

Abstract
Cilia are important cellular organelles for signaling and motility and are constructed via intraflagellar transport (IFT). RabL2 is a small GTPase that localizes to the basal body of cilia via an interaction with the centriolar protein CEP19 before downstream association with the IFT machinery, which is followed by initiation of IFT. We reconstituted and purified RabL2 with CEP19 or IFT proteins to show that a reconstituted pentameric IFT complex containing IFT81/74 enhances the GTP hydrolysis rate of RabL2. The binding site on IFT81/74 that promotes GTP hydrolysis in RabL2 was mapped to a 70‐amino‐acid‐long coiled‐coil region of IFT81/74. We present structural models for RabL2‐containing IFT complexes that we validate in vitro and in cellulo and demonstrate that Chlamydomonas IFT81/74 enhances GTP hydrolysis of human RabL2, suggesting an ancient evolutionarily conserved activity. Our results provide an architectural understanding of how RabL2 is incorporated into the IFT complex and a molecular rationale for why RabL2 dissociates from anterograde IFT trains soon after departure from the ciliary base.
Keywords: Cilium, GAP, GTPase, intraflagellar transport, RabL2
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Membranes & Trafficking; Structural Biology
The IFT‐B1 complex inactivates RabL2, promoting its dissociation from anterograde intraflagellar transport (IFT) trains for correct ciliogenesis.
Introduction
Cilia are slender organelles found on the surface of cells where they serve important functions in motility, sensory reception, and signaling (Rosenbaum & Witman, 2002). Cilia are believed to be ancient organelles present on the last eukaryotic common ancestor and are conserved from unicellular organisms such as the green algae Chlamydomonas reinhardtii (Cr), a key model organism for ciliary studies, to humans (Dutcher, 2014). Cilium formation is a multistep process that involves docking of a centriole at the plasma membrane (Sorokin, 1962), vesicular transport from the Golgi to the base of the cilium (Knödler et al, 2010; Vetter et al, 2015; Quidwai et al, 2021), and construction of the ciliary axoneme (Avasthi & Marshall, 2013). The elongation of the ciliary axoneme requires intraflagellar transport (IFT), the bi‐directional trafficking of large proteinaceous particles along the axonemal microtubules to deliver cargo for ciliary assembly (Kozminski et al, 1993, 1995; Pedersen & Rosenbaum, 2008). IFT is dependent on kinesin and dynein molecular motors as well as the large multi‐subunit IFT complex that mediates the interaction with ciliary cargoes (Kozminski et al, 1995; Hou et al, 2004; Bhogaraju et al, 2013; Taschner & Lorentzen, 2016a,b).
IFT complexes organize into IFT‐A and IFT‐B subcomplexes that accumulate at the ciliary base (Cole et al, 1998; Deane et al, 2001). The IFT‐B complex can be further subdivided into IFT‐B1 and IFT‐B2 subcomplex (Taschner et al, 2016). IFT‐A and IFT‐B complexes polymerize into linear assemblies known as IFT trains that are sandwiched between the ciliary axoneme and membrane (Kozminski et al, 1993, 1995; Pigino et al, 2009). Several recent publications use a combination of structural modeling in AlphaFold (Jumper et al, 2021), X‐ray crystallography and cryoelectron microscopy to elucidate the structure of the IFT‐B complex (Petriman et al, 2022), the IFT‐A complex (Hesketh et al, 2022; McCafferty et al, 2022; Meleppattu et al, 2022), and anterograde IFT trains in situ (Lacey et al, 2023). Anterograde IFT trains associate with ciliary cargo, such as axonemal components, and move from the ciliary base to the tip to deliver these cargoes (Bhogaraju et al, 2013; Lechtreck, 2022). Elegant cryo‐electron tomography (cryo‐ET) work has shown that anterograde IFT‐B trains organize into 6 nm linear repeat structures, whereas the IFT‐A trains have a 11 nm repeat (Jordan et al, 2018) resulting in an approximate 2:1 ratio for IFT‐B:IFT‐A complexes in accordance with mass spectrometry results (Lechtreck et al, 2009). At the ciliary tip, the kinesin motor dissociates from the IFT trains and diffuses back to the ciliary base in Chlamydomonas (Engel et al, 2009). The remaining components of the IFT trains are believed to partly break up before reassembling into dynein‐driven retrograde IFT trains that have a different ultrastructure (Stepanek & Pigino, 2016; Jordan et al, 2018). Except for the kinesin motor, these retrograde trains are thought to consist of the same IFT subunits as anterograde IFT trains (Chien et al, 2017). In C. elegans, tracking experiments show that IFT‐A and IFT‐B components have different dwelling times at the ciliary tip, suggesting that IFT trains are broken into separate IFT complexes (Mijalkovic et al, 2018). However, recent work on Chlamydomonas show that IFT‐A, IFT‐B, and IFT dynein subcomplexes stay associated through the switch from anterograde to retrograde IFT at the ciliary tip (Wingfield et al, 2021).
How do IFT proteins and complexes accumulate at the ciliary base for the initiation of anterograde IFT? Several studies using photobleaching of fluorescently tagged IFT subunits have addressed the mechanisms of IFT protein delivery to the base of the cilium. In Trypanosomes, experiments with GFP‐tagged IFT52 suggested that most IFT material at the ciliary base originates from recycled IFT trains with only a smaller part coming from the cytoplasm (Buisson et al, 2013). However, studies of IFT protein dynamics in vertebrate multiciliated cells show that IFT subcomplexes are preassembled in the cytoplasm and recruited to the ciliary base through a diffusion‐to‐capture mechanism (Hibbard et al, 2021). This result agrees with the observation that IFT46 depends on an interaction with IFT52, both subunits of the IFT‐B1 complex, for basal body localization in Chlamydomonas (Lv et al, 2017). A comprehensive study in Chlamydomonas uncovered that whereas IFT‐A and motor proteins are recruited to the ciliary base from the cytoplasm, IFT‐B proteins are both recruited from the cytoplasm as well as from “re‐used” retrograde IFT trains (Wingfield et al, 2017). Anterograde IFT cargo such as tubulin and IFT dynein are loaded onto anterograde IFT trains shortly before departure (Wingfield et al, 2017).
The mechanism of IFT train assembly at the ciliary base was also addressed by cryo‐ET in a recent seminal study demonstrating that IFT trains assemble in a sequential manner at the base of the cilium (van den Hoek et al, 2022). IFT train assembly appears to occur first through polymerization of IFT‐B followed by IFT‐A polymerization and lastly association of IFT motors (van den Hoek et al, 2022). Photobleaching experiments in Chlamydomonas show that IFT and motor proteins recover at different rates (3–10 s) with IFT43, IFT20, and IFT54 requiring about 9 s for full recovery (Wingfield et al, 2017). This result suggests that the timescale of IFT train assembly at the ciliary base is in the order of seconds, and is followed by injection into the cilium via an avalanche‐like mechanism (Ludington et al, 2013; Wingfield et al, 2017; van den Hoek et al, 2022).
Although the process of IFT initiation at the base of the cilium is not well understood, several lines of evidence suggest that the IFT‐B complex plays a crucial role. The IFT‐B polymers appear to form first and subsequently serve as a scaffold for the remaining IFT train components (van den Hoek et al, 2022). Furthermore, tomographic reconstructions show that the IFT‐B complex contacts to the kinesin‐II motor is required for initiating and driving anterograde IFT (Jordan et al, 2018), an interaction that likely occurs through the IFT88/52/57/38 heterotetramer (Funabashi et al, 2018). The IFT‐B1 complex contains the two small GTPases IFT22 and IFT27 (Taschner & Lorentzen, 2016a, 2016b). Small GTPases regulate many cellular processes by cycling between an inactive GDP‐bound conformation and an active GTP‐bound conformation that interacts with downstream effectors (Wittinghofer & Vetter, 2011). Activation through GDP➔GTP exchange is promoted by guanine nucleotide exchange factors (GEFs), whereas inactivation through GTP hydrolysis is promoted by GTPase‐activating proteins (GAPs). IFT27 (aka Rab‐like 4 [RabL4]) associates with IFT25 to form a heterodimer (Qin et al, 2007; Wang et al, 2009; Bhogaraju et al, 2011) and was initially suggested to play a role in IFT initiation (Wang et al, 2009). However, several subsequent studies have shown that IFT25/27 is dispensable for anterograde IFT but is instead required for the ciliary export of the BBSome complex and associated retrograde cargoes including sonic hedgehog signaling factors in mammals (Keady et al, 2012; Eguether et al, 2014; Liew et al, 2014; Dong et al, 2017) and phospholipase D in Chlamydomonas (Lechtreck et al, 2009, 2013). Recently, it was shown in Chlamydomonas that IFT25/27 promotes BBSome reassembly at the ciliary tip to facilitate ciliary exit of associated cargoes (Sun et al, 2021). IFT22 (aka RabL5) was initially discovered in Chlamydomonas (Wang et al, 2009) where it regulates the cellular levels of IFT proteins (Silva et al, 2012). However, IFT22 does not appear to be required for IFT initiation but, together with BBS3, is involved in recruiting the BBSome to the ciliary base (Xue et al, 2020). In Caenorhabditis elegans, mutation of IFT22 also does not affect ciliogenesis or IFT (Schafer et al, 2006; Inglis et al, 2009). In contrast, IFT22 in Trypanosoma brucei does appear to be required for proper ciliogenesis as IFT22 knockdown results in a retrograde IFT phenotype characterized by short cilia full of IFT material (Adhiambo et al, 2009; Wachter et al, 2019). However, the retrograde IFT phenotype of IFT22 knockdown cells suggests that IFT22, like IFT27, is not required for IFT initiation.
More recently, a third GTPase, RabL2, was shown to associate with the IFT‐B complex and regulate IFT initiation and cilium formation (Kanie et al, 2017; Nishijima et al, 2017). RabL2 is required for proper ciliogenesis in both Chlamydomonas (Nishijima et al, 2017) and in mammalian retinal pigment epithelial (RPE) cells (Kanie et al, 2017). However, RabL2 is dispensable for ciliogenesis in mice, and a mouse knockin bearing a GTP‐locked RabL2 allele displays no alteration of IFT frequency, velocity, or processivity (Duan et al, 2021). Mutations in RabL2 cause ciliopathies including male infertility because of defects in the assembly of cilia of sperm cells (Lo et al, 2012; Ding et al, 2020). Furthermore, RabL2 controls the ciliary localization of G‐protein‐coupled receptors (GPCRs) in primary cilia, suggesting a conserved role in the assembly/function of both motile and primary cilia (Dateyama et al, 2019). This agrees with the evolutionary conservation of RabL2 in ciliated species and the lack of RabL2 in non‐ciliated eukaryotes (Eliáš et al, 2016). RabL2 is recruited to the basal body of cilia via an interaction with the centriolar protein CEP19 (Jakobsen et al, 2011) and subsequently handed over to the IFT‐B complex prior to initiation of IFT at the ciliary base. Knockout of CEP19 or RabL2 significantly reduces the number of IFT trains in cilia suggesting that RabL2 helps control the injection of IFT trains into cilia (Kanie et al, 2017). Wild‐type (WT) RabL2 was shown to dissociate from IFT trains shortly after departure from the ciliary base, whereas a GTP‐locked RabL2 variant (Q80L in human RabL2) stays associated with IFT trains and accumulates in cilia (Kanie et al, 2017; Duan et al, 2021). It was furthermore shown that the S35N RabL2 mutant unable to bind GTP does not rescue ciliogenesis defects of RabL2‐knockout cells (Kanie et al, 2017). A recent study in mice suggested that the main function of RabL2 is not IFT initiation but rather the regulation of ciliary export of the BBSome complex and associated cargoes (Duan et al, 2021). This notion was based on observations that BBSome components and cargoes accumulate in RabL2 mutants unable to hydrolyze GTP (Duan et al, 2021). In any case, there is ample evidence that the nucleotide state of RabL2 is important for its ciliary function.
Here, we present a comprehensive biochemical analysis of RabL2 and the association with CEP19 and the IFT‐B1 complex. We show that the IFT complex, rather than CEP19, functions as a GAP that stimulates GTP hydrolysis to inactivate RabL2 and demonstrate that this activity is conserved from Chlamydomonas to human. Mutant IFT complexes that fail to recruit RabL2 have significantly lower IFT particle injection rates and ciliogenesis defects in mammalian cells. Structural predictions allow us to present architectural models of how RabL2 is recruited by CEP19 to the ciliary base and subsequently incorporated into the IFT complex. Our data suggest that RabL2 incorporation into the IFT complex is followed by stimulation of GTP hydrolysis, which in turn inactivates RabL2 to trigger its dissociation from IFT trains.
Results
CEP19 has high affinity for RabL2‐GTP but is not a GAP for RabL2
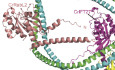
RabL2 was previously shown to locate to the basal bodies of cilia via an interaction with the protein CEP19 (Kanie et al, 2017). We purified WT and GTP‐locked Q83L mutant CrRabL2 (Appendix Fig S1A–D) and demonstrated that RabL2 does not carry over nucleotides during purification (Appendix Fig S1E and F), which is consistent with the reported low micromolar affinity of RabL2 for GTP/GDP nucleotides (Kanie et al, 2017). In addition, we purified CrCEP19 as a C‐terminal truncation encompassing residues 1–208 (CrCEP191‐208, Appendix Fig S1G and H). The C‐terminal 40 residues of CrCEP19 are predicted to be intrinsically disordered and were thus omitted in the CEP191‐208 construct (Appendix Fig S1I and J). The interaction between RabL2 and CEP19 was studied by size exclusion chromatography (SEC) and isothermal titration calorimetry (ITC). The results show that GTP‐bound RabL2 co‐purifies with CEP191‐208 on SEC at an elution volume that is significantly shifted when compared to CEP191‐208 suggesting the formation of a stable complex with RabL2 (Fig 1A). Furthermore, this result shows that RabL2/CEP19 complex formation only requires the N‐terminal 208 residues of CEP19 (Fig 1A and B). To obtain quantitative data on the affinities between RabL2 and CEP19, GTP‐ or GDP‐bound RabL2 was titrated with CEP191‐208 in ITC experiments. The results show that the CrRabL2‐GTP/ CEP191‐208 complex has a dissociation constant (Kd) of 0.28 μM (Fig 1C). Interestingly, GDP‐bound RabL2 still associates with CEP19, but with a K d of 12.7 μM for the CrRabL2‐GDP/CEP191‐208 complex (Fig 1D). The affinity of CEP19 for RabL2‐GDP is thus 45 times lower than for RabL2‐GTP. This result aligns with the understanding that CEP19 discerns the nucleotide state of RabL2, demonstrating a clear preference for the GTP‐bound state over the GDP‐bound state. Given that CEP19 preferably associates with the GTP‐bound state of RabL2, we tested if CEP19 functions as a GAP for RabL2 using GTPase assays with CrRabL2 alone or in complex with CrCEP191‐208. The results of the GAP assay show that CEP19 does not have any stimulating effect on the GTP hydrolysis rate of RabL2, demonstrating that CEP19 is not a GAP for RabL2 (Fig 1E).
Figure 1. GTP‐ and GDP‐dependent RabL2‐CEP19 complex formation.
-
ASEC profile that shows the co‐purification of CrCEP191‐208 and CrRabL2 in the presence of GTP. The elution volume of CrCEP19‐RabL2 is significantly shifted compared to the volumes of CEP19 or RabL2 alone, demonstrating the formation of a complex.
-
BCoomassie‐stained SDS–PAGE of the SEC fractions highlighted in (A) with a horizontal dashed line on top.
-
C, DITC measurements of purified CEP19 titrated with CrRabL2 in the presence of GTP (C) or GDP (D).
-
EGTPase assays of 250 μM RabL2 alone or in complex with 250 μM CEP191‐208 measuring the release of inorganic phosphate upon GTP hydrolysis as a function of time (1 mM GTP was added to each experiment). Each experiment was carried out in three technical replicates, curves are averages of these triplicates, and the error bars indicate standard deviation of measurements for every 100 s. Inorganic phosphate was used as the positive control and GTP in buffer as the negative control.
-
FSurface representation of the AlphaFold predicted structure for the complex between CrCEP19108‐194 (green color) and CrRabL2 (red‐salmon color).
-
GSEC profile (top) and Coomassie‐stained SDS gel (bottom) of the complex between RabL2 and a minimal binding region of CEP19 (residues 107–195) demonstrating a direct interaction.
There are currently no experimentally determined structures available for RabL2 or CEP19 proteins. We thus carried out structural modeling of the CrRabL2/CEP19 complex structure using AlphaFold multimer (Jumper et al, 2021; preprint: Evans et al, 2022; Fig 1F and Appendix Fig S1I and J). With exception of the N‐terminal 20 residues and the C‐terminal 40 residues, the structural model for CrRabL2 was predicted with very high confidence (predicted local distance difference test (pLDDT) score > 90) encompassing the entire core GTPase fold (Appendix Fig S1I). CrCEP19 is mostly predicted to fold into four α‐helices interspaced by long loop regions likely to represent intrinsically disordered regions (Appendix Fig S1I). However, two helices (residues 183–194 and 120–137) and two regions without secondary structure (residues 108–119 and 138–165) of CrCEP19 are predicted with very high confidence and form close contacts with RabL2 (Fig 1F and Appendix Fig S1I). The low predicted aligned error (PAE) between residues in these regions of CrCEP19 and CrRabL2 residues (Appendix Fig S1J) suggests that they are involved in CEP19/RabL2 complex formation (Fig 1F). These regions of CEP19 are predicted to encircle RabL2 forming a crown‐like structure (Fig 1F). Interestingly, residues 120–137 of CrCEP19 form an α‐helix that lines the nucleotide‐binding pocket of RabL2 and thus likely sense the nucleotide state of RabL2 to provide increased affinity for GTP‐bound RabL2. The remaining RabL2‐interacting parts of CEP19 (residues 140–165 and 183–194) likely provide nucleotide‐independent interactions that allow complex formation of CEP19 with RabL2‐GDP. To validate the structural model shown in Fig 1F and confirm the CEP19 minimal binding region for RabL2, CrRabL2/CEP19107‐195 was reconstituted and co‐purified by SEC demonstrating the formation of a stable complex (Fig 1G). The data presented in Fig 1 and Appendix Fig S1 allow us to conclude that residues 107–195 of CEP19 constitute a minimal binding region that prefers the GTP‐bound state of RabL2 but does not stimulate the GTP hydrolysis by RabL2.
