

Abstract
Long noncoding RNAs (lncRNAs) influence the transcription of gene networks in many cell types, but their role in tumor‐associated macrophages (TAMs) is still largely unknown. We found that the lncRNA ADPGK‐AS1 was substantially upregulated in artificially induced M2‐like human macrophages, macrophages exposed to lung cancer cells in vitro, and TAMs from human lung cancer tissue. ADPGK‐AS1 is partly located within mitochondria and binds to the mitochondrial ribosomal protein MRPL35. Overexpression of ADPGK‐AS1 in macrophages upregulates the tricarboxylic acid cycle and promotes mitochondrial fission, suggesting a phenotypic switch toward an M2‐like, tumor‐promoting cytokine release profile. Macrophage‐specific knockdown of ADPGK‐AS1 induces a metabolic and phenotypic switch (as judged by cytokine profile and production of reactive oxygen species) to a pro‐inflammatory tumor‐suppressive M1‐like state, inhibiting lung tumor growth in vitro in tumor cell‐macrophage cocultures, ex vivo in human tumor precision‐cut lung slices, and in vivo in mice. Silencing ADPGK‐AS1 in TAMs may thus offer a novel therapeutic strategy for lung cancer.
Keywords: Long noncoding RNAs, Lung cancer, Metabolic remodeling, Tumor microenvironment, Tumor‐associated macrophages
Subject Categories: Cancer, Immunology, Respiratory System
Expression of the long noncoding RNA ADPGK‐AS1 results in a phenotypic switch to the tumor‐promoting M2‐like identity in human macrophages.
Introduction
Lung cancer remains the leading cause of cancer death worldwide for both men and women, with a 5‐year survival rate of ≤ 15% (Siegel et al, 2020). Histologically, lung cancer is categorized as either small‐cell or nonsmall‐cell lung carcinoma, accounting for 15 and 85% of cases, respectively (Travis et al, 2013, 2015). Several options are available for treatment, including chemotherapy, radiotherapy, and immunotherapy targeting particular immune‐system components to activate the antitumor response (Seebacher et al, 2019). For example, immune checkpoint inhibitors, such as cytotoxic T lymphocyte‐associated antigen 4 (CTL‐4) and programmed cell death protein 1 (PD1), have been incorporated into immune‐based therapies for different cancer types (Topalian et al, 2012; Ock et al, 2017). However, lung cancer is usually diagnosed at late stages, and the current standard therapies rarely lead to a positive outcome (Brambilla & Gazdar, 2009) and indeed, the majority of patients remain unresponsive (Tan et al, 2020).
The tumor microenvironment (TME), which consists of various immune cells together with the extracellular matrix, endothelial cells, and fibroblasts, influences cancer biology, including the response to immune checkpoint blockade, and is highly associated with patient survival (Fridman et al, 2012, 2017). TAMs are the most abundant cells in the TME and, based on their immune responsiveness and metabolic profile, can have two distinct activation states (Zheng et al, 2017). TAMs that inhibit tumor growth, migration, and angiogenesis are known as pro‐inflammatory M1‐like TAMs, whereas anti‐inflammatory M2‐like TAMs promote tumor growth, invasion, angiogenesis, and metastasis. These two phenotypes differ in their expression of marker genes and metabolic profiles (Galvan‐Pena & O'Neill, 2014). The M1‐like macrophages, which may be induced by lipopolysaccharide (LPS) and/or interferon‐ɣ (IFNɣ), are characterized by high expression of inflammatory markers, such as interleukin (IL‐) 8, IL‐12, or tumor necrosis factor‐alpha (TNFα), and low expression of IL‐10, macrophage mannose receptor (CD206), or colony‐stimulating factor 1 receptor (CSF‐1R). On the other hand, M2‐like macrophages, such as those induced by IL‐4 in vitro, have low expression of IL‐8, IL‐12, and TNFα, whereas the expression of IL‐10, CD206, and CSF‐1R is highly upregulated. Several metabolomics studies have shown that M1‐like macrophages mainly employ aerobic glycolysis (Tannahill et al, 2013; Mills et al, 2016), whereas M2‐like macrophages present with highly active mitochondria, enhanced fatty acid oxidation, and oxidative phosphorylation (OXPHOS) (Vats et al, 2006; Huang et al, 2014). Thus, understanding how TAMs are activated and regulated within the TME is extremely important for understanding cancer biology and harnessing TAMs for future anti‐cancer strategies.
During recent decades, long noncoding RNAs (lncRNAs) have emerged from being considered “transcriptional noise” to being crucial regulators of various cellular processes. They represent a vast and diverse group of ncRNAs longer than 200 nucleotides and can interact with RNA, DNA, and proteins, which enables them to regulate the transcriptional and post‐transcriptional processes through various mechanisms. LncRNAs were found to participate in diverse cellular processes ranging from normal development to disease contexts, such as cancer progression (Karger et al, 2021). RNA‐based therapies targeting deregulated lncRNAs in diseases such as cancer may represent a promising approach, particularly for patients who are refractory to other treatment options. RNA therapeutics used in vivo include small interfering RNAs (siRNAs), antisense oligonucleotides (ASOs) such as a small hairpin RNA (shRNA), or the locked nucleic acid (LNA) GapmeR, which can trigger RNase H‐mediated RNA degradation, or CRISPR/Cas9‐mediated genome editing (Hu et al, 2020; Lee & Mendell, 2020; Martinez‐Lage et al, 2020; Quemener et al, 2020).
Contrary to most other RNA transcripts, lncRNAs are expressed in a highly tissue‐specific and cell type‐specific manner (Statello et al, 2021). Few lncRNAs that act specifically in macrophages have hitherto been described, and those that are known to affect cell proliferation, differentiation, activation status, metabolic signaling, tissue infiltration, and cell–cell interactions with cancer cells. For example, the noncoding antisense RNA guanine nucleotide‐binding protein, alpha stimulating (GNAS), also known as GNAS‐AS1, is highly upregulated in tumor‐promoting M2‐like macrophages, resulting in enhanced expression of anti‐inflammatory cytokines and tumor progression (Sun & Xu, 2019; Liu et al, 2020). In contrast, growth arrest‐specific 5 (GAS5) is expressed in M1‐like macrophages, thereby promoting a pro‐inflammatory phenotype (Wang et al, 2020).
The lncRNA ADPGK‐AS1 is a tumor‐associated RNA in different cancer types, and it promotes tumor cell proliferation and migration (Song et al, 2018; Luo et al, 2019; Yang et al, 2019; Jiang & Wang, 2020). Additionally, the upregulation of ADPGK‐AS1 in gastric cancer seems to correlate negatively with patient survival (Huang & Yang, 2019). The role of ADPGK‐AS1 in macrophages of the TME, however, remains largely unknown. Here we report that ADPGK‐AS1 regulates mitochondrial metabolism and a phenotypic transition of TAMs in lung cancer, and experimental manipulation of its expression allows switching between tumor‐promoting and tumor‐suppressive TAMs. ADPGK‐AS1 was found to localize to mitochondria, where it could bind mitochondrial ribosomal proteins, ultimately resulting in the alteration of TAM metabolism.
Results
The lncRNA ADPGK‐AS1 is upregulated in TAMs in association with mitochondria
To explore ADPGK‐AS1‐mediated regulation of cellular processes during macrophage activation, human peripheral blood mononuclear cell (PBMC)‐derived macrophages were first stimulated with LPS/IFNɣ (to promote an M1‐like state) or IL‐4 (to promote an M2‐like state). Then, whole‐cell transcriptomics was used to analyze the M1 and M2 phenotypes. RNA sequencing results revealed 18,032 expressed genes, including 4,545 noncoding genes (Fig 1A). Noncoding genes were further subdivided into lncRNAs, of which 407 were differentially expressed genes, and other noncoding RNAs, such as microRNAs (miRNAs) or small nucleolar RNAs (snoRNAs) (Fig 1A). LncRNAs upregulated in M2‐like macrophages (Fig 1B) were validated via quantitative PCR (qPCR) (Fig 1C). Candidates for further investigation were selected through various criteria, such as protein‐coding probability, RNA length, histone marks at the promotor region, or available publications (Fig EV1A). Six candidates, including ADPGK‐AS1, RP4‐644L1.2, RP11‐184M15.1, LINC01800 (AC007880.1), RP1‐80N2.1, and KB‐1991G5.1 were then further analyzed regarding their protein‐coding ability and expression pattern in various cancer cell lines. ADPGK antisense RNA 1 (ADPGK‐AS1) was found to be predominantly expressed as transcript variant 1 (Fig EV1B and C) and showed nonprotein‐coding ability in both in vitro transcription and translation (Fig EV1D and E). In addition, ADPGK‐AS1 was expressed at various levels among different cancer cells and tissues, being abundant in white blood cells and in particular in macrophages and their activated phenotypes (both M1 and M2; Fig EV1F and G). Additionally, ADPGK‐AS1 was more abundant in fluorescence‐activated cell sorting (FACS) sorted macrophages (TAMs) from lung cancer patients compared to macrophages obtained from healthy lung tissue (NMs) (Fig 1D), as well as IL‐4 stimulated PBMCs and THP1‐derived M2‐like macrophages (Fig EV1H and I). These findings suggest that ADPGK‐AS1 plays an important role in TAMs and possibly lung cancer progression. Therefore, we chose ADPGK‐AS1 for further characterization.
Figure 1.

lncRNA ADPGK‐AS1 is upregulated in TAMs in association with an increase in mitochondrial abundance
-
APIE charts of transcripts in M1‐like (stimulated with LPS/IFNɣ) and M2‐like (stimulated with IL‐4) macrophages categorized into coding and noncoding transcripts based on Ensembl gene types and the respective differentially expressed genes (DEGs) between M1 and M2 from RNA sequencing analysis.
-
BHeatmap of top the 40 upregulated lncRNAs in M2‐like macrophages (red) compared with M1‐like macrophages (blue).
-
CValidation of upregulated lncRNAs (ΔCT) in PBMC‐derived M2‐like macrophages (stimulated with IL‐4 for 24 h, M2). n = 6 biological replicates, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 compared with M1‐like macrophages (LPS/IFNɣ‐stimulated for 24 h, M1).
-
DExpression analysis of ADPGK‐AS1 in TAMs sorted from healthy lung tissue (normal macrophages, NMs) or lung tumor tissue (TAMs). n = 7 biological replicates, ***P ≤ 0.001 compared with NMs.
-
ERepresentative images of fluorescence in‐situ hybridization of ADPGK‐AS1 (magenta) and nuclei (blue) in IL‐4 stimulated M2‐like macrophages. β‐actin served as the cytoplasmic control and MALAT1 as the nuclear control. Scale bar = 10 μm.
-
FRNA expression analysis of ADPGK‐AS1, 47S ribosomal RNA (nuclear control), and β‐Tubulin (cytoplasmic control) in IL‐4‐stimulated M2‐like macrophages after subcellular fractionation. n = 3 independent experiments. The data represent the percentage of total detected RNA.
-
GVolcano plot showing the protein‐interactome of ADPGK‐AS1 after RNA pulldown compared to a control RNA. n = 5 biological replicates. Red dots represent ADPGK‐AS1‐interacting MRPs (MRPL15, MRPL35).
-
HCostaining for ADPGK‐AS1 (magenta) and the mitochondrial marker TOM20 (yellow) in IL‐4‐stimulated M2‐like macrophages. Scale bar = 10 μm (left), 5 μm (right).
-
IFluorescence intensity profile across costained macrophages (see white line) for ADPGK‐AS1 (purple) and TOM20 (orange).
-
JSubcellular fractionation of IL‐4‐stimulated M2‐like macrophages and isolation and analysis of ADPGK‐AS1 abundance in the cytoplasmic and mitochondrial fractions The mitochondrial encoded RNA mtATP6 served as the mitochondrial control and β‐tubulin as the cytoplasmic control. n = 3 independent experiments, the data represent the percentage of total detected RNA.
Figure EV1. Selection and characterization of ADPGK‐AS1 .

-
ASchematic overview of criteria for choosing appropriate deregulated lncRNA candidates based on RNA‐seq data for M1‐like and M2‐like macrophages.
-
BExon/intron structure of the three annotated transcript variants of ADPGK‐AS1 (lower panel) with RNA sequencing coverage (sashimi plot) from our sequencing of M2 macrophages of the same genomic location (upper panel), split by read per strand (m2_f=plus strand, m2_r=minus strand).
-
C5′ and 3′ sequences of ADPGK‐AS1 identified by 5′/3’ RACE are compared with the sequence of ADPGK‐AS1 transcript variant 1.
-
DCoding probability and coding potential score using the cDNA‐sequence of ADPGK‐AS1, with β‐actin as an example of a protein‐coding sequence and MALAT1 as an example of a noncoding transcript.
-
ETranscription and subsequent translation of ADPGK‐AS1 to exclude the coding function of proteins > 10 kDa. The asterisk denotes the encoded protein in the positive control (PC) and negative control (NC).
-
FRNA expression analysis of ADPGK‐AS1 in different cell lines and macrophage phenotypes. n = 6. Data was represented as mean ± SEM, **P ≤ 0.01 compared to healthy cell line B2B.
-
GRNA expression of ADPGK‐AS1 in an Illumina Seq dataset (Source: UCSC genome browser, July 2021).
-
H, IRNA expression analysis of ADPGK‐AS1 in PBMC‐derived (left panel) and THP1‐derived macrophages (right panel), either left untreated (M0) or activated to the M1‐like phenotype by stimulation with IFNɣ and LPS or to the M2‐like phenotype by stimulation with IL‐4. n = 4. (PBMC‐derived macrophages) and n = 6 (THP1‐derived macrophages) independent experiments, mean ± SEM, student's unpaired t‐test. *P ≤ 0.05, ***P ≤ 0.001 compared with M0.
-
JRNA‐pulldown validation of ADPGK‐AS1 using RNA isolated from streptavidin beads of ADPGK‐AS1 or control samples. n = 4 independent experiments.
-
KRepresentative immunoblot of MRPL15, MRPL35 localization after subcellular fractionation of macrophages into cytoplasmic and mitochondrial fractions. β‐actin served as the cytoplasmic control, succinate dehydrogenase (SDHA) as the mitochondrial control, ERp72 and protein disulfide isomerase (PDI) as endoplasmic reticulum (ER) controls, and Lamin A/C as the nuclear control. n = 3 independent experiments.
Source data are available online for this figure.
Fluorescence in situ hybridization (FISH) and subcellular fractionation revealed predominant cytoplasmic localization of ADPGK‐AS1 in macrophages (Fig 1E and F). To identify proteins that interact with ADPGK‐AS1, mass spectrometry analyses of proteins from ADPGK‐AS1 pulldown assays were performed (Fig EV1J). The results revealed that ADPGK‐AS1 could bind two mitochondrial ribosomal proteins (MRPs), namely MRPL35 and MRPL15 (Fig 1G; Appendix Table S1). Interestingly, costaining of macrophages for ADPGK‐AS1 and the mitochondrial marker TOM20 revealed overlapping staining, indicating that ADPGK‐AS1 can enter the mitochondria (Fig 1H and I). Subcellular fractionation of M2‐like macrophages followed by RNA expression analysis revealed that ~ 30% of ADPGK‐AS1 resided in the mitochondrial fraction and ~ 70% in the cytoplasm (Figs 1J and EV1K). Taken together, these results suggest that ADPGK‐AS1 plays a role in the mitochondrial aspects of the M2‐like macrophage phenotype.
MRPL35 is abundant in M2‐like TAMs and is associated with poor survival in lung cancer patients
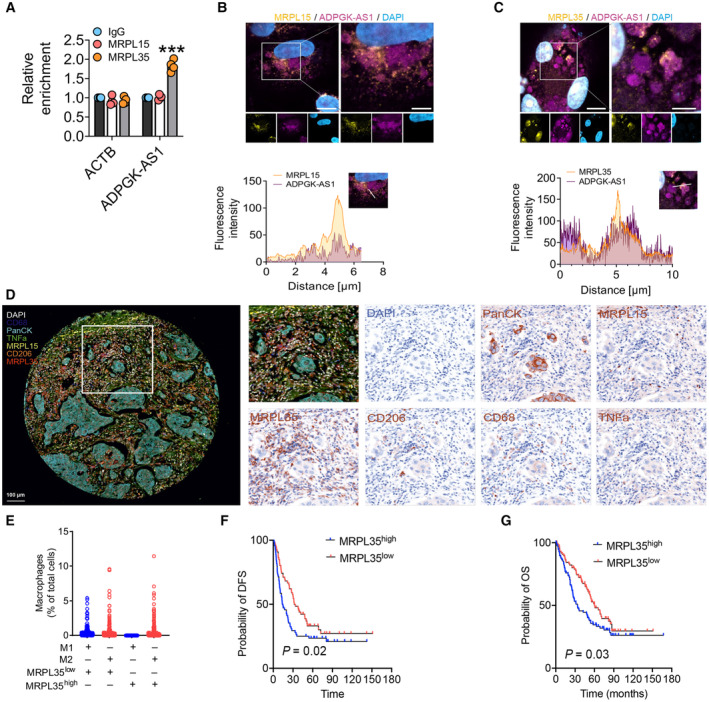
To determine whether MRPs interact with ADPGK‐AS1, we performed RNA immunoprecipitation (RIP) with antibodies targeting MRPL15 and MRPL35, followed by RNA isolation and RT‐qPCR for ADPGK‐AS1. This analysis demonstrated that ADPGK‐AS1 was significantly enriched for MRPL35 (Fig 2A). Costaining of ADPGK‐AS1 and MRPs (MRPL35 and MRPL15; Fig 2B and C) also showed colocalization of ADPGK‐AS1 with MRPL35. We also analyzed MRPL15 and MRPL35 in tissue microarrays (TMAs) of lung cancer patients by multiplex immunofluorescence imaging (Fig 2D). Interestingly, MRPL35 was expressed mainly in M2‐like TAMs (CD68+CD206+TNFα−) and not at all in M1‐like TAMs (CD68+CD206−TNFα+) (Fig 2E). Besides macrophages, a few other cell types of the tumor microenvironment expressed a low level of MRPL35, and MRPL35 was not detected in the tumor‐cell area (PanCK+). Additionally, we assessed the disease‐free survival (DFS) and overall survival (OS) of patients with high or low expression of MRPL35 in M2 TAMs. Patients with high MRPL35 expression had significantly decreased DFS and OS (Fig 2F and G), suggesting that an abundance of MRPL35 in M2‐like TAMs promotes lung cancer progression.
