

Abstract
Biotin-thiamine responsive basal ganglia disease is an inborn error of metabolism caused by mutations in SLC19A3, encoding a transporter of thiamine across the plasma membrane. We report a novel mutation identified in the homozygous state in a patient with typical brain MRI changes. In addition, this patient had markedly elevated CSF pyruvate, a low lactate-to-pyruvate molar ratio, and an abnormal pyruvate peak at 2.4 ppm on brain magnetic resonance spectroscopy. Using aggregated exome sequencing data, we calculate the carrier frequency of mutations in SLC19A3 as 1 in 232 individuals in the general population, for an estimated prevalence of the disease of approximately 1 in 215,000 individuals. The disease is thus more frequent than previously recognized, and the presence of a pyruvate peak on spectroscopy could serve as an important diagnostic clue.
Keywords: biotin-thiamine responsive basal ganglia disease, magnetic resonance spectroscopy, pyruvate, SLC19A3, thiamine metabolism dysfunction syndrome 2, thiamine transporter-2 deficiency
1 |. INTRODUCTION
Biotin-thiamine responsive basal ganglia disease (BTBGD), also known as thiamine transporter-2 deficiency, or thiamine metabolism dysfunction syndrome 2 (OMIM #607483) is an autosomal recessive condition caused by biallelic mutations in SLC19A3. The disease was initially named biotin-responsive basal ganglia disease, but once it was recognized that the culprit gene encodes a thiamine transporter and not a biotin transporter, its nomenclature was changed to BTBGD (Alfadhel et al., 2013). This inborn error of metabolism presents with neurologic dysfunction and associated bilateral basal ganglia lesions on brain imaging, both of which can worsen intermittently. The severity of the disease is quite variable, ranging from Leigh syndrome in infancy (Haack et al., 2014), to Wernicke’s encephalopathy in later life (Kono et al., 2009). Since its initial description in 1998 (Ozand et al., 1998), there have been 116 affected individuals reported in the medical literature, and 21 mutations in SLC19A3 have been described so far. Here, we report a new patient with typical neurologic and brain imaging findings of BTBGD, and an additional pyruvate peak on brain magnetic resonance spectroscopy (MRS) at 2.4 ppm—present during an acute crisis, but absent at baseline. The patient was found to have a previously unreported homozygous mutation in SLC19A3. Given the rarity of the condition, the prevalence of BTBGD remains unknown. We calculated its prevalence based on next generation sequencing data collected in the Exome Aggregation Consortium (ExAC), a cohort of 60,706 exomes (Lek et al., 2016) obtained from unrelated individuals, not ascertained for BTBGD. The calculated prevalence demonstrates that BTBGD remains likely underdiagnosed.
2 |. PATIENT AND METHODS
2.1 |. Clinical report
The patient was born at 41 weeks’ gestation after an uneventful pregnancy and delivery. Over the course of the first 2 years of life, she was noted to be delayed, as she did not sit up unsupported until 10 months of age, and did not start walking until 2 years of age. A neurological exam obtained at 2 years and 5 months old revealed truncal hypotonia with appendicular hypertonia, with a wide-based stiff gait and hyperreflexia of lower extremities without clonus. Further recommended workup included a normal chromosome microarray, plasma amino acids, urine organic acids, and plasma acylcarnitine profile. Lactate concentration was 18.4 mg/dl (2.04 mmol/L, reference range: 9–16 mg/dl), while pyruvate was normal at 0.84 mg/dl (0.095 mmol/L, ref: 0.3–1.5 mg/dl), for a lactate-to-pyruvate molar ratio of 21 (normal: 10–20). Baseline brain magnetic resonance imaging (MRI) and MRS were performed at 2 years and 6 months (see Neuroimaging section below), and an electroencephalogram performed at 2 years and 8 months was normal.
At 3 years and 1 month the patient was admitted with a 1-week history of intermittent subjective fever and emesis 4–5 times a day, followed by a 1-day history of decreased alertness and gait instability. Her lactate and pyruvate levels were normal one day after hospital admission, with a lactate of 1.5mmol/L (ref: 1–2.4), a pyruvate of 0.87 mg/dl (0.099mmol/L), and a molar ratio of 15.2.A lumbar puncture on hospital day 4 was unrevealing; neurotransmitters were sent showing low CSF tetrahydrobiopterin at 10 nmol/L (ref: 18–50), but normal neopterin at 20 nmol/L (ref: 7–65) and normal 5-methyltetrahydrofolate at 79 nmol/L (ref: 40–150). The CSF pyruvate was markedly elevated at >3 mg/dl (>0.34 mmol/L; ref: 0.54–1.7 mg/dl), lactate 2.8 mmol/L (ref: 1.11–2.44), and a molar ratio of <8.5, which was reported as compatible with pyruvate dehydrogenase deficiency. CSF amino acid concentrations were unremarkable, including a normal alanine level of 19.5 umol/L (ref: 12.6–34.7). On hospital day 5 the patient developed hypoxemia and impending respiratory failure requiring intubation. She also had moderately decreased left ventricular systolic function with severe septal hypokinesis, with a fraction of shortening of 17.3 % (ref: 28–40%) and an ejection fraction of 45.7 % (ref: 55–70%), requiring pressors. At this point, her lactate levels peaked at 13.78 mmol/L, and a new brain MRI with MRS was obtained that same day (see below).
She had a 7-week hospital stay, during which she developed other complications including seizures, responsive to levetiracetam. She eventually required a tracheostomy, and was discharged on minimal ventilator settings. Of note, a dual genome sequencing panel—including all 37 mitochondrial genes and over 100 nuclear genes encoding mitochondrial proteins—was obtained during this admission, and no variants were found.
When seen for follow-up at the age of 3 years and 5 months, the patient remained non-verbal. After the diagnosis of BTBGD was made via whole exome sequencing, she was placed on thiamine 750 mg/day (25 mg/kg/d) and biotin 30 mg/d (1 mg/kg/d), with no further metabolic decompensations by the age of 5 years and 1 month.
2.2 |. Neuroimaging
Brain MRI and MRS were initially performed at 2 years and 6 months of life on a 3T magnet (General Electric, Milwaukee, WI). The following pulse sequences of the brain were acquired: sagittal spoiled gradient echo (SPGR) T1WI, axial T2WI, axial T2 FLAIR, axial susceptibility weighted angiography (SWAN), coronal fat-saturated T2WI, and axial diffusion tensor imaging (DTI) with seven non-collinear directions of encoding. Single voxel MRS was performed with a 2 × 2 × 2 cm voxel over the left cerebral deep gray nuclei (TR 1500; TE 35 and 288 msec).