Reconstitution of RabL2‐containing IFT‐B1 complexes
RabL2 in the GTP‐bound state was previously shown to associate with the IFT complex via IFT81/74 (Kanie et al, 2017; Nishijima et al, 2017). Recently, visual immunoprecipitation experiments with full‐length IFT81 and different IFT74 truncations showed that the binding site for RabL2 is located on the IFT74/81 heterodimer N‐terminally to the IFT27/25 heterodimer (Zhou et al, 2022). We have recombinantly expressed and purified Chlamydomonas and human heterohexameric IFT‐B1 complexes containing RabL2 (IFT81/74/27/25/22/RabL2, IFT‐B1 hexamer, see Fig 2A–C). In humans, RabL2 is represented by two nearly identical paralogs, namely RabL2A and RabL2B (Wong et al, 1999). As these are only differentiated by three amino acids and are functionally equivalent in the rescue of the ciliogenesis defect of the RABL2A;RABL2B double‐knockout cells (Kanie et al, 2017), we used the RabL2B paralog for reconstitution of IFT‐B1 complexes. We were unable to express the human IFT‐B1 hexamer in Escherichia coli but did succeed in purification using insect cells as a eukaryotic expression system, although the yield obtained was much lower than for the Chlamydomonas counterpart using E. coli as an expression system.
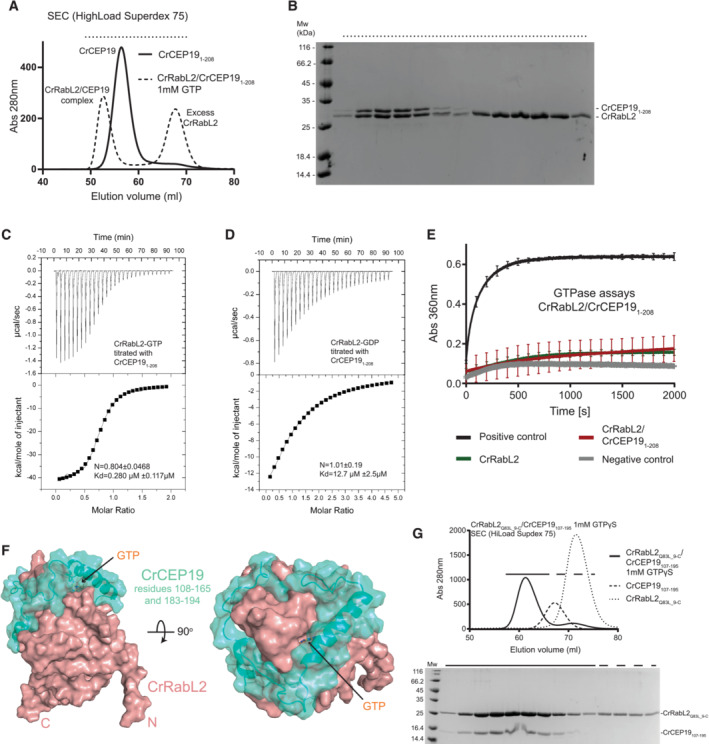
Figure 2. Purification and GTPase activity of IFT‐B1 complexes.
-
ASEC profile of CrRabL2Q83L co‐purification with CrIFT81/74/27/25/22 (top) in the presence of non‐hydrolyzable GTP homolog, GTPγS. Coomassie‐stained SDS gel of SEC fraction highlighted by dashed lines (bottom).
-
BSEC profile for the purification of the HsIFT81/74/27/25/22/RabL2 complex in the presence of GTPγS (top) and the corresponding Coomassie‐stained SDS gel (bottom).
-
CSchematic representation of the IFT‐B1 tetramer, pentamer, and hexamer.
-
DSEC profile of the incubation of the CrIFT‐B1 pentamer in the presence of CrRabL2 and GTP for 3 h at room temperature. The gel at the bottom shows that CrRabL2 does not stay associated with the IFT‐B1 pentamer under these conditions.
-
E–HGTPases assays with the indicated proteins following the release of inorganic phosphate upon GTP hydrolysis as a function of time. Each experiment was carried out in three technical replicates. Concentrations for experiments in (E) were 55 μM CrIFT‐B1 pentamer/CrRabL2, 60 μM CrIFT‐B1 pentamer, 70 μM CrIFT‐B1 tetramer, and 1 mM GTP. Concentrations for experiments in (G) were 60 μM protein and 30 μM GTP. The curves represent averages and error bars indicate standard deviation of measurements for every 100 s for panel (E) and every 200 s for panel (G). (F, H) Quantification of the reaction rates (arbitrary units of absorbance [Abs] per second [s]) for each experiment shown in panels (E) calculated using linear regression of the first 500 s and (H) using single exponential fit, and agreement with the fit to the curves in (E, G) are indicated by the error bars and R 2‐value.
In the presence of the non‐hydrolyzable GTP analog GTPγS, stable hexameric IFT‐B1 complexes containing Chlamydomonas RabL2Q83L or human WT RabL2B could be purified by SEC (Fig 2A and B). Interestingly, when WT CrRabL2 and GTP were mixed with the IFT‐B1 complex and incubated for 3 h at room temperature, RabL2 no longer associated with the IFT‐B1 complex, perhaps suggesting hydrolysis of GTP over time (Fig 2D). In the Chlamydomonas complex containing IFT74ΔN and IFT25ΔC, all three small GTPases of the IFT‐B1 complex (IFT27, IFT22, and RabL2) are present in apparent stoichiometric amounts, suggesting that their association with IFT‐B1 is not mutually exclusive and that each GTPase has a unique binding site within the IFT‐B1 complex (Fig 2A). The human hexameric IFT‐B1 complex contains full‐length subunits resulting in the co‐migration of IFT27 and IFT25 on the SDS gel (Fig 2B). As shown by Kanie et al, we observe that RabL2 association with the IFT‐B complex (IFT81/74/27/25/22, IFT‐B1 pentamer, see Fig 2C) is completely dependent on GTP as RabL2 does not co‐purify with IFT‐B1 in the absence of a non‐hydrolyzable GTP analog such as GTPγS (Appendix Fig S2A). In agreement with previous publications, IFT22 and IFT27 do not require the addition of GTP to associate with the IFT‐B1 complex (Appendix Fig S2A; Taschner et al, 2014; Wachter et al, 2019). Quantitative data on the affinity of CrRabL2 for the CrIFT‐B1 complex were obtained from ITC experiments revealing a Kd of 0.59 μM for the RabL2‐GTPγS‐bound IFT‐B1 complex (Appendix Fig S2B). No binding was observed between RabL2‐GDP and IFT‐B1, suggesting that the affinity is at least two orders of magnitudes lower than that for GTP‐bound RabL2 (Appendix Fig S2C). These experiments verify that the IFT‐B complex associates only with the active GTP‐bound conformation of RabL2 and indicate GTP hydrolysis over time resulting in dissociation of RabL2 from the IFT‐B1 complex.
The IFT‐B1 complex is a GAP for RabL2 but not for IFT27 or IFT22
Given that IFT81/74 is the binding platform for the three small GTPases IFT22, IFT27, and RabL2, we asked if IFT81/74 functions as a GAP for one or more of the small GTPases. In addition to the CrIFT‐B1 pentamer and hexamer (Fig 2A and Appendix Fig S2A), we purified the Chlamydomonas IFT81/74ΔN/27/25ΔC complex (IFT‐B1 tetramer), which contains IFT27 but lacks both RabL2 and IFT22 (Appendix Fig S2D). This IFT‐B1 tetramer does not require the addition of GTP for IFT27 to stay associated with the complex. Additionally, we showed in a previous study that IFT81/74/22 complexes co‐purify with GTP although GTP is not mandatory for association of IFT22 with the IFT complex (Wachter et al, 2019). In contrast, RabL2 alone does not co‐purify with GTP (Appendix Fig S1E and F) and requires the addition of GTP to form a complex with IFT81/74 (Fig 2A and B and Appendix Fig S2A; Kanie et al, 2017). Interestingly, while we observed that RabL2 incubated with the IFT‐B1 pentamer and GTPγS resulted in co‐purification of an IFT‐B1 hexamer on SEC (Fig 2A), the incubation of RabL2 with GTP and the IFT‐B1 pentamer results in separate elution peaks for RabL2 and the IFT‐B1 pentamer demonstrating that an IFT‐B1 hexamer was not formed (Fig 2D). Given that the intrinsic GTP hydrolysis rate of RabL2 is very low (Fig 1E), this finding could indicate that GTP was hydrolyzed during incubation of RabL2 with the IFT‐B1 pentamer, perhaps suggesting an increased rate of GTP hydrolysis when RabL2 associates with the IFT‐B complex.
To analyze the potential GAP function of IFT‐B1 complexes, GTPase assays were carried out with purified complexes to measure GTP hydrolysis rates. Initial assays with the IFT‐B1 tetramer, pentamer, or hexamer demonstrated that tetrameric and pentameric complexes without RabL2 do not have GTPase activity above background levels when using 1 mM GTP in the assay (Fig 2E). This result suggests that IFT81/74 is not a GAP for IFT22 or IFT27. In contrast, the hexameric IFT‐B1 complex containing RabL2 displayed robust GTP hydrolysis activity with a reaction rate about sixfold higher than what was observed for IFT complexes lacking RabL2 (Fig 2E and F). These data indicate that incorporation of RabL2 into the IFT complex activates the GTP hydrolysis in RabL2. Alternatively, RabL2 could act as a GAP towards IFT27 or IFT22 in context of the IFT‐B1 complex. To further analyze the GTP hydrolysis activity of the IFT‐B1 complex and distinguish between these two possibilities, GTPase assays were repeated using WT or RabL2Q83L catalytic mutant‐reconstituted IFT‐B1 hexamers (Fig 2G and H). Under the conditions of the assay (single turnover kinetics using 30 μM GTP), WT RabL2 in context of the hexameric IFT‐B1 complex has ninefold higher GTPase activity than WT RabL2 alone (Fig 2G and H). Adjusting for the low basal GTPase activity of IFT27 and IFT22 within the IFT‐B1 hexamer, the IFT‐B1 complex increases the reaction rate of RabL2 by approximately sevenfold under the conditions of the assay in Fig 2G.
Several protein families within the superfamily of small GTPases rely on a catalytic glutamine from the switch II region for GTP hydrolysis (Pai et al, 1990; Seewald et al, 2002). This catalytic glutamine is conserved in most Rab proteins including RabL2 (Q83 in CrRabL2 and Q80 in HsRabL2B) but is not conserved in most IFT27 or IFT22 sequences (Bhogaraju et al, 2011). Mutation of this catalytic glutamine to leucine (CrRabL2Q83L) is thus expected to abolish GTP hydrolysis in RabL2. Indeed, the hexameric IFT‐B1 complex containing the RabL2Q83L mutant has the GTPase activity reduced to background levels observed for the IFT‐B1 pentamer without RabL2 (Fig 2G and H). This result shows that the increase in GTPase activity of hexameric compared to pentameric or tetrameric IFT‐B1 complexes is a result of GTP hydrolysis in the active site of RabL2 and confirms that Q83 is important for catalysis. It is noteworthy that the GTPase activity observed for the IFT‐B1 pentamer, when compared to background levels, can be recapitulated by the IFT27/25 complex suggesting that the low level of GTP hydrolysis observed for the IFT‐B1 pentamer can be attributed to IFT27 rather than IFT22 (Fig 2G and H). Furthermore, this result verifies that the intrinsic GTPase activity of IFT27/25 is not increased in context of the IFT‐B1 complex. We conclude that the IFT‐B complex is a GAP for RabL2. Association of GTP‐bound RabL2 with the IFT‐B complex will thus lead to increased GTP hydrolysis to inactivate RabL2 resulting in the subsequent dissociation of GDP‐bound RabL2 from the IFT‐B complex.
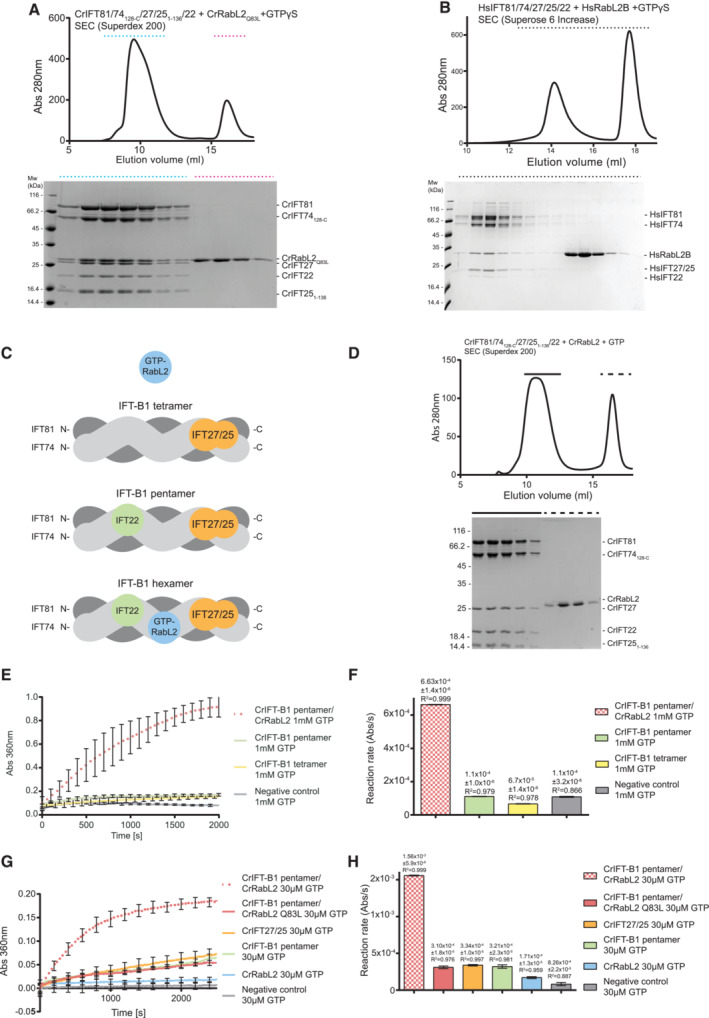
A minimal IFT81460 ‐533/74460‐532 complex binds RabL2 and stimulates GTPase activity
To biochemically map the binding site for RabL2 on IFT81/74, we reconstituted and purified Chlamydomonas complexes harboring truncated IFT81 and IFT74 proteins (Appendix Fig S3). Removing the most N‐terminal 150 residues of both IFT81 and IFT74 did not impact the ability of IFT27/25 or RabL2 to co‐purify with IFT81/74, confirming that the N‐termini of IFT81 or IFT74 are not required for complex formation with RabL2 or IFT27/25 (Appendix Fig S3A). However, deleting the 150 C‐terminal residues (IFT81133‐475/74132‐475) disrupts binding of RabL2 while retaining the ability to associate with IFT22 (Appendix Fig S3B). Importantly, a minimal complex containing the last three coiled‐coil segments of IFT81460‐C/74460‐C retains the ability to associate with both RabL2 and IFT27/25 (Appendix Fig S3C). Finally, we show that the most C‐terminal residues following the last coiled‐coil segment of IFT81/74 are not required for binding of RabL2 or IFT27/25 (Appendix Fig S3D). These experiments biochemically map the binding region for CrRabL2 to a coiled‐coil segment between residues 460–623 of CrIFT81 and residues 460–615 of CrIFT74. Further trimming of the C‐termini of IFT81 and IFT74 resulted in a predicted coiled‐coil segment of about 70 residues (IFT81460‐533/74460‐532) that co‐purifies with RabL2‐GTPγS to yield a stable complex on SEC (Fig 3A). Furthermore, pull‐down experiments with RabL2 demonstrated that IFT81460‐533/74460‐532 is sufficient to recapitulate RabL2 binding in the presence of GTPγS but not in the presence of GDP (Fig 3B and C). These results show that a short 70‐residue coiled‐coil fragment of IFT81/74 constitutes a minimal RabL2‐binding region. The fact that IFT81460‐533/74460‐532 discriminates between GTP‐ and GDP‐bound RabL2 conformations suggests that complex formation involves the GTPase switch regions of RabL2.
Figure 3. Mapping of a minimal CrIFT81460‐533/CrIFT74460‐532 complex that binds RabL2 and activates GTP hydrolysis.
-
ASEC profile showing that a minimal CrIFT81460‐533/CrIFT74460‐532 complex co‐purifies with CrRabL2Q83L in the presence of GTPγS (left). The right panel displays the Coomassie‐stained SDS gel of SEC fractions (horizontal top dashed line).
-
BAn N‐terminal hexa‐histidine‐tagged CrIFT81460‐533/74460‐532 complex interacts with untagged CrRabL2Q83L in a GTPγS‐dependent manner in pull‐down assays.
-
CSchematic of all CrIFT81/74 truncations used in this study for CrRabL2‐binding assays. The presence of CrIFT27/251‐136 and/or CrIFT22 in a complex with the CrIFT81/74 variants as well as their ability to bind CrRabl2 are indicated.
