐ and SPRTN‐depleted egg extracts, as indicated. At the indicated time points, plasmid was recovered under stringent conditions, the DNA was digested and released proteins were separated by SDS–PAGE. HMCES was detected by blotting. As expected, depletion of SPRTN alone resulted in a strong stabilization of HMCES‐DPCs. Depletion of REV1 alone did not stabilize HMCES‐DPCs, consistent with our data indicating that SPRTN represents the dominant mechanism for HMCES‐DPC resolution in egg extract. Surprisingly, when combined with SPRTN depletion, REV1 depletion partially suppressed the accumulation of HMCES‐DPCs. Superficially, this result is contrary to our expectations based on data presented in Fig 6. However, we interpret the result to indicate when the HMCES‐DPC is maintained at an ssDNA/dsDNA junction due to inefficient TLS, residual SPRTN or another protease can eventually degrade the HMCES‐DPC. Therefore, while these data do not provide evidence for HMCES‐DPC reversal during ICL repair in egg extract, they do reinforce the need for alternative removal mechanisms for HMCES‐DPCs present in dsDNA that are refractory to proteolysis.
-
KIn parallel with the reactions shown in (J), pICL‐lacO AP was replicated in the indicated egg extracts supplemented with [α‐32P]dCTP. Replication intermediates were separated on a native agarose gel and visualized by autoradiography. SC, supercoiled. OC, open circular. Consistent with a TLS defect upon Rev1 depletion, we observed accumulation of gapped, circular plasmids in replication gels, implying that the HMCES‐DPC is maintained at an ssDNA‐dsDNA junction.
Data information: Data in (H) represent the mean of three independent experiments ± SD.
Translesion synthesis across HMCES‐DPCs triggers their release
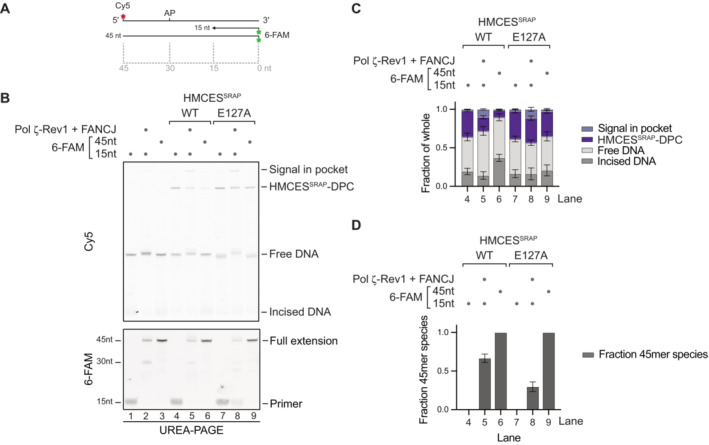
The fact that HMCES‐DPCs form specifically in ssDNA contexts (Fig 2A and B) (Mohni et al, 2019) leads to the question as to how HMCES‐DPCs are transferred to dsDNA, where release could occur. Translesion synthesis (TLS) polymerases can extend nascent strands across intact HMCES‐DPCs, as has been observed in Xenopus egg extracts and in vitro (Yaneva et al, 2023). Thus, we tested whether TLS across an HMCES‐DPC triggers reversal. We placed HMCESSRAP‐WT or ‐E127A‐DPCs in template DNA downstream of a primer (Fig 6A) and added TLS polymerase Pol ζ‐Rev1 and the helicase FANCJ, which promotes TLS across intact DPCs through unfolding of the crosslinked protein adduct (Yaneva et al, 2023). These assays were analysed using UREA–PAGE, which allows separation of the template strand and the extended primer. As a control, we annealed a complementary 45mer reverse oligonucleotide, mimicking full extension. Indeed, extension of the primer by Pol ζ‐Rev1 appeared to trigger release of HMCES‐DPCs, as evidenced by a loss of WT‐DPCs but not of E127A‐DPCs (Fig 6B, compared lanes 4 and 5 and lanes 7 and 8, and Fig 6C); the assessment of DPC release was complicated by a fraction of the Cy5 signal remaining in the gel pocket in the presence of Pol ζ‐Rev1‐FANCJ (Fig 6B, lane 5 and 8). DPC release was more pronounced when the complementary 45mer oligonucleotide was annealed to the template (Fig 6B and C, lane 6), which is in line with the fact that TLS did not extend all primers across the DPC (Fig 6B, 6‐FAM scan and Fig 6D, lane 5). Of note, we observed that extension of the primer was less efficient in templates containing a HMCESSRAP‐E127A‐DPC (Fig 6B and D, compare lanes 5 and 8). Thus, we cannot exclude that reversal of some DPCs occurs prior to TLS‐dependent extension. These results suggest that DPC reversal can be triggered by physiological processes that transfer HMCES‐DPCs from ssDNA into dsDNA.
Figure 6. Translesion synthesis across an HMCES‐DPC can trigger reversal in vitro .
-
A, BPrimer extension assay using Pol ζ‐Rev1. Fluorescently labelled primer‐template substrates containing an AP site at the indicated position were incubated alone or in the presence of HMCESSRAP‐WT or ‐E127A, recombinant human FANCJ and Pol ζ‐Rev1 as indicated for 2 h at 37°C prior to separation by denaturing UREA–PAGE. (A) Model of oligonucleotides. (B) Cy5 scan and 6‐FAM scan of denaturing UREA–PAGE.
-
CQuantification of Cy5 signals shown in (B).
-
DQuantification of 6‐FAM signals shown in (B).
Data information: Bar graphs in (C) and (D) show the mean of four independent experiments ± SD for lanes 4 to 7 and the mean of three independent experiments ± SD for lanes 8 and 9. One replicate was excluded for these conditions for technical reasons.
Source data are available online for this figure.
Discussion
In this study, we found that HMCES‐DPCs do not necessarily require proteolytic repair because they feature a built‐in release mechanism. Our data suggest a model in which auto‐release of HMCES‐DPCs occurs in two distinct steps (Fig 7). First, the conserved Glu127 residue (with a minor contribution of His210) catalyses the reversal of the crosslink between HMCES' active site cysteine and the AP site, as also observed in other recent work (Paulin et al, 2022). Second, the cysteine either re‐crosslinks or HMCES dissociates from DNA resulting in release of the AP site. The decision between these two options appears to be determined by binding strength to the underlying DNA. HMCES binds tightly to ssDNA‐dsDNA junctions, which favours re‐crosslinking over release. In contrast, HMCES binds poorly to dsDNA, resulting in release. Thus, this model explains how HMCES can protect AP sites at ssDNA‐dsDNA junctions against incisions by AP endonucleases, while promoting AP site cleavage within dsDNA.
Figure 7. Model of AP site protection by coordinated formation and release of HMCES‐DPCs.
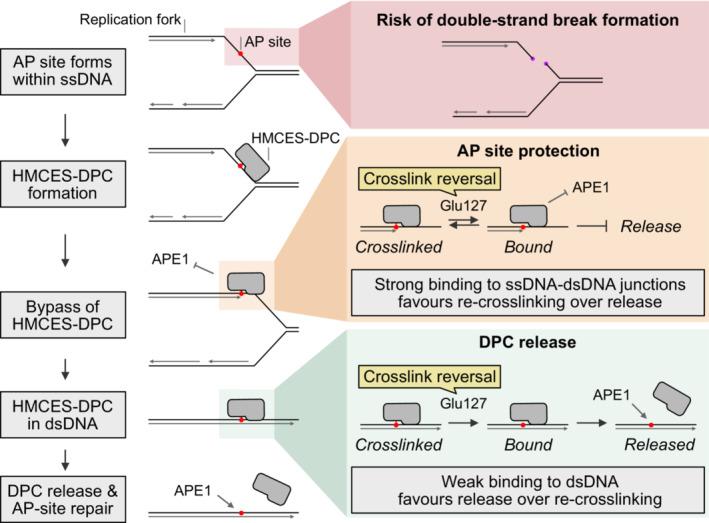
AP sites within ssDNA at the replication fork are dangerous because they can lead to the formation of toxic DNA double‐strand breaks. The conserved protein HMCES covalently crosslinks to AP sites in ssDNA to prevent strand scission.
