

ABSTRACT
Human cytomegalovirus (HCMV) is a beta herpesvirus that persists indefinitely in the human host through a latent infection. The polycistronic UL133–UL138 gene locus of HCMV encodes genes regulating latency and reactivation. While UL138 is pro-latency, restricting virus replication in CD34+ hematopoietic progenitor cells (HPCs), UL135 overcomes this restriction and is required for reactivation. By contrast, UL136 is expressed with later kinetics and encodes multiple proteins with differential roles in latency and reactivation. Like UL135, the largest UL136 isoform, UL136p33, is required for reactivation from latency in HPCs; viruses failing to express either protein are unresponsive to reactivation stimuli. Furthermore, UL136p33 is unstable, and its instability is important for the establishment of latency, and sufficient accumulation of UL136p33 is a checkpoint for reactivation. We hypothesized that stabilizing UL136p33 might overcome the requirement of UL135 for replication. We generated recombinant viruses lacking UL135 that expressed a stabilized variant of UL136p33. Stabilizing UL136p33 did not impact the replication of the UL135 mutant virus in fibroblasts. However, in the context of infection in HPCs, stabilization of UL136p33 strikingly compensated for the loss of UL135, resulting in increased replication in CD34+ HPCs and in humanized NOD-scid IL2Rγcnull (huNSG) mice. This finding suggests that while UL135 is essential for replication in HPCs, it functions largely at steps preceding the accumulation of UL136p33, and that stabilized expression of UL136p33 largely overcomes the requirement for UL135. Taken together, our genetic evidence indicates an epistatic relationship between UL136p33 and UL135, whereby UL135 may initiate events early in reactivation that drive the accumulation of UL136p33 to a threshold required for productive reactivation.
IMPORTANCE
Human cytomegalovirus (HCMV) is one of nine human herpesviruses and a significant human pathogen. While HCMV establishes a lifelong latent infection that is typically asymptomatic in healthy individuals, its reactivation from latency can have devastating consequences in the immunocompromised. Defining viral genes important in the establishment of or reactivation from latency is important to defining the molecular basis of latent and replicative states and in controlling infection and CMV disease. Here we define a genetic relationship between two viral genes in controlling virus reactivation from latency using primary human hematopoietic progenitor cells and humanized mouse models.
KEYWORDS: cytomegalovirus, herpesvirus, latency, reactivation, UL135, UL136
INTRODUCTION
The molecular programs by which herpesviruses persist in a latent state or reactivate for replication are poorly understood. DNA herpesviruses are a robust model for elucidating mechanisms of viral latency because of the complex virus–host interactions that allow the virus to “sense” and “respond” to changes in host biology. Human cytomegalovirus (HCMV) is a large ~236 kb double-stranded DNA beta herpesvirus with a global seroprevalence of approximately 60% to 99% based on geographic and socioeconomic factors (1 - 3). Infection is typically asymptomatic in immunocompetent individuals. Once latency is established, subclinical sporadic reactivation and virus shedding can occur throughout the lifetime of the host (4, 5). For immunocompromised individuals with inadequate cellular immunity, reactivation from latency can result in morbidity and mortality, particularly in stem cell and solid organ transplant recipients (4 - 8). Further, congenital CMV infection can cause significant neurodevelopmental abnormalities, affecting approximately 1 in 150 babies born in the United States (9 - 15). HCMV infection may enhance immune responses in young adults but may be a driver in age-related pathologies and altered T cell homeostasis (16 - 18). Therefore, understanding the viral and host mechanisms underpinning HCMV persistence and reactivation is critical to developing strategies to control its reactivation and associated pathologies.
HCMV exhibits broad intra-host tropism. HCMV replicates productively in human primary fibroblasts and in epithelial and endothelial cells. HCMV latency has been best characterized in hematopoietic progenitor cells (HPCs; CD34+ HPCs) and cells of the myeloid lineage (19). During latency, viral genomes are maintained, viral gene expression is restricted, and no new viral progeny are made. HCMV reactivates in response to viral and host cues, including cellular stress, inhibition of PI3K/Akt signaling, differentiation, or steroid treatment to re-initiate viral gene expression (4, 20 - 26). Viruses have evolved complex gene networks to sense and respond to multiple environmental stimuli that can feedback to impact viral gene expression (19, 27, 28).
The ~15 kb ULb’ region of the HCMV genome spans UL133 to UL150 and encodes genes important to the regulation of immune responses, viral persistence, and dissemination (4, 29 - 33). The ULb’ region is present in low-passage strains and clinical isolates but lost during serial passage of the virus in fibroblasts, thus partly or entirely lacking in most laboratory-adapted strains. While largely dispensable for replication in fibroblasts, these genes undoubtedly play important roles in other contexts of infection in the host (34 - 37). We have characterized a 3.6 kb polycistronic gene locus within the ULb’ region encoding four genes, UL133, UL135, UL136, and UL138, collectively referred to as the UL133–UL138 locus (19, 38). On the whole, the UL133–UL138 locus is suppressive to viral replication in HPCs (34, 35). Using recombinant viruses containing disruptions of a single gene or combinations of genes in this locus, we have defined genetic phenotypes for each gene with respect to the establishment of or reactivation from latency in hematopoietic cells. UL138 is pro-latency in that genetic disruption of UL138 allows HCMV to replicate in the absence of a reactivation stimulus and results in increased viral yields relative to the parental wild-type (WT) virus (34, 39). Conversely, UL135 is pro-replication and necessary for reactivation from latency (40). UL135 functions, in part, to overcome the suppressive effect of UL138 for reactivation. Both UL135 and UL138 are expressed early, prior to the onset of viral DNA synthesis. UL135 also has an epistatic relationship with the viral serine–threonine protein kinase, UL97, whereby UL135 function confers a heightened requirement for UL97 for viral DNA synthesis and viral gene expression in a productive infection (41). Although UL97 is the only HCMV-encoded kinase, it is remarkably dispensable for replication in the laboratory-adapted strains lacking UL135 (41, 42).
UL136 is a complex gene that encodes five proteins, referred to as isoforms, resulting from alternative transcription initiation start sites (43). UL136 is expressed with early to late expression kinetics (43, 44), as maximal expression requires viral DNA synthesis, in contrast to other UL133–UL138 locus genes that are expressed with delayed early kinetics. UL136 and its protein isoforms are dispensable for replication in fibroblasts, but important for replication in microvascular endothelial cells. Further, UL136 proteins have roles in both latency and reactivation. The UL136-33kDa (UL136p33) and UL136-26kDa (UL136p26) membrane-associated isoforms are required for reactivation, phenocopying a loss of UL135 (44). The UL136-25kDa (UL136p25) isoform has context-dependent roles, where disruption of this isoform results in a more replicative virus in HPCs infected in vitro, but a virus that fails to reactivate humanized mice (44). The UL136-23kDa (UL136p23) and UL136-19kDa (UL136p19) isoforms, which have not been separated genetically, are soluble and pro-latency. Similar to UL138, a virus that cannot express the UL136p23/19 isoforms replicates robustly in hematopoietic cells in the absence of a reactivation stimulus (43, 44). Given these early to late expression kinetics and the differential roles in latency and reactivation of the UL136 isoforms, we postulate that UL136 functions to toggle the balance between a UL138-dominant latent state and a UL135-dominant reactivated state in response to host cues.
We have recently defined an important regulatory checkpoint for controlling accumulation of the UL136p33 isoform for reactivation. UL136p33 is unstable relative to other UL136 isoforms (45). We have further shown that the host E3 ubiquitin ligase, an inducible degrader of low-density lipoprotein receptor (IDOL, also known as MYLIP), targets UL136p33 for rapid proteasomal degradation (45). When UL136p33 is stabilized, HCMV is more replicative in the absence of a reactivation stimulus in CD34+ HPCs. IDOL is highly expressed in undifferentiated hematopoietic cells, but is downregulated sharply upon differentiation. Consistent with a role for IDOL-mediated regulation of UL136p33 for latency, induction of IDOL during infection in CD34+ HPCs restricts replication, whereas knockdown of IDOL increases virus gene expression in hematopoietic models. This work demonstrates the importance of UL136p33 instability for the establishment of latency and defines a key cellular pathway regulating UL136p33 levels and HCMV fate decisions with regard to entry into and exit from latency (45).