-
DGTPase assay using CrRabL2 and a minimal IFT81/74 complex show stimulation of GTP hydrolysis. Concentrations for each experiment were 60 μM protein and 30 μM GTP. Each experiment was done in three technical replicates; curves represent the averages with error bars representing standard deviations each 200 s.
-
EQuantification of GTPase reaction rates (arbitrary units of absorbance [Abs] per second [s]) using single exponential fit (D); error bars and R 2‐value indicate the agreement of the fit to the curves in (D).
To test if IFT81460‐533/74460‐532 is sufficient to stimulate the GTPase activity of RabL2, GTPase assays were carried out with IFT81460‐533/74460‐532 alone or the mixture of RabL2 and IFT81460‐533/74460‐532. The results show that IFT81460‐533/74460‐532 in the absence of RabL2 does not stimulate hydrolysis of GTP but in the presence of RabL2, the GTP hydrolysis rate is increased by approximately threefold when compared to RabL2 alone (Fig 3D and E). Although significant, this threefold increase in GTPase activity is less than the sevenfold increased activity observed with longer IFT81/74 constructs in context of the IFT‐B1 pentamer (Fig 2G and H). A likely explanation for the lower activity could be that parts of the IFT81/74 complex other than the 70‐residue coiled‐coil region are required for the optimal positioning of residues involved in GTP hydrolysis. Alternatively, it could be that IFT81460‐533/74460‐532 in isolation does not adopt a perfectly productive conformation to allow for the full stimulation of GTPase activity. In any case, IFT81460‐533/74460‐532 increases the GTPase activity of RabL2 and likely constitutes the main high‐affinity binding site for RabL2 within the IFT‐B complex.
Structural modeling of RabL2‐containing IFT‐B1 complexes
To obtain structural insights into the binding of RabL2 to IFT‐B1, we carried out structural modeling using AlphaFold (Jumper et al, 2021). Previously determined crystal structures are published for the IFT27/25 complex (Bhogaraju et al, 2011) and the N‐terminal parts of IFT81/74 in complex with GTP‐bound IFT22 (Wachter et al, 2019). However, no experimental structures are available for the C‐terminal parts of IFT81/74 that we mapped as the binding site for RabL2. Structural modeling was carried out using the original AlphaFold2 algorithm (Jumper et al, 2021) as well as the later published AlphaFold multimer (preprint: Evans et al, 2022) using IFT25/27, the C‐terminal parts of IFT81/74 and full‐length RabL2 for both human and Chlamydomonas complexes (Fig 4A and B). The structural models were consistently predicted with high confidence and low error in relative positioning of subunits within the complex (Appendix Fig S4). Both models of the human and Chlamydomonas complexes reveal a horseshoe‐shaped architecture of IFT81C/74C with conserved binding sites for RabL2 and IFT25/27 (Fig 4A and B).
Figure 4. AlphaFold predicted structures of IFT81/74/27/25/RabL2 complexes.
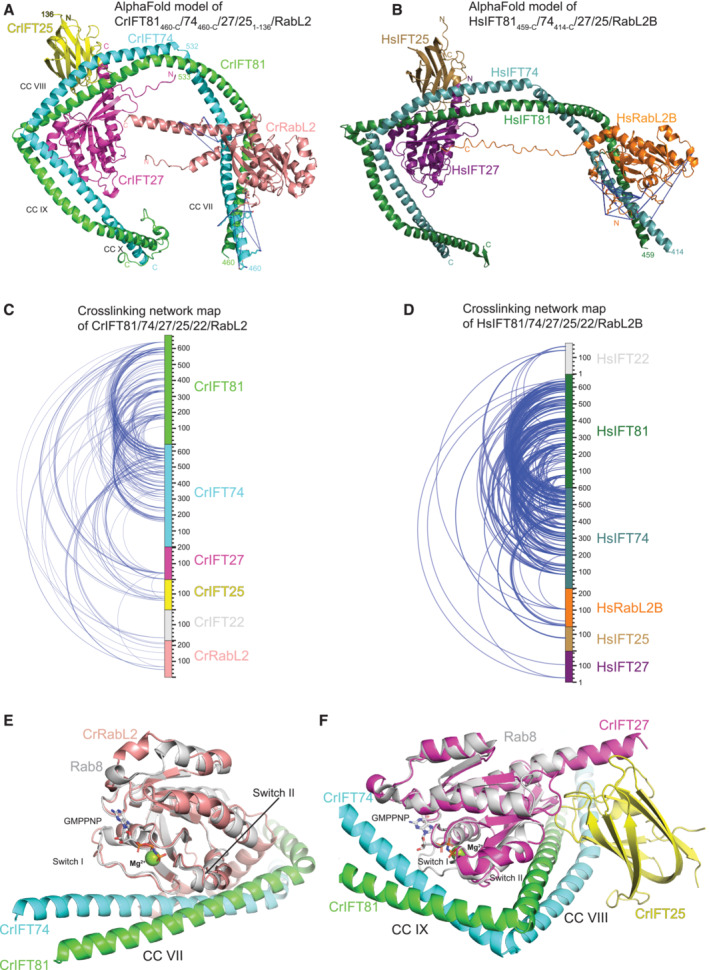
-
AAlphaFold predicted model of the Chlamydomonas IFT81460‐C/74460‐C/27/251‐136/RabL2 complex. The MS‐identified crosslinks between RabL2 and IFT81/74 are shown as blue dashed lines going between contributing reactive side chains displayed as sticks.
-
BAlphaFold predicted model of human IFT81459‐C/74414‐C/27/25/RabL2B with labeled intermolecular crosslinks as in (A).
-
CIntermolecular crosslinking pairs identified by MS of recombinant Chlamydomonas IFT81/74128‐C/27/251‐136/22/RabL2 protein complex.
-
DIntermolecular crosslinking pairs identified by MS of recombinant human IFT81/74/27/25/22/RabL2B protein complex.
-
EStructural model of CrIFT81/74/RabL2 highlighting the binding of the RabL2 switch I and II regions to coiled‐coil CC VII of IFT81/74. The non‐hydrolyzable GTP analog GppNHp is shown as sticks and the Mg2+ ion as a sphere after superpositioning of a Rab8 structure (pdb code 4LHW) onto CrRabL2.
-
FStructural model of IFT81/74/25/27 with Rab8 superposed onto IFT27 highlighting the switch regions.
The binding site for RabL2 on IFT81/74 was further confirmed by chemical crosslinking coupled to mass spectrometry (XL‐MS). Both human and Chlamydomonas IFT‐B1 hexamers (Fig 2A and B) were chemically crosslinked using the amine‐ and hydroxy‐specific homo‐bifunctional and MS‐cleavable crosslinker disuccinimidyl dibutyric urea (DSBU) with a crosslinking space arm of 12.5 Å (Iacobucci et al, 2018). MS‐cleavable crosslinkers can be cleaved in the mass spectrometer yielding two linear peptides, which are subsequently identified. Protein complexes were crosslinked by incubation with 0.25 mM DSBU and then digested with both LysC and trypsin. After digestion and to increase their identification rates, crosslinked peptides were enriched by strong cation exchange chromatography (SCX) and then subjected to MS/MS analysis. Using MeroX software (Götze et al, 2015), we identified 137 intra‐ and 211 intermolecular crosslinks at a false discovery rate (FDR) of 1% for the Chlamydomonas IFT‐B1 hexamer of which 34 belong to RabL2 (Fig 4C). For the human IFT‐B1 hexamer, 229 intra‐ and 333 intermolecular crosslinks were identified, of which 25 belong to RabL2 (Fig 4D). The crosslinking data are consistent with our recently published structural analysis of the Chlamydomonas IFT‐B complex in the absence of RabL2 (Petriman et al, 2022). For a more comprehensive analysis, the intermolecular crosslinking pairs formed between RabL2 and IFT81 or IFT74 were mapped on the AlphaFold predicted structures (Fig 4A and B; Movies EV1 and EV2 and Dataset EV1 and EV2). The MS/MS crosslinking data are consistent for human and Chlamydomonas complexes and show that reactive residues in RabL2 mainly crosslink to residues of IFT74 with fewer crosslinks to IFT81 (Movies EV1 and EV2). Most of the reactive side chains of the RabL2‐IFT74 and RabL2‐IFT81 crosslinking pairs are located close to each other within 25 Å with only three crosslinking pairs formed 36‐43 Å apart. Human RabL2B crosslinks along a 49‐Å‐long surface formed by coil‐coils of IFT74428‐458 and IFT81467‐500, whereas the Chlamydomonas counterpart crosslinks only to the IFT74460‐506 helix comprising a 64‐Å‐long surface. Although RabL2 and IFT25/27 are placed close in the structural models (Fig 4A and B), no direct interactions are observed and RabL2 displays only one chemical crosslink to IFT27 and none to IFT25. In summary, the chemical crosslinking results provide a validation of the structural predictions of the RabL2‐bound IFT‐B1 complex and corroborate a conserved binding site for RabL2 on IFT81/74.
In the structural model shown in Fig 4E, the RabL2 switch regions are highlighted and non‐hydrolyzable GTP analog GppNHP and Mg2+ are modeled based on the crystal structure of Rab8 (PDB code 4LHW, Guo et al, 2013). Interestingly, although GTP was not part of the structural modeling, the switch regions of RabL2 adopt a conformation very similar to that of other Rab GTPases bound to GTP or GTP analogs (Fig 4E). The structural prediction shows that the IFT81/74‐RabL2 complex is mainly formed through interactions of the switch regions of RabL2 with IFT74, with fewer contacts between RabL2 and IFT81 (Fig 4E). We note that this binding mode is consistent with IFT81/74 being an effector of RabL2. In the structural model, IFT81/74 does not insert any residues into the active site of RabL2 and no amino acid sidechain of IFT81/74 is closer than 10 Å from the GTPase site of RabL2. This indicates that the GAP activity of IFT81/74 toward RabL2 does not utilize the insertion of one or more residues in trans into the active site of RabL2.
The superpositioning of nucleotide‐bound Rab8 onto IFT27 is shown in Fig 4F and illustrates that the IFT25/27 complex forms an extended interface with coiled‐coil CCVIII and CCIX of IFT81/74. IFT25/27 appears to interact with IFT81/74 through both nucleotide‐dependent interactions as well as nucleotide‐independent interactions mediated by the central β‐sheet of IFT27 and the jelly‐roll fold of IFT25 to CCVIII of IFT81/74 (Fig 4F). This interaction mode is consistent with the nucleotide‐independent co‐purification of IFT25/27 with the IFT‐B1 complex shown in Appendix Fig S2A. Interestingly, both RabL2 and IFT25/27 appear to associate with regions of IFT81/74 containing sharp bends in the coiled‐coil structure (Fig 4E and F), perhaps suggesting an important role of the small GTPases in structuring the IFT‐B1 complex.
Chlamydomonas IFT81/74 binds human RabL2 to stimulate GTP hydrolysis
All subunits of the core IFT machinery, including IFT81, IFT74, and RabL2, are conserved between Chlamydomonas and human (van Dam et al, 2013), which is quite astonishing given the more than 1 B years of evolution separating the two species (Dutcher et al, 2012). CrRabL2 and HsRabL2 share 49% identity at the amino acid level. The structural models of Chlamydomonas and human RabL2 in complex with IFT81/74 (Fig 4A and B) suggest a common binding site for RabL2 on the IFT81/74 complex. To assess the conservation of the RabL2‐binding site on IFT81/74, the ConSurf server (Glaser et al, 2003; Landau et al, 2005) was used to plot the amino acid conservation onto the surface of the structural model (Fig 5A and B). The conservation plot reveals that the residues of IFT81/74 involved in binding RabL2 are highly conserved across RabL2‐containing ciliated species (Fig 5B). Interestingly, in C. elegans, where RabL2 is absent, the RabL2‐binding site on IFT81/74 is poorly conserved and partially missing (Appendix Fig S5). The surface of RabL2 engaging in interactions with IFT81/74 is also well conserved albeit less so than the IFT81/74 surface (Fig 5B).
Figure 5. RabL2 interacts with IFT81/74 through a conserved interface.
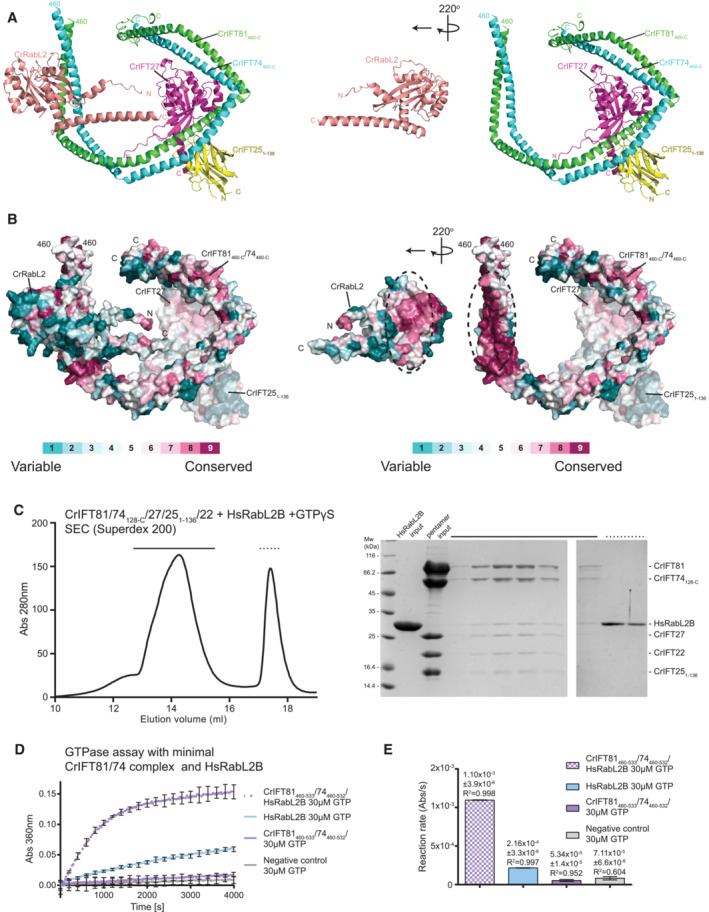
-
AThe predicted CrIFT81460‐C/74460‐C/27/251‐136/RabL2 structure (left) is computationally “opened up” by rotating RabL2 220° around the y‐axis (right) to visualize the binding interface between RabL2 and IFT81/74.
-
BSurface conservation map of the IFT81/74/RabL2 complex in the same position as in (A). Amino acid conservation is indicated according to the color code.
-
CSEC profile showing that human RabL2B co‐purifies with Chlamydomonas IFT‐B1 pentamer (left). Coomassie‐stained SDS gel of SEC fractions highlighted with dashed lines on the SEC profile (right).
-
DGTPase assay showing that the minimal IFT81/74 complex from Chlamydomonas induces GTP hydrolysis in human RabL2B. Concentrations for each experiment was 60 μM protein and 30 μM GTP. Each experiment was done in three technical replicates; curves represent the averages with error bars representing standard deviations each 200 s.
-
EQuantification of the reaction rates of the GTPase reaction based on single exponential fit of curves in (D); error bars and R 2 indicate the agreement of the fit.
To address the highly conserved IFT81/74‐RabL2 interface biochemically, we incubated the IFT‐B1 pentamer from Chlamydomonas with human RabL2 in the presence of GTPγS and carried out SEC. The result shows that human RabL2 indeed co‐purifies with the Chlamydomonas IFT‐B1 pentamer, demonstrating a direct physical interaction (Fig 5C). Co‐migration of subunits as a complex on the Superdex 200 column, in our experience, indicates a relatively strong interaction with a K d in the single‐digit μM range or lower (Vetter et al, 2015). The high degree of conservation of surface areas of the structural model shown in Fig 5B thus translates into a biochemically conserved interaction between RabL2 and IFT81/74 across species (Fig 5C). These results raise the question of whether the mechanism of RabL2 GTP hydrolysis activation by IFT81/74 is also conserved from Chlamydomonas to human. To test this, the Chlamydomonas IFT81460‐533/74460‐532 complex, constituting the minimal binding site for RabL2, was incubated with human RabL2B and GTP in a GTPase assay. The results show that Chlamydomonas IFT81460‐533/74460‐532 increases the reaction rate of GTP hydrolysis by fivefold when compared to human RabL2B alone (Fig 5D and E). Interestingly, the intrinsic hydrolysis rate of human RabL2B is three times higher than that of GTP in buffer and approximately 1.3 times higher than the intrinsic GTP hydrolysis rate of CrRabL2 (compare Figs 5E and 3E). However, the fivefold stimulation of GTPase activity of HsRabL2 by Chlamydomonas IFT81460‐533/74460‐532 shows that the ability to stimulate GTP hydrolysis of RabL2 is conserved between IFT81/74 from Chlamydomonas and human. The GAP activity of the IFT‐B1 complex, which inactivates RabL2 and dissociates it from the IFT trains, is thus likely to be an ancient mechanism involved in IFT initiation that is conserved across ciliated RabL2‐containing organisms.
An IFT74 point‐mutant deficient in RabL2 binding hampers with ciliogenesis and injection of IFT particles into cilia of mammalian cells
To further validate the structural model of RabL2‐bound IFT81/74 complexes (Fig 4) and obtain a structure‐guided point mutant for functional studies, we mutated a conserved threonine residue of CrIFT74 to arginine (IFT74T484R). T484 of IFT74 is in the interface with RabL2 and located close to switch I of RabL2 (Fig 6A). Mutation to an arginine is predicted to sterically break up complex formation with RabL2 while still allowing for IFT74 and IFT81 to associate (Fig 6A). Pull‐down experiments confirm that CrRabL2 does not associate with the CrIFT81/74T484R mutant complex (Fig 6B). Consistent with this result, the IFT81/74T484R mutant complex cannot stimulate the activation of GTPase activity in RabL2 (Fig 6C).