HMCES‐DPCs efficiently protect AP sites in ssDNA and at ssDNA‐dsDNA junctions against spontaneous or enzymatic incisions. While the active site constantly cycles between a crosslinked and a non‐crosslinked state (catalysed by Glu127), HMCES affinity to the underlying DNA is high which favours re‐crosslinking over dissociation. In dsDNA, affinity is low which favours release of HMCES over re‐crosslinking and thereby enables APE1 endonuclease to initiate AP site repair in dsDNA.
An important question arises regarding the mechanisms that transfer HMCES‐DPCs from ssDNA to dsDNA to create the conditions for DPC release. Two settings seem plausible: one, TLS polymerases extend nascent strands across intact HMCES‐DPCs with the help of FANCJ (Fig 6; Yaneva et al, 2023); two, nascent strands may be extended upon template switching, which would avoid the need for TLS and thus ensure error‐free repair (Mohni et al, 2019; Mehta et al, 2020; Srivastava et al, 2020). Notably, in either case, extension of the nascent strand past the protein adduct would prevent DPC proteolysis by SPRTN, which requires the presence of an ssDNA‐dsDNA junction in close proximity to the protein adduct to become activated (Larsen et al, 2019; Reinking et al, 2020). The relative importance of release, which has the added benefit of recycling the enzyme, versus proteolytic repair of HMCES‐DPCs, is an interesting future question. In Xenopus egg extracts, SPRTN‐dependent proteolysis appeared to be the dominant mechanism for the resolution of HMCES‐DPCs forming during replication‐coupled ICL repair (Fig EV4). However, template switching does not occur under these conditions (Semlow et al, 2022), but is the preferred mechanism to fill in DNA gaps during replication in mammalian cells (Tirman et al, 2021).
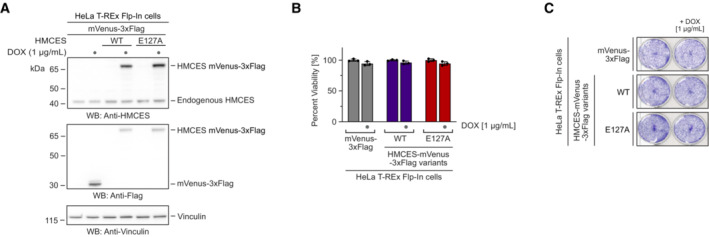
In mammalian cells, HMCESE127A has been reported to complement the sensitivity to AP site‐inducing drugs caused by an HMCES knock‐out (Srivastava et al, 2020) and to protect AP sites during somatic hypermutation (Wu et al, 2022), which is in line with this variant's ability to form DPCs. We did not observe toxicity in human cells upon overexpression of HMCESE127A (Fig EV5A–C), which may indicate that proteolytic repair fully compensates for the lack of DPC release. Both proteases targeting HMCES‐DPCs, SPRTN and the proteasome are essential for cell viability, which prohibits the analysis of HMCESE127A toxicity in the absence of DPC proteolysis in cells. Thus, the effects of defective auto‐release remain unclear. However, human cells expressing HMCESR98E are sensitive to ionizing radiation (Mohni et al, 2019), which together with our data indicate that increased DPC release compromises HMCES' ability to protect cells against AP sites in ssDNA. The precise regulation of the HMCES‐DPC auto‐release mechanism identified in this study emphasizes the need to restrict DPC formation to an absolute minimum.
Figure EV5. Cell viability is not affected upon overexpression of reversal‐defective HMCES variants. Related to Fig 7 .
-
A–CHeLa T‐REx Flp‐In cells expressing the indicated doxycycline‐inducible HMCES variants with a C‐terminal mVenus‐3xFlag‐tag were grown in the presence of 1 μg/ml doxycycline, as indicated. Expression levels were analysed by Western blotting (A). Cell viability was determined using AlamarBlue cell viability assay (B), or crystal violet staining (C).
Data information: Data in (B) represent the mean ± SD of three technical replicates normalized to the mean of untreated control (mVenus‐DOX).
Materials and Methods
Protein expression and purification
HMCESSRAP
An open reading frame containing human HMCESSRAP (amino acids 1–270) was codon optimized for bacterial expression and cloned in frame with a C‐terminal His6‐tag in a pNIC plasmid. HMCESSRAP‐variants (‐C2S, ‐R98E, ‐E127A, ‐H210A, ‐R98E/E127A) were generated by introducing point mutations using the Q5 site‐directed mutagenesis kit (New England BioLabs), following manufacturer's instructions. Mutations were confirmed by Sanger sequencing. BL21 (DE3) Escherichia coli cells were transformed with the corresponding plasmids for protein expression. Cells were grown in terrific broth (TB) medium at 37°C to OD600 0.7. Protein expression was induced by addition of 0.5 mM isopropyl‐ β‐D‐thiogalactoside (IPTG) for 4 h. Cells were harvested, snap‐frozen in liquid nitrogen and pellets were stored at −80°C. For protein purification, cells were resuspended in buffer A (50 mM HEPES/KOH pH 7.8, 500 mM KCl, 5 mM MgCl2, 30 mM imidazole, 10% glycerol, 0.1% IGEPAL, 0.04 mg/ml Pefabloc SC, cOmplete EDTA‐free protease inhibitor cocktail tablets and 1 mM tris(2‐carboxyethyl)phosphine hydrochloride (TCEP)) and lysed by sonication. All subsequent steps were carried out at 4°C. Cell lysates were incubated with benzonase nuclease (45 U/ml lysate) for 30 min before cell debris was removed by centrifugation at 18,000 g for 30 min. Cleared and filtered supernatants were applied to 3 ml Ni‐NTA Agarose (QIAGEN) equilibrated in buffer B (20 mM HEPES/KOH pH 7.8, 500 mM KCl, 5 mM MgCl2, 30 mM imidazole, 10% glycerol and 1 mM TCEP). Next, beads were washed with 15 column volumes (CV) of buffer B before protein was eluted in 2 CV of buffer C (20 mM HEPES/KOH pH 7.8, 500 mM KCl, 5 mM MgCl2, 300 mM imidazole, 10% glycerol and 1 mM TCEP). The elution was concentrated to 2 ml using a 10 kDa molecular weight cut‐off Amicon ultra centrifugal filter prior to loading on a HiLoad® 16/600 Superdex® 200 pg column equilibrated in buffer D (20 mM HEPES/KOH pH 7.8, 150 mM KCl, 5 mM MgCl2, 10% glycerol and 1 mM TCEP) for size exclusion chromatography. Eluted protein fractions were collected and concentrated with a 10 kDa molecular weight cut‐off Amicon Ultra centrifugal filter. Concentrated protein was aliquoted, snap‐frozen and stored at −80°C. Removal of the N‐terminal methionine was confirmed by mass spectrometry.
YedK
An open reading frame containing bacterial YedK was cloned in frame with a C‐terminal His6‐tag in a pNIC plasmid. YedK‐variant (−E105A) was generated by introducing a point mutation using the Q5 site‐directed mutagenesis kit (New England BioLabs), following manufacturer's instructions. Mutation was confirmed by Sanger sequencing. Purification was as described above for HMCESSRAP.
HMCESFL
For full‐length HMCES (HMCESFL), the open reading frame was codon optimized and cloned in a pNIC plasmid in frame with a C‐terminal TwinStrep‐ZB‐tag. Recombinant HMCES protein was expressed and purified using a protocol for purification of SPRTN (Reinking et al, 2020), with small modifications to some buffers. Cell pellets were resuspended in buffer A (20 mM HEPES/KOH pH 7.5, 500 mM KCl, 5 mM MgCl2, 30 mM imidazole, 10% glycerol, 0.1% IGEPAL, 0.04 mg/ml Pefabloc SC, cOmplete EDTA‐free protease inhibitor cocktail tablets and 1 mM TCEP). For washing steps, buffer B (20 mM HEPES/KOH pH 7.5, 500 mM KCl, 5 mM MgCl2 and 1 mM TCEP) was used. Protein was eluted from Strep‐Tactin®XT Superflow® high‐capacity cartridges with buffer B containing 50 mM Biotin and from HiTrap Heparin HP affinity columns in buffer C (20 mM HEPES/KOH pH 7.5, 1 M KCl, 5 mM MgCl2 and 1 mM TCEP). For size exclusion chromatography and storage of the protein, buffer D (20 mM HEPES/KOH pH 7.8, 150 mM KCl, 5 mM MgCl2, 10% glycerol and 1 mM TCEP) was used. Removal of the N‐terminal methionine was confirmed by mass spectrometry.