The independent requirements for UL135 and UL136p33 for reactivation from latency along with their sequential temporal expression pattern suggest an epistatic relationship between UL135 and UL136 genes for controlling latency and reactivation. We hypothesized that stabilizing UL136p33 might obviate the need for UL135 for replication in hematopoietic cells if, for example, UL135 initiated events in infection that resulted in the accumulation of UL136p33. To explore the possible interdependence between UL135 and UL136p33 for HCMV reactivation and replication, we generated UL136 recombinant viruses where UL136p33 was stabilized in the presence or absence of UL135. We demonstrate here that stabilizing UL136p33 does not compensate for the modest defects resulting from the loss of UL135 in fibroblasts. However, in a latent infection, stabilization of UL136p33 strikingly compensates for the loss of UL135, resulting in enhanced virus replication (a failure to establish latency) and genome amplification. Importantly, this phenotype is recapitulated in humanized NSG (huNSG) mice. Taken together, our evidence suggests an epistatic relationship between UL136p33 and UL135 for controlling HCMV reactivation from latency, where UL135 is dispensable for replication in CD34+ HPCs when UL136p33 expression is ensured.
RESULTS
Stabilizing UL136p33 modestly alters viral gene expression in a productive infection
We have previously shown that UL135 exhibits early expression kinetics, while UL136 isoforms exhibit early to late expression kinetics in a productive infection in fibroblasts (43) and that UL136p33 is unstable and its accumulation is required for reactivation (45). Further, as UL135 and UL136p33 are both required for reactivation from latency, we hypothesized an epistatic relationship whereby UL135 function may drive aspects of infection required for the accumulation of UL136p33 at late times. If true, then stabilizing UL136p33 might overcome the requirement for UL135 for replication. UL136p33 is stabilized by the substitution of four lysine residues for arginine, UL136mycΔK→R. To investigate the possible relationship between UL135 and UL136p33, we generated recombinant bacterial artificial chromosomes (BAC) by substituting UL136 variants encoding the C-terminal myc epitope-tagged version of UL136, UL136 myc, or the stabilized version of UL136, UL136mycΔK→R, into a previously characterized ∆UL135STOP bacterial artificial chromosome (BAC) where UL135 expression is disrupted by the insertion of stop codons (43, 45). The myc epitope tag is required to detect UL136 as we have been unsuccessful in generating antibodies to UL136 despite multiple attempts. Further, while UL136 is important for infection in hematopoietic cells as defined by the phenotypes associated with disruption of specific isoforms or combinations of isoforms (43, 44), UL136 is expressed at such low levels in CD34+ HPCs that we have not been able to detect protein even with the myc epitope tag. A schematic of the recombinant viruses used in this study is shown in Fig. 1A. Whole-genome sequencing was performed to ensure mutations were located at the desired positions on the viral genome and that recombination did not affect other regions of the genome (Fig. 1B). In addition, a schematic of the specific nucleotides that were mutated is specified for each recombinant virus (Fig. 1C).
Fig 1.

Schematic and whole-genome sequencing of viruses used in this study. (A) Schematic of changes within the UL133–UL138 locus in TB40/E recombinant viruses indicated. Parental (WT) UL136myc expresses UL136 fused in frame with a C-terminal myc epitope tag. All recombinant viruses used in this study are derived from this virus and express the myc epitope-tagged version of UL136 for protein detection. UL136mycΔ33kDa contains stop codon substitutions for 5’ AUGs to disrupt the expression of UL136p33. UL136mycΔK→R contains arginine substitutions for all four lysine residues in UL136 (amino acid positions 4, 20, 25, and 113). ΔUL135STOP/ΔUL136myc contains stop codon substitutions for 5’ AUGs to abrogate synthesis of UL135 protein (amino acid positions 1, 27, and 97). ΔUL135STOP/ΔUL136mycΔK→R combines the stop codon substitutions in UL135 with the lysine to arginine substitutions in UL136 described above. * indicates stop codon substitutions for methionine codons. (B) Whole-genome sequencing and alignment of parental (TB40/E) and recombinant HCMV bacterial artificial chromosomes (BACs) was performed to identify alterations in each recombinant virus. Thick blue lines represent each recombinant virus as labeled, and mutations in each genome are denoted by black dots. (C) Zoomed in pictograph of UL135 and UL136 genes with specific nucleotide changes evident in sequencing. Single-nucleotide polymorphisms (SNPs) are boxed in red; multiple nucleotide polymorphisms (MNPs) are boxed in green; and insertions (INS) are boxed in blue. Mutations for each recombinant virus are compared back to the WT TB40/E-5 parental virus.
We first characterized viral gene expression during infection in MRC-5 fibroblasts. Fibroblasts support only a replicative infection and are a foundational model in the field for understanding HCMV replication. While UL133–UL138 locus genes have important functions for replication and latency in hematopoietic cells, characterization in fibroblasts is important in defining the biology of these genes in HCMV infection and providing critical context for interpreting the functions and phenotypes of these genes apparent in hematopoietic cells. Fibroblasts were infected with UL136 myc, UL136mycΔK→R, ΔUL135STOP/UL136myc, or ΔUL135STOP/UL136mycΔK→R at an MOI of 1 and viral protein accumulation was analyzed over a time course (Fig. 2A). As expected, UL135 protein was not detected in ΔUL135STOP/UL136myc and ΔUL135STOP/UL136mycΔK→R infections. Further, as previously reported, at a high MOI, proteins representative immediate early, early, and late (IE1/2, UL44, pp28, and pp150, respectively) viral gene expression follows the standard gene expression cascade in the absence of UL135 (40). Stabilizing UL136p33 (UL136 myc∆K→R) resulted in increased p33 protein levels, but decreased levels of middle UL136 isoforms, particularly p25, relative to UL136 myc infection (Fig. 2A and B). Further, stabilizing UL136p33 had little to no effect on early or late protein expression, represented by UL44 and pp150, respectively, relative to UL136 myc infection (Fig. 2A and B). We detect no significant differences in IE or early gene expression between the mutant viruses. Although, as previously reported (40), viruses lacking UL135 may produce modestly diminished levels of IE and early proteins in infection. Levels of the pp150 late protein were diminished in viruses lacking UL135 and this was not rescued by the stabilization of UL136p33. With respect to the UL136 isoforms, UL136p33 levels were elevated in UL136 myc∆K→R infection, but only when UL135 was present (Fig. 2A and C). We previously described the diminished accumulation of middle UL136 isoforms during infection with UL136 myc ∆K→R (45). These results suggest that UL135 is necessary for maximal accumulation of UL136p33 at late times, while increased expression of UL136p33 diminishes middle UL136 isoform accumulation, some of which are pro-latency (43, 44).
Fig 2.

Stabilizing UL136p33 modestly alters viral gene expression in a productive infection. (A) MRC-5 lung fibroblast cells were infected with UL136 myc, UL136mycΔK→R, ΔUL135STOP/UL136myc, or ΔUL135STOP/UL136mycΔK→R recombinant viruses at MOI of 1. Lysates were collected over a time course of infection and immunoblotted using antibodies specific to the myc epitope tag (UL136 isoforms), UL135 (UL135 isoforms), UL44, IE1&2 (clone 3H4), pp28, and pp150. Tubulin was used as a loading control. Representative blots are shown. (B and C) Protein levels quantified over multiple independent experiments using Image Studio Lite quantification software. All protein bands are normalized to tubulin. Bars represent the averages of at least three independent experiments with standard deviation shown. Significance is calculated using a two-way ANOVA with Tukey’s multiple comparison tests. *P < 0.01; **P < 0.001; ***P < 0.0001.
Stabilization of UL136p33 does not rescue viral yields associated with disruption of UL135 in productive infection
We previously have shown that disruption of UL136p33 alone or stabilization of UL136p33 does not affect virus yields resulting from a productive infection in fibroblasts (43, 45). Further, we have previously reported that disruption of UL135 results in a modest viral yield reduction in fibroblasts (40). To determine if stabilizing UL136p33 could rescue viral replication of ∆UL135 STOP, we performed a multi-step growth curve (MOI 0.02) in fibroblasts infected with UL136 myc, UL136mycΔK→R, ΔUL135STOP/UL136myc, or ΔUL135STOP/UL136mycΔK→R recombinant viruses (Fig. 3A). While UL136myc∆K→R replicates with similar titers and yields as the parental virus, stabilization of UL136p33 did not compensate for the disruption of UL135; ΔUL135STOP/UL136mycΔK→R replicated with similar kinetics and yields as ΔUL135STOP/UL136myc.