Figure 6. T438R mutant of IFT74 exhibits cilium formation defect at least partially associated with a failure in the regulation of RabL2.
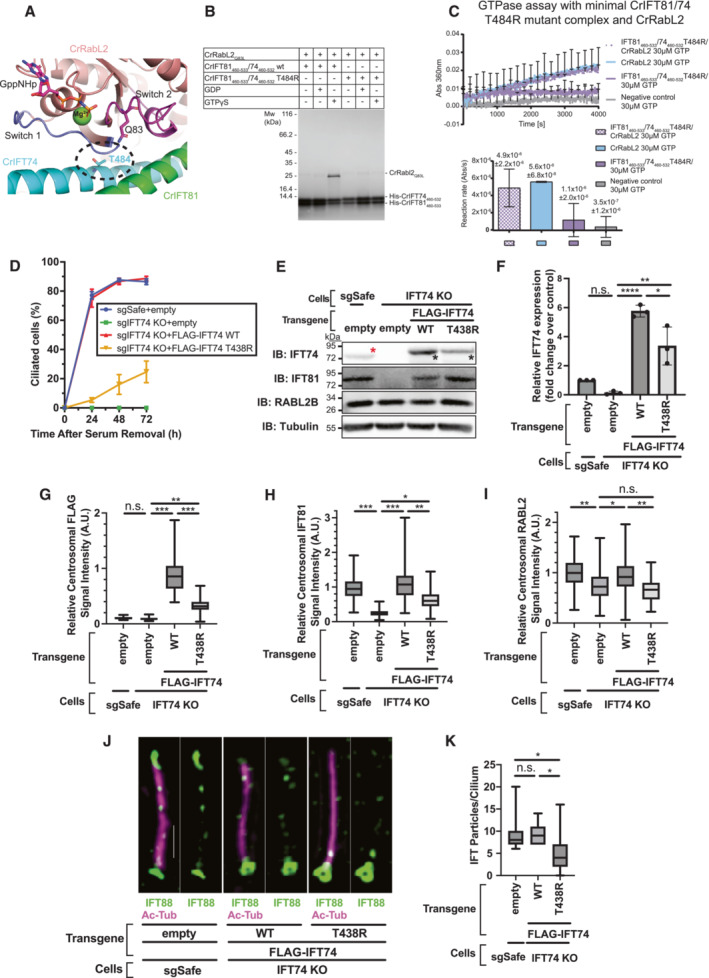
-
AZoom in on the interface between CrRabL2 switch regions and CrIFT81/74 in the structural model from Fig 4. The catalytic Q83 from CrRabL2 and a prominent interface residue from IFT74 (T484, encircled) are shown as sticks.
-
BAffinity pull‐down of CrRabL2 with His‐tagged WT or mutant CrIFT74T484R/81 complex in the presence or absence of nucleotides.
-
CGTPase assay using CrRabL2 and a minimal CrIFT81/74T484R mutant complex. Concentrations for each experiment were 120 μM for CrIFT81/74T484R, 60 μM for CrRabL2, and 30 μM GTP. Each experiment was done in three technical replicates. Quantification of the reaction rates of the GTPase reaction based on single exponential fit of curves with error bars and R 2 indicating the agreement of the fit.
-
DTime course of cilium formation assay in control (sgSafe) and IFT74‐knockout (KO) retinal pigment epithelial (RPE) cells stably expressing empty vector or indicated N‐terminally FLAG‐tagged transgenes. The indicated cells were fixed after 0, 24, 48, and 72 h of serum starvation. The fixed cells were stained with α‐ARL13B, α‐Ac‐Tub (to mark cilia), and α‐CEP170 (to mark centrioles), and imaged by wide‐field microscopy. Data are averaged from three experimental replicates. Error bars represent ± SEM. Detailed statistics are available in Appendix Fig S6B.
-
EImmunoblot (IB) analysis of expression of IFT74, IFT81, RabL2B, and α‐tubulin in control (sgSafe) or IFT74 KO RPE cells stably expressing empty vector, FLAG‐IFT74, or FLAG‐IFT74 T438R. Representative images from three experimental replicates are shown. The cells were grown to confluence (without serum starvation), lysed, and analyzed by immunoblot. Molecular weights (kDa) estimated from a protein marker are indicated. Red asterisk: endogenous IFT74; black asterisks: FLAG‐tagged IFT74.
-
FQuantification of the immunoblots of IFT74 shown in (E). Data are combined from three independent experiments. Error bars represent ± SD. Statistics were obtained through one‐way analysis of variance (ANOVA) with Tukey's multiple‐comparison test. Quantification of IFT81 and RabL2 is shown in Appendix Fig S6C and D.
-
G–IBox plots showing centrosomal signal intensity of FLAG (G), IFT81 (H), or RabL2 (I) in the cells described in (D) serum starved for 24 h. The relative fluorescence signal intensity compared with the average of the control (IFT74 KO + FLAG‐IFT74 WT for (G) and sgSafe + empty for (H) and (I)) is shown. Data are combined from three experimental replicates. Statistics obtained through nested one‐way ANOVA with Šídák's multiple‐comparison test. (G) The number of mother centrioles analyzed in each sample per replicate are as follows: 78, 77, 78 (sgSafe + empty); 102, 95, 101 (IFT74 KO + empty); 54, 77, 85 (IFT74 KO + FLAG‐IFT74 WT); 49, 83, 99 (IFT74 KO + FLAG‐IFT74 T438R), (H) The number of mother centrioles analyzed in each sample per replicate are: 93, 137, 138 (sgSafe + empty); 109, 133, 193 (IFT74 KO + empty); 62, 146, 131 (IFT74 KO + FLAG‐IFT74 WT); 41, 143, 133 (IFT74 KO + FLAG‐IFT74 T438R), (I) The number of mother centrioles analyzed in each sample per replicate are: 64, 101, 106 (sgSafe + empty); 84, 105, 129 (IFT74 KO + empty); 52, 96, 121 (IFT74 KO + FLAG‐IFT74 WT); and 73, 92, 102 (IFT74 KO + FLAG‐IFT74 T438R).
-
JRepresentative 3D‐structured illumination microscopy images of IFT88 in control (sgSafe) or IFT74 KO cells stably expressing indicated transgenes serum starved for 72 h. The images were created by maximum‐intensity z‐projection. Scale: 1 μm.
-
KBox plots showing the quantification of number of IFT88 particles per cilium in the experiment described in (J). Data are combined from three experimental replicates: The number of cilia analyzed in each sample per replicate are as follows: 20, 20, 18 (sgSafe + empty); 20, 20, 18 (IFT74 KO + WT); and 20, 20, 19 (IFT74 KO + FLAG‐IFT74 WT). The number of IFT particles in individual cells in each experiment is available from Appendix Fig S7A, D, and G. Statistics obtained through nested one‐way ANOVA with Tukey's multiple‐comparison test. Box plots show the five‐number summary of a set of data: including (from the bottom to the top) the minimum value, first (lower) quartile, median, third (upper) quartile, and maximum value, respectively. Asterisks denote *: 0.01 ≤ P < 0.05, **: P < 0.01, ***: P < 0.001, ****: P < 0.0001, n.s.: not significant. Statistical significance was calculated using GraphPad Prism 9 software.
We next sought to test the functional significance of disrupting the RabL2‐binding site on IFT‐B1 by introducing the IFT74 T484R mutation in mammalian cells. We generated retinal pigment epithelial (RPE)‐hTERT cells lacking IFT74 using the CRISPR‐Cas system (Jinek et al, 2012). We confirmed that a single‐cell clone of the knockout cells has a biallelic 7 bp deletion in IFT74 gene, resulting in a premature stop codon at residue 70 (Appendix Fig S6A). We then re‐introduced FLAG‐tagged IFT74 WT or the T438R mutant (Homo sapiens equivalent of Chlamydomonas reinhardtii T484R) in the IFT74‐knockout cells via lentiviral transduction. Upon serum withdrawal, control cells (RPE‐BFP‐Cas9 sgSafe) formed primary cilia over 24 h, and almost all cells completed cilium formation between 24 and 48 h (Fig 6D), as previously described (Kanie et al, 2017). In contrast, IFT74‐knockout cells expressing non‐targeting guide RNA (sgSafe) completely failed to form cilia even after prolonged serum starvation (Fig 6D and Appendix Fig S6B), confirming that IFT74 is indispensable for cilium biogenesis. Re‐expression of WT IFT74 almost fully rescued the ciliation defect of IFT74‐knockout cells, whereas the knockout cells expressing T438R variant of IFT74 exhibited a notable delay in initiating cilium formation (Fig 6D and Appendix Fig S6B). Immunoblot analysis showed that the T438R mutant did not express as strongly as wild type (Fig 6E and F), which may partially explain the ciliation defect caused by the mutant. Analysis of centrosomal localization of FLAG‐tagged IFT74 revealed that the WT protein accumulates at the ciliary base, consistent with the localization of other IFT components (Cole et al, 1998; Pazour et al, 1999; Deane et al, 2001; Kanie et al, 2017). IFT74T438R localizes to the mother centriole but with significantly lower efficiency (Fig 6G), consistent with the lower level of expression (Fig 6E and F). Nevertheless, the mutant was able to fully rescue the stability defect of IFT81 (Appendix Fig S6C), and partially rescue the centrosomal localization defect of IFT81 (Fig 6H).
Next, we tested if the IFT74T438R mutation prevented localization of RabL2 to basal bodies as the Chlamydomonas reinhardtii IFT81/74T484R mutant failed to associate with RabL2 in vitro (Fig 6B). We previously showed that CEP19 is primarily important to capture GTP‐bound RabL2 at the ciliary base, as RabL2 was not found at the centriole in CEP19‐knockout cells (Kanie et al, 2017). We also showed that a GTP‐locked form of RabL2 can bypass the requirement of CEP19 for its localization, as GTP‐locked RabL2 can bind to IFT‐B complex (Kanie et al, 2017). Consistent with our previous finding, centriolar RabL2 localization was partially but significantly reduced in IFT74 KO cells (Fig 6I). Re‐expression of WT IFT74 rescued the phenotype, whereas the centriolar localization of RabL2 was diminished to the same extent as the knockouts when T438R was introduced (Fig 6I). This result supports that the T438R mutant cannot bind to RabL2 in cellulo, which is consistent with our in vitro results (Fig 6B). The expression level of RabL2 was unaffected by expression of either wild type or T438R IFT74 (Appendix Fig S6D). Furthermore, examination of IFT particles via 3D‐structured illumination microscopy in cells that were serum starved for 72 h (a time point where T438R can partially rescue the ciliation defect of IFT74 KO, Fig 6D) revealed that the number of IFT particles inside the cilium was dramatically reduced when the T438R mutant was introduced into IFT74‐knockout cells (Fig 6J and K). This phenotype is reminiscent of RABL2‐knockout cells (Kanie et al, 2017). The reduction in IFT88 particles may partially derive from the reduction in the centriolar pool of IFT88 (Appendix Fig S6E and F), but it is unlikely that the 20% decrease in the centriolar IFT88 can explain the strong reduction in the number of the particles (Fig 6K). Given that the number of IFT particles inside the cilium is correlated with cilia length (Dentler, 2005), we excluded the possibility that the reduction is due to the decrease in cilia length in the knockout cells expressing IFT74T438R by examining both cilia length and the particle number per μm of cilium (Appendix Fig S6G and H). Collectively, our cell biological experiments showed that the IFT74T438R mutant exhibits a defect in IFT particle injection and a kinetic defect in ciliogenesis, which is at least partially attributable to the loss of binding to RabL2.
Discussion
Here, we provide structural models for how RabL2 associates with CEP19 and the IFT‐B1 complex. We show that the IFT‐B1 complex stimulates GTP hydrolysis of RabL2 and map the functional binding site to a short coiled‐coil segment of the IFT81/74 heterodimer. The RabL2 GTPase activity is increased by about sevenfold when incorporated into the IFT‐B1 complex. The intrinsic GTPase activity of small GTPases is typically very low and not compatible with biological timeframes of the processes that they regulate (Cherfils & Zeghouf, 2013), which is also the case for RabL2 (Figs 1E and 2G and H). GAPs are thus required to stimulate GTP hydrolysis and inactivate the GTPase at the appropriate time and location in the cell. The typical rate enhancement by GAPs is three to five orders of magnitude in vitro, ensuring almost instantaneous GTP hydrolysis upon complex formation between GTPase and its GAP (Scheffzek & Ahmadian, 2005). The increase in the GTP hydrolysis rate can, however, be a little as 10 times as seen for Sar1 during the dynamic assembly and disassembly of COPII (Antonny & Schekman, 2001). Sar1‐GTP initiates coat assembly by associating with vesicles budding from the ER, triggering the subsequent recruitment of Sec23/24 followed by Sec13/31 to complete COPII coat formation. The assembled COPII coat is a GAP for Sar1 that, mainly through Sec23 but assisted by Sec31 (which results in an additional 2–10× enhancement), activates GTP hydrolysis by Sar1 thus initiating coat disassembly (Bi et al, 2002, 2007). The relatively low activation rate of Sec23/31 toward Sar1 of only one order of magnitude likely reflects the timescale of COPII coat assembly/disassembly, which is in the order of seconds (Antonny & Schekman, 2001). Given that IFT train assembly at the ciliary base likely takes 3–10 s before injection into the cilium (Wingfield et al, 2017), the relatively low sevenfold activation of the reaction rate for GTP hydrolysis in RabL2 by the IFT complex appears to be biologically meaningful. We note that a much higher GAP activity of the IFT complex toward RabL2 would result in premature dissociation of RabL2 from IFT trains, which would be unproductive. On the other hand, a much lower GAP activity would likely result in too slow GTP hydrolysis in RabL2 and would result in continued association of RabL2 with IFT trains and faulty retrograde transport of BBSomes and associated cargoes as exemplified by the HsRabL2 Q80L mutant defective in GTP hydrolysis (Duan et al, 2021).
The wide range of catalytic activation mechanisms of GTP hydrolysis of small GTPases is mirrored by a high degree of structural and functional diversity among different GAPs. Many GAPs do, however, function by inserting one or more residues into the active site of the small GTPase to promote catalysis (Scheffzek & Ahmadian, 2005; Mishra & Lambright, 2016). The archetypical RasGAP functions by inserting an arginine finger into the active site of Ras to neutralize the build‐up of negative charge of the transition state (Pan et al, 2006; Scheffzek & Shivalingaiah, 2019). Other GTPase families such as Rho and Arl/Arf also rely on an arginine finger supplied in trans. Interestingly, Rap1GAP works by inserting a catalytic asparagine thumb into the active site of Rap1 (Daumke et al, 2004). Rab proteins often rely on GAPs of the TBC domain‐containing family where both an arginine finger and a glutamine, replacing the catalytic glutamine of switch II, are inserted into the active site of the Rab protein (Pan et al, 2006). For this reason, mutation of the switch II glutamine in some Rabs is not sufficient to create a constitutively active Rab as exemplified by Rab33, which associate with the dual‐finger RabGAP RUTBC1 (Nottingham et al, 2011; Cherfils & Zeghouf, 2013). In the structural models of the IFT81/74‐RabL2 complexes presented in Fig 4, the IFT81/74 complex mainly associates with the switch regions of RabL2 but do not appear to insert any residues into the GTP‐binding active site. However, it is worth noting that the structural models of RabL2 in complex with IFT74/81 depicted in Fig 4 do leave room for a yet unidentified protein to bind and potentially insert residues into the GTPase site of RabL2, further stimulating the GTPase hydrolysis. In any case, there are several examples of GAPs that activate GTP hydrolysis of small GTPases without inserting residues directly into the active site. MnmE and dynamin family GTPases were shown to use K+/Na+ cations instead of a catalytic arginine (Mishra & Lambright, 2016). In addition, the structure of Ran bound to RanGAP and RanBP1 shows that the Ran protein itself provides the catalytic machinery without the insertion of residues from RanGAP into the active site (Seewald et al, 2002). Instead, RanGAP and RanBP1 appear to activate Ran via an allosteric effect that stabilizes the switch regions including switch II, which contain the catalytic glutamine. Given the structural model in Fig 4, it appears likely that the IFT‐B1 complex stimulates the GTP hydrolysis activity of RabL2 through an allosteric effect that stabilizes a catalytically competent conformation of the switch regions and active site of RabL2. We show that the mechanism of GTP hydrolysis in RabL2 does rely on a classical catalytic switch II glutamine as the Q83L mutation abolishes the enhanced GTPase activity of a RabL2Q83L‐containing IFT‐B1 hexamer (Fig 2G and H). The detailed unraveling of the catalytic mechanism of RabL2 and GAP function of the IFT‐B1 complex awaits detailed experimental and structural elucidation.