FANCJ
Human FANCJ‐WT followed by a C‐terminal TEV‐cleavage site and TwinStrep‐ZB tag was expressed and purified from Hi5 cells as described previously (Yaneva et al, 2023) with minor modifications. Briefly, cells were lysed in 200 ml of lysis buffer (50 mM Tris–HCl pH 8, 500 mM KCl, 0.1% Triton X‐100, 10 mM MgCl2, smDNAse nuclease, 0.04 mg/ml Pefabloc SC, cOmplete EDTA‐free protease inhibitor cocktail tablets and 1 mM TCEP) with a Dounce homogenizer (25×). The cleared lysate was loaded on a 5 ml Strep‐Tactin XT 4Flow cartridge. The column was washed with five column volumes (CV) of wash buffer (50 mM Tris–HCl pH 8, 150 mM KCl, 1 mM TCEP) and proteins were eluted with strep elution buffer (50 mM Tris–HCl pH 8, 150 mM KCl, 50 mM Biotin and 1 mM TCEP). Fractions were pooled and loaded on a 1 ml HiTrap Heparin HP affinity column equilibrated in wash buffer, and eluted in heparin elution buffer (50 mM Tris–HCl pH 8, 1 M KCl and 1 mM TCEP). Fractions were pooled, diluted down to 500 mM KCl and the Z‐basic‐TwinStrep tag was removed over night with the addition of His‐TEV protease. Next, the protein sample was loaded on Superdex 200 Increase 10/300 GL column equilibrated in equilibration buffer (50 mM Tris–HCl pH 8, 200 mM KCl, 10% glycerol and 1 mM TCEP). Eluted proteins were concentrated with 10 kDa cut‐off Amicon ultra centrifugal filters before snap‐freezing in liquid nitrogen and storage at ‐80°C.
Pol ζ‐Rev1
Pol ζ and Rev1 were purified as described previously (Kochenova et al, 2017).
Biotinylated LacI
Biotinylated LacI was purified as described previously (Dewar et al, 2015). Briefly, pET11a[LacR‐Avi] and pBirAcm (Avidity) vectors were transformed into T7 express‐competent cells. LacI and biotin ligase expression was induced with 1 mM IPTG in Luria–Bertani (LB) medium supplemented with 50 μM biotin for 2 h at 37°C. Cells were harvested, snap frozen and stored at −80°C. Cell pellets were lysed in lysis buffer (50 mM Tris–HCl pH 7.5, 5 mM EDTA, 100 mM NaCl, 1 mM DTT, 10% sucrose, cOmplete protease inhibitors, 0.2 mg/ml lysozyme and 0.1% Brij 58) for 30 min at room temperature (RT). Lysates were centrifuged at 21,300 g for 1 h at 4°C. Pellets containing chromatin‐bound LacI were then suspended in 50 mM Tris–HCl pH 7.5, 5 mM EDTA, 1 M NaCl, 30 mM IPTG, 1 mM DTT and LacI was released from DNA by sonication followed by addition of polymin P to 0.03–0.06% (w/v) at 4°C. Biotinylated LacI was precipitated with 37% ammonium sulphate, pelleted by centrifugation and then suspended in 50 mM Tris–HCl pH 7.5, 1 mM EDTA, 2.6 M NaCl, 1 mM DTT and cOmplete protease inhibitors. Biotinylated LacI was then bound to SoftLINK avidin, washed with 50 mM Tris–HCl pH 7.5, 1 mM EDTA, 2.6 M NaCl, 1 mM DTT, cOmplete protease inhibitors and eluted with 50 mM Tris–HCl pH 7.5, 100 mM NaCl, 1 mM EDTA, 5 mM biotin and 1 mM DTT. Pooled fractions containing biotinylated LacI were buffer exchanged into 50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA and 1 mM DTT using an Amicon ultra‐0.5 ml 3 kDa molecular weight cut‐off filter unit. Biotinylated LacI was aliquoted, snap frozen, and stored at −80°C.
USP2‐cc
To purify USP2‐catalytic core (USP2‐cc), pH10E USP‐cc plasmid was transformed into BL21 (DE3) E. coli cells. Expression was induced with 0.5 mM IPTG in LB medium for 16 h at 18°C. Cells were pelleted and lysed in lysis buffer E (20 mM Tris pH 8.0, 300 mM NaCl, 10 mM imidazole, 10% glycerol, 1% Triton X‐100, 1 mg/ml lysozyme, cOmplete EDTA‐free protease inhibitor cocktail tablets, 5 mM 2‐β‐mercaptoethanol (BME) and 10 U/ml benzonase (Sigma, 70746‐3)). Cell lysates were cleared by centrifugation at 18,000 g for 20 min. His‐tagged Usp2‐cc was bound to Ni‐NTA Agarose (QIAGEN) equilibrated in buffer F (20 mM Tris pH 8.0, 300 mM NaCl, 10 mM imidazole, 10% glycerol, 0.05% Triton X‐100 and 5 mM BME) for 4 h at 4°C, washed three times with buffer F and then eluted with buffer G (20 mM Tris pH 8.0, 300 mM NaCl, 300 mM imidazole, 10% glycerol, 0.05% Triton X‐100 and 1 mM TCEP). Eluted protein was dialyzed in dialysis buffer H (20 mM Tris pH 8.0, 150 mM NaCl, 10% glycerol and 1 mM TCEP). Protein was then aliquoted, snap frozen and stored at −80°C.
xl‐HMCES
An open reading frame containing Xenopus laevis HMCES (amino acid 2–336) was cloned in frame with an N‐terminal His10‐ubiquitin (Ub) to generate pHUE‐xl.HMCES plasmid (Catanzariti et al, 2004). pHUE‐xl.HMCES(E129A) plasmid was generated by inverse PCR using primer pairs (5’‐CAG GAC GGT GAA AAA CAA CCG TAC‐3′/5’‐GCG TTT CCA TGC ATA GAA CCC GTC C‐3′). All constructs were confirmed by Sanger sequencing and transformed into ArcticExpress (DE3)‐competent cells for protein expression. Cells were grown in LB medium at 37°C to OD600 0.6. Protein expression was induced by addition of 0.5 mM IPTG for 24 h. Cells were harvested, washed once with PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 and 1.8 mM KH2PO4), snap‐frozen and stored at −80°C. For protein purification, cells were resuspended in buffer E, incubated on ice for 30 min and briefly sonicated. Cell debris was removed by centrifugation at 18,000 g for 20 min. Cleared supernatants were applied to Ni‐NTA Agarose (QIAGEN) equilibrated in buffer F. Next, beads were washed thrice with 15 CV of buffer F and the protein was eluted with 4 CV of buffer G. The eluted protein was dialyzed against buffer H. The His‐tagged proteins were incubated overnight at 4°C with His10‐USP2‐cc (molar ratio 1/100) to cleave the His10‐Ub tag from the N‐terminus of HMCES. The cleavage reaction mixtures were incubated with 1 ml prewashed Ni‐NTA agarose to remove His10‐Ub, His10‐USP2‐cc, and uncleaved His10‐Ub‐HMCES. HMCES in the flowthrough was further purified by anion exchange chromatography using mono Q50/5 GL column (Cytiva). Samples were eluted over a gradient of 150 to 100 mM NaCl. Fractions containing proteins were pooled and concentrated using Amicon Ultra‐15 centrifugal filter unit with 10 kDa molecular weight cut‐off. Protein was aliquoted, snap‐frozen and stored at −80°C.
xl‐HMCES‐3xFlag
To purify xl‐HMCES‐3xFlag and xl‐HMCES(E129A)‐3xFlag, a DNA sequence encoding 3xFlag was inserted downstream of xl‐HMCES and xl‐HMCES(E129A) in pHUE backbone plasmid to generate pHUE‐xl.HMCES‐3xFlag and pHUE‐xl.HMCES (E129A)‐3xFlag, respectively. Correct sequences were confirmed by Sanger sequencing followed by transformation of plasmids into ArcticExpress (DE3)‐competent cells. Xl‐HMCES‐3xFlag and xl‐HMCES(E129A)‐3xFlag proteins were expressed and purified as described above for xl‐HMCES and xl‐HMCES(E129A).
