

Abstract
A critical step in preserving protein homeostasis is the recognition, binding, unfolding, and translocation of protein substrates by six AAA-ATPase proteasome subunits (ATPase Associated with various cellular Activities) termed PSMC1–6, which are required for degradation of proteins by 26S proteasomes. Here, we identified fifteen de novo missense variants in the PSMC3 gene encoding the AAA-ATPase proteasome subunit PSMC3/Rpt5 in twenty-three unrelated heterozygous patients with an autosomal dominant form of neurodevelopmental delay and intellectual disability. Expression of PSMC3 variants in mouse neuronal cultures led to altered dendrite development and deletion of the PSMC3 fly ortholog Rpt5 impaired reversal learning capabilities in fruit flies. Structural modeling as well as proteomic and transcriptomic analyses of T cells derived from patients with PSMC3 variants implicated the PSMC3 variants in proteasome dysfunction through disruption of substrate translocation, induction of proteotoxic stress and alterations in proteins controlling developmental and innate immune programs. The proteostatic perturbations in T cells from patients with PSMC3 variants correlated with a dysregulation in type I interferon (IFN) signaling in these T cells, which could be blocked by inhibition of the intracellular stress sensor, protein kinase R (PKR). These results suggest that proteotoxic stress activated PKR in patient-derived T cells resulting in a type I IFN response. The potential relationship among proteosome dysfunction, type I IFN production and neurodevelopment suggest new directions in our understanding of pathogenesis in some neurodevelopmental disorders.
INTRODUCTION
Proteasomes are large multi-protein complexes whose structure is adapted to the function of regulated protein degradation, thereby controlling many cellular processes (1, 2). Together with the autophagosomal-lysosomal system, proteasomes maintain protein homeostasis by counterbalancing the synthesis of new proteins by the translational machinery (3–6). The proteasome is part of the ubiquitin-proteasome system (UPS) which counts numerous enzymes acting upstream of the proteasome (7, 8). Aged or unstructured proteins are preliminary ubiquitin-tagged by the ubiquitination machinery through a cascade of enzymatic reactions for degradation by the 26S proteasome (9, 10). The 26S proteasome consists of two parts, the 20S core proteolytic particle and the 19S regulatory particle which caps the 20S particle at one or both ends (11, 12). Polyubiquitinated proteins are recognized by the 19S regulatory particle which comprises two parts: a base and a lid. In the base, four regulatory particle non-ATPase (Rpn) subunits (Rpn1, Rpn2, Rpn10 and Rpn13; encoded by the proteasome genes PSMD2, PSMD1, PSMD4 and ADRM1, respectively) ensure recognition and capture of ubiquitin-modified substrates (13, 14). The lid contains eight additional non-ATPase subunits Rpn3, Rpn5–9, Rpn12, and Rpn15 which serve as scaffolds for binding of other subunits (15, 16) and the deubiquitinating enzyme Rpn11 (encoded by PSMD14) (17). Six AAA-ATPase subunits (Rpt1–6), encoded by the genes PSMC1–6 in the base use the energy provided by ATP hydrolysis to unfold and translocate the substrate into the barrel-shaped 20S proteolytic core particle by gate opening. The 20S complex comprises heptameric α- and β-rings with an α1–7β1–7β1–7 α1–7 architecture encoded by proteasome alpha subunit PSMA1-7 or beta subunit genes PSMB1-7. The β-ring may be subjected to variations, thereby giving rise to two major proteasome isoforms, namely standard- and immunoproteasomes. Standard proteasomes typically contain the catalytic β1, β2 and β5 subunits with caspase-, trypsin- and chymotrypsin-like activities, respectively (18, 19). In immunoproteasomes, the β1, β2 and β5 subunits are replaced by the inducible β1i, β2i and β5i subunits, encoded by the PSMB9, PSMB10 and PSMB8 genes, respectively (20). Although standard proteasomes are expressed in virtually all types of tissues, the expression of the inducible β-subunits is restricted to immune cells and non-immune cells exposed to type I or II interferons (IFNs) (21, 22).
Pathogenic variants in proteasome subunit genes cause rare proteasomopathies with a broad spectrum of symptoms (23, 24). So far, with the exception of the PSMB1 (β6) subunit (25), all pathogenic variants related to the 20S core particle have been shown to provoke immune dysregulation. Indeed, several genes encoding β-subunits (PSMB4, PSMB8, PSMB9, PSMB10), α-subunits (PSMA3) or assembly chaperone genes (proteasome maturation protein POMP, proteasome assembly chaperone Pac2 gene PSMG2) of the 20S proteasome complex have been involved in autosomal recessive proteasome-associated autoinflammatory syndromes (PRAAS) typically characterized by persistent type I IFN signaling (26–33). By contrast, genetic disorders involving genes of the 19S regulatory particle such as the Stankiewicz-Isidor syndrome (STISS, MIM: 617516) caused by truncating variants of PSMD12 (also referred to as Rpn5) are neurodevelopmental polymalformative syndromes (34) with subclinical activation of type I IFN signaling (35, 36) These observations place both PRAAS and STISS in the category of type I interferonopathies, a recent family of genetically determined rare autoinflammatory syndromes with dysregulated type I IFN signaling that includes Aicardi-Goutières syndrome and familial chilblain lupus (37, 38). Clinically, these diseases are complex, demonstrating multiple organ involvement (often brain and skin), encompassing a broad range of phenotypes and being associated with high morbidity and mortality (39). In this work, we identified fifteen dominant de novo variants in the PSMC3 gene coding for the AAA-ATPase PSMC3/Rpt5. These rare missense variants were detected in twenty-three individuals presenting with neurodevelopmental delay (NDD), intellectual disability (ID) or both, together with various congenital malformations. Together, our data highlight interferonopathy as a potential contributor to the pathogenesis of NDD/ID in patients carrying loss-of-function variants in subunits of the 19S proteasome regulatory particle and identify protein kinase R (PKR) as a major player in disease pathogenesis.
RESULTS
Identification of PSMC3 variants
The first PSMC3 variant was detected in Patient #2, a female newborn presenting with severe cardiac, gastrointestinal, inflammatory and immune issues. Whole-exome sequencing (WES) highlighted the de novo nonsynonymous c.523A>G p.(M175V) variant (GenBank ID: NM_002804.4) which was absent in any public variant databases (gnomAD, >246,000 chromosomes; NHLBI Exome Variant Server, >13,000 alleles; Bravo, 125,568 alleles) and predicted to be pathogenic by bioinformatics programs including SIFT, PolyPHen-2n CADD, REVEL, and Metadome as well as all programs compiled by MobiDetails (40). Our overall strategy described in the Materials and Methods allowed us to identify a total of 15 distinct rare de novo missense PSMC3 variants in 23 unrelated children presenting with a syndrome characterized by neurodevelopmental delay (NDD) and various congenital anomalies (Table 1).
Table 1.
Main characteristics of the PSMC3 de novo variants¥ identified in the patients included in the study.
| Chromosomal localization (Chr11/GRCh37) | cDNA change ¶ | Protein change | Variant database † | CADD Phred score (v1.6.) | Meta-dome | Mobi-Details ‡ | Effect on neuronal development | Effect on PSMC3 stability | Effect on proteasome assembly | Effect on mitophagy | IFN signature | Number of patientss with the variant |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| g.47445677G>A | c.511C>T | R171W | rs775517283 | 27.3 | I | 45364 | improvement | Decrease | ND | ND | ND | 1 |
| g.47445665T>C | c.523A>G | M175V | Absent | 23.4 | I | 45365 | ND | none | ND | ND | ND | 1 |
| g.47444430G>A | c.686C>T | P229L | Absent | 25.5 | I | 195979 | ND | ND | ND | ND | ND | 1 |
| g.47444406G>A | c.710C>T | A237V | Absent | 26.9 | HI | 45366 | ND | Decrease | ND | ND | ND | 1 |
| g.47444234T>C | c.775A>G | M259V | Absent | 23.4 | I | 45367 | ND | none | ND | ND | ND | 1 |
| g.47444233A>G | c.776T>C | M259T | Absent | 24.7 | I | 45368 | ND | Decrease | ND | ND | ND | 1 |
| g.47444227A>G | c.782T>C | I261T | Absent | 24.6 | I | 45369 | ND | Decrease | ND | ND | ND | 6 |
| g.47444225C>T | c.784G>A | G262R | Absent | 26.8 | I | 45370 | ND | none | none | increase | moderate | 1 |
| g.47444203C>G | c.806G>C | R269P | Absent | 24 | I | 45371 | ND | Decrease | ND | 1 | ||
| g.47444150C>G | c.859G>C | E287Q | Absent | 26.7 | I | 45372 | ND | Decrease | ND | 1 | ||
| g.47442253G>A | c.910C>T | R304W | rs1363348500 | 31 | I | 45373 | impairment | none | impairment | increase | strong/very strong | 4 |
| g.47442253G>C | c.910C>G | R304G | Absent | 28.2 | I | 45374 | ND | none | ND | 1 | ||
| g.47442248C>A | c.915G>T | E305D | Absent | 23.1 | I | 45375 | impairment | none | impairment | increase | moderate | 1 |
| g.47442234A>G | c.929T>C | M310T | Absent | 26.2 | HI | 45376 | ND | none | ND | 1 | ||
| g.47440729C>T | c.1147G>A | E383K | Absent | 27.8 | N | 45377 | impairment | none | ND | 1 |
All variants presented are de novo, except variant c.775A>G that is suspected de novo.
RefSeq transcript used for PSMC3 is NM_002804.4; † gnomAD V3, dbSNP v154, ClinVar v20210828; ‡ The access to detailed predictions for variant XXXXX (5364, 45365…) is as follows: https://mobidetails.iurc.montp.inserm.fr/MD/api/variant/XXXXX/browser/
ND: not determined; IFN: interferon; Metadome: I= intolerent; HI= highly intolerent; N= neutral
As shown in Fig. 1, most of the PSMC3 substitutions were localized in the AAA domain that was predicted to be intolerant to variations (Fig. S1). Two distinct regions of the AAA domain were particularly prone to substitutions. The first hotspot was centered on the recurrent variant c.910C>T p.(R304W) detected in four unrelated children and encompassed variants c.910C>G p.(R304G), c.915G>T p.(E305D) and c.929T>C p.(M310T) (Fig. 1). The second region, enriched in rare variants [c.775A>G p.(M259V), c.776T>C p.(M259T), c.782T>C p.(I261T) -seen six times-, c.784G>A p.(G262R) and c.806G>C p.(R269P)], was more N-terminally located (Fig. 1). All thirteen affected residues were highly conserved across species from mammalians down to fission yeast (Fig. 1). One major phenotypic hallmark of all individuals carrying PSMC3 variants was the predominance of neurodevelopmental or neuropsychiatric symptoms (Table S1). In more detail, apart from Patient #2, all affected children exhibited developmental delay (22/22; 100%) characterized by speech delay (19/19; 100%) alone or with intellectual disability (16/18; 89%) as well as motor delay (15/19; 79%). Brain magnetic resonance imaging highlighted frequent anomalies (11/15; 73%), whereas the occurrence of abnormal behavior (9/18; 50%) and seizures (5/21; 24%) varied. Nine of 19 (47%) individuals experienced growth failure, most with feeding difficulties (8/18; 44%). Malformations were frequently observed in the skeleton [11/15; 73%; scoliosis, acetabular dysplasia, brachymetatarsy), heart (10/18; 56%; ventricular or septal defects, patent ductus arteriosus, pulmonary hypertension and atresia), kidney (4/15; 27%; horseshoe shape, pelvicalyceal dilatation, nephrocalcinosis, and multi-cystic dysplastic kidney), and head (microcephaly in 6/17 (35%); relative to severe macrocephaly in 2/16 (13%)]. Tumors were noted in 2/19 (11%) individuals (craniopharyngioma and neuroblastoma). Hearing loss was detected in 9/19 individuals (47%) and labeled as sensorineural in two and conductive in one of them, respectively. Most of the children (18/20; 90%) displayed dysmorphic facial features (Fig. S2), including tall or broad forehead (7/19; 37%), thin upper lip with down-turned corners of mouth (6/19; 32%), abnormal palate (5/19; 265/19; 26%), epicanthal folds (5/19; 26%), and orofacial clefts (2/19; 10%). Computational analysis of facial morphology by GestaltMatcher (41) revealed that facial dysmorphism among the patients carrying PSMC3 variants was rather heterogeneous with similarities only observed between patients carrying identical variants (Fig. S2).
Fig. 1: Distribution of the de novo heterozygous PSMC3/Rpt5 variants identified in patients.
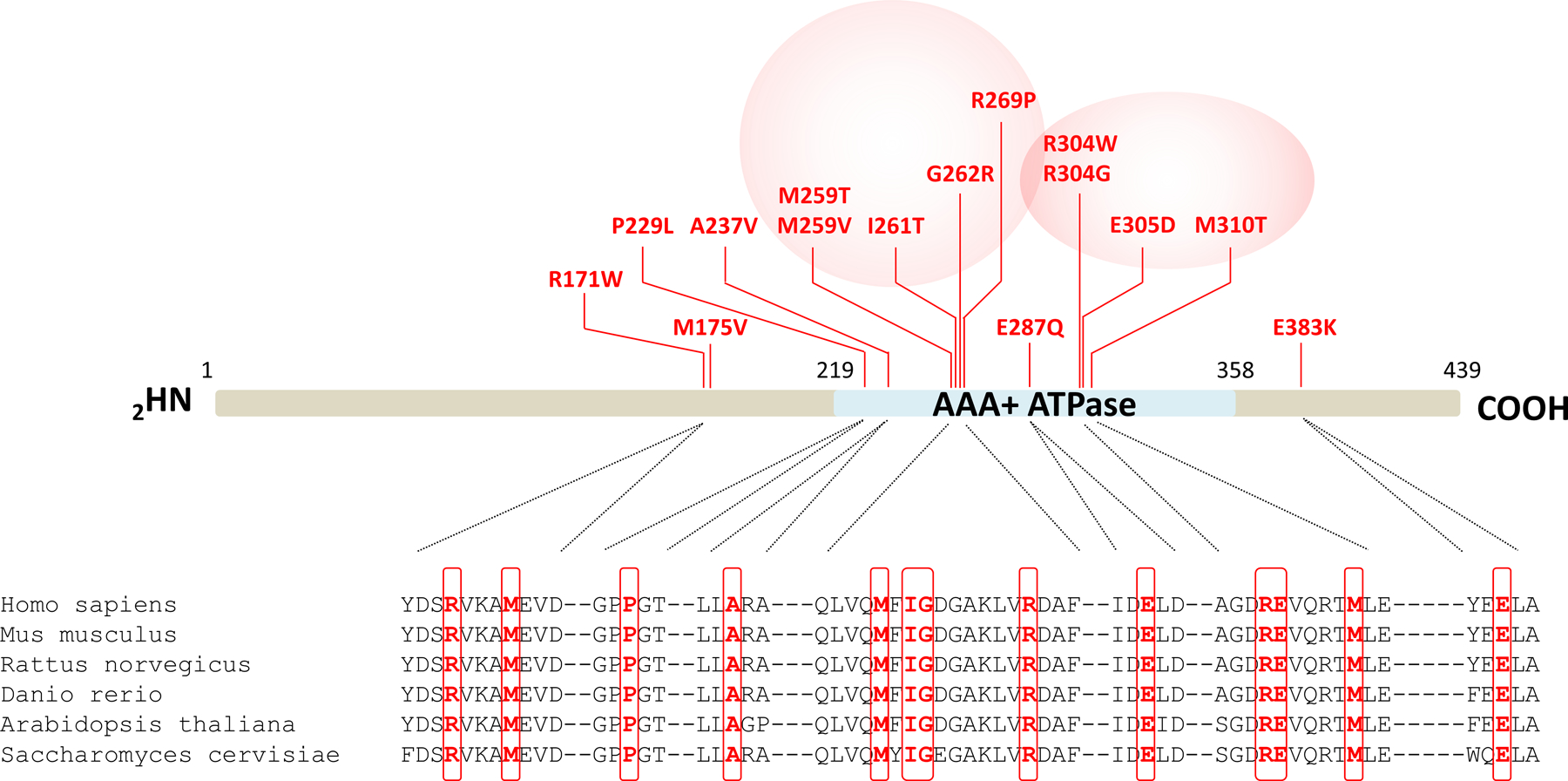
Shown are the locations of the fifteen NDD-causing missense variants (indicated in red) along the PSMC3/Rpt5 protein. The AAA-ATPase domain of the PSMC3/Rpt5 proteasome subunit of the 19S regulatory particle is depicted in blue. Pink circles indicate the presence of variants hotspots. Shown is also a sequence alignment of regions immediately adjacent to the amino acids subjected to missense substitutions. Comparison of the PSMC3/Rpt5 primary structure across six eukaryotic organisms indicates the high conservation of the missense variant residues identified in NDD/ID patients which are highlighted by red boxes.
Silencing of the PSMC3 Drosophila ortholog Rpt5 in adult flies fails to reverse stimulus contingencies
Given the neuronal nature of the phenotype of patients carrying PSMC3 variants, we next sought to address the potential involvement of PSMC3 in cognitive function by evaluating the learning performance of Drosophila melanogaster fruit flies with a RNAi knockdown of Rpt5 (Drosophila ortholog of human PSMC3) expression by two different siRNAs, namely Rpt532422 and Rpt553886 targeting Rpt5 transcripts at two different sites. To this end, we used a standard conditioning of odor-avoidance paradigm, in which animals were exposed to two different odors (3-octanol, OCT or 4-methylcyclohexanol, MCH), only one of which resulted in the simultaneous application of a foot-shock (OCT+, MCH−), as previously described (42) (Fig. 2A and B). Silencing of Rpt5 using Rpt5 RNAi under control of the neuron specific embryonic lethal abnormal visual system (Elav) promoter resulted in no significant differences in learning performance for Rpt532422 (WT vs. Elav:RPT532422, P=0.6435, N=4) or Rpt553886 (WT vs. Elav:RPT553886, P=0.5282, N=6) siRNAs (Fig. 2C). We next determined the reversal learning performance of Rpt5-silenced flies by training with an initial odor shock pairing (OCT+, MCH−) immediately followed by training with a reversed odor shock pairing (OCT−, MCH+) (Fig. 2B, lower panel). Reversal learning performance was significantly poorer with pan-neuronal Rpt5 RNAi expression of Rpt532422 (WT vs. Elav:RPT532422, P<0.0001, N=4) or Rpt553886 (WT vs. Elav:RPT553886, P=0.0022, N=6) siRNAs. The three control groups once again did not significantly differ from each other (Fig. 2C). These data suggest that PSMC3 ortholog Rpt5 appears as a prerequisite for the changes in learned associations in Drosophila melanogaster.