Figure 2. ADPGK‐AS1 interacts with MRPL35, and increased expression of MRPL35 in M2‐like TAMs is negatively associated with the survival of lung cancer patients.
-
AValidation of the ADPGK‐AS1‐interacting protein MRPL35 by RNA immunoprecipitation (RIP) followed by qPCR for ADPGK‐AS1. n = 4 independent experiments; ***P ≤ 0.001 compared with IgG control.
-
B, CCostaining for ADPGK‐AS1 (magenta) and MRPL35 or MRPL15 (yellow) with respective fluorescence intensity across costained macrophages (see white line) for ADPGK‐AS1 and MRPL35 or MRPL15. Scale bar = 10 μm (left), 5 μm (right).
-
DMultiplex immunofluorescence staining of TMAs of lung cancer tissues for CD68 (dark blue), PanCK (cyan), TNFα (green), MRPL15 (yellow), CD206 (orange), MRPL35 (red), and DAPI (white). n = 200 biological replicates.
-
ENumber of M1‐like or M2‐like macrophages based on TMAs data for lung cancer patients with either high (above median) or low (below median) expression of MRPL35.
-
F, GKaplan–Meier curve estimate of disease‐free survival (DFS) or overall survival (OS) of patients with high (above median) or low (below median) expression of MRPL35 in M2‐like macrophages. n = 200 biological replicates.
ADPGK‐AS1 overexpression alters the regulation of mitochondrial metabolism
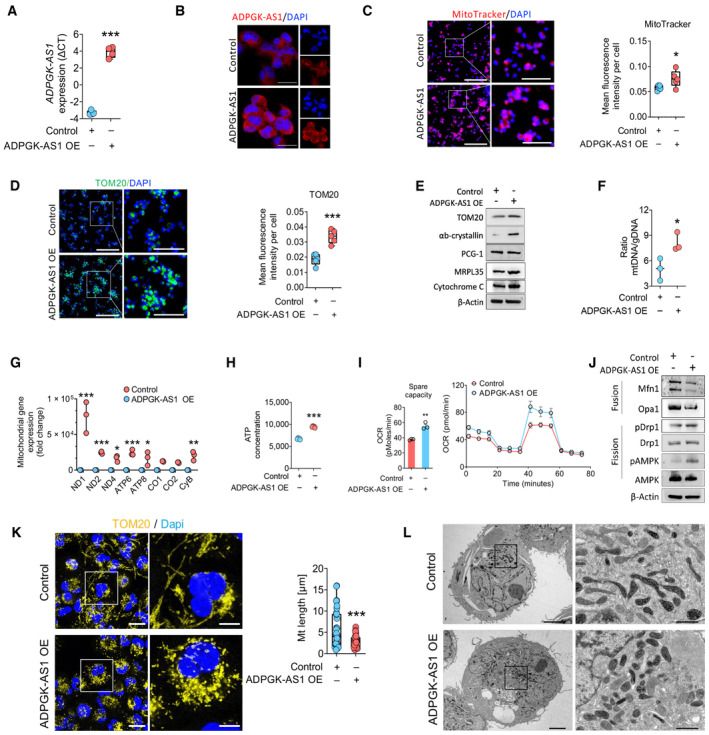
To further assess the function of ADPGK‐AS1 in cellular energy production and mitochondrial metabolism, we generated a stable ADPGK‐AS1 overexpression (OE) line in macrophages, and ADPGK‐AS1 OE was confirmed by qPCR and FISH (Fig 3A and B). Interestingly, ADPGK‐AS1 OE led to an increased number of mitochondria, as observed by staining the macrophages with MitoTracker and TOM20 (Fig 3C and D). Additionally, upregulated expression was observed for the three mitochondria‐associated proteins TOM20, cytochrome C, and αb‐crystalline, as well as for MRPL35 (Fig 3E). Mitochondrial DNA (mtDNA) was then isolated, and its ratio to genomic DNA, as well as a PCR analysis of the mitochondrial genes NADH–ubiquinone oxidoreductase (ND1, ND2, ND4), ATP synthase membrane subunits (ATP6, ATP8), cytochrome C oxidase (CO1, CO2), and cytochrome B (CyB) further confirmed the increased number of mitochondria in ADPGK‐AS1 OE macrophages (Fig 3F and G). In addition, the measurement of ATP production (Fig 3H) and oxygen consumption by the seahorse method revealed enhanced spare capacity and oxygen consumption rate (OCR) in the seahorse assay (Fig 3I). Additionally, mitochondrial translational activity was seemingly elevated in ADPGK‐AS1 OE macrophages (Appendix Fig S1A–C). Furthermore, we observed downregulation of mitochondrial fusion markers (Mfn1 and Opa1) and increased levels of mitochondrial fission marker via upregulation of phosphorylated dynamin‐related protein‐1 (pDrp1 at Ser616), and the phosphorylation of AMPK (pAMPK) was observed (Fig 3J). Notably, the upregulation of the mitochondrial fission markers upon ADPGK‐AS1 OE was reversed upon ADPGK‐AS1 knockdown (Fig EV2A). In addition, confocal microscopy images of TOM20‐stained and transmission electron microscopy images of macrophages confirmed an increase in the numbers of smaller, fragmented mitochondria in ADPGK‐AS1 OE macrophages (Fig 3K and L). Overall, these findings suggest that ADPGK‐AS1 plays a fundamental role in mitochondrial dynamics in macrophages, including metabolic regulation and mitochondrial fragmentation.
Figure 3. ADPGK‐AS1 overexpression alters the regulation of mitochondrial metabolism.
-
AExpression of ADPGK‐AS1 lncRNA (ΔCT) in ADPGK‐AS1 overexpressing (OE) THP1 cells. n = 4 independent experiments. ***P ≤ 0.001 compared with control.
-
BRepresentative FISH images of ADPGK‐AS1 (red) and nuclei (blue) in ADPGK‐AS1 OE THP1 cells. Scale bar = 25 μm.
-
C, DRepresentative images and quantification of mitochondria by MitoTracker (red) and TOM20 (green) in THP1 control and ADPGK‐AS1 OE cells. Nuclei were stained with DAPI (blue). Scale bar = 200 μm (left), 100 μm (right, magnification).
-
ERepresentative immunoblot for mitochondrial protein expression (TOM20, αb‐crystallin, PCG‐1, MRPL15, MRPL35, cytochrome C) in THP1 control and ADPGK‐AS1 OE cells. n = 4 independent experiments.
-
FRatio of mitochondrial DNA (mtDNA) to genomic DNA (gDNA) content in THP1 control and ADPGK‐AS1 OE cells. n = 3 independent experiments. *P ≤ 0.05 compared with control.
-
GGene expression analysis (fold change) of mitochondrially encoded genes (ND1, ND2, ND4, ATP6, ATP8, CO1, CO2, CyB) after isolation of mtDNA from THP1 control and ADPGK‐AS1 OE cells. n = 3 independent experiments. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 compared with control.
-
H, IATP production (n = 5) and full oxygen consumption plot (n = 8) by the seahorse assay in THP1 control and ADPGK‐AS1 OE cells. **P ≤ 0.01, ***P ≤ 0.001 compared with control.
-
JRepresentative immunoblot of proteins involved in fission and fusion processes of mitochondria (Mfn1, Opa1, Drp1, pAMPK, AMPK). n = 4 independent experiments.
-
KRepresentative confocal fluorescence images and quantification of mitochondrial shape with the mitochondrial marker TOM20 (yellow) in THP1 control and ADPGK‐AS1 OE cells. Nuclei were stained with DAPI (blue). n = 35 cells, Scale bar = 10 μm (left), 5 μm (magnification, right).
-
LTransmission electron microscopy images of mitochondrial shapes in THP1 control and ADPGK‐AS1 OE cells. n = 4 cells, Scale bar = 5 μm (left), 1 μm (right, magnification).
Source data are available online for this figure.
Figure EV2. ADPGK‐AS1 enhances the mitochondrial respiratory chain and stimulates the activities of TCA cycle enzymes.

-
ARepresentative immunoblot of mitochondrial fission (DRP1, pDRP1, AMPK, pAMPK) and fusion (Mfn1) marker markers and β‐actin in THP1 control and ADPGK‐AS1 OE macrophages transfected with a siRNA against ADPGK‐AS1 or a negative control. n = 3 independent experiments.
-
BRepresentative immunoblot of pyruvate dehydrogenase (PDH) and β‐actin in THP1 control and ADPGK‐AS1 OE macrophages. n = 3 independent experiments.
-
CLDH enzyme activity in ADPGK‐AS1 OE macrophages compared to control cells. n = 3 independent experiments, mean ± SEM, student's unpaired t‐test, *P ≤ 0.05, compared with control.
-
D–GEnzymatic activity by staining and quantification of succinate dehydrogenase (SDH) and isocitrate dehydrogenase (IDH1) in THP1 control and ADPGK‐AS1 OE macrophages. scale bar = 100 μm n = 4 independent experiments, mean ± SEM, student's unpaired t‐test, **P ≤ 0.01, compared with control.
-
HADPGK‐AS1 expression in PBMC‐derived macrophages polarized to the M1‐like (M1, +LPS/IFNɣ for 24 h), M2‐like (M2, +IL‐4 for 24 h) or M0 (unstimulated) state and then transfected with antisense LNA GapmeRs against ADPGK‐AS1 or a negative control. n = 6 biological replicates, mean ± SEM, one‐way ANOVA, ***P ≤ 0.001, compared with M2 control.
-
ILC–MS/MS analysis of deregulated metabolites of PBMC‐derived macrophages polarized to the M2‐like state (M2, +IL‐4 for 24 h) and then transfected with antisense LNA GapmeRs against ADPGK‐AS1 or a negative control. Results represent the percentage of the ratio of lysate to supernatant. n = 4 biological replicates, mean ± SEM, student's unpaired t‐test, **P ≤ 0.01, ***P ≤ 0.001 compared with the GapmeR control.
Source data are available online for this figure.
ADPGK‐AS1 impacts the macrophage metabolic state of macrophages
We also examined the role of ADPGK‐AS1 in metabolic signaling in macrophages. Metabolome analysis of ADPGK‐AS1 OE macrophages revealed increased levels of metabolites of the TCA cycle, such as malate, α‐ketoglutarate (αKG), and 2‐hydroxyglutarate (2‐HG), in ADPGK‐AS1 OE macrophages, whereas lactate levels were decreased (Fig 4A). These results indicated an increase in the activity of mitochondrial metabolic pathways, which was confirmed by an increase in pyruvate dehydrogenase (PDH) protein levels (Fig EV2B), a decrease in lactate dehydrogenase (LDH) activity (Fig EV2C), and an increase in succinate dehydrogenase (SDH) and isocitrate dehydrogenase 1 (IDH1) activities (Fig EV2D–G). Analysis of the metabolome in ADPGK‐AS1‐deficient M2‐like macrophages (Fig EV2H) revealed effects opposite to those of ADPGK‐AS1 OE (Fig EV2I). Furthermore, ADPGK‐AS1 OE increased mitochondrial membrane potential as determined by the accumulation of JC‐1 aggregates (Fig 4B–D), which was rescued by ADPGK‐AS1 and MRPL35 knockdown. Furthermore, MitoSOX staining revealed decreased ROS levels in ADPGK‐AS1 OE macrophages, which increased again after the knockdown of ADPGK‐AS1 and MRPL35 (Fig 4E). Similarly, in the seahorse assay, the increased OCR in ADPGK‐AS1 OE macrophages was reduced after ADPGK‐AS1 knockdown as well as after MRPL35 knockdown (Fig 4F). These data suggested that ADPGK‐AS1 OE macrophages have highly active mitochondria, increased levels of TCA cycle metabolites, increased activities of TCA cycle enzymes, and reduced LDH activity. In summary, the results indicated that ADPGK‐AS1 functions through MRPL35 to determine the macrophage metabolic state.
Figure 4. ADPGK‐AS1 expression has an impact on macrophage metabolic state.
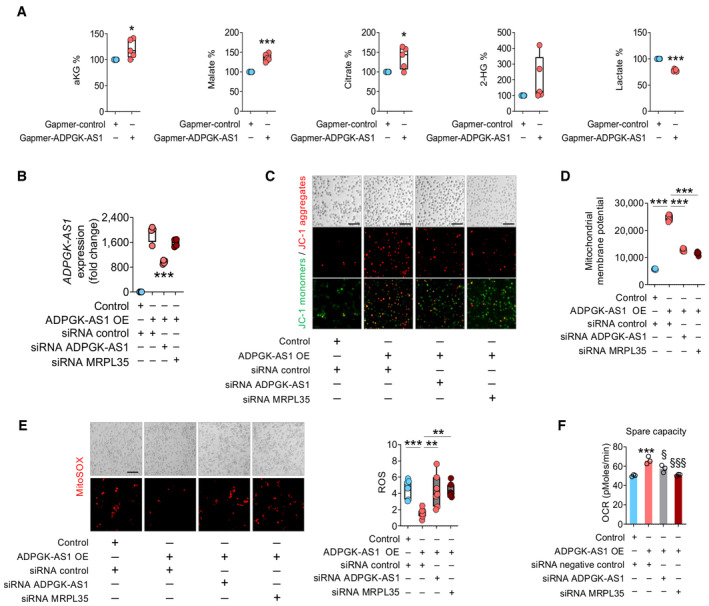
- LC–MS/MS analysis of deregulated metabolites in cell lysates of THP1 control and ADPGK‐AS1 OE macrophages. Results represent the percentage of the ratio of lysate to supernatant. n = 5 biological replicates. *P ≤ 0.05, ***P ≤ 0.001 compared with control.
- Expression analysis of ADPGK‐AS1 and MRPL35 (fold change) in THP1 control and ADPGK‐AS1 OE macrophages transfected with a siRNA against ADPGK‐AS1, MRPL35, or a negative control. n = 4 independent experiments. ***P ≤ 0.001 compared with the ADPGK‐AS1 OE siRNA control.
- Representative images of JC‐1 monomers (green) and aggregates (red) in THP1 control and ADPGK‐AS1 overexpressing macrophages transfected with a siRNA against ADPGK‐AS1, MRPL35, or a negative control. Scale bar = 100 μm.
- Mitochondrial membrane potential measured at 590 nm (JC‐1 aggregates) in THP1 control and ADPGK‐AS1 OE macrophages transfected with a siRNA against ADPGK‐AS1, MRPL35, or a negative control. n = 5 independent experiments. ***P ≤ 0.001 compared with control.
- Representative images (left panel) and quantification (right panel) of ROS accumulation in THP1 control and ADPGK‐AS1 OE cells transfected with a siRNA against ADPGK‐AS1, MRPL35, or a negative control. n = 7. ***P ≤ 0.001 compared to control.
- Spare capacity (oxygen consumption rate) measured with the seahorse assay in THP1 control and ADPGK‐AS1 OE cells transfected with a siRNA against ADPGK‐AS1, MRPL35, or a negative control. n = 3 independent experiments. ***P ≤ 0.001 compared with control, § P ≤ 0.05, §§§ P ≤ 0.001 compared with ADPGK‐AS1 OE siRNA negative control.
Macrophages upregulate ADPGK‐AS1 and adapt their metabolic signaling pathways after cross talking with tumor cells
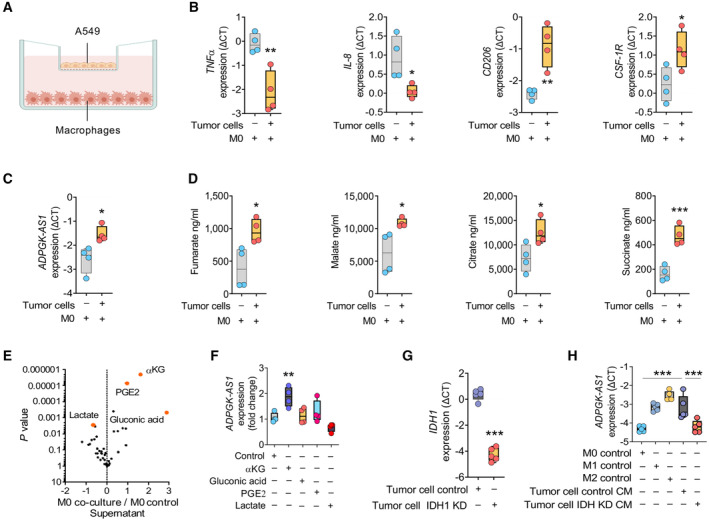
To further characterize the role of ADPGK‐AS1 in macrophages and the associated changes we identified in macrophage metabolism in the context of lung cancer, ADPGK‐AS1 expression in macrophages was examined after indirect coculture with the lung adenocarcinoma cell line A549 (Fig 5A). After coculture, the M1‐like macrophage marker genes (TNFα and IL‐8) were downregulated in the macrophages, whereas the M2‐like macrophage marker genes (CD206 and CSF‐1R) were upregulated. This demonstrated that the cocultured macrophages inherited a tumor‐promoting phenotype (Fig 5B). Cocultured macrophages with A549 cells also increased ADPGK‐AS1 expression in comparison with macrophage controls (Fig 5C), indicating that ADPGK‐AS1 is indeed associated with M2‐like TAMs, with a potential impact on disease development. Metabolome analysis of lysates of cocultured macrophages revealed upregulation of TCA cycle metabolites (Fig 5D), similar to that in macrophages overexpressing ADPGK‐AS1.
