

Abstract
GATA2 is a transcription factor critical for hematopoiesis. Germline mutations in GATA binding protein 2 (GATA2) led to haploinsufficiency, severe cytopenias of multiple cell lineages, susceptibility to infections and strong propensity to develop myelodysplastic syndrome, and acute myeloid leukemia. Mechanisms of progressive cytopenias remain unclear. MicroRNA (miRNA) represents a unique mechanism of post-transcriptional gene regulation. In this study, miRNA profiles were evaluated and eight miRNAs were found to be differentially expressed (≥2-fold, P ≤ 0.05) in patient-derived cell lines (N = 13) in comparison to controls (N = 10). miR-9, miR-181a-2–3p, miR-181c, miR-181c-3p, miR-486–3p, and miR-582 showed increased expression, whereas miR-223 and miR-424–3p showed decreased expression. Cell death assays indicated that miR-181c potently induces cell death in lymphoid (Ly-8 and SP-53) and myeloid (HL-60) cell lines. miR-181c was predicted to target myeloid cell leukemia (MCL)1, which was confirmed by transfection assays, resulting in significantly reduced MCL1 mRNA and decreased live cell numbers. Bone marrow analysis of 34 GATA2 patients showed significantly decreased cellularity, CD34-positive cells, monocytes, dendritic cells, NK cells, B cells, and B cell precursors in comparison to healthy controls (N = 29; P < 0.001 for each), which was accompanied by decreased levels of MCL1 (P < 0.05). GATA2 expression led to significant repression of miR-181c expression in transfection experiments. Conversely, knockdown of GATA2 led to increased miR-181c expression. These findings indicate that miR-181c expression is increased and MCL1 levels decreased in GATA2 deficiency cells, and that GATA2 represses miR-181c transcription. Increased miR-181c may contribute to elevated cell death and cytopenia in GATA2 deficiency potentially through down-regulation of MCL1.
Keywords: AML, apoptosis, bone marrow failure, GATA2, immunodeficiency, MCL1, MDS, miR-181c, miRNA
1 |. INTRODUCTION
GATA2 deficiency is caused by germline heterozygous mutations in GATA binding protein 2 (GATA2) resulting in haploinsufficiency. GATA2 encodes a transcription factor containing two zinc finger domains and is required for hematopoiesis and maintenance of hematopoietic stem cells.1–5 Known targets of GATA2 include p53, PU.1, RUNX1, and LMO2.2,6,7 Patients with several overlapping syndromes, including monocytopenia/mycobacterium avium complex syndrome,8 Dendritic cell, monocyte, B and natural killer lymphoid deficiency (DCML deficiency),9 Emberger’s syndrome,10 and familial myelodysplastic syndrome/acute myeloid leukemia (MDS/AML),11 were previously found to harbor germline GATA2 mutations with an autosomal dominant pattern of inheritance. They are now referred to as one disease termed GATA2 deficiency.12 GATA2 deficiency is associated with progressive cytopenias, bone marrow hypocellularity, severe immunodeficiency, sensoneurial hearing loss, lymphedema, abnormal myeloid differentiation, and increased propensity to develop bone marrow failure, MDS, AML, or chronic myelomonocytic leukemia.13–16 Hematopoietic stem cell transplantation is the only therapy that cures myeloid malignancy and restores the immune system.17 Although some functions of GATA2 in hematopoiesis have been characterized, molecular mechanisms of cytopenias, including the loss of monocytes, B cells, and NK cells, and disease progression are not well understood.
MicroRNAs (miRNAs) are evolutionarily conserved small noncoding RNAs that employ a post-transcriptional gene regulation mechanism that has been shown to play a role in development, differentiation, and tumorigenesis.18–20 miRNAs regulate gene expression by binding to the 3′ untranslated region (3′UTR) of target messenger RNAs, leading to mRNA degradation or translational repression.18–20 Studies have demonstrated the ability of miRNA expression profiles to discriminate between specific types of cancer and the normal corresponding host tissue, as well as to discriminate between subclassifications of tumors,21,22 and to serve as markers for disease prognosis and progression.23 Many reports have implicated miRNAs in hematopoiesis and hematologic malignancies.24–29 No study to date has reported the role that miRNAs may play in GATA2 deficiency. Identifying miRNAs that contribute to bone marrow failure in GATA2 deficiency may provide insights into novel therapeutic avenues for this syndrome, as well as expand our understanding of abnormal hematopoiesis in this disease.
In this study, using miRNA microarrays and real-time PCR, we found that several miRNAs were differentially expressed in patient-derived GATA2 deficiency cell lines. Among them, miR-181c expression was significantly increased and potently induced apoptosis in lymphoid and myeloid cell lines. Here we provide evidence that GATA2 represses miR-181c expression, and thus in GATA2 deficiency, miR-181c is derepressed and expressed in high enough levels to exert a biologic effect on the cells. Additionally, we show that transcripts from the anti-apoptotic myeloid cell leukemia-1 gene (MCL1) are targeted by miR-181c and associated with cell death. Reduction of MCL1 is observed in GATA2 deficiency patient bone marrow cells. These findings have implications for understanding loss of hematopoietic cell populations, and disease manifestations in GATA2 deficiency.
2 |. MATERIALS AND METHODS
2.1 |. Cell culture
EBV-immortalized healthy control (HC) and GATA2 deficient B cell lines, OCI-Ly-8 (generously provided by Dr. Mark Minden, Ontario Cancer Institute, Toronto, Ontario, Canada),30 HL-60, K562, and SP5331 cells were grown in RPMI 1640 medium; and HEK293 cells were grown in DMEM with 5% CO2. EBV-immortalized B cells of 10 HCs and 13 GATA2 deficient patients were used in the study. GATA2 mutation status among patient samples: two samples with +9.5 regulatory region mutation (c.1017+572C > T), one with 2033 bp deletion (c.1–200_871+527del2033), one with T354M (c.1061C > T) mutation, two samples with R396W (c.1186C > T) mutation, and five with R398W (c.1192C > T) mutation.
2.2 |. Bone marrow samples
Bone marrow samples from GATA2 patients (n = 34) and healthy donors (n = 29) were obtained with consent from patients enrolled on clinical trial protocols (NIAID 13-I-0157 and NCI 13-C-1032) that were approved by institutional review boards at our institution, and in accordance with the Declaration of Helsinki. Bone marrow mononuclear cells were separated by centrifugation using Corning Lymphocyte Separation Medium (Fisher Scientific, Waltham, MA, USA) and were viably frozen. Bone marrow cellularity was assessed on H&E stains of bone marrow core biopsies (n = 34). Bone marrow images were captured on an Olympus BX41 microscope equipped with a DP74 Olympus camera and Olympus cellSens Entry software.
2.3 |. Bone marrow flow cytometry
BM aspirates were stained using a panel of antibodies including: Kappa, Lambda, CD5, CD19, CD34, CD20, CD45, CD23, CD10, CD11c, CD22, CD14, CD16, CD13, CD117, CD11b, CD38, HLA DR, CD42b, CD61, CD15, CD7, CD71, CD2, CD36, CD64, CD33, CD123, CD14, CD57, CD3, CD56, CD7, CD8, CD4, and CD138. Analysis was performed using a fluorescence-activated cell sorter (FACS) Canto II Analyzer (BD Biosciences, San Jose, CA, USA) equipped with three lasers and eight fluorescent detectors. Briefly, cells were stained with the appropriate antibodies for 15–20 min. Red blood cells were lysed with BD FACS lysing solution, and cells were washed with PBS containing 1% albumin. Subsequently, the cells were fixed in 1% paraformaldehyde solution, and 1 × 105 events were acquired using FACSDiva (BD Biosciences). The list mode files were analyzed with FCS Express v6 (De Novo Software, Glendale, CA, USA).
2.4 |. RNA isolation
RNA was isolated from EBV-immortalized B cell lines of HCs and GATA2 deficient patients, bone marrow mononuclear cells of healthy donor or GATA2 patients, or transfected cell lines using the miRNeasy Mini Kit (Qiagen, Germantown, MD, USA). RNA was eluted in water. RNA concentration was determined using NanoDrop 2000 instrument (Thermo Scientific, Wilmington, DE, USA).
2.5 |. Agilent human miRNA array
Total RNA (100 ng) and spike-in controls were cyanine 3-pCp-labeled using the Agilent miRNA labeling kit according to the manufacturer’s protocol. The labeled RNA was purified with Micro Bio-Spin 6 columns (BioRad, Hercules, CA, USA), dried and redissolved in hybridization buffer and hybridized on Agilent high density human miRNA array (v.3, Rel.12, with 851 human miRNAs on the array) at 56°C and 20 rpm for 20 h. After washing, array images were scanned on Agilent’s G2505C scanner and processed using Agilent’s Feature Extraction software (v.10.7.3.1). Array images were processed using Agilent’s Feature Extraction software.
2.6 |. Plasmid construction
pcDNA6.2-EmGFP-mir9 was a gift from Lynn Hudson (Addgene plasmid # 22741, Addgene, Cambridge, MA, USA).32 GFP-tagged miR-181c, or miR-223 were cloned into pcDNA-6.2 GW/EmGFP miR-9 at the Xho I and BamH I sites by replacing miR-9 hairpin with miR-181c or miR-223 hairpin using the cold fusion cloning kit (System Biosciences, Mountain View, CA, USA). The empty control plasmid was constructed by removing miR-9 from pcDNA-6.2 GW/EmGFP at the Xho I and Sal I sites. The luciferase reporter plasmid was cloned by inserting a 414 bp fragment containing the putative miR-181c binding site in the 3′UTR of MCL1 into the Sac I and Xho I sites of the pmirGLO luciferase reporter vector (Promega, Madison, WI, USA). The mutated form of luciferase reporter plasmid was obtained by using the site-directed mutagenesis kit (Agilent, Santa Clara, CA, USA) deleting the putative miR-181c binding site in the 3′UTR of MCL1. All the cloning and mutagenesis were verified by Sanger Sequencing. Primers for cloning, mutagenesis and sequencing are included in Supporting Information Table S2.