Fig. 2: Pan-neuronal RNAi-mediated knockdown of PSMC3/Rpt5 results in normal learning performance but defective reversal learning performance.
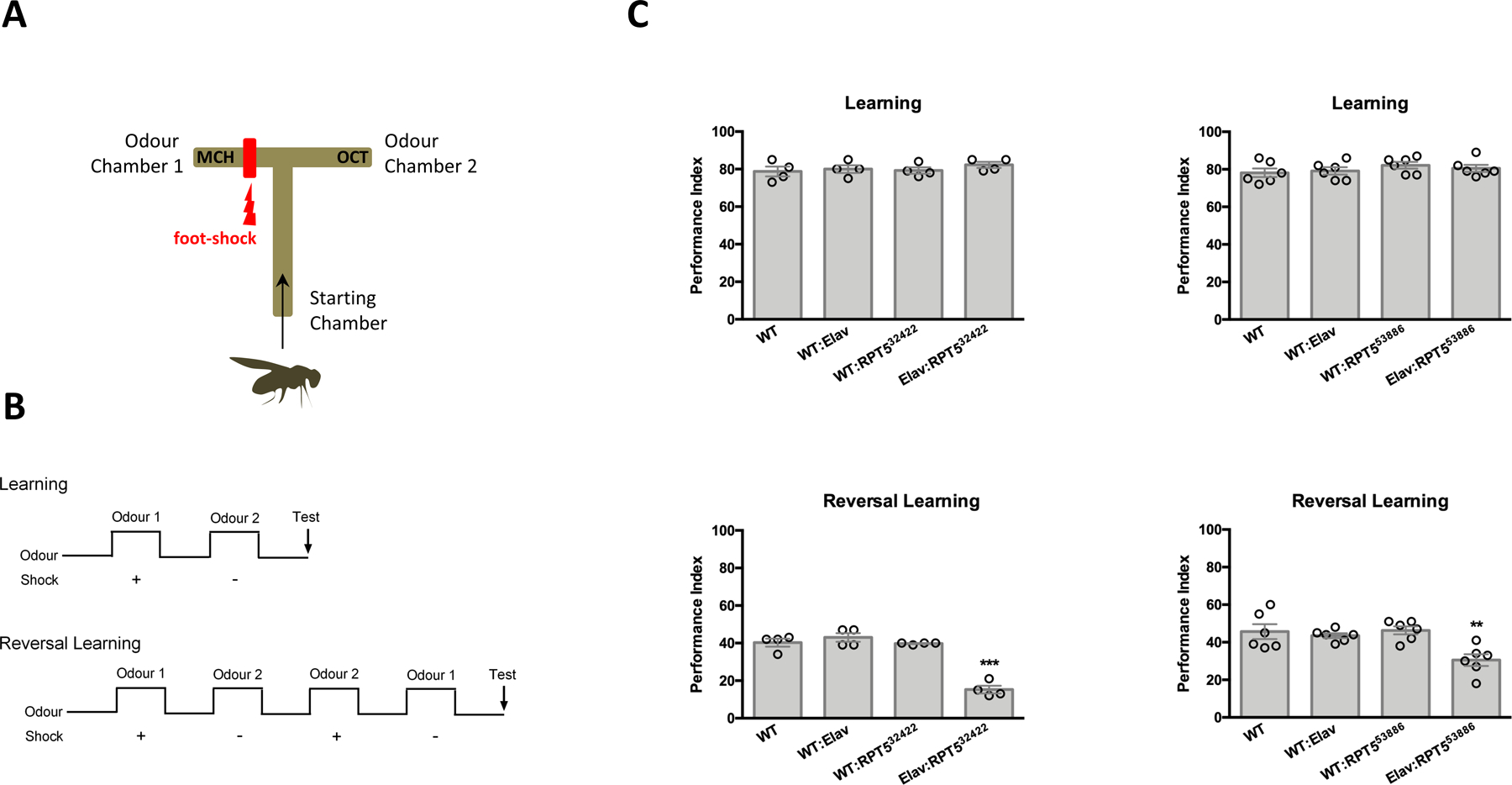
A. Shown is an illustration of a T-maze used for conditioning of odor-avoidance in Drosophila. Flies were trained to avoid one particular odor chamber that was associated with a foot-shock (in this example, odor chamber 1). B. The time course of the learning and reversal learning protocols used in these experiments is illustrated. Reversal learning was assessed by reversing odor shock pairing (4-methylcyclohexanol, MCH−; 3-octanol, OCT+), as indicated. C. Upper left: Shown is the learning performance index of wildtype flies (WT), flies expressing elav, flies with full expression of Rpt532422 RNAi (WT: Rpt532422) and flies with pan-neuronal expression of Rpt532422 RNAi (Elav: Rpt532422) (P=0.6435, N=4). Upper right: Shown is the learning performance index of wildtype flies (WT), flies exressing elav (WT:Elav), flies with full expression of Rpt553886 RNAi (WT: Rpt553886) and flies with pan-neuronal expression of Rpt553886 RNAi (Elav: Rpt553886) (P=0.5282, N=6; Upper right). Lower left: Shown is the reversal learning performance of all group described in C, Upper left. (P<0.0001, N=4). Lower right: Shown is the reversal learning performance index for all groups descripted in C, Upper right (P=0.0022, N=6). Statistical analysis was performed using ANOVA and then Tukey tests in JMP (SAS).**, P<0.01; ***, P<0.001.
Ectopic expression of PSMC3/Rpt5 or its variants differentially impact neuronal development
In view of the negative impact of PSMC3/Rpt5 gene silencing on reversal learning, we next asked whether PSMC3 was involved in the regulation of hippocampal neuron dendritic development. We therefore ectopically expressed wild-type PSMC3/Rpt5 in murine primary hippocampal neurons prior to neurite length quantification, as previously described (43). As shown in Fig. S3, expression of wild-type PSMC3/Rpt5 at an early developmental time point in vitro [Day In Vitro (DIV) 3] resulted in significantly reduced neurite length of the neurons (empty vector vs PSMC3 WT, p=0.0089). These results suggested that ectopic expression of wild-type PSMC3/Rpt5 may be detrimental to neurite outgrowth. We next sought to determine whether the different PSMC3/Rpt5 variants identified in patients with NDD/ID behaved differently compared to wild-type PSMC3/Rpt5 in neurons when ectopically expressed. Expression of the R304W, E305D and E383L PSMC3/Rpt5 variants resulted in similar neuronal morphological changes as seen with wild-type PSMC3/Rpt5 (Fig. S3). By contrast, expression of the M175V variant did not affect neuronal morphology when compared to the empty vector control, and showed significant improvement when compared to wild-type PSMC3/Rpt5 (Fig. S3). The positive effects exerted by the M175V PSMC3/Rpt5 variant on neurite length and arborization are intriguing but do not reflect an increased ability of the mutant subunit to incorporate into 19S-capped proteasome complexes (Fig. S4). In addition, these observations do not preclude a milder pathogenicity of this variant, because the morphological changes seen are not necessarily beneficial for neurite outgrowth. Taken together these results suggest that PSMC3/Rpt5 participates in the regulation of neurite development and that any alteration of this gene might affect this process positively or negatively.
PSMC3 gene variants differentially affect PSMC3/Rpt5 steady-state protein expression
Because missense variants may cause haploinsufficiency by affecting mRNA and/or protein turnover, we next sought to determine the impact of the identified PSMC3 variants on PSMC3/Rpt5 steady-state protein expression. To this end, thirteen of the PSMC3 variants were expressed in the SHSY5Y neuroblastoma cell line with a fused N-terminal hemagglutinin (HA) tandem repeat prior to Western-blot analysis. As shown in Fig. 3A, the four PSMC3/Rpt5 variants carrying the R171W, A237V, M259V and I261T mutations exhibited lower PSMC3/Rpt5 protein expression than their wild-type counterpart. Densitometric analysis of the HA-PSMC3 bands (Fig. 3A, lower left panel) and of PSMC3 bands (Fig. 3A, lower right panel) emerging from these constructs revealed that PSMC3/Rpt5 protein expression was reduced by about 90% when compared to wild-type HA-PSMC3 (Fig. 3A). However, all PSMC3 variants generated equivalent amounts of HA-PSMC3 transcripts in SHSY5Y cells in a 24-h plasmid-driven expression, as determined by RT-PCR and densitometric quantification (Fig. 3B), thereby indicating that reduced protein expression was due to increased protein turnover, decreased translation efficiency, or both. To determine whether these effects could be caused by non-pathogenic PSMC3 mutations as well, we next analyzed the steady-state expression of three HA-tagged single nucleotide polymorphisms (SNPs) PSMC3/Rpt5 variants (I77N, I291V and P355L) reported in the Genome Aggregation Database (gnomAD). As shown in Fig. S5, the three investigated SNPs behaved similarly to their wild-type counterpart, suggesting that decreased PSMC3/Rpt5 protein expression may be a specific feature of some pathogenic variants.
Fig. 3: PSMC3/Rpt5 protein variants do not behave similarly at the molecular level.
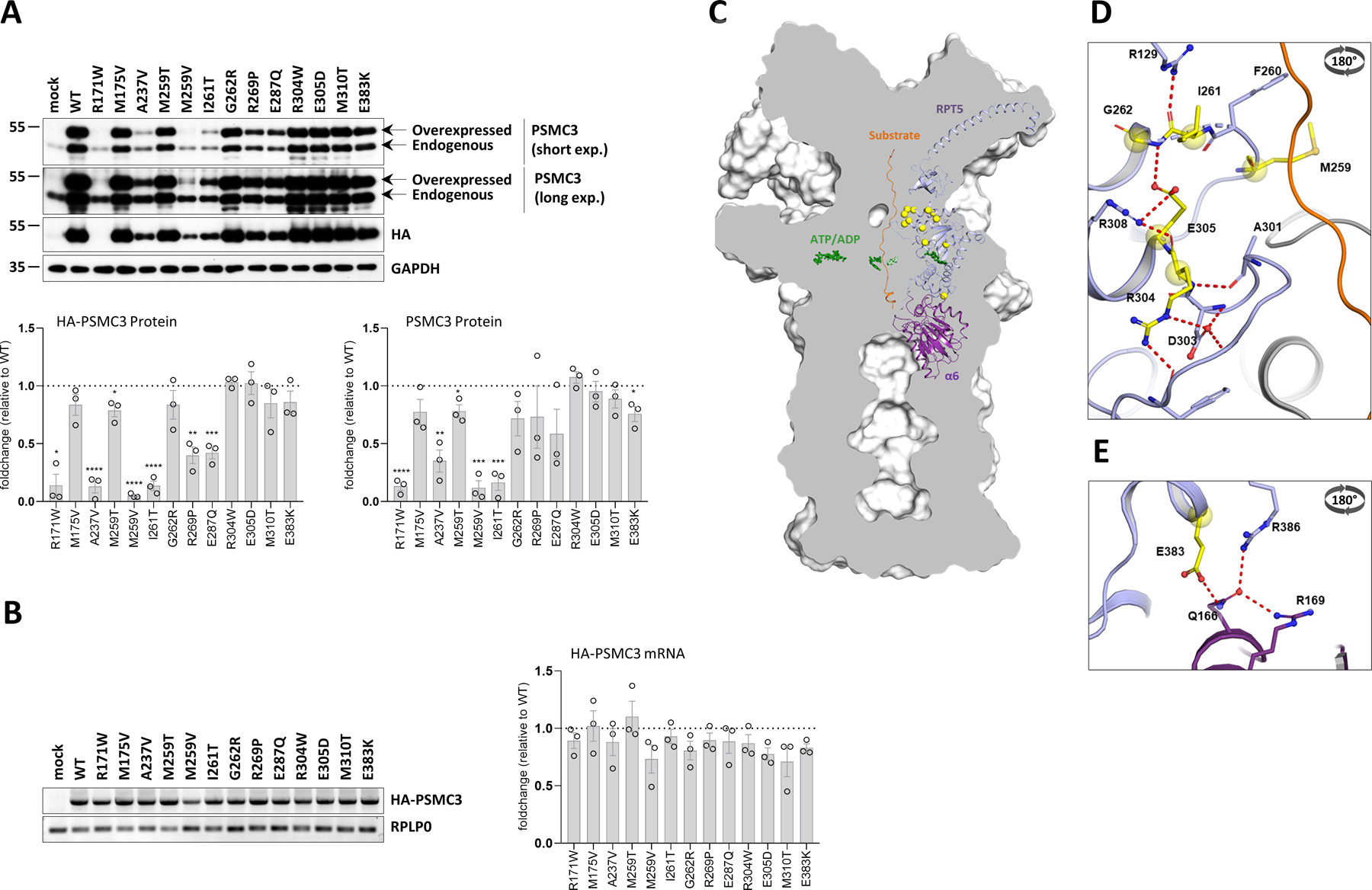
A. Top: SHSY5Y cells were transfected with HA-tagged PSMC3 mutants for 24 h before protein extraction and Western-blotting using antibodies specific for PSMC3/Rpt5 and HA, as indicated. Non-transfected and mock-transfected cells served as negative controls. Equal protein loading was ensured by probing the membranes with an anti-α-tubulin monoclonal antibody (two exposure times are shown). Arrows indicate overexpressed and endogenous PSMC3/Rpt5. Shown is one representative experiment out of three. Bottom: quantification of HA-tagged and untagged PSMC3/Rpt5 proteins in transfected SHSY5Y cells by densitometry. Data are presented as protein foldchanges to wild-type (WT) PSMC3/Rpt5 proteins whose densitometry measurements were set to 1 (gridline) after normalization with GAPDH. Shown are mean values ± SEM from three independent experiments. Statistical significance was assessed by unpaired Student’s test (*p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001). B. Left: SHSY5Y cells that were transfected with HA-PSMC3 variants were subjected to total RNA extraction followed by semi-quantitative RT-PCR using primer located in PSMC3 and the polyadenylation signal of the pcDNA3.1/myc-HIS expression vector (BGH). Equal loading between the samples was ensured by amplifying the RPL0 gene, as indicated. Right: quantification of HA-tagged PSMC3 transcripts in transfected SHSY5Y cells by densitometry. Data are presented as mRNA foldchanges to wild-type (WT) HA-PSMC3 mRNA whose densitometry measurements were set to 1 (gridline) after normalization with RPLP0. Shown are mean values ± SEM from three independent experiments. C. A sliced surface view of the 26S proteasome (grey) was superimposed with a cartoon representation of the subunit PSMC3/RPT5 (blue) and PSMB1/α6 (purple) as well as the substrate (orange). The ATP/ADP molecules of the AAA-ATPase ring are shown as green sticks, while the positions of the investigated missense variants are indicated as bright yellow spheres. D. Detailed representation of the missense variants in the loop region of the N-terminal α/β domain. The residues affected by these variants are involved in a polar interaction network close to the substrate tunnel (view rotated by 180° around the x-axis). E. Close up view on the RPT5-α6 interface affected by the E383L variant. Residues affected by this variant are shown as bright yellow balls and sticks with atoms coloured by polarity (oxygen in red, nitrogen in blue and sulphur in dark yellow, view rotated by 180° around the x-axis).
Ectopic expression of HA-PSMC3/Rpt5 fusion proteins in SHSY5Y cells was accompanied by expression of untagged PSMC3/Rpt5 (Figs. 3A and S5). One potential explanation for this observation implies a partial destruction of the HA epitope, as previously described (44). This assumption is, however, not supported by the fact that such phenomenon was not observed when other proteasomes AAA-ATPases such as PSMC5/Rpt6 were N-terminally tagged with the same HA epitope (Fig. S6). Rather, it may be that the increased pools of untagged PSMC3/Rpt5 detected in cells expressing HA-PSMC3/Rpt5 might reflect a stabilization of endogenous PSMC3/Rpt5. Overexpressed and endogenous PSMC3/Rpt5 proteins might undergo self-association resulting in the formation of homomers protecting them from degradation. To address this point, HA-PSMC3/Rpt5 fusion proteins were pulled down from SHSY5Y cells using HA antibody followed by Western-blot analysis for PSMC3/Rpt5. As shown in Fig. S7, HA-PSMC3 coprecipitated with untagged PSMC3/Rpt5, thereby confirming physical interaction between overexpressed and endogenous PSMC3/Rpt5. However, although the regulation of PSMC3/Rpt5 subunit in response to concentration changes is potentially interesting, the observation that this feature does not vary between wild-type and stable PSMC3 variants suggests that this is not relevant to disease pathogenesis. Altogether, these data showed that the PSMC3 missense variants identified in patients with NDD/ID differentially impact protein expression and stability.
