

Abstract
Hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC) has recently been associated with a single heterozygous p.D249N mutation in TUBB4A. We describe two novel mutations in this gene. A p.C239F mutation was found in one of the originally described H-ABC patients, for whom we provide follow-up 11 years after the original publication. The second novel mutation, p.R262H, was found in a patient with a typical clinical presentation for H-ABC, but with a novel neuroimaging phenotype, given the absence of atrophy of the putamen and caudate nucleus despite 7 years of follow-up. The recent recognition of TUBB4A mutations as the underlying etiology of H-ABC will likely lead to the identification of subtler clinical and neuroimaging presentations of this disorder, like in our third patient. Thus mutations in this gene should be suspected in any patient with hypomyelination, regardless of the long-term presence of neostriatal atrophy.
Keywords: hypomyelination with atrophy of the basal ganglia and cerebellum, TUBB4A, tubulin beta 4
INTRODUCTION
Hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC, OMIM 612438) was initially delineated by its progressive neuroimaging findings and neurological deterioration. H-ABC is a leukodystrophy characterized by diffuse central hypomyelination, atrophy of the caudate, putamen, and cerebellum (particularly the vermis), and normal thalamus and globus pallidus. The clinical features include extrapyramidal and pyramidal manifestations with ataxia. Most patients present with cognitive deficit of variable severity and onset. Since its first description of seven patients in 2002 [van der Knaap et al., 2002], 23 additional patients have been reported [Mercimek-Mahmutoglu et al., 2005; Wakusawa et al., 2006; Matta and Ribas, 2007; Mercimek-Mahmutoglu and Stockler-Ipsiroglu, 2007; van der Knaap et al., 2007; Wakusawa et al., 2007; Narumi et al., 2011; Simons et al., 2013]. More recently, it was discovered that H-ABC is associated with mutations in TUBB4A, encoding for tubulin beta 4A [Simons et al., 2013]. All 11 patients described in that publication had the same heterozygous c.745G>A (p.D249N) mutation, which was a de novo change in all patients studied, except in one family where somatic mosaicism was detected in the clinically unaffected mother.
Here, we report on a follow-up to one of the original seven patients, and describe two novel mutations in TUBB4A. We expand the neuroimaging phenotype of the disease, and review the literature based on the 32 patients published.
PATIENTS AND METHODS
Patient 1
This female corresponds to Patient 5 in the original paper describing H-ABC [van der Knaap et al., 2002]. She never acquired language. Her motor deterioration started in infancy, and she never achieved independent ambulation. At age 7.5 years, she underwent bilateral adductor tenotomies, hamstring and psoas release in order to ameliorate her lower extremity spasticity. At 7 years 9 months she was diagnosed with oropharyngeal swallowing dysfunction after experiencing two episodes of aspiration pneumonia. She underwent a gastrostomy tube placement at 8 years. At age 13 she had an episode of respiratory arrest secondary to tracheomalacia. A tracheostomy was performed at 14 years 11 months, and she remained ventilator-dependent since. At last follow-up at age 24, she remained bed-bound and required continuous tube feedings. She was non-verbal, did not track objects, and showed marked exotropia with nonspecific, random, horizontal eye movements. Muscle tone showed diffuse rigidity despite oral administration of baclofen, and she had contractures of multiple joints. Deep tendon reflexes were 2+ throughout, with the exception of ankle reflexes that were absent bilaterally, along with bilateral Babinski reflexes.