The data presented here combined with previously published results allow us to propose a refined model for RabL2 functions in ciliary trafficking (Fig 7). RabL2 is recruited at the ciliary base, most likely in the GTP‐bound form, via an interaction with the centriolar protein CEP19 (Fig 7A). RabL2‐GTP is then handed over to and incorporated into IFT trains through an interaction with the C‐terminal part of the IFT81/74 subcomplex. Interestingly, the effect of CEP19 knockout on ciliogenesis can in part be rescued by overexpressing WT RabL2 and fully rescued by overexpressing RabL2 Q80L in RPE cells (Kanie et al, 2017). This result suggests that RabL2 can be recruited to the ciliary base through a direct interaction with the IFT complex. The main function of CEP19 in IFT initiation could thus be to concentrate the RabL2 protein at the base of the cilium. In agreement with this, CEP19 knockout in mice is not embryonic lethal but results in morbidly obese and hyperphagic mice (Shalata et al, 2013). Interestingly, a homoallelic nonsense mutation in CEP19 gene leads to premature truncation at residue R82 manifested by obesity, decreased sperm count, fatty liver, heart problems. and intellectual disability has been documented in humans (Shalata et al, 2013). We note that the human CEP19 protein sequence used by Shalata et al, 2013, has four additional N‐terminal residues (MYMG) as compared to the CEP19 sequence available in Uniport (https://www.uniprot.org/uniprot/Q96LK0). Our data suggest that this CEP19R82* mutant lacks the entire RabL2‐binding site (Fig 1) and is thus unable to recruit RabL2 to the ciliary base, which provide a molecular mechanism for the disease phenotype. Following recruitment of RabL2 by CEP19 at the ciliary base, RabL2‐GTP is incorporated into IFT trains, which may prime these trains for initiation of anterograde transport (Fig 7B). Indeed, we show that IFT particles unable to bind RabL2 (IFT74T438R) have lower injection rates of IFT material into cilia and display ciliogenesis defects (Fig 6D and K). The molecular mechanism through which RabL2 stimulates IFT initiation remains unknown, but it might involve a conformational change within the IFT‐B complex that is necessary for its association with the anterograde kinesin‐II motor. It is well documented that the phosphorylation of the kinesin‐II motor within the cilia of Chlamydomonas disrupts its interaction with IFT‐B, an essential step for initiating retrograde IFT from the ciliary tip to the base (Liang et al, 2014).
Figure 7. Model for RabL2 function in ciliary trafficking.
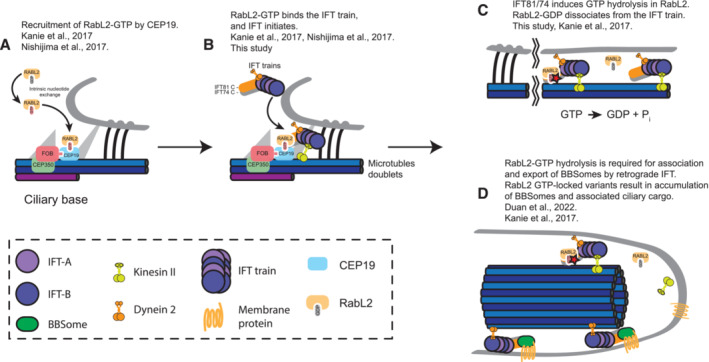
-
AFollowing activation of RabL2‐GDP by intrinsic nucleotide exchange, the active GTP‐bound RabL2 is recruited to the ciliary base by CEP19.
-
BRabL2‐GTP is then handed over to the IFT train through an interaction with a stretch of 70 residues within the coiled‐coil domain of IFT81/74, thereby initiating IFT.
-
CGTP hydrolysis in RabL2 is stimulated by IFT81/74, which converts the active GTP‐bound RabL2 into its inactive GDP‐bound form. The inactive RabL2‐GDP then dissociates from the anterograde IFT train.
-
DGTP hydrolysis in RabL2 is required for the export of BBSomes and associated cargo by retrograde IFT. Mutations that prevent GTP hydrolysis in RabL2 result in the continued association of RabL2‐GTP with IFT trains. This, in turn, prevents the export of, and results in the accumulation of, BBSomes and BBSome‐associated ciliary cargo.
Shortly after departure of IFT trains from the ciliary base, the GAP activity of IFT‐B1 toward RabL2 induces GTP hydrolysis and dissociation of RabL2‐GDP from the anterograde train (Fig 7C). This dissociation of RabL2 from the anterograde IFT trains appears to be required for the proper formation of BBSome‐containing retrograde IFT trains (Fig 7D). The HsRabL2Q80L mutant, where the IFT‐B1 complex is unable to promote GTP hydrolysis in RabL2, does rescue ciliogenesis of RabL2 knockouts (Kanie et al, 2017). However, the HsRabL2Q80L mutant accumulates in cilia together with the BBSome protein BBS4 and the GPCR GPR161, which is not the case in WT or S35N‐mutant RabL2 (Kanie et al, 2017). This ciliary accumulation of the BBSome and its GPCR cargoes in RabL2Q80L cells suggests that persistent RabL2‐GTP association with IFT‐B prevents BBSome association with retrograde IFT trains (Duan et al, 2021). The molecular basis for this regulation is not known, but the continued association of RabL2Q80L‐GTP with IFT trains might prevent the recruitment of BBSomes to retrograde IFT trains via IFT27/25 although a direct interaction between IFT25/27 and the BBSome has not been shown (Eguether et al, 2014). We note that RabL2 is located close to IFT27/25 in the C‐terminal region of the IFT81/74 subcomplex (Fig 4) and the continued association of RabL2 with the IFT complex may thus hamper with IFT27/25‐mediated BBSome recruitment to retrograde IFT trains. It is important to note that the functions of RabL2 in IFT initiation and in regulation of BBSome association with IFT trains (Fig 7D) are not mutually exclusive. The molecular mechanisms of how RabL2‐GTP promotes IFT initiation and how dissociation of RabL2‐GDP promotes the switch to retrograde IFT could involve conformational changes in the coiled‐coil segments of the IFT74/81 complex but remain to be elucidated in future studies.
Materials and Methods
Cloning and expression of CEP19 and RabL2 in E. coli
The gene encoding C. reinhardtii CEP191‐208 was subcloned from the pGEX‐6P‐1‐CrCEP19 to the pEL‐K vector. The genes encoding CrRabL2 and HsRabL2B were subcloned from the pGEX‐6P‐1‐CrRabL2 and pGEX‐6P‐1‐HsRabL2B to the pEL‐A vector. Protein encoding genes were amplified using forward primer encoding a 5′‐His(6)‐TEV overhang allowing cloning of a N‐terminal His(6)‐TEV‐tag and reverse primer to allow cloning into the pEL‐K vector. The DNA fragments pEL‐K, the amplified insert, and the His(6)‐TEV‐tag were assembled by Gibson Assembly (Gibson et al, 2009). CrRabL2Q83L mutant was constructed by PCR mutagenesis using pEL‐A‐His(6)‐TEV‐CrRabL2 as template. The CrRabL2Q83L‐Δ2‐8 truncation was constructed by mutagenesis using pEL‐A‐His(6)‐TEV‐CrRabL2Q83L as a template. The primers were constructed to omit 24 bp of CrRabL2Q83L starting from the second codon. The protein expressed from this plasmid is hereafter termed CrRabL2Q83L‐9‐C. CrCEP19107‐195 truncation was constructed by PCR mutagenesis using pEL‐K‐His(6)‐TEV‐CrCEP191‐208 as template. The forward primer omitted the first 318 bp of the gene encoding CrCEP191‐208 and the reverse primer omitted the last 39 bp. The forward primer encoding a 5′‐His(6)‐TEV overhangs allowing cloning of a N‐terminal His(6)‐TEV‐tag and reverse primer to allow cloning into the pEL‐K vector. Plasmids were transformed into E. coli BL21 (DE3) cells by heat shock at 42°C for 40 s. Heat shocked cells were plated on LB agar plates supplemented with appropriate antibiotics and incubated overnight.
For recombinant expression of CrRabL2, CrRabL2Q83L, CrRabL2Q83L‐9‐C, HsRabL2B, CrCEP191‐208, and CrCEP19107‐195, an overnight preculture was used to inoculate 6 L Terrific broth (TB) medium supplemented with antibiotics. Expression cultures were grown at 37°C until OD600 reached 1.2, at this point the temperature was lowered to 18°C, and 0.5 mM of isopropyl β‐D‐1‐thiogalactopyroside (IPTG) was added after 1.5 h. Expression cultures were incubated at 18°C for 18 h after induction and the cells were harvested by centrifugation (rotor F9‐6x1000lex) at 7822 RCF (relative centrifugal force) at 4°C for 12 min.
Purification of CEP19 and RabL2 proteins
Escherichia coli cells with overexpressed CEP19 or RabL2 protein were resuspended in lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 10% [v/v] glycerol, 5 mM MgCl2, 20 mM imidazole, and 5 mM β‐mercaptoethanol [BME]) containing 1 mM phenylmethanesulfonyl fluoride (PMSF) and lysed by sonication using the Sonopuls (Bandelin) fitted with VS 70T probe in two cycles of 10 min at 40% amplitude and of 1/1 s pulses. The cell lysate was centrifuged (rotor A27‐8x50) at 69028 RCF at 4°C for 30 min and the cleared lysate was aspirated and 1 μl DNase was added (ThermoFisher). Purification of the CrRabL2, CrRabL2Q83L, CrRabL2Q83L‐9‐C, HsRabL2B, CrCEP191‐208, and CrCEP19107‐195 was performed by loading the cleared lysate onto an IMAC cOmplete His‐Tag 5 ml column (Roche) pre‐equilibrated in five column volumes (CV) of lysis buffer. The column was after loading washed with 5CV of wash buffer (50 mM Tris pH 7.5, 1 M NaCl, 10% [v/v] glycerol, 5 mM MgCl2, 20 mM imidazole, and 5 mM BME), followed by equilibration in 5CV of lysis buffer. Elution was performed on an Äkta primer (GE Healthcare) using a gradient from 0 to 100% Ni‐elution buffer (50 mM Tris pH 7.5, 150 mM NaCl, 10% [v/v] glycerol, 5 mM MgCl2, 600 mM imidazole, and 5 mM BME). Peak fractions were pooled and 1 mg of TEV (Tobacco Etch Virus) protease was added followed by overnight dialysis at 4°C against dialysis buffer (20 mM Tris pH 7.5, 50 mM NaCl, 10% [v/v] glycerol, 5 mM MgCl2, and 1 mM DTT) to cleave off the affinity tags. The dialyzed sample was loaded onto the cOmplete His‐Tag column pre‐equilibrated in 5CV of low‐salt buffer (20 mM Tris pH 7.5, 50 mM NaCl, 10% [v/v] glycerol, 5 mM MgCl2, and 5 mM BME), and for RabL2 proteins, the flow through loaded onto a HiTrap Q HP 5 ml anion exchange column (GE Healthcare) pre‐equilibrated in low‐salt buffer. Elution was performed on the Äkta prime with gradient going from 0 to 100% high‐salt buffer (20 mM Tris pH 7.5, 1 M NaCl, 10% [v/v] glycerol, 5 mM MgCl2, and 5 mM BME). For CrCEP191‐208 and CrCEP19107‐195, the flow through was loaded onto an anion exchange MonoQ column (GE Healtcare) pre‐equilibrated in low‐salt buffer. Elution was performed with a 0–40% high‐salt buffer gradient on the Äkta purifier (GE Healtcare). Following purification by anion exchange, the protein sample was loaded onto the HiLoad 16/600 Superdex 75 (GE Healtcare) on the Äkta purifier pre‐equilibrated in SEC buffer (10 mM HEPES pH 7.5, 150 mM NaCl, 5 mM MgCl2, and 1 mM DTT). Note that all CrCEP19 purification buffers were prepared without MgCl2.
Expression and purification of Chlamydomonas IFT‐B1 complexes
Recombinant expression of Chlamydomonas IFT‐B1 complexes was achieved through co‐expression from plasmids in E. coli BL21 (DE3) cells. The CrIFT81/74128‐C/27/251‐136/22 complex was expressed from pEC‐A‐His(6)‐TEV‐CrIFT81, pEC‐K‐His(6)‐TEV‐CrIFT74128‐C, pEC‐S‐His(6)‐TEV‐CrIFT251‐136, pEC‐Cm‐CrIFT27, and pEC‐A‐CrIFT22. The CrIFT81152‐C/74150‐C/27/251‐136 complex was expressed from pEC‐A‐His(6)‐TEV‐CrIFT81152‐C, pEC‐K‐His(6)‐TEV‐CrIFT74150‐C, pEC‐S‐His(6)‐TEV‐CrIFT251‐136, and pEC‐Cm‐CrIFT27. The CrIFT81133‐475/74132‐475/22 complex was expressed from pEC‐A‐His(6)‐TEV‐CrIFT81133‐475, pEC‐K‐His(6)‐TEV‐CrIFT74132‐475, and pEC‐A‐CrIFT22. The CrIFT81460‐C/74460‐C/27/251‐136 complex was expressed from pEL‐A‐His(6)‐CrIFT81460‐C, pEL‐K‐His(6)‐Strep‐TEV‐CrIFT74460‐C, and pEC‐S‐His(6)‐TEV‐CrIFT251‐136‐RBS‐CrIFT27. The CrIFT81460‐623/74460‐615/27/251‐136 complex was expressed from pEL‐A‐His(6)‐CrIFT81460‐623, pEL‐K‐His(6)‐TEV‐CrIFT74460‐615, and pEC‐S‐His(6)‐TEV‐CrIFT251‐136‐RBS‐CrIFT27. The CrIFT81460‐533/74460‐532 complex was expressed from pEL‐A‐His(6)‐CrIFT81460‐533 and pEL‐K‐His(6)‐TEV‐CrIFT74460‐532. For recombinantly expression of IFT‐B1 complexes, an overnight preculture was used to inoculate 6LTB medium supplemented with antibiotics. Expression cultures were grown at 37°C until OD600 reached 0.8, the temperature was lowered to 18°C, and 0.5 mM of IPTG was added after 1.5 h. Expression cultures were incubated at 18°C for 18 h after induction. Lysing cells expressing CrIFT81/74128‐C/27/251‐136 allowed for the purification of the tetrameric IFT‐B1 complex, co‐lysing these with cells expressing IFT22 and/or CrRabL2 yields pentameric or hexameric IFT‐B1 complexes, respectively. Harvest and sonication of cells were performed as described above for RabL2 and CEP19. Cleared lysate was loaded onto the cOmplete His‐Tag column pre‐equilibrated in lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 10% [v/v] glycerol, 20 mM imidazole, 1 mM MgCl2, and 5 mM BME), followed by 5CV wash buffer (50 mM Tris pH 7.5, 1 M NaCl, 10% [v/v] glycerol, 20 mM imidazole, 1 mM MgCl2 and 5 mM BME). The column was washed with low‐salt buffer (50 mM Tris pH 7.5, 75 mM NaCl, 10% [v/v] glycerol, 1 mM MgCl2, and 5 mM BME). Elution was performed directly from the cOmplete His‐tag column and onto the HiTrap Hp Q anion column with Ni‐elution buffer (50 mM Tris pH 7.5, 75 mM NaCl, 10% [v/v] glycerol, 600 mM imidazole, 1 mM MgCl2, and 5 mM BME). The elution was collected and loaded onto the HiLoad 16/600 Superdex 200 (GE Healthcare) equilibrated in SEC buffer (10 mM HEPES pH 7.5, 150 mM NaCl, 1 mM MgCl2, and 1 mM DTT).
Cloning, expression, and purification of the human IFT81/74/27/25/22 complex from SF21 insect cells
The genes encoding the HsIFT81/74/27/25/22 were ordered from VectorBuilder® with codon optimized for expression in insect cells. For further affinity purification, DNA sequences encoding both a deca‐histidine tag followed by a TEV cleavage site at the N‐terminus of IFT81 and a hexa‐histidine tag at the N‐terminus of IFT74 followed by Strep‐II tag and TEV cleavage site were included. Each gene was flanked by an individual promoter and terminator and one copy of each of the five genes was assembled in one pFastBac™ Dual vector. The construct containing all five expression cassettes, named pNAP‐AG‐HsIFT81/74/27/25/22, was used for the HsIFT81/74/27/25/22 complex expression in insect cells. The bacmids packing, virus generating, and expression procedure are the same as that reported previously (Taschner & Lorentzen, 2016a, 2016b).
The infected cells containing the recombinantly expressed HsIFT81/74/27/25/22 were harvested by centrifugation, resuspended in one volume of lysis buffer (50 mm Tris–HCl, pH 7.5, 150 mm NaCl, 1 mM MgCl2, 10% glycerol, 5 mM BME, 1× complete protease inhibitor mixture [Roche], and 10 μg/ml DNase I), and lysed by 50× strokes in a Dounce tissue homogenizer. The cell debris was cleared by centrifugation at 20,000 g for 30 min at 4°C and the supernatant was filtered through a 5 μm filter. The supernatant was then loaded onto a pre‐equilibrated TALON HiTrap column with a peristaltic pump by recirculation at a 5 ml/min flow rate. Four washing steps were performed: (i) only lysis buffer; (ii) lysis buffer supplemented with 40 mM imidazole pH 7.5; 3. high‐salt buffer (50 mm Tris–HCl, pH 7.5, 1 m NaCl, 1 mM MgCl2, 10% glycerol, and 5 mm BME); and (iv) low‐salt buffer (50 mm Tris–HCl, pH 7.5, 75 mM NaCl, 1 mM MgCl2, 10% glycerol, and 5 mm BME). The HsIFT81/74/27/25/22 complex was eluted from the TALON beads with a lysis buffer supplemented with 300 mM imidazole pH 7.5. Elutions were concentrated with a 100 kDa cutoff Amicon Ultra filter and loaded onto a Superose 6 Increase SEC column with a 500 μl injection loop. Samples were collected from each SEC fraction, migrated on SDS–PAGE, and visualized by staining the gel with 1% of Coomassie brilliant blue. The SEC fractions that contained all proteins within HsIFT81/74/27/25/22 complex were combined and concentrated. Hexameric IFT‐B1 complex was obtained by mixing purified HsIFT81/74/27/25/22 with HsRabL2 and performing SEC on a Superose 6 Increase column.