Structural modeling predicts that PSMC3 substitutions affect inter and intra-molecular interactions between proteasome subunits
We next attempted to predict the structural consequences of each of the fourteen PSMC3 substitutions by assessing the localization of the mutated residues in the human 26S proteasome structure generated by Dong et al. (Protein Data Bank, PDB-entry code: 6MSK) (45). Most of the affected amino acids emerged within the N-terminal α/β domain of PSMC3/Rpt5 with five residues (G262, I261, M259, R304 and E305) residing in two loops adjacent to the substrate tunnel pointing towards the center of the AAA-ATPase ring (Figs. 3C and D). Specifically, on one loop, G262 was fixed by a main chain hydrogen bond to E305, thereby promoting flexibility of the preceding loop containing M259. Besides, E305 itself was held through a salt bridge by R308 with its preceding residue R304 involved in a polar network stabilizing the neighbouring loop (Fig. S8). Because these six residues stabilized or were part of the tertiary structure of the loops, any alteration of these amino acids is predicted to affect substrate trafficking, as well as interactions with other AAA-ATPase subunits. As shown in Fig. S8, the A237V variant was more difficult to classify and did not reveal itself structurally at first sight. However, one cannot exclude that the slight increase in residue size at position 237 might lead to structural changes. The overexpression assays in SHSY5Y cells suggest that such substitution does affect sidechain packing and protein stability (Fig. 3A). The E383L missense variant was the only substitution lying within the C-terminal α-helical domain of PSMC3/Rpt5 adjacent to the 19S-20S interface. E383 held Q166 from the PSMA1/α6 subunit for polar interactions with both of the R169 and R386 residues (Fig. 3E). Changing the negatively charged E383 to a positively charged K383 is therefore predicted to disrupt such hydrogen bond network and affect the association of the 19S complex with the 20S core particle. E287 was also located in close proximity to the ATP binding site (Fig. S8) and its substitution with Q287 presumably generated additional polar bonds with N333 likely to affect ATP binding, hydrolysis, or both. R171 was positioned at the PSMC3/Rpt5-PSMC2/Rpt1 interface and was part of a polar network involving the neighboring D169 and Q258 residues (Fig. S9). As such, the substitution of positively charged arginine to hydrophobic tryptophan at this position is predicted to disrupt these interactions, and a fortiori to affect the contact between the two subunits (Fig. S9). The change of M175to V175 resulted in the loss of a polarized thiol group and hydrophilic environment which was likely to destabilize the tertiary structure of this protein region (Fig. S9). Taken together, these data suggest that the complex 26S proteasome structure may be strongly affected by the identified PSMC3 missense variants, even though an incorporation of dysfunctional PSMC3/Rpt5 subunit with no detrimental effects of proteasome structure cannot be fully excluded.
PSMC3 variants differentially impact proteasome assembly
To further address the pathogenicity of the PSMC3 variants, T cells from Patients #13, #18 and #21 were analyzed for their proteasome contents. The intracellular expression of the α7 proteasome subunits and PA28-α did not vary between relative controls (father, mother or both) and index cases (Fig. 4A). Likewise, the abundance of the 19S subunit PSMD12/Rpn5 in mutant T cells was comparable to that detected in their control counterparts. T cells expanded from controls and patients exhibited two prominent PSMC3/Rpt5 immunoreactive bands migrating at about 50 and 40 kDa (Fig. 4A). Although the upper band running at ~50 kDa corresponded to PSMC3/Rpt5 expected size, the nature of the lower band running at ~40 kDa was unclear. It is, however, unlikely that this band may be non-specific, because it was detectable in T cells using another anti-PSMC3/Rpt5 antibody or in other cell types (Fig. S10). This additional species might reflect a shorter, as yet undescribed PSMC3/Rpt5 isoform or a processed form arising from the PSMC3/Rpt5 full-length protein. Nevertheless, the unchanged protein expression profile of PSMC3/Rpt5 between controls and patients indicates that none of these phenomena is affected by any of the PSMC3 variants. Likewise, none of PSMC3 variant T cells showed reduced expression of the PSMC3/Rpt5 full-length protein, suggesting that proteasome dysfunction in these affected individuals was not caused by haploinsufficiency.
Fig. 4: PSMC3/Rpt5 protein variants lead to proteasome assembly defects in patient T cells.
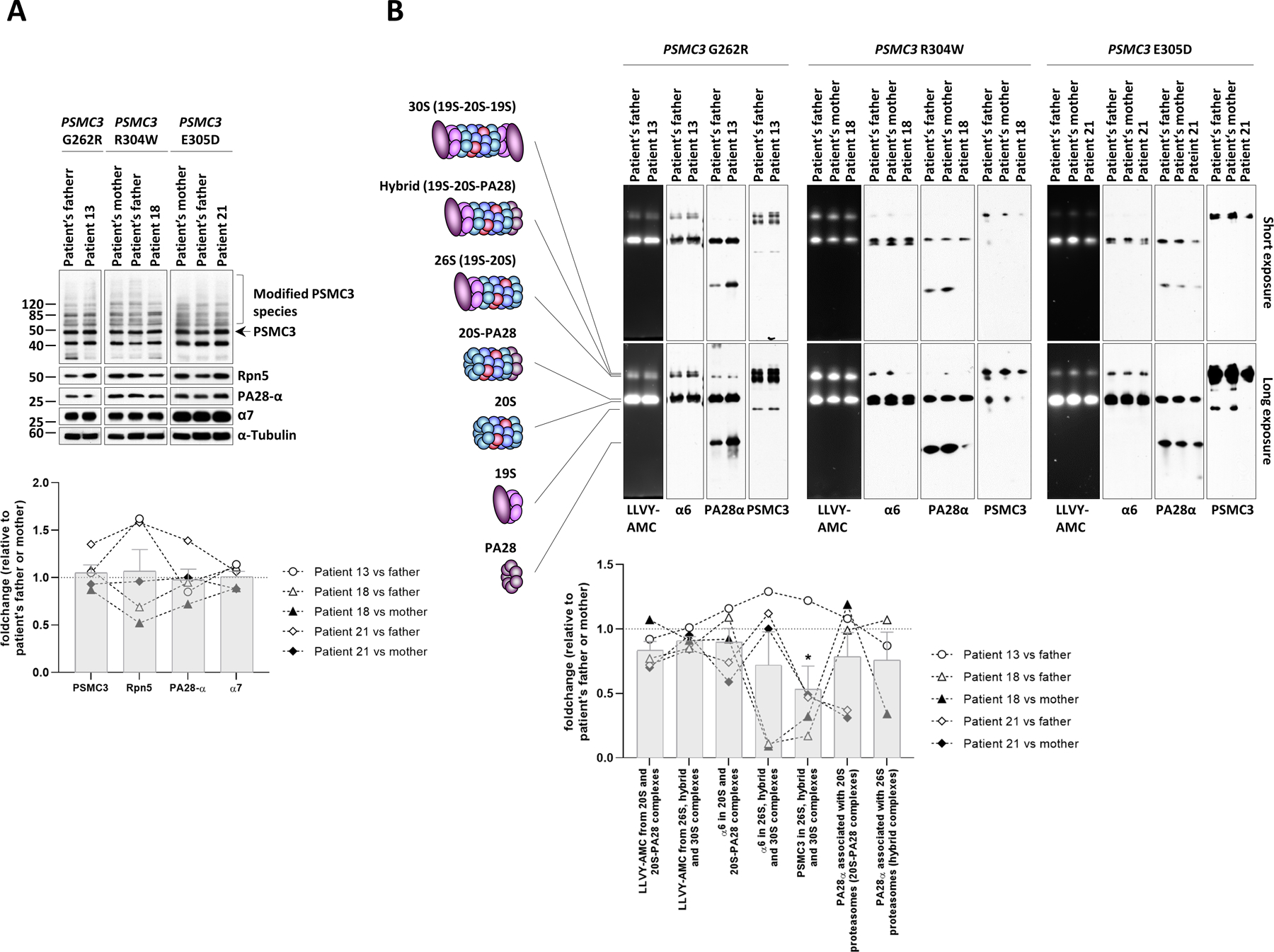
A. Top: five to twenty micrograms of radio-immunoprecipitation assay (RIPA) lysates from T cells isolated from Patients #13, #18 and #21 as well as related controls (index case’s father and/or mother) were separated by SDS-PAGE followed by Western-blotting using antibodies directed against PSMC3/Rpt5, PSMD12/Rpn5, PA28-α and α7, as indicated. Equal protein loading was ensured by probing the membrane with an anti-α-tubulin antibody. Arrow indicates full-length PSMC3/Rpt5 migrating at the predicted size of about 50 kDa. High molecular weight (HMW) modified PSMC3/Rpt5 species are marked by a bracket. Bottom: quantification of the Western blots by densitometry. Data are presented as foldchanges in Patients #13, #18 and #21 vs their father, mother or both, whose densitometric measurements were set to 1 (gridline), as indicated. Columns indicate the foldchange mean values ± SEM calculated from the five normalizations. B. Top: twenty micrograms of T-cell lysates from patients #13, #18 and #21 and their parents (mother and/or father) were separated by 3–12% native-PAGE. Proteasome chymotrypsin-like activity was assessed in gels using the LLVY-AMC fluorogenic peptide, as indicated. Gels were subsequently subjected to Western-blotting using antibodies specific for α6, PSMC3/Rpt5 and PA28-α, as indicated. The schematic to the left depicts the proteasome complexes (30S, hybrid, 26S, 20S-PA28 and 20S) and free regulators (19S and PA28) detected by the three antibodies. Bottom: quantification of the LLVY-AMC fluorescent signals and α6, PA28α and PSMC3 immunoreactive bands in 20S (short exposure), 20S-PA28 (short exposure), 26S (long exposure), hybrid (long exposure) and/or 30S (long exposure) proteasome complexes by densitometry, as indicated. Data are presented as activity (LLVY-AMC) and protein (α6, PA28α and PSMC3) foldchanges in Patients #13, #18 and #21 vs their father and/or mother whose densitometric measurements were set to 1 (gridline), as indicated. Columns indicate the foldchange mean values ± SEM calculated from the five normalizations. Statistical significance was assessed by unpaired Student’s test (*p<0.05).
Next, T cells from affected individuals and relative controls were analyzed for proteasome complex formation and activity by in-gel fluorescence followed by Western blotting on native-PAGE. As shown in Fig. 4B, the chymotrypsin-like activity of the 26S and 20S proteasome complexes (LLVY-AMC) was reduced for the majority of comparisons between patients and controls. Subsequent Western-blot analysis revealed that the decreased 20S activity observed in Patient #21 was associated with a decreased pool of 20S-PA28 complexes, as determined by reduced band intensity for the α6 and PA28 proteins. The amounts of unbound PA28α/β complexes in T cells were increased in Patient #13 and decreased in Patient #18 (Fig. 4B). The reasons for these contrasting data between these two patients are unclear but might reflect distinct abilities to compensate for proteasome dysfunction. It should be noted, however, that, in contrast to PA28-bound proteasomes whose amounts did not change in these patients, free PA28 α/β are not equipped with protease activity and as such have no impact on intracellular proteolysis. Densitometric analysis of the PSMC3/Rpt5 signals revealed impaired subunit incorporation into 26S, hybrid and 30S proteasome complexes in patients when compared to controls (P=0.0329) (Fig. 4B, bottom). Diminished expression of the α6 and PSMC3/Rpt5 subunits was observed in the 26S proteasomes of Patient #18 (Fig. 4B), indicating that the decline in 26S activity detected in this patient was likely to be attributed to decreased amounts of 26S complexes. Likewise, the PSMC3/Rpt5 contents in 26S complexes were also reduced in Patient #21 (Fig. 4B). Unexpectedly 20S and 26S proteasome pools of Patient #13 did not substantially vary when compared to those of the related control (Fig. 4B). The minor effects exerted by the G262R substitution in Patient #13 may be partially explained by the fact that G262 is surrounded by a large amount of empty space (Fig. S11), thereby allowing this region of the PSMC3/Rpt5 protein to accommodate mutations to some extent. Nevertheless, these data indicated that both of the R304W and E305D PSMC3/Rpt5 variants affect 20S proteasome assembly, 26S proteasome assembly, or both in individuals with NDD/ID, whereas the G262R variant has little impact in this process (Table 1).
Quantitative proteomics identifies cellular pathways affected by PSMC3 loss-of-function in patients with NDD/ID
To better understand the cellular consequences of PSMC3 loss-of-function, we next performed a mass spectrometry-based comparative analysis of the T-cell proteomes of Patient #17 and Patient #21 (R304W and E305D, respectively) to that of their relative controls. As shown in Fig. 5, our data identified a protein signature consisting of seventeen ribosomal proteins of the small 40S (RPS) or large 60S (RPL) ribosomal subunits that were specifically upregulated in both investigated patients. This suggests that mRNA translation is a major affected pathway upon PSMC3 loss-of-function, a notion which is further supported by the fact that components of the mRNA processing machinery such as CUG-binding protein Elav-Like Family Member 1 (CELF1), a member of the LSm family of RNA-binding proteins (LSM1), RNA Polymerase II CTD Phosphatase (SSU72) and Inosine Triphosphatase (ITPA) were also differentially expressed between patients and controls (Fig. 5). Other notable proteins whose abundances varied in patients carrying PSMC3 variants included components of the immune system such as MX Dynamin Like GTPase 1 (MX1) –a typical IFN-stimulated gene product– and the α-chain of the IL3 receptor (IL3RA). These proteins were regulated in opposite directions with both patients exhibiting higher amounts of MX1 but reduced amounts of IL3RA (Fig. 5). Our analysis further revealed that PSMC3 loss-of-function was also associated with increased protein expression of the H1.5 and H1.2 linker histone H1 variants, a finding that is in line with a role of the UPS in chromatin regulation (46). Protein set enrichment analysis uncovered that most of the significant (P<0.05) proteomic changes between control and patients carrying PSMC3 variants were related to mRNA metabolism and translation (Fig. S12), confirming that protein synthesis was dysregulated in these patients. Other differentially expressed proteins found to be enriched in T cells with PSMC3 variants belonged to the category of viral processes (Fig. S12), unveiling a potential relationship between proteasome loss-of-function and innate immunity. Collectively, these data suggested that patients with PSMC3 variants exhibit alterations in basic cellular processes including mRNA translation, immune signaling and chromatin remodeling.
Fig. 5: Proteomic signatures of patients carrying PSMC3/Rpt5 variants.
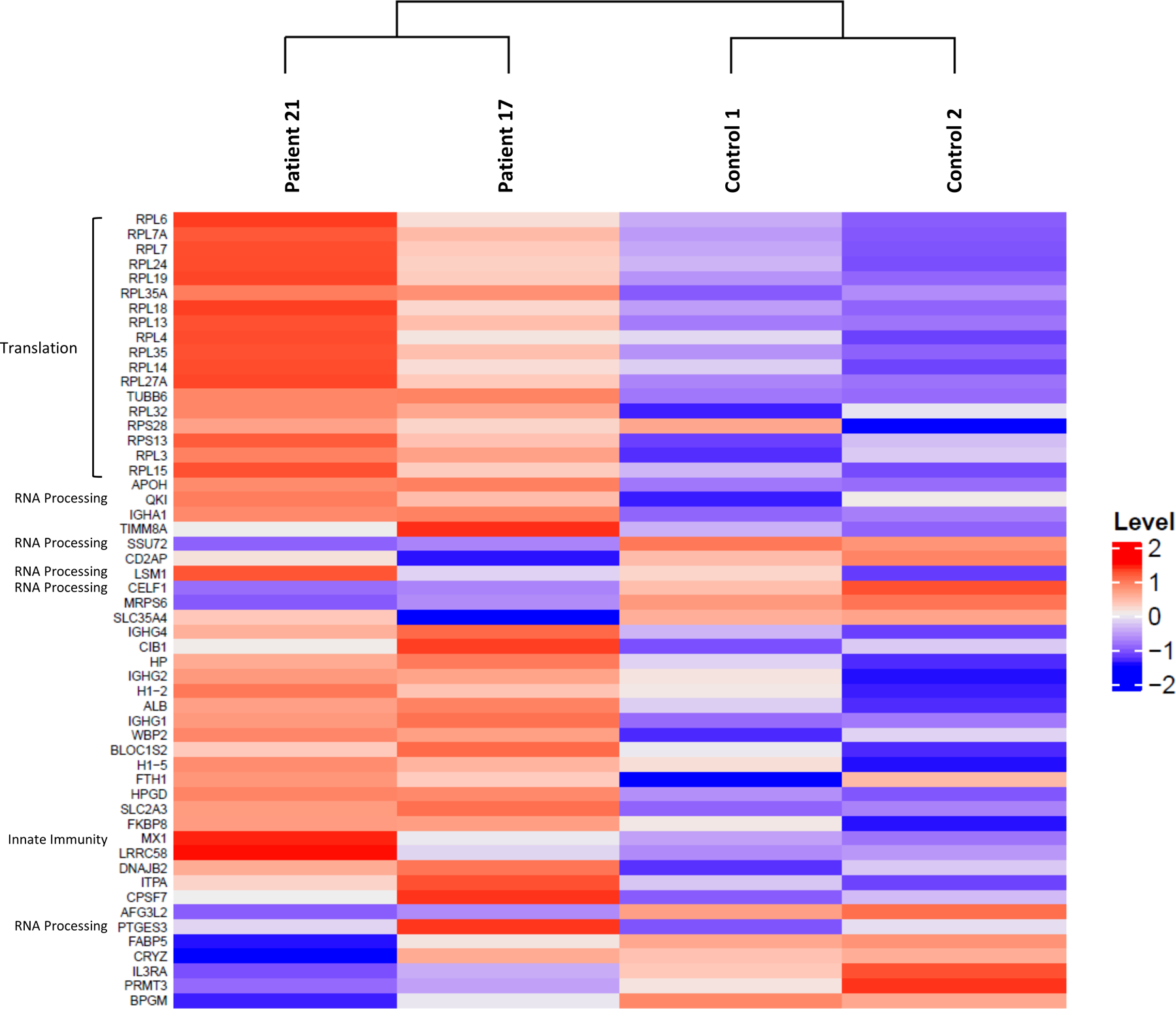
Heatmap cluster analysis showing the similarities in the protein expression profiles of the Patients #17 and #21 (carrying the R304W and E305D PSMC3 variants, respectively) compared to their related controls (father and/or mother of the proband), as indicated. The heatmap indicates the normalized and scale expression value of proteins in the individual samples. Only the differentially expressed proteins with an absolute value of log2 fold-change greater than 2 were selected for the clustering analysis.