A follow-up brain MRI was performed at age 3 years and 2 months (3T MRI; General Electric) with similar sequences. Axial arterial spin-labeling (ASL) perfusion images and coronal high resolution T2WI through the hippocampi were also acquired. Single voxel MRS was performed with a 2 × 2 × 2 cm voxel over the left cerebral deep gray nuclei (TR 1500; TE 144 msec) and left parietal white matter (TR 1500; TE 35 and 144 msec).
2.3 |. Pyruvate dehydrogenase enzyme activity
Pyruvate dehydrogenase (PDH) complex activity was assayed at 3 years and 9 months in fresh isolated blood lymphocytes by measuring the decarboxylation of 1-14C-pyruvate in the presence of thiamine pyrophosphate, NAD+ and coenzyme A—both after activation with dichloroacetate and inactivation with fluoride—as previously described (Kerr et al., 1987; Sheu, Hu, & Utter, 1981).
2.4 |. Whole exome sequencing (WES)
WES was obtained at a commercial lab at the age of 3 years and 9 months. Briefly, fragmented DNA was ligated to paired-end adapters. Target enrichment was achieved as previously described (Bainbridge et al., 2011), sequencing was performed on an Illumina HiSeq platform (Illumina Inc, San Diego, CA), and short reads were aligned using the Burrows–Wheeler transform (Li & Durbin, 2010). Data analysis and interpretation was performed by using the Mercury analysis pipeline.
2.5 |. Calculation of prevalence
We queried all published SLC19A3 mutations in the literature, as well as performed a gene search of the Human Gene Mutation Database (HGMD®) (Stenson et al., 2014). The pathogenicity of these variants was assessed by reviewing publications with clinical and/or functional data. To be considered pathogenic, the variant had to be reported in a patient with a classical phenotype who had biallelic variants in SLC19A3.
The Exome Aggregation Consortium (ExAC) was used to obtain the allele frequencies of the SLC19A3 pathogenic variants, and it also allowed us to identify potentially pathogenic variants that may be predicted damaging to SLC19A3 function, but that have not been described int the medical literature. ExAC is a database of 121,412 alleles from 60,706 unrelated individuals (Lek et al., 2016), including 33,370 non-Finnish European individuals, 8,256 South Asians, 5,789 individuals of Latino descent, 5,203 individuals of African or African-American descent, 4,327 East Asians, 3,307 persons of Finnish heritage, and 454 people of other ethnicity. In order to perform a bioinformatic analysis of variant potential pathogenicity, we queried ExAC for canonical splice site, missense, frameshift, nonsense (stop gain), and stop loss mutations. None of the cohorts or consortia in ExAC includes patients ascertainedfor the presence of BTBGD, so we considered them to be unbiased with respect to variation in the SLC19A3 gene. Variants were excluded if: (1) they had a minor allele frequency of >1% in any population; (2) they were found in sites covered in fewer than 80% of individuals, as this may indicate a low-quality site; (3) they were only present in a non-canonical transcript; and (4) they were in an untranslated region. Variants that passed these filters were evaluated for their potential to alter protein function by using in silico prediction models: PolyPhen-2 (Adzhubei et al., 2010), SIFT (Kumar, Henikoff, & Ng, 2009), and the Combined Annotation-Dependent Depletion (CADD) score (Kircher et al., 2014). Unpublished variants predicted to be benign by one or more in silico models were not considered for the calculation of prevalence. For likely pathogenic variants, such as frameshift, stop-gain (non-sense), and canonical splice site mutations, only the CADD Phred-scaled score was provided.
The carrier frequency was calculated as the number of individuals carrying an SLC19A3 variant known or predicted to alter protein function, divided by the total number of individuals ascertained. The prevalence of the disease was calculated based on the carrier frequency and/or the allele frequency using Hardy–Weinberg equilibrium. The Hardy–Weinberg principle establishes that p2 + 2pq + q2 = 1, with p being the reference allele frequency, q being the minor allele frequency, 2pq the carrier frequency, and q2 the frequency of the disease.
3 |. RESULTS
3.1 |. Neuroimaging findings
Brain MRI and MRS at 2 years and 6 months (Figure 1a–d): small hyperintense lesions were present in the putamen and right caudate head that demonstrated T1 and T2 prolongation, partial T2 FLAIR signal suppression, and facilitated diffusion without mass effect, consistent with chronic areas of encephalomalacia, and necrosis. Trace susceptibility was present in associated putaminal lesions, likely representing hemosiderin from prior hemorrhage. Brain parenchyma was otherwise normal in signal intensity for age. Bilateral hippocampal dysgenesis was also noted; the hippocampi were globular and dysmorphic in appearance. MRS of the left basal ganglia demonstrated normal metabolic ratios for age. No abnormal pyruvate or lactate was detected.
FIGURE 1.