Patient 2
He was born at 37 weeks gestation following an induction for oligohydramnios. There were no developmental concerns until age 14 months when he was not yet able to walk. He never achieved unsupported ambulation, but by age 2 years he was able to walk with a gait trainer. Language development at 2 years was normal. At age 4, he developed motor regression and although he was still able to sit unsupported until age 6, he lost that ability soon afterwards. Since age 6 years, he had progressively worsening choreiform movements as well as dystonic posturing. From age 5, he developed progressive decline in speech and became effectively nonverbal by age 10. His comprehension, however, is nearly intact, even at the most recent visit at age 13. During his last follow-up he was noticed to have increased tone and rigidity, more prominent on the right side of his body, hyperreflexia with deep tendon reflexes 3+ throughout, bilateral ankle clonus more prominent on the right side, and bilateral Babinski responses. He showed dysmetria on the left more than on the right, and was noticed to have choreiform movements of all extremities at rest, as well as oral dyskinesia. Over the years, he had an extensive battery of tests including normal karyotype, chromosomal microarray, plasma amino acids, urine organic acids, carnitine levels, very long chain fatty acids, lactate, transferrin isoelectric focusing, lysosomal enzyme panel, urine sialic acid, vitamin E levels, alpha-feto protein, EMG, muscle biopsy with normal light and electron microscopy findings, as well as electron transport chain enzymology. He had normal sequencing and deletion/duplication analysis of the PLP1 gene, as well as normal sequencing of the genes responsible for Friedreich’s ataxia, DRPLA, and spinocerebellar ataxias 1–3, 6–8, 10, and 17. He had normal CSF neurotransmitters, lactate and pyruvate, but initially CSF amino acids showed elevation of glycine to 101 μmol/L. Sequencing of the three genes responsible for non-ketotic hyper-glycinemia was undertaken (AMT, GLDC, and GCSH) but no mutations were found, and a repeat lumbar puncture revealed a CSF glycine of 10 μmol/L, suggesting that the first specimen was contaminated with blood. He was treated with levodopa/carbidopa and trihexyphenidyl without benefit, and is currently on baclofen for his increased muscle tone, with questionable effect.
Patient 3
Patient 3 was diagnosed by prenatal ultrasound with congenital heart disease. This was postnatally confirmed to be a transposition of the great vessels with atrial septal defect and patent ductus arteriosus. He underwent cardiac repair soon after birth. During infancy he had poor weight gain, and at 15 months was diagnosed with celiac disease based on an intestinal biopsy. He had nystagmus during infancy, and at age 1 year he was diagnosed with global developmental delay, truncal hypotonia and spasticity of the limbs. At age 3 years he had an electroretinogram that revealed a moderate reduction in the combined cone- and rod-mediated responses, with primarily a decrease in the B-wave. At age 5 he underwent a gastrostomy tube placement, given increasing swallowing difficulties and multiple episodes of aspiration pneumonia. By age 7 he was still able to sit unsupported for a few seconds, but he lost this ability the following year. Due to worsening scoliosis of up to 95 degrees, he underwent spinal fusion at age 10. Over the years his physical exam revealed truncal hypotonia, deep tendon reflexes 2+ throughout, except in the ankles, where sustained clonus could be elicited bilaterally, and downgoing plantar reflexes. The appendicular tone was decreased, but he had been receiving regular Botox injections for years. Cerebellar testing was not possible due to the severe cognitive defect. At the last follow-up at age 11, he was nonverbal, with very poor head control, and bed-bound with spontaneous but non-purposeful movements of the extremities. Pertinent testing included a normal karyotype, SNP microarray, plasma amino acid levels, very long chain fatty acids, creatine kinase, lactate, pyruvate, gastrin levels, plasma CoQ10 levels. Urine studies revealed normal organic acids, sialic acid, mucopolysaccharides, and oligosaccharides. A conjunctival biopsy showed no inclusions on electron microscopy. Sequencing and deletion/duplication analysis of the PLP1 gene, and sequencing of GJC2, POLG, TYMP, and RRM2B were also unremarkable.
Neuroimaging Analysis
All available magnetic resonance (MR) images were qualitatively evaluated for morphological abnormalities and signal changes. For each patient, two brain MR studies were available. For Patient 1, the MRI studies were performed at age 11 months and 9.8 years, respectively, for Patient 2 at ages 2.7 years and 8.2 years, respectively, and for Patient 3 at ages 2 years and 8.7 years, respectively. For Patient 2, a spine MR study was available at age 2.7 years.
Molecular Genetics Analysis
Patient 1 underwent Sanger sequencing; the relevant portion of TUBB4A was PCR-amplified from genomic DNA. Bidirectional sequence was obtained on an ABI3730XL sequencer (Applied Biosystems, Foster City, CA) and analyzed with Mutation Surveyor (Softgenetics, State College, PA). Patients 2 and 3 underwent whole-exome sequencing on a clinical basis. In Patient 2, a pre-capture library was prepared by fragmentation of genomic DNA followed by adapter ligation and PCR amplification. Exome-in-solution capture was performed (VCRome 2.1, Roche Nimblegen, Madison, WI), and the enriched targets were sequenced in an Illumina HiSeq platform (Illumina, Inc, San Diego, CA). Data analysis and interpretation were performed by using the Mercury analysis pipeline. In Patient 3, exome capture was performed by using the SureSelect Target Enrichment System (Agilent Technologies, Santa Clara, CA), and the final libraries were sequenced in an Illumina HiSeq 2000. Sequences were aligned and variants called were generated using CASAVA (Illumina, Inc), while variants were annotated using the Ambry Variant Analyzer tool (Ambry Genetics, Aliso Viejo, CA).