PSMC3 variants cause proteotoxic stress in patient T cells
Proteasome dysfunction has been shown to be accompanied by accumulation of ubiquitin-protein conjugates and proteotoxic stress (24). As shown in Fig. 6A, all four investigated patients exhibited typical features of unbalanced protein homeostasis, as evidenced by increased accumulation of ubiquitin-modified species when compared to their respective related controls using immunoblotting and densitometric analysis. Proteotoxic stress is known to induce the unfolded protein response and integrated stress response (UPR and ISR, respectively) (24, 47). To address this point, we quantified the expression of the glucose-regulated protein 94 chaperone protein (GRP94, the heat shock protein 90kDa in the ER) whose upregulation is understood to be a major hallmark of the UPR (48). The expression of GRP94 was increased in all patients (Fig. 6A). However, the activation of the UPR was only partial, because the phosphorylation and activation status of two other UPR markers, namely serine/threonine-protein kinase/endoribonuclease inositol-requiring enzyme 1 α (IRE1) and eukaryotic translation initiation factor (eIF2)α, was not changed between controls and patients carrying PSMC3 variants (Fig. 6A). The failure to detect increased phosphorylated eIF2α, although the upstream kinase PKR was consistently activated in all patients (Fig. 6A), can be explained by upregulation of both eIF2α phosphatases GADD34 (growth arrest and DNA damage-inducible protein 34) and CReP (constitutive repressor of eIF2α phosphorylation) across all four patients. Immunoblotting and densitometric analysis revealed that T cells of the patients carrying PSMC3 variants were also endowed with increased protein expression of microtubule-associated protein 1A/1B-light chain 3 phosphatidylethanolamine conjugate LC3b-II (Fig. 6B), suggesting that the inability of these cells to eliminate ubiquitin-protein aggregates cells via their 26S proteasomes triggers a compensatory mechanism mediated by activation of the autophagy system. Consistently, the mitochondrial proteins PINK1 (PTEN-induced putative kinase protein 1) and Bnip3L/NIX (BCL2/adenovirus E1B 19 kDa protein-interacting protein 3-like/NIP3-like protein X) were found to be decreased in the patients carrying PSMC3 variants (Fig. 6B, Table 1), supporting the notion that selective autophagic processes including mitophagy were activated upon PSMC3 disruption. Because proteasome impairment typically results in the release of the TCF11/Nrf1 (transcription factor 11/nuclear factor, erythroid derived 2, like 1) protein from the ER membrane (49, 50), we next sought to determine the TCF11/Nrf1 processing pattern in NDD/ID affected individuals. However, no differences in the TCF11/Nrf1 processing pattern could be detected between controls and patients carrying PSMC3 variants (Fig. 6B), suggesting that PSMC3 variants associated with NDD/ID do not lead to activation of the TCF11/Nrf1 signaling pathway.
Fig. 6: T cells from patients carrying PSMC3/Rpt5 variants exhibit signs of protein homeostasis perturbations and alterations of the UPR, ISR and autophagy/mitophagy pathways.
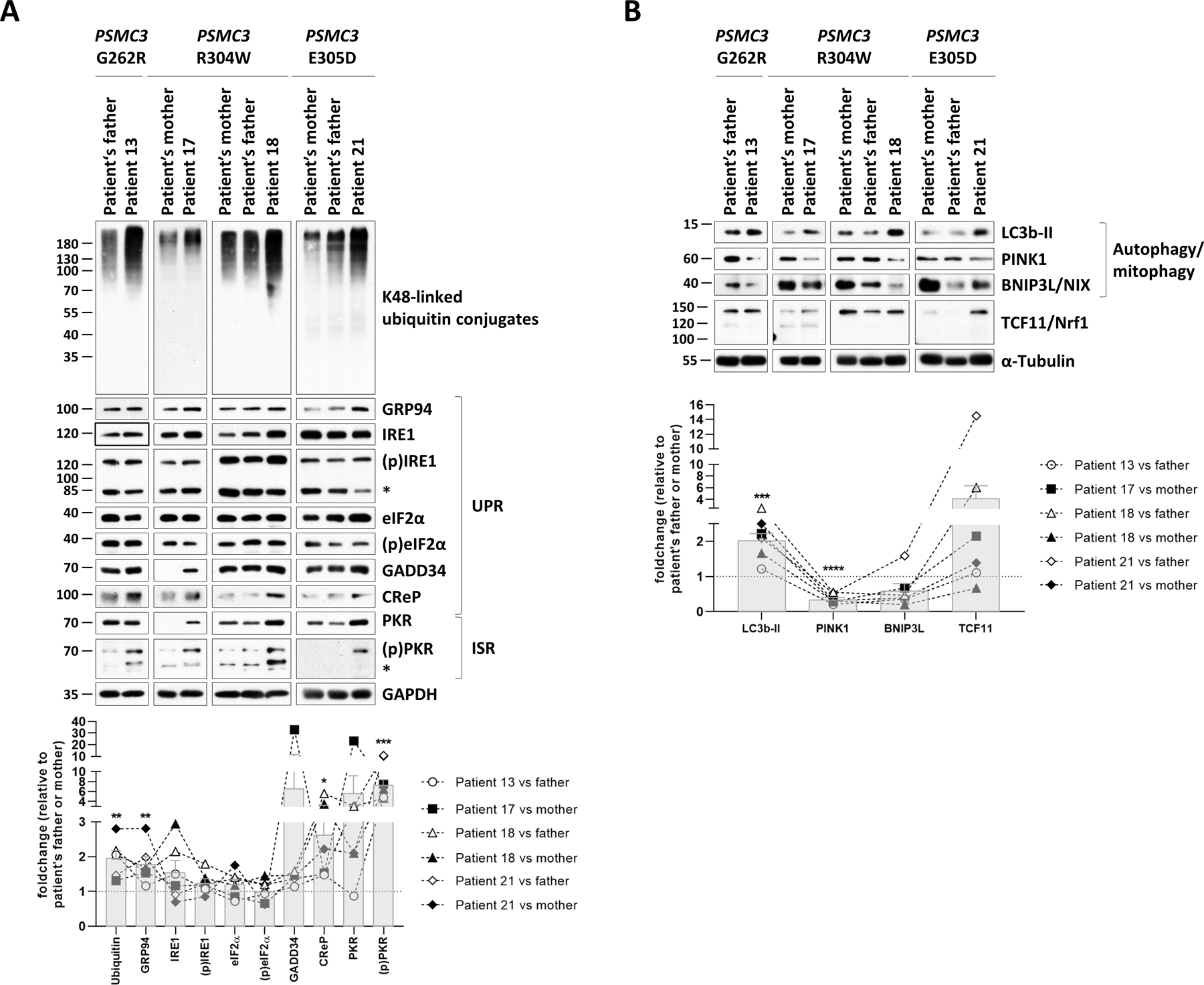
A. Top: five to twenty micrograms of RIPA lysates from T cells isolated from Patients #13, #17, #18 and #21 as well as related controls (PSMC3 index case’s father and/or mother) were separated by SDS-PAGE followed by Western-blotting using antibodies directed against K48-linked ubiquitin-modified proteins, GRP94, IRE1, phospho-IRE1, eIF2α, phospho-eIF2α, GADD34, CReP, PKR, phospho-PKR and GAPDH (loading control), as indicated. Bottom: Shown is the quantification of the Western-blots by densitometry. Data are presented as foldchanges in Patients #13, #17, #18 and #21 vs their father and/or mother whose densitometric measurements were set to 1 (gridline), as indicated. Columns indicate the foldchange mean values ± SEM calculated from the six normalizations. Statistical significance was assessed by unpaired Student’s test (*p<0.05, **p<0.01 and ***p<0.001). B. Top: RIPA-cell lysates from Patients #13, #17, #18 and #21 as well as their related controls (index case’s father and/or mother) were subjected to SDS-PAGE/Western-blotting using antibodies specific for LC3b, PINK1, BNIP3L and α-tubulin (loading control), as indicated. Bottom: quantification of the Western-blots by densitometry. Data are presented as protein foldchanges in Patients #13, #17, #18 and #21 vs their father and/or mother whose densitometric measurements were set to 1 (gridline), as indicated. Columns indicate the foldchange mean values ± SEM of the six normalizations. Statistical significance was assessed by unpaired Student’s test (***p<0.001 and ****p<0.0001).
T cells with PSMC3 variants exhibit a type I IFN signature
Because proteasome loss-of-function results in the generation of a typical type I IFN response in PRAAS patients (30), we next sought to determine whether alterations in the PSMC3 gene would induce type I IFN signatures as well. To this end, we undertook a comparative examination of the mRNA expression of 750 predefined immunologically relevant genes in T cells from patients carrying PSMC3 variants and relative controls (father and/or mother) using the NanoString® nCounter platform. A total of 30 differentially expressed genes could be identified including 11 IFN-stimulated genes (ISGs) that were specifically upregulated in all patients carrying PSMC3 variants (Fig. 7A), suggesting that PSMC3 loss-of-function is associated with type I IFN responses. The transcriptomic analysis of control and patient T cells further revealed that PSMC3 disruption resulted in the upregulation of genes of the notch signaling pathway such as NOTCH2 (neurogenic locus notch homolog protein 2) and JAG2 (protein jagged-2) involved in developmental pathways including neurodevelopment (51) (Fig. 7A). To validate the type I IFN gene signature revealed by our omics profiling, we next evaluated the expression of seven ISG (IFI27, IFI44L, IFIT1, ISG15, MX1, RSAD2 and IFI44) in samples of these four families with PSMC3 missense variants. As shown in Fig. 7B, all four affected children’s samples (Patients #13, #17, #18 and #21) exhibited much higher ISG expression than their parents (father and/or mother; Table 1). Calculation of the fold change median of the seven ISG revealed that a significant increase was detected in all PSMC3 index cases when compared to their respective controls (Patient #13 vs father, p=0.0148; Patient #17 vs mother, p=0.0008; Patient #18 vs father, p=0.0002, Patient #18 vs mother, p=0.0011; Patient #21 vs father, p=0.0012 and Patient #21 vs mother, p=0.0142) (Fig. 7B). The strongest type I IFN signature was observed in Patient #18 whose ISG were upregulated by approximately 40- or 10-fold when compared to the father or mother, respectively. Patients #13, #17 and #21 exhibited a milder type I IFN induction characterized by a 2-to 6-fold increase in ISG transcripts than their respective controls. Among the seven ISGs tested, IFIT1, and IFI44L were the genes which underwent the most pronounced upregulation in all four affected individuals.
Fig. 7: T cells from patients with PSMC3/Rpt5 missense variants exhibit a typical type I interferon (IFN) signature.
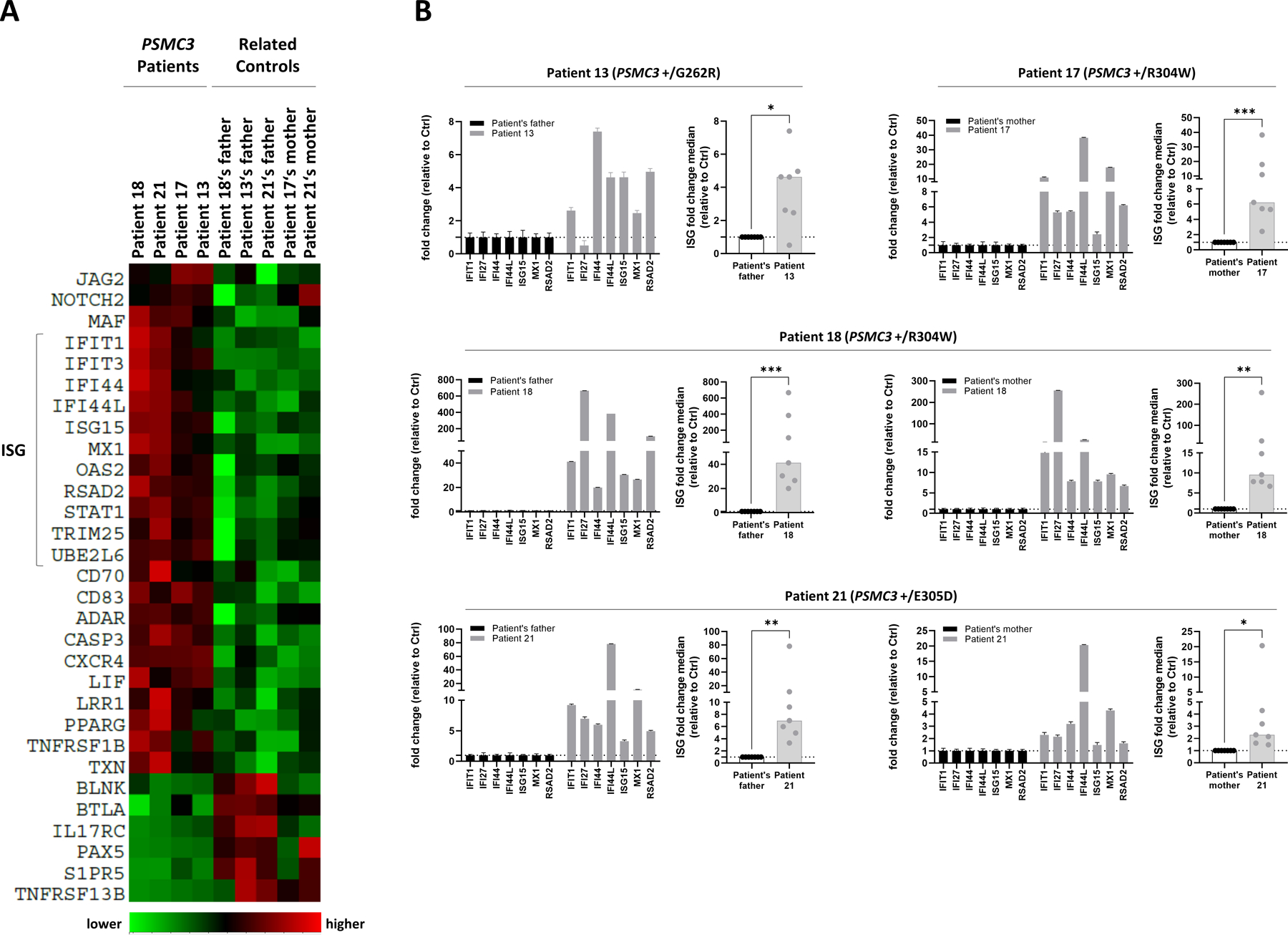
A. Heat map clustering of gene expression in T cells isolated from patients carrying a PSMC3 variant and their relative controls (father and/or mother). Each column represents one individual patient or related control and each row represents one gene. Clustering of genes and samples was carried out by centred Pearson correlation. Colour indicates normalized counts of each transcript, with green representing higher expression and red relatively lower expression. B. Gene expression of seven typical IFN-stimulated genes (IFIT1, IFI27, IFI44, IFI44L, ISG15, MX1 and RSAD2) was assayed by RT-qPCR on T cells derived from Patients #13, #17, #18 and #21 as well as their respective controls (the index case’s father and/or mother). Expression levels were normalized to GAPDH and relative quantifications (RQ) are presented as fold change over controls. Shown is also the median fold expression of the seven ISGs over relative controls. Statistical significance was assessed by ratio paired t test where *indicates p<0.05, ** indicates p<0.01 and *** indicates p<0.001.
The type I IFN signature generated in PSMC3 mutant T cells is PKR-dependent
We next calculated and compared the IFN scores of both patients carrying PSMC3 variants and their related controls to those of T cells isolated from six healthy donors. As shown in Fig. 8A, three of the related controls had an IFN score slightly above the cut-off value of 2.466 defined by Rice et al. to be abnormal (52). However, the IFN scores of all related and unrelated controls remained significantly lower than those of the four tested patients (P=0.0845 and P=0.0044), thereby confirming that these PSMC3 variants were associated with enhanced type I IFN signaling. A second independent assessment of Patient #18 at 14 months after enrollment revealed that this patient still exhibited a very high type I IFN score (Fig. S13). Because all three inducible immunoproteasome subunits β1i (PSMB9), β2i (PSMB10) and β5i (PSMB8) are encoded by genes typically stimulated by type I and II IFN (20), we next asked whether PSMC3 loss-of-function was accompanied by a switch from standard proteasomes to immunoproteasomes. As illustrated in Fig. S14, the steady-state expression of these subunits in T cells did not substantially change between control and patients, as determined by Western-blotting. This may be due to the known fact that T cells, as immune cells, express high amounts of immunoproteasomes, a feature that renders any further protein upregulation of the inducible subunits very difficult (53).
Fig. 8: T cells from patients carrying PSMC3/Rpt5 variants exhibit high protein kinase R (PKR)-dependent type I IFN scores.