Generation of HMCES‐DPCs
Crosslinking reactions with different HMCES variants were carried out in 10 μl reactions containing 8.02 μl reaction buffer (20 mM HEPES/KOH pH 7.5, 50 mM KCl, 10 mM MgCl2, 2 mM TCEP and 0.1 mg/ml BSA), 0.5 μl HMCESFL/HMCESSRAP (prediluted to 40 μM in purification buffer D), 1 μl Cy5‐labelled forward oligonucleotide (prediluted to 10 μM in DPC dilution buffer—50 mM HEPES/KOH pH 7.5, 100 mM KCl, 10% glycerol and 0.4 mg/ml BSA) and 0.48 μl UDG (New England BioLabs), adding up to final concentrations of 2 μM HMCESFL/HMCESSRAP, 1 μM DNA and 0.1 U/μl UDG (New England Biolabs). Reactions were incubated for 1 h at 37°C. Crosslinking reactions with YedK and xl‐HMCES (Fig EV3C and D) were carried out identically as described above, except that the 10 μl reactions contained 1.5 μl YedK/xl‐HMCES (prediluted to 13.2 μM in purification buffer D) and 7.02 μl reaction buffer. As a standard and if not stated otherwise, a 30mer oligonucleotide containing a central dU (5′‐Cy5‐CCC AAA AAA AAA AAdU AAA AAA AAA AAA CCC‐3′) was used for crosslinking. For generation of different DNA structures, 1 μl of corresponding reverse oligonucleotides (diluted to 12 μM in nuclease‐free H2O) were added to the crosslinking reaction and incubated for 2 min at 37°C before the temperature was decreased by 1°C/min until 20°C was reached to allow annealing of the reverse oligo. For ssDNA samples, a non‐complementary reverse oligo was added (5′‐AAA CCC CCC CCC CCA CCC CCC CCC AAA‐3′); for ssDNA‐dsDNA junction samples, a 15mer reverse oligo was added (5′‐GGG TTT TTT TTT TTT‐3′); and for dsDNA samples, a 30mer reverse oligo was added (5′‐GGG TTT TTT TTT TTT ATT TTT TTT TTT GGG‐3′). Reverse oligonucleotides were annealed prior to crosslinking for experiments shown in Fig 2A and C. For experiments shown in Figs 3A, EV2, 4, 5, EV3C and D, and EV4I, reverse oligonucleotides were annealed after crosslinking.
HMCES‐DPC formation assays
For the experiments shown in Fig EV1B, the indicated HMCES variants were prediluted to 64, 32, 16, 8, 4, 2, 1 and 0.5 μM in purification buffer D prior to crosslinking. 0.5 μl of the predilutions were added to the crosslinking reactions as described above resulting in final HMCES concentrations of 3.2, 1.6, 0.8, 0.4, 0.2, 0.1, 0.05 and 0.025 μM. Otherwise, crosslinking reactions were carried out as described above. Reactions were stopped by addition of 5.5 μl LDS sample buffer and boiling for 1 min at 95°C. Samples were resolved on 4–12% SDS–PAGE gels. Gels were photographed using a BioRad Chemidoc MP system using appropriate filter settings for Cy5 fluorescence. Crosslinking was quantified using ImageJ by measuring the relative fraction of Cy5 signal in the DPC band.
For the experiments shown in Fig 2A and C, crosslinking reactions were set up as described above. Incubation and annealing of reverse oligonucleotides were performed in the absence of HMCES to generate desired DNA structures (ssDNA, ssDNA‐dsDNA junction and dsDNA) before incubation with HMCES. For experiments in Fig 2C, a different 30mer oligonucleotide containing a central dU (5′‐Cy5‐CCC CCG GAA AAA AAdU AAA AAA AAG GCC CCC‐3′) was used and annealed with a 15mer reverse oligonucleotide (5′‐Fam‐GGG GGC CTT TTT TTT‐3′) for ssDNA‐dsDNA junction, two 15mer reverse oligonucleotide (5′‐Fam‐GGG GGC CTT TTT TTT‐3′ and 5′‐TTT TTT TTC CGG GGG‐3′) for dsDNA containing a nick, a 15mer reverse oligonucleotide and a 10mer reverse oligonucleotide (5′‐Fam‐GGG GGC CTT TTT TTT‐3′ and 5′‐T TTC CGG GGG‐3′) for dsDNA containing a 4 nt‐gap and a 30mer reverse oligonucleotide (5′‐GGG GGC CTT TTT TTT TTT TTT TTC CGG GGG‐3′) for dsDNA. Following annealing, 0.5 μl HMCES (prediluted to 40 μM in purification buffer D) was added to the reactions. Reactions were incubated for 0, 1, 5, 10, 20, or 30 min at 37°C before being stopped by addition of 5.5 μl LDS sample buffer and boiling for 1 min at 95°C. Samples were frozen in liquid nitrogen and stored at −80°C. Before resolving samples on 4–12% SDS–PAGE gels, samples were boiled again at 95°C for 30 s. Gels were photographed using a BioRad Chemidoc MP system using appropriate filter settings for Cy5 fluorescence. Quantification was performed using ImageJ by measuring the relative fraction of Cy5 signal in the DPC band.
HMCES‐DPC release assays
The experiments shown in Fig 1E and G indicated that HMCES variants were crosslinked to a 30mer oligonucleotide containing a central dU or dT (5′‐Cy5‐CCC AAA AAA AAA AAdU/dT AAA AAA AAA AAA CCC‐3′) as described above. In parallel, crosslinking reactions containing a 6‐FAM‐labelled 30mer oligonucleotide also containing a central dU (5′‐6‐FAM‐CCC AAA AAA AAA AAdU AAA AAA AAA AAA CCC‐3′) with 0.5 μl purification buffer D instead of protein were prepared and incubated at 37°C for 1 h as well. To inactive non‐crosslinked HMCES, reactions containing the Cy5‐oligonucleotide were incubated for 5 min at 60°C. In the following step, reactions containing the Cy5‐oligonucleotide and HMCES were mixed 1:1 with reactions containing the 6‐FAM‐labelled oligonucleotide and incubated for 2 h at 37°C. To stop reactions, 11 μl of LDS sample buffer was added and reactions were boiled for 1 min at 95°C before analysis on 4–12% SDS–PAGE gels. Gels were photographed using a BioRad Chemidoc MP system using appropriate filter settings for Cy5 and 6‐FAM fluorescence. 6‐FAM‐ and Cy5‐DPC formation was quantified using ImageJ.
For experiments shown in Figs 3A, EV2C and EV4G, indicated HMCES variants were crosslinked to an ssDNA oligonucleotide, as described above. Afterwards, corresponding reverse oligonucleotides were annealed as described above. Following annealing, 1 μl of the crosslinking reaction was added to 9 μl of master mix, resulting in a final buffer composition of 17.1 mM HEPES, 85.6 mM KCl, 3.1% glycerol, 5.5 mM TCEP, 2 mM MgCl2 and 0.1 mg/ml BSA. Reactions were either stopped directly after annealing (0 h) or after 1, 2, 4, or 18 h incubation at 37°C by addition of 5.5 μl LDS sample buffer. For experiments shown in Fig EV4G, corresponding samples were incubated at 20°C after annealing. Reactions were boiled for 1 min at 95°C before being frozen in liquid nitrogen and stored at −80°C. Samples were boiled at 95°C for 30 s before being resolved on 4–12% SDS–PAGE gels. Gels were photographed using a BioRad Chemidoc MP system using appropriate filter settings for Cy5 fluorescence. Quantification was performed using ImageJ by measuring the relative fraction of Cy5 signal in the DPC band.