RESULTS
Neuroimaging Findings
Hypomyelination was found in all patients and was generalized in Patients 1 (Fig. 1B,D) and 2 (Fig. 1F,H), but focal and milder in Patient 3. In Patient 3, the genu and splenium of the corpus callosum as well as the posterior limbs of the internal capsule appeared to be partially myelinated at age 2 years (Fig. 1J) and no progression in myelination occurred by the time of his last neuroimaging follow-up at age 8.7 years (Fig. 1L). In Patients 1 and 3, progressive loss of white matter with secondary cerebral atrophy and enlargement of the lateral ventricles occurred between the MR studies (Fig. 1D,L), while the amount of white matter appeared to remain stable over time in Patient 2 (Fig. 1H). In Patient 1, putamina were already absent on the first MR study at age 11 months (Fig. 1B), while progressive atrophy of the putamina was seen in Patient 2 (Fig. 1F,H). In Patients 1 and 2, progressive atrophy of the head of the caudate was noted (Fig. 1B,D,F,H). In Patient 3, however, the size of the putamina and heads of the caudate remained stable between first and follow-up MR studies (Fig. 1J,L). Additionally, the basal ganglia had a rather globular appearance in Patient 3. At follow-up, we found mild atrophy of the thalami in Patients 1 and 2 (Fig. 1D,H), while the size of the globi pallidi was normal in all patients. At follow-up, we saw cerebellar atrophy in all patients (Fig. 1C,G,K). On the first MRI, the morphology of the cerebellum was normal in Patient 1 (Fig. 1A), while the cerebellum was mildly hypoplastic in Patient 2 (Fig. 1E) and the anterior vermis was mildly atrophic in Patient 3 (Fig. 1I). A mild pontine atrophy was seen on the follow-up MR images of Patient 1 (Fig. 1C). At age 2.7 years, spine MRI was performed in Patient 2 and showed normal size and signal of the spinal cord.
FIG. 1.
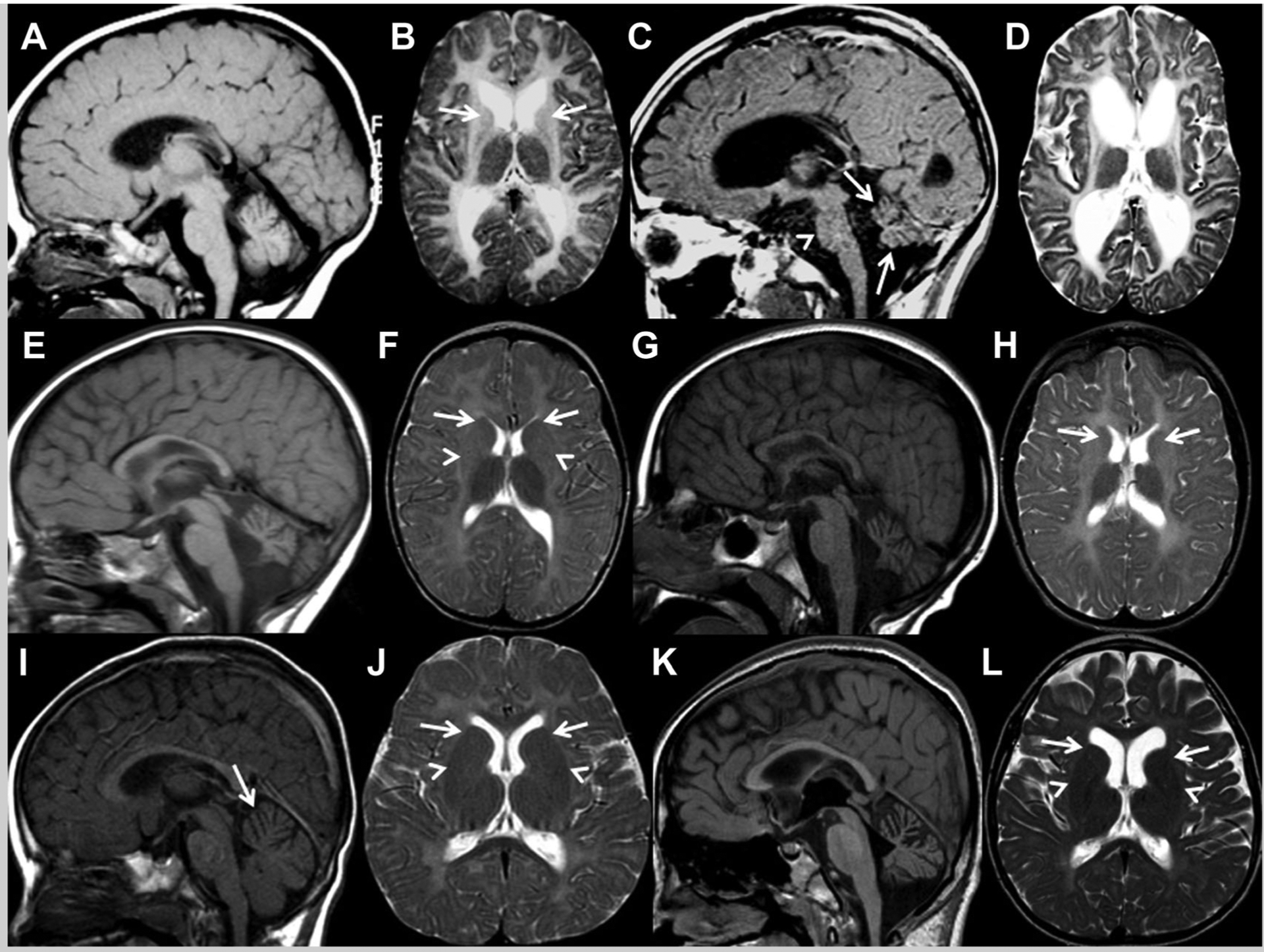