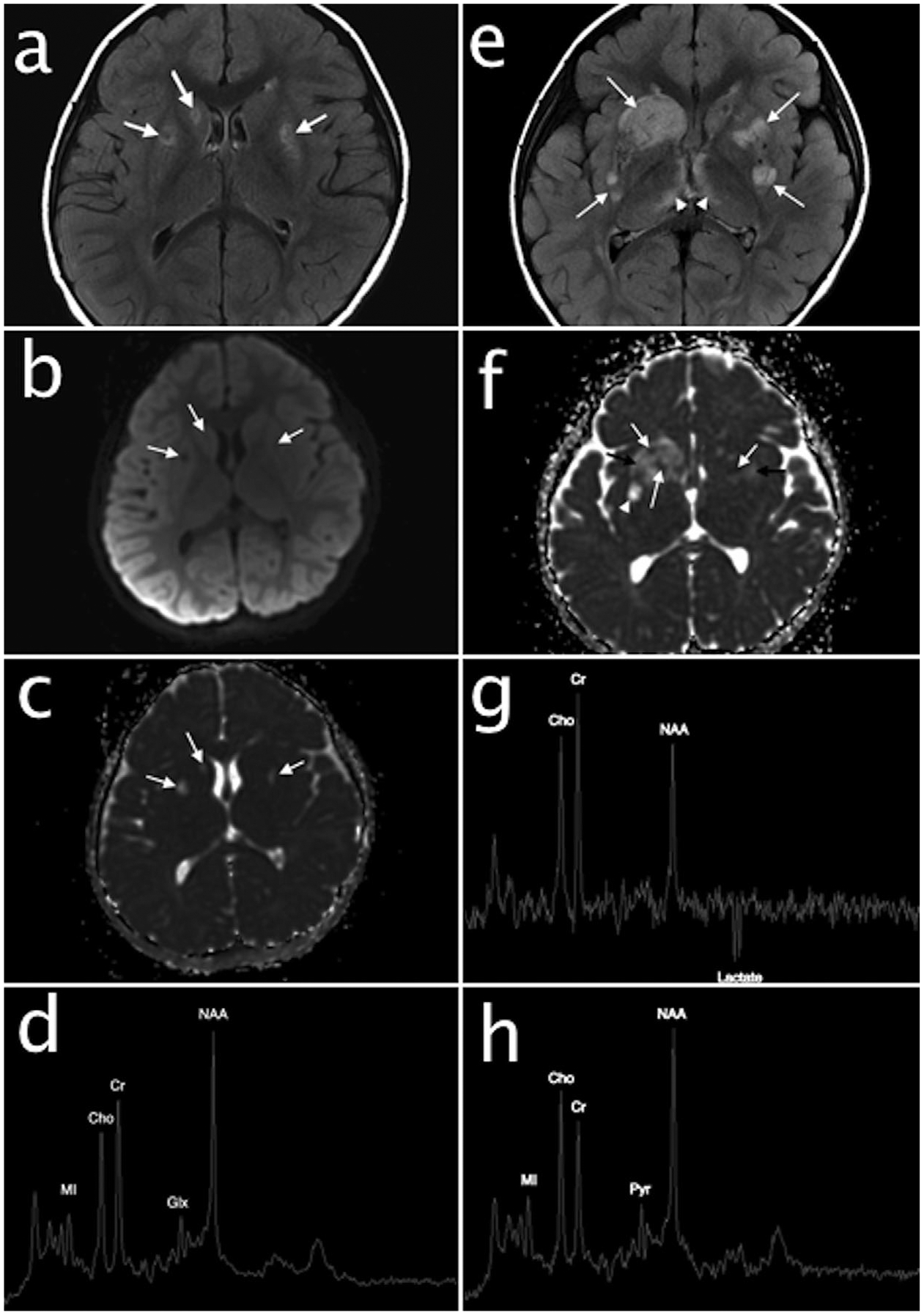
Baseline brain MRI and MRS performed at 2 years and 6 months. Axial T2 FLAIR image (a) and diffusion weighted images (b and c) demonstrates hyperintense lesions in the putamen and right caudate head with partial FLAIR signal suppression (arrows, a) and facilitated diffusion (arrows, b and c) consistent with areas of chronic encephalomalacia and necrosis. Short echo single voxel MRS (d) over the basal ganglia shows normal metabolic ratios; no abnormal pyruvate or lactate are present. Follow-up brain MRI and MRS at 3 years and 2 months. Axial T2 FLAIR (e) demonstrates acute on chronic cerebral deep gray nuclear lesions with new hyperintense lesions in the striatum (arrows) and medial thalami (arrowheads). Mixed diffusion abnormalities in the lesions (arrows, f) represent a combination of cytotoxic edema (restricted diffusion, white arrows), vasogenic edema (facilitated diffusion with mass effect, black arrows), and encephalomalacia/necrosis (facilitated diffusion without mass effect, arrowhead). Intermediate echo single voxel MRS (g) over the basal ganglia reveals an inverted lactate doublet at 1.3 ppm consistent with anaerobic metabolism and a decreased NAA to creatine ratio consistent with neuronal loss. Short echo MRS (h) over the left parietal white matter demonstrates an unusually prominent peak at 2.4 ppm consistent with pyruvate (Pyr). NAA, N-acetylaspartate; Cho, choline; Cr, creatine; MI, myoinositol
Brain MRI and MRS at 3 years and 2 months (Figure 1e–h): multiple new brain lesions intervally developed in the striatum, thalami, hypothalami, midbrain, pons, and medulla oblongata. The new lesions in the striatum and thalami demonstrated hyperintense signal on T2WI, mass effect, and mixed restricted and facilitated diffusion representing a combination of cytotoxic and vasogenic edema. The remainder of the brain was normal in signal. MRS of the left basal ganglia revealed moderately decreased NAA to creatine ratio, consistent with neuronal loss, and a moderate lactate peak. MRS of the left parietal white matter demonstrated an elevated peak at 2.4 ppm, consistent with pyruvate. Although protons associated with glutamine and glutamate (Glx) co-resonate in this location, adjacent peaks associated with these protons were not elevated and there was no elevation Glx alpha proton at 3.8 ppm to support glutamine and/or glutamate elevation.
3.2 |. PDH enzyme testing
The presence of a pyruvate peak on brain MRS had previously been reported in a case of PDH deficiency (Zand et al., 2003). This, in addition to the elevated CSF pyruvate—above the limit of quantitation—with a decreased lactate-to-pyruvate ratio raised suspicion for PDH deficiency. Pyruvate dehydrogenase complex activity in lymphocytes was thus obtained, but it revealed an activity of 3.28 nmol/min/mg protein (ref: 0.98–2.72), relatively high in reference to concurrent and prior controls, corresponding to 201% of the control mean. The assay showed good activation and inactivation after pre-incubation with dichloroacetate and fluoride, respectively. The activity of dihydrolipoamide dehydrogenase (E3) was within the reference range, and the ratio of PDC/E3 was 3.6 (ref: 1.41–3.55), also above the reference range at 156%. Thus, the activity was relatively high both relative to total cell protein and to the activity of the internal mitochondrial reference enzyme, E3.
3.3 |. Whole exome sequencing
A homozygous variant of unknown significance was detected in the SLC19A3 gene (NM_025243.3:c.416T>A; p.Val139Glu). Both parents were noted to be heterozygous for this change. This variant is exceedingly rare, as it was not found in ExAC. It is also well conserved through evolution (see Supplementary Figure S1), and prediction models suggest this change is probably damaging (Polyphen-2) or deleterious (SIFT), with a Phred-scaled CADD score of 27.1. In addition, a novel de novo heterozygous frameshift variant was detected in the DDX3X gene (NM_001356.3:c.14_17delCAGT; p.Ala5Glyfs*14), associated with X-linked dominant intellectual disability (OMIM#300958) (Snijders Blok et al., 2015).
3.4 |. Calculation of prevalence
The allele count for known pathogenic variants was 19 (see Table 1), while the allele count for predicted pathogenic variants was 243 (see Table 2), for a total of 262 variants found in 121,412 alleles. The allele frequency is thus 0.216%, or 1 in 463, for a carrier frequency of 1 in 232 individuals in the general population. The estimated prevalence of the disease is thus calculated as 1 in 214,744 individuals.