Figure 5. Macrophages upregulate ADPGK‐AS1 and adapt their metabolic signaling pathways after crosstalk with tumor cells.
- Schematic of the experimental design of indirect co‐culture of macrophages with A549 cells using a transwell system.
- mRNA expression analysis (ΔCT) of macrophage markers (TNFα, IL‐8, CD206, CSF‐1R) in cocultured PBMC‐derived macrophages with tumor cells or macrophage control (M0). n = 4 biological replicates. *P ≤ 0.05, **P ≤ 0.01 compared with M0.
- Expression of ADPGK‐AS1 lncRNA in macrophages cocultured with tumor cells (A549) or macrophage controls (M0). n = 4 biological replicates. *P ≤ 0.05, compared with M0.
- Mass spectrometry analysis of TCA cycle metabolites (fumarate, malate, citrate, and succinate) in macrophages cocultured with tumor cells (A549) and macrophage control (M0). n = 4 biological replicates. *P ≤ 0.05, **P ≤ 0.01 compared with M0.
- Volcano plot showing deregulated metabolites in the culture medium of a macrophage‐tumor cell (A549)‐cocultures compared with macrophage control medium (M0). n = 6 biological replicates. Red dots represent highly regulated metabolites (αKG, PGE2, gluconic acid, and lactate).
- RNA expression analysis of ADPGK‐AS1 in macrophages treated with metabolites (αKG, gluconic acid, PGE2, lactate) for 24 h. n = 4 independent experiments. **P ≤ 0.01 compared with M0 control.
- Expression of IDH 1 mRNA in A549 cells transfected with siRNA specific for IDH 1 (IDH KD) or a negative control. n = 5 independent experiments. ***P ≤ 0.001 compared with the A549 control.
- RNA expression analysis of ADPGK‐AS1 in THP1 macrophages stimulated to have an M1‐like or M2‐like phenotype, untreated (M0) or treated with conditioned medium (CM) of A549 cells transfected with a siRNA specific for IDH (IDH KD) or negative control. n = 5 biological replicates. ***P ≤ 0.001, compared with tumor cell control CM.
Next, we analyzed the culture medium of the macrophage‐A549 indirect coculture system to determine whether any secreted metabolites could be responsible for the induction of ADPGK‐AS1 expression in the macrophages. The results revealed significantly higher concentrations of αKG, gluconic acid, and prostaglandin E2 in the coculture medium, whereas lactate concentration was lower (Fig 5E). Treatment of macrophages with the identified metabolites showed that only αKG significantly induced ADPGK‐AS1 expression (Fig 5F). To further analyze whether αKG could be involved in signaling upstream of macrophage ADPGK‐AS1 expression, we used A549 cells with a siRNA specific for IDH1, which is the enzyme mainly responsible for αKG production (Fig 5G). Macrophages that were treated with conditioned medium (CM) of IDH1 knockdown A549 cells had reduced induction of ADPGK‐AS1 expression compared with macrophages treated with A549 control‐CM (Fig 5H), thereby supporting a potential role for αKG in ADPGK‐AS1 expression. Overall, these results suggest that ADPGK‐AS1 expression in macrophages is induced as a consequence of crosstalk between macrophages and tumor cells, with tumor‐cell secreted αKG playing a major role. These results confirmed the induction of a tumor‐promoting M2 macrophage phenotype and a related metabolic switch.
ADPGK‐AS1 promotes a switch in macrophage phenotype from M1 like to M2‐like, with an impact on cancer cell biology
Against the background of upregulated ADPGK‐AS1 in cancer cell cocultured macrophages and TAMs from patients with lung cancer, we hypothesized that ADPGK‐AS1‐induced changes in macrophages could impact the biology of adjacent cancer cells. We therefore examined macrophage activation marker genes in ADPGK‐AS1 OE macrophages (Fig 6A and B). M1‐like marker genes, such as TNFα, IL‐8, and CXCL10 were downregulated, whereas M2‐like marker genes, such as CSF‐1R, CD206, IL‐10, ALOX15, TGFβ and IL‐1Ra, were upregulated. CM from ADPGK‐AS1 OE macrophages did not affect tumor‐cell proliferation (Figs 6C and EV3A), but it reduced the capacity of tumor cells to undergo apoptosis and increased tumor‐cell migration compared with tumor cells incubated with macrophage control‐CM (Figs 6D and E, and EV3B and C). These results demonstrated that ADPGK‐AS1 OE in macrophages is sufficient to induce a tumor‐promoting M2‐like phenotype. Interestingly, inhibition of translation in ADPGK‐AS1 OE macrophages led to rescue of the M2‐like phenotype by upregulation of M1‐like marker genes (TNFα and IL‐8) and downregulation of M2‐like marker genes (CSF‐1R and CD206, Appendix Fig S1D), further indicating a connection between ADPGK‐AS1, mitochondrial dynamics, and macrophage polarization. Thus, the downregulation of ADPGK‐AS1 might serve as a novel approach to the development of lung cancer treatments. To examine this possibility, we isolated primary PBMC‐derived macrophages, activated them to generate an M1‐like or M2‐like phenotype (Sarode et al, 2020b), and subsequently silenced ADPGK‐AS1 by LNA GapmeRs (Fig 6F). This approach resulted in the reversal of the effects of ADPGK‐AS1 OE. M1‐like macrophage markers, such as TNFα, IL‐8, CD80, and CCR7, were upregulated in M2‐like ADPGK‐AS1 knockdown (KD) macrophages, whereas expression of CSF‐1R, CD206, IL‐10, and TGFβ were downregulated (Fig 6G and H). CM from M2‐like ADPGK‐AS1 GapmeR macrophages did not affect tumor cell proliferation, but increased tumor cell apoptosis and decreased migration capacity in comparison with tumor cells incubated with control CM (Figs 6I–K, and EV3D–F). To further demonstrate that ADPGK‐AS1 function is a consequence of its mitochondrial localization, we ruled out that it functions via ADP‐dependent glucokinase (ADPGK), that is, its corresponding protein‐coding gene (Fig EV3G). Indeed, the cellular abundance of ADPGK was elevated in macrophages overexpressing ADPGK‐AS1 or treated with GapmeR (Fig EV3H and I). However, a luciferase assay‐based analysis did not reveal any direct regulation of the ADPGK promoter by ADPGK‐AS1 (Fig EV3J). Additionally, the treatment of macrophages with 8‐bromo‐AMP, an established inhibitor of ADPGK (Grudnik et al, 2018), did not have a similar effect on macrophage activation as ADPGK‐AS1 knockdown (Fig EV3K), including impact on tumor‐cell functions (Fig EV3L). Finally, we investigated whether ADPGK‐AS1 OE or knockdown could also affect the M1‐like macrophage phenotype. Although the expression of M1 marker genes increased after ADPGK‐AS1 knockdown in M1‐like macrophages, the expression of M2 macrophage marker genes was not affected; moreover, there was no effect on tumor cell proliferation, apoptosis, or migration compared with the M1 control, suggesting that the observed effect on the modulation of ADPGK‐AS1 expression was specific to M2‐like macrophages (Appendix Figs S2 and S3). These results demonstrated that ADPGK‐AS1 plays a crucial role in macrophage activation and phenotypic state, with a consequent impact on tumor cell apoptosis and migration.
Figure 6. ADPGK‐AS1 regulates macrophage activation state and influences tumor‐cell apoptosis and migration.

-
A, BmRNA expression analysis (ΔCT) of M1 (TNFα, IL‐8, CXCL10) and M2 (CSF‐1R, CD206, IL‐10, ALOX15, TGFβ, IL‐1Ra) macrophage marker genes in THP1 control and ADPGK‐AS1 OE cells. n = 4 independent experiments. *P ≤ 0.05, ***P ≤ 0.001 compared with control.
-
CProliferation of A549 cells treated with conditioned medium (CM) of THP1 control and ADPGK‐AS1 OE macrophages. n = 5 independent experiments.
-
DApoptosis of A549 cells treated with CM from THP1 control and ADPGK‐AS1 OE macrophages. n = 5 independent experiments. ***P ≤ 0.001 compared with control.
-
EMigration (right panel) of A549 cells treated with CM from THP1 control or ADPGK‐AS1 OE macrophages. Representative membrane images are shown in the left panel. n = 5 independent experiments. ***P ≤ 0.001 compared with control. Scale bar = 400 μm.
-
FRNA expression (ΔCT) analysis of ADPGK‐AS1 in PBMC‐derived M1‐like (M1) and M2‐like (M2) macrophages transfected with antisense LNA GapmeRs specific for ADPGK‐AS1 or a negative control. n = 5 biological replicates. *P ≤ 0.05 compared with M2 control.
-
G, HmRNA expression analysis (ΔCT) of M1 (TNFα, IL‐8, CD80, CCR7) and M2 (CSF‐1R, CD206, IL‐10, TGFβ) macrophage marker genes in primary M1‐like (M1) and M2‐like (M2) macrophages transfected with antisense LNA GapmeRs specific for ADPGK‐AS1 or negative control. n = 5 biological replicates, *P ≤ 0.05, **P ≤ 0.01, compared to M2 control.
-
IProliferation of tumor cells (A549) treated with CM of PBMC‐derived M1‐like (M1) and M2‐like (M2) macrophages transfected with antisense LNA GapmeRs specific against ADPGK‐AS1 or a negative control. n = 5.
-
JApoptosis of tumor cells (A549) treated with CM from PBMC‐derived M1‐like (M1) or M2‐like (M2) macrophages transfected with antisense LNA GapmeRs specific for ADPGK‐AS1 or a negative control. n = 5 biological replicates. **P ≤ 0.01, compared with M2 control.
-
KMigration (right panel) of tumor cells (A549) treated with CM from PBMC‐derived M1‐like (M1) or M2‐like (M2) macrophages transfected with antisense LNA GapmeRs specific for ADPGK‐AS1 or a negative control. Representative membrane images are shown in the left panel. n = 5 biological replicates. *P ≤ 0.05, compared with M2 control. Scale bar = 400 μm.
Figure EV3. ADPGK‐AS1 has antitumor activity in vitro, and its function is not regulated via ADPGK.

-
A–CProliferation (n = 5), apoptosis (n = 5), and migration (n = 3) (left panel) with representative membrane images (right panel) of H1650 cells treated with CM from THP1 control or ADPGK‐AS1 OE macrophages. The n values represent independent experiments, mean ± SEM, student's unpaired t‐test, *P ≤ 0.05, ***P ≤ 0.001, compared with control. Scale bar = 400 μm.
-
D–FProliferation (n = 5), apoptosis (n = 5), and migration (n = 3) (left panel) with representative membrane images (right panel) of H1650 cells treated with CM from primary M1‐like (M1) or M2‐like (M2) macrophages transfected with antisense LNA GapmeRs specific for ADPGK‐AS1 or a negative control. The n values represent biological replicates, mean ± SEM, one‐way ANOVA, **P ≤ 0.01, ***P ≤ 0.001, compared with M2 control. Scale bar = 400 μm.
-
GSchematic diagram of the genomic region of the ADPGK and ADPGK‐AS1 genes.
-
HmRNA expression analysis of ADPGK in THP1 control and ADPGK‐AS1 OE macrophages. n = 4 technical replicates, mean ± SEM, student's unpaired t‐test, *P ≤ 0.05, compared with control.
-
IExpression of ADPGK mRNA in primary M1‐like (M1) and M2‐like (M2) macrophages transfected with antisense LNA GapmeRs specific for ADPGK‐AS1 or a negative control. n = 6 technical replicates, mean ± SEM, one‐way ANOVA, *P ≤ 0.05, compared with M2 control.
-
JLuciferase‐based assay for analyzing activation of the ADPGK promotor in THP1 control and ADPGK‐AS1 OE cells. n = 6 technical replicates, mean ± SEM, student's unpaired t‐test.
-
KRNA expression of ADPGK‐AS1, ADPGK, and macrophage activation markers IL‐8 and CD206 in macrophages treated with different concentrations of ADPGK inhibitor 8‐Br‐AMP. n = 6 technical replicates, mean ± SEM, one‐way ANOVA, *P ≤ 0.05, compared with M2 control.
-
LMigration of tumor cells (A549) treated with CM of M1‐like or M2‐like macrophages treated with 8‐Br‐AMP. n = 6 technical replicates, mean ± SEM, one‐way ANOVA, *P ≤ 0.05, compared with M1.
ADPGK‐AS1 knockdown inhibits tumor growth in vivo
To further understand the effect of ADPGK‐AS1 on macrophage biology and tumor growth and progression in vivo, we coinjected immunosuppressed mice (NOD‐scid IL2Rgammanull mice; NSG mice) with A549 human lung cancer cells along with macrophages transfected with a vector for ADPGK‐AS1 knockdown (or negative control vector) and monitored tumor growth (note that ADPGK‐AS1 does not have an annotated ortholog in mice; Appendix Fig S4A and B). Tumor growth was enhanced in control mice as well as in mice injected with M2‐like control macrophages (Fig 7A–C). In contrast, mice coinjected with M1‐like macrophages exhibited reduced tumor growth as well as reduced tumor weight. Co‐injection of mice with M2‐like macrophages and ADPGK‐AS1 knockdown macrophages resulted in reduced tumor growth in mice as well as low‐weight tumors compared with mice in the M2‐like control macrophage group. Consistently, the analysis of the proliferation marker (Ki67) and apoptosis marker (cleaved caspase 3) using immunofluorescence staining confirmed these observations. The results revealed elevated levels of Ki67 in M2‐like control coinjected mice but relatively low levels of Ki67 in M1‐like control and M2‐like ADPGK‐AS1 KD mice (Fig 7C and D). Also, M1‐like control and M2‐like ADPGK‐AS1 knockdown mice showed intense staining for cleaved caspase 3, whereas M2‐like control tumors had relatively low levels of this marker. LDH activity, which is also often used as a clinical marker for malignant transformation in patients with cancer (Forkasiewicz et al, 2020), was substantially higher in tumors from M2‐like control coinjected animals compared with the tumors of the ADPGK‐AS1 knockdown group (Fig 7E). This result highlighted the tumor‐promoting function of M2‐like macrophages. RNA isolation and expression analysis of these tumors revealed decreased expression of ADPGK‐AS1 in the M2‐like ADPGK‐AS1 KD group (Fig 7F). Analysis of the ADPGK‐AS1 interacting partner MRPL35 revealed higher levels of MRPL35 in tumors derived from coinjected A549 cancer cells and M2‐like control macrophages, but reduced MRPL35 levels for the M1 control as well as the M2‐like ADPGK‐AS1 knockdown group (Fig 7G). Overall, these results demonstrated that increased ADPGK‐AS1 expression in TAMs correlated with increased tumor growth and progression in vivo.
Figure 7. ADPGK‐AS1 knockdown inhibits tumor growth in vivo .
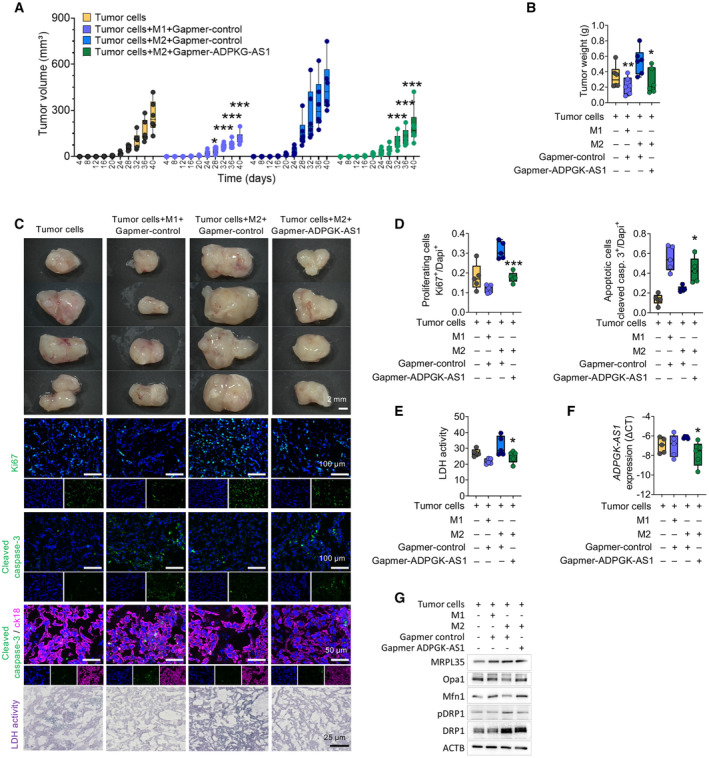
- Time‐dependent tumor growth (size, mm3) analysis within 40 days after subcutaneous coinjection with M1‐like or M2‐like macrophages transfected with antisense LNA GapmeRs specific for ADPGK‐AS1 or a negative control together with A549 tumor cells or A549 cells alone as a control. Each dot represents an animal. n = 6 biological replicates. ***P ≤ 0.001 compared with the group of tumor cells coinjected with M2 control macrophages.