A: Midsagittal T1- and (B) axial T2-weighted images of patient 1 at the age of 11 months show diffuse T2-hyperintense signal of the white matter, very small and T2-hyperintense caudate nuclei (arrows) and normal size of cerebellum, thalami and globi pallidi, while the putamina are completely atrophic and not visible. C: Midsagittal T1- and (D) axial T2-weighted follow-up images at the age of 9.8 years demonstrate atrophy of the cerebellum (arrows), unchanged diffuse T2-hyperintense signal of the white matter consistent with hypomyelination, decreased amount of white matter leading to cerebral atrophy and enlargement of the lateral ventricles, progressive atrophy of the head of the caudate nuclei (not visible anymore), mild reduction in size of the thalami, normal size of globi pallidi, and mild pontine atrophy (arrowhead). E: Midsagittal T1- and (F) axial T2-weighted images of Patient 2 at the age of 2.7 years reveal cerebellar atrophy superimposed on a small (hypoplastic) cerebellar vermis, mild atrophy of caudate nuclei (arrows) and putamina (arrowheads) that lack their normal intermediate signal intensity, and normal size of thalami and globi pallidi. G: Midsagittal T1- and (H) axial T2-weighted follow-up images at the age of 8.2 years show unchanged atrophy of the cerebellum and diffuse T2-hyperintense signal of the white matter consistent with hypomyelination, stable amount of white matter, progressive atrophy and loss of intermediate signal intensity of caudate nuclei (arrows) and putamina, subtle decrease in size of the thalami and normal size of globi pallidi. I: Midsagittal T1- and (J) axial T2-weighted images of patient 3 at age 2 years demonstrate diffuse T2-hyperintense signal of the cortical and subcortical white matter, T2-hypointense signal in the posterior limb of the internal capsule as well as genu and splenium of the corpus callosum, and normal signal intensity and size of thalami, normal signal intensity, but a rather globular appearance of the basal ganglia (arrows and arrowheads in J), and mild atrophy of the anterior vermis (arrow in I). K: Midsagittal T1- and (L) axial T2-weighted follow-up images at age 8.7 years reveal progressive cerebellar atrophy, unchanged signal intensity of the white matter consistent with hypomyelination, decreased amount of white matter with secondary cerebral atrophy and ventricular enlargement, and unchanged size and signal of basal ganglia (arrows and arrowheads) and thalami.