Fig 3.

Stabilization of UL136p33 does not rescue viral yields associated with disruption of UL135 in productive infection. (A) Primary MRC-5 cells were infected with WT UL136 myc, UL136mycΔK→R, ΔUL135STOP/UL136myc, or ΔUL135STOP/UL136mycΔK→R recombinant viruses, and a multistep (MOI of 0.02) growth curve was performed. Cells and culture supernatant were collected at the indicated time points, and virus titers were measured by TCID50. Data points represent the averages from three independent experiments and error bars represent standard deviations. A two-way ANOVA with Tukey’s multiple comparison tests were performed to determine statistical significance for each mutant infection relative to the WT UL136myc infection. *P < 0.05 for WT UL136myc compared to ΔUL135STOP/UL136myc and ΔUL135 STOP/UL136mycΔK→R at 8 dpi and **P < 0.01 for WT UL136myc compared to ΔUL135STOP/UL136myc and ΔUL135STOP/UL136mycΔK→R at16 dpi. (B) Total DNA was isolated from primary MRC-5 cells infected at an MOI of 1 with WT UL136 myc, UL136mycΔK→R, ΔUL135STOP/UL136myc, or ΔUL135STOP/UL136mycΔK→R recombinant viruses at 6 and 72 hpi. The total number of viral genomes were quantified using qPCR with primers to β2.7kb relative to the cellular RNaseP gene. The viral genomes present in the cell at 6 hpi represent the input genome copy number. Averages of duplicate measurements from three independent experiments with standard deviation bars are shown for each virus at each time point. A two-way ANOVA with Tukey’s multiple comparison tests were performed to determine statistical significance values for each mutant infection relative to its 6 hpi time point (***, P < 0.001 and ****, P < 0.0001).
We also analyzed viral genomes amplified in cells infected with WT UL136 myc, UL136mycΔK→R, ΔUL135STOP/UL136myc, or ΔUL135 STOP/UL136mycΔK→R recombinant viruses at an MOI of 1 (Fig. 3B). Total viral genomes were quantified using real-time quantitative polymerase chain reaction (qPCR) with primers specific to sequences encoding the β2.7 region of the genome (46) at 6 and 72 hours post-infection (hpi). Viral genomes were amplified to similar levels in each infection, indicating that viral genome synthesis is not impacted by the loss of UL135 or the stabilization of UL136p33. The replication defect observed for ∆UL135 STOP infection in fibroblasts is due to late-phase defects that were not rescued by the stabilization of UL136p33.
Increased UL136p33 concentration does not enhance viral gene expression separate from viral DNA synthesis
Maximal expression of UL136 requires viral DNA synthesis (39, 40, 43). To examine the relationship between UL136 and viral gene expression around viral DNA synthesis, we analyzed the accumulation of viral proteins in fibroblasts infected with UL136 myc or UL136mycΔK→R and treated with phosphonoacetic acid (PAA) to inhibit HCMV genome synthesis or a vehicle control (Fig. 4A). As previously shown, PAA treatment diminishes the accumulation of UL136 isoforms (43) and the canonical late protein, pp28, as well as amplification of IE2 at 48–72 hpi (43, 47, 48). While UL136mycΔK→R increased levels of UL136p33, late-phase UL136p33 accumulation (48–72 hpi) is limited by PAA relative to the vehicle control (Fig. 4A). Quantification of UL136p33, UL135, IE2 and pp28 protein levels is shown in Fig. 4B. Stabilization of UL136p33 did not rescue late-phase IE2 or pp28 gene expression in infected cells where viral genome synthesis and entry into late phase was blocked by PAA (Fig. 4B). Further, stabilization of UL136p33 does not fully compensate for the onset of viral genome amplification and late-phase events in driving maximal UL136p33 accumulation.
Fig 4.

Increased UL136p33 concentration does not enhance viral gene expression separate from viral DNA synthesis. (A) MRC-5 cells were infected with WT UL136 myc or UL136mycΔK→R, at an MOI of 1 and treated with PAA (50 µg/mL) or ethanol as the vehicle control at the time of infection. The drug was refreshed at 24 and 48 hpi to account for decay. Lysates were collected and immunoblotted at the indicated time points for viral proteins using antibodies described in Table 1. The tubulin antibody was used as a loading control. Representative blots are shown. (B) Multiple independent experiments were quantified. Data points represent the averages from three independent experiments, and error bars represent standard deviations. A two-way ANOVA with Tukey’s multiple comparison tests were performed to determine the statistical significance values for each infection relative to PAA treatment at each time point. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Stabilizing UL136p33 does not direct middle UL136 isoforms for proteasomal degradation
We have previously shown that UL136p33 is targeted for rapid proteasomal turnover by a host E3 ubiquitin ligase, IDOL (45). We next wanted to ask if the loss of middle UL136 isoforms when UL136p33 is stabilized (Fig. 2) is due to proteasomal degradation. To investigate this, we infected cells with either UL136 myc or UL136mycΔK→R and treated cells with MG132 or vehicle 6 h prior to collecting lysates at 24 hpi (Fig. 5A). All UL136 isoforms are quantified over multiple independent replicates in Fig. 5B. As observed earlier, UL136p33 levels increased with MG132 treatment (45). Somewhat surprisingly, proteasomal inhibition also increased levels of UL136mycΔK→R, indicating that in addition to IDOL-dependent targeting to the proteasome, UL136 may also be targeted for ubiquitin-independent, proteasome-dependent turnover. Ubiquitin-independent, proteasome-dependent turnover has been reported as a mechanism by which the HCMV tegument protein pp71 targets host factors (49, 50), including the retinoblastoma protein and Daxx for destruction, as well as other viral or cellular oncoproteins (51 - 53). More work is required to fully investigate this possibility.
Fig 5.

Stabilizing UL136p33 does not direct the middle isoforms of UL136 for proteasomal degradation. MRC-5 fibroblasts were infected with either UL136 myc or UL136mycΔK→R. 6 h prior to lysate collection, infected cells were treated with 20 µM MG132 to inhibit the proteasome or vehicle control. Lysates were collected at 24 hpi and immunoblotted for myc to detect protein isoforms and tubulin (as a loading control) with antibodies described in Table 2. Three independent biological replicates were used to calculate statistical significance. Statistical significance was calculated through a two-way ANOVA with Tukey’s multiple comparison tests for each isoform as follows. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
There are four lysine residues in UL136p33 (K4, K20, K25, and K113) and only K113 is shared by the UL136 isoforms smaller than p33. Like UL136p33, p25 and p23 are also rescued by MG132 treatment, indicating that they are targeted to the proteosome. MG132 has no effect on p25 levels in the K→R mutant virus, suggesting that ubiquitination of K113 is required for its turnover. However, we have shown that IDOL does not target p25 (45) and so may be targeted by a distinct E3 ubiquitin ligase. UL136p23 expressed from the K→R mutant virus is rescued by MG132-treatment, indicating that it may be targeted for proteasome-dependent degradation, independently of ubiquitination at K113. UL136p26 levels were not affected by MG132, although UL136p26 levels were reduced in the K→R mutant virus infection, suggesting that this protein is not targeted for proteasomal degradation but that the stabilization of UL136p33 negatively affects the expression of p26. Lastly, UL136p19 was not affected by K→R substitution or by proteasomal inhibition, suggesting that UL136p19 is not regulated by the proteasome or UL136p33. Taken together, these results indicate that UL136p33 negatively impacts the expression of p26 and p25 through non-proteasomal degradation. Heightened UL136p33 levels likely downregulate transcription of smaller isoforms. However, differential transcriptional regulation of the UL136 isoforms is difficult to assess since UL136 isoforms are expressed from overlapping transcripts generated by alternative transcriptional start sites (43).