For the experiments shown in Fig 4, indicated HMCESSRAP variants were crosslinked as described above. Afterwards, corresponding reverse oligonucleotides were annealed to create an ssDNA‐dsDNA junction, while a non‐complimentary oligonucleotide was added in ssDNA conditions. HMCESFL was prediluted to 20 μM in competition buffer (150 mM KCl, 50 mM HEPES and 10% glycerol). The final assay was carried out in a reaction volume of 10 μl with 1 μl of the crosslinking reaction and 1 μl of prediluted HMCESFL, in a final buffer composition of 17.1 mM HEPES, 85.6 mM KCl, 3.1% glycerol, 5.5 mM TCEP, 2 mM MgCl2 and 0.1 mg/ml BSA. Reactions were incubated for 0, 1, 2, 3, 4 or 5 h at 37°C before being stopped by addition of 5.5 μl LDS sample buffer. The reactions were boiled for 1 min at 95°C before being frozen in liquid nitrogen and stored at −80°C. After thawing, samples were boiled at 95°C for 30 s before being resolved on 4–12% SDS–PAGE gels. Gels were photographed using a BioRad Chemidoc MP system, using appropriate filter settings for Cy5 fluorescence. Quantification was done using ImageJ, by measuring the relative Cy5 signals of HMCESFL‐DPCs and HMCESSRAP‐DPCs.
APE1 incision assays
The experiments shown in Fig 5 indicated HMCESSRAP variants were crosslinked to a 30mer oligonucleotide containing a central dU as described above. Reverse oligonucleotides were annealed to generate an ssDNA‐dsDNA junction or dsDNA. After annealing, 1 μl of the HMCESSRAP‐DNA crosslinking reaction and 0.5 μl of APE1 (New England BioLabs) were added to 8.5 μl final reaction buffer, bringing final concentrations to 17.1 mM HEPES, 85.6 mM KCl, 3.1% glycerol, 5.5 mM TCEP, 2 mM MgCl2 and 0.1 mg/ml BSA. For experiments shown in Fig EV3C and D, reactions were prepared similarly except that 1.5 μl prediluted YedK‐WT or ‐E105A and xl‐HMCES‐WT or ‐E129A were added with the crosslinking reaction with 7.02 μl reaction buffer, as described before. To samples not containing APE1, 0.5 μl APE1 buffer was added (10 mM Tris–HCl, 50 mM NaCl, 1 mM DTT, 0.05 mM EDTA, 200 μg/ml BSA and 50% glycerol, pH 8). Reactions were either stopped directly (0 h) or after 1 or 18 h of incubation at 37°C by the addition of 5.5 μl LDS sample buffer. Reactions were boiled for 1 min at 95°C before being frozen in liquid nitrogen and stored at −80°C. Samples were boiled again at 95°C for 30 s after thawing before being resolved on 4–12% SDS–PAGE gels. Gels were photographed using a BioRad Chemidoc MP system using appropriate filter settings for Cy5 fluorescence.
DNA‐binding assays
Electrophoretic mobility shift assays
HMCESSRAP‐WT and variants were prediluted to 40, 10 and 2.5 μM in purification buffer D. Binding reactions were carried out in 10 μl with 0.5 μl of HMCESSRAP dilutions, 1 μl of 1 μM Cy5‐labelled 30mer dT‐oligonucleotide and 8.5 μl reaction buffer (20 mM HEPES/KOH pH 7.5, 50 mM KCl, 10 mM MgCl2, 2 mM TCEP and 0.1 mg/ml BSA). Reactions were incubated for 20 min on ice before addition of 4 μl 6× Orange G‐loading dye. Samples were then resolved at 4°C on 6% native PAGE gels using 0.5× TBE as running buffer. Gels were photographed using a BioRad Chemidoc MP system using appropriate filter settings for Cy5 fluorescence.
Fluorescence polarization
HMCESSRAP‐WT and variants were prediluted to 200 μM, 40 μM, 8 μM, 1.6 μM, 0.32 μM, 1.28 nM and 0.256 nM in purification buffer D. Binding was carried out in 50 μl final volume with 5 μl of HMCESSRAP predilutions, 5 μl of 250 nM Cy5‐labelled 30mer dT‐oligonucleotide and 40 μl of reaction buffer (20 mM HEPES/KOH pH 7.5, 50 mM KCl, 10 mM MgCl2, 2 mM TCEP and 0.1 mg/ml BSA). Binding reactions were incubated for 20 min on ice before 10 μl of the reactions were pipetted into a 384‐well microplate (Greiner Bio‐One). Fluorescence polarization was measured using a Tecan Spark multimode microplate reader using appropriate filter settings for Cy5 fluorescence.
Primer extension assay
Primer extension assays were used to analyse HMCESSRAP reversibility following bypass by TLS polymerases Pol ζ‐Rev1. HMCESSRAP‐WT‐DPCs and ‐E127A‐DPCs were crosslinked as described above to a 45mer forward Cy5‐labelled oligonucleotide containing a dU at position 30 (5′‐CY5‐ACC AGT GCC TTG CT[U] GGA CAT CTT TGC CCA CCT GCA GGT TCA CC‐3′). To generate an AP site, 0.1 U/μl UDG (New England Biolabs) was added to the crosslinking reaction. After 1 h incubation at 37°C, either a 15mer 6‐FAM‐labelled primer (5′‐6‐FAM‐GGG TGA ACC TGC AGG‐3′) or a corresponding 45mer 6‐FAM labelled oligonucleotide (5′‐6‐FAM‐GGG TGA ACC TGC AGG TGG GCA AAG ATG TCC AAG CAA GGC ACT GGT‐3′) to generate dsDNA was annealed as described above. FANCJ‐dependent primer extension with Pol ζ‐Rev1 was performed as described previously (Yaneva et al, 2023). For the final reaction, 1 μl of the HMCESSRAP‐DPC reaction, 100 nM FANCJ, 25 nM Pol ζ and 40 nM Rev1 were mixed in a final volume of 10 μl and the following conditions: 17.1 mM HEPES/KOH pH 7.5, 5.5 mM Tris–HCl pH 8.0, 70.6 mM KCl, 2.5 mM NaCl, 6.6% glycerol, 5 mM TCEP, 2 mM ATP, 0.2 mM dNTPs, 2.9 mM MgCl2 and 0.1 mg/ml BSA. Reactions were incubated for 2 h at 37°C and stopped by the addition of 10 μl UREA‐loading buffer (8 M UREA, 15% Ficoll). The reactions were then boiled for 10 min at 95°C and resolved on denaturing 12% UREA–PAGE gels (12% acrylamide, 8 M UREA and 1×TBE) at 60°C in 1× TBE running buffer. Gels were photographed using a BioRad Chemidoc MP system using appropriate filter settings for Cy5 and 6‐FAM fluorescence. Quantification was performed using ImageJ, by measuring the relative Cy5 signals (Fig 5C) or the relative 6‐FAM signals of the 45mer species (Fig 5D).
Generation of cell lines
HeLa T‐REx Flp‐In cells were provided by Cell Services, The Francis Crick Institute, and grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% (v/v) foetal bovine serum (FBS). For doxycycline‐inducible overexpression of HMCES variants, the coding sequence of HMCES was amplified from cDNA using Q5 Master Mix (M0544, NEB) before being shuttled into p221 plasmid using BP clonase (11789100, Thermo Fisher). Next, the E127A mutation was introduced with Q5® Site‐Directed Mutagenesis Kit Protocol (E0554, NEB) and both sequences were subcloned into pcDNA5/FRT/TO‐mVenus‐3xFlag‐Gateway (Addgene, #40999) using LR clonase (11791020, Thermo Fisher) before generation of stable cell lines using the T‐REx Flp‐In system (Thermo Fisher) according to manufacturer's instructions. Briefly, HeLa T‐REx Flp‐In cells were grown to 50% confluency in six‐well plates prior to transfection of pOG44 (1.8 μg) and the respective pcDNA5‐FRT/TO plasmids (0.2 μg, containing HMCES‐WT‐mVenus‐3xFlag, HMCES‐E127A‐mVenus‐3xFlag or mVenus‐3xFlag (the gateway recombination cassette was deleted) using Lipofectamine 2000 (Invitrogen). Sixteen hours after transfection, cells were selected in 150 μg/ml hygromycin B (Fisher Scientific)‐containing DMEM media for 10 days.