Molecular Findings
All patients were found to have heterozygous mutations in TUBB4A (see Table I). The mutations in Patients 2 and 3 were confirmed by Sanger sequencing. All three changes occurred in a highly conserved amino acid position (see Fig. 2A). In Patient 2, parental Sanger sequencing did not reveal the p.D249N mutation, suggesting that this change arose de novo. In Patient 3, familial Sanger sequencing confirmation and segregation analysis of the p.R262H mutation revealed that the mother, father and unaffected sister do not carry the mutation, indicating a de novo occurrence, although parental germline mosaicism cannot be excluded.
TABLE I.
Information Regarding Mutations Found in Our Cohort
| Frequency | In silico prediction | |||||||
|---|---|---|---|---|---|---|---|---|
| Chromosomal location (GRCh37/hg19) | cDNA change | Protein change | dbSNP139 | NHLBI EVSa | 1000 Genomes | PolyPhen-2 Humvarb | PROVEANc (cutoff <2.5) | |
| Patient 1 | chr19:g.6495794 | c.716G>T | p.C239F | Absent | Absent | Absent | Probably damaging: 0.971 | Deleterious: –7.983 |
| Patient 2 | chr19:g.6495765 | c.745G>A | p.D249N | Absent | Absent | Absent | Probably damaging: 0.999 | Deleterious: –4.341 |
| Patient 3 | chr19:g.6495725 | c.785G>A | p.R262H | Absent | Absent | Absent | Benign: 0.155 | Deleterious: –4.261 |
EVS: Exome Variant Server (NHLBI Exome Sequencing Project).
FIG. 2.

Multiple sequence alignment and Sanger sequencing of the two novel mutations. A: Multiple protein sequence alignment over the changed amino acids is shown from human to fish. B: Chromatogram of the novel p.C239F mutation. C: Chromatogram of the novel p.R262H mutation.
DISCUSSION
H-ABC is a hypomyelinating leukodystrophy defined by the peculiar neuroimaging pattern [van der Knaap et al., 2002]. The discovery of the underlying genetic etiology enables a better characterization of the full phenotypic spectrum of this disease. In this article, we expand the clinical and neuroimaging phenotype of H-ABC and report on two novel mutations in TUBB4A.
In reviewing all 30 patients reported to date (see Supplementary Table SI), we found that about two-thirds are males, two-thirds had their first symptoms before age 2 years, and also in two thirds the motor development was delayed. Indeed, the most common initial complaints were motor in 61% of patients, with about 1 in 4 presenting with visual/oculomotor signs, and another 1/4 presenting with speech complaints; hypotonia was the first sign in about 1 in 5 patients, and gait instability in 1 in 10. Spasticity was present in 100% of patients, while ataxia was present in 87%, choreoathetosis in 45%, dystonia in 88%, rigidity in 69%, and dysarthria in 93%; nystagmus in 33%, decreased vision or poor visual tracking in 27%, sensorineural hearing loss in 14%, and seizures in 18%. Tremors, microcephaly and short stature were present in roughly half. The neurological and cognitive phenotypes of our patients match the spectrum reported so far.
Patient 3 has involvement outside the central nervous system including congenital heart defects, celiac disease and retinal abnormalities. Systemic involvement has not been reported in other patients with H-ABC. Congenital heart defects and celiac disease are common and a chance association is likely. Additionally, TUBB4A is not highly expressed in heart and gastrointestinal tract, while it is highly expressed in the brain, especially in the cerebellum, putamen and white matter [Leandro-García et al., 2010; Hersheson et al., 2013]. It remains unclear whether the abnormal ERG findings in Patient 3 are related to his TUBB4A mutation.