- Tumor weight at 40 days after coinjection with M1‐like (M1) or M2‐like (M2) macrophages transfected with antisense LNA GapmeRs specific for ADPGK‐AS1 or a negative control together with A549 tumor cells or A549 cells alone as a control. Each dot represents a mouse. n = 6 biological replicates. *P ≤ 0.05, compared with tumor cells.
- Representative brightfield images of tumors (scale bar = 2 mm) and fluorescence images (Ki67, cleaved caspase 3, scale bar = 100 μm; cleaved caspase 3 (green) and cytokeratin 18 (magenta), scale bar = 50 μm) and LDH enzyme activity (scale bar = 25 μm) after coinjection with M1‐like (M1) or M2‐like (M2) macrophages transfected with antisense LNA GapmeRs specific for ADPGK‐AS1 or negative control (ctrl) together with A549 tumor cells alone as a control.
- Quantification of Ki67+ cells and cleaved caspase 3+ of coinjected tumors. n = 5 biological replicates. **P ≤ 0.01, ***P ≤ 0.001 compared with M2 control.
- Quantification of LDH enzyme activity in coinjected tumors. n = 5 biological replicates. *P ≤ 0.05 compared with M2 control.
- RNA expression analysis of ADPGK‐AS1 in coinjected tumor tissues. n = 5 biological replicates. *P ≤ 0.05, compared with M2 control.
- Representative immunoblot of the mitochondrial‐related proteins MRPL35, Opa1, Mfn1, pDRP1, and DRP1 about β‐actin in isolated tumor tissue. n = 3 biological replicates.
Source data are available online for this figure.
ADPGK‐AS1 influences human lung tumor progression ex vivo
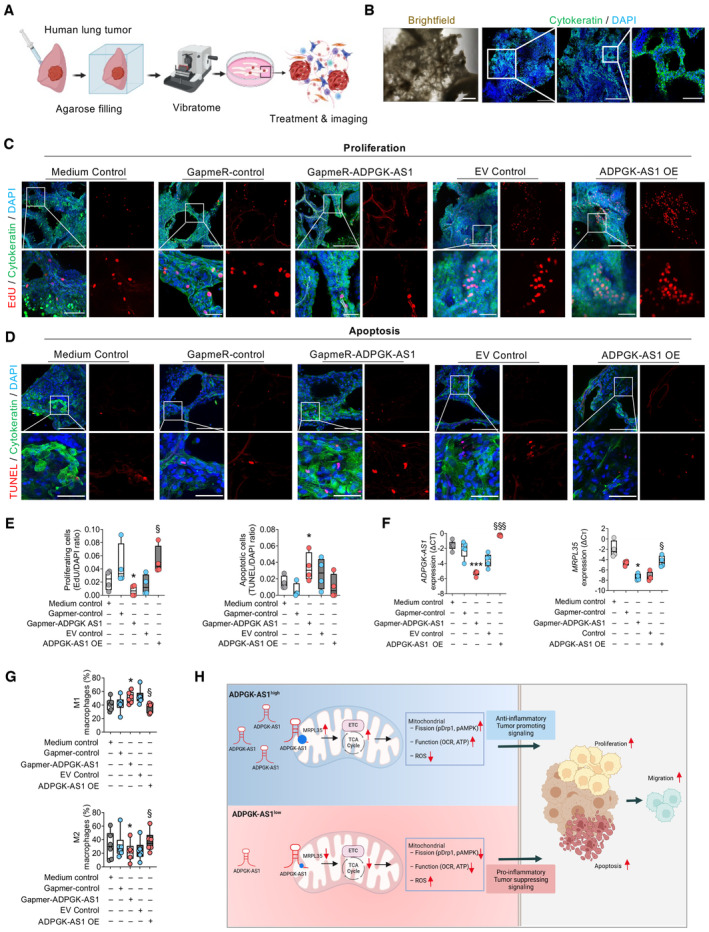
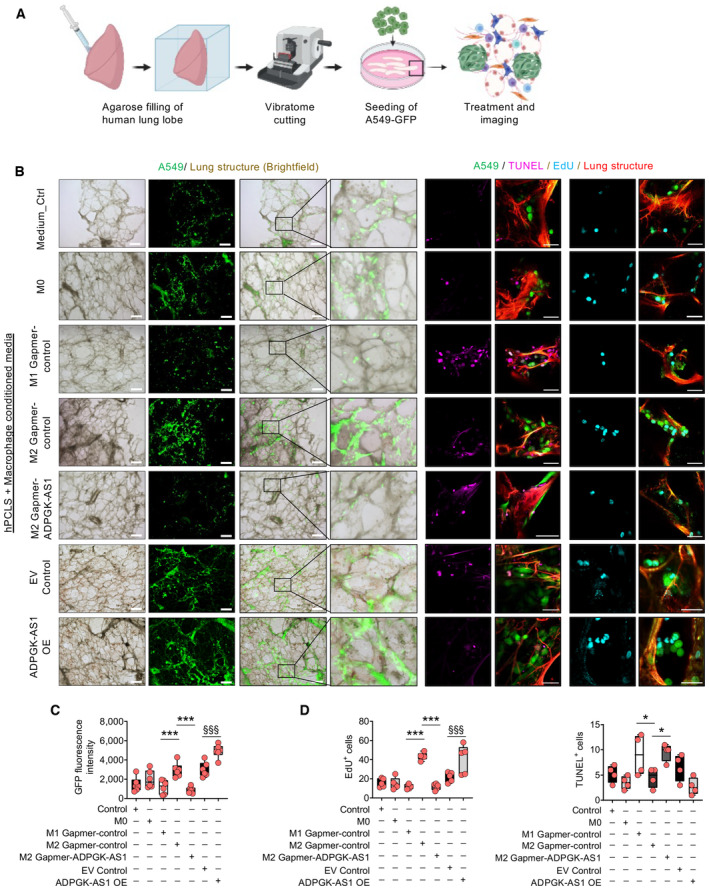
Next, ADPGK‐AS1 was analyzed via an ex vivo method, namely using precision‐cut lung slices (PCLS). We extended this method to human tumor PCLS (tPCLS), which allowed us to culture and directly treat the preserved human lung cancer tissue (Fig 8A and B). For this approach, we performed antisense LNA GapmeR‐mediated KD of ADPGK‐AS1 or added ADPGK‐AS1 OE macrophages to tPCLS. We also stained for markers of proliferation (EdU) and apoptosis (TUNEL) together with cytokeratin and DAPI. The tPCLS were then analyzed with confocal imaging (Fig 8C and D). EdU/DAPI and TUNEL/DAPI ratios revealed reduced cell proliferation and increased apoptosis in ADPGK‐AS1 KD tPCLS compared with cells in the negative control slices. Conversely, the presence of ADPGK‐AS1 OE macrophages led to increased proliferation and decreased apoptosis (Fig 8E). Notably, we were able to detect successful downregulation versus overexpression of ADPGK‐AS1 and MRPL35 on RNA expression in the tPCLS tissue (Fig 8F). Furthermore, multispectral flow cytometry analysis of the immune cell infiltrates of the tPCLS after ADPGK‐AS1 knockdown or overexpression (Appendix Fig S5A) revealed an increase of M1‐like macrophages after ADPGK‐AS1 knockdown and a decrease after ADPGK‐AS1 OE (Fig 8G). In contrast, the numbers of M2‐like macrophages decreased after knockdown, whereas they increased after ADPGK‐AS1 OE. Notably, there was no significant change in the abundance of other immune cell populations such as CD8+, CD4+, Treg cells, B cells, monocytes, or neutrophils (Appendix Fig S5B). In addition, these results could be reproduced with healthy PCLS with the addition of GFP‐labeled A549 cells and culture in macrophage CM, i.e., to mimic lung tumor growth (Fig EV4A–D). Furthermore, we ruled out the possibility that the observed effects were due to the effect of ADPGK‐AS1 on cancer cells by using in vitro proliferation, apoptosis, and migration assays for A549 cells transfected with antisense LNA GapmeRs (Fig EV5A–D). Additionally, we repeated the human PCLS experiment with A549‐GFP cells and again carried out a knockdown of ADPGK‐AS1; however, there was no effect (Fig EV5E). Similarly, no change in functional assays or human PCLS experiments was seen with A549 ADPGK‐AS1 OE cells (Fig EV5F–J). In summary, these results demonstrated that high expression of ADPGK‐AS1 in TAMs could drive the M2‐like macrophage phenotype and contribute to lung tumor growth, whereas ADPGK‐AS1 knockdown could enhance the M1‐like macrophage phenotype and reduce lung tumor progression ex vivo.
Figure 8. ADPGK‐AS1 influences human lung tumor progression ex vivo .
-
ASchematic overview of the preparation of precision‐cut lung slices (PCLS) from a human tumor‐bearing lung lobe.
-
BRepresentative brightfield and fluorescence images of tumor PCLS stained with cytokeratin (green) to visualize the tumor area. Nuclei were stained with DAPI (blue). Scale bar (left to right) = 500 μm, 1 mm, 500, and 100 μm.
-
C, DRepresentative fluorescence images of PCLS with proliferative (EdU+) and apoptotic (TUNEL+) cells (red) and nuclear dye (DAPI+, blue) in the tumor area (cytokeratin+, green). Scale bar = 250 μm (c, upper panel), 150 μm (d, upper panel), and 50 μm (c and d lower panel, magnification).
-
EQuantification of proliferative (EdU+) and apoptotic (TUNEL+) cells (red) about total cell number (DAPI+, blue) in the tumor area (Cytokeratin+, green). n = 4 independent experiments. *P ≤ 0.05, ***P ≤ 0.001 compared with control GapmeR, §§ P ≤ 0.001 compared with empty vector (EV) control.
-
FRNA expression analysis of ADPGK‐AS1 and MRPL35 in tumor PCLS treated with antisense LNA GapmeRs specific for ADPGK‐AS1 or a negative control or seeded with THP1 control or ADPGK‐AS1 OE cells. n = 5 independent experiments. *P ≤ 0.05, ***P ≤ 0.001 compared with control GapmeR, § P ≤ 0.05, §§§ P ≤ 0.001 compared with EV control.
-
GMultispectral flow cytometry analysis of M1‐like and M2‐like macrophage populations in tumor PCLS treated with antisense LNA GapmeRs specific for ADPGK‐AS1 or a negative control or seeded with THP1 control or ADPGK‐AS1 OE cells. n = 6 biological replicates. *P ≤ 0.05 compared with the Gapmer negative control, § P ≤ 0.05 compared with the EV control.
-
HProposed mechanism: M2‐like macrophages express high levels of ADPGK‐AS1 with translocation of the transcript into the mitochondria, leading to an increased abundance of TCA cycle metabolites, increased mitochondrial activity, and induction of mitochondrial fission, thereby enhancing the number of mitochondria in these macrophages. Subsequently, macrophages exhibit increased anti‐inflammatory and tumor‐promoting signaling, leading to enhanced lung tumor cell proliferation and migration. Knockdown of ADPGK‐AS1 in macrophages has the reverse effect, with an increase in pro‐inflammatory signaling and upregulated ROS, leading to lung tumor cell apoptosis. These results suggest the potential for using ADPGK‐AS1 as a therapeutic target in lung cancer via the regulation of the TAM phenotype.
Figure EV4. ADPGK‐AS1 influences lung cancer cell proliferation and apoptosis ex vivo in normal lung PCLS.
-
ASchematic overview and experimental design of precision‐cut lung slices (PCLS) using healthy (nontumor) lung lobes and subsequent seeding with A549‐GFP cells.
-
BRepresentative images of healthy PCLS (red, autofluorescence) with A549‐GFP (green), apoptotic cells (TUNEL+, magenta), and proliferative cells (EdU+, cyan). Scale bar = 500 μm (brightfield images), 50 μm (fluorescence images on the right).
-
C, DQuantification of GFP+ tumor cells (A549, n = 6), EdU+ (proliferative, n = 5), and TUNEL+ (apoptotic, n = 4) cells on healthy PCLS treated with CM (medium control, M0, M1, or M2 macrophages transfected with antisense LNA GapmeRs specific for ADPGK‐AS1) or negative control, THP1 control (EV), and ADPGK‐AS1 overexpressing (OE) macrophages. The n values represent biological replicates, mean ± SEM, one‐way ANOVA, *P ≤ 0.05, ***P ≤ 0.001, compared with M2 GapmeR‐control, §§§ P ≤ 0.001 compared with EV control.
Figure EV5. ADPGK‐AS1 knockdown has no effect on A549 cells.

-
A–DProliferation (n = 5), apoptosis (n = 5), migration (transwell, n = 4), and wound healing (n = 5) assays of tumor cells (A549) transfected with antisense LNA GapmeRs against ADPGK‐AS1 or a negative control. The n values represent independent experiments, mean ± SEM, one‐way ANOVA. ns, nonsignificant.
-
EHuman healthy PCLS seeded with A549‐GFP cells transfected with antisense LNA GapmeRs against ADPGK‐AS1 or a negative control with quantification of A549‐GFP‐positive cells after 48 h. n = 5 independent experiments, mean ± SEM, student's unpaired t‐test. ns, nonsignificant, scale bar = 500 μm.
-
F–IProliferation (n = 6), apoptosis (n = 4), migration (transwell, n = 4), and wound healing (n = 4) assays of tumor cells (A549) control and ADPGK‐AS1 OE cells. The n values represent independent experiments, mean ± SEM, one‐way ANOVA. ns, nonsignificant.
-
JHuman healthy PCLS seeded with A549‐GFP transfected with the ADPGK‐AS1 OE plasmid or an empty vector control with quantification of A549‐GFP positive cells after 48 h. n = 5 independent experiments, mean ± SEM, student's unpaired t‐test. ns, nonsignificant scale bar = 500 μm.
Discussion
In the present study, we provide strong evidence that the lncRNA ADPGK‐AS1 (i) is associated with macrophage mitochondria and interacts with specific mitochondrial ribosomal proteins; (ii) regulates mitochondrial metabolism, energy production, and signaling; and (iii) determines the phenotypic M1‐like versus M2‐like state. We also demonstrate that high levels of lncRNA ADPGK‐AS1 in TAMs promote lung tumor growth, and knockdown of lncRNA ADPGK‐AS1 in TAMs suppresses lung tumor growth as demonstrated in in vitro, ex vivo, and in vivo studies. TAMs have been known to be crucial players during tumor development and progression. Several studies have shown that TAM density correlates with poor prognosis in lung cancer patients (Ruffell & Coussens, 2015; Schmall et al, 2015; Zheng et al, 2020). Macrophages can dynamically adapt their phenotype to either a more pro‐inflammatory and anti‐tumorigenic (M1‐like) or an anti‐inflammatory tumor‐promoting (M2‐like) phenotype. This plasticity is orchestrated by various cytokines, chemokines, and metabolites within the TME (Ley, 2017; Li et al, 2020; Sarode et al, 2020a). Understanding the molecular mechanisms underlying this plasticity is important for the development of new immunotherapeutic strategies. LncRNAs can influence different cellular processes through various signaling pathways, and many lncRNAs, such as Metastasis‐Related Lung Adenocarcinoma Transcript 1 (MALAT1), are known to be commonly deregulated in various cancer types (Ji et al, 2003; Yang et al, 2015; Jin et al, 2017; Wu et al, 2018). In the present study, we analyzed macrophage phenotype‐associated regulation of lncRNA transcripts and were able to identify lncRNA ADPGK‐AS1 as being strongly upregulated in tumor‐promoting M2‐like macrophages. ADPGK‐AS1 has been previously described to be highly expressed in several cancer types and cell lines, such as colon cancer, pancreatic cancer, and breast cancer (Song et al, 2018; Yang et al, 2019; Jiang & Wang, 2020). Mechanistically, ADPGK‐AS1 has been suggested to act as an RNA sponge for microRNAs such as miR‐525 and miR‐3,196 in the cytoplasm, leading to reduced tumor‐cell migration, e.g., via regulating the epithelial‐to‐mesenchymal transition.