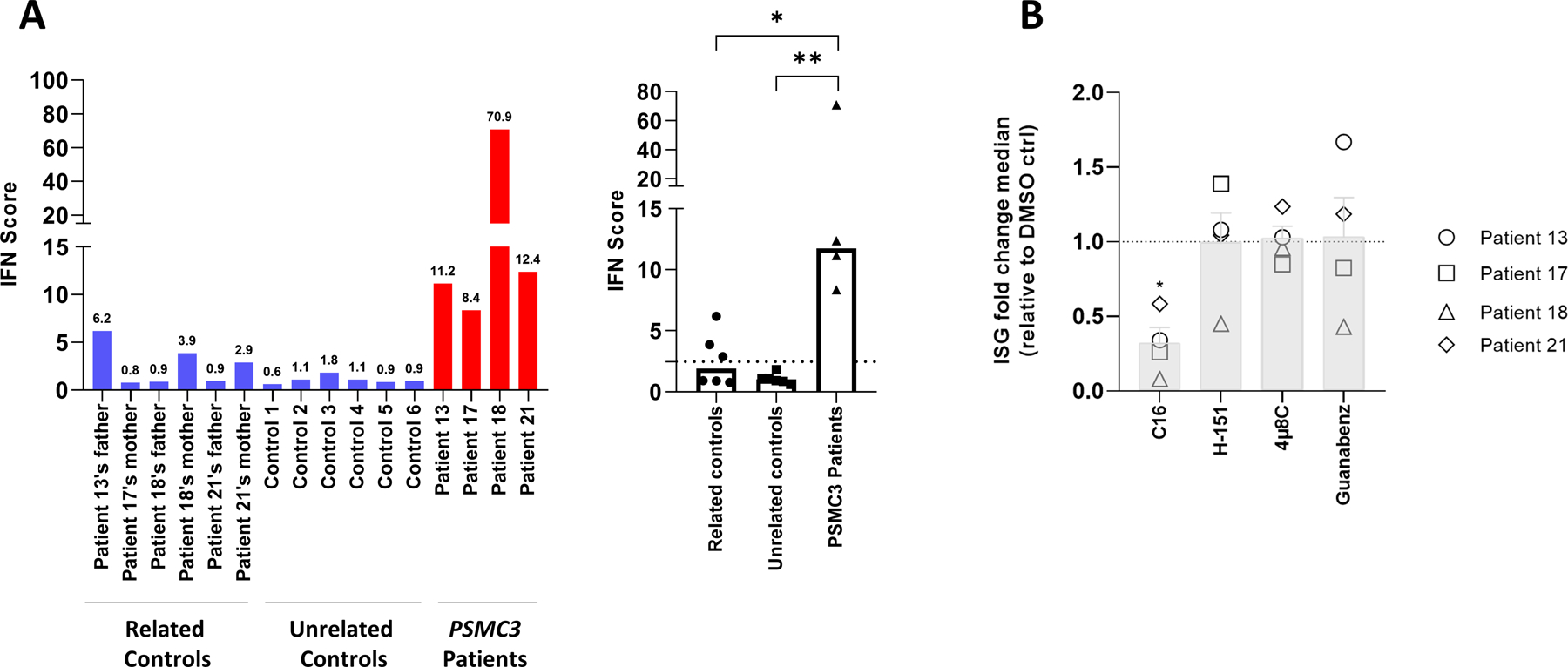
A. left panel: IFN scores for Patients #13, #17, #18 and #21 and related controls, as well as for six unrelated controls (1 to 6) were calculated as the median of the relative quantifications of the seven ISGs over a single calibrator control. Shown are the IFN scores of each sample (left panel) and the sample groups, namely parents, unrelated healthy donors and patients carrying PSMC3 variants, as indicated. right panel: Box plot of concatenated data. Statistical significance was assessed by unpaired t test where *indicates p<0.05 and ** indicates p<0.001. B. T cells isolated from individuals carrying PSMC3 variants were subjected to a 6-h treatment with DMSO (vehicle), C16 (500 nM), H-151 (2 µM), 4µ8C (100 µM) or Guanabenz (50 µM) inhibitors before RNA extraction and RT-qPCR for expression analysis of IFI27, IFI44L, IFIT1, ISG15, RSAD2, IFI44, OASL and MX1. Transcript expression was normalized to GAPDH and data are presented as the foldchange median values of the eight ISG relative to DMSO (gridline) for each patient in each treatment. Columns indicate the foldchange mean values ± SEM of the patient group (n=4) for each treatment. Statistical significance was assessed by ratio paired t test where * indicates p<0.05.
We next attempted to unravel the mechanisms by which type I IFN responses were initiated in patients carrying PSMC3 loss-of-function variants. Having shown that all four affected patients exhibited marked alterations in the mitophagy, UPR and ISR pathways (Fig. 6), we postulated that the dysregulation of either one of these pathways might act as a danger signal triggering innate immunity. To address this point, we inhibited key players of the UPR and/or ISR including PKR, IRE1 and GADD34 (24) by treating patient T cells with the C16, 4µ8C or Guanabenz inhibitors, respectively. In addition, the potential implication of mitophagy-mediated mitochondrial DNA in this process was investigated by treating the cells with H-151, an inhibitor of the cytosolic DNA sensor STING (stimulator of interferon genes protein) (54). Of the four inhibitors used, only C16, targeting PKR, could substantially reduce the type I IFN response associated with PSMC3 variants in these patients (Fig. 8B). These data thus suggest PKR as a sensor of proteasome dysfunction triggering a type I IFN signature in individuals carrying PSMC3 variants.
DISCUSSION
In this study, we identified fifteen missense variants in the PSMC3 gene in twenty-three unrelated individuals with NDD/ID (Fig. 1, Tables 1 and S1) and showed that the 19S AAA-ATPase proteasome subunit PSMC3/Rpt5 is a critical protein for the development of the central nervous system (CNS). This notion is in line with previous reports showing that conditional inactivation of other 19S proteasome subunits (Psmc2/Rpt2 and Psmc4/Rpt3) in mice results in severe neuronal phenotypes with features of neurodegeneration and locomotor dysfunction (55, 56). Recently, a homozygous deep intronic variant creating a cryptic exon in the PSMC3 gene was linked to a familial recessive neurosensory syndrome (57). However, given their distinct modes of inheritance and pathogenesis, this recessive disorder observed in a single family and the dominant variants, which we describe are likely two different clinical entities with only partial overlap of clinical and molecular disease phenotypes (57). This dichotomy between dominant and recessive disorders may, nevertheless, be too reductive, because our data show that the dominant PSMC3 variants do not necessarily exert the same effects on proteasome expression, assembly, or both (Figs. 3A, 4B and Table 1). These variations make it difficult to classify dominant PSMC3 variants according to their impact on cellular function without any in vitro functional studies.
There are several limitations to this study. Given the limited number of biological samples investigated, a correlation between genotype and cellular phenotype could not be established. In this regard it should also be noted that the gene silencing in the Drosophila model does not completely reflect disease biology patients, given that gene product is still available. We can furthermore not fully exclude a recruitment bias in our patient data set.
Cognitive flexibility is an important aspect of typical brain function which allows adaptation to both physical and social environmental changes (58, 59). This may be assessed by evaluating reversal learning performance, a process which was initially identified in Drosophila models (60) and whose dysfunction has been associated with the pathogenesis of various neuropsychiatric disorders (61–65). Although Rpt5 gene-silencing in flies had no discernible effect on learning performance, it led to compromised reversal learning (Fig. 2). Although proteasomes have been shown to regulate long-term potentiation (LTP) (66, 67), their involvement in reversal learning has not previously been shown. Our data identify PSMC3/Rpt5 as a key regulator of this process whose molecular landscape was initially limited to a few molecules related to the cytoskeleton and GABAergic system (68–70). One cannot exclude that Rpt5 gene-silencing in Drosophila may result in global depletion of 26S complexes which in turn impairs reversal learning. However, some of the missense mutations identified in this study led to decreased PSMC3/Rpt5 expression (Fig. 3A), reduced incorporation into 26S complexes (Fig. 4B), or both suggesting that a shortage of the PSMC3/Rpt5 subunit may indeed reflect disease pathogenesis. The mechanisms by which proteasomes regulate reversal learning are unclear but may imply the degradation of specific neuronal proteins to control some synaptic connections and “reset” learning associations. It is tempting to speculate that one of these substrates could be Arc (activity-regulated cytoskeleton-associated protein), a key mediator of synaptic plasticity whose persistent expression has recently been shown to interfere with the reversal learning process (71). Additional evidence in favour of a critical role of PSMC3/Rpt5 in behavioural flexibility emerged from our experiments in primary hippocampal neurons showing that ectopic expression of PSMC3/Rpt5 affected dendrite growth (Fig. S3). The observation that the R304W, E305D and E383L PSMC3 variants did not differ from their wild-type counterpart in this process is intriguing but may have several explanations. First, wild-type PSMC3 overexpression may exert ceiling effects that overshadow any potential detrimental impact of the PSMC3 variants on neuronal morphology. Secondly, it may be that PSMC3 variants, with the exception of M159V, have no substantial effect on neuronal morphology. The reasons why the M159V PSMC3 variant behaves differently are unclear but these findings might be explained by distinct half-life, intracellular localization or post-translational modifications. One could also argue that the adverse effects of PSMC3/Rpt5 on this process might be due to extra-proteasome functions as a consequence of an excess of “free” subunits following transfection. However, our investigations on patient T cells showed that PSMC3 missense variants were associated with an increased accumulation of ubiquitin-modified proteins (Fig. 6A), suggesting that these alterations give rise to proteasome loss-of-function variants associated with perturbed protein homeostasis. Future functional studies involving a larger number of biological samples are required to evaluate a possible correlation between PSMC3 variants and the extent of intracellular proteolysis dysfunction as a prerequisite for predicting disease severity.
Our proteomic analysis revealed that T cells from patients carrying PSMC3 variants were enriched with ribosomal proteins such as RPL4, RPL6, RPL7A and RPL7 (Fig. 5 and Table 2). These proteins may be specifically targeted for degradation and their accumulation may occur as a consequence of impaired intracellular protein clearance. Consistent with this notion, proteasome inhibition has been recently shown to result in the aggregation of ubiquitin-modified ribosomal proteins (72). Our data therefore support the recent view that ribosome dysregulation defines a key feature of NDD/ID phenotypes (73, 74) and the concept of translational arrest upon proteotoxic stress via the action of eIF2α kinases (Fig. 6A).
Table 2.
List of proteins enriched in Patients 17 and 21 versus their controls (mother and father/mother, respectively)
| PG.Protein Groups | Protein name | Gene name | Mut/Co|signal_log2_ratio | Mut/Co|raw_p_value | Mut/Co|adjusted_p_value |
|---|---|---|---|---|---|
| P02749 | Beta-2-glycoprotein 1 | APOH | 3,90875773 | 3,9233E-05 | 0,01752392 |
| P01861 | Immunoglobulin heavy constant gamma 4 | IGHG4 | 3,41081457 | 3,9313E-07 | 0,0003951 |
| Q02878 | 60S ribosomal protein L6 | RPL6 | 3,09519982 | 8,1085E-05 | 0,02173071 |
| P62424 | 60S ribosomal protein L7a | RPL7A | 2,97520531 | 2,4946E-06 | 0,00143263 |
| P18124 | 60S ribosomal protein L7 | RPL7 | 2,90834996 | 5,5264E-05 | 0,01815176 |
| P83731 | 60S ribosomal protein L24 | RPL24 | 2,78639617 | 5,8732E-05 | 0,01815176 |
| P16403 | Histone H1.2 | H1–2 | 2,72232694 | 4,4042E-05 | 0,0177049 |
| P02768 | Serum albumin | ALB | 2,64236248 | 3,3722E-18 | 1,3556E-14 |
| P36578 | 60S ribosomal protein L4 | RPL4 | 2,50022766 | 3,276E-07 | 0,0003951 |
| P01876 | Immunoglobulin heavy constant alpha 1 | IGHA1 | 2,32067402 | 3,007E-05 | 0,01511011 |
| P20591 | Interferon-induced GTP-binding protein Mx1 | MX1 | 2,20162951 | 6,3215E-05 | 0,01815176 |
| Q6P2Q9 | Pre-mRNA-processing-splicing factor 8 | PRPF8 | 1,33941179 | 1,5549E-06 | 0,00125012 |
| Q01469 | Fatty acid-binding protein 5 | FABP5 | -2,29088412 | 5,7962E-05 | 0,01815176 |
One key finding is the observation that patients with PSMC3 variants generate a type I IFN gene signature (Figs. 7 and 8A). Although it is well-established that proteasome loss-of-function variants cause interferonopathies in patients with PRAAS (27–33), it was only recently that pathogenic mutations in the 19S regulatory particle subunit PSMD12/Rpn5 were reported to engage constitutive type I IFN signaling in patients with this NDD disorder (35, 36) sharing similarities with the patients described in this manuscript. Although patients with PRAAS and individuals with NDD/ID and PSMD12 or PSMC3 variants carry genomic alterations that affect the same multi-subunit enzyme (26S proteasome), their clinical phenotypes do not entirely overlap. For instance, patients with NDD/ID and PSMC3 variants did not develop recurrent fever, lipodystrophy and/or skin lesions which are usually detected in patients with PRAAS (Table 1). One could argue that such differences may reflect distinct localizations of the affected subunits within the 26S proteasome complex, thereby suggesting that alterations of the 19S regulatory particle promote the generation of NDD/ID, whereas those of the 20S core particle or assembly chaperones favor the development a PRAAS phenotype. This assumption is however challenged by the fact that PSMB1/β6 variants of the 20S core particle lead to the acquisition of neuronal phenotype very similar to that observed in patients with NDD/ID and PSMC3 variants (25). The lack of systemic autoinflammation in patients with NDD/ID and PSMC3 variants mounting a constitutive type I IFN response may seem surprising at first sight, but it is not totally unexpected, since this inconsistency is found in other NDDs including Aicardi-Goutières (75, 76) and Down syndromes (77, 78). This is particularly well exemplified in Down syndrome patients who, like patients with NDD/ID and PSMC3 dominant variants, exhibit a constitutive activation of type I IFN signaling (79, 80). To what extent type I IFN actively contributes to the pathogenesis of these disorders remains to be fully determined, even though a growing body of evidence supports the notion that IFN has detrimental effects on CNS function (81–84) as well as stem cell function and differentiation (85, 86). Because proteasome dysfunction typically engages stress responses involving compensatory mechanisms such as autophagy (87), ISR and UPR (24, 88), we reasoned that the type I IFN response detected in patients with PSMC3 variants might be triggered by sustained activation of either one of these pathways. As anticipated, high expression of autophagy and ER stress markers were detected in affected individuals, as evidenced by increased expression of the LC3-II and GRP94 proteins (Fig. 6A and B). Both PINK1 and NIX mitochondrial proteins were found to be decreased in affected individuals (Fig. 6B), suggesting that PSMC3 variants increase autophagy-driven elimination of mitochondria (mitophagy). This observation supports the growing consensus that mitochondrial dysfunction is a key determinant of the pathogenesis of neurodevelopment (89) and the cause of interferonopathies (90). The activation status of PKR, a protein of the ISR that intersects with the UPR (91), was substantially increased in all investigated patients carrying the PSMC3 variant. Both ISR and UPR have the ability to counterbalance proteotoxic stress by inducing a global translational arrest via eIF2α phosphorylation. This is accompanied by concomitant accumulation of non-translated mRNAs, the formation of stress granules recruiting different RNA species and RNA processing enzymes, and IRE-1-dependent mRNA decay (RIDD) (92). Although PKR typically responds to viral double stranded RNA (93), it also may undergo activation under sterile conditions upon different stresses including ER-stress involving PKR-associated activator (PACT) and its modulator TAR RNA-binding protein 2 (TRBP), a protein required for micro-RNA biogenesis (94–96). Both PACT and TRBP were observed to be increased in patient cells along with several RNA-processing factors. Our inhibition experiments suggested PKR as the inducer of type I IFN in these patients (Fig. 8B). The mechanisms by which PSMC3 variants activate PKR in affected individuals remain unclear, but our data open the possibility that PKR may sense a broader spectrum of danger signals than initially assumed, including perturbations of protein homeostasis. This concept is in line with the observation that activated PKR was found in the CNS of patients with neurodegenerative diseases (97–100) and that neurodegeneration is associated with neuroinflammation (101, 102). Altogether, our work demonstrates that heterozygous PSMC3 dominant variants result in a neurodevelopmental syndrome associated with a specific type I IFN gene signature and suggests treatment options targeting type I IFN signaling or PKR.
MATERIALS AND METHODS
Study design
The aim of the study was to determine the involvement of the human PSMC3 gene in a neurodevelopmental disorder hitherto unreported. Affected individuals were recruited and data were collected on an ongoing basis. Affected individuals were identified via data sharing platform GeneMatcher (103) and direct requests in variants databases whose access was authorized to the University of Washington School of Medicine. Clinical and molecular data were provided by the referring geneticists following the patients. Facial recognition by GestaltMatcher (41) was used to measure the similarities of the facial phenotypes between affected individuals (Fig. S2). Prediction of variant pathogenicity was done using the bioinformatics programs indicated in Table 1, and their frequency was determined in public variant databases (gnomAD (104), Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA (URL: http://evs.gs.washington.edu/EVS/) [03/2023 accessed]; Bravo powered by TOPMed Freeze 8 on GRCh38 (105)). The effects of the PSMC3 variants on proteasome function, protein homeostasis, proteotoxic stress sensors (UPR, ISR), autophagy/mitophagy and inflammation status (type I IFN) were studied in vitro by western-blotting, NanoString® analysis and qPCR using T cells expanded from whole-blood samples of patients (n=4) and their parents (n=6) who agreed to provide blood specimens. The first available T-cell samples (patients n=2, related controls n=2) were used for proteomics analyses to explore deregulated biological pathways. The impact of the variants on neuronal function was assessed in two models: 1. Upstream Activation Sequence (UAS)-Rpt5 RNAi Drosophila melanogaster lines targeting psmc3 for evaluating the effect of Rpt5 knockdown on behavior (Fig. 2); 2. primary hippocampal neuronal cultures overexpressing cDNA constructs containing the first identified variants (n=4) for evaluating their effects on neuronal morphology (Fig. S3). Details about data replication are provided in the figure legends. Experimenters were not blinded.