TABLE 1.
Known pathogenic variants in SLC19A3
| In silico prediction | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Genomic coordinate (GRCh37/hg19) | cDNA (NM_025243.3) | Protein (NP_079519.1) | dbSNP ID | Type of mutation | Polyphen-2 | SIFT | CADD Phred | Allele number | Allele count | Allele frequency | Reference |
| chr2:228,568,440–228,613,489 | – | – | – | Promoter deletion | – | – | – | – | – | – | Flønes et al. (2016) |
| chr2:228567015 | c.20C>A | p.Ser7* | – | Stop-gain | – | – | 35 | – | – | – | Gerards et al. (2013) |
| chr2:228566967 | c.68G>T | p.Gly23Val | rs121917882 | Missense | Probably damaging | Deleterious | 28.1 | 121372 | 1 | 0.0008239% | Kevelam et al. (2013); Pérez-Dueñas et al. (2013); Zeng et al. (2005) |
| chr2:228566960 | c.74dupT | p.Ser26Leufs*19 | – | Frameshift | – | – | 35 | – | – | – | Debs et al. (2010); Sremba, Chang, Elbalalesy, Cambray-Forker, & Abdenur, (2014) |
| chr2:228566953–228566954 | c.81_82dupGA | p.Met28Argfs*2 | – | Frameshift | – | – | 29.6 | 121374 | 2 | 0.001648% | Sremba et al. (2014) |
| chr2:228566905 | c.130A>G | p.Lys44Glu | rs137852957 | Missense | Possibly damaging | Deleterious | 25.5 | – | – | – | Kono et al. (2009); Miyajima and Kono (2010) |
| chr2:228564094 | C.337T>C | p.Tyr113His | rs145999922 | Missense | Probably damaging | Deleterious | 25.5 | 121402 | 7 | 0.005766% | Flønes et al. (2016) |
| chr2:228563967 | c.464C>T | p.Ser155Leu | – | Missense | Probably damaging | Deleterious | 27.3 | – | – | – | Kohrogi et al. (2015) |
| chr2:228563924 | c.507C>G | p.Tyr169* | – | Stop-gain | – | – | 35 | – | – | – | Kevelam et al. (2013) |
| chr2:228563914 | c.517A>G | p.Asn173Asp | – | Missense | Probably damaging | Tolerated | 18.23 | – | – | – | Fassone et al. (2013) |
| chr2:228563904 | c.527C>A | p.Ser176Tyr | – | Missense | Probably damaging | Deleterious | 22.3 | 121400 | 1 | 0.00082% | Kevelam et al. (2013) |
| chr2:228563890 | C.541T>C | p.Ser181Pro | – | Missense | Probably damaging | Tolerated | 22.1 | 121404 | 11 | 0.00906% | Flønes et al. (2016); Kevelam et al. (2013) |
| chr2:228563506–228563536 | c.895_925del31 | p.Val299Profs*11 | – | Frameshift | – | – | 35 | – | – | – | Kevelam et al. (2013) |
| chr2:228563473 | c.958G>C | p.Glu320Gln | – | Missense | Probably damaging | Deleterious | 29.7 | – | – | – | Kono et al. (2009); Miyajima and Kono (2010); Yamada et al. (2010) |
| chr2:228560811 | c.980–14A>G | – | rs200542114 | Exon 4 skipping | – | – | 0.223 | 120582 | 3 | 0.002488% | Debs et al. (2010) |
| chr2:228560795 | c.982delG | p.Ala328Leufs*10 | – | Frameshift | – | – | 35 | – | – | – | Haack et al. (2014) |
| chr2:228560623 | c.1154T>G | p.Leu385Arg | rs563607795 | Missense | Probably damaging | Deleterious | 33 | 121338 | 1 | 0.0008241% | Kevelam et al. (2013) |
| chr2:228552882–228553023 | r.1173_1314del | p.Gln393* | – | Exon 5 deletion | – | – | – | – | – | – | Kevelam et al. (2013) |
| chr2:228553000 | c.1196A>T | p.Asn399Ile | – | Missense | Benign | Deleterious | 24 | – | – | – | Kohrogi et al. (2015) |
| chr2:228552932 | c.1264A>G | p.Thr422Ala | rs121917884 | Missense | Benign | Deleterious | 23.5 | – | – | – | Alfadhel et al. (2013); Distelmaier et al. (2014); Zeng et al. (2005) |
| chr2:228552272 | c.1332C>G | p.Ser444Arg | – | Missense | Possibly damaging | Deleterious | 26.3 | – | – | – | Kevelam et al. (2013) |
TABLE 2.