Cell viability assay
To analyse cell viability of HeLa T‐REx Flp‐In cells overexpressing HMCES‐WT‐mVenus‐3xFlag or HMCES‐E127A‐mVenus‐3xFlag, cells were counted and seeded in 12‐well plates (10,000 cells per well) with DMEM −/+ 1 μg/ml doxycycline in technical triplicates and incubated for 4 days. HeLa T‐REx Flp‐In cells overexpressing mVenus‐3xFlag were included as a control. After this, cell viability was assessed by AlamarBlue assay (Sigma, R7017‐1G, 0.04% in PBS assay concentration). One well of each condition was harvested and analysed by western blotting using anti‐HMCES antibody (Santa Cruz, #sc‐514238), anti‐Flag‐M2 antibody (Sigma, #F3165) and anti‐Vinculin antibody (Santa Cruz, #sc‐73614). Plates were afterwards stained with 0.5% crystal violet and scanned.
Preparation of oligonucleotide duplexes with AP‐ICL
To generate the AP‐ICL‐containing oligonucleotide duplex, the complementary 5′‐phosphorylated oligonucleotides (AP‐ICL top: 5′‐GCA CCT TCC GCT CdUT CTT TC‐3′ and AP‐ICL bottom: CCC TGA AAG AAG AGC GGA AG) heated for 5 min at 95°C in 30 mM HEPES‐KOH pH 7.0, 100 mM NaCl and annealed by cooling at 1°C per min to 18°C. The annealed duplex was treated with uracil glycosylase (NEB) in 1 × UDG buffer for 2 h at 37°C followed by phenol/chloroform extraction and ethanol precipitation. The oligo duplex was suspended in 50 mM HEPES‐KOH pH 7.0, 100 mM NaCl and then incubated at 37°C for 5 days to allow for crosslink formation. Crosslinked DNA duplex was separated on a 20% polyacrylamide, 1 × Tris‐borate‐EDTA (TBE) and 8 M urea gel, and the crosslinked product was excised from the gel and eluted into TE (pH 8.0) buffer. Eluted DNA was concentrated by adding 4.5 times volume of 1‐butanol, extracted with phenol:choloroform:isoamyl alcohol (25:24:1; pH 8.0) and precipitated with ethanol. The AP‐ICL DNA oligo was then suspended in 10 mM Tris–HCl (pH 8.5) and stored at −80°C.
Preparation of crosslink DNA construct pICL‐lacO AP
pICL‐lacO AP was prepared as described previously (Semlow et al, 2016). Briefly, the backbone plasmid (with 48 lacO repeats) was incubated with BbsI in NEBuffer 2.1 for 24 h at 37°C followed by phenol/chloroform extraction. The digested plasmid was further purified using a HiLoad 16/60 Superdex 200 column, which was equilibrated in TE pH 8.0 buffer. Fractions containing the linearized plasmid were pooled, precipitated in ethanol and dissolved in 10 mM Tris–HCl pH 8.5. The abasic site interstrand crosslink (ICLAP)‐containing duplexes were ligated into the linearized plasmid backbone using T4 DNA ligase (NEB). The ligated plasmid was dialysed into TE pH 8.0 buffer and concentrated using an Amicon Ultra‐15 10 kDa molecular weight cut‐off filter unit. The covalently closed circular plasmids were further extracted using the CsCl ethidium bromide method. Ethidium bromide was then removed from DNA by mixing with equal volume of saturated isobutanol. The purified pICL‐lacO AP was then dialyzed into TE pH 8.0, concentrated, snap frozen and stored at −80°C.
Xenopus egg extracts
Unfertilized eggs were collected from female Xenopus laevis frogs (Xenopus1, Cat# 4280; age > 2 years). Sperm chromatin was prepared from male Xenopus laevis frogs (Xenopus1, Cat# 4290, age > 1 year). All animal work was performed at Caltech and by the IACUC (Protocol IA20‐1797, approved 28 May 2020). The institution has an approved Animal Welfare Assurance (no. D16‐00266) from the NIH Office of Laboratory Animal Welfare. The high‐speed supernatant (HSS) and nucleoplasmic extracts (NPE) were prepared from Xenopus laevis eggs as described previously (Semlow et al, 2022). Briefly, six adult female X. laevis frogs were induced to produce eggs by injection with 500 IU hCG. Eggs were collected and dejellied in 1 l of 2.2% (w/v) cysteine, pH 7.7. Dejellied eggs were then washed with 2 l of 0.5× Marc's modified Ringer's solution (2.5 mM HEPES‐KOH, pH 7.8, 50 mM NaCl, 1 mM KCl, 0.25 mM MgSO4, 1.25 mM CaCl2 and 0.05 mM EDTA) followed by 1 l of egg lysis buffer (ELB; 10 mM HEPES‐KOH, pH 7.7, 50 mM KCl, 2.5 mM MgCl2 and 250 mM sucrose) supplemented with 1 mM DTT and 50 μg/ml cycloheximide. Eggs were then packed and crushed in the presence of 5 μg/ml aprotinin, 5 μg/ml leupeptin and 2.5 μg/ml cytochalasin B by centrifugation at 20,000 g for 20 min at 4°C. The soluble extract layer (the low‐speed supernatant (LSS)) was collected and supplemented with 50 μg/ml cycloheximide, 1 mM DTT, 10 μg/ml aprotinin, 10 μg/l leupeptin and 5 μg/ml cytochalasin B. LSS was centrifuged in thin‐walled ultracentrifuge tubes at 260,000 g for 90 min at 2°C in a TLS 55 rotor. Supernatant (HSS) was collected, aliquoted, snap‐frozen in liquid nitrogen and stored at −80°C. To prepare NPE, LSS was prepared from eggs collected from 20 female X. laevis frogs as described above. LSS was then supplemented with 50 μg/ml cycloheximide, 1 mM DTT, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 5 μg/ml cytochalasin B and 3.3 μg/ml nocodazole, and centrifuged at 20,000 g for 15 min at 4°C. After removing lipids, the clarified cytoplasmic fraction was collected and supplemented with ATP‐regenerating mix (2 mM ATP, 20 mM phosphocreatine and 5 μg/ml phosphokinase) and 4,400 demembranated X. laevis sperm chromatin/μl to initiate nuclei formation. After ~90 min incubation at RT, reaction mixture was centrifuged for 3 min at 18,000 g at 4°C. The nuclei layer was collected from the top of the tubes and centrifuged at 260,000 g for 30 min at 2°C. The lipid layer was removed and the NPE fraction was collected, aliquoted, snap frozen in liquid nitrogen and stored at − 80°C.
Immunodepletions
Immunodepletions of SPRTN and REV1 were performed as described (Semlow et al, 2022). Briefly, protein A Sepharose fast flow beads were washed in 1× PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 and 1.8 mM KH2PO4) and then incubated with an appropriate volume of antibodies overnight at 4°C. For SPRTN depletion, three volumes of polyclonal SPRTN anti‐serum (Pocono Rabbit Farm and Laboratory rabbit 31053) were used for each volume of beads. For REV1 depletion, one volume of polyclonal REV1 C‐terminus anti‐serum (Pocono Rabbit Farm and Laboratory rabbit 1010) or one volume of polyclonal REV1 N‐terminus anti‐serum (Pocono Rabbit Farm and Laboratory rabbit 714) was used for each volume of beads. Equivalent volumes of rabbit pre‐immune serum were incubated with beads for mock depletions. The beads were then washed twice with PBS, once with ELB, twice with ELB supplemented with 500 mM NaCl and thrice with ELB. HSS and NPE were immunodepleted by three rounds of incubation at 4°C for 60 min with protein A Sepharose‐bound antibodies (five volume extract per volume of beads). In the case of REV1 depletion, two rounds of depletion were performed using the N‐terminal antibody and one round was performed using the C‐terminal antibody. Extracts were centrifuged for 30 s at 622 g in an S‐24‐11‐AT rotor using an Eppendorf 5430R centrifuge and the supernatants were collected.