The characteristic neuroimaging findings in H-ABC include diffuse hypomyelination and progressive caudate, putaminal and cerebellar atrophy [van der Knaap et al., 2002]. Patient 3 in this report presented with a fairly typical clinical phenotype, but his neuroimaging findings were unusual for H-ABC. Putamina and caudate nuclei remained normal in size over 7 years and the degree of hypomyelination was milder than typically seen in H-ABC. Thus, it is likely that the recent identification of the genetic etiology for H-ABC will reveal novel and subtler presentations of this disorder, thus broadening the clinical and neuroimaging phenotype. We suggest that H-ABC should be considered in the differential diagnosis of hypomyelination, even in the absence of marked putaminal atrophy as in Patient 3. In Patient 3, the basal ganglia had a rather globular appearance. Prominence of the basal ganglia has been reported in several patients with mutations in other tubulin genes, such as TUBA1A and TUBB2B [Cushion et al., 2013]. In contrast to Patient 3 in this article, basal ganglia in patients with mutations in TUBA1A and TUBB2B are dysmorphic, with absence of the internal capsule. Patient 2 had a normal spine MRI at age 2.7 years, when cortical white matter already showed hypomyelination. The normal size and MR signal of the spinal cord, the first reported spinal imaging in this disorder, suggests that the hypomyelination may be confined to the brain, but further study is needed.
The causal gene for H-ABC has been recently elucidated. A single, de novo mutation was reported in 11 patients with H-ABC [Simons et al., 2013]. Patient 2 in our cohort carries this common c.745G>A mutation. This mutation is located in the T7 loop of the protein, which corresponds to the GTP nucleotide binding domain, implicated in polymerization-dependent activation of GTPase activity [Lowe and Amos, 1999]. The TUBB4A mutations identified in Patients 1 and 3 have not been reported previously in H-ABC. The pR262H mutation is located in the second domain, in a loop between the H8 and S10 helices, while the p. D249N is located in the core H7 helix that connects the nucleotide binding domain with the second domain [Lowe et al., 2001]. Compared to mutations in Patients 1 and 2, the p R262H mutation in Patient 3 seems to result in a similar clinical, but milder neuroimaging phenotype. A larger cohort and functional studies are needed to confirm this observation.
Tubulin beta 4A, encoded by TUBB4A, is the major tubulin isotype in brain, where it represents 46% of all beta-tubulins [Leandro-García et al., 2010]. Thus, it is not surprising that mutations in this gene cause two neurologic disorders: dystonia type 4 (OMIM 128101) [Hersheson et al., 2013] and H-ABC [Simons et al., 2013], both associated with extrapyramidal signs. In dystonia type 4, the onset of dystonia, however, occurs later than in H-ABC, cognitive function is usually preserved, and neuroimaging reveals normal findings [Lohmann et al., 2013]. The phenotypic spectrum associated with mutations in TUBB4A is thus wide and most likely represents a continuum.
In conclusion, we describe novel mutations in TUBB4A leading to H-ABC. The expansion of the neuroimaging phenotype described here leads us to consider alterations in TUBB4A in any patient with central hypomyelination, even in the absence of neostriatal atrophy in young children.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank the patients and their families for their kind cooperation.
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of this article at the publisher’s web-site.
Exome Variant Server (NHLBI Exome Sequencing Project, Seattle,WA), http://evs.gs.washington.edu/EVS/
Mercury pipeline, https://github.com/dsexton2/Mercury-Pipeline
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Genome Browser, http://genome.ucsc.edu/
1000 Genomes, ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/
REFERENCES