Human participants
All affected participants were initially referred for unexplained developmental delay (DD) and/or intellectual disability (ID) together with various congenital malformations. They underwent extensive clinical examination by at least one clinical geneticist participating in the study. Routine genetic testing was performed whenever clinically relevant, including copy number variation (CNV) analysis by high-resolution array-based comparative genomic hybridization (aCGH). Because these tests failed to establish the diagnosis of a specific disease, trio-based whole exome sequencing (WES) was performed on a diagnostic or research setting whenever parental samples were available. Because disorders associated with PSMC3 are very rare, the reported patient cohort is representative of cases identified worldwide over the past six years and is not, for example, a selection of a subgroup of a larger patient population. This study was approved by the CHU de Nantes ethics committee (Research Programme “Génétique Médicale DC-2011–1399). Probands 2, 5–10, 12–14, 16–18, and 20–23 were enrolled by one of the participating centers (Washington University in St. Louis, University Medical Centre of Utrecht, University Hospital Center of Nantes, Arnold Palmer Hospital, Seattle Children’s Hospital, St. Luke’s Hospital, Health San Antonio, Vanderbilt University Medical Center, Sydney Medical School, Children’s Hospital of Philadelphia, Technical University of Munich, Pitié-Salpêtrière University Hospital, Children’s Hospital of Orange County, Ambry Genetics, Geisinger Medical Center, Guy’s & St Thomas’ NHS Foundation Trust, McGill University Health Centre, Nottingham University Hospitals NHS Trust, Hôpital Universitaire Necker-Enfants Malades, University Children’s Hospital, Salzburger Landeskliniken (SALK) and Paracelsus Medical University) after approval of genetic studies by local ethics committees. Written informed consent was obtained from all study participants, including probands and healthy parents. All affected individuals were initially referred for unexplained developmental delay (DD) and/or intellectual disability (ID) together with various congenital malformations. They underwent extensive clinical examination by at least one expert clinical geneticist. Individuals 1, 4, 11, 15 and 19 participated in ‘Simons Foundation Powering Autism Research’ (SPARK) or ‘Deciphering Developmental Disorders’ initiative; clinical information about individuals 1, 3 and 14 was retrieved by members of the University of Washington School of Medicine in agreement with SPARK and DDD.
Cell culture
Peripheral blood mononuclear cells (PBMC) used in this paper were isolated from blood draws from patients and related healthy controls (father and/or mother of the proband). Briefly, PBMC were isolated by PBMC spin medium gradient centrifugations (pluriSelect), washed three times with PBS, frozen in FBS with 10% DMSO and stored in liquid nitrogen for further use. In some experiments, collected PBMC were expanded in U-bottom 96-well plates together with feeder cells using RPMI 1640 supplemented with 10% human AB serum (both purchased from PAN-Biotech GmbH) in the presence of 150 U/ml IL-2 (Miltenyi Biotec) and 1 µg/µl L-PHA (Sigma) following the procedure of Fonteneau et al. (106). After 3–4 weeks of culture, resting T cells were washed and frozen as dry pellets for further use.
SDS-PAGE and western-blot analysis
Cell pellets from resting T cell isolated from patients and related controls were lysed in equal amounts of standard RIPA buffer (50 mM Tris pH 7.5, 150 mM NaCl, 2 mM EDTA, 1 mM N-ethylmaleimide, 10 µM MG-132, 1% NP40, 0.1% SDS) and separated by 10 or 12.5% SDS-PAGE before transfer to PVDF membranes (200V for 1h). After blocking (20-min exposure to 1X Roti®-Block at room temperature), membranes were probed with relevant primary antibodies overnight at 4°C under shaking. The anti-α6 (clone MCP20), anti-α7 (clone MCP72), anti-β1 (clone MCP421), anti-PSMC2 (BML-PW8315) and anti-PSMC3 (BML-PW8310) primary antibodies were purchased from Enzo Life Sciences. Primary antibodies specific for TCF11/Nrf1 (clone D5B10), ubiquitin (clone D9D5), GAPDH (clone 14C10), PINK1 (clone D8G3), BNIP3L/NIX (clone D8G3), LC3b (#2775), eIF2α (#9722), phospho-eIF2α (ser51, #119A11) were obtained from Cell Signaling Technology. The anti-PSMD12 antibody (clone H3) was a purchase from Santa Cruz Biotechnology Inc. The anti-PA28-α (K232/1) is laboratory stock and was used in previous studies (31). Antibodies directed against β5 (ab3330), α-Tubulin (clone DM1A) and phospho-PKR (Thr446, clone E120) were purchased from Abcam. Following incubation with primary antibodies, membranes were washed three times with PBS/0.2% Tween and subsequently incubated with anti-mouse or –rabbit HRP conjugated secondary antibodies (1/5.000) for 1h at room temperature. Proteins were then visualized using an enhanced chemiluminescence detection kit (ECL) (Biorad).
RNA isolation, reverse-transcription and PCR analysis
Total RNA was isolated from resting T cells using the kit from Analytic Jena AG following the manufacturer’s instructions. For subsequent real-time PCR, 100–500 ng of the isolated total RNA was reverse transcribed using the M-MLV reverse transcriptase (Promega). Quantitative PCR was performed using the Premix Ex Taq™ (probe qPCR purchased from TaKaRa) and in duplicates to determine the mRNA expression of each IFN-stimulated gene (ISG) using FAM-tagged TaqMan™ Gene Expression Assays obtained from Thermo Fisher Scientific according to the manufacturer’s instructions. TaqMan™ probes used in this study for ISG quantification included IFI27, IFI44L, IFIT1, ISG15, RSAD2, IFI44, MX1, OASL1, CXCL9 and CXCL10. The cycle threshold (Ct) values for target genes were converted to values of relative expression using the relative quantification (RQ) method (2-∆∆Ct). Target gene expression was calculated relative to Ct values for the GAPDH control housekeeping gene.
Statistical analyses
Figures were created with PyMOL v. 2.0 (pymol.org) using the human 26S proteasome structure. Data are typically median or mean ± SEM and analyzed by unpaired and pair ratio t-test between two groups. Neuronal morphology data was analyzed using a One-Way analysis of variance (ANOVA), followed by a Tukey’s post-hoc test for multiple comparisons. All charts and statistical analyses were generated using GraphPad Prism version 8. A p-value <0.05 was considered significant.
Supplementary Material
OVERLINE: NEURODEVELOPMENTAL DISORDERS.
One Sentence Summary:
PSMC3 variants associated with neurodevelopmental disorders lead to a type I interferon response by T cells that can be blocked by protein kinase R inhibition.
Editor’s summary:
Insights into pathogenic proteasome variants
Pathogenic proteasome gene variants are associated with a broad spectrum of diseases. Ebstein and colleagues identified fifteen de novo missense variants in the PSMC3 proteasome gene in patients with neurodevelopmental delay. Expression of PSMC3 variants in mouse neuronal cultures led to altered dendrite development, and deletion of the fly PSMC3 ortholog resulted in learning deficits in the fruit flies. Proteasome dysfunction in T cells from patients with PSMC3 variants led to upregulation of proteotoxic markers and production of interferon type 1 that could be alleviated by inhibition of protein kinase R. These findings implicate PSMC3 pathogenic variants in neurodevelopmental disorders and suggest a therapeutic strategy for patients carrying these missense mutations. ---DN
Acknowledgments:
The sequencing for Patient #3 was performed under the Care4Rare Canada Consortium funded by Genome Canada and the Ontario Genomics Institute (OGI-147), the Canadian Institutes of Health Research, Ontario Research Fund, Genome Alberta, Genome British Columbia, Genome Quebec, and Children’s Hospital of Eastern Ontario Foundation. The Deciphering Developmental Disorder (DDD) study presents independent research commissioned by the Health Innovation Challenge Fund [grant number HICF-1009–003]. This study makes use of DECIPHER (http://www.deciphergenomics.org), which is funded by the Wellcome Trust. The authors are grateful to R. Beyer and A. Brandenburg for excellent technical assistance.
Funding:
This work was supported by German Research Foundation Grants SFBTR167, RGT2719-PRO project B4 (to EK), E-Rare project GENOMIT (Austrian Science Fund FWF, I4695-B) to JAM, US National Institutes of Health (NIH) grants (R01MH101221) and a grant from the Simons Foundation (SFARI #608045) to EEE. EEE is an investigator of the Howard Hughes Medical Institute. This work was also supported, in part, by “the Fundamental Research Funds for the Central Universities” starting fund (BMU2022RCZX038) to TW.
Footnotes
List of Supplementary Materials
Materials and Methods
Competing interests: Authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials. Drosophila lines are available from the Bloomington Drosophila Stock Center. Research Resource Identifiers (RRID) are provided in the Supplementary Materials and Methods. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD041182. Data sets from transcriptomics analysis (NanoString) has been deposited to the GEO repository under accession number GSE228675. Plasmids generated in this study can be provided by E.K. under a Universitaetsmedizin Greifswald Material Transfer agreement. Human cells derived from Patients #13, #17, #18 and #21 are stored in the University Medicine Greifswald Biobank. Availability of patient’s samples is restricted depending on patient’s consents for follow-up analysis.
References and notes
- 1.Tanaka K, Mizushima T, Saeki Y. The proteasome: molecular machinery and pathophysiological roles. Biol Chem 393, 217–34 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Collins GA, Goldberg AL. The Logic of the 26S Proteasome. Cell 169, 792–806 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hetzer MW, Toyama BH, Protein homeostasis: live long, won’t prosper. Nat Rev Mol Cell Biol 14, 55–61 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goloubinoff P. Mechanisms of protein homeostasis in health, aging and disease. Swiss Med Wkly 146, w14306 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Seifert U, Bialy LP, Ebstein F, Bech-Otschir D, Voigt A, Schröter F, Prozorovski T, Lange N, Steffen J, Rieger M, Kuckelkorn U, Aktas O, Kloetzel P-M, Krüger E. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell 142, 613–24 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Krüger E, Kloetzel PM. Immunoproteasomes at the interface of innate and adaptive immune responses: Two faces of one enzyme. Curr Opin Immunol 24, 77–83 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem 70, 503–33 (2001). [DOI] [PubMed] [Google Scholar]
- 8.DeMartino GN, Gillette TG. Proteasomes: Machines for All Reasons. Cell 129, 659–62 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Pickart CM, Fushman D. Polyubiquitin chains: Polymeric protein signals. Curr Opin Chem Biol 8, 610–6 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Pickart CM. Back to the Future with Ubiquitin. Cell 116, 181–90 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Tanaka K. The proteasome: Overview of structure and functions. Proc Jpn Acad Ser B Phys Biol Sci 85, 12–36 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dahlmann B. Mammalian proteasome subtypes: Their diversity in structure and function. Arch Biochem Biophys 591, 132–40 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Tomko RJ, Funakoshi M, Schneider K, Wang J, Hochstrasser M. Heterohexameric Ring Arrangement of the Eukaryotic Proteasomal ATPases: Implications for Proteasome Structure and Assembly. Mol Cell 38, 393–403 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith DM, Chang SC, Park S, Finley D, Cheng Y, Goldberg AL. Docking of the Proteasomal ATPases’ Carboxyl Termini in the 20S Proteasome’s α Ring Opens the Gate for Substrate Entry. Mol Cell 27, 731–44 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greene ER, Dong KC, Martin A. Understanding the 26S proteasome molecular machine from a structural and conformational dynamics perspective. Curr Opin Struct Biol 61, 33–41 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greene ER, Goodall EA, de La Peña AH, Matyskiela ME, Lander GC, Martin A. Specific lid-1 base contacts in the 26S proteasome control the conformational switching required for substrate degradation. Elife 8, e49806 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verma R, Aravind L, Oania R, McDonald WH, Yates JR, Koonin E. v., Deshaies RJ. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 298, 611–615 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Kisselev AF, van der Linden WA, Overkleeft HS. Proteasome inhibitors: An expanding army attacking a unique target. Chem Biol 19, 99–115 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finley D, Chen X, Walters KJ. Gates, Channels, and Switches: Elements of the Proteasome Machine. Trends Biochem Sci 41, 77–93 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ebstein F, Kloetzel P-M, Krüger E, Seifert U. Emerging roles of immunoproteasomes beyond MHC class I antigen processing. Cell Mol Life Sci 69, 2543–58 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strehl B, Seifert U, Krüger E, Heink S, Kuckelkorn U, Kloetzel PM. Interferon-γ, the functional plasticity of the ubiquitin-proteasome system, and MHC class I antigen processing. Immunol Rev 207, 19–30 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Shin EC, Seifert U, Urban S, Truong KT, Feinstone SM, Rice CM, Kloetzel PM, Rehermann B. Proteasome activator and antigen-processing aminopeptidases are regulated by virus-induced type I interferon in the hepatitis C virus-infected liver. J Interferon Cytokine Res 27, 985–90 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Brehm A, Krüger E. Dysfunction in protein clearance by the proteasome: impact on autoinflammatory diseases. Semin Immunopathol 37, 323–333 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Ebstein F, Poli Harlowe MC, Studencka-Turski M, Krüger E. Contribution of the Unfolded Protein Response (UPR) to the Pathogenesis of Proteasome-Associated Autoinflammatory Syndromes (PRAAS). Front Immunol 10, 2756 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ansar M, Ebstein F, Özkoç H, Paracha SA, Iwaszkiewicz J, Gesemann M, Zoete V, Ranza E, Santoni FA, Sarwar MT, Ahmed J, Krüger E, Bachmann-Gagescu R, Antonarakis SE. Biallelic variants in PSMB1 encoding the proteasome subunit β6 cause impairment of proteasome function, microcephaly, intellectual disability, developmental delay and short stature. Hum Mol Genet 29, 1132–1143 (2021). [DOI] [PubMed] [Google Scholar]
- 26.Agarwal AK, Xing C, Demartino GN, Mizrachi D, Hernandez MD, Sousa AB, Martínez De Villarreal L, dos Santos HG, Garg A. PSMB8 Encoding the β5i Proteasome Subunit Is Mutated in Joint Contractures, Muscle Atrophy, Microcytic Anemia, and Panniculitis-Induced Lipodystrophy Syndrome. Am J Hum Genet 87, 866–72 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arima K, Kinoshita A, Mishima H, Kanazawa N, Kaneko T, Mizushima T, Ichinose K, Nakamura H, Tsujino A, Kawakami A, Matsunaka M, Kasagi S, Kawano S, Kumagai S, Ohmura K, Mimori T, Hirano M, Ueno S, Tanaka K, Tanaka M, Toyoshima I, Sugino H, Yamakawa A, Tanaka K, Niikawa N, Furukawa F, Murata S, Eguchi K, Ida H, Yoshiura KI. Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proc Natl Acad Sci U S A 108, 14914–14919 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kitamura A, Maekawa Y, Uehara H, Izumi K, Kawachi I, Nishizawa M, Toyoshima Y, Takahashi H, Standley DM, Tanaka K, Hamazaki J, Murata S, Obara K, Toyoshima I, Yasutomo K. A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J Clin Invest 121, 4150–60 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y, Ramot Y, Torrelo A, Paller AS, Si N, Babay S, Kim PW, Sheikh A, Lee CCR, Chen Y, Vera A, Zhang X, Goldbach-Mansky R, Zlotogorski A. Mutations in PSMB8 Cause CANDLE Syndrome with Evidence of Genetic and Phenotypic Heterogeneity. Arthritis Rheum 64, 895–907 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brehm A, Liu Y, Sheikh A, Marrero B, Omoyinmi E, Zhou Q, Montealegre G, Biancotto A, Reinhardt A, de Jesus AA, Pelletier M, Tsai WL, Remmers EF, Kardava L, Hill S, Kim H, Lachmann HJ, Megarbane A, Chae JJ, Brady J, Castillo RD, Brown D, Casano AV, Gao L, Chapelle D, Huang Y, Stone D, Chen Y, Sotzny F, Lee CCR, Kastner DL, Torrelo A, Zlotogorski A, Moir S, Gadina M, McCoy P, Wesley R, Rother K, Hildebrand PW, Brogan P, Krüger E, Aksentijevich I, Goldbach-Mansky R. Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J Clin Invest 125, 4196–211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poli MC, Ebstein F, Nicholas SK, de Guzman MM, Forbes LR, Chinn IK, Mace EM, Vogel TP, Carisey AF, Benavides F, Coban-Akdemir ZH, Gibbs RA, Jhangiani SN, Muzny DM, Carvalho CMB, Schady DA, Jain M, Rosenfeld JA, Emrick L, Lewis RA, Lee B, Zieba BA, Küry S, Krüger E, Lupski JR, Bostwick BL, Orange JS. Heterozygous Truncating Variants in POMP Escape Nonsense-Mediated Decay and Cause a Unique Immune Dysregulatory Syndrome. Am J Hum Genet 102, 1126–1142 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Jesus AA, Brehm A, VanTries R, Pillet P, Parentelli AS, Montealegre Sanchez GA, Deng Z, Paut IK, Goldbach-Mansky R, Krüger E. Novel Proteasome Assembly Chaperone mutations in PSMG2/PAC2, cause the autoinflammatory interferonopathy, CANDLE/PRAAS4. J Allergy Clin Immunol 143, 1939–1943 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarrabay G, Méchin D, Salhi A, Boursier G, Rittore C, Crow Y, Rice G, Tran TA, Cezar R, Duffy D, Bondet V, Boudhane L, Broca C, Kant BP, VanGijn M, Grandemange S, Richard E, Apparailly F, Touitou I. PSMB10, the last immunoproteasome gene missing for PRAAS. J Allergy Clin Immunol 145, 1015–1017.e6 (2020). [DOI] [PubMed] [Google Scholar]