Importantly, our data provides the first evidence that ADPGK‐AS1 in TAMs and M2‐like macrophages is enriched in mitochondria and modulates mitochondrial metabolism, dynamics, and phenotype, thereby impacting tumor progression. Several cytoplasmic lncRNAs have been reported to influence mitochondrial metabolism. For example, the cytoplasmic lncRNAs (Cerox1, Tug1, and Uca1) have been proposed to regulate mitochondrial bioenergetics and functions by indirectly influencing mitochondrial‐related genes or miRNAs (Long et al, 2016; Li et al, 2017; Sirey et al, 2019; Wang et al, 2022). In addition, the cytoplasmic lncRNA stem and progenitor enrichment required for hematopoietic differentiation (Spehd) was shown to be required for hematopoiesis and effective differentiation of myeloid progenitors with functional oxidative phosphorylation (Delas et al, 2019). Also, lncRNA Survival Associated Mitochondrial Melanoma Specific Oncogenic Non‐Coding RNA (SAMMSON) was shown to interact with p32, thereby regulating mitochondrial homeostasis and metabolism, especially in cancer cells (Leucci et al, 2016). Few studies, however, have posited intra‐mitochondrial functions for nuclear‐encoded lncRNAs with a hitherto unresolved import mechanism. Zhao et al (2019) discovered that MALAT1 is normally enriched in the nucleus but is also found in mitochondria collected from HepG2 cells. MALAT1‐deficient HepG2 cells produced less ATP and impaired cell invasion capacity, suggesting a role of this lncRNA in mitochondrial metabolism. To date, few nuclear‐encoded mitochondria‐associated lncRNAs have been reported to be upregulated in cancer cell lines (Long et al, 2016; Li et al, 2017; Sirey et al, 2019; Gambi et al, 2022; Wang et al, 2022). In addition, the existence and functional relevance of these lncRNAs in macrophages are completely unknown. To the best of our knowledge, ADPGK‐AS1 is one of the few lncRNAs that has been shown to translocate to mitochondria in macrophages in the context of tumors, as demonstrated by the results from our FISH, RNA pulldown, RNA immunoprecipitation (RIP), subcellular fractionation, and colocalization studies.
Mitochondria contain a small genome that is transcribed and further translated via mitochondrial ribosomal proteins (MRPs) (Taanman, 1999). MRPs were recently shown to be involved in the translation of mitochondrial genes, regulation of oxidative phosphorylation, and induction of apoptosis (Huang et al, 2020).
Interestingly, we found that ADPGK‐AS1 OE in macrophages resulted in increased numbers of mitochondria and hence a greater abundance of mitochondrial DNA as well as an increased abundance of MRPs and components of the electron transport chain (ETC), such as cytochrome C. MRPL35, which we identified as an interaction partner of ADPGK‐AS1, was previously shown to play a key role in coordinating the synthesis and assembly of cytochrome C in yeast, thereby directly regulating the rate of oxidative phosphorylation (Box et al, 2017). Consistent with these findings, elevated MRPL35 and cytochrome C levels in our ADPGK‐AS1 OE macrophages were associated with increased mitochondrial respiration. Deregulation of MRPs can lead to mitochondrial metabolic disorders or cellular dysfunction, and several MRPs are associated with various cancer types. Notably, MRPL35 was reported to be a prognostic marker for colorectal cancer (Sotgia et al, 2017; Zhang et al, 2019). Interestingly, our results show that MRPL35 was found to be highly expressed in the TAMs of lung cancer patients, all of which were M2‐like TAMs. These data, together with the knowledge of upregulated expression of ADPGK‐AS1 in TAMs, suggest an important role for MRPL35 and ADPGK‐AS1 in TAMs. In addition, high MRPL35 levels in TAMs of lung cancer patients were correlated with shorter disease‐free and overall survival, thus suggesting a central role for ADPGK‐AS1 in the plasticity and mitochondrial function of TAMs via MRPL35 with an impact on lung cancer biology.
It has recently become clear that mitochondria are highly mobile organelles that have an impact on the metabolism, division, differentiation, and death of cells. Interestingly, both the mitochondrial fusion markers mitofusin 1 (Mfn1) and optic atrophy 1 (Opa1) (El‐Hattab et al, 2018; Altieri, 2019; Xie et al, 2020) were found to be downregulated in cells with high ADPGK‐AS1 expression. Further, mitochondrial fission‐associated proteins Drp1 and AMPK (Herzig & Shaw, 2018) become activated in cells overexpressing ADPGK‐AS1. Mitochondrial dynamics are very important in quality control, that is, regulation of mitophagy and maintenance of metabolic homeostasis. Although mitochondrial dynamics are known to be connected with their physiological functions, the dynamics still need to be fully understood in the context of the immune response (Wai & Langer, 2016; Gao et al, 2017). Notably, we found that high ADPGK‐AS1 expression upregulated TCA cycle activity and increased mitochondrial membrane potential.
Thus, ADPGK‐AS1 alters macrophage metabolism toward enhanced oxidative phosphorylation (OXPHOS) linked with the anti‐inflammatory tumor‐promoting M2‐like phenotype (Kelly & O'Neill, 2015). Additionally, elevated ROS levels in macrophages are known to be important in host defense against pathogens and are usually upregulated in M1‐like macrophages (Tan et al, 2016; Deng et al, 2019). Thus, the downregulation of ROS noted upon ADPGK‐AS1 overexpression further fits into the switch toward an M2‐like phenotype.
Unexpectedly, the mitochondrial ROS reduction in ADPGK‐AS1 OE macrophages is concomitant with mitochondrial fission or fragmentation, which suggests that ROS production can be independent of mitochondrial morphology under ADPGK‐AS1 overexpressing conditions. In support of this idea, it has been previously shown that increasing mitochondrial mass is not correlated with ROS production under inflammatory conditions (Widdrington et al, 2018). Along the same line, Yu et al (2006) showed that increased ROS production under high glucose conditions did not lead to mitochondrial fragmentation. Considering the functional role of MRPs in the mitochondrial translation‐morphology axis (Cheong et al, 2020), the increase in M1 marker genes along with the downregulation of M2 marker genes after treatment with puromycin in ADPGK‐AS1 OE macrophages suggests that the mitochondrial translational machinery may be involved in the M2‐like macrophage/TAM phenotype as well as in mitochondrial fission driven by ADPGK‐AS1 OE in macrophages. However, we cannot rule out the possibility that ADPGK‐AS1‐associated cytoplasmic translational machinery has an impact on macrophage markers because puromycin can also inhibit translational machinery in the cytoplasmic compartment. Further studies are needed to elucidate the cytoplasmic functions of ADPGK‐AS1 in macrophages and other immune cells.
Notably, the phenotype of increased mitochondrial membrane potential, higher oxygen consumption rate, and decreased ROS in ADPGK‐AS1 OE macrophages could be rescued by either ADPGK‐AS1 or MRPL35 knockdown, further confirming that this function of ADPGK‐AS1 might be executed through MRPL35. In addition, increased ROS, as seen by MRPL35 knockdown, has already been shown in colorectal cancer cells (Sotgia et al, 2017; Zhang et al, 2019). Further, the cause‐and‐effect relationship between mitochondrial morphology, OCR, and ROS seems to be cell‐specific, as it has been shown that embryonic stem cells have a higher basal OCR than trophoblast stem cells, despite their similar mitochondrial morphology (Choi et al, 2020). However, exactly how MRPs regulate the interplay between mitochondrial morphology, energy, and ROS production still needs to be further elucidated, especially in immune cells.
Furthermore, we wanted to know if this regulation definitely plays a role in the context of lung cancer‐associated macrophages. Therefore, we used coculture experiments to facilitate crosstalk between macrophages and lung cancer cells to generate TAMs. These experiments revealed that ADPGK‐AS1 was upregulated in the TAMs; and its expression is correlated with elevated expression of M2‐like macrophage markers and enhanced concentrations of the TCA metabolites fumarate, malate, and citrate. “Teaching” macrophages adjacent to tumor cells to acquire an M2‐like tumor‐promoting phenotype thus reflects the macrophage changes induced by ADPGK‐AS1 overexpression. The central role of ADPGK‐AS1 in driving an M2‐like macrophage/TAM phenotype with an impact on cancer biology is further supported by a series of in vitro, in vivo, and ex vivo experiments. In artificially polarized macrophages, high expression of ADPGK‐AS1 was shown to enhance M2‐like macrophage activation, which was linked with decreased apoptosis and increased migration in tumor‐cell functional assays. Downregulation of ADPGK‐AS1 showed the complete opposite effect, that is, increased expression of inflammatory M1‐like markers while M2‐like marker genes were downregulated. Furthermore, the inhibition of ADPGK‐AS1 in M2‐like macrophages induced tumor‐cell apoptosis and reduced tumor‐cell migration. Due to the lack of a mouse homolog, we used highly immunosuppressed NSG mice to develop a humanized in vivo tumor model for further investigation of the biological significance of ADPGK‐AS1 in the cancer context. Indeed, the knockdown of ADPGK‐AS1 in M2‐like macrophages in these studies significantly attenuated tumor growth by induction of apoptosis and reduction of tumor size. Additionally, the interaction partner MRPL35 was reduced in tumors that developed under the influence of macrophages with an ADPGK‐AS1 knockdown. Finally, we aimed to characterize the therapeutic potential of ADPGK‐AS1 blockade in a human tissue system. To this end, we established the ex vivo method of tumor PCLS, which enabled us to directly culture and treat human lung cancer tissue/cells. Here, inhibition of ADPGK‐AS1 again led to reduced tumor progression with an increase in tumor‐cell apoptosis and decreased proliferation, together with a switch of macrophage markers toward a M1‐like phenotypic state. In addition, by investigating the immune cell profile, we were able to detect a phenotypic switch in macrophages, whereas no other immune cells were affected, suggesting a specific influence of ADPGK‐AS1 on macrophages. Macrophages have emerged as promising targets in cancer immunotherapies because macrophages account for up to 50% of infiltrating immune cells in the TME (Duan & Luo, 2021).
In conclusion, our findings strongly support the notion that ADPGK‐AS1 possesses a crucial regulatory role in determining the TAM phenotype in the lung cancer microenvironment. Therapeutically targeting this lncRNA, for example, through antisense oligonucleotides, may thus represent a novel approach for the treatment of several pathologies associated with macrophage deregulation, in particular lung cancer.
Materials and Methods
Cell lines
Human cell lines A549, BEAS‐2B, H1650, HEK293‐T, and THP1 were purchased from the American Type Culture Collection (ATCC, Manassas, USA) with cell‐culture conditions per ATCC guidelines. A549 and HEK293‐T cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum (FCS) and 1% penicillin/streptomycin (P/S). H1650 cells were cultured in RPMI 1640 medium, supplemented with 10% FCS and 1% P/S. THP1 cells were cultured in RPMI 1640 medium supplemented with 10% FCS, 1% P/S, and 5% HEPES. Media and supplements were purchased from Gibco (Texas, USA). All cell lines were tested for mycoplasma using the LookOut® Mycoplasma PCR Detection kit (Merck, Darmstadt, Germany).
Generation macrophages
Primary human macrophages were generated starting with PBMCs isolated from the buffy‐coat fraction of blood samples obtained from the blood bank of the Universities Giessen and Marburg Lung Center (UGMLC) through Ficoll density gradient centrifugation as previously described (Schmall et al, 2015; Sarode et al, 2020b). PBMCs were seeded on 6‐well plates (Sarstedt, Nümbrecht, Germany) and coverslips (Neuvitro, #H‐18‐1.5‐PDL). The PBMCs were cultured for 1 h in RPMI 1640 medium supplemented with 1% P/S. Afterward, nonadherent cells were removed, and the remaining PBMCs were cultured in macrophage medium (RPMI 1640 medium with 2% human serum, and 1% P/S) for 7 days to allow monocyte‐to‐macrophage differentiation. The density of macrophages was roughly 1 × 105 cells per well in six‐well plates.
Regarding THP1 monocytes, we treated THP1 cells with 10 ng/ml of phorbol 12‐myristate‐12 acetate (PMA) for 24 h and changed to a PMA‐free medium while allowing them to differentiate into macrophages for another 24 h. Further, we activated and polarized macrophages to the M1 phenotype by stimulating M0 macrophages with LPS (100 ng/ml, Sigma) and Interferon γ (IFNɣ) (100 U/ml, Roche, Basel, Switzerland). PBMC‐derived macrophages were activated to the M2 phenotype by stimulating M0 macrophages with IL‐4 (20 ng/ml; VWR, Radnor, USA) for 24 h, while THP1 macrophages were activated to M2 by stimulation of M0 macrophages with IL‐4 and IL‐13 (20 ng/ml) for 24 h.
Coculture
Macrophages were cocultured with A549 cells using a transwell system with a 6‐well layout (Sarstedt, Nümbrecht, Germany). First, 5 × 105 A549 cells were seeded into transwells with a pore size of 0.4 μm and allowed to attach to the membrane. After 20 min, transwell inserts were placed in the 6‐well plates containing macrophages, with subsequent incubation for 24 h. Further, A549 and macrophages were seeded separately as controls. After incubation, the CM was harvested, centrifuged, and stored at −80°C. Additionally, each cell type was harvested separately for RNA or protein isolation.
Transfection with siRNA and antisense LNAGapmeR
Macrophages or A549 cells were transfected with different siRNAs and/or antisense LNA GapmeRs using the HiPerFect Transfection Reagent (Qiagen, Hilden, Germany) in an Opti‐MEM serum‐free medium. Silencer Select siRNA and negative controls were obtained from ThermoFisher Scientific (#4392420, ADPGK‐AS1 ID; n513852; #4390824 IDH ID: s7119, negative control #4390843; #4392421, MRPL35 ID s27942), and antisense LNA GapmeRs (#339511, ADPGK‐AS1 #LG00304486‐DDA (CAGATAGCAGGTGACA), negative control #339516) were obtained from Qiagen (Hilden, Germany). Transfected cells were cultured in the appropriate medium for 24 h.
Stable transfection
Stable overexpression of ADPGK‐AS1 and stable overexpression in THP1 cells was achieved using lentiviral vectors (pLV‐puro‐CMV) containing cDNA for a specific lncRNA that was purchased from VectorBuilder (Chicago, USA). An empty vector was used as a negative control. Lentiviral particles were produced by cotransfection of HEK293T cells with a lentiviral plasmid (psPAX2; Addgene, Watertown, USA) and packaging plasmid (pMD2.G; Addgene, Watertown, USA) using TurboFect transfection reagent (Fermentas, Waltham, USA). The medium containing the viral particles was collected after 48–72 h of transfection. THP1 cells were cultured in 6‐well plates and transduced with lentivirus in the presence of 6 μg/ml polybrene (Merck, Darmstadt, Germany). Thereafter, 24 h after transfection, the medium was replaced with a fresh medium containing puromycin (Gibco, Texas, USA).
Proliferation and apoptosis assay
Tumor cells (A549 or H1650) were seeded at 5,000 cells/well in a 96‐well plate and incubated in the appropriate growth medium for 24 h, followed by serum starvation for 24 h. After starvation, the cells were incubated for another 24 h in with different CM, such as M0 − M2 macrophages, THP1 control, and ADPGK‐AS1 OE macrophages, for another 24 h. The following day, proliferation was assessed using a bromodeoxyuridine (BrdU) cell proliferation assay kit, and apoptosis was assessed using an ELISA‐based cell death detection kit (both from Roche, Basel, Switzerland), according to the manufacturer's protocol.
Migration assay
The migratory capacity of tumor cells (A549 and H1650) was quantified using a Boyden Chamber transwell assay or wound healing assay. For the transwell assay, we seeded 5 × 104 cells/100 μl medium in the upper part of cell culture inserts with a 0.8 μm pore size (BD Biosciences, San Jose, USA), and the inserts were placed in a 24‐well companion plate containing 700 μl per well of CM, such as M0, M1, M2, and THP1 control as well as ADPGK‐AS1 OE macrophages. A549 cells were incubated for 6 h and H1650 cells overnight at 37°C. The transwell inserts were subsequently washed with 1x phosphate‐buffered saline (PBS). Inserts were wiped from the inside to remove nonmigrated cells and placed in methanol for fixation, followed by crystal violet staining for 10 min. After washing with distilled water, each membrane was mounted on a slide with Pertex (Medite GmbH, Burgdorf, Switzerland). Slides were scanned using the NanoZoomer 2.0‐HT digital slide scanner C9600 (Hamamatsu Photonics, Japan). The number of migrated cells was quantified using ImageJ software (National Institutes of Health, Bethesda, USA), as previously described by (Schmall et al, 2015). For the wound healing assay, 30,000 A549 cells per well were seeded in 96‐well image lock plates (Essen Bioscience, Hertfordshire, UK). On the next day, wells were scratched using a wound maker (Sartorius, Göttingen, Germany) and cells were treated with 100 μl/well CM. Wound healing was imaged and analyzed using the IncuCyte® SX5 (Sartorius, Göttingen, Germany) with the respective software.
RNA isolation and cDNA synthesis, for quantitative PCR
Total RNA was extracted from cell pellets by lysing in either in TRIzol or QIAzol (Qiagen, Hilden, Germany), followed by purification using the RNeasy Mini kit (Qiagen, Hilden, Germany). The RNA was then reverse transcribed to yield complementary DNA (cDNA) using the high‐capacity cDNA reverse transcription kit (Applied Biosystems, Waltham, USA) according to the manufacturer's instructions. Also, quantitative PCR (qPCR) was performed using the SYBR Green PCR Master Mix and the StepOne real‐time PCR System (Applied Biosystems, Waltham, USA). Intron‐spanning human primers were designed using sequence information obtained from the National Center for Biotechnology Information (NCBI) database, and purchased from Sigma‐Aldrich (Taufkirchen, Germany). Expression was determined using the ΔCT method. The CT‐values were normalized to the housekeeping gene encoding hypoxanthine‐guanine phosphoribosyl transferase (HPRT1) using the equation ΔCT = CTreference − CTtarget. The primer sequences used in this study are shown in AppendixTable S2.