Predicted pathogenic variants in SLC19A3
| Genomic coordinate (GRCh37/hg19) | cDNA (NM_025243.3) | Protein (NP_079519.1) | dbSNP ID | Type of mutation | Polyphen-2 | SIFT | CADD Phred | Allele number | Allele count | Allele frequency (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| chr2:228566999 | c.36G>C | p.Trp12Cys | – | Missense | Probably damaging | Deleterious | 32 | 121308 | 1 | 0.0008243 |
| chr2:228566979 | C.56T>C | p.Leu19Pro | rs142553867 | Missense | Probably damaging | Deleterious | 27.9 | 121356 | 2 | 0.001648 |
| chr2:228566967 | c.68G>A | p.Gly23Asp | rs121917882 | Missense | Probably damaging | Deleterious | 26.9 | 121372 | 1 | 0.0008239 |
| chr2:228566935 | c.100T>G | p.Phe34Val | rs201603239 | Missense | Probably damaging | Deleterious | 29.8 | 121370 | 3 | 0.002472 |
| chr2:228566923 | c.112T>C | p.Tyr38His | rs368305641 | Missense | Possibly damaging | Deleterious | 25.9 | 121356 | 2 | 0.001648 |
| chr2:228566922 | c.113A>G | p.Tyr38Cys | – | Missense | Probably damaging | Deleterious | 25 | 121358 | 1 | 0.000824 |
| chr2:228566883 | c.150 + 2T>C | – | – | Canonical splice | – | – | 23.3 | 121304 | 1 | 0.0008244 |
| chr2:228564282 | c.151–2A>T | – | – | Canonical splice | – | – | 23.2 | 120730 | 1 | 0.0008283 |
| chr2:228564259 | c.172G>T | p.Val58Phe | rs200412423 | Missense | Probably damaging | Deleterious | 23.8 | 121114 | 1 | 0.0008257 |
| chr2:228564244 | c.187T>C | p.Tyr63His | rs144817990 | Missense | Probably damaging | Deleterious | 24.3 | 121286 | 14 | 0.01154 |
| chr2:228564225 | c.206C>T | p.Pro69Leu | – | Missense | Probably damaging | Deleterious | 25.6 | 121356 | 1 | 0.000824 |
| chr2:228564223 | c.208G>C | p.Val70Leu | – | Missense | Probably damaging | Deleterious | 25.7 | 121356 | 2 | 0.001648 |
| chr2:228564216 | c.215T>C | p.Val72Ala | – | Missense | Possibly damaging | Deleterious | 25.4 | 121382 | 1 | 0.0008238 |
| chr2:228564214 | c.217C>T | p.Leu73Phe | rs373943165 | Missense | Possibly damaging | Deleterious | 25.2 | 121384 | 2 | 0.001648 |
| chr2:228564189 | c.242C>T | p.Pro81Leu | – | Missense | Probably damaging | Deleterious | 27.5 | 121402 | 2 | 0.001647 |
| chr2:228564177 | C.254T>C | p.Leu85Ser | – | Missense | Possibly damaging | Deleterious | 22.7 | 121404 | 1 | 0.0008237 |
| chr2:228564126 | c.305G>A | p.Gly102Glu | – | Missense | Probably damaging | Deleterious | 31 | 121412 | 1 | 0.0008236 |
| chr2:228564112 | c.319C>A | p.Gln107Lys | – | Missense | Probably damaging | Deleterious | 27.7 | 121410 | 1 | 0.0008237 |
| chr2:228564098 | c.333C>A | p.Phe111Leu | – | Missense | Probably damaging | Deleterious | 26.5 | 121398 | 1 | 0.0008237 |
| chr2:228564087 | C.344T>C | p.Met115Thr | – | Missense | Possibly damaging | Deleterious | 23.5 | 121394 | 1 | 0.0008238 |
| chr2:228564085 | c.346G>A | p.Val116Ile | – | Missense | Possibly damaging | Deleterious | 25.2 | 121388 | 1 | 0.0008238 |
| chr2:228564082 | c.349A>G | p.Thr117Ala | – | Missense | Possibly damaging | Deleterious | 23.2 | 121370 | 1 | 0.0008239 |
| chr2:228564058 | c.373G>C | p.Ala125Pro | – | Missense | Probably damaging | Deleterious | 29.8 | 121342 | 5 | 0.004121 |
| chr2:228564054 | c.377A>C | p.Tyr126Ser | – | Missense | Probably damaging | Deleterious | 28.5 | 121334 | 1 | 0.0008242 |
| chr2:228564050 | c.381A>G | p.Ilel27Met | – | Missense | Probably damaging | Deleterious | 20.1 | 121330 | 1 | 0.0008242 |
| chr2:228564048 | c.383A>G | p.Tyr128Cys | – | Missense | Probably damaging | Deleterious | 26 | 121330 | 15 | 0.01236 |
| chr2:228564021 | c.410A>C | p.Gln137Pro | – | Missense | Probably damaging | Deleterious | 25.9 | 121288 | 2 | 0.001649 |
| chr2:228564021 | c.410dup | p.Arg138GlufsTer87 | – | Frameshift | – | – | 33 | 121272 | 2 | 0.001649 |
| chr2:228564021 | c.410A>G | p.Gln137Arg | – | Missense | Possibly damaging | Tolerated | 23 | 121288 | 1 | 0.0008246 |
| chr2:228564019 | c.412A>G | p.Arg138Gly | rs201318094 | Missense | Possibly damaging | Deleterious | 24.9 | 121268 | 1 | 0.0008246 |
| chr2:228564006 | c.425A>G | p.Tyr142Cys | – | Missense | Possibly damaging | Deleterious | 25.1 | 121226 | 1 | 0.0008249 |
| chr2:228563965 | c.466G>A | p.Val156Met | – | Missense | Probably damaging | Deleterious | 25.3 | 121336 | 1 | 0.0008242 |
| chr2:228563949 | C.482T>C | p.Leu161Ser | – | Missense | Probably damaging | Deleterious | 24.4 | 121352 | 1 | 0.000824 |
| chr2:228563928 | c.503C>G | p.Ser168Trp | rs139611208 | Missense | Probably damaging | Deleterious | 24.2 | 121382 | 1 | 0.0008238 |
| chr2:228563911 | c.520G>T | p.Val174Phe | rs59736804 | Missense | Possibly damaging | Deleterious | 26.4 | 121396 | 1 | 0.0008238 |
| chr2:228563889 | c.542C>T | p.Ser181Phe | – | Missense | Possibly damaging | Deleterious | 21.8 | 121404 | 1 | 0.0008237 |
| chr2:228563839 | c.592T>C | p.Phe198Leu | rs370725962 | Missense | Probably damaging | Deleterious | 29.2 | 121332 | 1 | 0.0008242 |
| chr2:228563838 | c.593T>C | p.Phe198Ser | – | Missense | Probably damaging | Deleterious | 28.6 | 121334 | 1 | 0.0008242 |
| chr2:228563832 | c.599A>G | p.His200Arg | – | Missense | Probably damaging | Deleterious | 24.5 | 121316 | 1 | 0.0008243 |
| chr2:228563646 | c.785T>C | p.Phe262Ser | – | Missense | Possibly damaging | Deleterious | 25 | 121402 | 4 | 0.003295 |
| chr2:228563614 | c.817T>G | p.Tyr273Asp | – | Missense | Probably damaging | Deleterious | 29.3 | 121404 | 1 | 0.0008237 |
| chr2:228563577 | c.854G>A | p.Trp285Ter | rs373198092 | Stop-gain | – | – | 40 | 121404 | 1 | 0.0008237 |
| chr2:228563538 | c.893A>G | p.Tyr298Cys | rs370104837 | Missense | Possibly damaging | Deleterious | 25.7 | 121326 | 2 | 0.001648 |
| chr2:228563515 | c.916A>T | p.Lys306Ter | – | Stop-gain | – | – | 37 | 121028 | 1 | 0.0008263 |
| chr2:228563476 | c.955G>A | p.Val319Ile | rs372511010 | Missense | Probably damaging | Deleterious | 34 | 119876 | 2 | 0.001668 |
| chr2:228560774 | c.1003G>C | p.Gly335Arg | – | Missense | Probably damaging | Deleterious | 34 | 121348 | 1 | 0.0008241 |