- 34.Küry S, Besnard T, Ebstein F, Khan TN, Gambin T, Douglas J, Bacino CA, Sanders SJ, Lehmann A, Latypova X, Khan K, Pacault M, Sacharow S, Glaser K, Bieth E, Perrin-Sabourin L, Jacquemont M-L, Cho MT, Roeder E, Denommé-Pichon A-S, Monaghan KG, Yuan B, Xia F, Simon S, Bonneau D, Parent P, Gilbert-Dussardier B, Odent S, Toutain A, Pasquier L, Barbouth D, Shaw CA, Patel A, Smith JL, Bi W, Schmitt S, Deb W, Nizon M, Mercier S, Vincent M, Rooryck C, Malan V, Briceño I, Gómez A, Nugent KM, Gibson JB, Cogné B, Lupski JR, Stessman HAF, Eichler EE, Retterer K, Yang Y, Redon R, Katsanis N, Rosenfeld JA, Kloetzel P-M, Golzio C, Bézieau S, Stankiewicz P, Isidor B. De Novo Disruption of the Proteasome Regulatory Subunit PSMD12 Causes a Syndromic Neurodevelopmental Disorder. Am J Hum Genet 100, 352–363 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Isidor B, Ebstein F, Hurst A, Vincent M, Bader I, Rudy NL, Cogne B, Mayr J, Brehm A, Bupp C, Warren K, Bacino CA, Gerard A, Ranells JD, Metcalfe KA, van Bever Y, Jiang YH, Mendelssohn BA, Cope H, Rosenfeld JA, Blackburn PR, Goodenberger ML, Kearney HM, Kennedy J, Scurr I, Szczaluba K, Ploski R, de Saint Martin A, Alembik Y, Piton A, Bruel AL, Thauvin-Robinet C, Strong A, Diderich KEM, Bourgeois D, Dahan K, Vignard V, Bonneau D, Colin E, Barth M, Camby C, Baujat G, Briceño I, Gómez A, Deb W, Conrad S, Besnard T, Bézieau S, Krüger E, Küry S, Stankiewicz P. Stankiewicz-Isidor syndrome: expanding the clinical and molecular phenotype. Genet Med 24, 179–191 (2022). [DOI] [PubMed] [Google Scholar]
- 36.Yan K, Zhang J, Lee PY, Tao P, Wang J, Wang S, Zhou Q, Dong M. Haploinsufficiency of PSMD12 Causes Proteasome Dysfunction and Subclinical Autoinflammation. Arthritis Rheumatol 74, 1083–1090 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.d’Angelo DM, di Filippo P, Breda L, Chiarelli F, Type I Interferonopathies in Children: An Overview. Front Pediatr 9,631329 (2021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Savic S, Coe J, Laws P. Autoinflammation: Interferonopathies and Other Autoinflammatory Diseases. J Invest Dermatol 142, 781–792 (2022). [DOI] [PubMed] [Google Scholar]
- 39.Crow YJ, Stetson DB. The type I interferonopathies: 10 years on. Nat Rev Immunol 22 (2022), doi: 10.1038/s41577-021-00633-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baux D, van Goethem C, Ardouin O, Guignard T, Bergougnoux A, Koenig M, Roux AF. MobiDetails: online DNA variants interpretation. Eur J Hum Genet 29, 356–360 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsieh TC, Bar-Haim A, Moosa S, Ehmke N, Gripp KW, Pantel JT, Danyel M, Mensah MA, Horn D, Rosnev S, Fleischer N, Bonini G, Hustinx A, Schmid A, Knaus A, Javanmardi B, Klinkhammer H, Lesmann H, Sivalingam S, Kamphans T, Meiswinkel W, Ebstein F, Krüger E, Küry S, Bézieau S, Schmidt A, Peters S, Engels H, Mangold E, Kreiß M, Cremer K, Perne C, Betz RC, Bender T, Grundmann-Hauser K, Haack TB, Wagner M, Brunet T, Bentzen HB, Averdunk L, Coetzer KC, Lyon GJ, Spielmann M, Schaaf CP, Mundlos S, Nöthen MM, Krawitz PM. GestaltMatcher facilitates rare disease matching using facial phenotype descriptors. Nat Genet 54, 349–357 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tully T, Quinn WG. Classical conditioning and retention in normal and mutant Drosophila melanogaster. J Comp Physiol A 157, 263–77 (1985). [DOI] [PubMed] [Google Scholar]
- 43.Küry S, van Woerden GM, Besnard T, Proietti Onori M, Latypova X, Towne MC, Cho MT, Prescott TE, Ploeg MA, Sanders S, Stessman HAF, Pujol A, Distel B, Robak LA, Bernstein JA, Denommé-Pichon AS, Lesca G, Sellars EA, Berg J, Carré W, Busk ØL, van Bon BWM, Waugh JL, Deardorff M, Hoganson GE, Bosanko KB, Johnson DS, Dabir T, Holla ØL, Sarkar A, Tveten K, de Bellescize J, Braathen GJ, Terhal PA, Grange DK, van Haeringen A, Lam C, Mirzaa G, Burton J, Bhoj EJ, Douglas J, Santani AB, Nesbitt AI, Helbig KL, Andrews M. v., Begtrup A, Tang S, van Gassen KLI, Juusola J, Foss K, Enns GM, Moog U, Hinderhofer K, Paramasivam N, Lincoln S, Kusako BH, Lindenbaum P, Charpentier E, Nowak CB, Cherot E, Simonet T, Ruivenkamp CAL, Hahn S, Brownstein CA, Xia F, Schmitt S, Deb W, Bonneau D, Nizon M, Quinquis D, Chelly J, Rudolf G, Sanlaville D, Parent P, Gilbert-Dussardier B, Toutain A, Sutton VR, Thies J, Peart-Vissers LELM, Boisseau P, Vincent M, Grabrucker AM, Dubourg C, Tan WH, Verbeek NE, Granzow M, Santen GWE, Shendure J, Isidor B, Pasquier L, Redon R, Yang Y, State MW, Kleefstra T, Cogné B, Petrovski S, Retterer K, Eichler EE, Rosenfeld JA, Agrawal PB, Bézieau S, Odent S, Elgersma Y, Mercier S. De Novo Mutations in Protein Kinase Genes CAMK2A and CAMK2B Cause Intellectual Disability. Am J Hum Genet 101, 768–788 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schembri L, Dalibart R, Tomasello F, Legembre P, Ichas F, de Giorgi F. The HA tag is cleaved and loses immunoreactivity during apoptosis. Nat Methods 4, 107–8 (2007) [DOI] [PubMed] [Google Scholar]
- 45.Dong Y, Zhang S, Wu Z, Li X, Wang WL, Zhu Y, Stoilova-McPhie S, Lu Y, Finley D, Mao Y. Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 565, 49–55 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shmueli MD, Sheban D, Eisenberg-Lerner A, Merbl Y. Histone degradation by the proteasome regulates chromatin and cellular plasticity. FEBS J 289, 3304–3316 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McConkey DJ, The integrated stress response and proteotoxicity in cancer therapy. Biochem Biophys Res Commun 482, 450–453 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marzec M, Eletto D, Argon Y. GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim Biophys Acta Mol Cell Res 1823, 774–87 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Radhakrishnan SK, Lee CS, Young P, Beskow A, Chan JY, Deshaies RJ. Transcription Factor Nrf1 Mediates the Proteasome Recovery Pathway after Proteasome Inhibition in Mammalian Cells. Mol Cell 38, 17–28 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steffen J, Seeger M, Koch A, Krüger E. Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol Cell 40, 147–58 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Kostyszyn B, Cowburn RF, Seiger Å, Kjældgaard A, Sundström E. Distribution of presenilin 1 and 2 and their relation to Notch receptors and ligands in human embryonic/foetal central nervous system. Brain Res Dev Brain Res 151, 75–86 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Rice GI, Melki I, Frémond ML, Briggs TA, Rodero MP, Kitabayashi N, Oojageer A, Bader-Meunier B, Belot A, Bodemer C, Quartier P, Crow YJ. Assessment of Type I Interferon Signaling in Pediatric Inflammatory Disease. J Clin Immunol 37, 123–132 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ebstein F, Lange N, Urban S, Seifert U, Krüger E, Kloetzel PM, Maturation of human dendritic cells is accompanied by functional remodelling of the ubiquitin-proteasome system. Int J Biochem Cell Biol 41, 1205–1215 (2009). [DOI] [PubMed] [Google Scholar]
- 54.Tesser A, Piperno GM, Pin A, Piscianz E, Boz V, Benvenuti F, Tommasini A. Priming of the cGAS‐STING‐TBK1 pathway enhances LPS‐induced release of type I interferons. Cells 10, 785 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bedford L, Hay D, Devoy A, Paine S, Powe DG, Seth R, Gray T, Topham I, Fone K, Rezvani N, Mee M, Soane T, Layfield R, Sheppard PW, Ebendal T, Usoskin D, Lowe J, Mayer RJ. Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and lewy-like inclusions resembling human pale bodies. J Neurosci 28, 8189–98 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tashiro Y, Urushitani M, Inoue H, Koike M, Uchiyama Y, Komatsu M, Tanaka K, Yamazaki M, Abe M, Misawa H, Sakimura K, Ito H, Takahashia R. Motor neuron-specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J Biol Chem 287, 42984–94 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kröll‐Hermi A, Ebstein F, Stoetzel C, Geoffroy V, Schaefer E, Scheidecker S, Bär S, Takamiya M, Kawakami K, Zieba BA, Studer F, Pelletier V, Eyermann C, Speeg‐Schatz C, Laugel V, Lipsker D, Sandron F, McGinn S, Boland A, Deleuze J, Kuhn L, Chicher J, Hammann P, Friant S, Etard C, Krüger E, Muller J, Strähle U, Dollfus H. Proteasome subunit PSMC3 variants cause neurosensory syndrome combining deafness and cataract due to proteotoxic stress. EMBO Mol Med 12, e11861 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reed P, Watts H, Truzoli R. Flexibility in young people with autism spectrum disorders on a card sort task. Autism 17, 162–71 (2013). [DOI] [PubMed] [Google Scholar]
- 59.Leung RC, Zakzanis KK. Brief report: Cognitive flexibility in autism spectrum disorders: A quantitative review. J Autism Dev Disord 44, 2628–45 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Tully T, Boyton S, Brandes C, Dura JM, Mihalek R, Preat T, Villella A. Genetic dissection of memory formation in Drosophila melanogaster. Cold Spring Harb Symp Quant Biol 55, 203–11 (1990). [DOI] [PubMed] [Google Scholar]
- 61.Swainson R, Rogers RD, Sahakian BJ, Summers BA, Polkey CE, Robbins TW. Probabilistic learning and reversal deficits in patients with Parkinson’s disease or frontal or temporal lobe lesions: Possible adverse effects of dopaminergic medication. Neuropsychologia 38, 596–612 (2000). [DOI] [PubMed] [Google Scholar]
- 62.Remijnse PL, Nielen MMA, van Balkom AJLM, Cath DC, van Oppen P, Uylings HBM, Veltman DJ. Reduced orbitofrontal-striatal activity on a reversal learning task in obsessive-compulsive disorder. Arch Gen Psychiatry 63, 1225–36 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Finger EC, Marsh AA, Mitchell DG, Reid ME, Sims C, Budhani S, Kosson DS, Chen G, Towbin KE, Leibenluft E, Pine DS, Blair JR. Abnormal ventromedial prefrontal cortex function in children with psychopathic traits during reversal learning. Arch Gen Psychiatry 65, 586–94 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leeson VC, Robbins TW, Matheson E, Hutton SB, Ron MA, Barnes TRE, Joyce EM. Discrimination Learning, Reversal, and Set-Shifting in First-Episode Schizophrenia: Stability Over Six Years and Specific Associations with Medication Type and Disorganization Syndrome. Biol Psychiatry 66, 586–93 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Izquierdo A, Jentsch JD. Reversal learning as a measure of impulsive and compulsive behavior in addictions. Psychopharmacology (Berl) 219, 607–20 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Smolen P, Baxter DA, Byrne JH, Paradoxical LTP maintenance with inhibition of protein synthesis and the proteasome suggests a novel protein synthesis requirement for early LTP reversal. J Theor Biol 457, 79–87 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dong C, Bach S. v., Haynes KA, Hegde AN. Proteasome modulates positive and negative translational regulators in long-term synaptic plasticity. J Neurosci 34, 3171–82 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wilhelmsson U, Pozo-Rodrigalvarez A, Kalm M, de Pablo Y, Widestrand Å, Pekna M, Pekny M. The role of GFAP and vimentin in learning and memory. Biol Chem 400, 1147–1156 (2019). [DOI] [PubMed] [Google Scholar]
- 69.Hausrat TJ, Muhia M, Gerrow K, Thomas P, Hirdes W, Tsukita S, Heisler FF, Herich L, Dubroqua S, Breiden P, Feldon J, Schwarz JR, Yee BK, Smart TG, Triller A, Kneussel M. Radixin regulates synaptic GABA A receptor density and is essential for reversal learning and short-term memory. Nat Commun 6, 6872 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.DePoy LM, Gourley SL. Synaptic Cytoskeletal Plasticity in the Prefrontal Cortex Following Psychostimulant Exposure. Traffic 16, 919–40 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wall MJ, Collins DR, Chery SL, Allen ZD, Pastuzyn ED, George AJ, Nikolova VD, Moy SS, Philpot BD, Shepherd JD, Müller J, Ehlers MD, Mabb AM, Corrêa SAL. The Temporal Dynamics of Arc Expression Regulate Cognitive Flexibility. Neuron 98, 1124–1132 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sung MK, Reitsma JM, Sweredoski MJ, Hess S, Deshaies RJ. Ribosomal proteins produced in excess are degraded by the ubiquitin-proteasome system. Mol Biol Cell 27, 2642–52 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hetman M, Slomnicki LP. Ribosomal biogenesis as an emerging target of neurodevelopmental pathologies. J Neurochem 148, 325–347 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bourque DK, Hartley T, Nikkel SM, Pohl D, Tétreault M, Kernohan KD, Dyment DA. A de novo mutation in RPL10 causes a rare X-linked ribosomopathy characterized by syndromic intellectual disability and epilepsy: A new case and review of the literature. Eur J Med Genet 61, 89–93 (2018). [DOI] [PubMed] [Google Scholar]
- 75.Crow YJ, Manel N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat Rev Immunol 15, 429–40 (2015). [DOI] [PubMed] [Google Scholar]
- 76.Livingston JH, Crow YJ. Neurologic phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1: Aicardi-Goutières syndrome and beyond. Neuropediatrics 47, 355–360 (2016). [DOI] [PubMed] [Google Scholar]
- 77.Araya P, Waugh KA, Sullivan KD, Núñez NG, Roselli E, Smith KP, Granrath RE, Rachubinski AL, Estrada BE, Butcher ET, Minter R, Tuttle KD, Bruno TC, Maccioni M, Espinosa JM. Trisomy 21 dysregulates T cell lineages toward an autoimmunity-prone state associated with interferon hyperactivity. Proc Natl Acad Sci U S A 116, 24231–24241 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Waugh KA, Araya P, Pandey A, Jordan KR, Smith KP, Granrath RE, Khanal S, Butcher ET, Estrada BE, Rachubinski AL, McWilliams JA, Minter R, Dimasi T, Colvin KL, Baturin D, Pham AT, Galbraith MD, Bartsch KW, Yeager ME, Porter CC, Sullivan KD, Hsieh EW, Espinosa JM. Mass Cytometry Reveals Global Immune Remodeling with Multi-lineage Hypersensitivity to Type I Interferon in Down Syndrome. Cell Rep 29, 1893–1908 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sullivan KD, Lewis HC, Hill AA, Pandey A, Jackson LP, Cabral JM, Smith KP, Liggett LA, Gomez EB, Galbraith MD, Degregori J, Espinosa JM. Trisomy 21 consistently activates the interferon response. Elife 5, e16220 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sullivan KD, Evans D, Pandey A, Hraha TH, Smith KP, Markham N, Rachubinski AL, Wolter-Warmerdam K, Hickey F, Espinosa JM, Blumenthal T. Trisomy 21 causes changes in the circulating proteome indicative of chronic autoinflammation. Sci Rep 7, 14818 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Akwa Y, Hassett DE, Eloranta M-L, Sandberg K, Masliah E, Powell H, Whitton JL, Bloom FE, Campbell IL. Transgenic Expression of IFN-α in the Central Nervous System of Mice Protects Against Lethal Neurotropic Viral Infection but Induces Inflammation and Neurodegeneration. J Immunol 161, 5016–26 (1998). [PubMed] [Google Scholar]