2.7 |. RNA interference
The GIPZ GATA2 lentiviral shRNA plasmids were purchased from GE Dharmacon (Lafayette, CO, USA). The mature antisense sequences of three shRNA constructs are: TCTCTACATAAAGTTGTC, TCTGAACAGGAACGAGCCT, and TGGACATCTTCCGGTTCCG.
2.8 |. Transient transfection
Transient transfection of Ly-8, HL-60, K562, and SP53 cells was performed using Lonza 4D-Nucleofector apparatus and Nucleofector solution SF (Lonza, Frederick, MD, USA) as per the manufacturer’s instructions. Briefly, 7.5 × 106 cells were transfected with 7.5 μg of plasmid DNA. After transfection, cells were added to prewarmed media and placed in a 37°C/5% CO2 incubator. Cells were harvested for sorting or RNA isolation 24 or 48 h after transfection.
2.9 |. Quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) was used to determine the expression level of miRNAs and genes. For miRNA quantitation, reverse transcription was performed from 100 ng of total RNA with TaqMan reverse transcription primers using High Capacity cDNA Reverse Transcription Kit on a GeneAmp PCR system 9700 instrument (Applied Biosystems, Foster City, CA, USA). TaqMan assays for miR-181c, miR-223, and miR-9 (Thermo Fisher, Waltham, MA, USA) were performed in triplicate on a StepOne Plus instrument (Applied Biosystems). Ribosome RNA 18S and small nuclear RNA (RNU44 and RNU48) were amplified as internal controls. The relative miRNA expression level was calculated as 2−deltaCt.
To determine the gene expression level, reverse transcription was performed using High Capacity cDNA Reverse Transcription Kit. qRT-PCR was performed using TaqMan universal PCR master mix and FAM dye-labeled TaqMan MGB probes (MCL1, p53, GUSB, HPRT, TFRC, and PIAA) on the StepOnePlus instrument (Foster City, CA, USA). Delta Ct value was calculated as Ct value of target gene probe subtracted by the geometric mean of Ct values of GUSB, TFRC, and HPRT probes. The relative gene expression level was calculated as 2−deltaCt.
2.10 |. Cell death detection ELISA and trypan blue staining
Apoptosis was measured using the Cell Death Detection ELISA Plus (Roche Applied Science, Indianapolis, IN, USA) according to manufacturer’s recommendations. Briefly, cells transfected with miR-181c, miR-223, or miR-9 were harvested 48 h after transfection. Cells were lysed and incubated for 30 min. Protein concentration was measured using the BioRad Protein Assay (BioRad). The immunoreagent master mix was then added to the samples and incubated at room temperature for 2 h. Samples were then washed and apoptosis was measured at 405 nm using the BioTek Synergy 4 (BioTek, Winooski, VT, USA). A negative control and a positive control were included.
Dead cell percentage was also calculated by counting dead cell numbers using trypan blue staining (Life Technologies, Grand Island, NY, USA) 60 h after cells were transfected with miR-181c, miR-223, or miR-9.
2.11 |. WST-1 assay
The colorimetric water soluble tetrazolium (WST)-1 assay (Millipore-Sigma, St. Louis, MO, USA) was used to quantify the cellular proliferation and viability according to manufacturer’s instructions. Cells were transfected with GFP control vector, miR-181c, miR-223, and miR-9 expression plasmids and cultured for 48 h. Then the cell proliferation reagent WST-1 was added and incubated for 4 h. The quantity of viable cells was measured at 440 nm using the BioTek Synergy 4.
2.12 |. Cell sorting by flow cytometry
GFP vector and miR-181c-GFP transfected cells were harvested 48 h after transfection. The dead cell clumps were filtered using the 40 μm nylon Cell Strainer (BD Falcon, Franklin Lakes, NJ, USA). Total GFP-positive live cells were sorted on a BD FACS Canto with BD FACSDiva software (BD Biosciences).
2.13 |. Luciferase reporter assay
The pmirGLO plasmids containing wild-type (GLO-MCL1-UTR) or mutated (GLO-MCL1-muUTR) MCL1 3′UTRs, and vector control and miR-181c expressing plasmids were cotransfected using Lipofectamine 3000 (Thermo Fisher, Carlsbad, CA, USA) into HEK293 cells. A luciferase reporter assay was performed with the Dual-Glo Luciferase Assay (Promega). Cells were rinsed with PBS and lysed with the passive lysis buffer 48 h after transfection. A fraction of lysate was used for RNA isolation and the expression of miR-181c and 18S internal control were quantitated by real-time RT-PCR with assays from Thermo Fisher as described earlier. The remaining lysate was used for luciferase quantitation using the protocol as recommended by Promega. Firefly and renilla luciferases were quantitated using Synergy 4 instrument with Gen5 software (BioTek). Relative luciferase level was calculated as firefly luciferase level normalized to renilla luciferase control level.
2.14 |. Statistical analysis
Data from Agilent miRNA array were analyzed using Agilent’s Gene-Spring GX (v.11.5.1) software. The array data were normalized to the data point of 75 percentile signal strength and to a set of spike-in and control probes on the array, respectively. The differences between the means of experimental groups were analyzed by 3-tailed Mann-Whitney rank sum tests. The miRNAs with significant P-value (P ≤ 0.05) and fold change (≥2-fold) in both normalization methods (75th percentile and control probes) were selected for further analysis.
The signal intensity values and fold changes presented in figures are from data normalized by control gene probes. Raw data and data normalized by control gene probes were deposited in the GEO database with accession number GSE51132. The differences between the means of experimental groups of normalized qRT-PCR assay data were also analyzed by 2-tailed Mann-Whitney rank sum tests. P-values of less than 0.05 were considered significant. Hierarchic clustering analysis was performed using JMP software v11 (SAS, Cary, NC, USA). Scatter plots were generated using Prism (GraphPad Software, La Jolla, CA, USA).
2.15 |. Data availability
Raw array data and data normalized by control gene probes were deposited in the GEO database with accession number GSE51132. All other data are contained within this article.
3 |. RESULTS
3.1 |. Differential expression of miRNAs in GATA2 deficient cell lines
To investigate whether miRNAs play a role in cell proliferation and death in GATA2 deficient cells, differential miRNA expression was assessed. RNA from EBV-immortalized B cell lines from 10 HCs and 13 patients with GATA2 deficiency was analyzed on a hybridization-based Agilent high density miRNA array platform containing probes for 851 human miRNAs. Requiring a two-fold or greater difference in mean values between controls and GATA2 deficient cells and P-value less than 0.05, eight miRNAs (miR-181a-2–3p, miR-181c, miR-181c-3p, miR-223, miR-424–3p, miR-486–3p, miR-582, and miR-9) were found to be significantly differentially expressed. Among them, six miRNAs (miR-181a-2–3p, miR-181c, miR-181c-3p, miR-486–3p, miR-582, and miR-9) were up-regulated and two miRNAs (miR-223 and miR-424–3p) were down-regulated in GATA2 deficient cells. The miR-181c-3p is complementary to miR-181c. Based on reported roles played by miRNAs in hematopoiesis, cell proliferation, and differentiation, and on our preliminary studies, we chose three miRNAs, miR-181c, miR-223, and miR-9, for further investigation (Table 1 and Supporting Information Table S1).
TABLE 1.
miR-181c, miR-223, and miR-9 were differentially expressed in GATA2 patient-derived EBV-immortalized B cells in comparison to EBV immortalized B cells from controls
| MicroRNA (miRNA) | Regulation | Array |
TaqMan |
||
|---|---|---|---|---|---|
| P-value | Fold change | P-value | Fold change | ||
| miR-181c | Up | 0.02 | 2.69 | 0.01 | 1.66 |
| miR-223 | down | 0.03 | −5.57 | 0.02 | −2.04 |
| miR-9 | Up | 0.03 | 9.52 | 0.04 | 2.07 |
3.2 |. miR-181c targets MCL1 and induces cell death
GATA2 deficiency is associated with progressive cytopenias involving multiple cell linages, including monocytes, B cells, NK cells, and dendritic cells, often progressing to pancytopenia with loss of myeloid cells, red cells, and platelets. To investigate the potential roles of differentially expressed miRNAs in the biology of GATA2 deficient cells, we evaluated whether miRNAs have an effect on cell proliferation or cell death. Expression plasmids of miR-181c, miR-223, and miR-9 were transfected into HL60 (promyelocytic leukemia [myeloid]), Ly-8 (transformed follicular lymphoma [lymphoid]), and SP53 (B cell lymphoma [lymphoid]) cell lines. A Cell Death ELISA assay, trypan blue staining/cell counting, and WST-1 assay were performed to measure changes in apoptosis induction, dead cell percentage, and cell viability. The Cell Death ELISA assay quantifies apoptosis by measuring histones and DNA of nucleosomes released to the cytoplasmic fraction of cell lysates using specific antibodies against DNA and histones. As shown in Figure 1A, relative to empty vector, a significant increase in apoptosis was observed upon expression of miR-181c, but not miR-223 or miR-9, in HL-60 (1.4-fold, P = 0.01), Ly-8 (1.6-fold, P = 0.04), and SP53 cells (2.4-fold, P < 0.01) 48 h after transfection (Fig. 1A). When cells from the same transfection experiments were stained by trypan blue and counted, a significant increase in the percentage of dead cells was observed in miR-181c transfected HL-60 (10-fold, P < 0.001), Ly-8 cells (18-fold, P < 0.001), and SP53 cells (2.6-fold, P < 0.001) 60 h after transfection. There was no significant increase in dead cells in miR-223 and miR-9 transfected HL-60 cells, whereas there was a slight increase in dead cells in miR-223 (2.8-fold, P = 0.03) and miR-9 (1.8-fold, P < 0.01) transfected Ly-8 cells and SP53 cells (1.4-fold, P < 0.01, and 1.4-fold, P < 0.01, respectively; Fig. 1B).
FIGURE 1. Expression of miR-181c induces apoptosis and inhibits cell proliferation.