Pull‐down assays
Samples containing purified Rabl2Q83L, IFT81460‐533/74460‐532, IFT81460‐533/74460‐532;T484R, and nucleotides (where indicated) were incubated for 1 h at 4°C in a 150 μl PD buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1 mM MgCl2, and 5 mm tris(2‐carboxyethyl)phosphine [TCEP]). Each sample was incubated with 30 μl pre‐equilibrated Ni2+‐NTA beads at 4°C for 1 h. The beads were recovered by low‐speed centrifugation (1,100 g) for 3 min and washed three times to remove the unbound proteins. The first washing step was performed with 500 μl of PD buffer and the second and the third washing with PD buffer supplemented with 20 mM imidazole pH 7.5. The histidine‐tagged IFT81460‐533/74460‐532 or IFT81460‐533/74460‐532;T484R was eluted from the beads by incubation with 50 μl PD buffer supplemented with 600 mM imidazole pH 7.5 for 10 min. The protein composition of each sample was evaluated on SDS–PAGE stained with Coomassie brilliant blue.
Isothermal titration calorimetry (ITC)
ITC was performed at 25°C using a VP‐ITC MicroCalorimeter (MicroCal, GE Healthcare). CrRabL2 and CrCEP19 were buffered in 10 mM HEPES pH 7.5, 150 mM NaCl, 5 mM MgCl2, 1 mM TCEP, and 1 mM GTP. A volume of 1.43 ml of 10 μM CrRabL2 was titrated with 100 μM CrCEP19 over 29 injections of 10 μl with a stirring speed of 312 rpm. Duration of each injection was 17.1 s with 200 s of spacing between individual injections. Titrations were performed in triplicates, and for each titration, a background curve consisting of titrant titrated into buffer was subtracted. The ITC data were analyzed with Origin 7 provided by MicroCal. The CrRabL2‐IFT‐B interaction was analyzed on a VP‐ITC MicroCalorimeter (MicroCal) instrument. A volume of 1.43 ml of IFT81152‐C/74150‐C/27/251‐136 supplemented with 100 μM GTPγS was titrated with a solution of 100 μM RabL2Q83L supplemented with 100 μM GTPγS over 29 injections of 10 μl with a stirring speed of 312 rpm. Three measurements were performed at 25°C in a buffer containing 20 mM HEPES pH 7.5, 150 mM NaCl, 1 mM MgCl2, and 0.5 mM TCEP. The obtained ITC data were analyzed using the Origin 7 software provided by MicroCal.
Protein complex prediction with AlphaFold multimer
For prediction of structures of RabL2 in complex with IFT81/74 or CEP19, we used a local installation of AlphaFold v2.1.0 (Jumper et al, 2021; preprint: Evans et al, 2022) as well as a modified version on Colab notebook (Mirdita et al, 2022). As input for AlphaFold, we used the relevant protein sequences or truncations from Chlamydomonas reinhardtii or Homo sapiens IFT81, IFT74, IFT27, IFT25, CEP19, and RabL2. All used sequences have > 500 homologs in available databases and all structural predictions shown in the figures have low PAE scores for the interacting regions indication a high degree of certainty in the relative positions of subunits within the complexes. All figures of protein structures were prepared using PyMOL v. 2.5 (Schrodinger LLC, https://pymol.org).
GTPase assays
The GTPase activity was measured using the EnzCheck Phosphate Assay Kit (ThermoFisher) at 22°C. GTPase assays were performed in 200 μl volumes in 96‐well plates and the release of inorganic phosphate (Pi) was monitored at 360 nm using the MultiSkan Go plate reader (ThermoFisher). Assays were performed in triplicate, and a reference data set containing reaction buffer substrate (MESG) and purine nucleoside phosphorylase (PNP) enzyme was subtracted from each data set. Negative controls contained reaction buffer, MESG, PNP enzyme, and GTP, and positive controls contained reaction buffer, MESG, PNP enzyme, and 100 μM Pi. Reaction components were mixed and incubated at 22°C for 15 min before GTP was added and the 96‐well plate was shaken for 2 s in the plate reader before measurement was started. The IFT‐B1 complex was incubated at 22°C for 6 h followed by buffer exchange to ensure that any co‐purified nucleotides were hydrolyzed and that any Pi was removed before the GTPase assays were performed. Reaction concentrations of CrRabl2 and CrRabL2/CrCEP19 (Fig 1E) were 250 μM (with 1 mM GTP) and concentrations of IFT‐B1 complexes were 55–70 μM (with 1 mM GTP) for Fig 2E. Reaction concentrations for Figs 2G, 3D and 5D were 60 μM CrIFT‐B1 complexes, 60 μM CrRabL2/HsRabL2, and 30 μM GTP. For Fig 6C, the concentrations were 120 μM for the minimal CrIFT81/74T484R complex and 60 μM for CrRabL2 (with 30 μM GTP). Quantification of reaction rates (arbitrary units of absorbance [Abs] per [s]) was calculated using linear regression for the first 500 s in Fig 2F, whereas single exponential fits were used to quantify the reactions rates for the single turnover experiments shown in Figs 2H, 3E, 5E and 6C. Fitting of GTPase data and calculation of reaction rates were done in the program GraphPad Prism 6.
DSBU crosslinking
Crosslinking experiments were performed using the MS‐cleavable crosslinker disuccinimidyl dibutyric urea (DSBU). The optimal concentration of DSBU was determined by titration and SDS–PAGE to allow only the formation of specific crosslinks. Reactions containing 20 μg of purified IFT‐B1 hexameric complexes dissolved in a buffer containing 20 mM HEPES pH 7.5, 150 mM NaCl, 1 mM MgCl2, and 0.5 mM TCEP were crosslinked for 30 min with various DSBU concentrations ranging from 0.1 to 1.53 mM (representing a molar excess ranging from 15 to 250 times), quenched by addition of 1 M Tris pH 8.8 and monitored on SDS–PAGE and Coomassie staining. A DSBU concentration of 0.25 mM per 6 μM of IFT‐B1 hexamer was chosen as an optimal crosslinking concentration for the data presented in this work.
Mass spectrometry analysis of DSBU‐crosslinked protein complexes
For the Chlamydomonas IFT‐B1 hexamer, 150 μg of the purified complex were crosslinked at 6 μM (1.33 mg/ml) and a final DSBU concentration of 0.25 mM for 30 min at room temperature (RT). The reaction was stopped by adding Tris–HCl pH 8.0 to a final concentration of 125 mM and incubated at RT for an additional 10 min. The sample was denatured and reduced with 6 M guanidine hydrochloride (Guan‐HCl) and 10 mM TCEP in 200 mM Tris–HCl pH 8.3 at 95°C for 5 min and stirred at RT for an additional 20 min. Cysteine residues were alkylated by incubation with 10 mM 2‐chloroacetamide (CAA) for 20 min in darkness. In‐solution digestion was performed by incubation with the endoprotease Lys‐C (enzyme:protein ratio 1:100) for 1 h followed by addition of 20 mM ammonium bicarbonate pH 8.0 to reduce the concentration of Guan‐HCl to 2 M. The protease trypsin was added at an enzyme:protein ratio of 1:50 and incubated at 37°C overnight. Digested peptides were acidified by adding 25% trifluoroacetic acid (TFA) to a final concentration of 1%.
For the human IFT‐B1 hexamer, 60 μg of the crosslinked complex was denatured, reduced and alkylated in lysis buffer containing 5% SDS, 10 mM TCEP, and 11 mM CAA in 100 mM Tris–HCl pH 8.5 for 10 min at 95°C. On‐bead protein digestion was performed following the protein aggregation capture (PAC) protocol (Batth et al, 2019) with some modifications. Crosslinked proteins were precipitated on 120 μg of MagResyn HILIC magnetic particles (Resyn Biosciences, Pretoria, Gauteng, South Africa) in 70% acetonitrile for 20 min and washed three times with 95% acetonitrile and two times with 70% ethanol. Magnetic particles were then incubated with Lys‐c in 50 mM ammonium bicarbonate pH 8.0 at a ratio of 1:200 (with respect to protein) for 1 h at RT followed by the addition of trypsin in the same buffer at a ratio of 1:100. Trypsin digestion was carried out overnight at 37°C and peptides were recovered by transferring the supernatant to a new tube and acidified with 1% TFA.
Digested and acidified peptides from both human and Chlamydomonas complexes were first purified with a Sep‐Pak tC18 cartridge (Waters Corporation). Briefly, the cartridge was conditioned by adding 50% and 100% acetonitrile and washed with 0.1% TFA prior to sample loading by gravity. Peptides were washed with 0.1% TFA and eluted using 50 and 80% acetonitrile in 0.1% TFA. Organic solvent was removed using a speedvac and one‐sixth or one‐tenth (Chlamydomonas or human complexes, respectively) of the sample was saved as the total peptide mixture for direct MS analysis. The rest of the sample was fractionated by cation exchange to enrich for the multiple‐charged crosslinked products as follows: Peptides were diluted in 4% phosphoric acid (H3PO4) and loaded into an Oasis MCX 1 cc Vac Cartridge (Waters Corporation) conditioned by adding methanol and 4% H3PO4. Peptides were recovered in four different fractions through stepwise elution in 0.5% acetic acid and increasing concentrations of ammonium acetate and methanol (Iacobucci et al, 2018). The flow through was collected and all fractions were desalted with a Sep‐Pak tC18 cartridge as described above.
Total peptide mixtures as well as fractions were analyzed on an Easy nanoLC system coupled directly to a Thermo Fisher Orbitrap Exploris mass spectrometer. Purified peptides were redissolved in 2% acetonitrile, 0.1% TFA, loaded onto a fused silica capillary column (75 μm ID, packed in‐house with Reprosil‐Pur C18, 1.9 μm reserved phase material), equilibrated with solvent A (0.1% formic acid), and separated with a linear gradient of 5–45% solvent B (0.1% formic acid and 95% acetonitrile). Mass spectra were acquired using data‐dependent acquisition. Top 15 ions with a charge state between 3 and 8 were selected for HCD fragmentation using stepped normalized collision energy (NCE) of 27, 30, and 33. Crosslinked peptides were identified using the Program MeroX version 2.0 (Götze et al, 2015). In the Mass Comparison settings, the precursor and fragment ion precisions were set to 5.0 and 10.0 ppm respectively, the S/N ratio to 1.5, and the minimum charge to 3. The RISEUP mode was used, maximum missing ions was set to 1, and the neutral loss of identified fragments was selected. In order to have a highly confident identification of the crosslinks, a prescore of 30 was applied, the FDR cut off was set to 1%, and the cRap database was included.
Lentiviral vector construction
pMCB306, a lentiviral vector containing loxP‐mU6 single‐guide RNA (sgRNA)‐puro resistance EGFP‐loxP cassette, and p293 Cas9‐blue fluorescent protein (BFP) vector were gifts from Prof. Michael Bassik. Lentivirus envelope and packaging vector, pCMV‐VSV‐G and pCMV‐dR8.2 dvpr, respectively, were gifts from Prof. Bob Weinberg (Addgene plasmid #8454 and #8455). A lentiviral vector containing single‐guide RNA (sgRNA) that targets IFT74 (sgIFT74) was generated by ligating 50 fmol oligonucleotides carrying sgRNA sequence into 25 ng of the pMCB306 vector digested with BstXI (R0113S, NEB) and BlpI (R0585S, NEB) restriction enzymes along with 0.25 μl of T4 ligase (M0202S, NEB) in 2.5 μl total reaction volume. Before ligation, 4 μM of forward and reverse oligonucleotides were annealed in 50 μl of annealing buffer (100 mM potassium acetate, 30 mM HEPES [pH 7.4], and 3 mM magnesium acetate) at room temperature following denaturation in the same buffer at 95°C for 5 min. The targeting sequence for sgIFT74 is 5′‐ATCGCCCTGTAACACAA‐3′.
Gateway cloning compatible lentiviral vector, pWPXLd/FLAG‐N/blast/DEST vector, was created by inserting FLAG/DEST/blasticidin resistance cassette into a second‐generation lentiviral vector, pWPXLd. pWPXLd/FLAG‐C/blast vector was created by inserting DEST/FLAG/blasticidin resistance cassette into pWPXLd vector. pWPXLd vector was a gift from Prof. Didier Trono (Addgene plasmid #12258). The lentiviral vectors were propagated in Stbl3‐competent cells to reduce unwanted recombination of long terminal repeat of the vectors. The gateway entry vector for H. sapiens IFT74 was created by fusion polymerase chain reaction (PCR) using pCMV‐SPORT6‐IFT74 (HsCD00339316) and pDNR‐LIB‐IFT74 (HsCD00340259, Harvard Plasmid) as templates, followed by BP recombination. The gateway entry vector for IFT74 T438R mutant was created via quick‐change mutagenesis using the entry vector for IFT74 described above. The quick‐change mutagenesis was performed by PCR with a complementary primer set (forward and reverse) that has a point mutation in the middle of the primers. Following the PCR, the PCR product was treated with 20 U of DpnI (R0176L, NEB) for 1 h at 37°C to eliminate the template and was then used to transform competent cells. Lentiviral vectors encoding N‐terminally FLAG‐tagged IFT74 (WT and T438R mutant) were created by LR recombination between the IFT74 entry vectors described above and pWPXLd/FLAG‐N/blast/DEST vector described above. The empty lentiviral vector used for control of the cell biological experiment was generated by LR recombination between pENTR1A vector without ccdB and the pWPXLd/FLAG‐C/blat vector described above.
Cell line, cell culture, transfection, and lentiviral expression
hTERT‐immortalized retinal pigment epithelial cells (hTERT RPE‐1) and 293T cells were grown in DMEM/F‐12 (12400‐024, Thermo Fisher Scientific) supplemented with 10% FBS (2461864RP, Gibco), 1× GlutaMax supplement (35050061, Gibco), and 100 U/ml penicillin–streptomycin (15140122, Thermo Fisher Scientific) at 37°C in 5% CO2. To induce cilium formation, cells were incubated in DMEM/F‐12 supplemented with 1× GlutaMax and 100 U/ml penicillin–streptomycin (serum‐free media). Both cell lines were authenticated via a short‐tandem repeat‐based test. The authentication was performed by MTCRO‐COBRE cell‐line authentication core of the University of Oklahoma Health Sciences Center. Mycoplasma negativity of the original cell lines (hTERT RPE‐1 and 293T) grown in antibiotics‐free media was confirmed by a PCR‐based test (G238, Applied Biological Materials).
Lentivirus carrying either gene of interest (IFT74 WT and T438R mutant) or sgIFT74 was produced by co‐transfecting 293T cells with 150 ng of pCMV‐VSV‐G, 350 ng of pCMV‐dR8.2 dvpr, and 500 ng of lentiviral transfer plasmids described above along with 3 μl of Fugene 6 (E2692, Promega) transfection reagent. Media were replaced 24 h after transfection to omit transfection reagent, and virus was harvested at 48 h post‐transfection. Virus was then filtered with a 0.45 μm PVDF filter (SLHV013SL, Millipore), mixed with fourfold volume of fresh media containing 12.5 μg/ml polybrene (TR‐1003‐G, Millipore), and was added to cells indicated in the figures. Following infection for 66 h, cells were selected with either 10 μg/ml puromycin (P9620, SIGMA‐Aldrich) or 10 μg/ml blasticidin (30‐100‐RB, Corning) for at least 10 days before subsequent analysis.
CRISPR knockout
hTERT RPE‐1 cells expressing BFP‐Cas9 were generated by infection with lentivirus carrying P293 Cas9‐BFP (a gift from Prof. Michael Bassik), followed by sorting BFP‐positive cells using FACSAria (BD). RPE‐BFP‐Cas9 cells were then infected with lentivirus carrying sgIFT74 in the pMCB306 vector to generate IFT74‐knockout cells. After selection with 10 μg/ml puromycin, cells were infected with adenovirus carrying Cre‐recombinase (1045N, Vector Biolabs) at a multiplicity of infection of 50 to remove the sgIFT74 puromycin resistance EGFP cassette. Ten days after adenovirus infection, GFP‐negative single cells were sorted using FACSAria. Single‐cell clones of IFT74‐knockout cells were expanded, and their knockout efficiency was determined by genomic PCR combined with inference of CRISPR edits (ICE) analysis (Conant et al, 2022).
Immunofluorescence
For wide‐field microscopy, cells were grown on acid‐washed 12 mm #1.5 round coverslips (72230‐10, Electron Microscopy Sciences) and fixed in 4% paraformaldehyde (15710, Electron Microscopy Sciences) in phosphate‐buffered saline (PBS) for 15 min at room temperature. After permeabilization with immunofluorescence (IF) buffer (3% bovine serum albumin [BP9703100, Fisher Scientific], 0.02% sodium azide [71448‐16, Fisher Scientific], and 0.1% IGEPAL [I8896‐100ML, SIGMA] in PBS) for 30 min at room temperature, cells were incubated with primary antibody in IF buffer for 4 h at room temperature, followed by rinsing with IF buffer five times. The samples were then incubated with fluorescent‐labeled secondary antibody in IF buffer for 1 h at room temperature, followed by rinsing with IF buffer five times. After nuclear staining with 4′,6‐diamidino‐2‐phenylindole (DAPI) (D9542‐1MG, SIGMA) in IF buffer at a final concentration of 0.5 μg/ml, coverslips were mounted with Fluoromount‐G (0100‐01, SouthernBiotech) onto glass slides (22‐339‐411, Epredia). Images were acquired on a Nikon Ti2 Live‐Cell Imaging Microscope equipped with Zyla 4.2 sCMOS camera (ZYLA‐4.2P‐USB3, ANDOR) and Sola light engine (SE 5‐LCR‐VB, Lumencor). 40× NA1.3 Plan‐Fluor objective lens (MRH00401, Nikon) was used for ciliation assays, and a 60× NA1.4 CFI Plan Apo Lambda objective lens (MRD01605, Nikon) was used for other analyses.