| chr2:228560744 | c.1033G>A | p.Gly345Arg | – | Missense | Probably damaging | Deleterious | 35 | 121392 | 2 | 0.001648 |
| chr2:228560725 | c.1052T>A | p.Val351Asp | – | Missense | Possibly damaging | Deleterious | 24.4 | 121402 | 1 | 0.0008237 |
| chr2:228560723 | c.1054T>G | p.Phe352Val | – | Missense | Possibly damaging | Deleterious | 25 | 121402 | 1 | 0.0008237 |
| chr2:228560707 | c.1070C>T | p.Ala357Val | – | Missense | Possibly damaging | Deleterious | 25.8 | 121398 | 7 | 0.005766 |
| chr2:228560689 | c.1088T>C | p.Met363Thr | – | Missense | Probably damaging | Deleterious | 26.4 | 121402 | 2 | 0.001647 |
| chr2:228560662 | c.1115G>A | p.Cys372Tyr | – | Missense | Probably damaging | Deleterious | 32 | 121390 | 3 | 0.002471 |
| chr2:228560623 | c.1154T>C | p.Leu385Pro | rs563607795 | Missense | Probably damaging | Deleterious | 33 | 121338 | 1 | 0.0008241 |
| chr2:228560615 | c.1162A>T | p.Thr388Ser | – | Missense | Probably damaging | Deleterious | 29 | 121322 | 1 | 0.0008243 |
| chr2:228560611 | c.1166T>C | p.Ile389Thr | rs149282475 | Missense | Possibly damaging | Deleterious | 29.6 | 121308 | 1 | 0.0008243 |
| chr2:228553024 | c.1173–1G>A | – | rs531876613 | Canonical splice | – | – | 26.3 | 119782 | 1 | 0.0008248 |
| chr2:228553013 | c.1183G>T | p.Ala395Ser | – | Missense | Probably damaging | Deleterious | 34 | 120494 | 1 | 0.0008299 |
| chr2:228553012 | c.1184C>T | p.Ala395Val | – | Missense | Possibly damaging | Deleterious | 34 | 120506 | 1 | 0.0008298 |
| chr2:228552992 | c.1204C>T | p.Arg402Cys | rs150523975 | Missense | Possibly damaging | Deleterious | 31 | 120836 | 11 | 0.009103 |
| chr2:228552992 | c.1204C>G | p.Arg402Gly | rs150523975 | Missense | Probably damaging | Deleterious | 34 | 120836 | 1 | 0.0008276 |
| chr2:228552991 | c.1205G>A | p.Arg402His | – | Missense | Possibly damaging | Deleterious | 34 | 120852 | 3 | 0.002482 |
| chr2:228552981 | c.1215G>T | p.Leu405Phe | – | Missense | Possibly damaging | Deleterious | 27.6 | 120918 | 1 | 0.000827 |
| chr2:228552961 | c.1235T>C | p.Phe412Ser | – | Missense | Probably damaging | Deleterious | 31 | 121022 | 1 | 0.0008263 |
| chr2:228552944 | c.1252C>G | p.Gln418Glu | rs368364131 | Missense | Probably damaging | Deleterious | 27.8 | 121048 | 83 | 0.06857 |
| chr2:228552934 | c.1262T>G | p.Met421Arg | – | Missense | Possibly damaging | Deleterious | 24.8 | 121042 | 1 | 0.0008262 |
| chr2:228552931 | c.1265C>T | p.Thr422Ile | – | Missense | Probably damaging | Deleterious | 28.5 | 121046 | 1 | 0.0008261 |
| chr2:228552920 | c.1276G>A | p.Val426Ile | – | Missense | Possibly damaging | Deleterious | 27.4 | 120990 | 3 | 0.00248 |
| chr2:228552905 | c.1291C>T | p.Leu431Phe | – | Missense | Probably damaging | Deleterious | 28.6 | 120926 | 1 | 0.000827 |
| chr2:228552881 | c.1314 + 1G>A | – | rs141957107 | Canonical splice | – | – | 25.8 | 120800 | 1 | 0.0008278 |
| chr2:228552279 | c.1325A>T | p.Tyr442Phe | – | Missense | Probably damaging | Deleterious | 32 | 120246 | 3 | 0.002495 |
| chr2:228552255 | c.1349C>T | p.Ala450Val | – | Missense | Possibly damaging | Deleterious | 33 | 121132 | 1 | 0.0008255 |
| chr2:228552255 | c.1349C>G | p.Ala450Gly | – | Missense | Possibly damaging | Deleterious | 29.5 | 121132 | 3 | 0.002477 |
| chr2:228552249 | c.1355T>C | p.Ile452Thr | rs140706520 | Missense | Possibly damaging | Deleterious | 26.8 | 121202 | 4 | 0.0033 |
| chr2:228552248 | c.1356delA | p.Ile452PhefsTer3 | rs140706520 | Frameshift | – | – | 34 | 121202 | 1 | 0.0008251 |
| chr2:228552233 | c.1371C>A | p.Ser457Arg | rs200054205 | Missense | Possibly damaging | Deleterious | 24.4 | 121328 | 3 | 0.002473 |
| chr2:228552129 | c.1475dup | p.Met492AsnfsTer27 | rs192876801 | Frameshift | – | – | 23.8 | 121118 | 1 | 0.0008256 |
4 |. DISCUSSION
In the present paper we describe a novel mutation in the SLC19A3 gene, which we deem pathogenic given the frequency, conservation and in silico prediction data, but more importantly because it is consistent with the patient’s clinical phenotype and characteristic brain MRI findings. BTBGD is a treatable genetic condition characterized by episodic encephalopathy, often triggered by febrile illness. It is caused by mutations in the SLC19A3 gene, encoding the human thiamine transporter 2 (hTHTR-2), a high-affinity thiamine transporter (Rajgopal, Edmondnson, Goldman, & Zhao, 2001), but not a biotin transporter (Subramanian, Marchant, & Said, 2006). It is thus intriguing that the disease was initially described as being responsive to biotin (Ozand et al., 1998), but this is likely related to the fact that biotin increases the expression of SLC19A3, and individuals with biotin deficiency will only show 33% gene expression as compared to controls, ranging from 16 to 70% (Vlasova, Stratton, Wells, Mock, & Mock, 2005). However, a recent open-label, prospective, comparative study showed that the combination of thiamine plus biotin was not superior to thiamine alone in terms of number of recurrences, neurologic sequelae or brain MRI findings, but it was associated with decreased duration of the acute crises (Tabarki et al., 2015). Indeed, it is known that normal cells exhibit upregulation of SLC19A3 expression under situations of stress, but this adaptive stress-induced upregulation of gene expression is lost in patients with BTBGD (Schänzer et al., 2014). Since about 48% of the intestinal transport of thiamine is mediated by hTHTR-2 (Said, Balamurugan, Subramanian, & Marchant, 2004), and because SLC19A3 is also expressed at the blood–brain barrier (Geier et al., 2013), large doses of thiamine should be administered in order to overcome the transport block. In addition, drugs that are known to inhibit hTHTR-2—such as metformin, famotidine, chloroquine, and verapamil—carry the theoretical risk of worsening the condition, and thus should be avoided in these patients (Liang et al., 2015).