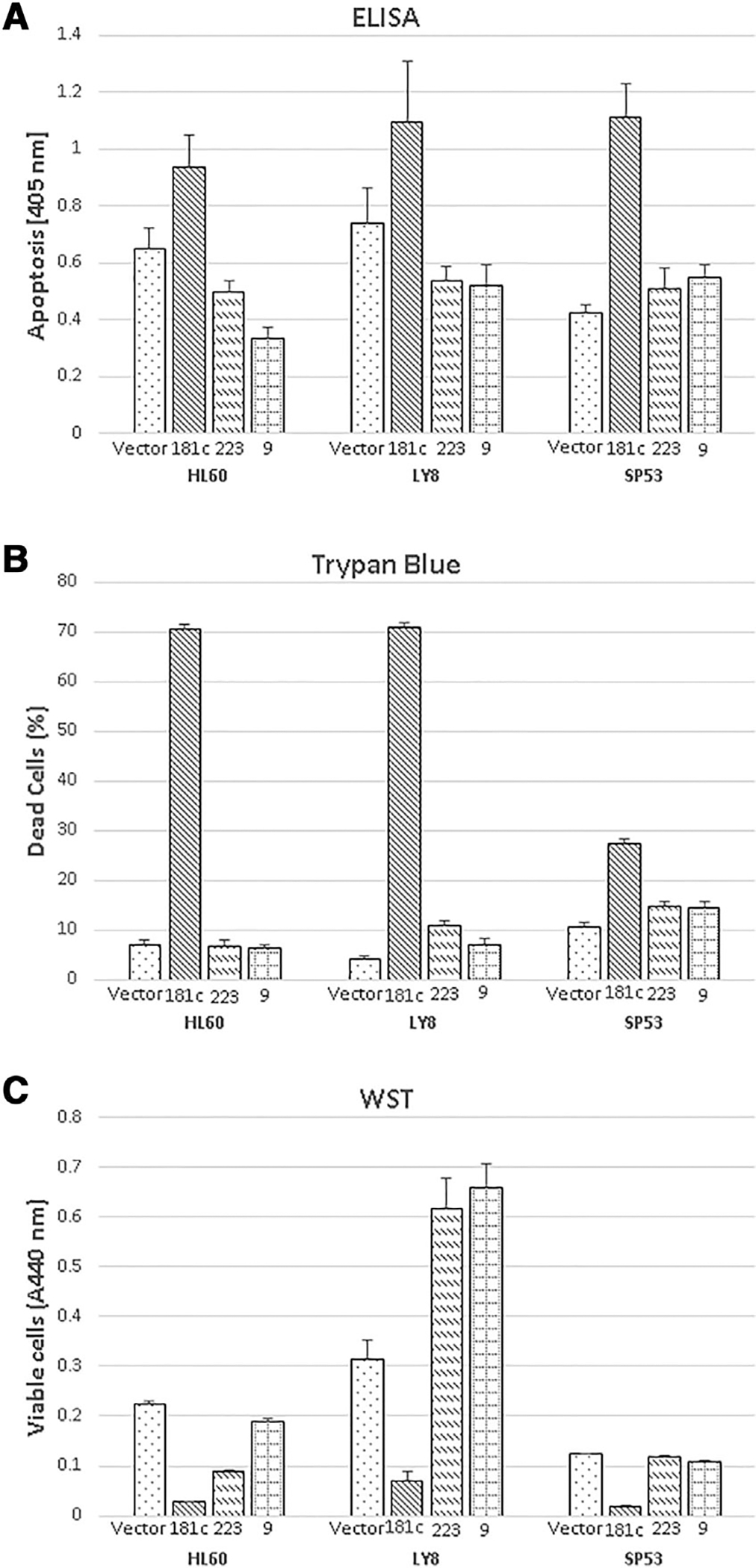
GFP control vector, miR-181c, miR-223, and miR-9 expression plasmids were transfected into HL-60, Ly-8, and SP53 cells. Three repeated experiments were performed in triplicate. Results of technical triplicates (mean and SD) are shown. The significance is shown as compared to vector control group. *, P < 0.05; **, P < 0.01; ***, P < 0.001. A. Apoptosis was determined by Cell Death Detection ELISA 48 h after transfection. B. Dead cell percentage was obtained by trypan blue staining 60 h (HL-60 and Ly-8) or 48 h (SP53) after transfection. C. Cell proliferation was assessed by WST-1 assay 48 h after transfection
To check the effect of these miRNAs on cell metabolism as an indicator of cell proliferation and viability, the WST-1 assay was performed. This colorimetric assay allows quantification of cell metabolism, cellular proliferation, and viability. The number of viable cells is quantified by measuring the cleavage of tetrazolium salts added to the culture medium. Cell proliferation was decreased 8.8-fold (P < 0.01) by miR-181c and 2.5-fold (P < 0.05) by miR-223 in HL-60 cells. Cell proliferation was decreased 4.4-fold (P < 0.01) by miR-181c and increased by miR-223 and miR-9 in Ly-8 cells. In SP-53 cells, cell proliferation was decreased 7.1-fold (P < 0.001) by miR-181c and only slightly by miR-223 and miR-9 (Fig. 1C). Taken together, these results demonstrated that, among miRNAs tested, miR-181c potently induces cell death and inhibits cell proliferation in lymphoid and myeloid cell lines.
To further investigate the potential function of differentially expressed miRNAs in GATA2 deficient cells, TargetScan analysis was performed for miR-181c, miR-223, and miR-9 to search for potential targets. Among thousands of predicted targets, some were reported to play important roles in hematopoiesis and in cell differentiation, proliferation, and cell death. The miR-181c putatively targets BCL2, MCL1, HOXA, and NLK; miR-9 was predicted to regulate BCL2, MYC, PRDM1/BLIMP1, BCL6, and RUNX1; and miR-223 was predicted to target LMO2, E2F1, FOXO1, and CEBPα/β. There was one site on the 3′UTR of MCL1 that was predicted by TargetScan analysis to bind to miR-181c (Fig. 2A). In order to confirm that miR-181c targets MCL1, the MCL1 3′ UTR fragment containing the putative wild-type and mutated miR-181c binding sites were subcloned into luciferase reporter plasmids. Real-time RT-PCR experiments showed that mir-181c was expressed in respective groups after transfection (Fig. 2B). Luciferase assay results indicated that transfection of miR-181c significantly reduced expression of the luciferase-MCL1–3′UTR (GLO-MCL1-UTR; 22% reduction, P < 0.01), but not the luciferase-mutated-MCL1–3′UTR (GLO-MCL1-muUTR; 9% increase, P < 0.01; Fig. 2C). These results demonstrate that miR-181c indeed targets MCL1.
FIGURE 2. miR-181c targets the 3′ untranslated region (3′UTR) of myeloid cell leukemia (MCL)1.
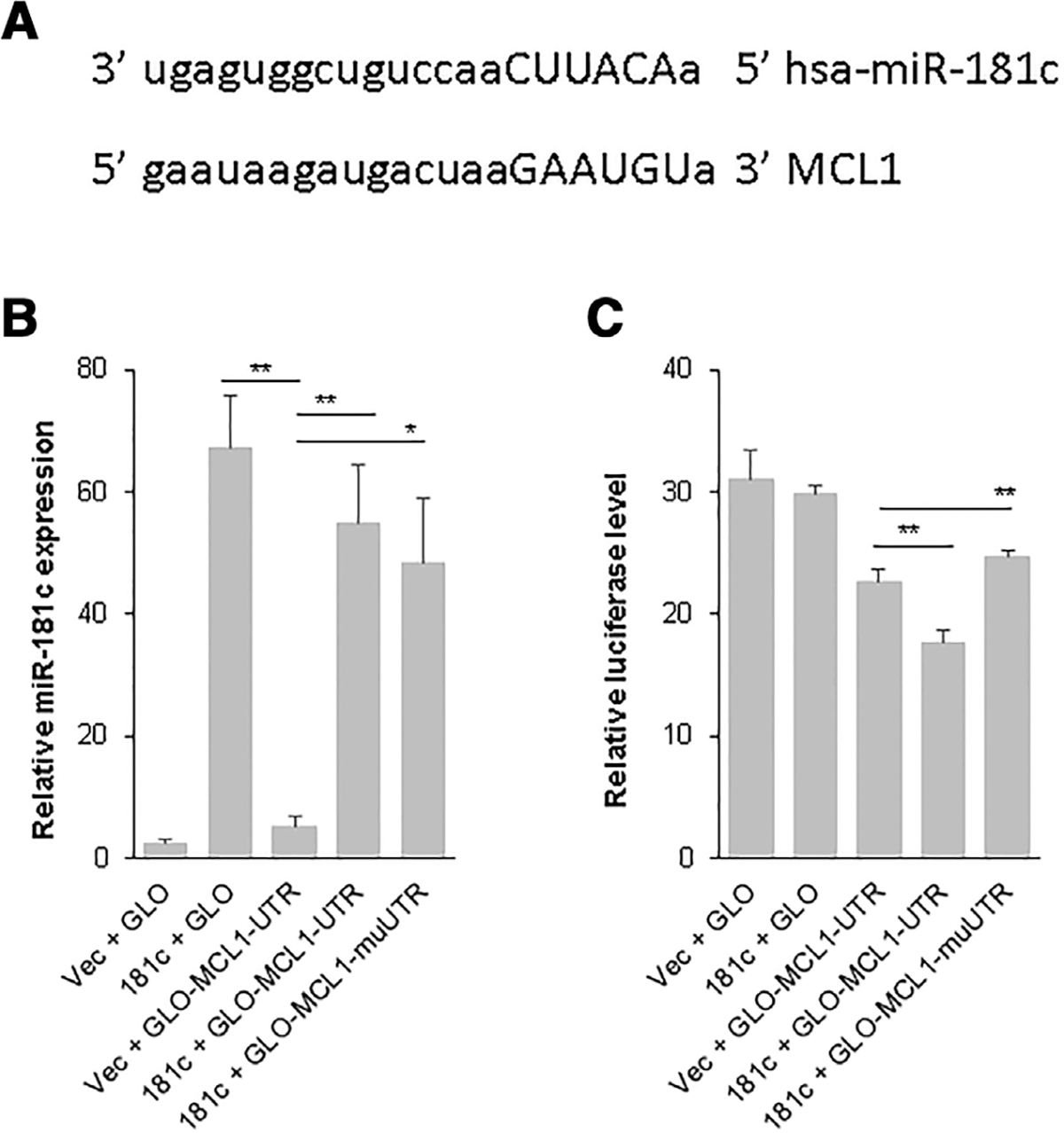
A. Putative miR-181c binding sites in the 3′UTRs of MCL1 mRNA as predicted by TargetScan. B. and C. Relative miR-181c expression (B) and relative luciferase reporter expression (C) after cotransfection with luciferase reporter plasmids containing putative wild-type (GLO-MCL1-UTR) and mutated (GLO-MCL1-muUTR) miR-181c binding sites in MCL1 3′UTR in HEK293 cells. Three repeated experiments were performed; results of technical triplicates of one set of experiments are shown. *, P < 0.05; **, P < 0.01
To further investigate the effect of miR-181c levels on MCL1 expression and apoptosis induction, GFP control vector and GFP-tagged miR-181c expressing plasmids were transfected into HL-60 cells. Live GFP-positive cells were sorted and collected using an FACS sorter. Real-time RT-PCR results indicated that there was a dramatic increase in miR-181c expression in GFP-positive HL-60 (5.1-fold, P = 0.04) cells 48 h after miR-181c transfection (Fig. 3A). A significant reduction in MCL1 mRNA (1.6-fold, P = 0.01) transcripts were observed in GFP-positive miR-181c-expressing HL-60 cells (Fig. 3B). When aliquots of same volume of transfected cells were counted for live GFP-positive HL-60 cells there was a 68% reduction of live HL-60 cells in miR-181c transfected versus GFP-vector transfected groups (Fig. 3C). Similarly, when miR-181c was transfected into SP53 cells, the MCL1 mRNA level was significantly reduced 48 h after transfection (Supporting Information Fig. S1). Taken together, these results demonstrated that miR-181c expression resulted in increased cell death by negatively regulating the anti-apoptotic protein MCL1.