For ciliation assay, cells were plated into a six‐well plate at a density of 2 × 105 cells/well and grown for 48 h. Cells were serum starved for 24, 48, or 72 h and fixed in 4% paraformaldehyde (PFA). After the permeabilization step, cells were stained with anti‐ARL13B (17711‐1‐AP, Proteintech), anti‐CEP170 (41‐3200, Invitrogen), and anti‐acetylated tubulin (Ac‐Tub) antibodies (sc‐23950, Santa Cruz Biotechnology), washed, and then stained with goat anti‐rabbit Alexa Fluor 488 (A11034, Thermo Scientific), goat anti‐mouse IgG1‐Alexa Fluor 568 (A‐21124, Invitrogen), and goat anti‐mouse IgG2b Alexa Fluor 647 (A‐21242, Invitrogen). All the images were captured by focusing on CEP170 without looking at a channel of the ciliary proteins to avoid selecting specific area based on the percentage of ciliated cells. The structures extending from the centrosome and positive for both Ac‐Tub and ARL13B with a length of more than 1 μm were counted as primary cilia. At least six images from different fields per sample were captured for typical analysis. Typically, at least 200 cells were analyzed per sample in each experiment. The percentage of ciliated cells was manually counted using Nikon Elements Software.
For other immunofluorescence staining, cells were plated into a six‐well plate at a density of 2 × 105 cells/well and grown for 48 h. Cells were serum starved for 24 h or 72 h and fixed in 4% PFA. Primary antibodies used for the staining are anti‐FLAG (F1804‐50UG, SIGMA), anti‐CEP164 (22227‐1‐AP, Proteintech), anti‐CEP170 (41‐3200, Invitrogen), anti‐IFT81 (11744‐1‐AP, Proteintech), anti‐IFT88 (13967‐1‐AP, Proteintech), and rabbit anti‐RABL2 (CA5834, third bleed, affinity purified) antibodies (Kanie et al, 2017). Secondary antibodies used for the staining are goat anti‐rabbit Alexa Fluor 488 (A11034, Thermo Scientific), goat anti‐mouse IgG1‐Alexa Fluor 568 (A‐21124, Invitrogen), and goat anti‐mouse IgG2b Alexa Fluor 647 (A‐21242, Invitrogen). All the images were captured by focusing on CEP164 or CEP170 without looking at other channels to avoid selecting specific area based on the fluorescent intensity of protein of interest (POI). At least six images from different fields per sample were captured for typical analysis. Exact number of cells analyzed in each replicate is available through Figure Legends.
For 3D‐structured illumination microscopy (SIM), cells were grown on acid‐washed 12 mm #1.5 round coverslips (72230‐10, Electron Microscopy Sciences), fixed, and stained as described above. DAPI staining was not included for 3D‐SIM samples. Coverslips were mounted with Fluoromount‐G (0100‐01, SouthernBiotech). Images were acquired on a Nikon N‐SIM‐E/STORM super‐resolution microscope equipped with a 100×/1.49 NA CFI SR HP Apo TIRF 100XC Oil objective lens (MRD01995, Nikon), LU‐N3 compact laser unit (248071, Nikon), and ORCA‐Flash4.0 V3 Digital CMOS camera (C13440, HAMAMATSU). Image stacks of appropriate z‐steps to cover entire cilium (typically 0.7 μm) were taken in 0.1 μm increments to ensure Nyquist sampling. Images were then computationally reconstructed and subjected to image registration by using Nikon NIS‐Elements software. To count the number of IFT88 particles, maximum‐intensity projection was implemented on the reconstructed 3D‐SIM images, and look‐up table (LUT) was set to optimal range to reduce cytoplasmic IFT88 signal. The number of IFT88 particles that co‐localize with Ac‐Tub in each cilium was manually counted. The cilia length was measured by drawing a polysegment line from the distal to the proximal end using the Ac‐Tub channel. We deliberately avoided taking images of very short cilia (< 2 μm) in IFT74 KO‐expressing FLAG‐IFT74 T438R as some of the cilia in this cell line were very short, which could result in lower number of IFT particles.
Immunoblot
Cells were plated in a six‐well plate and grown to confluence. Cells were lysed in IGEPAL CA‐630 lysis buffer (25 mM HEPES buffer pH 7.4, 150 mM NaCl, and 0.3% IGEPAL® CA‐630 [9002‐93‐1, SIGMA]) containing 1× Halt™ protease and phosphatase inhibitor cocktail (100×) (78440, Thermo Fisher Scientific). Following clarification of the lysate by centrifugation at 15,000 g for 10 min at 4°C, the samples were mixed with 4× Laemmli sample buffer and 2‐mercaptoethanol (M3148, SIGMA). This ensured the final samples contained 50 mM Tris–HCl, 2% SDS, 10% glycerol, 0.005% brilliant blue, and 2.5% 2‐mercaptoethanol. The samples were then incubated at 95°C for 5 min. Proteins were separated in either 8% or 12% acrylamide gel in 1× SDS–PAGE running buffer (25 mM Tris base, 192 mM glycine, and 0.1% SDS) and transferred onto an Immobilon™‐FL PVDF transfer membrane (IPFL85R, EMD Millipore) in Towbin buffer (25 mM Tris and 192 mM glycine, pH 8.3). Membranes were incubated in TrueBlack® WB blocking buffer (23013T, Biotium) for 30 min at room temperature, and then probed overnight at 4°C with the appropriate primary antibody diluted in the TrueBlack® WB antibody diluent (23013B, Biotium). Next, the membrane was washed 3× 5 min in TBST buffer (20 mM Tris, 150 mM NaCl, and 0.1% Tween 20, pH 7.5) at room temperature, and incubated with the appropriate CF® antibodies (Biotium) diluted in the antibody diluent for 30 min at room temperature. After washing three times in TBST buffer, the membrane was scanned on an Odyssey Fc Imaging System (LI‐COR) and proteins were detected at wavelengths 700 and 800 nm. Primary antibodies used for immunoblotting were anti‐IFT74 (27334‐1‐AP, Proteintech Group), anti‐IFT81 (11744‐1‐AP, Proteintech Group), anti‐RABL2B antibody (CA5834 (in house), affinity purified), and anti‐α‐Tubulin antibody (sc‐32293, Santa Cruz Biotechnology). Secondary antibodies used for immunoblotting were CF®770 goat anti‐rabbit IgG (H + L), highly cross‐adsorbed (20078‐1, Biotium), and CF®680 goat anti‐mouse IgG (H + L), highly cross‐adsorbed (20065‐1, Biotium).
Experimental replicates
The term “replicates” used in this paper indicates that the same cell lines were plated at different dates for each experiment. In most cases, cell lines were thawed from liquid nitrogen at different dates, and immunostaining was performed at different dates among the replicates. For GTPase assays, all curves are the average of three technical replicates using the same batch of purified proteins for each replicate.
Quantification of fluorescent intensity and statistical analysis
The fluorescent intensity was measured with 16‐bit multicolor stack images acquired at 60× magnification (NA1.4) by using Image J software. To measure the fluorescent intensity of FLAG, IFT81, and IFT88, channels containing CEP170 (or CEP164) and the POI were individually extracted into separate images. A rolling ball background subtraction with a rolling ball radius of 5 pixels was implemented for both CEP170 (or CEP164) and the POI to perform local background subtraction. The mask for both CEP170 (or CEP164) and the POI was created by setting the lower threshold to the minimum level that covers only centrosome. Each mask was then combined by converting the two masks to a stack followed by z‐projection and then dilating the mask until the two masks were merged. After eroding the dilated masks several times, the fluorescent intensity of the POI was measured via “analyze particles” command with optimal size and circularity. The size and circularity are optimized for individual POI to detect most of the centrosomes in the image without capturing non‐centrosomal structure. Outliers (likely non‐centrosomal structure) were then excluded from the data using the ROUT method with a false discovery rate of 1% using GraphPad Prism 9 software. Fluorescent intensity of RABL2 was measured similarly to other proteins but using a single CEP170 mask because RABL2 shows high cytoplasmic signal, which is occasionally detected as a centrosomal mask.
For the immunoblot, the membrane was scanned in a LI‐COR Odyssey FC imager (Licor, USA) using the Image Studio software (Licor, USA) and analyzed with the ImageJ software. The obtained images were converted to 8‐bit format. Each band was individually selected and circumscribed with the rectangular ROI (region of interest) selection and quantified using “Plot Lanes” function. Data were acquired as arbitrary area values.
Statistical analysis
For ciliation assay, percentage of ciliated cells in each group from three experimental replicates were compared with that in every other group using two‐way analysis of variance (ANOVA) with Tukey's multiple‐comparison test. To test whether the difference in the signal intensity is statistically significant between groups, centrosomal signal intensity of POI from three experimental replicates was compared by using nested one‐way ANOVA with Šídák's multiple‐comparison test. For IFT particle quantification, the number of IFT88 particles from three experimental replicates were compared by using one‐way ANOVA (Appendix Fig S7A, D and G) or nested one‐way ANOVA (Fig 6K) with Tukey's multiple‐comparison test. Cilia length and IFT particles/μm of cilium were also analyzed through one‐way ANOVA (Appendix Fig S7B, C, E, F, H, and I) or nested one‐way ANOVA (Appendix Fig S6E and F) with Tukey's multiple‐comparison test. For all the statistics used in this paper, asterisks denote *: 0.01 ≤ P < 0.05, **: P < 0.01, ***: P < 0.001, n.s.: not significant. Statistical significance was calculated by using GraphPad Prism 9 software.
Author contributions
Esben Lorentzen: Conceptualization; data curation; formal analysis; supervision; funding acquisition; validation; investigation; visualization; methodology; project administration; writing – review and editing. Niels Boegholm: Conceptualization; data curation; formal analysis; supervision; validation; investigation; visualization; methodology; writing – original draft; project administration; writing – review and editing. Narcis A Petriman: Conceptualization; data curation; formal analysis; supervision; funding acquisition; validation; investigation; visualization; methodology; writing – original draft; project administration; writing – review and editing. Jiaolong Wang: Investigation; methodology; writing – original draft; writing – review and editing. Marta Loureiro‐López: Data curation; formal analysis; investigation; methodology; writing – original draft. Jens S Andersen: Conceptualization; data curation; formal analysis; supervision; funding acquisition; writing – original draft; project administration. Tomoharu Kanie: Formal analysis; supervision; funding acquisition; investigation; methodology; writing – original draft; project administration; writing – review and editing. Beibei Liu: Investigation; methodology. Peter K Jackson: Conceptualization; writing – original draft. Roy Ng: Investigation; methodology. Miren Itxaso Santiago Vela: Investigation; methodology.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix S1
Movie EV1
Movie EV2
Dataset EV1
Dataset EV2
Acknowledgements
We would like to thank Maximilian Stoetzel for contributing to preliminary experiments on RabL2 complexes and Anni Christensen and Kathrine Kjærgaard Frederiksen for technical assistance with protein purification. We acknowledge Michael Taschner for initial work on subcloning RabL2 constructs and for carefully reading this manuscript. We also thank Jesper Lykkegaard Karlsen for assistance with running AlphaFold and the EMCC computing facility at Aarhus University for computing time. This work was funded by grants from the Novo Nordisk Foundation (grant number NNF15OC00114164), the Independent Research Fund Denmark (grant no: 1026‐00016B), and the Carlsberg Foundation (grant no: CF22‐0971) to EL. NAP was supported by a postdoc fellowship from the European Commission (H2020, Grant Agreement number 888322). MLL and JSA were supported by the Independent Research Fund Denmark (grant no: 8021‐00425B) to JSA. We also thank Dr. David Sherry and Ms. Megan Stiles for their technical advice on the 3D‐SIM and Nikon Ti2 live cell imaging microscope experiments. The Nikon N‐SIM‐E/STORM super‐resolution microscope is supported by a Large Equipment Grant from the Oklahoma Center for Adult Stem Cell Research (OCASCR) and the OUHSC Department of Cell Biology. The Nikon Ti2 live‐cell imaging microscope is supported by an Equipment Grant from the Presbyterian Health Foundation (PHF, grant no: GRF00006006) and the OUHSC Department of Cell Biology. The cell authentication service performed by MTCRO‐COBRE cell line authentication core of the University of Oklahoma Health Science Center was supported partly by P20GM103639 of the National Institute of Health (NIH) and the National Cancer Institute Grant P30CA225520 of NIH.
The EMBO Journal (2023) 42: e111807
Data availability
The structural models are available in ModelArchive as ma‐4eif4 (https://www.modelarchive.org/doi/10.5452/ma‐4eif4) and ma‐4ct9w (https://www.modelarchive.org/doi/10.5452/ma‐4ct9w). The source data for Figs 1, 2, 3, 5, and 6 are deposited in BioImage Archives with the accession number S‐BSST1153 (https://www.ebi.ac.uk/biostudies/studies/S‐BSST1153). The raw images of the 3D structured illumination data shown in Fig 6J are deposited in BioStudies with the accession number S‐BIAD817 (https://www.ebi.ac.uk/biostudies/bioimages/studies/S‐BIAD817).