Replication reactions
For replication reactions, licensing was conducted by incubating HSS with 15 ng/μl pICL‐lacO AP plasmid in the presence of 3 μg/ml nocodazole, 20 mM phosphocreatine, 2 mM ATP and 5 μg/ml creatine phosphokinase with or without 0.1 μM 3,000 Ci/mmol [α‐32P] dCTP for 30 min at RT. Replication was then initiated by mixing two volumes of NPE mix (50% (v/v) NPE, 20 mM phosphocreatine, 2 mM ATP, 5 μg/ml creatine phosphokinase and 13.5 mM DTT in ELB) with one volume of licensing mix. Replication reactions were additionally supplemented with 0.2 or 2 μM Xl‐HMCES‐3xFlag, as indicated. 32P‐radiolabeled reactions were quenched by adding 1 μl of replication reaction to 6 μl of replication stop buffer (8 mM EDTA, 0.13% phosphoric acid, 10% ficoll, 5% SDS, 0.2% bromophenol blue and 80 mM Tris–HCl, pH 8.0) at the indicated time points followed by digestion with proteinase K (2.5 mg/ml) for 60 min at 37°C. Replication products were resolved on 0.8% agarose gels and visualized by phosphor imaging using a GE Healthcare Typhoon FLA 9500 (FujiFilm FLA 9500 user interface v.1.1). Images were analysed using Image Lab v.6.4.0 (Bio‐Rad).
Plasmid pulldowns and immunoblotting
Plasmid pulldowns were performed as described previously (Semlow et al, 2022). Briefly, streptavidin‐coupled magnetic Dynabeads (10 μl per pull down) were washed thrice with bead wash buffer 1 (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA pH 8.0 and 0.02% Tween‐20). Dynabeads were then incubated with biotinylated LacI (0.4 pmol per 1 μl of beads) at RT for 60 min. The beads were then washed four times with pulldown buffer (20 mM Tris pH 7.5, 150 mM NaCl, 2 mM EDTA pH 8 and 0.5% IGEPAL‐CA630). Eight microlitres replication reaction was quenched into 400 μl of pulldown buffer and stored on ice. Samples were then incubated with 10 μl of LacI‐coated streptavidin Dynabeads at 4°C for 30 min on a rotating wheel. The beads were washed thrice with wash buffer 2 (20 mM Tris–HCl [pH 7.5], 1.5 M NaCl, 2 mM EDTA and 0.5% IGEPAL‐CA630) and then twice with Benzonase equilibration buffer (20 mM Tris–HCl [pH 7.5], 20 mM NaCl, 2 mM MgCl2 and 0.02% Tween‐20). Beads were then suspended in 7.5 μl of benzonase buffer (20 mM Tris–HCl [pH 8.0], 20 mM NaCl, 2 mM MgCl2 and 0.02% Tween‐20) containing 12 U benzonase and incubated for 1 h at 37°C. 7.5 μl of 2 × Laemmli loading buffer was added and the samples were incubated at 95°C for 5 min. The supernatants were collected and resolved on a 10% Criterion TGX precast midi protein gel (Bio‐Rad), and transferred to polyvinyl difluoride membranes (Thermo Scientific). Membranes were blocked with 5% dried milk in PBST for 60 min at room temperature, rinsed several times with PBST and incubated with primary antibodies, as indicated, overnight at 4°C. Endogenous and rHMCES‐3xFlag proteins were detected by immunoblotting with affinity‐purified HMCES SRAP domain antiserum (1:5,000 dilution in PBST; Pocono Rabbit Farm and Laboratory rabbit 38,389). SPRTN was detected using SPRTN antiserum (Pocono Rabbit Farm and Laboratory rabbit 31053; 1:5,000 dilution in PBST). REV1 was detected using REV1 C‐terminus antiserum (Pocono Rabbit Farm and Laboratory rabbit 1010; 1:5,000 dilution in PBST). Membranes were washed thrice with PBST for 10 min at room temperature, incubated with goat anti‐rabbit secondary antibody peroxidase conjugate (1:20,000 dilution in PBST; Jackson ImmunoResearch Laboratories Inc., 111‐035‐003) for 30 min at room temperature, and then washed thrice with PBST for 10 min at room temperature. Blots were imaged with chemiluminescence using SuperSignal West Dura Extended Duration Substrate (Thermo Scientific) using a Bio‐Rad ChemiDoc Imaging System (user interface v.2.4) and analysed using Image Lab v.6.4.0 (Bio‐Rad).
Quantification and statistical analysis
Statistical details of each experiment (including the exact value of n, what n represents and precision measures) can be found in the figure legends.
Resource availability
Materials availability
All plasmids and cell lines are available on request from the corresponding author.
Author contributions
Maximilian Donsbach: Conceptualization; investigation; writing – original draft; writing – review and editing. Sophie Dürauer: Conceptualization; investigation; writing – original draft; writing – review and editing. Florian Grünert: Investigation; writing – review and editing. Kha T Nguyen: Investigation; writing – review and editing. Richa Nigam: Investigation; writing – review and editing. Denitsa Yaneva: Investigation; writing – review and editing. Pedro Weickert: Investigation; writing – review and editing. Rachel Bezalel‐Buch: Resources. Daniel R Semlow: Supervision; funding acquisition; investigation; writing – review and editing. Julian Stingele: Conceptualization; supervision; funding acquisition; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank T. Fröhlich for mass spectrometry analyses of recombinant HMCES protein and T. Mackens‐Kiani for help with preparing Fig 1C. Research in the lab of DRS is supported by NIH grant no. GM129422 and an award from the Shurl and Kay Curci Foundation. Research in the lab of JS is supported by the European Research Council under the European Union's Horizon 2020 research and innovation programme (grant agreement number 801750), by the Alfried Krupp Prize for Young University Teachers awarded by the Alfried Krupp von Bohlen und Halbach Foundation, the European Molecular Biology Organization (YIP4644), the Vallee Foundation and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) (Project ID 213249687—SFB 1064 and Project‐ID 393547839—SFB 1361). NIH grant GM118129 to Peter M. Burgers supported RB‐B. Open Access funding enabled and organized by Projekt DEAL. Open Access funding enabled and organized by Projekt DEAL.
The EMBO Journal (2023) 42: e113360
Data availability
Original gel images of all main figures are provided as source data. Original gel images of extended view figures will be shared by the corresponding author upon request. This study did not generate original code. Any additional information required to reanalyse the data reported in this paper is available from the corresponding author upon request.