FIGURE 3. miR-181c expression results in decreased myeloid cell leukemia (MCL)1 level and increased cell death.
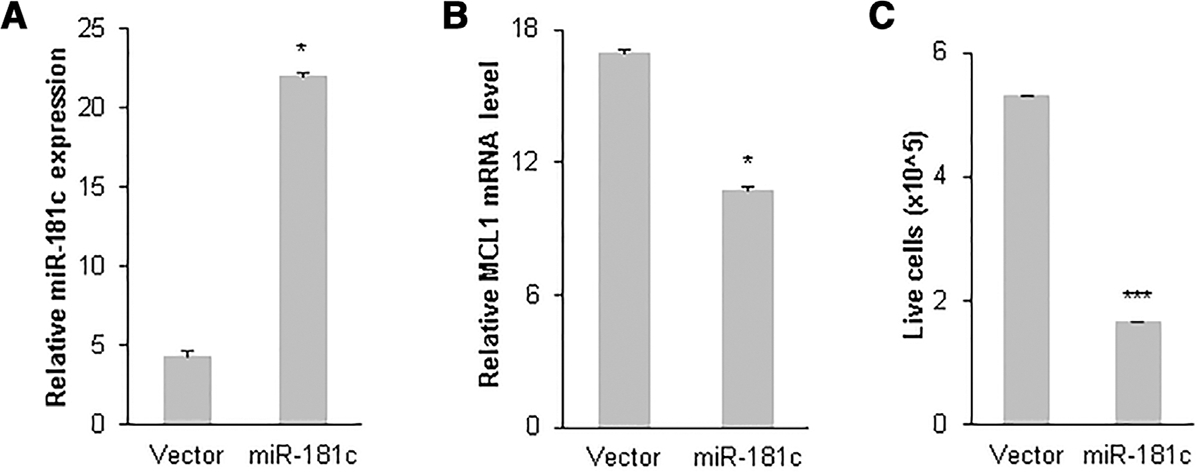
GFP-tagged miR-181c expression plasmid was transfected into HL-60 cells. GFP-positive cells were sorted and collected by FACS sorter 48 h after transfection. Increased miR-181c level (A) and decreased MCL1 mRNA level (B) were observed by quantitative real-time PCR (qRT-PCR) in GFP-positive HL-60 cells 48 h after transfection. Aliquots of same volume of transfected cells were counted for live GFP-positive HL-60 cells (C). Mean and SD of triplicate experiments are shown. **, P < 0.01
MCL1 level is decreased in bone marrow mononuclear cells from GATA2 deficiency patients with hypocellular marrow and loss of hematopoietic lineages.
Bone marrow cellularity and flow cytometric quantification of bone marrow hematopoietic lineages were assessed in 34 pretransplant GATA2 deficiency patients in comparison to 16 HCs. The median age of the GATA2 patients was 27.5 yr (range 10–66 yr) and the median age of the HCs was 39.5 yr (range 23–66 yr). All GATA2 patients had low peripheral blood counts with varying degrees of neutropenia, anemia, thrombocytopenia, lymphopenia, and monocytopenia (Supporting Information Table S3). The majority of the GATA2 patients (n = 26) were diagnosed with MDS; the remaining eight had abnormal hypocellular marrows not meeting criteria for MDS and were diagnosed with GATA2 deficiency associated bone marrow and immunodeficiency disorder. Bone marrow cellularity was significantly decreased in the GATA2 patients relative to HCs (median cellularity of 26% vs. 45%, P = 0.0002; Fig. 4A, Supporting Information Tables S4 and S5), despite the overall younger median age of the GATA2 cohort. Flow cytometry analysis of marrow aspirates demonstrated disproportionate and significant loss of the following populations in GATA2 patients in comparison to HCs (Fig. 4B–F, Supporting Information Tables S4 and S5): CD34-positive cells (median 0.32% vs. 0.9%, P < 0.001); monocytes based on expression of CD14 and CD64 (median 0.33% vs. 3.95%, P < 0.001); plasmacytoid dendritic cells (pDCs) based on expression of CD123 and HLA-DR (median 0.00% vs. 0.22%, P < 0.001); NK cells based on expression of CD56 and CD7 (not shown) and negativity for CD3 within lymphocytes (median 2.17% vs. 9.91%, P < 0.001); mature B cells based on expression of CD20 and CD19 (not shown) and negativity for CD10 within lymphocytes (median 2.39% vs. 12.42%, P < 0.001); and B cell precursors based on expression of CD10 and CD19 (not shown) within lymphocytes (median 0.01% vs. 5.84%, P < 0.001).
FIGURE 4. Loss of hematopoietic cell lineages in the bone marrow of GATA2 deficiency patients.
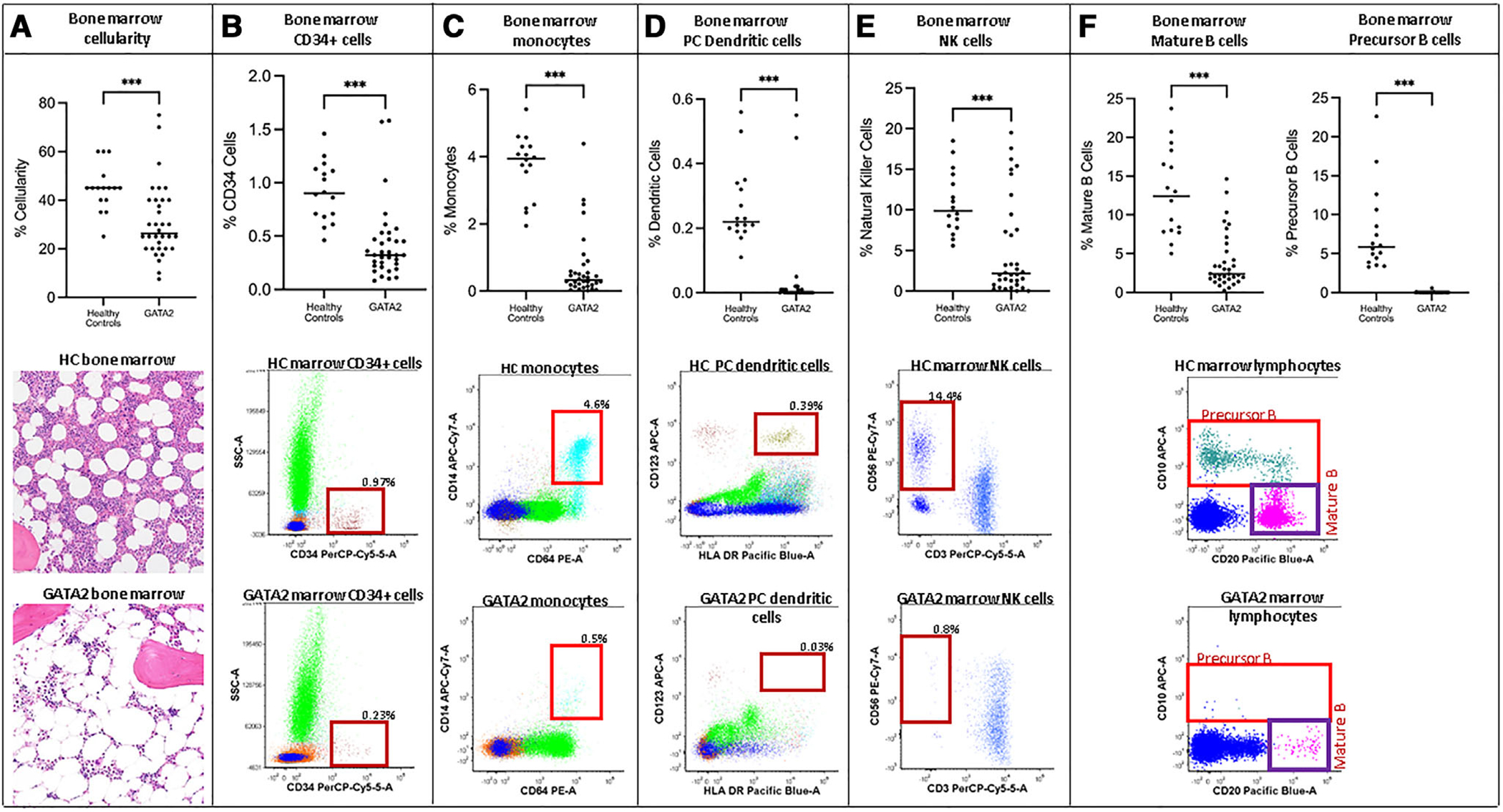
Assessment of bone marrow cellularity and flow cytometric analysis of marrow from 34 patients with GATA2 deficiency and 16 healthy controls (HCs) were performed. Bone marrow cellularity was significantly decreased in GATA2 patients (A) as shown on scatter plot and in representative images of bone marrow biopsies from GATA2 patient and HC (200×, H&E stain). Flow cytometric analysis of marrow showed CD34-positive cells are significantly decreased in GATA2 patients (B), as are monocytes (C), plasmacytoid (PC) dendritic cells (D), NK cells (E), precursor B cells and mature B cells (F). Scatter plots with cell population values from GATA2 and HC marrows are displayed on top and representative flow cytometry plots from GATA2 and HC are displayed beneath with the indicated populations highlighted within boxes on the plots. ***, P < 0.001
We then measured the level of MCL1 mRNA in BM mononuclear cells of all 34 GATA2 patients in comparison to 29 HCs (16 HCs noted earlier plus an additional 13 HCs). The qRT-PCR results demonstrated that median MCL1 mRNA levels were significantly decreased (2.5-fold, P = 0.019) in GATA2 patients (n = 34) bone marrow mononuclear cells prior to bone marrow transplantation (pre-BMT) compared with that of HCs (n = 29; Fig. 5A and Supporting Information Table S5). We then compared the MCL1 mRNA levels in 11 pairs of pre- and post-BMT bone marrow samples of GATA2 patients. It was found that MCL1 mRNA levels were also significantly lower (1.7-fold, P < 0.05) in the mononuclear cells of pre-BMT GATA2 patients (n = 11) than in post-transplant GATA2 wild-type donor bone marrow cells (Fig. 5B). These findings demonstrate loss of bone marrow cellularity and cell lineages (monocytes, B cells, B cell precursors, NK cells, and pDCs) in the marrow of GATA2 patients, and decreased levels of MCL1 in comparison to HCs. Patients who underwent BMT were transplanted with donors that were screened for GATA2 mutations and were negative. Importantly, patients who underwent BMT showed increased overall expression of MCL1 in the marrow associated with marrow reconstitution by normal donor hematopoiesis. These findings suggest that cell loss in GATA2 deficiency may be related to decreased MCL1 expression.