The prevalence of the disease was previously unknown, but it is reportedly very rare based on the fact that there have been only 116 case reports published in the literature to date. We sought to generate an accurate estimate of disease prevalence based on the carrier frequencies of pathogenic and potentially pathogenic variants in a large cohort that was not selected for the presence of BTBGD, and should thus be free of ascertainment bias. We found a surprisingly high birth prevalence of about 1 in 215,000 individuals. It should be noted that this estimated prevalence corresponds to all forms of the disease, regardless of its severity—ranging from early infantile lethal Leigh syndrome to late-onset Wernicke-like encephalopathy. Based on this calculated prevalence, there should be about 1,500 individuals with this disease in the United States alone; the fact that the number of patients is much lower indicates that the disease remains largely underdiagnosed.
Since the disease is treatable but underdiagnosed, new diagnostic clues for BTBGD would be welcome. We describe the presence of a pyruvate peak on brain MRS during an episode of acute encephalopathy. It should be noted that pyruvate dehydrogenase is one of the three main mitochondrial enzymes that require thiamine as a cofactor. The presence of a high CSF pyruvate, low CSF lactate-to-pyruvate molar ratio, and high intracerebral pyruvate as assessed by brain MRS all point toward a deficiency of PDH in vivo. The measured activity of PDH in vitro was likely not decreased because it is measured in the presence of added thiamine pyrophosphate (TPP); in fact, the PDH enzyme activity in our patient was elevated. It should be noted that pyruvate and succinate both peak around 2.4 ppm on brain MRS (Chawla, Kumar, & Gupta, 2004), and thus a diagnosis of succinate dehydrogenase (SDH) deficiency should be entertained when such a peak is found (Helman et al., 2016). However, when a tall 2.4 ppm peak is found, SDH deficiency can be readily excluded by a normal urine organic acid analysis, while PDH deficiency can be excluded by normal—or even increased—enzyme activity in cells in the presence of TPP.
It is also interesting to note that our patient was found to have two separate conditions, BTBGD and X-linked dominant intellectual disability due to DDX3X mutations. Such blended phenotypes resulting from two unrelated genetic conditions coexisting in the same individual were found in 4.6% of positive cases in a study of 2,000 patients in whom clinical exome sequencing was performed (Yang et al., 2014).
In summary, we identified a novel mutation in SLC19A3, causative of biotin-thiamine responsive basal ganglia disease. Based on the calculated prevalence of the disease, it is very likely that the condition remains underdiagnosed. A pyruvate peak identified by brain MRS during episodes of acute encephalopathy might aid in the identification and earlier diagnosis of this treatable inborn error of metabolism.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank the patient and her family for their kind cooperation.
Footnotes
URLs: ExAC, http://exac.broadinstitute.org/; Mercury pipeline, https://github.com/dsexton2/Mercury-Pipeline; Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
CONFLICTS OF INTEREST
The authors report no conflict of interest.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of this article at the publisher’s web-site.
REFERENCES
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, … Sunyaev SR (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfadhel M, Almuntashri M, Jadah RH, Bashiri FA, Rifai MT, Al Shalaan H, & Al-Twaijri W (2013). Biotin-responsive basal ganglia disease should be renamed biotin-thiamine-responsive basal ganglia disease: A retrospective review of the clinical, radiological and molecular findings of 18 new cases. Orphanet Journal of Rare Diseases, 8, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge MN, Wang M, Wu Y, Newsham I, Muzny DM, Jefferies JL, … Gibbs RA (2011). Targeted enrichment beyond the consensus coding DNA sequence exome reveals exons with higher variant densities. Genome Biology, 12, R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla S, Kumar S, & Gupta RK (2004). Marker of parasitic cysts on in vivo proton magnetic resonance spectroscopy: Is it succinate or pyruvate? Journal of Magnetic Resonance Imaging, 20, 1052–1053. [DOI] [PubMed] [Google Scholar]
- Debs R, Depienne C, Rastetter A, Bellanger A, Degos B, Galanaud D, … Sedel F (2010). Biotin-responsive basal ganglia disease in ethnic Europeans with novel SLC19A3 mutations. Archives of Neurology, 67, 126–130. [DOI] [PubMed] [Google Scholar]
- Distelmaier F, Huppke P, Pieperhoff P, Amunts K, Schaper J, Morava E, … Karenfort M (2014). Biotin-responsive Basal Ganglia disease: A treatable differential diagnosis of leigh syndrome. JIMD Reports, 13, 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassone E, Wedatilake Y, DeVile CJ, Chong WK, Carr LJ, Rahman S (2013). Treatable Leigh-like encephalopathy presenting in adolescence. BMJ Case Reports, 2013, 200838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flønes I, Sztromwasser P, Haugarvoll K, Dölle C, Lykouri M, Schwarzlmüller T, … Tzoulis C (2016). Novel SLC19A3 promoter deletion and allelic silencing in biotin-thiamine-responsive basal ganglia encephalopathy. PLoS ONE, 11, e0149055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geier EG, Chen EC, Webb A, Papp AC, Yee SW, Sadee W, & Giacomini KM (2013). Profiling solute carrier transporters in the human blood-brain barrier. Clinical Pharmacology and Therapeutics, 94, 636–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerards M, Kamps R, van Oevelen J, Boesten I, Jongen E, de Koning B, … Smeets H (2013). Exome sequencing reveals a novel Moroccan founder mutation in SLC19A3 as a new cause of early-childhood fatal Leigh syndrome. Brain: A Jounal of Neurology, 136, 882–890. [DOI] [PubMed] [Google Scholar]
- Haack TB, Klee D, Strom TM, Mayatepek E, Meitinger T, Prokisch H, & Distelmaier F (2014). Infantile Leigh-like syndrome caused by SLC19A3 mutations is a treatable disease. Brain Neurology Journal, 137, e295. [DOI] [PubMed] [Google Scholar]