Stabilizing UL136p33 rescues viral replication in the absence of UL135 in CD34+ HPCs
Both UL135 and UL136p33 are required for reactivation from latency (40, 44). Further, stabilization of UL136p33 results in virus that produces increased frequencies of infectious centers in CD34+ HPCs in the absence of a reactivation stimulus (45). This result indicates that the instability of UL136p33 is important for the establishment of latency. We next wanted to determine if stabilizing UL136p33 could compensate for a loss of UL135 in driving reactivation from latency in CD34+ HPCs. We infected CD34+ HPCs with WT UL136myc, UL136mycΔ33kDa (TB40/E UL136myc containing a stop codon disruption of the UL136p33 isoform) (44), UL136mycΔK→R, and ΔUL135STOP/UL136mycΔK→R recombinant viruses. Infected HPCs (CD34+/GFP+) were purified by FACS and seeded into long-term bone marrow culture over a stromal cell support. At 10 dpi, the infected cell culture was split. Half the culture (live cells) was seeded by limiting dilution onto permissive fibroblast monolayers in a cytokine-rich medium to stimulate differentiation and reactivation (Reactivation). The other half of the culture was lysed and seeded in parallel on top permissive fibroblast monolayers by limiting dilution to determine infectious centers present prior to reactivation (Pre-Reactivation control) (54) (Fig. 6A). While we were unable to produce high enough titer stocks of the ΔUL135STOP/UL136myc virus sufficient for infection of CD34+ HPCs for all replicates, we have previously shown that ∆UL135 STOP viruses fail to reactivate, similarly to the UL136mycΔ33kDa infection (40, 55). As previously reported, the UL136mycΔK→R infection resulted in a virus that replicated in CD34+ HPCs in the absence of a replication stimulus (45), indicating an inability to establish latency. Intriguingly, ΔUL135STOP/UL136mycΔK→R infection resulted in a virus that was equally as replicative as UL136mycΔK→R. These data indicate that when UL136p33 is stabilized, the loss of UL135 has little or no consequence for replication in hematopoietic cells. We also analyzed viral genome copy number at 10 dpi in two independent experiments using two independent donors (Fig. 6B). Consistent with the infectious centers measurements, viral genome levels were equivalent to or greater in the ΔUL135STOP/UL136mycΔK→R infection relative to UL136mycΔK→R infection. Taken together, these findings suggest that stabilization of UL136p33 can rescue the defect in replication associated with the loss of UL135 in CD34+ HPCs and suggest cell type-dependent functions or mechanisms for the UL133/8 locus genes.
Fig 6.
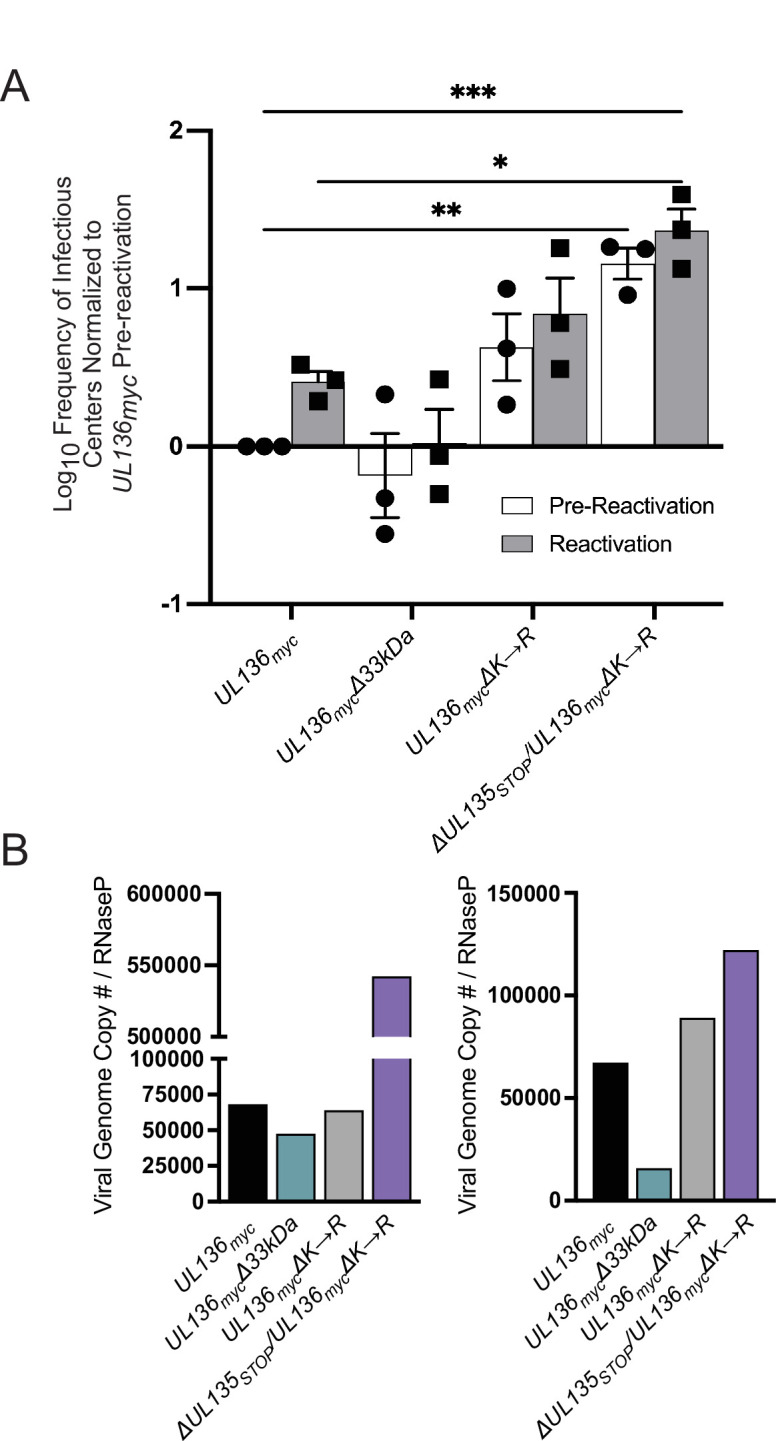
Stabilizing UL136p33 rescues viral replication in the absence of UL135 in CD34+ HPCs. (A) CD34+ HPCs were infected with WT UL136myc, UL136mycΔ33kDa, UL136mycΔK→R, and ΔUL135STOP/UL136mycΔK→R at an MOI of 2. At 24 hpi, CD34+/GFP+ (infected cells) were sorted and seeded into long-term bone marrow culture. After 10 days in culture, parallel populations of either mechanically lysed cells or live cells were plated onto fibroblast monolayers in cytokine-rich media. 14 days later, GFP+ wells were scored, and the frequency of infectious centers was determined by extreme limiting dilution analysis. The mechanically lysed population defines the quantity of virus present prior to reactivation (pre-reactivation; white bar). The live-cell population defines the quantity of virus present after reactivation (reactivation; gray bar). The frequency was normalized to WT UL136myc pre-reactivation, and the average of three independent experiments is shown. Statistical significance was calculated using a two-way ANOVA with Tukey’s multiple comparison tests. *, P < 0.05; **, P < 0.01; ***, P < 0.001). (B) Total DNA was isolated from CD34+ HPCs infected with WT UL136myc, UL136mycΔ33kDa, UL136mycΔK→R, and ΔUL135STOP/UL136mycΔK→R at an MOI of 2 at 10 dpi. The number of viral genomes relative to the level of RNaseP expression was quantified by qPCR using β2.7kb RNA gene- and RNaseP-specific primers. Two biological replicates from two independent cell donors are shown.
Stabilizing UL136p33 compensates for the loss of UL135 for viral replication in huNSG mice
We next wanted to analyze the activity of these recombinant viruses in NOD-scid IL2Rγcnull (huNSG) mice. We have previously demonstrated that UL136p33 is necessary for reactivation of HCMV post-G-CSF stimulation in huNSG mice engrafted with CD34+ HPCs (44). While the huNSG mouse model has typically recapitulated results from our in vitro CD34+ HPC experimental latency model, the one notable exception is for the UL136 myc∆25 kDa virus, which fails to reactivate in huNSG mice, but is more replicative in CD34+ HPCs infected in vitro (44). huNSG mice were sublethally irradiated and engrafted with human CD34+ HPCs. After CD34+ engraftment, mice were injected with human fibroblasts infected with WT UL136myc, UL136mycΔ33kDa, UL136mycΔK→R, and ΔUL135STOP/UL136mycΔK→R recombinant viruses. At 4 wk post-infection, five mice from each group of 10 were treated with G-CSF and AMD-3100 to induce stem cell mobilization and HCMV reactivation. At 1 wk post-mobilization, viral genome load was assessed in spleen and liver tissues from treated and untreated mouse groups to evaluate amplification of viral genomes and dissemination of infected cells to organs, as described previously (44).