FIGURE 5. Median myeloid cell leukemia (MCL)1 expression levels were decreased in GATA2 patient bone marrow mononuclear cells in comparison to healthy controls (HCs) and post-transplant donor marrow.
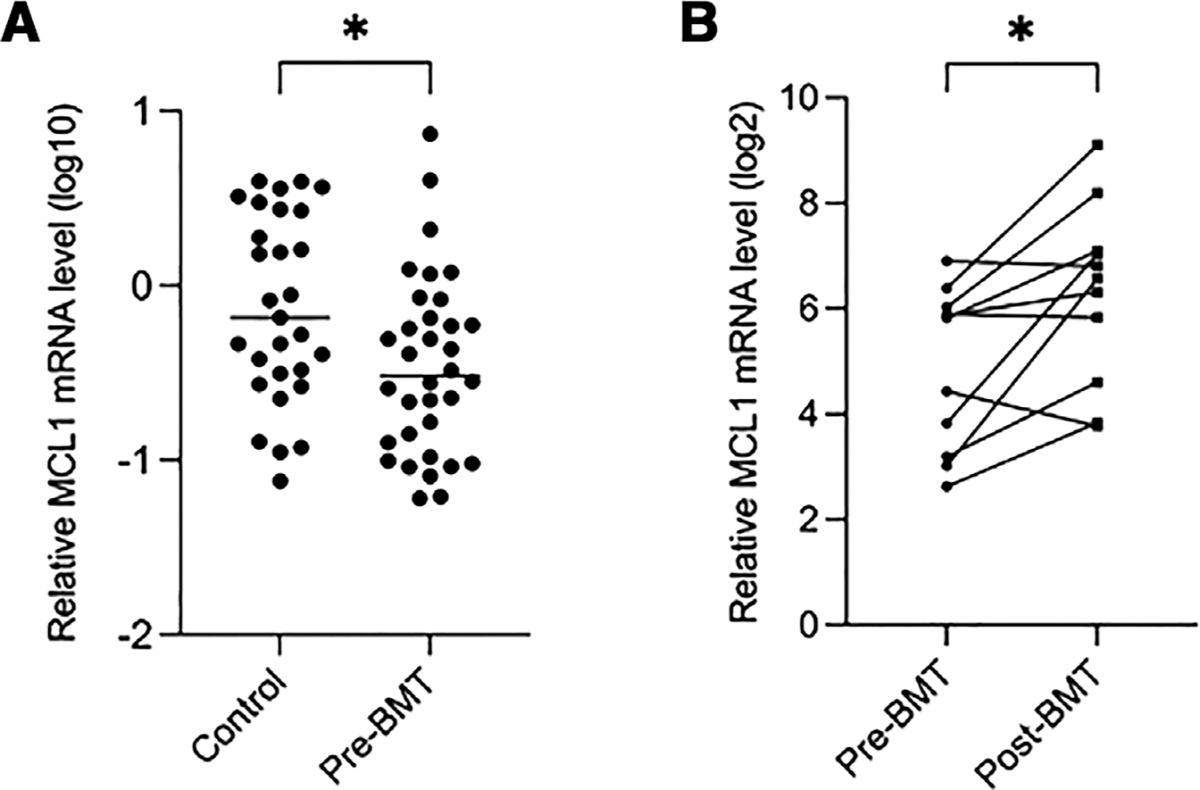
Real-time RT-PCR results showing the relative MCL1 mRNA level in bone marrow mononuclear cells of HCs (n = 29) and GATA2 patients prior to bone marrow transplantation (pre-BMT; n = 34) (A), and in paired pre- and post-BMT GATA2 patient samples (n = 11) (B). *, P < 0.05
3.3 |. miR-181c expression is repressed by transcription factor GATA2
To investigate the role of GATA2 on miR-181c expression, GATA2 expression plasmids were transiently transfected into HL-60, Ly-8, and SP53 cells. Real-time RT-PCR assays were performed to determine the transcription of miR-181c, GATA2, and the known GATA2 target gene, p53, 48 h after transfection. As shown in Figure 5A, in HL-60 cells, GATA2 transcription was greatly increased (4000-fold, P < 0.01) after transfection. Increased GATA2 expression resulted in significant inhibition of miR-181c transcription (3.5-fold, P < 0.01). When the transcription of the known GATA2 target gene, p53, was evaluated, it was found that p53 expression was slightly activated (1.1-fold, P = 0.02) by GATA2 (Fig. 6A). Similarly, when GATA2 was transiently expressed in Ly-8 cells, increased GATA2 transcription was observed (2253-fold, P < 0.01), miR-181c transcription was significantly inhibited (1.5-fold, P = 0.02), and p53 expression was significantly activated (2.3-fold, P = 0.02) by GATA2 48 h after transfection (Fig. 6B). A similar pattern of miR-181c repression by GATA2 was also observed in SP53 cells transiently transfected by GATA2 (Fig. 6C).
FIGURE 6. GATA2 expression inhibits miR-181c expression.
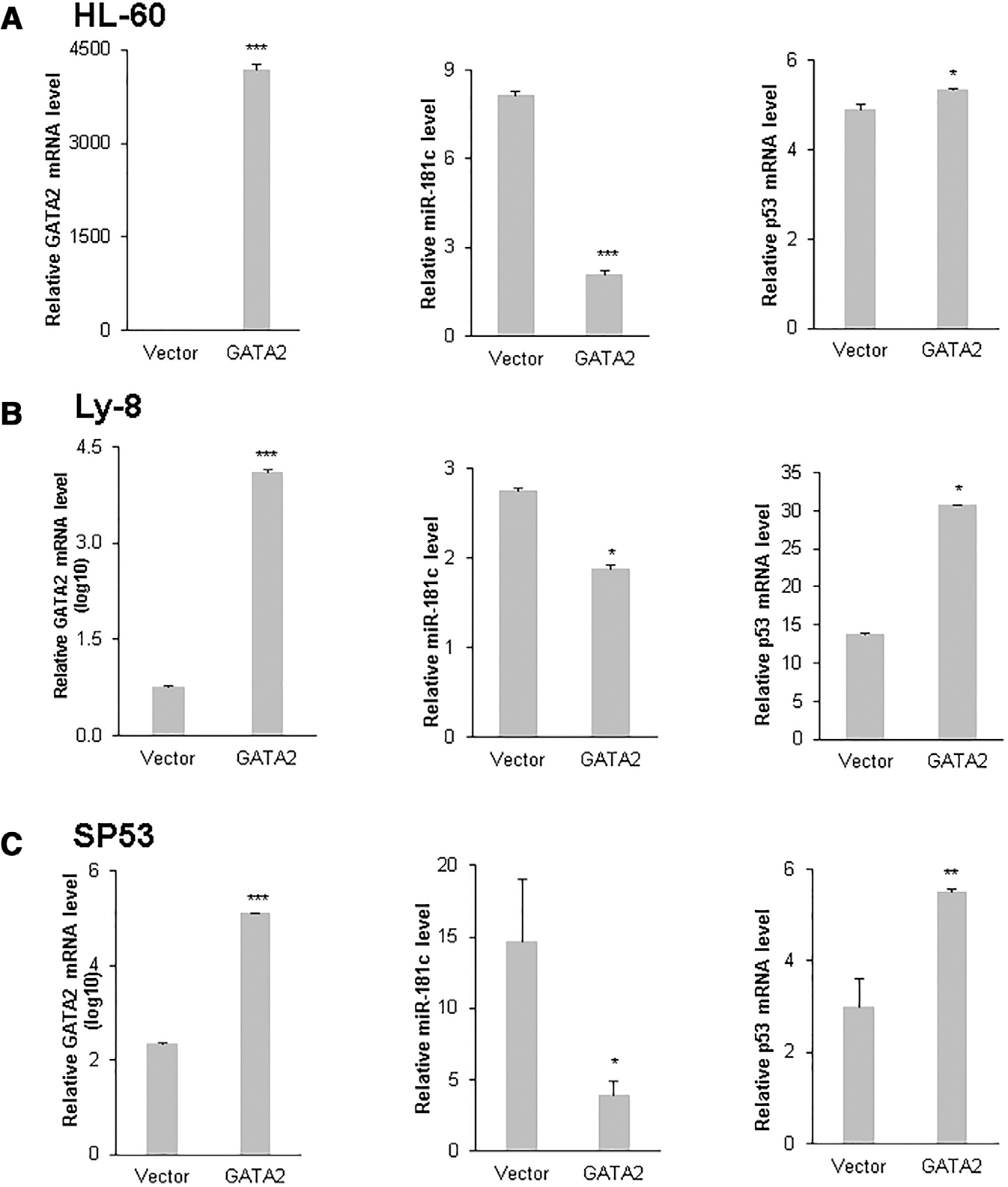
Vector and GATA2 expression plasmids were transfected into HL-60 (A), Ly-8 (B) and SP53 (C) cells. Quantitative RT-PCR for mRNA level of GATA2, miR-181c, and p53 was performed 48 h after transfection. Mean and SD of triplicate experiments are shown. *, P < 0.05; **, P < 0.01; ***, P < 0.001
To further confirm the inhibitory effect of GATA2 on miR-181c transcription, RNA interference experiments were performed in the human B cell lymphoma cell line SP53 and the human erythroleukemic cell line K562. The scrambled control vector and lentiviral vectors expressing short hairpin RNA against GATA2 (shGATA2) were transfected into SP53 and K562 cell lines; real-time PCR results demonstrated that endogenous GATA2 was effectively knocked down by short-hairpin RNA against GATA2 (shGATA2) in SP53 cells (5.9-fold, P < 0.05; Fig. 7A), and knockdown of GATA2 expression resulted in a significant increase in miR-181c transcription (2.4-fold, P < 0.001; Fig. 7B). Similarly, in K562 cells, shGATA2 effectively knocked down endogenous GATA2 expression (2.4-fold, P < 0.05; Fig. 7C), and this resulted in a significant increase in miR-181c transcription (1.8-fold, P < 0.05; Fig. 7D). Taken together, these results demonstrated that transcription factor GATA2 represses the transcription of miR-181c. In GATA2 deficient cells, reduced GATA2 expression resulted in increased levels of miR-181c.