- Helman G, Caldovic L, Whitehead MT, Simons C, Brockmann K, Edvardson S, … van der Knaap MS (2016). Magnetic resonance imaging spectrum of succinate dehydrogenase-related infantile leukoencephalopathy. Annals of Neurology, 79, 379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr DS, Ho L, Berlin CM, Lanoue KF, Towfighi J, Hoppel CL, … Patel MS (1987). Systemic deficiency of the first component of the pyruvate dehydrogenase complex. Pediatric Research, 22, 312–318. [DOI] [PubMed] [Google Scholar]
- Kevelam SH, Bugiani M, Salomons GS, Feigenbaum A, Blaser S, Prasad C, … van der Knaap MS (2013). Exome sequencing reveals mutated SLC19A3 in patients with an early-infantile, lethal encephalopathy. Brain: A Journal of Neurology, 136, 1534–1543. [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, & Shendure J (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics, 46, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohrogi K, Imagawa E, Muto Y, Hirai K, Migita M, Mitsubuchi H, … Endo F (2015). Biotin-responsive basal ganglia disease: a case diagnosed by whole exome sequencing. Journal of Human Genetics, 60, 381–385. [DOI] [PubMed] [Google Scholar]
- Kono S, Miyajima H, Yoshida K, Togawa A, Shirakawa K, & Suzuki H (2009). Mutations in a thiamine-transporter gene and Wernicke’s-like encephalopathy. The New England Journal of Medicine, 360, 1792–1794. [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, & Ng PC (2009). Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature Protocols, 4, 1073–1081. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, … Exome Aggregation Consortium. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Chien H-C, Yee SW, Giacomini MM, Chen EC, Piao M, … Giacomini KM (2015). Metformin is a substrate and inhibitor of the human thiamine transporter, THTR-2 (SLC19A3). Molecular Pharmaceutics, 12, 4301–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, & Durbin R (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England), 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyajima H, & Kono S (2010). [Familial Wernicke’s-like encephalopathy]. Rinshō Shinkeigaku = Clinical Neurology, 50, 855–857. [DOI] [PubMed] [Google Scholar]
- Ozand PT, Gascon GG, Al Essa M, Joshi S, Al Jishi E, Bakheet S, & Dabbagh O (1998). Biotin-responsive basal ganglia disease: A novel entity. Brain Neurology Journal, 121(Pt 7), 1267–1279. [DOI] [PubMed] [Google Scholar]
- Pérez-Dueñas B, Serrano M, Rebollo M, Muchart J, Gargallo E, Dupuits C, & Artuch R (2013). Reversible lactic acidosis in a newborn with thiamine transporter-2 deficiency. Pediatrics, 131, e1670–e1675. [DOI] [PubMed] [Google Scholar]
- Rajgopal A, Edmondnson A, Goldman ID, & Zhao R (2001). SLC19A3 encodes a second thiamine transporter ThTr2. Biochimica et Biophysica Acta, 1537, 175–178. [DOI] [PubMed] [Google Scholar]
- Said HM, Balamurugan K, Subramanian VS, & Marchant JS (2004). Expression and functional contribution of hTHTR-2 in thiamin absorption in human intestine. American Journal of Physiology Gastrointestinal and Liver Physiology, 286, G491–G498. [DOI] [PubMed] [Google Scholar]
- Schänzer A, Döring B, Ondrouschek M, Goos S, Garvalov BK, Geyer J, … Hahn A (2014). Stress-induced upregulation of SLC19A3 is impaired in biotin-thiamine-responsive basal ganglia disease. Brain Pathology (Zurich, Switzerland), 24, 270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu KF, Hu CW, & Utter MF (1981). Pyruvate dehydrogenase complex activity in normal and deficient fibroblasts. Journal of Clinical Investigation, 67, 1463–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijders Blok L, Madsen E, Juusola J, Gilissen C, Baralle D, Reijnders MRF, … Kleefstra T (2015). Mutations in DDX3X are a common cause of unexplained intellectual disability with gender-Specific effects on wnt signaling. American Journal of Human Genetics, 97, 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sremba LJ, Chang RC, Elbalalesy NM, Cambray-Forker EJ, & Abdenur JE (2014). Whole exome sequencing reveals compound heterozygous mutations in SLC19A3 causing biotin-thiamine responsive basal ganglia disease. Molecular Genetics and Metabolism Reports, 1, 368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, & Cooper DN (2014). The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Human Genetics, 133, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian VS, Marchant JS, & Said HM (2006). Biotin-responsive basal ganglia disease-linked mutations inhibit thiamine transport via hTHT R2: Biotin is not a substrate for hTH TR2. American Journal of Physiology Cell Physiology, 291, C851–C859. [DOI] [PubMed] [Google Scholar]
- Tabarki B, Alfadhel M, AlShahwan S, Hundallah K, AlShafi S, & AlHashem A (2015). Treatment of biotin-responsive basal ganglia disease: Open comparative study between the combination of biotin plus thiamine versus thiamine alone. European Journal of Paediatric Neurology, 19, 547–552. [DOI] [PubMed] [Google Scholar]
- Vlasova TI, Stratton SL, Wells AM, Mock NI, & Mock DM (2005). Biotin deficiency reduces expression of SLC19A3, a potential biotin transporter, in leukocytes from human blood. Journal of Nutrition, 135, 42–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Miura K, Hara K, Suzuki M, Nakanishi K, Kumagai T, … Wakamatsu N (2010). A wide spectrum of clinical and brain MRI findings in patients with SLC19A3 mutations. BMC Medical Genetics, 11, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, … Eng CM (2014). Molecular findings among patients referred for clinical whole-exome sequencing. JAMA, 312, 1870–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zand DJ, Simon EM, Pulitzer SB, Wang DJ, Wang ZJ, Rorke LB, … Berry GT (2003). In vivo pyruvate detected by MR spectroscopy in neonatal pyruvate dehydrogenase deficiency. AJNR American Journal of Neuroradiology, 24, 1471–1474. [PMC free article] [PubMed] [Google Scholar]
- Zeng W-Q, Yamani E. Al-., Acierno JS, Slaugenhaupt S, Gillis T, MacDonald ME, … Gusella JF (2005). Biotin-responsive basal ganglia disease maps to 2q36.3 and is due to mutations in SLC19A3. American Journal of Human Genetics, 77, 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.