As previously demonstrated, G-CSF mobilization of UL136 myc-infected humanized mice resulted in increased viral genomes detected in the spleen and liver relative to unmobilized mice, consistent with reactivation of virus replication (Fig. 7). By contrast, UL136 myc∆33 kDa failed to reactivate and similar levels of genomes were measured in G-CSF/AMD-3100-treated as compared to untreated huNSG mice infected with either UL136 myc or UL136mycΔ33kDa. The viral genome copy number in the unmobilized and mobilized mice infected with UL136mycΔK→R was equivalent to the G-CSF/AMD-3100-mobilized UL136 myc infection, indicating a failure to establish latency (replication in the absence of a stimulus). This phenotype recapitulates the UL136mycΔK→R phenotype in CD34+ HPCs infected in vitro (Fig. 6). Strikingly, the ΔUL135STOP/UL136mycΔK→R virus also recapitulated our in vitro studies with increased viral genome levels in the spleen and liver regardless of G-CSF/AMD-3100 treatment compared to mice infected with UL136mycΔK→R. Taken together, these results support an epistatic relationship between UL135 and UL136, whereby stabilization of UL136p33 compensates for the loss of UL135 in replication following a reactivation stimulus. Further, these data suggest a restrictive or deleterious consequence of UL135 function in reactivation and replication and that these defects can be overcome by robust expression of UL136p33.
Fig 7.

Stabilizing UL136p33 compensates for a loss of UL135 for viral replication in huNSG mice. Humanized NSG mice were injected with fibroblasts infected with either HCMV WT UL136myc, UL136mycΔ33kDa, UL136mycΔK→R, and ΔUL135STOP/UL136mycΔK→R (n = 10 per group). At 4 wk post-infection, half of the mice were treated with G-CSF and AMD-3100 to induce cellular mobilization and promote HCMV reactivation. Control mice were left untreated. At 1 wk post-mobilization, mice were euthanized, and tissues were collected. Total DNA was extracted using DNAzol, and HCMV viral load was determined by qPCR on 1 µg of total DNA prepared from spleen or liver tissue. Error bars represent standard error of the mean between average DNA copies from two or four tissue sections, respectively, for individual animals. All samples were compared by a two-way ANOVA with Tukey’s multiple comparison tests within experimental groups (nonmobilized [-G-CSF] versus mobilized [+G-CSF] for each virus and between all virus groups for both nonmobilized and mobilized conditions). Statistical significance where **, P < 0.01 and ***, P < 0.001.
DISCUSSION
Viruses use complex gene-regulatory networks to coordinate major infection checkpoints. To direct the transition from latency to reactivation, herpesviruses must sense and respond to environmental cues and integrate multiple cellular states to exit the latent state for replication. HCMV has a large coding capacity of more than 170 genes for regulating infection in multiple cell types with the possibility of a much-expanded array of open reading frames (56). Many genes encode multiple proteins (referred to as isoforms), as is the case for UL136, UL44, UL99, and UL112-113, through alternative transcriptional and/or translational start sites within a single gene (34, 43, 57 - 61). Other herpesviruses, such as Kaposi’s sarcoma-associated herpesvirus (KSHV) and herpes simplex type-1 (HSV-1), also have genes that encode for multiple protein isoforms with differential spatiotemporal expression, for example, ORF50 and UL12, respectively (62, 63). While the full potential of protein isoforms encoded by herpesviruses remains to be defined, genes encoding multiple proteins allow for coordinated regulation of effectors to dictate infection outcomes. Protein isoforms encoded from a single gene may have synergistic or opposing roles (e.g., dominant negative) in dictating patterns of infection in a context-dependent manner (4). Further, epistatic interactions of viral genes in the same locus (or gene circuit) add to the complexity of HCMV biology. This work investigates the role of UL136, specifically the instability of the UL136p33 isoform, and its epistatic relationship with UL135 in driving replication. This question is significant to understanding the complex, context-dependent interactions between virus–virus and virus–host factors that influence the “decision” to enter and maintain latency or to reactivate from latency.
Both UL135 and UL136p33 are important for reactivation from latency (40, 44). UL135-mutant viruses exhibit a modest defect for replication and production of progeny virus in fibroblasts (34, 40), whereas disruption or stabilization of UL136p33 had no impact on replication in fibroblasts (43, 45). UL135 and UL136 are also important for replication in endothelial cells (64). While stabilizing UL136p33 did not compensate for the loss of UL135 in late gene expression or virus yields during infection in fibroblasts (Fig. 2 and 3), stabilization of UL136p33 strikingly compensated for the loss of UL135 in hematopoietic cells infected in vitro and in humanized mice, resulting in increased virus replication in HPCs in the absence of a stimulus for reactivation (Fig. 6 and 7). This indicates distinct cell type-dependent roles for UL135 and UL136p33 for replication. These findings suggest that while UL135 is important to the initiation of or commitment to replication, accumulation of UL136p33 represents a subsequent threshold for replication. Once UL136p33 accumulation is ensured, in this case experimentally by lysine to arginine substitution, the additional loss of UL135 does not confer a disadvantage for replication and reactivation in experimental HPC models of latency. A limitation in the interpretation of these findings is our inability to acquire enough cells infected with the UL135-mutant virus expressing UL136myc for latency assays. UL135-mutant viruses are very difficult to reconstitute from transfection of infectious BAC clones and exhibit a defect for reactivation, although virus stocks once achieved have only a modest defect for replication in fibroblasts. Further work is required to determine phenotypes associated with these viruses during infection in endothelial or epithelial cells. It is possible that stabilization of UL136p33 may complement the replication defect associated with loss of UL135 during infection in endothelial cells.
In collaboration with the Kamil lab, we previously defined an epistatic relationship between UL135 and UL97 in promoting late-phase viral gene expression, viral DNA synthesis, and viral replication in a productive infection (41). In these studies, UL97, the only HCMV-encoded kinase, was remarkably dispensable for replication in the AD169 laboratory-adapted strain (42). However, the loss of UL97 conferred defects in replication in fibroblasts in low-passage strains containing the ULb’ region of the viral genome that is lost from laboratory-adapted strains. Analysis of recombinant viruses containing mutations in the UL133–UL138 locus genes identified UL135 as the gene that conferred a requirement for UL97 for virus infection. In other words, UL97 compensated for deleterious effects on infection due to the action UL135, such inhibition of UL97 only conferred defects on virus replication when UL135 was expressed (41). Similarly, the recombinant UL135 STOP/UL136myc∆K→R virus replicates efficiently without a reactivation stimulus and to enhanced levels compared to the parental virus or a virus disrupted for expression of UL136p33, particularly in the humanized mouse model (Fig. 6 and 7). Taken together with the UL97/UL135 relationship, the present study suggests that UL135 functions, while important for reactivation, pose deleterious consequences for virus replication and other viral genes function to mitigate those effects. The requirement for UL135 in replication in hematopoietic cells was dispensable when UL136p33 was stabilized, suggesting that UL135 function is important in stimulating activities in infection that result in the accumulation of UL136p33. The temporal expression and relative accumulation of UL135 and UL136 gene products to required thresholds are undoubtedly important for the progression to successful reactivation.
While UL135 is expressed with early kinetics, UL136 and UL97 are expressed with early/late or leaky late kinetics and their maximal accumulation requires viral DNA synthesis and entry into late phase (43, 65). These kinetics and the instability of UL136p33 suggest a model whereby latency depends both on UL138 and the instability UL136p33 (Fig. 8). Reactivation from latency, by contrast, depends on the expression of UL135, which functions, at least in part, to overcome the suppressive effects of UL138 (40, 66) and to drive infection for the eventual accumulation of UL136p33. At the same time, UL97 expression may compensate for deleterious effects of UL135 (41). As UL136p33 accumulation is further fortified by the initiation of viral DNA synthesis (Fig. 4), this indicates a second checkpoint (following UL135 expression) in the commitment to reactivation and replication (Fig. 8). This model suggests a stepwise progression toward reactivation where thresholds must be met to pass checkpoints that would otherwise block reactivation and maintain latency. Much remains to be understood about the UL135-UL136 interaction and the mechanisms through which they function to control latency-reactivatation decisons. While UL135 and UL136 may physically interact, it is as likely that they co-regulate cellular pathways.
Fig 8.