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. 2010. A method and server for predicting damaging missense mutations. Nat Methods 7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. 2012. Predicting the functional effect of amino acids substitutions and indels. PLoS ONE 7: e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushion TD, Dobyns WB, Mullins JG, Stoodley N, Chung SK, Fry AE, Hehr U, Gunny R, Aylsworth AS, Prabhakar P, Uyanik G, Rankin J, Rees MI, Pilz DT. 2013. Overlapping cortical malformations and mutations in TUBB2B and TUBA1A. Brain 136:536–548. [DOI] [PubMed] [Google Scholar]
- Hersheson J, Mencacci NE, Davis M, MacDonald N, Trabzuni D, Ryten M, Pittman A, Paudel R, Kara E, Fawcett K, Plagnol V, Bhatia KP, Medlar AJ, Stanescu HC, Hardy J, Kleta R, Wood NW, Houlden H. 2013. Mutations in the autoregulatory domain of β-tubulin 4a cause hereditary dystonia. Ann Neurol 73:546–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leandro-García LJ, Leskela S, Landa I, Montero-Conde C, Lopez-Jimenez E, Leton R, Cascon A, Robledo M, Rodriguez-Antona C. 2010. Tumoral and tissue-specific expression of the major human beta-tubulin isotypes. Cytoskeleton 67:214–223. [DOI] [PubMed] [Google Scholar]
- Lohmann K, Wilcox RA, Winkler S, Ramirez A, Rakovic A, Park JS, Arns B, Lohnau T, Groen J, Kasten M, Bruggermann N, Hagenah J, Schmidt A, Kaiser FJ, Kumar KR, Zschiedrich K, Alvarez-Fischer D, Altenmuller E, Ferbert A, Lang AE, Muchau A, Kostic V, Simonyan K, Agzarian M, Ozelius LJ, Langeveld AP, Sue CM, Tijssen MA, Klein C. 2013. Whispering dysphonia (DYT4 dystonia) is caused by a mutation in the TUBB4 gene. Ann Neurol 73:537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe J, Amos LA. 1999. Tubulin-like protofilaments in Ca2+ -induced FtsZ sheets. EMBO J 18:2364–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe J, Li H, Downing KH, Nogales E. 2001. Refined structure of alpha beta-tubulin at 3.5 A resolution. J Mol Biol 313:1045–1057. [DOI] [PubMed] [Google Scholar]
- Matta AP, Ribas MC. 2007. Hypomyelination with atrophy of the basal ganglia and cerebellum: Case report. Arq Neuropsiquiatr 65:161–163. [DOI] [PubMed] [Google Scholar]
- Mercimek-Mahmutoglu S, van der Knaap MS, Baric I, Prayer D, Stoeckler-Ipsiroglu S. 2005. Hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC). Report of a new case. Neuropediatrics 36:223–226. [DOI] [PubMed] [Google Scholar]
- Mercimek-Mahmutoglu S, Stockler-Ipsiroglu S. 2007. Cerebral folate deficiency and folinic acid treatment in hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC) syndrome. Tohoku J Exp Med 211:95–96. [DOI] [PubMed] [Google Scholar]
- Narumi Y, Shiihara T, Yoshihashi H, Sakazume S, van der Knaap MS, Nishimura-Tadaki A, Matsumoto N, Fukushima Y. 2011. Hypomyelination with atrophy of the basal ganglia and cerebellum in an infant with Down syndrome. Clin Dysmorphol 20:166–167. [DOI] [PubMed] [Google Scholar]
- Simons C, Wolf NI, McNeil N, Caldovic L, Devaney JM, Takanohashi A, Crawford J, Ru K, Grimmond SM, Miller D, Tonduti D, Schmidt JL, Chudnow RS, van Coster R, Lagae L, Kisler J, Sperner J, van der Knaap M, Schiffman R, Taft R, Vanderver A. 2013. A de novo mutation in the beta-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am J Hum Genet 92:767–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Knaap MS, Naidu S, Pouwels PJ, Bonavita S, van Coster R, Lagae L, Sperner J, Surtees R, Schiffmann R, Valk J. 2002. New syndrome characterized by hypomyelination with atrophy of the basal ganglia and cerebellum. AJNR Am J Neuroradiol 23:1466–1474. [PMC free article] [PubMed] [Google Scholar]
- van der Knaap MS, Linnankivi T, Paetau A, Feigenbaum A, Wakusawa K, Haginoya K, Kohler W, Henneke M, Dinopoulos A, Grattan-Smith P, Brockmann K, Schiffmann R, Blaser S. 2007. Hypomyelination with atrophy of the basal ganglia and cerebellum: Followup and pathology. Neurology 69:166–171. [DOI] [PubMed] [Google Scholar]
- Wakusawa K, Haginoya K, Kitamura T, Togashi N, Ishitobi M, Yokoyama H, Higano S, Onuma A, Nara T, Iinuma K. 2006. Effective treatment with levodopa and carbidopa for hypomyelination with atrophy of the basal ganglia and cerebellum. Tohoku J Exp Med 209:163–167. [DOI] [PubMed] [Google Scholar]
- Wakusawa K, Uematsu M, Tsuchiya S, Haginoya K, Blau N. 2007. The cerebrospinal fluid level of 5-methylterahydrofolate in a Japanese boy with hypomyelination with atrophy of the basal ganglia and cerebellum. Tohoku J Exp Med 213:373. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.