- 82.Campbell IL, Krucker T, Steffensen S, Akwa Y, Powell HC, Lane T, Carr DJ, Gold LH, Henriksen SJ, Siggins GR. Structural and functional neuropathology in transgenic mice with CNS expression of IFN-alpha. Brain Res 835, 46–61 (1999). [DOI] [PubMed] [Google Scholar]
- 83.Kavanagh D, McGlasson S, Jury A, Williams J, Scolding N, Bellamy C, Gunther C, Ritchie D, Gale DP, Kanwar YS, Challis R, Buist H, Overell J, Weller B, Flossmann O, Blunden M, Meyer EP, Krucker T, Evans SJW, Campbell IL, Jackson AP, Chandran S, Hunt DPJ. Type I interferon causes thrombotic microangiopathy by a dose-dependent toxic effect on the microvasculature. Blood 128, 2824–2833 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Goldmann T, Zeller N, Raasch J, Kierdorf K, Frenzel K, Ketscher L, Basters A, Staszewski O, Brendecke SM, Spiess A, Tay TL, Kreutz C, Timmer J, Mancini GM, Blank T, Fritz G, Biber K, Lang R, Malo D, Merkler D, Heikenwälder M, Knobeloch K, Prinz M. USP 18 lack in microglia causes destructive interferonopathy of the mouse brain. EMBO J 34, 1612–29 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Eggenberger J, Blanco-Melo D, Panis M, Brennand KJ, ten Oever BR. Type I interferon response impairs differentiation potential of pluripotent stem cells. Proc Natl Acad Sci U S A 116, 1384–1393 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yu Q, Katlinskaya Y. v., Carbone CJ, Zhao B, Katlinski K. v., Zheng H, Guha M, Li N, Chen Q, Yang T, Lengner CJ, Greenberg RA, Johnson FB, Fuchs SY. DNA-Damage-Induced Type I Interferon Promotes Senescence and Inhibits Stem Cell Function. Cell Rep 11, 785–797 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhu K, Dunner K, McConkey DJ. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 29, 451–62 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Studencka-Turski M, Çetin G, Junker H, Ebstein F, Krüger E. Molecular Insight Into the IRE1α-Mediated Type I Interferon Response Induced by Proteasome Impairment in Myeloid Cells of the Brain. Front Immunol 10, 2900 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Khacho M, Harris R, Slack RS. Mitochondria as central regulators of neural stem cell fate and cognitive function. Nat Rev Neurosci 20, 34–48 (2019). [DOI] [PubMed] [Google Scholar]
- 90.Yang K, Huang R, Fujihira H, Suzuki T, Yan N. N-glycanase NGLY1 regulates mitochondrial homeostasis and inflammation through NRF1. J Exp Med 215, 2600–2616 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sadler AJ, Williams BRG. Structure and function of the protein kinase R. Curr Top Microbiol Immunol 316, 253–92 (2007). [DOI] [PubMed] [Google Scholar]
- 92.Reid DW, Chen Q, Tay ASL, Shenolikar S, Nicchitta C. v.. The unfolded protein response triggers selective mRNA release from the endoplasmic reticulum. Cell 158, 1362–1374 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Meurs E, Chong K, Galabru J, Thomas NSB, Kerr IM, Williams BRG, Hovanessian AG. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 62, 379–90 (1990). [DOI] [PubMed] [Google Scholar]
- 94.Benkirane M, Neuveut C, Chun RF, Smith SM, Samuel CE, Gatignol A, Jeang KT. Oncogenic potential of TAR RNA binding protein TRBP and its regulatory interaction with RNA-dependent protein kinase PKR. EMBO Journal 16, 611–24 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Patel RC, Sen GC. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO Journal 17 (1998), doi: 10.1093/emboj/17.15.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Siwecka N, Rozpędek-Kamińska W, Wawrzynkiewicz A, Pytel D, Diehl JA, Majsterek I. The structure, activation and signaling of IRE1 and its role in determining cell fate. Biomedicines 9, 156 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Deture MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener 14, 32 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gal-Ben-Ari S, Barrera I, Ehrlich M, Rosenblum K. PKR: A kinase to remember. Front Mol Neurosci 11, 480 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Marchal JA, Lopez GJ, Peran M, Comino A, Delgado JR, García-García JA, Conde V, Aranda FM, Rivas C, Esteban M, Garcia MA. The impact of PKR activation: From neurodegeneration to cancer. FASEB J 28, 1965–74 (2014). [DOI] [PubMed] [Google Scholar]
- 100.Chukwurah E, Farabaugh KT, Guan BJ, Ramakrishnan P, Hatzoglou M. A tale of two proteins: PACT and PKR and their roles in inflammation. FEBS J 288, 6365–6391 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Muzio L, Viotti A, Martino G. Microglia in Neuroinflammation and Neurodegeneration: From Understanding to Therapy. Front Neurosci 15, 742065 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ahmed MM, Johnson NR, Boyd TD, Coughlan C, Chial HJ, Potter H. Innate Immune System Activation and Neuroinflammation in Down Syndrome and Neurodegeneration: Therapeutic Targets or Partners? Front Aging Neurosci 13, 718426 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 36, 928–930 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, Aguilar Salinas CA, Ahmad T, Albert CM, Ardissino D, Atzmon G, Barnard J, Beaugerie L, Benjamin EJ, Boehnke M, Bonnycastle LL, Bottinger EP, Bowden DW, Bown MJ, Chambers JC, Chan JC, Chasman D, Cho J, Chung MK, Cohen B, Correa A, Dabelea D, Daly MJ, Darbar D, Duggirala R, Dupuis J, Ellinor PT, Elosua R, Erdmann J, Esko T, Färkkilä M, Florez J, Franke A, Getz G, Glaser B, Glatt SJ, Goldstein D, Gonzalez C, Groop L, Haiman C, Hanis C, Harms M, Hiltunen M, Holi MM, Hultman CM, Kallela M, Kaprio J, Kathiresan S, Kim BJ, Kim YJ, Kirov G, Kooner J, Koskinen S, Krumholz HM, Kugathasan S, Kwak SH, Laakso M, Lehtimäki T, Loos RJF, Lubitz SA, Ma RCW, MacArthur DG, Marrugat J, Mattila KM, McCarroll S, McCarthy MI, McGovern D, McPherson R, Meigs JB, Melander O, Metspalu A, Neale BM, Nilsson PM, O’Donovan MC, Ongur D, Orozco L, Owen MJ, Palmer CNA, Palotie A, Park KS, Pato C, Pulver AE, Rahman N, Remes AM, Rioux JD, Ripatti S, Roden DM, Saleheen D, Salomaa V, Samani NJ, Scharf J, Schunkert H, Shoemaker MB, Sklar P, Soininen H, Sokol H, Spector T, Sullivan PF, Suvisaari J, Tai ES, Teo YY, Tiinamaija T, Tsuang M, Turner D, Tusie-Luna T, Vartiainen E, Watkins H, Weersma RK, Wessman M, Wilson JG, Xavier RJ, Neale BM, Daly MJ. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R, Taliun SAG, Corvelo A, Gogarten SM, Kang HM, Pitsillides AN, LeFaive J, been Lee S, Tian X, Browning BL, Das S, Emde AK, Clarke WE, Loesch DP, Shetty AC, Blackwell TW, Smith A. v., Wong Q, Liu X, Conomos MP, Bobo DM, Aguet F, Albert C, Alonso A, Ardlie KG, Arking DE, Aslibekyan S, Auer PL, Barnard J, Barr RG, Barwick L, Becker LC, Beer RL, Benjamin EJ, Bielak LF, Blangero J, Boehnke M, Bowden DW, Brody JA, Burchard EG, Cade BE, Casella JF, Chalazan B, Chasman DI, Chen YDI, Cho MH, Choi SH, Chung MK, Clish CB, Correa A, Curran JE, Custer B, Darbar D, Daya M, de Andrade M, DeMeo DL, Dutcher SK, Ellinor PT, Emery LS, Eng C, Fatkin D, Fingerlin T, Forer L, Fornage M, Franceschini N, Fuchsberger C, Fullerton SM, Germer S, Gladwin MT, Gottlieb DJ, Guo X, Hall ME, He J, Heard-Costa NL, Heckbert SR, Irvin MR, Johnsen JM, Johnson AD, Kaplan R, Kardia SLR, Kelly T, Kelly S, Kenny EE, Kiel DP, Klemmer R, Konkle BA, Kooperberg C, Köttgen A, Lange LA, Lasky-Su J, Levy D, Lin X, Lin KH, Liu C, Loos RJF, Garman L, Gerszten R, Lubitz SA, Lunetta KL, Mak ACY, Manichaikul A, Manning AK, Mathias RA, McManus DD, McGarvey ST, Meigs JB, Meyers DA, Mikulla JL, Minear MA, Mitchell BD, Mohanty S, Montasser ME, Montgomery C, Morrison AC, Murabito JM, Natale A, Natarajan P, Nelson SC, North KE, O’Connell JR, Palmer ND, Pankratz N, Peloso GM, Peyser PA, Pleiness J, Post WS, Psaty BM, Rao DC, Redline S, Reiner AP, Roden D, Rotter JI, Ruczinski I, Sarnowski C, Schoenherr S, Schwartz DA, Seo JS, Seshadri S, Sheehan VA, Sheu WH, Shoemaker MB, Smith NL, Smith JA, Sotoodehnia N, Stilp AM, Tang W, Taylor KD, Telen M, Thornton TA, Tracy RP, van den Berg DJ, Vasan RS, Viaud-Martinez KA, Vrieze S, Weeks DE, Weir BS, Weiss ST, Weng LC, Willer CJ, Zhang Y, Zhao X, Arnett DK, Ashley-Koch AE, Barnes KC, Boerwinkle E, Gabriel S, Gibbs R, Rice KM, Rich SS, Silverman EK, Qasba P, Gan W, Abe N, Almasy L, Ament S, Anderson P, Anugu P, Applebaum-Bowden D, Assimes T, Avramopoulos D, Barron-Casella E, Beaty T, Beck G, Becker D, Beitelshees A, Benos T, Bezerra M, Bis J, Bowler R, Broeckel U, Broome J, Bunting K, Bustamante C, Buth E, Cardwell J, Carey V, Carty C, Casaburi R, Castaldi P, Chaffin M, Chang C, Chang YC, Chavan S, Chen BJ, Chen WM, Chuang LM, Chung RH, Comhair S, Cornell E, Crandall C, Crapo J, Curtis J, Damcott C, David S, Davis C, de las Fuentes L, DeBaun M, Deka R, Devine S, Duan Q, Duggirala R, Durda JP, Eaton C, Ekunwe L, el Boueiz A, Erzurum S, Farber C, Flickinger M, Fornage M, Frazar C, Fu M, Fulton L, Gao S, Gao Y, Gass M, Gelb B, Geng XP, Geraci M, Ghosh A, Gignoux C, Glahn D, Gong DW, Goring H, Graw S, Grine D, Gu CC, Guan Y, Gupta N, Haessler J, Hawley NL, Heavner B, Herrington D, Hersh C, Hidalgo B, Hixson J, Hobbs B, Hokanson J, Hong E, Hoth K, Hsiung CA, Hung YJ, Huston H, Hwu CM, Jackson R, Jain D, Jhun MA, Johnson C, Johnston R, Jones K, Kathiresan S, Khan A, Kim W, Kinney G, Kramer H, Lange C, Lange E, Lange L, Laurie C, LeBoff M, Lee J, Lee SS, Lee WJ, Levine D, Lewis J, Li X, Li Y, Lin H, Lin H, Lin KH, Liu S, Liu Y, Liu Y, Luo J, Mahaney M, Make B, Manson JA, Margolin L, Martin L, Mathai S, May S, McArdle P, McDonald ML, McFarland S, McGoldrick D, McHugh C, Mei H, Mestroni L, Min N, Minster RL, Moll M, Moscati A, Musani S, Mwasongwe S, Mychaleckyj JC, Nadkarni G, Naik R, Naseri T, Nekhai S, Neltner B, Ochs-Balcom H, Paik D, Pankow J, Parsa A, Peralta JM, Perez M, Perry J, Peters U, Phillips LS, Pollin T, Becker JP, Boorgula MP, Preuss M, Qiao D, Qin Z, Rafaels N, Raffield L, Rasmussen-Torvik L, Ratan A, Reed R, Regan E, Reupena MS, Roselli C, Russell P, Ruuska S, Ryan K, Sabino EC, Saleheen D, Salimi S, Salzberg S, Sandow K, Sankaran VG, Scheller C, Schmidt E, Schwander K, Sciurba F, Seidman C, Seidman J, Sherman SL, Shetty A, Sheu WHH, Silver B, Smith J, Smith T, Smoller S, Snively B, Snyder M, Sofer T, Storm G, Streeten E, Sung YJ, Sylvia J, Szpiro A, Sztalryd C, Tang H, Taub M, Taylor M, Taylor S, Threlkeld M, Tinker L, Tirschwell D, Tishkoff S, Tiwari H, Tong C, Tsai M, Vaidya D, VandeHaar P, Walker T, Wallace R, Walts A, Wang FF, Wang H, Watson K, Wessel J, Williams K, Williams LK, Wilson C, Wu J, Xu H, Yanek L, Yang I, Yang R, Zaghloul N, Zekavat M, Zhao SX, Zhao W, Zhi D, Zhou X, Zhu X, Papanicolaou GJ, Nickerson DA, Browning SR, Zody MC, Zöllner S, Wilson JG, Cupples LA, Laurie CC, Jaquish CE, Hernandez RD, O’Connor TD, Abecasis GR. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fonteneau JF, Larsson M, Somersan S, Sanders C, Münz C, Kwok WW, Bhardwaj N, Jotereau F, Generation of high quantities of viral and tumor-specific human CD4+ and CD8+ T-cell clones using peptide pulsed mature dendritic cells. J Immunol Methods 258, 111–126 (2001). [DOI] [PubMed] [Google Scholar]
- 107.Bolduc FV, Bell K, Cox H, Broadie KS, Tully T. Excess protein synthesis in Drosophila Fragile X mutants impairs long-term memory. Nat Neurosci 11, 1143–5 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tully T, Quinn WG. Classical conditioning and retention in normal and mutant Drosophila melanogaster. J Comp Physiol A 157, 263–77 (1985). [DOI] [PubMed] [Google Scholar]
- 109.Tully T, Boyton S, Brandes C, Dura JM, Mihalek R, Preat T, Villella A. Genetic dissection of memory formation in Drosophila melanogaster. Cold Spring Harb Symp on Quant Biol 55, 203–11 (1990). [DOI] [PubMed] [Google Scholar]
- 110.Palma Medina LM, Becker AK, Michalik S, Yedavally H, Raineri EJM, Hildebrandt P, Salazar MG, Surmann K, Pförtner H, Mekonnen SA, Salvati A, Kaderali L, Van Dijl JM, Völker U. Metabolic Cross-talk between Human Bronchial Epithelial Cells and Internalized Staphylococcus aureus as a Driver for Infection. Mol Cell Proteomics 18; 892–908 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Suomi T, Elo LL. Enhanced differential expression statistics for data-independent acquisition proteomics. Sci Rep 7, 5869 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Phipson B, Lee S, Majewski IJ, Alexander WS, Smyth GK. ROBUST HYPERPARAMETER ESTIMATION PROTECTS AGAINST HYPERVARIABLE GENES AND IMPROVES POWER TO DETECT DIFFERENTIAL EXPRESSION. Ann Appl Stat 10, 946–963 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Perez-Riverol Y, Bai J, Bandla C, García-Seisdedos D, Hewapathirana S, Kamatchinathan S, Kundu DJ, Prakash A, Frericks-Zipper A, Eisenacher M, Walzer M, Wang S, Brazma A, Vizcaíno JA. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res 50, D543–D552 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hsieh TC, Bar-Haim A, Moosa S, Ehmke N, Gripp KW, Pantel JT, Danyel M, Mensah MA, Horn D, Rosnev S, Fleischer N, Bonini G, Hustinx A, Schmid A, Knaus A, Javanmardi B, Klinkhammer H, Lesmann H, Sivalingam S, Kamphans T, Meiswinkel W, Ebstein F, Krüger E, Küry S, Bézieau S, Schmidt A, Peters S, Engels H, Mangold E, Kreiß M, Cremer K, Perne C, Betz RC, Bender T, Grundmann-Hauser K, Haack TB, Wagner M, Brunet T, Bentzen HB, Averdunk L, Coetzer KC, Lyon GJ, Spielmann M, Schaaf CP, Mundlos S, Nöthen MM, Krawitz PM. GestaltMatcher facilitates rare disease matching using facial phenotype descriptors. Nat Genet 54, 349–357 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Knaus A, Pantel JT, Pendziwiat M, Hajjir N, Zhao M, Hsieh TC, Schubach M, Gurovich Y, Fleischer N, Jäger M, Köhler S, Muhle H, Korff C, Møller RS, Bayat A, Calvas P, Chassaing N, Warren H, Skinner S, Louie R, Evers C, Bohn M, Christen HJ, van den Born M, Obersztyn E, Charzewska A, Endziniene M, Kortüm F, Brown N, Robinson PN, Schelhaas HJ, Weber Y, Helbig I, Mundlos S, Horn D, Krawitz PM. Characterization of glycosylphosphatidylinositol biosynthesis defects by clinical features, flow cytometry, and automated image analysis. Genome Med 10, 3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Marbach F, Rustad CF, Riess A, Đukić D, Hsieh TC, Jobani I, Prescott T, Bevot A, Erger F, Houge G, Redfors M, Altmueller J, Stokowy T, Gilissen C, Kubisch C, Scarano E, Mazzanti L, Fiskerstrand T, Krawitz PM, Lessel D, Netzer C. The Discovery of a LEMD2-Associated Nuclear Envelopathy with Early Progeroid Appearance Suggests Advanced Applications for AI-Driven Facial Phenotyping. Am J Hum Genet 104, 749 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Oti M, Huynen MA, Brunner HG. Phenome connections. Trends Genet 24, 103–106 (2008). [DOI] [PubMed] [Google Scholar]
- 118.Gurovich Y, Hanani Y, Bar O, Nadav G, Fleischer N, Gelbman D, Basel-Salmon L, Krawitz PM, Kamphausen SB, Zenker M, Bird LM, Gripp KW. Identifying facial phenotypes of genetic disorders using deep learning. Nat Med 25, 60–64 (2019). [DOI] [PubMed] [Google Scholar]
- 119.Küry S, van Woerden GM, Besnard T, Proietti Onori M, Latypova X, Towne MC, Cho MT, Prescott TE, Ploeg MA, Sanders S, Stessman HAF, Pujol A, Distel B, Robak LA, Bernstein JA, Denommé-Pichon AS, Lesca G, Sellars EA, Berg J, Carré W, Busk ØL, van Bon BWM, Waugh JL, Deardorff M, Hoganson GE, Bosanko KB, Johnson DS, Dabir T, Holla ØL, Sarkar A, Tveten K, de Bellescize J, Braathen GJ, Terhal PA, Grange DK, van Haeringen A, Lam C, Mirzaa G, Burton J, Bhoj EJ, Douglas J, Santani AB, Nesbitt AI, Helbig KL, Andrews MV, Begtrup A, Tang S, van Gassen KLI, Juusola J, Foss K, Enns GM, Moog U, Hinderhofer K, Paramasivam N, Lincoln S, Kusako BH, Lindenbaum P, Charpentier E, Nowak CB, Cherot E, Simonet T, Ruivenkamp CAL, Hahn S, Brownstein CA, Xia F, Schmitt S, Deb W, Bonneau D, Nizon M, Quinquis D, Chelly J, Rudolf G, Sanlaville D, Parent P, Gilbert-Dussardier B, Toutain A, Sutton VR, Thies J, Peart-Vissers LELM, Boisseau P, Vincent M, Grabrucker AM, Dubourg C, Tan WH, Verbeek NE, Granzow M, Santen GWE, Shendure J, Isidor B, Pasquier L, Redon R, Yang Y, State MW, Kleefstra T, Cogné B, Petrovski S, Retterer K, Eichler EE, Rosenfeld JA, Agrawal PB, Bézieau S, Odent S, Elgersma Y, Mercier S, De Novo Mutations in Protein Kinase Genes CAMK2A and CAMK2B Cause Intellectual Disability. Am J Hum Genet 101, 768–788 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Goslin K, Banker G. Rapid changes in the distribution of GAP-43 correlate with the expression of neuronal polarity during normal development and under experimental conditions. J Cell Biol 110, 1319–31 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.