Western blotting
Cells (THP1 control, ADPGK‐AS1 OE macrophages, and PBMC‐derived macrophages) were lysed in RIPA lysis buffer containing protease and phosphatase inhibitors. Lysates were then cleared via 15 min of centrifugation. Proteins in lysates were separated using 10–15% polyacrylamide gels and transferred to nitrocellulose membranes (Bio‐Rad, Hercules, USA). Each membrane was then blocked with 5% milk in Tris‐buffered saline and Tween 20 (TBS‐T) for 1 h. followed by incubation with a primary antibody overnight at 4°C on a rotating platform. After washing with TBS‐T, each membrane was incubated with a secondary antibody conjugated with horseradish peroxidase. Bound protein‐antibody conjugates were detected using an enhanced chemiluminescence detection system (Biozym, Germany). The details of the used antibodies are shown in AppendixTable S3.
Cloning
An empty vector control for ADPGK‐AS1 OE was produced by digesting the plasmid pLV‐CMV‐ADPGK‐AS1 plasmid (#VB181004‐1001qb, VectorBuilder, Chicago, USA) with NotI and PacI to cut out the ADPGK‐AS1 cDNA sequence. The empty plasmid backbone was incubated with T4 DNA polymerase (NEB, Frankfurt Main, Germany) to generate blunt ends, then phosphorylated using the T4 polynucleotide kinase (PNK, NEB, Frankfurt Main, Germany), and ligated with T4 DNA ligase (NEB, Frankfurt Main, Germany). For cloning of the ADPGK‐AS1 cDNA sequence into vector pcDNA 3.1+, PCR was carried out with primers flanking the ADPGK‐AS1 cDNA sequence containing restriction sites for Acc65I and NotI (forward primer: ATATTAGGTACCAAAAGTACAAAGAAAGGAGGTAGTGTC, Reverse Primer: ATTTGCGGCCGCCTTTTACACTTGTTCATTTTTTA) using plasmid pLV‐CMV‐ADPGK‐AS1 as a template. The purified PCR product (insert), as well as plasmid pCDNA 3.1+ cleaved with Acc65I and NotI (vector), were ligated with a molar ratio of vector to insert of 1:2–1:5 using the T4 DNA ligase (NEB, Frankfurt Main, Germany). For the luciferase assay, the ADPGK promotor region was amplified by PCR using genomic DNA as a template and primers designed to flank the promotor region, containing restriction sites for XhoI and HindIII (forward primer: ATATTACTCGAGTGGCTCAGTCCCCTCTGGGTGCCA, reverse primer: ATTTAAGCTTCTAGCCCGCGCCTCTTCCGGGCTC). The pGL3 basic plasmid (Promega, Madison, USA) was cleaved with XhoI and HindIII (NEB, Frankfurt Main, Germany), and the purified PCR product (insert) was ligated into the restricted pGL3 plasmid (vector) as described. Generated plasmids were transformed in competent E. coli (10‐beta, NEB, Frankfurt Main, Germany) and purified using the plasmid mini or maxi kit (Qiagen, Hilden, Germany).
Luciferase assay
Macrophages were cotransfected in six‐well plates using a firefly luciferase construct containing the ADPGK promotor and a Renilla luciferase construct. After cotransfection, cells were incubated for 24 h. Luciferase activities were quantified with the dual‐luciferase reporter assay system (Promega, Madison, USA), according to the manufacturer's protocol, by using a luminescence plate reader (Tecan infinite M200 PRO, Tecan Group, Männedorf, Switzerland). The ratio of the Firefly luciferase signal to the Renilla luciferase signal was calculated as previously described by (Sarode et al, 2020b).
Immunofluorescence staining
For immunofluorescence staining, we seeded 200,000 THP1‐derived macrophages on cover slips, using frozen tissue sections and fresh precision‐cut lung slices (PCLS). Tissue sections were dried at room temperature (RT), then fixed with acetone or methanol at −20°C for 15 min. Cells were washed with PBS and subsequently fixed in 4% paraformaldehyde (PFA) for 10 min at room temperature, whereas PCLS were fixed with 1% paraformaldehyde (PFA) at 4°C overnight. After fixation, slides, cells, and tissues were permeabilized with 0.3% Triton X‐100/PBS (cells and slides for 10 min, PCLS for 30 min), and then blocked in 5% bovine serum albumin for 1 h. The samples were washed with 1x PBS three times for 5 min each and incubated with primary antibodies at 4°C overnight. Samples were then washed with 1x PBS for 5 min and incubated with a fluorochrome‐conjugated secondary antibody for 1 h at room temperature. The samples were washed again with 1x PBS for 5 min and stained the nuclei with 4′,6‐diamidino‐2‐phenylindole (DAPI) for 15 min at room temperature. Subsequently, each sample was mounted with either DAKO tissue mounting medium (Agilent, CA, USA) or ProLong Glass mounting medium (Invitrogen, Waltham, USA). Staining was visualized via either wide‐field microscopy (Keyence, Osaka, Japan) or confocal microscopy (Zeiss LSM 710, Leica SP8). Details of the antibodies used in this study are shown in AppendixTable S3.
Fluorescence in situ hybridization (FISH)
RNA FISH was used to visualize the subcellular localization of RNA transcripts within a cell. For this purpose, custom LNA detection probes for ADPGK‐AS1 (Qiagen, #339500, LCD0165475‐BKP), MALAT1 (Qiagen, #339500, LCD0161992‐BKP), β‐actin (Exiqon, #300500. 247370‐1), and a nonbinding probe (Qiagen, #339500, LCD0172658‐BKP) were designed and modified at the 5′‐end with the red fluorescence dyeTYE‐665. Macrophages were grown on coverslips and before staining, rinsed with 1x PBS and fixed in 4% PFA for 7 min at room temperature. The fixative was removed by washing three times with 1x PBS for 5 min. Cells were permeabilization was then performed in 0.5% Triton X‐100, supplemented with RNase inhibitor (Applied Biosystems, Waltham, USA), on ice for 10 min. Subsequently, coverslips were washed with 1x PBS for 5 min and rinsed once with 2X SSC buffer (Gibco, Texas, USA). The hybridization mixture containing the fluorescence probe was heated to 80°C for 90 s before use. Hybridization was subsequently performed overnight at 37°C in a humid chamber (Slide Moat, Boekel Scientific, Netherlands). After incubation, cells were washed four times with 2X SSC buffer with 50% Formamide for 20 min at 37°C: Coverslips were then mounted in mounting medium containing DAPI. Slides were stored at 4°C or −20°C for long‐term storage. Fluorescence signals were analyzed, and representative images were taken with a ZEISS Imager Z1 or Leica SP8 confocal microscope.
RNA pulldown
For RNA pulldown, RNAs encoded in pcDNA3.1+ plasmids were linearized with ScaI (single cutter not present in the ADPGK‐AS1 cDNA sequence) before in vitro transcription. This was done using the HiScribe T7 quick, high‐yield RNA synthesis kit (New England Biolabs, Frankfurt, Germany). Transcribed RNA was isolated and biotinylated using a 3′‐end biotin labeling Kit (Pierce, Waltham, USA) according to the manufacturer's instructions. RNA was again isolated, and 3 μg of biotinylated RNA was resuspended in the RNA folding buffer to be heated up to 90°C for 2 min. Samples were then incubated on ice for 3 min and then for 25 min at room temperature to allow the correct folding of the RNA. RNA pulldown was performed by incubating biotinylated RNA in macrophage whole cell lysate in a head‐over‐tail rotator at 4°C overnight. The formed RNA‐protein complexes were crosslinked via exposure to UV light for 90 s. Streptavidin beads were equilibrated with dilution buffer, and then they were added to the samples for further incubation for 1 h at 4°C in a head‐over‐tail rotator for pulldown of the crosslinked RNA‐protein complexes. The beads were washed four times for 10 min each in dilution buffer at 4°C. The final washing step was carried out in dilution buffer without the addition of detergents or proteinase inhibitors, and beads were transferred into a fresh tube. Samples were centrifuged and the supernatants discarded, and beads were frozen at −80°C for subsequent mass spectrometry. For controls, either 1.5x SDS loading buffer was added to the beads to elute proteins for western blot analysis or TRIzol/QIAzol (Qiagen, Hilden, Germany) was added for RNA isolation and expression analysis via qPCR. The mass spectrometry proteomics data were deposited to the ProteomeXchange Consortium via the PRIDE (Perez‐Riverol et al, 2019) partner repository with the dataset identifier PXD030605. (Username: reviewer_pxd030605@ebi.ac.uk, Password: 6RQg2uMc). A list of the top 50 detected proteins in the pulldown is shown in AppendixTable S1.
Subcellular fractionation
Macrophages were grown on 10‐cm plates and harvested by scraping and centrifugation before the experiment. For subcellular fractionation to obtain cytoplasmic and nuclear fractions, cells were washed with cold 1x PBS and centrifuged shortly at high speed. Each cell pellet was resuspended in a weak lysis buffer (10 mM HEPES pH 7.9 with 10 mM KCl, 0.1 mM EDTA, and 0.1 mM EGTA), and incubated on ice for 15 min. After the addition of 10% Nonidet P‐40 (NP‐40), each sample was briefly mixed vigorously and centrifuged for 10 min at 1,699 g. The supernatant containing the cytoplasmic fraction was transferred into a new tube and lysed in TRIzol. The pellet containing the nuclei was washed twice with buffer A and then also lysed in a half volume TRIzol that was used for a cytoplasmatic fraction. For fractionation of the cytoplasm and mitochondrial fraction, the mitochondrial isolation Kit (Abcam, #ab110170) was performed using a 2.0‐ml dounce homogenizer (VWR, Radnor, USA) according to the manufacturer's instructions. For subsequent qPCRs, the same volume of the fractions was used. The calculation of the respective percentage of total gene expression was done by summarizing the calculated relative expression in both compartments. Several control genes were used to verify successful fractionation.
Seahorse analysis
To measure oxygen consumption rate (OCR), a 96‐well Seahorse XF cell culture plate (Agilent, Santa Clara, USA) was seeded with 80,000 THP1 cells per well in RPMI medium containing PMA. After 24 h, the medium was replaced with fresh medium without PMA and again incubated for 24 h. Afterwards, OCR was analyzed using the Agilent Seahorse XF Analyzer.
ATP determination
For the determination of ATP production, the ATP Determination Kit (ThermoFisher Scientific, Waltham, USA) was used according to the manufacturer's instructions. Further, 1 × 106 THP1 cells were seeded into 6‐well plates and differentiated into macrophages through PMA treatment. A total of 90 μl of THP1 macrophage whole cell extract was mixed with a 10 μl reaction solution containing D‐luciferin and firefly luciferase. Subsequently, luminescence was measured using a fluorescence plate reader (Tecan Infinite M200 PRO).
Mitochondrial membrane potential
THP1 cells 1 × 106 per well were seeded into 6‐well plates and differentiated into macrophages before the analysis of mitochondrial membrane potential using the JC‐1 mitochondrial membrane potential assay Kit (Abcam, Cambridge, UK) according to the manufacturer's instructions. Cells were collected, washed in 1x dilution buffer, and resuspended in JC‐1 working solution. After 30 min of incubation at 37°C in the dark, cells were washed with 1x dilution buffer and resuspended in 1x supplemented dilution buffer. A total of 200,000 THP1 cells per well were seeded into a dark 96‐well plate and analyzed with a fluorescent plate reader (Tecan Infinite M200 PRO). Excitation and emission wavelengths were 535 ± 17.5 nm and 530 ± 15 nm, respectively. Afterward, cells were transferred into a transparent well plate that was shortly centrifuged, and JC‐1 staining was analyzed with a fluorescence microscope (Keyence, Osaka, Japan).
MitoSOXROS staining
Reactive oxygen species (ROS) formation in THP1 macrophages was determined using the MitSOX™ red mitochondrial superoxide indicator (Invitrogen, Waltham, USA) according to the manufacturer's instructions. THP1 macrophages were grown on cover slips and treated with a 5 μM MitoSOX reagent working solution for 10 min at 37°C. Subsequently, coverslips were washed three times with warm 1x PBS and imaged using a fluorescence microscope (Keyence, Osaka, Japan).
Enzyme activity assays
Enzyme activity assays and staining of tissue sections were carried out as described by (Miller et al, 2017). Specific buffers were prepared as follows: For the LDH or IDH assays, the buffer was 0.1 Tris‐maleate buffer pH 7.5; for the SDH activity assay, it was 0.1 M Tris‐HCl buffer pH 8.0. In the enzyme‐specific buffers, 10% polyvinyl alcohol was dissolved at 60°C with stirring until the mixture was clear. Enzyme‐specific assay media were prepared (including negative control reactions) with the addition of 200 mM sodium oxamate (LDH inhibition), 100 mM oxaloacetic acid (IDH inhibition), and 250 mM malonic acid (for SDH inhibition). An assay medium containing the enzyme‐specific substrates was applied to cover the whole tissue section or cell slide. Enzyme reactions were carried out at RT for around 15 min or until high staining was visible, and stopped by the removal of the incubation medium and washing with PBS. Tissue sections or cell slides were either mounted directly or used following antibody staining.
Metabolome measurement via LC–MS/MS
Cell samples were lysed in ice‐cold 85% methanol (10 μl/mg) with two freeze‐thaw cycles. Medium samples were directly processed. The homogenate or culture medium was centrifuged (15,000 g, 5 min, 4°C). An equal volume of supernatant was collected, an isotope‐labeled internal standard mix was added, and the samples were evaporated to dryness in a Concentrator Plus (Eppendorf, Wesseling‐Berzdorf, Germany). Samples were reconstituted in 50 μl of water +0.5% formic acid, transferred to autosampler vials, and subsequently analyzed via liquid chromatography coupled to tandem mass spectrometry (LC–MS/MS). Negative ionization ESI‐LC MS/MS was performed on an Agilent 1290 Infinity LC system (Agilent, Waldbronn, Germany) coupled to a QTrap 5500 mass spectrometer (Sciex, Darmstadt, Germany). Ion source parameters were as follows: CUR 30 psi, CAD medium, Ion Spray Voltage ‐ 4,500 V, TEM 400°C, GS1 45 psi, GS2 25 psi. TCA metabolites were identified with authentic standards and/or via retention time, elution order from the column, and 1–2 transitions. For quantification, specific MRM transitions for every compound were normalized to appropriate isotope‐labeled internal standards. Reversed‐phase LC separation was performed by using a Waters Acquity UPLC HSS T3 column (150 mm × 2.1 mm, 1.8 μm). The column oven temperature was 40°C, and the autosampler was 6°C. Injection volume was 2.5 μl. Solvent A consisted of water containing 0.5% formic acid, and solvent B consisted of 100% methanol containing 0.5% formic acid. Compounds were eluted with a flow rate of 0.4 ml/min and with the following 10 min gradient: 2% B for 1.5 min, a 3 min gradient to 100% B, and a cleaning and equilibration step.
Preparation of precision‐cut lung slices (PCLS)
All the tissues were obtained from the Department of Thoracic Surgery at the University Clinics of Giessen and Marburg (UKGM) and provided by the UGMLC Giessen Biobank. The study protocol for tissue donation was approved by the ethics committee (“Ethik Kommission am Fachbereich Humanmedizin der Justus Liebig Universität Giessen”) of the University Hospital Giessen (Giessen, Germany) in accordance with national law and with “Good Clinical Practice/International Conference on Harmonization” guidelines. Written informed consent was obtained from each patient or the patient's next of kin (reference AZ 58/15).
Lobes of the human lung (healthy or tumor‐bearing) were filled through an open bronchus with 2.5% low melting agarose (Sigma, Missouri, USA) in PBS: DMEM/F12 medium 1:1 (v/v) containing 1% penicillin–streptomycin. After inflation of the whole lobe, the entry opening was closed, and the lobe was placed on ice to cool down and solidify the agarose. The filled lung lobe was cut into smaller pieces and embedded in 4% agarose, then cut into 300–400 μm thick sections using a VT1200S vibratome (Leica, Wetzlar, Germany). Tumor PCLS was transfected with antisense LNA GapmeRs as previously described and seeded with THP1 cells. On healthy PCLS, A549‐GFP cells were seeded, and allowed to settle down on the tissue. After 30 min, macrophage CM was added and incubated for 24 h. Tumors and healthy PCLS were fixed in 1% PFA at 4°C overnight before IF staining. The detection of proliferating cells was done using the Click‐IT™ EdU cell proliferation Kit (Invitrogen, Waltham, USA), while the detection of apoptotic cells was done using In‐situ cell death detection Kit TMR rot (Roche, Basel, Switzerland), according to the manufacturer's instructions. Stained PCLS were imaged using the Leica SP8 confocal microscope. For flow cytometry, PCLS were dissociated into a single cell suspension by using the MACS tumor dissociation kit and gentleMACS dissociator (Milteny Biotec, Bergisch Gladbach, Germany).
Animal experiment
All mice were maintained under specific pathogen‐free conditions and handled according to the guidelines of the European Union Commission on Laboratory Animals. Female, 16 weeks old, NOD.Cg‐Prkdc scid Il2rgtm1Wjl/SzJ (NSG) mice were obtained from Jackson Laboratory (Bar Harbor, USA). All the animal experiments were performed at the Max Planck Institute for Heart and Lung Research (Bad Nauheim, Germany) and were approved by local authorities (Regierungspräsidium Darmstadt, Hessen, Germany, Proposal No. B2/1225). NSG mice were subcutaneously coinjected with 1 × 106 A549 cells and 1 × 106 polarized and transfected macrophages in a 200 μl total volume. On day 40, mice were euthanized and the tumors harvested as previously described (Schmall et al, 2015).