Model for UL135 and UL136p33 epistasis for coordinating reactivation of HCMV. We have previously shown that viral factors, such as UL138, function in a pro-latency manner in the establishment of latency, whereas UL135 and UL136p33 function in a pro-reactivation manner. We have further demonstrated that the instability of UL136p33, driven by the IDOL ubiquitin E3 ligase, is also important for the establishment of latency. Reactivation cues stimulate increased expression of UL135 relative to UL138 and increased accumulation of UL136p33 through two mechanisms: (i) the loss of IDOL and (ii) increased UL136p33 synthesis following commitment to viral DNA synthesis. Because ensuring the accumulation of UL136p33 through its stabilization precludes the need for UL135 for replication in HPCs, we propose the existence of checkpoints in the decision to reactivate where UL135 initiates reactivation and subsequent accumulation of UL136p33 is required to meet a threshold in the commitment to reactivation. Created with BioRender.com.
Cellular factors are critical to HCMV latency through, at least in part, their impact on viral gene expression and proteins. We have previously shown that UL138 expression is driven by binding of the host EGR-1 transcription factor to sites just upstream of UL136 (67). As EGR-1 is highly expressed in HPCs, these cells are primed to express UL138, and the ratio of transcripts encoding UL138 to those encoding UL135 is high. We have also shown that UL136p33 is maintained at low levels by the host E3 ubiquitin ligase, IDOL, which is also highly expressed in undifferentiated hematopoietic cells (45). Both EGR-1 and IDOL expression are sharply downregulated by differentiation (45, 67), a well-appreciated trigger of HCMV reactivation (4, 68). Therefore, differentiation is expected to increase the ratio of UL135 transcripts relative to UL138 transcripts (67) and increase the accumulation of UL136p33, which is further amplified by the onset of viral DNA synthesis and entry into late phase. Additional work is required to understand how UL135 functions to promote the progression of infection toward replication and the accumulation of UL136p33. UL135 is well-appreciated for its role in modulating host signaling and cytoskeletal organization, and these roles could impact the expression of UL136 indirectly (55, 66, 69).
Interactions (either physical or by differentially impacting the same processes in infection) between UL136 isoforms or with other UL133/8 locus proteins represent important questions for further investigation. We have shown that while UL136p33 and p26 are important for reactivation, UL136p23/p19 are suppressive to replication for latency (44). The antagonistic or synergistic roles of the UL136 protein isoforms may allow the virus to navigate the commitment to reactivation or the maintenance of latency. The decision to maintain latency or reactivate at the point of viral DNA synthesis may depend on competition between or the differential accumulation of UL136 isoforms (44).
We have demonstrated here that stabilization of UL136p33 leads to a reduction in the protein levels of the middle UL136 isoforms (Fig. 2, 4, and 5), which may aid in allowing HCMV to commit to reactivation and replication. It will be important to define both transcriptional and posttranslational mechanisms regulating UL136 isoform accumulation. UL136 isoforms are synthesized from unique transcripts derived from alternative transcriptional start sites and may be regulated in a context-dependent manner (43). The promoter elements driving UL136 isoform expression have not been characterized. We have shown previously that HCMV reactivation from latency stimulates major immediate early gene expression from alternative promoter sequences embedded within intron A of the MIE transcriptional unit, while the MIEP apparently remains silent (47). In addition, KSHV Orf50 encodes four different protein isoforms of the replication and transcription activator protein, RTA, that temporally regulate each other’s expression for driving the viral gene expression cascade post-reactivation (62). This study demonstrated specifically that induction of the upstream Orf50 N5 promoter that produces RTA isoform four suppresses expression of RTA isoform one through transcriptional interference. Further work is required to define the promoters and transcription factors governing UL136 isoform expression and their impact on one another. Beyond transcriptional regulation, some isoforms are targeted for proteasomal degradation (p33, p25, and p23), while others are not (p26 and p19). Of those targeted for proteasomal degradation, p23 appears to be targeted by ubiquitin-independent mechanisms since K→R substitution had no effect on its turnover. Finally, differential regulation of distinct UL136 protein isoforms may reflect their distinct subcellular localization (44).
This study highlights the complexity of the UL133–UL138 gene network and its phenotypes depending on the context of infection. The ability of a virus to move between bistable infection states—latency or replication—requires that the virus can “sense” host cues, filter and utilize fluctuations (“noise”) to respond appropriately to the host cues to regulate infection fate decisions. UL136p33 may function in an ultrasensitive manner to control the switch for reactivation upon differentiation of HPCs—rapid accumulation of UL136p33 to a threshold to tip the system to reactivation (70). Going forward, it will be important to define how expression from the UL133–UL138 locus is regulated and if UL136 isoforms help the virus stabilize or filter noise to understand HCMV fate decisions.
MATERIALS AND METHODS
Cells
Human primary embryonic lung fibroblasts (MRC-5, purchased from ATCC; Manassas, VA) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) and supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES, 1 mM sodium pyruvate, 2 mM L-alanyl-glutamine, 0.1 mM nonessential amino acids, 100 U/mL penicillin, and 100 µg/mL streptomycin. The Institutional Review Board approved a protocol to obtain human cells from bone marrow transplant waste at the University Medical Center at the University of Arizona. Specimens were de-identified and provided as completely anonymous samples. CD34+ HPCs were isolated and cultured as previously described (34, 54). Briefly, CD34+ HPCs were isolated using the CD34 MicroBead Kit (MACS, Miltenyi Biotec, San Diego, CA). Pure populations of CD34+ HPCs were subsequently cultured in MyeloCult H5100 (Stem Cell Technologies, Cambridge, MA) supplemented with hydrocortisone, 100 U/mL penicillin, and 100 µg/mL streptomycin and maintained in long-term co-culture with M2-10B4 and Sl/Sl murine stromal cell lines (kind gift from Stem Cell Technologies on behalf of D. Hogge, Terry Fox Laboratory, University of British Columbia, Vancouver, BC, Canada) (34, 54). All cells were maintained at 37°C with 5% CO2.
Recombinant viruses
The TB40/E BAC was engineered to express the green fluorescent protein (GFP) driven by the SV40 early promoter located in the intergenic region in between US34 and TRS1 genes as a marker for infection (34, 71). All recombinant viruses were created using a two-step, positive/negative selection approach that leaves no trace of the recombination process (39, 72, 73). Three recombinant viruses used in this study were previously engineered—UL136 myc,UL136mycΔ33kDa, and UL136mycΔK→R—as described previously (43, 45). Two new recombinant viruses were generated for this study—ΔUL135 STOP/UL136myc (to detect the UL136 isoforms when UL135 is disrupted) and ΔUL135 STOP/UL136mycΔK→R (where UL136p33 is stabilized when UL135 is disrupted). We previously generated a UL135stop UL136 <GalK > intermediate virus that serves as the base BAC for the two new recombinant viruses created in this study (64). The “UL136myc” and “UL136mycΔK→R” sequence mutations were PCR amplified off of BACs previously made and characterized with these desired mutations using the following primers: UL138 (139 bp) Rev and UL135 (854 bp) Fwd (43, 45). The sequences for the primers used for this amplification are listed in Table 1. The PCR-amplified products were then recombined into the UL135stop UL136 <GalK > BAC as described previously (43, 64). BAC integrity was tested by enzyme digest fragment analysis and sequencing of the UL136 region using the same primers described in 1. All BAC genomes were maintained in SW102 Escherichia coli and viral stocks were propagated by transfecting 15–20 μg of each BAC genome, along with 2 µg of a plasmid encoding UL82 (pp71) into 5 × 106 MRC-5 fibroblasts, allowed to propagate in MRC-5 fibroblasts and then subsequently purified and stored as previously described to generate the parent (P0) virus (39). To grow the next generation of each virus (P1), the infectious inoculum from the P0 virus was added to MRC-5 fibroblasts that were passaged into roller bottles and cultured for anywhere from 8 to 30 days depending on the recombinant virus being made. Viruses were subsequently purified and stored as previously described (39). Virus titers were determined by 50% tissue culture infectious dose (TCID50) on MRC-5 fibroblasts.
TABLE 1.