References
- Abbotts R, Wilson DM 3rd (2017) Coordination of DNA single strand break repair. Free Radic Biol Med 107: 228–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amidon KM, Eichman BF (2020) Structural biology of DNA abasic site protection by SRAP proteins. DNA Repair (Amst) 94: 102903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhargava V, Goldstein CD, Russell L, Xu L, Ahmed M, Li W, Casey A, Servage K, Kollipara R, Picciarelli Z et al (2020) GCNA preserves genome integrity and fertility across species. Dev Cell 52: 38–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biayna J, Garcia‐Cao I, Alvarez MM, Salvadores M, Espinosa‐Carrasco J, McCullough M, Supek F, Stracker TH (2021) Loss of the abasic site sensor HMCES is synthetic lethal with the activity of the APOBEC3A cytosine deaminase in cancer cells. PLoS Biol 19: e3001176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgermann N, Ackermann L, Schwertman P, Hendriks IA, Thijssen K, Liu JC, Lans H, Nielsen ML, Mailand N (2019) SUMOylation promotes protective responses to DNA‐protein crosslinks. EMBO J 38: e101496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catanzariti AM, Soboleva TA, Jans DA, Board PG, Baker RT (2004) An efficient system for high‐level expression and easy purification of authentic recombinant proteins. Protein Sci 13: 1331–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar JM, Budzowska M, Walter JC (2015) The mechanism of DNA replication termination in vertebrates. Nature 525: 345–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokshin GA, Davis GM, Sawle AD, Eldridge MD, Nicholls PK, Gourley TE, Romer KA, Molesworth LW, Tatnell HR, Ozturk AR et al (2020) GCNA interacts with Spartan and Topoisomerase II to regulate genome stability. Dev Cell 52: 53–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duxin JP, Dewar JM, Yardimci H, Walter JC (2014) Repair of a DNA‐protein crosslink by replication‐coupled proteolysis. Cell 159: 346–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halabelian L, Ravichandran M, Li Y, Zeng H, Rao A, Aravind L, Arrowsmith CH (2019) Structural basis of HMCES interactions with abasic DNA and multivalent substrate recognition. Nat Struct Mol Biol 26: 607–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenova OV, Bezalel‐Buch R, Tran P, Makarova AV, Chabes A, Burgers PM, Shcherbakova PV (2017) Yeast DNA polymerase zeta maintains consistent activity and mutagenicity across a wide range of physiological dNTP concentrations. Nucleic Acids Res 45: 1200–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima Y, Machida Y, Palani S, Caulfield TR, Radisky ES, Kaufmann SH, Machida YJ (2020) FAM111A protects replication forks from protein obstacles via its trypsin‐like domain. Nat Commun 11: 1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokan HE, Bjoras M (2013) Base excision repair. Cold Spring Harb Perspect Biol 5: a012583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen NB, Gao AO, Sparks JL, Gallina I, Wu RA, Mann M, Raschle M, Walter JC, Duxin JP (2019) Replication‐coupled DNA‐protein crosslink repair by SPRTN and the proteasome in Xenopus egg extracts. Mol Cell 73: 574–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessel D, Vaz B, Halder S, Lockhart PJ, Marinovic‐Terzic I, Lopez‐Mosqueda J, Philipp M, Sim JC, Smith KR, Oehler J et al (2014) Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nat Genet 46: 1239–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskey RS, Kim MS, Baker DJ, Childs B, Malureanu LA, Jeganathan KB, Machida Y, van Deursen JM, Machida YJ (2014) Spartan deficiency causes genomic instability and progeroid phenotypes. Nat Commun 5: 5744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskey RS, Flatten KS, Sieben CJ, Peterson KL, Baker DJ, Nam HJ, Kim MS, Smyrk TC, Kojima Y, Machida Y et al (2017) Spartan deficiency causes accumulation of Topoisomerase 1 cleavage complexes and tumorigenesis. Nucleic Acids Res 45: 4564–4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta KPM, Lovejoy CA, Zhao R, Heintzman DR, Cortez D (2020) HMCES maintains replication fork progression and prevents double‐strand breaks in response to APOBEC deamination and abasic site formation. Cell Rep 31: 107705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohni KN, Wessel SR, Zhao R, Wojciechowski AC, Luzwick JW, Layden H, Eichman BF, Thompson PS, Mehta KPM, Cortez D (2019) HMCES maintains genome integrity by shielding abasic sites in single‐strand DNA. Cell 176: 144–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulin KA, Cortez D, Eichman BF (2022) The SOS response‐associated peptidase (SRAP) domain of YedK catalyzes ring opening of abasic sites and reversal of its DNA‐protein cross‐link. J Biol Chem 298: 102307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price NE, Johnson KM, Wang J, Fekry MI, Wang Y, Gates KS (2014) Interstrand DNA‐DNA cross‐link formation between adenine residues and abasic sites in duplex DNA. J Am Chem Soc 136: 3483–3490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinking HK, Kang HS, Gotz MJ, Li HY, Kieser A, Zhao S, Acampora AC, Weickert P, Fessler E, Jae LT et al (2020) DNA structure‐specific cleavage of DNA‐protein crosslinks by the SPRTN protease. Mol Cell 80: 102–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semlow DR, Zhang J, Budzowska M, Drohat AC, Walter JC (2016) Replication‐dependent unhooking of DNA interstrand cross‐links by the NEIL3 glycosylase. Cell 167: 498–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semlow DR, MacKrell VA, Walter JC (2022) The HMCES DNA‐protein cross‐link functions as an intermediate in DNA interstrand cross‐link repair. Nat Struct Mol Biol 29: 451–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serbyn N, Noireterre A, Bagdiul I, Plank M, Michel AH, Loewith R, Kornmann B, Stutz F (2020) The aspartic protease Ddi1 contributes to DNA‐protein crosslink repair in yeast. Mol Cell 77: 1066–1079 [DOI] [PubMed] [Google Scholar]
- Srivastava M, Su D, Zhang H, Chen Z, Tang M, Nie L, Chen J (2020) HMCES safeguards replication from oxidative stress and ensures error‐free repair. EMBO Rep 21: e49123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele J, Schwarz MS, Bloemeke N, Wolf PG, Jentsch S (2014) A DNA‐dependent protease involved in DNA‐protein crosslink repair. Cell 158: 327–338 [DOI] [PubMed] [Google Scholar]
- Stingele J, Bellelli R, Alte F, Hewitt G, Sarek G, Maslen SL, Tsutakawa SE, Borg A, Kjaer S, Tainer JA et al (2016) Mechanism and regulation of DNA‐protein crosslink repair by the DNA‐dependent metalloprotease SPRTN. Mol Cell 64: 688–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele J, Bellelli R, Boulton SJ (2017) Mechanisms of DNA‐protein crosslink repair. Nat Rev Mol Cell Biol 18: 563–573 [DOI] [PubMed] [Google Scholar]
- Thompson PS, Cortez D (2020) New insights into abasic site repair and tolerance. DNA Repair (Amst) 90: 102866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PS, Amidon KM, Mohni KN, Cortez D, Eichman BF (2019) Protection of abasic sites during DNA replication by a stable thiazolidine protein‐DNA cross‐link. Nat Struct Mol Biol 26: 613–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirman S, Quinet A, Wood M, Meroni A, Cybulla E, Jackson J, Pegoraro S, Simoneau A, Zou L, Vindigni A (2021) Temporally distinct post‐replicative repair mechanisms fill PRIMPOL‐dependent ssDNA gaps in human cells. Mol Cell 81: 4026–4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretyakova NY, Groehler A 4th, Ji S (2015) DNA‐protein cross‐links: formation, structural identities, and biological outcomes. Acc Chem Res 48: 1631–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz B, Popovic M, Newman JA, Fielden J, Aitkenhead H, Halder S, Singh AN, Vendrell I, Fischer R, Torrecilla I et al (2016) Metalloprotease SPRTN/DVC1 orchestrates replication‐coupled DNA‐protein crosslink repair. Mol Cell 64: 704–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Bao H, Chen L, Liu Y, Li Y, Wu B, Huang H (2019) Molecular basis of abasic site sensing in single‐stranded DNA by the SRAP domain of E. coli yedK. Nucleic Acids Res 47: 10388–10399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weickert P, Stingele J (2022) DNA‐protein crosslinks and their resolution. Annu Rev Biochem 91: 157–181 [DOI] [PubMed] [Google Scholar]
- Wilson DM 3rd, Takeshita M, Grollman AP, Demple B (1995) Incision activity of human apurinic endonuclease (Ape) at abasic site analogs in DNA. J Biol Chem 270: 16002–16007 [DOI] [PubMed] [Google Scholar]
- Wu L, Shukla V, Yadavalli AD, Dinesh RK, Xu D, Rao A, Schatz DG (2022) HMCES protects immunoglobulin genes specifically from deletions during somatic hypermutation. Genes Dev 36: 433–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaneva D, Sparks JL, Donsbach M, Zhao S, Weickert P, Bezalel‐Buch R, Stingele J, Walter JC (2023) The FANCJ helicase unfolds DNA‐protein crosslinks to promote their repair. Mol Cell 83: 43–56 [DOI] [PMC free article] [PubMed] [Google Scholar]