Hematoxylin and eosin staining
Frozen sections on slides were thawed for 10 min on RT and then fixed for 10 min in acetone or methanol at −20°C. After additional 10 min at RT, slides were washed for 5 min in distilled water and then stained with hematoxylin (Invitrogen, Waltham, USA). Slides were washed and placed in 96% ethanol for 1 min before staining with an eosin solution (Richard‐Allan Scientific, Kalamazoo, USA) for 4 min. Slides were then rinsed with water and sequentially dehydrated through immersion in 96% ethanol, 99.8% ethanol, and xylol. They were subsequently mounted with coverslips using Entellan (Sigma‐Aldrich, Taufkirchen, Germany) and mounting medium (Merck Millipore, Darmstadt, Germany).
Coding probability and in vitro translation
To estimate the coding potential of a lncRNA, cDNA sequences were obtained using the NCBI database and analyzed using the coding potential calculator on the EMBnet website (https://www.embnet.org/wp/resources/coding‐potential‐calculator/) as well as a protein‐coding assessment tool (https://wlcb.oit.uci.edu/cpat/). Plasmids containing the cDNA of the respective lncRNA were used for in vitro transcription and translation. The Promega TnT quick‐coupled transcription/translation system and transcend nonradioactive translation detection system were used for in vitro transcription and translation of each full‐length lncRNA from the T7 promoter of a pcDNA3.1+ plasmid, per the manufacturer's instructions. Following the completion of the transcription/translation reaction, 1 μl of the reaction product was added to 15 μl of SDS sample buffer, heated for 2 min to 100°C to denature the proteins, and then loaded onto a 4–12% 1‐mm 15‐W NuPage SDS–polyacrylamide gel (Life Technologies). Protein products labeled with the biotinylated transcend tRNA were detected with streptavidin antibodies and western blue reagents per the manufacturer's instructions. The luciferase T7 control DNA plasmid supplied with the TnT quick‐coupled transcription/translation system was used as a positive control.
RNA‐seq analysis
RNA was isolated from M1 and M2 polarized PBMC‐derived macrophages using the miRNeasy micro kit (Qiagen, Hilden, Germany). This was combined with on‐column DNase digestion (DNase Set, Qiagen) to avoid genomic DNA contamination. RNA integrity was analyzed with a BioAnalyzer 2100 (Agilent, Santa Clara, USA) and a LabChip GX Touch 24 (PerkinElmer). We used total RNA (2 μg) for Truseq Stranded mRNA Library library preparation following the low sample protocol, and sequencing was performed with the NextSeq500 instrument (Illumina) using v2 chemistry with a 2 × 75‐bp single‐end setup.
The resulting raw reads were assessed for quality, adapter content, and duplication rates with FastQC (Andrews S. 2010, FastQC: a quality control tool for high‐throughput sequence data, available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Trimmomatic version 0.39 was employed to trim reads having a quality value less than Q15 with a window of 5 nucleotides (Bolger et al, Trimmomatic: a flexible trimmer for Illumina sequence data). Only reads longer than 15 nucleotides were cleared for further analyses. Trimmed and filtered reads were aligned versus the Ensembl human genome version hg38 (GRCh38) using STAR 2.7.9a with the parameters “‐‐outFilterMismatchNoverLmax 0.1 ‐‐outFilterMatchNmin 20 ‐‐alignIntronMax 200000” (Dobin et al, STAR: ultrafast universal RNA‐seq aligner). The number of reads aligning to genes was counted with the featureCounts 2.0.2 tool from the Subread package (Liao et al, featureCounts: an efficient general‐purpose program for assigning sequence reads to genomic features). Only reads mapping at least partially inside exons were admitted and aggregated per gene. Reads overlapping multiple genes or aligning to multiple regions were excluded. A combined raw count matrix was produced and batch corrected per donor (batch 1: *_1, batch 2: *_2, batch 3: *_3) using CountClust (Dey et al, 2017), Visualizing the structure of RNA‐seq expression data using the grade of membership models, PLoS Genetics). The batch‐corrected matrix was used for differential expression analysis using DESeq2 version 1.30.1 (Love et al, Moderated estimation of fold change and dispersion for RNA‐Seq data with DESeq2). Differentially expressed genes (DEG) of M2/M1 were analyzed at a log2 fold change of > 0.59 or < −0.59 and a Benjamini‐Hochberg adjusted P‐value of < 0.05. The Ensemble annotation was enriched with UniProt data (release 08.03.2018) based on Ensembl gene identifiers (Activities at the Universal Protein Resource (UniProt).
5′/3′ rapid amplification of cDNA ends (RACE)
The SMARTer® RACE 5′/3′ kit (Takara Bio, Kusatsu, Japan) was used to identify the transcript variant and 5′ and 3′ end of the ADPGK‐AS1 cDNA according to the manufacturer's protocol. First, the total RNA of PBMC‐derived M2‐like macrophages was used to synthesize cDNA for 5′‐ or 3′‐RACE with the specific provided 5′‐ or 3′‐Primers. Next, the cDNA was diluted and used as a template for the RACE PCR using universal primers as well as primers specific for the ADPGK‐AS1 sequence containing a sequence for the in‐fusion cloning into the pRACE vector (5′‐Primer: GATTACGCCAAGCTTGGGCACACAGATAGCAGGTGACAGA, 3′‐Primer: GATTACGCCAAGCTTCAGGGTTCTGGATGAAGGGAGGGGT). PCR products were loaded on an agarose gel, and fitting bands were cut and purified by NucleoSpin Gel and the PCR Clean‐Up Kit. Purified PCR products were used for in‐fusion cloning into a linearized pRACE vector, which was subsequently transformed into the provided Stellar Competent bacteria. Transformed cells were plated on an LB plate containing 100 μg/ml ampicillin. On the next day, individual clones were picked from the plate, and plasmids were isolated with a GenElute™ Plasmid Miniprep‐kit (Merck, Darmstadt, Germany). To determine the presence of the RACE insert, plasmids were tested by restriction digestion with EcoRI and HindIII (NEB, Massachusetts, USA) and subsequently sent for sanger sequencing (Eurofins Genomics, Ebersberg, Germany).
RNA immunoprecipitation (RIP)
PBMC‐derived macrophages were grown on 6‐cm dishes and polarized to an M2‐like phenotype by stimulation with IL‐4 (20 ng/ml) for 24 h. Macrophages were washed once with 1× cold PBS and crosslinked with 1% formaldehyde for 5 min. The reaction was quenched with glycine (125 mM) on ice for 5 min. Cells were scraped in 1× PBS and collected by centrifugation at 1,000 rpm at 4°C for 5 min. Cells were lysed in lysis buffer (50 mM Tris‐HCl pH 7.4, 100 mM NaCl, 1% NP‐40, 0.1% SDS, 0.5% Sodium deoxycholate, proteinase inhibitors), and 10% was saved as input. Four micrograms of anti‐MRPL35, anti‐MRPL15, or anti‐IgG antibodies were coupled with 50 μl of Protein A Dynabeads (ThermoFisher Scientific, Waltham, USA) in lysis buffer and washed once with high salt buffer (1 M NaCl) and twice with lysis buffer. The antibody‐coupled beads were added to the cell lysate and rotated for 1 h at 4°C. Beads were collected by centrifugation (1,000 rpm, 2 min) and washed three times with high salt buffer (4°C, 10 min, rotator) and then washed twice with buffer PNK (350 mM Tris‐HCl pH 6.5, 50 mM MgCl2, 5 mM DTT). For elution, all wash buffer was removed, and RNA isolation was performed with QIAzol (Qiagen, Hilden, Germany) as described under RNA isolation.
Multiplex immunofluorescence staining
Tissue microarrays (TMAs) were constructed from paraffin blocks of selected lung specimens. The Ethical Committee of the University Hospital Munich, Lungbiobank Heidelberg, and the Biobank Platform of the German Center for Lung Research in Germany approved the collection and analysis of all samples (AZ 58/15) in accordance with the national laws and the Good Clinical Practice/International Conference on Harmonization guidelines. Informed consent was obtained from all patients (Weigert et al, 2022). TMA lung cancer tissue sections were stained with Opal 7‐Color Automation IHC kits (Akoya Bioscience) in the BOND‐RX Multiplex IHC Stainer (Leica). Each section was put through 6 sequential rounds of staining, which included blocking in 5% BSA followed by incubation with primary antibodies as follows, MRP‐L35 (Invitrogen, PA5‐38964, 1:100), MRP‐L15 (G Biosciences, ITT2848, 1:200), TNFα (Abcam, ab6671, 1:500), pCK (DAKO, ab7753, 1:200), CD206 (Cell Signaling, 91992, 1:250), CD68 (DAKO, M087601‐2, 1:100), corresponding secondary HRP‐conjugated antibodies, and Opal fluorophores as described (Strack et al, 2020; Zheng et al, 2020). Nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI) contained in the Opal 7‐Color Automation IHC Kits, and slides were mounted with Fluoromount‐G (Southern Biotech). Imaging was performed with the PhenoImager HT (Akoya Bioscience), and images were analyzed using the phenotyping application of the inForm software V2.5 (Akoya Bioscience).
Multispectral flow cytometry
Single‐cell suspensions of tPCLS were generated as described above. Single‐cell suspensions were blocked with FcR blocking reagent (Miltenyi Biotec) in 0.5% PBS‐BSA for 20 min, stained with fluorochrome‐conjugated antibodies, and analyzed on a FACSSymphony A5SE flow cytometer (BD Biosciences). Live single cells were identified by FSC and SSC characteristics. The data were analyzed using FlowJo V10 (TreeStar). All antibodies and secondary reagents were titrated to determine optimal concentrations. Comp‐Beads (BD) were used for single‐color compensation to create multicolor compensation matrices. For gating, fluorescence minus one control was used. The instrument calibration was controlled daily using Cytometer Setup and Tracking Beads (BD Biosciences). The following antibodies were used: CD3‐BUV805 (#612896, BD Biosciences), CD4‐BB630 (#562316, BD Biosciences), CD8‐BV650 (#743067, BD Biosciences), CD14‐PerCP‐Cy5.5 (#561116, BD Biosciences), CD15‐BUV805 (#742057, BD Biosciences), CD16‐BV650 (#563692, BD Biosciences), CD19‐APC‐H7 (#560252, BD Biosciences), CD25‐PE‐Cy7 (#557741, BD Biosciences), CD33‐BV510 (#563257, BD Biosciences), CD45‐AF700 (#368514, BD Biosciences), CD80‐BV711 (#740801, BD Biosciences), CD206‐PE/Cy7 (#321124, BioLegend), CD326‐FITC (#324203, BioLegend), HLA‐DR‐APC/Fire750 (#307658, BioLegend), MerTK‐BV421 (#367603, BioLegend).
Analysis of mitochondrial protein synthesis
THP1 control or ADPGK‐AS1 OE macrophages (1 × 106) were grown in 6‐well plates and treated with O‐propargyl‐puromycin (OP‐puro, Jena Bioscience, Jena, Germany) as previously described (D'Andrea et al, 2016; Delaunay et al, 2022). Cells were incubated with 50 μM OP‐Puro for 1 h and 200 nM MitoTracker (red, ThermoFisher Scientific, Waltham, USA) was added 30 min before cell collection. Cells were rinsed with 1x PBS, scraped, and collected by centrifugation (1,000 rpm, 5 min). Mitochondrial isolation was carried out with the mitochondrial isolation kit (Abcam, #ab110170) according to the manufacturers' instructions. The extracted organelles were fixed with 1% PFA for 15 min on ice in the dark. After fixation, samples were washed in PBS and permeabilized in PBS supplemented with 3% FCS and 0.1% saponin (Sigma‐Aldrich) for 5 min at RT. To conjugate OP‐puro to a fluorochrome, an azide‐alkyne cycloaddition was performed for 30 min at room temperature in the dark. For this, the Click‐iT Cell Reaction Buffer Kit (ThermoFisher Scientific) and 5 μM of Alexa Fluor 488‐Azide (ThermoFisher Scientific) were used. To remove Excel reagents and reduce the background signal, the cells were washed twice with PBS supplemented with 3% FCS and 0.1% saponin. Finally, all samples were mounted on a slide, and the fluorescence signal was imaged with a confocal microscope (Leica SP8).
Transmission electron microscopy
The TEM preparation was performed as described in (Madela et al, 2014). Sixty nanometer ultra‐thin sections were obtained using a Leica UC7 and collected on copper grids. After post‐staining with uranyl acetate and lead citrate, the ultra‐thin sections were examined with a JEM‐1400 Plus transmission electron microscope (Jeol, Japan), operated at an accelerating voltage of 120 kV. Digital images were recorded with an EM‐14800Ruby Digital CCD camera unit.
Statistical analysis
All data were analyzed using Prism 6.0 and 9.0 (GraphPad Software), and statistical comparisons between the two groups were made with the unpaired sample student's t‐test. Comparisons of more than two groups utilized the one‐way analysis of variance followed by Tukey's post hoc test. Data were represented as mean ± SEM.
Author contributions
Annika Karger: Data curation; formal analysis; validation; investigation; visualization; methodology; writing – original draft; writing – review and editing. Siavash Mansouri: Data curation; formal analysis; methodology; writing – review and editing. Matthias, S. Leisegang: Resources; data curation; software; formal analysis; investigation; methodology; writing – review and editing. Andreas Weigert: Resources; data curation; formal analysis; validation; investigation; visualization; methodology; writing – review and editing. Stefan Günther: Resources; software; formal analysis; methodology; writing – review and editing. Carsten Kuenne: Software; formal analysis; methodology; writing – review and editing. Ilka Wittig: Software; formal analysis; investigation; methodology; writing – review and editing. Sven Zukunft: Data curation; methodology; writing – review and editing. Stephan Klatt: Data curation; methodology; writing – review and editing. Blerina Aliraj: Data curation; methodology; writing – review and editing. Laura, V Klotz: Resources; formal analysis; methodology; writing – review and editing. Hauke Winter: Formal analysis; methodology; project administration; writing – review and editing. Poornima Mahavadi: Resources; formal analysis; methodology; writing – review and editing. Ingrid Fleming: Resources; methodology; writing – review and editing. Clemens Ruppert: Resources; methodology; writing – review and editing. Biruta Witte: Resources; methodology; writing – review and editing. Ibrahim Alkoudmani: Resources; methodology; writing – review and editing. Stefan Gattenlöhner: Resources; methodology; writing – review and editing. Friedrich Grimminger: Resources; funding acquisition; methodology; writing – review and editing. Werner Seeger: Conceptualization; resources; supervision; funding acquisition; writing – review and editing. Soni, Savai Pullamsetti: Conceptualization; resources; formal analysis; supervision; funding acquisition; methodology; writing – original draft; project administration; writing – review and editing. Rajkumar Savai: Conceptualization; resources; data curation; formal analysis; supervision; funding acquisition; investigation; methodology; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
PDF+
Source Data for Figure 3
Source Data for Figure 7
Acknowledgments
The authors thank Yanina Knepper, Jeanette Knepper, Dr. Rajender Nandigama, Dr. Sascha Seidel, Margarete Mijatovic, Dr. Sebastian Konzok, and the Imaging Platform of the MPI for Heart and Lung Research for excellent technical assistance. Work in the laboratories of RS was supported by the Max Planck Society, the Institute for Lung Health (ILH), and the German Center for Lung Research (DZL). BMBF (KMU‐innovative‐22: miRTumorProst): 031B0768C, Deutsche Forschungsgemeinschaft (DFG, German Research Foundation): SA 1923/7‐1, SFB1213 (Project A10N*); the Excellence Cluster Cardio‐Pulmonary Institute (EXC 2026: Cardio Pulmonary Institute (CPI), project 390649896), and the State of Hesse (LOEWE iCANx, Project A6 and Area C). Work in the laboratories of SSP was supported by the Max Planck Society, the German Center for Lung Research (DZL). European Research Council (ERC) Consolidator Grant (#866051 to SSP), Deutsche Forschungsgemeinschaft (DFG, German Research Foundation): SFB1213 (Project A01 and A05); the Excellence Cluster Cardio‐Pulmonary Institute (EXC 2026: Cardio Pulmonary Institute (CPI), project 390649896); and the State of Hesse (LOEWE iCANx, Project B4, B5, and Area C). Work in the laboratories of MSL and IW was supported by the DFG Transregio TRR267 (Projekt‐ID 403584255 – TRR 267, TP A04, TP Z02). Open Access funding enabled and organized by Projekt DEAL. Open Access funding enabled and organized by Projekt DEAL.
The EMBO Journal (2023) 42: e111620
Data availability
The RNA‐seq data were deposited in the Gene Expression Omnibus archive (accession number GSE195440: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE195440). The mass spectrometry proteomics data were deposited to the ProteomeXchange Consortium via the PRIDE (Perez‐Riverol et al, 2019) partner repository with the dataset identifier PXD030605 (http://www.ebi.ac.uk/pride/archive/projects/PXD030605).