Primers used in this study for BAC recombineering
| Primer name | Primer sequence |
|---|---|
| UL135 (bp854) Forward | 5′-CGGAGCCGACCACGC TGCCTATCG-3′ |
| UL138(bp 139) Reverse | 5′-GCCAGCGGTAGCTCAAAAACATGCGC-3′ |
Whole-genome next-generation sequencing and computational analysis
Seven recombinant HCMV BAC genome DNAs were extracted from SW102 E. coli propagating each HCMV genome using a BAC miniprep DNA isolation method. DNA was then sent to SeqCenter, LLC for short read Illumina whole-genome sequencing using an Illumina NextSeq 2000 (200 Mbp sequencing package—https://www.seqcenter.com/dna-sequencing/). Demultiplexing, quality control, and adapter trimming were performed with bcl-convert (v3.9.3). The single-nucleotide polymorphisms (SNPs) were identified using a python package called SNIPPY (https://github.com/tseemann/snippy, accessed June 23, 2022). Analysis is based on the BWA-mem/freebayes pipeline that identifies the mutations between a haploid reference genome and the next-generation sequence reads. The package was implemented with default parameters on the University of Arizona High Performance Computing (HPC) environment, dedicating 80 GB RAM for the job. SNIPPY was able to identify SNPs, multi-nucleotide polymorphisms (MNPs), complex mutations as well as indels.
Immunoblotting
Immunoblotting was performed as previously described (39, 43). Briefly, 50 µg of protein lysate were separated on 12% bis-tris gels by electrophoresis and transferred to 0.45 μm polyvinylidene difluoride (Immobilon-FL, Millipore) membranes. Proteins were detected using epitope- or protein-specific antibodies and fluorescently conjugated secondary antibodies using the Odyssey infrared imaging system (Li-Cor). All antibodies were used as listed in Table 2. Where indicated, cells were treated with 50–100μg of PAA, 1–50μM MG132, or vehicle control. We are grateful for the gift of antibodies from Drs. Thomas Shenk, William Britt, and John Purdy.
TABLE 2.
List of antibodies used in this study
| Antigen | Antibody | Concentration | Animal | Source |
|---|---|---|---|---|
| myc epitope-M | 9B11 | 1:1,000 | Mouse | Cell Signaling |
| myc epitope-R | 71D10 | 1:1,000 | Rabbit | Cell Signaling |
| myc epitope-R | 1:1,000 | Rabbit | Proteintech | |
| alpha-tubulin | DM1A | 1:2,000 | Mouse | Sigma |
| UL138 | 1:500 | Rabbit | Open Biosystems | |
| UL135 | use at 2 ug/mL | Rabbit | Open Biosystems | |
| IE1&2 | 3H4 | 1:250 | Mouse | Gift, Dr. Thomas Shenk through John Purdy |
| pp150 | 1:15 | Mouse | Gift from Dr. William Britt | |
| pp28 (UL99) | 10B4-29 | 1:50 | Mouse | Gift, Dr. Thomas Shenk |
| UL44 | 10D8 | 1:12,000 | Mouse | Virusys |
Viral growth curves
Quantification of infectious virus produced by fibroblasts was determined by infecting MRC-5s at an MOI of 0.02 and subsequently collecting cells and medium over a 16-day infection time course. Virus titers were determined by TCID50 in MRC-5 fibroblasts.
Quantification of viral genomes in fibroblasts
Total DNA was isolated from ~6 × 105 to 7 × 105 MRC-5-infected fibroblasts using the Zymo Duet RNA/DNA isolation Kit (Zymo Research). Viral genomes were quantitated by quantitative PCR (qPCR) using the LightCycler 480 1X SYBR Green master mix (Roche) according to the manufacturer’s instructions. Primers specific to the noncoding b2.7 RNA gene were used. To determine the number of viral genomes present, viral DNA copy numbers were quantified relative to a BAC standard curve normalized to a cellular housekeeping gene RNase P as previously described (47).
Infectious centers assay
CD34+ HPCs, isolated from human cord blood, were used to assess latency and reactivation of HCMV in vitro as previously described (34, 54). Briefly, CD34+ HPCs were infected at an MOI of 2 for 20 h after which a pure population (>97%) of infected (GFP+) CD34+ cells were isolated via FACS (FACSAria, BD Biosciences Immunocytometry Systems, San Jose, CA, USA) using a phycoerythrin-conjugated CD34-specific antibody (BD Biosciences) and propidium iodide to exclude dead cells. These cells were cultured in transwells above irradiated (4,000 rads, 137Cs gammacell-40 irradiator type B, Atomic Energy of Canada LTD, Ottawa, Canada) M2-10B4 and Sl/Sl stromal cells for 10 days. The frequency of the production of infectious centers was measured using an extreme limiting dilution assay as described previously (34, 54). The frequency of infectious centers, based on the number of GFP+ cells at 14 days post-plating, was calculated using ELDA, extreme limiting dilution analysis software (http://bioinf.wehi.edu.au/software/elda/) (74).
Engraftment and infection of huNSG mice
All animal studies were carried out in strict accordance with the recommendations of the American Association for Accreditation of Laboratory Animal Care (AAALAC). The protocol was approved by the Institutional Animal Care and Use Committee (protocol 0922) at the Vaccine and Gene Therapy Institute (OHSU). NOD-scid IL2Rγcnull mice were maintained at a pathogen-free facility at Oregon Health and Science University in accordance with procedures approved by the Institutional Animal Care and Use Committee. Both sexes of animals were used. Humanized mice were generated as previously described (44, 75). The animals (12–14 wk post-engraftment) were treated with 1 mL of 4% thioglycolate (Brewer’s medium; BD) via intraperitoneal (IP) injection to recruit monocytes/macrophages). After 24 h, mice were infected with HCMV TB40/E-UL136myc or UL136myc mutant-infected fibroblasts (approximately 105 PFU per mouse) via IP injection. A control group of engrafted mice was mock infected using uninfected fibroblasts. The virus was reactivated as previously described (44, 75).
Quantitative PCR for viral genomes in huNSG mice
Total DNA was extracted from approximately 1 mm2 sections of mouse spleen or liver using the DNAzol kit (Life Technologies), and processed as previously described (44). Primers and a probe-recognizing HCMV UL141 were used to quantify HCMV genomes (probe = CGAGGGAGAGCAAGTT; forward primer = 5′GATGTGGGCCGAGAATTATGA and reverse primer = 5′ATGGGCCAGGAGTGTGTCA). The reaction was initiated using TaqMan Fast Advanced Master Mix (Applied Biosystems) activated at 95°C for 10 min followed by 40 cycles (15 s at 95°C and 1 min at 60°C) using a StepOnePlus TaqMan PCR machine. Results were analyzed using ABI StepOne software.
ACKNOWLEDGMENTS
This study was supported by the National Institutes of Allergy and Infectious Diseases/National Institutes of Health (NIAID/NIH) grants AI079059 and AI127335 to F.G. and AI127335 awarded to P.C.
We are grateful for the support of the Flow Cytometry and Human Immune Monitoring Shared Resource, supported through the Cancer Center Support Grant P30 CA023074 awarded to the University of Arizona. We are grateful to Pierce Longmire for assistance with the SRA sequence submission.
We thank Drs. James Alwine (University of Pennsylvania) and Lynn Enquist (Princeton University) for critical discussions on this work. We are grateful to Dr. Emmanuel Katsanis for his assistance in coordinating medical waste use in these studies.
Footnotes
visualization
Contributor Information
Felicia Goodrum, Email: fgoodrum@arizona.edu.
Anna Ruth Cliffe, University of Virginia, Charlottesville, Virginia, USA .
DATA AVAILABILITY
Raw viral genome sequences read have been deposited in the Sequence Read Archive (SRA) (BioProject ID: PRJNA926319).