References
- Adhiambo C, Blisnick T, Toutirais G, Delannoy E, Bastin P (2009) A novel function for the atypical small G protein Rab‐like 5 in the assembly of the trypanosome flagellum. J Cell Sci 122: 834–841 [DOI] [PubMed] [Google Scholar]
- Antonny B, Schekman R (2001) ER export: public transportation by the COPII coach. Curr Opin Cell Biol 13: 438–443 [DOI] [PubMed] [Google Scholar]
- Avasthi P, Marshall W (2013) Ciliary secretion: switching the cellular antenna to ‘transmit’. Curr Biol 23: R471–R473 [DOI] [PubMed] [Google Scholar]
- Batth TS, MaximAX T, Rüther P, Gonzalez‐Franquesa A, Prabhakar BS, Bekker‐Jensen S, Deshmukh AS, Olsen JV (2019) Protein aggregation capture on microparticles enables multipurpose proteomics sample preparation. Mol Cell Proteomics 18: 1027–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhogaraju S, Taschner M, Morawetz M, Basquin C, Lorentzen E (2011) Crystal structure of the intraflagellar transport complex 25/27. EMBO J 30: 1907–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhogaraju S, Engel BD, Lorentzen E (2013) Intraflagellar transport complex structure and cargo interactions. Cilia 2: 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi X, Corpina RA, Goldberg J (2002) Structure of the Sec23/24–Sar1 pre‐budding complex of the COPII vesicle coat. Nature 419: 271–277 [DOI] [PubMed] [Google Scholar]
- Bi X, Mancias JD, Goldberg J (2007) Insights into COPII coat nucleation from the structure of Sec23•Sar1 complexed with the active fragment of Sec31. Dev Cell 13: 635–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson J, Chenouard N, Lagache T, Blisnick T, Olivo‐Marin J‐C, Bastin P (2013) Intraflagellar transport proteins cycle between the flagellum and its base. J Cell Sci 126: 327–338 [DOI] [PubMed] [Google Scholar]
- Cherfils J, Zeghouf M (2013) Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev 93: 269–309 [DOI] [PubMed] [Google Scholar]
- Chien A, Shih SM, Bower R, Tritschler D, Porter ME, Yildiz A (2017) Dynamics of the IFT machinery at the ciliary tip. Elife 6: e28606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole DG, Diener DR, Himelblau AL, Beech PL, Fuster JC, Rosenbaum JL (1998) Chlamydomonas Kinesin‐II–dependent Intraflagellar Transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory neurons. J Cell Biol 141: 993–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant D, Hsiau T, Rossi N, Oki J, Maures T, Waite K, Yang J, Joshi S, Kelso R, Holden K et al (2022) Inference of CRISPR Edits from Sanger Trace Data. CRISPR J 5: 123–130 [DOI] [PubMed] [Google Scholar]
- van Dam TJP, Townsend MJ, Turk M, Schlessinger A, Sali A, Field MC, Huynen MA (2013) Evolution of modular intraflagellar transport from a coatomer‐like progenitor. Proc Natl Acad Sci USA 110: 6943–6948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dateyama I, Sugihara Y, Chiba S, Ota R, Nakagawa R, Kobayashi T, Itoh H (2019) RABL2 positively controls localization of GPCRs in mammalian primary cilia. J Cell Sci 132: jcs224428 [DOI] [PubMed] [Google Scholar]
- Daumke O, Weyand M, Chakrabarti PP, Vetter IR, Wittinghofer A (2004) The GTPase‐activating protein Rap1GAP uses a catalytic asparagine. Nature 429: 197–201 [DOI] [PubMed] [Google Scholar]
- Deane JA, Cole DG, Seeley ES, Diener DR, Rosenbaum JL (2001) Localization of intraflagellar transport protein IFT52 identifies basal body transitional fibers as the docking site for IFT particles. Curr Biol 11: 1586–1590 [DOI] [PubMed] [Google Scholar]
- Dentler W (2005) Intraflagellar transport (IFT) during assembly and disassembly of Chlamydomonas flagella . J Cell Biol 170: 649–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X, Fragoza R, Singh P, Zhang S, Yu H, Schimenti JC (2020) Variants in RABL2A causing male infertility and ciliopathy. Hum Mol Genet 29: 3402–3411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong B, Wu S, Wang J, Liu Y‐X, Peng Z, Meng D‐M, Huang K, Wu M, Fan Z‐C (2017) Chlamydomonas IFT25 is dispensable for flagellar assembly but required to export the BBSome from flagella. Biol Open 6: 1680–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan S, Li H, Zhang Y, Yang S, Chen Y, Qiu B, Huang C, Wang J, Li J, Zhu X et al (2021) Rabl2 GTP hydrolysis licenses BBSome‐mediated export to fine‐tune ciliary signaling. EMBO J 40: e105499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutcher SK (2014) The awesome power of dikaryons for studying flagella and basal bodies in Chlamydomonas reinhardtii . Cytoskeleton 71: 79–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutcher SK, Li L, Lin H, Meyer L, Giddings TH Jr, Kwan AL, Lewis BL (2012) Whole‐genome sequencing to identify mutants and polymorphisms in Chlamydomonas reinhardtii . G3 (Bethesda) 2: 15–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguether T, San Agustin JT, Keady BT, Jonassen JA, Liang Y, Francis R, Tobita K, Johnson CA, Abdelhamed ZA, Lo CW et al (2014) IFT27 links the BBSome to IFT for maintenance of the ciliary signaling compartment. Dev Cell 31: 279–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliáš M, Klimeš V, Derelle R, Petrželková R, Tachezy J (2016) A paneukaryotic genomic analysis of the small GTPase RABL2 underscores the significance of recurrent gene loss in eukaryote evolution. Biol Direct 11: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel BD, Ludington WB, Marshall WF (2009) Intraflagellar transport particle size scales inversely with flagellar length: revisiting the balance‐point length control model. J Cell Biol 187: 81–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans R, O'Neill M, Pritzel A, Antropova N, Senior A, Green T, Žídek A, Bates R, Blackwell S, Yim J et al (2022) Protein complex prediction with AlphaFold‐Multimer. bioRxiv 10.1101/2021.10.04.463034 [PREPRINT] [DOI]
- Funabashi T, Katoh Y, Okazaki M, Sugawa M, Nakayama K (2018) Interaction of heterotrimeric kinesin‐II with IFT‐B–connecting tetramer is crucial for ciliogenesis. J Cell Biol 217: 2867–2876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R‐Y, Venter JC, Hutchison CA, Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343–345 [DOI] [PubMed] [Google Scholar]
- Glaser F, Pupko T, Paz I, Bell RE, Bechor‐Shental D, Martz E, Ben‐Tal N (2003) ConSurf: identification of functional regions in proteins by surface‐mapping of phylogenetic information. Bioinformatics 19: 163–164 [DOI] [PubMed] [Google Scholar]
- Götze M, Pettelkau J, Fritzsche R, Ihling CH, Schäfer M, Sinz A (2015) Automated assignment of MS/MS cleavable cross‐links in protein 3D‐structure analysis. J Am Soc Mass Spectrom 26: 83–97 [DOI] [PubMed] [Google Scholar]
- Guo Z, Hou X, Goody RS, Itzen A (2013) Intermediates in the guanine nucleotide exchange reaction of Rab8 protein catalyzed by guanine nucleotide exchange factors Rabin8 and GRAB*. J Biol Chem 288: 32466–32474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesketh SJ, Mukhopadhyay AG, Nakamura D, Toropova K, Roberts AJ (2022) IFT‐A structure reveals carriages for membrane protein transport into cilia. Cell 185: 4971–4985.e16 [DOI] [PubMed] [Google Scholar]
- Hibbard JVK, Vazquez N, Satija R, Wallingford JB (2021) Protein turnover dynamics suggest a diffusion‐to‐capture mechanism for peri‐basal body recruitment and retention of intraflagellar transport proteins. Mol Biol Cell 32: 1171–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Hoek H, Klena N, Jordan MA, Alvarez Viar G, Righetto RD, Schaffer M, Erdmann PS, Wan W, Geimer S, Plitzko JM et al (2022) In situ architecture of the ciliary base reveals the stepwise assembly of intraflagellar transport trains. Science 377: 543–548 [DOI] [PubMed] [Google Scholar]
- Hou Y, Pazour GJ, Witman GB (2004) A dynein light intermediate chain, D1bLIC, Is required for retrograde intraflagellar transport. Mol Biol Cell 15: 4382–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobucci C, Götze M, Ihling CH, Piotrowski C, Arlt C, Schäfer M, Hage C, Schmidt R, Sinz A (2018) A cross‐linking/mass spectrometry workflow based on MS‐cleavable cross‐linkers and the MeroX software for studying protein structures and protein–protein interactions. Nat Protoc 13: 2864–2889 [DOI] [PubMed] [Google Scholar]
- Inglis PN, Blacque OE, Leroux MR (2009) Chapter 14 – Functional genomics of intraflagellar transport‐associated proteins in C. elegans . In Methods in Cell Biology, King SM, Pazour GJ (eds), pp 267–304. Cambridge, MA: Academic Press; [DOI] [PubMed] [Google Scholar]
- Jakobsen L, Vanselow K, Skogs M, Toyoda Y, Lundberg E, Poser I, Falkenby LG, Bennetzen M, Westendorf J, Nigg EA et al (2011) Novel asymmetrically localizing components of human centrosomes identified by complementary proteomics methods. EMBO J 30: 1520–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual‐RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan MA, Diener DR, Stepanek L, Pigino G (2018) The cryo‐EM structure of intraflagellar transport trains reveals how dynein is inactivated to ensure unidirectional anterograde movement in cilia. Nat Cell Biol 20: 1250–1255 [DOI] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A et al (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596: 583–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanie T, Abbott KL, Mooney NA, Plowey ED, Demeter J, Jackson PK (2017) The CEP19‐RABL2 GTPase complex binds IFT‐B to initiate intraflagellar transport at the ciliary base. Dev Cell 42: 22–36.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keady BT, Samtani R, Tobita K, Tsuchya M, San Agustin JT, Follit JA, Jonassen JA, Subramanian R, Lo CW, Pazour GJ (2012) IFT25 links the signal‐dependent movement of hedgehog components to intraflagellar transport. Dev Cell 22: 940–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knödler A, Feng S, Zhang J, Zhang X, Das A, Peränen J, Guo W (2010) Coordination of Rab8 and Rab11 in primary ciliogenesis. Proc Natl Acad Sci USA 107: 6346–6351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozminski KG, Johnson KA, Forscher P, Rosenbaum JL (1993) A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc Natl Acad Sci USA 90: 5519–5523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozminski KG, Beech PL, Rosenbaum JL (1995) The Chlamydomonas kinesin‐like protein FLA10 is involved in motility associated with the flagellar membrane. J Cell Biol 131: 1517–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey SE, Foster HE, Pigino G (2023) The molecular structure of IFT‐A and IFT‐B in anterograde intraflagellar transport trains. Nat Struct Mol Biol 30: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, Ben‐Tal N (2005) ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res 33: W299–W302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechtreck K (2022) Cargo adapters expand the transport range of intraflagellar transport. J Cell Sci 135: jcs260408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechtreck K‐F, Johnson EC, Sakai T, Cochran D, Ballif BA, Rush J, Pazour GJ, Ikebe M, Witman GB (2009) The Chlamydomonas reinhardtii BBSome is an IFT cargo required for export of specific signaling proteins from flagella. J Cell Biol 187: 1117–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechtreck KF, Brown JM, Sampaio JL, Craft JM, Shevchenko A, Evans JE, Witman GB (2013) Cycling of the signaling protein phospholipase D through cilia requires the BBSome only for the export phase. J Cell Biol 201: 249–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Pang Y, Wu Q, Hu Z, Han X, Xu Y, Deng H, Pan J (2014) FLA8/KIF3B phosphorylation regulates kinesin‐II interaction with IFT‐B to control IFT entry and turnaround. Dev Cell 30: 585–597 [DOI] [PubMed] [Google Scholar]
- Liew GM, Ye F, Nager AR, Murphy JP, Lee JS, Aguiar M, Breslow DK, Gygi SP, Nachury MV (2014) The intraflagellar transport protein IFT27 promotes BBSome exit from cilia through the GTPase ARL6/BBS3. Dev Cell 31: 265–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo JCY, Jamsai D, O'Connor AE, Borg C, Clark BJ, Whisstock JC, Field MC, Adams V, Ishikawa T, Aitken RJ et al (2012) RAB‐Like 2 has an essential role in male fertility, sperm intra‐flagellar transport, and tail assembly. PLoS Genet 8: e1002969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludington WB, Wemmer KA, Lechtreck KF, Witman GB, Marshall WF (2013) Avalanche‐like behavior in ciliary import. Proc Natl Acad Sci USA 110: 3925–3930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv B, Wan L, Taschner M, Cheng X, Lorentzen E, Huang K (2017) Intraflagellar transport protein IFT52 recruits IFT46 to the basal body and flagella. J Cell Sci 130: 1662–1674 [DOI] [PubMed] [Google Scholar]
- McCafferty CL, Papoulas O, Jordan MA, Hoogerbrugge G, Nichols C, Pigino G, Taylor DW, Wallingford JB, Marcotte EM (2022) Integrative modeling reveals the molecular architecture of the intraflagellar transport A (IFT‐A) complex. Elife 11: e81977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meleppattu S, Zhou H, Dai J, Gui M, Brown A (2022) Mechanism of IFT‐A polymerization into trains for ciliary transport. Cell 185: 4986–4998.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijalkovic J, van Krugten J, Oswald F, Acar S, Peterman EJG (2018) Single‐molecule turnarounds of intraflagellar transport at the C. elegans ciliary tip. Cell Rep 25: 1701–1707.e2 [DOI] [PubMed] [Google Scholar]
- Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M (2022) ColabFold: making protein folding accessible to all. Nat Methods 19: 679–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra AK, Lambright DG (2016) Invited review: small GTPases and their GAPs. Biopolymers 105: 431–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishijima Y, Hagiya Y, Kubo T, Takei R, Katoh Y, Nakayama K (2017) RABL2 interacts with the intraflagellar transport‐B complex and CEP19 and participates in ciliary assembly. Mol Biol Cell 28: 1652–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nottingham RM, Ganley IG, Barr FA, Lambright DG, Pfeffer SR (2011) RUTBC1 protein, a Rab9A effector that activates GTP hydrolysis by Rab32 and Rab33B proteins. J Biol Chem 286: 33213–33222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai EF, Krengel U, Petsko GA, Goody RS, Kabsch W, Wittinghofer A (1990) Refined crystal structure of the triphosphate conformation of H‐ras p21 at 1.35 A resolution: implications for the mechanism of GTP hydrolysis. EMBO J 9: 2351–2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Eathiraj S, Munson M, Lambright DG (2006) TBC‐domain GAPs for Rab GTPases accelerate GTP hydrolysis by a dual‐finger mechanism. Nature 442: 303–306 [DOI] [PubMed] [Google Scholar]
- Pazour GJ, Dickert BL, Witman GB (1999) The DHC1b (DHC2) isoform of cytoplasmic dynein is required for flagellar assembly. J Cell Biol 144: 473–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen LB, Rosenbaum JL (2008) Chapter Two Intraflagellar Transport (IFT): role in ciliary assembly, resorption and signalling. In Current Topics in Developmental Biology, pp 23–61. Cambridge, MA: Academic Press; [DOI] [PubMed] [Google Scholar]
- Petriman NA, Loureiro‐López M, Taschner M, Zacharia NK, Georgieva MM, Boegholm N, Wang J, Mourão A, Russell RB, Andersen JS et al (2022) Biochemically validated structural model of the 15‐subunit intraflagellar transport complex IFT‐B. EMBO J 41: e112440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigino G, Geimer S, Lanzavecchia S, Paccagnini E, Cantele F, Diener DR, Rosenbaum JL, Lupetti P (2009) Electron‐tomographic analysis of intraflagellar transport particle trains in situ. J Cell Biol 187: 135–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H, Wang Z, Diener D, Rosenbaum J (2007) Intraflagellar transport protein 27 is a small G protein involved in cell‐cycle control. Curr Biol 17: 193–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quidwai T, Wang J, Hall EA, Petriman NA, Leng W, Kiesel P, Wells JN, Murphy LC, Keighren MA, Marsh JA et al (2021) A WDR35‐dependent coat protein complex transports ciliary membrane cargo vesicles to cilia. Elife 10: e69786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum JL, Witman GB (2002) Intraflagellar transport. Nat Rev Mol Cell Biol 3: 813–825 [DOI] [PubMed] [Google Scholar]
- Schafer JC, Winkelbauer ME, Williams CL, Haycraft CJ, Desmond RA, Yoder BK (2006) IFTA‐2 is a conserved cilia protein involved in pathways regulating longevity and dauer formation in Caenorhabditis elegans . J Cell Sci 119: 4088–4100 [DOI] [PubMed] [Google Scholar]
- Scheffzek K, Ahmadian MR (2005) GTPase activating proteins: structural and functional insights 18 years after discovery. Cell Mol Life Sci CMLS 62: 3014–3038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffzek K, Shivalingaiah G (2019) Ras‐specific GTPase‐activating proteins—structures, mechanisms, and interactions. Cold Spring Harb Perspect Med 9: a031500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seewald MJ, Körner C, Wittinghofer A, Vetter IR (2002) RanGAP mediates GTP hydrolysis without an arginine finger. Nature 415: 662–666 [DOI] [PubMed] [Google Scholar]
- Shalata A, Ramirez MC, Desnick RJ, Priedigkeit N, Buettner C, Lindtner C, Mahroum M, Abdul‐Ghani M, Dong F, Arar N et al (2013) Morbid obesity resulting from inactivation of the ciliary protein CEP19 in humans and mice. Am J Hum Genet 93: 1061–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva DA, Huang X, Behal RH, Cole DG, Qin H (2012) The RABL5 homolog IFT22 regulates the cellular pool size and the amount of IFT particles partitioned to the flagellar compartment in Chlamydomonas reinhardtii . Cytoskeleton 69: 33–48 [DOI] [PubMed] [Google Scholar]
- Sorokin S (1962) Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J Cell Biol 15: 363–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanek L, Pigino G (2016) Microtubule doublets are double‐track railways for intraflagellar transport trains. Science 352: 721–724 [DOI] [PubMed] [Google Scholar]
- Sun W‐Y, Xue B, Liu Y‐X, Zhang R‐K, Li R‐C, Xin W, Wu M, Fan Z‐C (2021) Chlamydomonas LZTFL1 mediates phototaxis via controlling BBSome recruitment to the basal body and its reassembly at the ciliary tip. Proc Natl Acad Sci USA 118: e2101590118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschner M, Lorentzen E (2016a) Recombinant reconstitution and purification of the IFT‐B core complex from Chlamydomonas reinhardtii . In Cilia: Methods and Protocols, Satir P, Christensen ST (eds), pp 69–82. New York, NY: Springer New York; [DOI] [PubMed] [Google Scholar]
- Taschner M, Lorentzen E (2016b) The intraflagellar transport machinery. Cold Spring Harb Perspect Biol 8: a028092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschner M, Kotsis F, Braeuer P, Kuehn EW, Lorentzen E (2014) Crystal structures of IFT70/52 and IFT52/46 provide insight into intraflagellar transport B core complex assembly. J Cell Biol 207: 269–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschner M, Weber K, Mourão A, Vetter M, Awasthi M, Stiegler M, Bhogaraju S, Lorentzen E (2016) Intraflagellar transport proteins 172, 80, 57, 54, 38, and 20 form a stable tubulin‐binding IFT‐B2 complex. EMBO J 35: 773–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter M, Stehle R, Basquin C, Lorentzen E (2015) Structure of Rab11–FIP3–Rabin8 reveals simultaneous binding of FIP3 and Rabin8 effectors to Rab11. Nat Struct Mol Biol 22: 695–702 [DOI] [PubMed] [Google Scholar]
- Wachter S, Jung J, Shafiq S, Basquin J, Fort C, Bastin P, Lorentzen E (2019) Binding of IFT22 to the intraflagellar transport complex is essential for flagellum assembly. EMBO J 38: e101251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Fan Z‐C, Williamson SM, Qin H (2009) Intraflagellar Transport (IFT) protein IFT25 Is a phosphoprotein component of IFT complex B and physically interacts with IFT27 in Chlamydomonas . PloS One 4: e5384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield JL, Mengoni I, Bomberger H, Jiang Y‐Y, Walsh JD, Brown JM, Picariello T, Cochran DA, Zhu B, Pan J et al (2017) IFT trains in different stages of assembly queue at the ciliary base for consecutive release into the cilium. Elife 6: e26609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield JL, Mekonnen B, Mengoni I, Liu P, Jordan M, Diener D, Pigino G, Lechtreck K (2021) In vivo imaging shows continued association of several IFT‐A, IFT‐B and dynein complexes while IFT trains U‐turn at the tip. J Cell Sci 134: jcs259010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittinghofer A, Vetter IR (2011) Structure‐function relationships of the G domain, a canonical switch motif. Annu Rev Biochem 80: 943–971 [DOI] [PubMed] [Google Scholar]
- Wong ACC, Shkolny D, Dorman A, Willingham D, Roe BA, McDermid HE (1999) Two novel human RAB genes with near identical sequence each map to a telomere‐associated region: the subtelomeric region of 22q13.3 and the ancestral telomere band 2q13. Genomics 59: 326–334 [DOI] [PubMed] [Google Scholar]
- Xue B, Liu Y‐X, Dong B, Wingfield JL, Wu M, Sun J, Lechtreck KF, Fan Z‐C (2020) Intraflagellar transport protein RABL5/IFT22 recruits the BBSome to the basal body through the GTPase ARL6/BBS3. Proc Natl Acad Sci USA 117: 2496–2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Qiu H, Castro‐Araya R‐F, Takei R, Nakayama K, Katoh Y (2022) Impaired cooperation between IFT74/BBS22–IFT81 and IFT25–IFT27/BBS19 causes Bardet‐Biedl syndrome. Hum Mol Genet 31: 1681–1693 [DOI] [PubMed] [Google Scholar]