FIGURE 7. Knockdown of endogenous GATA2 results in increased miR-181c expression.
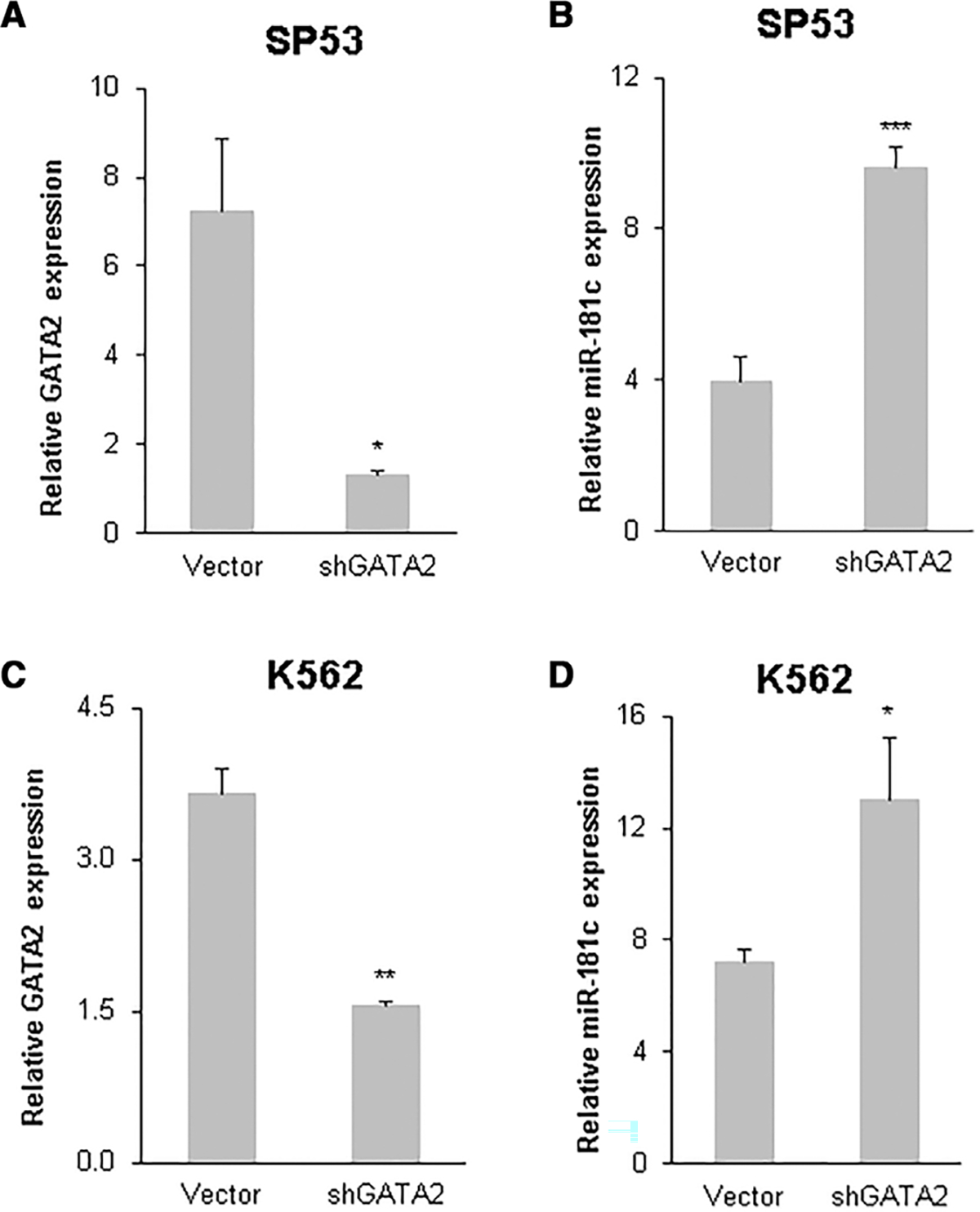
The endogenous GATA2 level was knocked down by RNA interference. Short hairpin RNA against GATA2 (shGATA2) plasmids were transfected into SP53 and K562 cells. Quantitative RT-PCR was performed 48 h after transfection for miR-181c and GATA2 in SP53 (A and B) and K562 (C and D) cells. Mean and SD of triplicate experiments are shown. *, P < 0.05; **, P < 0.01
4 |. DISCUSSION
GATA2 deficiency is manifested by loss of multiple cellular linages in the bone marrow (CD34+ cells, monocytes, B cells, NK cells, and pDCs) leading to bone marrow failure and immunodeficiency. Subcloning and functional testing of three miRNAs from miRNA profiling in GATA2 deficiency cell lines indicated that miR-181c induces cell death in lymphoid and myeloid cell lines studies. This prompted us to further study the potential mechanisms of miR-181c regulation by GATA2 and its role in cell survival in GATA2 deficient environment. In experiments with myeloid and lymphoid cell lines, and together with our unsuccessful attempts to establish miR-181c stable cell lines (in contrast, vector control stable cell lines could be easily established), we observed that miR-181c is likely a potent cell death inducer in these cell lines. We did measure a lower percentage of dead cells in SP53 cells in comparison to HL60 and LY8 cells, which could be due to several factors including robust growth state of SP53 cells, efficiency of transfection, or relatively higher level of endogenous anti-apoptotic proteins. Our limited ability to quantify cell death induction by mir-181c in this study could have been impacted by poor transfection efficiency in suspended cells, the amount of DNA added, or the timing of the assay. Similarly, the slight increase in p53 expression in HL-60 cells, compared with relatively stronger increase in p53 in Ly-8 and SP53 cells, could be due to the difference of background p53 expression levels and levels of transcription cofactors of GATA2 in these cell lines.
The wide range of MCL1 expression levels in the bone marrow of GATA2 patients could be due to diverse genetic background and/or physiologic conditions. Patients from the same family, with the same GATA2 mutation, often display different spectrums of disease manifestations, severity, and rate of disease progression, the etiology of which is not entirely understood.
Even though cytopenia is one of hallmark symptoms of GATA2 deficiency, it is also highly variable and may even be completely absent in asymptomatic carriers. GATA2 expression, methylation, mutation status, and acquired somatic mutations may lead to variable disease manifestations in patients. Thus, apoptosis levels—the level of expression of pro-survival/anti-apoptotic BCL2 family of proteins (BCL-2, BCL-xL, MCL-1, BCL-W, and BFL1) and proteins of pro-apoptotic effectors (BAK, BAX, BOK), BH3-only activators (BIM, BID, and PUMA), and sensitizers (NOXA, BAD, bone marrow failure, BIK, and Hrk) in vivo in GATA2 patients could be variable. Mechanisms of cell loss, cell death, and cytopenia in GATA2 deficiency remain to be elucidated. In this study, we provided evidence that the aberrant expression of miR-181c and MCL1, and possibly BCL2, may contribute to cell death and cytopenia in GATA2 deficiency.