References
- Altieri DC (2019) Mitochondrial dynamics and metastasis. Cell Mol Life Sci 76: 827–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Box JM, Kaur J, Stuart RA (2017) MrpL35, a mitospecific component of mitoribosomes, plays a key role in cytochrome c oxidase assembly. Mol Biol Cell 28: 3489–3499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla E, Gazdar A (2009) Pathogenesis of lung cancer signalling pathways: roadmap for therapies. Eur Respir J 33: 1485–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong A, Archambault D, Degani R, Iverson E, Tremblay KD, Mager J (2020) Nuclear‐encoded mitochondrial ribosomal proteins are required to initiate gastrulation. Development 147: dev188714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Seo BJ, La H, Yoon SH, Hong YJ, Lee JH, Chung HM, Hong K, Do JT (2020) Comparative analysis of the mitochondrial morphology, energy metabolism, and gene expression signatures in three types of blastocyst‐derived stem cells. Redox Biol 30: 101437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Andrea A, Gritti I, Nicoli P, Giorgio M, Doni M, Conti A, Bianchi V, Casoli L, Sabo A, Mironov A et al (2016) The mitochondrial translation machinery as a therapeutic target in Myc‐driven lymphomas. Oncotarget 7: 72415–72430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delas MJ, Jackson BT, Kovacevic T, Vangelisti S, Munera Maravilla E, Wild SA, Stork EM, Erard N, Knott SRV, Hannon GJ (2019) lncRNA spehd regulates hematopoietic stem and progenitor cells and is required for multilineage differentiation. Cell Rep 27: 719–729.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaunay S, Pascual G, Feng B, Klann K, Behm M, Hotz‐Wagenblatt A, Richter K, Zaoui K, Herpel E, Munch C et al (2022) Mitochondrial RNA modifications shape metabolic plasticity in metastasis. Nature 607: 593–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, Shi F, Zhou Z, Sun F, Sun MH, Sun Q, Chen L, Li D, Jiang CY, Zhao RZ et al (2019) M1 macrophage mediated increased reactive oxygen species (ROS) influence wound healing via the MAPK signaling in vitro and in vivo . Toxicol Appl Pharmacol 366: 83–95 [DOI] [PubMed] [Google Scholar]
- Dey KK, Hsiao CJ, Stephens M (2017) Visualizing the structure of RNA-seq expression data using grade of membership models. PLoS Genet 13: e1006599. 10.1371/journal.pgen.1006599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Z, Luo Y (2021) Targeting macrophages in cancer immunotherapy. Signal Transduct Target Ther 6: 127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Hattab AW, Suleiman J, Almannai M, Scaglia F (2018) Mitochondrial dynamics: biological roles, molecular machinery, and related diseases. Mol Genet Metab 125: 315–321 [DOI] [PubMed] [Google Scholar]
- Forkasiewicz A, Dorociak M, Stach K, Szelachowski P, Tabola R, Augoff K (2020) The usefulness of lactate dehydrogenase measurements in current oncological practice. Cell Mol Biol Lett 25: 35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman WH, Pages F, Sautes‐Fridman C, Galon J (2012) The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 12: 298–306 [DOI] [PubMed] [Google Scholar]
- Fridman WH, Zitvogel L, Sautes‐Fridman C, Kroemer G (2017) The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol 14: 717–734 [DOI] [PubMed] [Google Scholar]
- Galvan‐Pena S, O'Neill LA (2014) Metabolic reprograming in macrophage polarization. Front Immunol 5: 420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambi G, Mengus G, Davidson G, Demesmaeker E, Cuomo A, Bonaldi T, Katopodi V, Malouf GG, Leucci E, Davidson I (2022) The LncRNA LENOX interacts with RAP2C to regulate metabolism and promote resistance to MAPK inhibition in melanoma. Cancer Res 82: 4555–4570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Li Y, Wang F, Huang T, Fan K, Zhang Y, Zhong J, Cao Q, Chao T, Jia J et al (2017) Mitochondrial dynamics controls anti‐tumour innate immunity by regulating CHIP‐IRF1 axis stability. Nat Commun 8: 1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grudnik P, Kaminski MM, Rembacz KP, Kuska K, Madej M, Potempa J, Dawidowski M, Dubin G (2018) Structural basis for ADP‐dependent glucokinase inhibition by 8‐bromo‐substituted adenosine nucleotide. J Biol Chem 293: 11088–11099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Shaw RJ (2018) AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 19: 121–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Zhong L, Weng Y, Peng L, Huang Y, Zhao Y, Liang XJ (2020) Therapeutic siRNA: state of the art. Signal Transduct Target Ther 5: 101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Yang H (2019) Upregulation of the long noncoding RNA ADPGK‐AS1 promotes carcinogenesis and predicts poor prognosis in gastric cancer. Biochem Biophys Res Commun 513: 127–134 [DOI] [PubMed] [Google Scholar]
- Huang SC, Everts B, Ivanova Y, O'Sullivan D, Nascimento M, Smith AM, Beatty W, Love‐Gregory L, Lam WY, O'Neill CM et al (2014) Cell‐intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol 15: 846–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Li H, Zhang H (2020) Abnormal expression of mitochondrial ribosomal proteins and their encoding genes with cell apoptosis and diseases. Int J Mol Sci 21: 8879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji P, Diederichs S, Wang W, Boing S, Metzger R, Schneider PM, Tidow N, Brandt B, Buerger H, Bulk E et al (2003) MALAT‐1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early‐stage non‐small cell lung cancer. Oncogene 22: 8031–8041 [DOI] [PubMed] [Google Scholar]
- Jiang HY, Wang ZJ (2020) ADPGK‐AS1 promotes the progression of colorectal cancer via sponging miR‐525 to upregulate FUT1. Eur Rev Med Pharmacol Sci 24: 2380–2386 [DOI] [PubMed] [Google Scholar]
- Jin Y, Feng SJ, Qiu S, Shao N, Zheng JH (2017) LncRNA MALAT1 promotes proliferation and metastasis in epithelial ovarian cancer via the PI3K‐AKT pathway. Eur Rev Med Pharmacol Sci 21: 3176–3184 [PubMed] [Google Scholar]
- Karger A, Nandigama R, Stenzinger A, Grimminger F, Pullamsetti SS, Seeger W, Savai R (2021) Hidden treasures: macrophage long non‐coding RNAs in lung cancer progression. Cancers 13: 4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly B, O'Neill LA (2015) Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res 25: 771–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Mendell JT (2020) Antisense‐mediated transcript knockdown triggers premature transcription termination. Mol Cell 77: 1044–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leucci E, Vendramin R, Spinazzi M, Laurette P, Fiers M, Wouters J, Radaelli E, Eyckerman S, Leonelli C, Vanderheyden K et al (2016) Melanoma addiction to the long non‐coding RNA SAMMSON. Nature 531: 518–522 [DOI] [PubMed] [Google Scholar]
- Ley K (2017) M1 Means Kill; M2 Means Heal. J Immunol 199: 2191–2193 [DOI] [PubMed] [Google Scholar]
- Li HJ, Sun XM, Li ZK, Yin QW, Pang H, Pan JJ, Li X, Chen W (2017) LncRNA UCA1 promotes mitochondrial function of bladder cancer via the MiR‐195/ARL2 signaling pathway. Cell Physiol Biochem 43: 2548–2561 [DOI] [PubMed] [Google Scholar]
- Li Y, He Y, Miao K, Zheng Y, Deng C, Liu TM (2020) Imaging of macrophage mitochondria dynamics in vivo reveals cellular activation phenotype for diagnosis. Theranostics 10: 2897–2917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SQ, Zhou ZY, Dong X, Guo L, Zhang KJ (2020) LncRNA GNAS‐AS1 facilitates ER+ breast cancer cells progression by promoting M2 macrophage polarization via regulating miR‐433‐3p/GATA3 axis. Biosci Rep 40: BSR20200626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J, Badal SS, Ye Z, Wang Y, Ayanga BA, Galvan DL, Green NH, Chang BH, Overbeek PA, Danesh FR (2016) Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J Clin Invest 126: 4205–4218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo XF, Wu XJ, Wei X, Wang AG, Wang SH, Wang JL (2019) LncRNA ADPGK‐AS1 regulated cell proliferation, invasion, migration and apoptosis via targeting miR‐542‐3p in osteosarcoma. Eur Rev Med Pharmacol Sci 23: 8751–8760 [DOI] [PubMed] [Google Scholar]
- Madela K, Banhart S, Zimmermann A, Piesker J, Bannert N, Laue M (2014) A simple procedure to analyze positions of interest in infectious cell cultures by correlative light and electron microscopy. Methods Cell Biol 124: 93–110 [DOI] [PubMed] [Google Scholar]
- Martinez‐Lage M, Torres‐Ruiz R, Puig‐Serra P, Moreno‐Gaona P, Martin MC, Moya FJ, Quintana‐Bustamante O, Garcia‐Silva S, Carcaboso AM, Petazzi P et al (2020) In vivo CRISPR/Cas9 targeting of fusion oncogenes for selective elimination of cancer cells. Nat Commun 11: 5060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller A, Nagy C, Knapp B, Laengle J, Ponweiser E, Groeger M, Starkl P, Bergmann M, Wagner O, Haschemi A (2017) Exploring metabolic configurations of single cells within complex tissue microenvironments. Cell Metab 26: 788–800.e6 [DOI] [PubMed] [Google Scholar]
- Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Dabritz JHM, Gottlieb E, Latorre I et al (2016) Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167: 457–470.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ock CY, Hwang JE, Keam B, Kim SB, Shim JJ, Jang HJ, Park S, Sohn BH, Cha M, Ajani JA et al (2017) Genomic landscape associated with potential response to anti‐CTLA‐4 treatment in cancers. Nat Commun 8: 1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47: D442–D450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quemener AM, Bachelot L, Forestier A, Donnou‐Fournet E, Gilot D, Galibert MD (2020) The powerful world of antisense oligonucleotides: from bench to bedside. Wiley Interdiscip Rev RNA 11: e1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffell B, Coussens LM (2015) Macrophages and therapeutic resistance in cancer. Cancer Cell 27: 462–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarode P, Schaefer MB, Grimminger F, Seeger W, Savai R (2020a) Macrophage and tumor cell cross‐talk is fundamental for lung tumor progression: we need to talk. Front Oncol 10: 324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarode P, Zheng X, Giotopoulou GA, Weigert A, Kuenne C, Gunther S, Friedrich A, Gattenlohner S, Stiewe T, Brune B et al (2020b) Reprogramming of tumor‐associated macrophages by targeting beta‐catenin/FOSL2/ARID5A signaling: a potential treatment of lung cancer. Sci Adv 6: eaaz6105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmall A, Al‐Tamari HM, Herold S, Kampschulte M, Weigert A, Wietelmann A, Vipotnik N, Grimminger F, Seeger W, Pullamsetti SS et al (2015) Macrophage and cancer cell cross‐talk via CCR2 and CX3CR1 is a fundamental mechanism driving lung cancer. Am J Respir Crit Care Med 191: 437–447 [DOI] [PubMed] [Google Scholar]
- Seebacher NA, Stacy AE, Porter GM, Merlot AM (2019) Clinical development of targeted and immune based anti‐cancer therapies. J Exp Clin Cancer Res 38: 156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A (2020) Cancer statistics, 2020. CA Cancer J Clin 70: 7–30 [DOI] [PubMed] [Google Scholar]
- Sirey TM, Roberts K, Haerty W, Bedoya‐Reina O, Rogatti‐Granados S, Tan JY, Li N, Heather LC, Carter RN, Cooper S et al (2019) The long non‐coding RNA Cerox1 is a post transcriptional regulator of mitochondrial complex I catalytic activity. Elife 8: e45051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Yu W, Lin S, Zhang M, Wang T, Guo S, Wang H (2018) LncRNA ADPGK‐AS1 promotes pancreatic cancer progression through activating ZEB1‐mediated epithelial‐mesenchymal transition. Cancer Biol Ther 19: 573–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotgia F, Fiorillo M, Lisanti MP (2017) Mitochondrial markers predict recurrence, metastasis and tamoxifen‐resistance in breast cancer patients: early detection of treatment failure with companion diagnostics. Oncotarget 8: 68730–68745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Statello L, Guo CJ, Chen LL, Huarte M (2021) Gene regulation by long non‐coding RNAs and its biological functions. Nat Rev Mol Cell Biol 22: 96–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack E, Rolfe PA, Fink AF, Bankov K, Schmid T, Solbach C, Savai R, Sha W, Pradel L, Hartmann S et al (2020) Identification of tumor-associated macrophage subsets that are associated with breast cancer prognosis. Clin Transl Med 10: e239. 10.1002/ctm2.239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Xu J (2019) TCF‐4 regulated lncRNA‐XIST promotes M2 polarization of macrophages and is associated with lung cancer. Onco Targets Ther 12: 8055–8062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taanman JW (1999) The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta 1410: 103–123 [DOI] [PubMed] [Google Scholar]
- Tan HY, Wang N, Li S, Hong M, Wang X, Feng Y (2016) The reactive oxygen species in macrophage polarization: reflecting its dual role in progression and treatment of human diseases. Oxid Med Cell Longev 2016: 2795090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S, Li D, Zhu X (2020) Cancer immunotherapy: pros, cons and beyond. Biomed Pharmacother 124: 109821 [DOI] [PubMed] [Google Scholar]
- Tannahill GM, Curtis AM, Adamik J, Palsson‐McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH et al (2013) Succinate is an inflammatory signal that induces IL‐1beta through HIF‐1alpha. Nature 496: 238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB et al (2012) Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med 366: 2443–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis WD, Brambilla E, Nicholson AG, Yatabe Y, Austin JHM, Beasley MB, Chirieac LR, Dacic S, Duhig E, Flieder DB et al (2015) The 2015 world health organization classification of lung yumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol 10: 1243–1260 [DOI] [PubMed] [Google Scholar]
- Travis WD, Brambilla E, Riely GJ (2013) New pathologic classification of lung cancer: relevance forclinical practice and clinical trials. J Clin Oncol 31: 992–1001 [DOI] [PubMed] [Google Scholar]
- Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, Chawla A (2006) Oxidative metabolism and PGC‐1beta attenuate macrophage‐mediated inflammation. Cell Metab 4: 13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T, Langer T (2016) Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab 27: 105–117 [DOI] [PubMed] [Google Scholar]
- Wang X, Li FY, Zhao W, Gao ZK, Shen B, Xu H, Cui YF (2020) Long non‐coding RNA GAS5 overexpression inhibits M2‐like polarization of tumour‐associated macrophages in SMCC‐7721 cells by promoting PTEN expression. Int J Exp Pathol 101: 215–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YW, Dong HZ, Tan YX, Bao X, Su YM, Li X, Jiang F, Liang J, Huang ZC, Ren YL et al (2022) HIF‐1alpha‐regulated lncRNA‐TUG1 promotes mitochondrial dysfunction and pyroptosis by directly binding to FUS in myocardial infarction. Cell Death Discov 8: 178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigert A, Zheng X, Nenzel A, Turkowski K, Gunther S, Strack E, Sirait‐Fischer E, Elwakeel E, Kur IM, Nikam VS et al (2022) Fibrocytes boost tumor‐supportive phenotypic switches in the lung cancer niche via the endothelin system. Nat Commun 13: 6078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widdrington JD, Gomez‐Duran A, Pyle A, Ruchaud‐Sparagano MH, Scott J, Baudouin SV, Rostron AJ, Lovat PE, Chinnery PF, Simpson AJ (2018) Exposure of monocytic cells to lipopolysaccharide induces coordinated endotoxin tolerance, mitochondrial biogenesis, mitophagy, and antioxidant defenses. Front Immunol 9: 2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Meng WY, Jie Y, Zhao H (2018) LncRNA MALAT1 induces colon cancer development by regulating miR‐129‐5p/HMGB1 axis. J Cell Physiol 233: 6750–6757 [DOI] [PubMed] [Google Scholar]
- Xie JH, Li YY, Jin J (2020) The essential functions of mitochondrial dynamics in immune cells. Cell Mol Immunol 17: 712–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Bai HS, Deng Y, Fan L (2015) High MALAT1 expression predicts a poor prognosis of cervical cancer and promotes cancer cell growth and invasion. Eur Rev Med Pharmacol Sci 19: 3187–3193 [PubMed] [Google Scholar]
- Yang J, Wu W, Wu M, Ding J (2019) Long noncoding RNA ADPGK‐AS1 promotes cell proliferation, migration, and EMT process through regulating miR‐3196/OTX1 axis in breast cancer. In Vitro Cell Dev Biol Anim 55: 522–532 [DOI] [PubMed] [Google Scholar]
- Yu T, Robotham JL, Yoon Y (2006) Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci USA 103: 2653–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Lu P, Yan L, Yang L, Wang Y, Chen J, Dai J, Li Y, Kang Z, Bai T et al (2019) MRPL35 is up‐regulated in colorectal cancer and regulates colorectal cancer cell growth and apoptosis. Am J Pathol 189: 1105–1120 [DOI] [PubMed] [Google Scholar]
- Zhao Y, Liu S, Zhou L, Li X, Meng Y, Li Y, Li L, Jiao B, Bai L, Yu Y et al (2019) Aberrant shuttling of long noncoding RNAs during the mitochondria-nuclear crosstalk in hepatocellular carcinoma cells. Am J Cancer Res 9: 999–1008 [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Turkowski K, Mora J, Brune B, Seeger W, Weigert A, Savai R (2017) Redirecting tumor‐associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget 8: 48436–48452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Weigert A, Reu S, Guenther S, Mansouri S, Bassaly B, Gattenlohner S, Grimminger F, Pullamsetti S, Seeger W et al (2020) Spatial density and distribution of tumor‐associated macrophages predict survival in non‐small cell lung carcinoma. Cancer Res 80: 4414–4425 [DOI] [PubMed] [Google Scholar]