A number of studies have implicated miRNAs in hematopoiesis and hematologic malignancies.24–29 Interestingly, several miRNAs identified in this study, for example, miR-181, miR-223, and miR-9, have been associated with hematopoietic cell differentiation and hematologic disease.24–26,33–35 miR-223 was reported to direct lineage choice of hematopoietic progenitors36 and attenuate hematopoietic cell proliferation.37 miR-223 deficiency increases eosinophil progenitor proliferation.38 miR-9 has been implicated in pediatric AML with t(8;21)39 and in myelopoiesis.40 Previously, we found that miR-9 potentially regulates germinal center B cell differentiation and lymphomagenesis by targeting BLIMP1 (PRDM1) in follicular lymphoma cells.29
The miR-181 family of miRNAs has been reported to play important roles in regulating apoptosis, tumorigenesis and hematopoietic progenitor cell differentiation, and malignancy.24–26 The miR-181 miRNAs are preferentially expressed in the B-lymphoid cells and they modulate hematopoietic lineage differentiation.41 The miR-181 family is overexpressed in AML, and has been suggested to be a potentially useful diagnostic marker and prognostic predictor in AML and MDS.33–35 The miR-181 miRNAs inhibit myeloid differentiation by targeting the expression of PRKCD, CTDSPL, and CAMKK1, affecting the p38/CEBPa and the RB pathways in AML.34 Down-regulation of miR-181 and up-regulation of a HOXA-PBX3 homeobox-gene signature is associated with adverse prognosis in patients with cytogenetically abnormal AML.42 Another study found that miR-181 family miRNAs were overexpressed in high-risk MDS specimens and their expression correlates with the survival of patients with low-risk MDS.43 Higher levels of miR-181a/b are associated with prolonged overall survival in patients with hematologic malignancies, but not with other cancers.44 The miR-181a/b were found to be down-regulated in CLL and gliomas and low expression of miR-181a/b was associated with poor prognosis or malignancy.45,46 One study profiling the aberrant expression of miRNAs in imatinib-resistant CML cells found that the expression of the miR-181 family (a–d) was significantly reduced in drug-resistant cells, and miR-181b modulated the imatinib-resistance by targeting MCL1.47 Other studies also found that miR-181a/b enhance apoptosis by targeting MCL1, BCL2, XIAP, and TCL1 proteins.45,46,48,49 Consistent with these findings, we found that miR-181c targets MCL1 in this study.
MCL1 is a member of the BCL2 family, and plays important roles in regulating human stem cell survival and self-renewal.50–52 MCL1 is an anti-apoptotic protein, binding and sequestering pro-apoptotic effector, activator, and sensitizer proteins and has been heavily studied.53,54 Inducible deletion of Mcl-1 during early lymphocyte differentiation led to apoptosis and rapid loss of T and B cell population.55 Systemic deletion of MCL-1 in murine embryonic stem (ES) cells resulted in embryonic lethality (Mcl-1 deficiency results in peri-implantation embryonic lethality).56 Tumor formation studies in mouse models indicated that the anti-apoptotic function of MCL-1 is essential in MYC-driven lymphomagenesis,57 AML, T cell lymphomas, and breast cancer.58,59 Deletion of MCL1 has been shown to cause apoptosis, loss of hematopoietic stem cells, and cytopenias.50,55 Recent studies using CRISPR/Cas9 genome-wide screening in a large number of tumor cell lines have further demonstrated solid tumors mainly depend on MCL1, and hematologic malignancies depend on both MCL-1 and BCL-2 for tumor cell survival (www.depmap.org).54,60 In light of these, we did not examine the function of MCL1 as an anti-apoptotic protein or its proven role of rescuing cells from cell death. Instead, in this study we focused on abnormal expression of miR-181c in GATA2 deficient environment, the potential regulation MCL1 by miR-181c, and its potential influence on cell survival in GATA2 deficient cells.
There are two putative miR-181c binding sites in the 3′UTR of BCL2 mRNA. Our preliminary data also indicated that miR-181c targets BCL2. However, it was challenging to measure changes in BCL2 for several reasons: (i) BCL2 levels were very high at baseline in the cell lines we used and (ii) BCL-2 expression is normally high in T cells in the bone marrow and T cells are generally preserved in GATA2 deficiency. Even though this study was centered on the expression and regulation of GATA2/miR-181c/MCL1, it is highly likely that BCL2 is regulated by miR-181c in a similar fashion as to MCL1, contributing to cell survival. Future studies on the role of miR-181c and BCL2 in cytopenic cell lineages of GATA2 deficiency may be warranted.
Our study showed aberrant expression of miRNAs in GATA2 deficient cells in comparison to controls. GATA2 has been reported to regulate miRNA expression in disease and cancer. In immature erythroid progenitors, GATA2 occupies miR-27a and miR-24 promoters, repressing their transcription; as erythropoiesis proceeds, GATA1 replaces GATA2 and activates miR-27a and miR-24 transcription, which then inhibits GATA2 translation, facilitating the replacement of GATA2 by GATA1 in a positive feedback manner.61 In another study, the PU.1 transcription factor, also a GATA2 target gene, was found to regulate monocyte/macrophage differentiation by activating miR-22 in AML.62 GATA2 gain- and loss-of-function experiments in human umbilical vein endothelial cells indicated that GATA2 overexpression leads to increased proangiogenic miR-126, which targets antiangiogenic SPRED1 and FOXO3a proteins. GATA2 silencing results in DNA hypomethylation of miR-221 promoter leading to induced expression of antiangiogenic miR-221, which targets proangiogenic ICAM1 and ETS1 proteins, potentially playing a role in vascular disease manifestations in GATA2 deficiency.63
One study reported that TGF-b/SMAD2/3 signaling directly regulates the transcription of pri-miR-181c/d in mouse embryonic stem cells.64 Others found that miR-181c was epigenetically silenced by promoter methylation in gastric cancer.65 The promoter region of miR-181c has not been defined and studied. Little is known regarding the regulation of miR-181c expression. GATA2 is a master transcriptional regulator, ChIP-Seq data66 from ENCODE/UCSC Genome Browser indicates that there is a GATA2-binding cluster 170 bp upstream of miR-181c. One report predicted that the putative transcriptional start site of miR-181c might be 9 kb upstream of the miR-181c precursor.67 We found that miR-181c was repressed by enforced GATA2 expression and was increased in cells derived from patients with GATA2 deficiency. Considering the complexity of miR-181c transcriptional start site and promoter regulation, miR-181c could be regulated by GATA2 directly or indirectly by GATA2 controlled transcription factors or promoter methylation mechanisms. Given that miR-181c is a potent inducer of cell death in the lymphoid and myeloid cell lines in this study, increased levels of miR-181c in the setting of GATA2 deficiency, combined with reduced MCL1, may contribute to cell loss and immune deficiencies associated with disease.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the NIH Division of Intramural Research of the NIH Clinical Center, National Institute of Allergy and Infectious Diseases, and the National Cancer Institute. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The authors thank Dr. Masanori Daibata (Kochi University Medical School, Kochi, Japan) for use of the SP53 cell line and Dr. Mark Minden (Ontario Cancer Institute, Toronto, Ontario, Canada) for generously providing the OCI-Ly-8 cell line.
Abbreviations:
- 3′UTR
3′ untranslated region
- AML
acute myeloid leukemia
- BMT
bone marrow transplantation
- GATA2
GATA binding protein 2
- HC
healthy control
- MCL1
myeloid cell leukemia-1 gene
- MDS
myelodysplastic syndrome
- miRNA
microRNA
- pDCs
plasmacytoid dendritic cells
- shGATA2
short hairpin RNA against GATA2
Footnotes
DISCLOSURES
The authors declare no conflicts of interest.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
REFERENCES
- 1.Tsai FY, Orkin SH. Transcription factor GATA-2 is required for proliferation/survival of early hematopoietic cells and mast cell formation, but not for erythroid and myeloid terminal differentiation. Blood. 1997;89:3636–3643. [PubMed] [Google Scholar]
- 2.Vicente C, Conchillo A, García-Sánchez MA, Odero MD. The role of the GATA2 transcription factor in normal and malignant hematopoiesis. Crit Rev Oncol Hematol. 2012;82:1–17. [DOI] [PubMed] [Google Scholar]
- 3.Jung M, Cordes S, Zou J, et al. GATA2 deficiency and human hematopoietic development modeled using induced pluripotent stem cells. Blood Adv. 2018;2:3553–3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson KD, Hsu AP, Ryu MJ, et al. Cis-element mutated in GATA2-dependent immunodeficiency governs hematopoiesis and vascular integrity. J Clin Invest. 2012;122:3692–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Menendez-Gonzalez JB, Vukovic M, Abdelfattah A, et al. Gata2 as a crucial regulator of stem cells in adult hematopoiesis and acute myeloid leukemia. Stem Cell Rep. 2019;13:291–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yaguchi T, Nakano T, Gotoh A, Nishizaki T. Adenosine promotes GATA-2-regulated p53 gene transcription to induce HepG2 cell apoptosis. Cell Physiol Biochem. 2011;28:761–770. [DOI] [PubMed] [Google Scholar]
- 7.Bonadies N, Göttgens B, Calero-Nieto FJ. The LMO2 −25 region harbours GATA2-dependent myeloid enhancer and RUNX-dependent T-lymphoid repressor activity. PloS One. 2015;10:e0131577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsu AP, Sampaio EP, Khan J, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood. 2011;118:2653–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dickinson RE, Griffin H, Bigley V, et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood. 2011;118:2656–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ostergaard P, Simpson MA, Connell FC, et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat Genet. 2011;43:929–931. [DOI] [PubMed] [Google Scholar]
- 11.Hahn CN, Chong CE, Carmichael CL, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43:1012–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spinner MA, Sanchez LA, Hsu AP, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood. 2014;123:809–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weinberg OK, Kuo F, Calvo KR. Germline predisposition to hematolymphoid neoplasia. Am J Clin Pathol. 2019;152:258–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ganapathi KA, Townsley DM, Hsu AP, et al. GATA2 deficiency-associated bone marrow disorder differs from idiopathic aplastic anemia. Blood. 2015;125:56–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calvo KR, Vinh DC, Maric I, et al. Myelodysplasia in autosomal dominant and sporadic monocytopenia immunodeficiency syndrome: diagnostic features and clinical implications. Haematologica. 2011;96:1221–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wlodarski MW, Collin M, Horwitz MS. GATA2 deficiency and related myeloid neoplasms. Semin Hematol. 2017;54:81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parta M, Shah NN, Baird K, et al. Allogeneic hematopoietic stem cell transplantation for GATA2 deficiency using a busulfan-based regimen. Biol Blood Marrow Transplant. 2018;24:1250–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ambros V The functions of animal microRNAs. Nature. 2004;431:350–355. [DOI] [PubMed] [Google Scholar]
- 20.Di Leva G, Croce CM. Roles of small RNAs in tumor formation. Trends Mol Med. 2010;16:257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iorio MV, Ferracin M, Liu CG, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. [DOI] [PubMed] [Google Scholar]
- 22.Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. [DOI] [PubMed] [Google Scholar]
- 23.Zuo Z, Calin GA, de Paula HM, et al. Circulating microRNAs let-7a and miR-16 predict progression-free survival and overall survival in patients with myelodysplastic syndrome. Blood. 2011;118:413–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramkissoon SH, Mainwaring LA, Ogasawara Y, et al. Hematopoietic-specific microRNA expression in human cells. Leuk Res. 2006;30:643–647. [DOI] [PubMed] [Google Scholar]
- 25.Merkerova M, Belickova M, Bruchova H. Differential expression of microRNAs in hematopoietic cell lineages. Eur J Haematol. 2008;81:304–310. [DOI] [PubMed] [Google Scholar]
- 26.Yu J, Wang F, Yang GH, et al. Human microRNA clusters: genomic organization and expression profile in leukemia cell lines. Biochem Biophys Res Commun. 2006;349:59–68. [DOI] [PubMed] [Google Scholar]
- 27.Starczynowski DT, Kuchenbauer F, Argiropoulos B, et al. Identification of miR-145 and miR-146a as mediators of the 5q- syndrome phenotype. Nat Med. 2010;16:49–58. [DOI] [PubMed] [Google Scholar]
- 28.Saleh LM, Wang W, Herman SE, et al. Ibrutinib downregulates a subset of miRNA leading to upregulation of tumor suppressors and inhibition of cell proliferation in chronic lymphocytic leukemia. Leukemia. 2017;31:340–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang W, Corrigan-Cummins M, Hudson J, et al. MicroRNA profiling of follicular lymphoma identifies microRNAs related to cell proliferation and tumor response. Haematologica. 2012;97:586–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang H, Blondal JA, Benchimol S, Minden MD, Messner HA. mutations, c-myc and bcl-2 rearrangements in human non-Hodgkin’s lymphoma cell lines. Leuk Lymphoma. 1995;19:165–171. [DOI] [PubMed] [Google Scholar]
- 31.Daibata M, Kubonishi I, Eguchi T, Yano S, Ohtsuki Y, Miyoshi I. The establishment of Epstein-Barr virus nuclear antigen-positive (SP-50B) and Epstein-Barr virus nuclear antigen-negative (SP-53) cell lines with t(11;14)(q13;q32) chromosome abnormality from an intermediate lymphocytic lymphoma. Cancer. 1989;64:1248–1253. [DOI] [PubMed] [Google Scholar]
- 32.Lau P, Verrier JD, Nielsen JA, Johnson KR, Notterpek L, Hudson LD. Identification of dynamically regulated microRNA and mRNA networks in developing oligodendrocytes. J Neurosci. 2008;28:11720–11730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pons A, Nomdedeu B, Navarro A, et al. Hematopoiesis-related microRNA expression in myelodysplastic syndromes. Leuk Lymphoma. 2009;50:1854–1859. [DOI] [PubMed] [Google Scholar]
- 34.Su R, Lin HS, Zhang XH, et al. MiR-181 family: regulators of myeloid differentiation and acute myeloid leukemia as well as potential therapeutic targets. Oncogene. 2015;34:3226–3239. [DOI] [PubMed] [Google Scholar]
- 35.Weng H, Lal K, Yang FF, Chen J. The pathological role and prognostic impact of miR-181 in acute myeloid leukemia. Cancer Genet. 2015;208:225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vian L, Di Carlo M, Pelosi E, et al. Transcriptional fine-tuning of microRNA-223 levels directs lineage choice of human hematopoietic progenitors. Cell Death Differ. 2014;21:290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun W, Shen W, Yang S, Hu F, Li H, Zhu TH. miR-223 and miR-142 attenuate hematopoietic cell proliferation, and miR-223 positively regulates miR-142 through LMO2 isoforms and CEBP-beta. Cell Res;20:1158–1169. [DOI] [PubMed] [Google Scholar]
- 38.Lu TX, Lim EJ, Besse JA, et al. MiR-223 deficiency increases eosinophil progenitor proliferation. J Immunol. 2013;190:1576–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emmrich S, Katsman-Kuipers JE, Henke K, et al. miR-9 is a tumor suppressor in pediatric AML with t(8;21). Leukemia. 2014;28:1022–1032. [DOI] [PubMed] [Google Scholar]
- 40.Senyuk V, Zhang Y, Liu Y, et al. Critical role of miR-9 in myelopoiesis and EVI1-induced leukemogenesis. Proc Natl Acad Sci U S A. 2013;110:5594–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–86. [DOI] [PubMed] [Google Scholar]
- 42.Li Z, Huang H, Li Y, et al. Up-regulation of a HOXA-PBX3 homeobox-gene signature following down-regulation of miR-181 is associated with adverse prognosis in patients with cytogenetically abnormal AML. Blood. 2012;119:2314–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sokol L, Caceres G, Volinia S, et al. Identification of a risk dependent microRNA expression signature in myelodysplastic syndromes. Br J Haematol. 2011;153:24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin S, Pan L, Guo S, et al. Prognostic role of microRNA-181a/b in hematological malignancies: a meta-analysis. PloS One. 2013;8:e59532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu DX, Zhu W, Fang C, et al. miR-181a/b significantly enhances drug sensitivity in chronic lymphocytic leukemia cells via targeting multiple anti-apoptosis genes. Carcinogenesis. 2012;33:1294–1301. [DOI] [PubMed] [Google Scholar]
- 46.Shi L, Cheng Z, Zhang J, et al. hsa-mir-181a and hsa-mir-181b function as tumor suppressors in human glioma cells. Brain Res. 1236:185–193. [DOI] [PubMed] [Google Scholar]
- 47.Zimmerman EI, Dollins CM, Crawford M, et al. Lyn kinase-dependent regulation of miR181 and myeloid cell leukemia-1 expression: implications for drug resistance in myelogenous leukemia. Mol Pharmacol. 2010;78:811–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ouyang YB, Lu Y, Yue S, Giffard RG. miR-181 targets multiple Bcl-2 family members and influences apoptosis and mitochondrial function in astrocytes. Mitochondrion. 2012;12:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bresin A, Callegari E, D’Abundo L, et al. miR-181b as a therapeutic agent for chronic lymphocytic leukemia in the Emicro-TCL1 mouse model. Oncotarget. 2015;6:19807–19818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Opferman JT, Iwasaki H, Ong CC, et al. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science. 2005;307:1101–1104. [DOI] [PubMed] [Google Scholar]
- 51.Campbell CJ, Lee JB, Levadoux-Martin M, et al. The human stem cell hierarchy is defined by a functional dependence on Mcl-1 for self-renewal capacity. Blood. 2010;116:1433–1442. [DOI] [PubMed] [Google Scholar]
- 52.Zhou P, Qian L, Kozopas KM, Craig RW. Mcl-1, a Bcl-2 family member, delays the death of hematopoietic cells under a variety of apoptosis-inducing conditions. Blood. 1997;89:630–643. [PubMed] [Google Scholar]
- 53.Senichkin VV, Streletskaia AY, Zhivotovsky B, Kopeina GS. Molecular comprehension of Mcl-1: from gene structure to cancer therapy. Trends Cell Biol. 2019;29:549–562. [DOI] [PubMed] [Google Scholar]
- 54.Bolomsky A, Vogler M, Köse MC, et al. MCL-1 inhibitors, fast-lane development of a new class of anti-cancer agents. J Hematol Oncol. 2020;13:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires anti-apoptotic MCL-1. Nature. 2003;426:671–676. [DOI] [PubMed] [Google Scholar]
- 56.Rinkenberger JL, Horning S, Klocke B, Roth K, Korsmeyer SJ. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev. 2000;14:23–27. [PMC free article] [PubMed] [Google Scholar]
- 57.Grabow S, Delbridge AR, Aubrey BJ, Vandenberg CJ, Strasser A. Loss of a single Mcl-1 allele inhibits MYC-driven lymphomagenesis by sensitizing Pro-B cells to. Apoptosis Cell Rep. 2016;14:2337–2347. [DOI] [PubMed] [Google Scholar]
- 58.Glaser SP, Lee EF, Trounson E, et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012;26:120–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grabow S, Delbridge AR, Valente LJ, Strasser A. MCL-1 but not BCL-XL is critical for the development and sustained expansion of thymic lymphoma in p53-deficient mice. Blood. 2014;124:3939–3946. [DOI] [PubMed] [Google Scholar]
- 60.Tsherniak A, Vazquez F, Montgomery PG, et al. Defining a cancer dependency map. Cell. 2017;170:564–576.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang F, Zhu Y, Guo L, et al. A regulatory circuit comprising GATA1/2 switch and microRNA-27a/24 promotes erythropoiesis. Nucleic Acids Res. 2014;42:442–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen C, Chen MT, Zhang XH, et al. The PU.1-modulated microRNA-22 is a regulator of monocyte/macrophage differentiation and acute myeloid leukemia. PLoS Genet. 2016;12:e1006259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hartmann D, Fiedler J, Sonnenschein K, et al. MicroRNA-based therapy of GATA2-deficient vascular disease. Circulation. 2016;134:1973–1990. [DOI] [PubMed] [Google Scholar]
- 64.Redshaw N, Camps C, Sharma V, et al. TGF-beta/Smad2/3 signaling directly regulates several miRNAs in mouse ES cells and early embryos. PloS One. 2013;8:e55186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hashimoto Y, Akiyama Y, Otsubo T, Shimada S, Yuasa Y. Involvement of epigenetically silenced microRNA-181c in gastric carcinogenesis. Carcinogenesis. 2010;31:777–784. [DOI] [PubMed] [Google Scholar]
- 66.The ENCODE Project Consortium: an integrated encyclopedia of DNA elements in the human genome. Nature. 20162;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chien CH, Sun YM, Chang WC, et al. Identifying transcriptional start sites of human microRNAs based on high-throughput sequencing data. Nucleic Acids Res. 2011;39:9345–9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw array data and data normalized by control gene probes were deposited in the GEO database with accession number GSE51132. All other data are contained within this article.