REFERENCES
- 1. Cannon MJ, Schmid DS, Hyde TB. 2010. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev Med Virol 20:202–213. doi: 10.1002/rmv.655 [DOI] [PubMed] [Google Scholar]
- 2. Korndewal MJ, Mollema L, Tcherniaeva I, van der Klis F, Kroes ACM, Oudesluys-Murphy AM, Vossen ACTM, de Melker HE. 2015. Cytomegalovirus infection in the Netherlands: seroprevalence, risk factors, and implications. J Clin Virol 63:53–58. doi: 10.1016/j.jcv.2014.11.033 [DOI] [PubMed] [Google Scholar]
- 3. Zuhair M, Smit GSA, Wallis G, Jabbar F, Smith C, Devleesschauwer B, Griffiths P. 2019. Estimation of the worldwide seroprevalence of cytomegalovirus: a systematic review and meta-analysis. Rev Med Virol 29:e2034. doi: 10.1002/rmv.2034 [DOI] [PubMed] [Google Scholar]
- 4. Goodrum F, Britt W, Mocarski ES. 2021. Cytomegalovirus. In Whelan SPJ, Freed EO, Enquist L (ed), Fields virology, 7th ed, vol 2. DNA Viruses, Wolters Kluwer, Philadelphia, Baltimore, New York, London, Hong Kong, Buenos Aires, Sydney, Tokyo. [Google Scholar]
- 5. Britt W. 2008. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr Top Microbiol Immunol 325:417–470. doi: 10.1007/978-3-540-77349-8_23 [DOI] [PubMed] [Google Scholar]
- 6. Boeckh M. 2011. Complications, diagnosis, management, and prevention of CMV infections: current and future. Hematology Am Soc Hematol Educ Program 2011:305–309. doi: 10.1182/asheducation-2011.1.305 [DOI] [PubMed] [Google Scholar]
- 7. Boeckh M, Geballe AP. 2011. Cytomegalovirus: pathogen, paradigm, and puzzle. J Clin Invest 121:1673–1680. doi: 10.1172/JCI45449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boeckh M, Leisenring W, Riddell SR, Bowden RA, Huang M-L, Myerson D, Stevens-Ayers T, Flowers MED, Cunningham T, Corey L. 2003. Late cytomegalovirus disease and mortality in recipients of allogeneic hematopoietic stem cell transplants: importance of viral load and T-cell immunity. Blood 101:407–414. doi: 10.1182/blood-2002-03-0993 [DOI] [PubMed] [Google Scholar]
- 9. Britt WJ. 2018. Maternal immunity and the natural history of congenital human cytomegalovirus infection. Viruses 10:405. doi: 10.3390/v10080405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cannon MJ. 2009. Congenital cytomegalovirus (CMV) epidemiology and awareness. J Clin Virol 46:S6–S10. doi: 10.1016/j.jcv.2009.09.002 [DOI] [PubMed] [Google Scholar]
- 11. Cannon MJ, Hyde TB, Schmid DS. 2011. Review of cytomegalovirus shedding in bodily fluids and relevance to congenital cytomegalovirus infection. Rev Med Virol 21:240–255. doi: 10.1002/rmv.695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Britt WJ. 2020. Human cytomegalovirus infection in women with preexisting immunity: sources of infection and mechanisms of infection in the presence of antiviral immunity. J Infect Dis 221:S1–S8. doi: 10.1093/infdis/jiz464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Anastasio ART, Yamamoto AY, Massuda ET, Manfredi AKS, Cavalcante JMS, Lopes BCP, Aragon DC, Boppana S, Fowler KB, Britt WJ, Mussi-Pinhata MM. 2021. Comprehensive evaluation of risk factors for neonatal hearing loss in a large Brazilian cohort. J Perinatol 41:315–323. doi: 10.1038/s41372-020-00807-8 [DOI] [PubMed] [Google Scholar]
- 14. Krstanović F, Britt WJ, Jonjić S, Brizić I. 2021. Cytomegalovirus infection and inflammation in developing brain. Viruses 13:1078. doi: 10.3390/v13061078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou YP, Mei MJ, Wang XZ, Huang SN, Chen L, Zhang M, Li XY, Qin HB, Dong X, Cheng S, Wen L, Yang B, An XF, He AD, Zhang B, Zeng WB, Li XJ, Lu Y, Li HC, Li H, Zou WG, Redwood AJ, Rayner S, Cheng H, McVoy MA, Tang Q, Britt WJ, Zhou X, Jiang X, Luo MH. 2022. A congenital CMV infection model for follow-up studies of neurodevelopmental disorders, neuroimaging abnormalities, and treatment. JCI Insight 7:e152551. doi: 10.1172/jci.insight.152551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Redeker A, Remmerswaal EBM, van der Gracht ETI, Welten SPM, Höllt T, Koning F, Cicin-Sain L, Nikolich-Žugich J, Ten Berge IJM, van Lier RAW, van Unen V, Arens R. 2017. The contribution of cytomegalovirus infection to immune senescence is set by the infectious dose. Front Immunol 8:1953. doi: 10.3389/fimmu.2017.01953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Griffiths SJ, Riddell NE, Masters J, Libri V, Henson SM, Wertheimer A, Wallace D, Sims S, Rivino L, Larbi A, Kemeny DM, Nikolich-Zugich J, Kern F, Klenerman P, Emery VC, Akbar AN. 2013. Age-associated increase of low-avidity cytomegalovirus-specific CD8+ T cells that re-express CD45RA. J Immunol 190:5363–5372. doi: 10.4049/jimmunol.1203267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wertheimer AM, Bennett MS, Park B, Uhrlaub JL, Martinez C, Pulko V, Currier NL, Nikolich-Žugich D, Kaye J, Nikolich-Žugich J. 2014. Aging and cytomegalovirus infection differentially and jointly affect distinct circulating T cell subsets in humans. J Immunol 192:2143–2155. doi: 10.4049/jimmunol.1301721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goodrum F. 2022. The complex biology of human cytomegalovirus latency. Adv Virus Res 112:31–85. doi: 10.1016/bs.aivir.2022.01.001 [DOI] [PubMed] [Google Scholar]
- 20. Hale AE, Collins-McMillen D, Lenarcic EM, Igarashi S, Kamil JP, Goodrum F, Moorman NJ. 2020. FOXO transcription factors activate alternative major immediate early promoters to induce human cytomegalovirus reactivation. Proc Natl Acad Sci U S A 117:18764–18770. doi: 10.1073/pnas.2002651117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Min C-K, Shakya AK, Lee B-J, Streblow DN, Caposio P, Yurochko AD. 2020. The differentiation of human cytomegalovirus infected-monocytes is required for viral replication. Front Cell Infect Microbiol 10:368. doi: 10.3389/fcimb.2020.00368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Krishna BA, Wass AB, O’Connor CM. 2020. Activator protein-1 transactivation of the major immediate early locus is a determinant of cytomegalovirus reactivation from latency. Proc Natl Acad Sci U S A 117:20860–20867. doi: 10.1073/pnas.2009420117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang H, Domma AJ, Goodrum FD, Moorman NJ, Kamil JP, Shenk T. 2022. The Akt forkhead box O transcription factor axis regulates human cytomegalovirus replication. mBio 13:e0104222. doi: 10.1128/mbio.01042-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hancock MH, Crawford LB, Perez W, Struthers HM, Mitchell J, Caposio P. 2021. Human cytomegalovirus UL7, miR-US5-1, and miR-UL112-3p inactivation of FOXO3a protects CD34+ hematopoietic progenitor cells from apoptosis. mSphere 6:e00986-20. doi: 10.1128/mSphere.00986-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Crawford LB, Kim JH, Collins-McMillen D, Lee B-J, Landais I, Held C, Nelson JA, Yurochko AD, Caposio P. 2018. Human cytomegalovirus encodes a novel FLT3 receptor ligand necessary for hematopoietic cell differentiation and viral reactivation. mBio 9:e00682-18. doi: 10.1128/mBio.00682-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goodrum F, Britt W, Mocarski ES. 2022. Howley P. M., Knipe D. M., Cohen J. L., and Damania B. A.. Cytomegalovirus. 7th ed. Vol. 2. Fields Virology: DNA Viruses, Wolters Kluwer. [Google Scholar]
- 27. McAdams HH, Arkin A. 1998. Simulation of prokaryotic genetic circuits. Annu Rev Biophys Biomol Struct 27:199–224. doi: 10.1146/annurev.biophys.27.1.199 [DOI] [PubMed] [Google Scholar]
- 28. Brophy JAN, Voigt CA. 2014. Principles of genetic circuit design. Nat Methods 11:508–520. doi: 10.1038/nmeth.2926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cha TA, Tom E, Kemble GW, Duke GM, Mocarski ES, Spaete RR. 1996. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J Virol 70:78–83. doi: 10.1128/JVI.70.1.78-83.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dolan A, Cunningham C, Hector RD, Hassan-Walker AF, Lee L, Addison C, Dargan DJ, McGeoch DJ, Gatherer D, Emery VC, Griffiths PD, Sinzger C, McSharry BP, Wilkinson GWG, Davison AJ. 2004. Genetic content of wild-type human cytomegalovirus. J Gen Virol 85:1301–1312. doi: 10.1099/vir.0.79888-0 [DOI] [PubMed] [Google Scholar]
- 31. Murphy E, Rigoutsos I, Shibuya T, Shenk TE. 2003. Reevaluation of human cytomegalovirus coding potential. Proc Natl Acad Sci U S A 100:13585–13590. doi: 10.1073/pnas.1735466100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Murphy E, Shenk T. 2008. Human cytomegalovirus genome. Curr Top Microbiol Immunol 325:1–19. doi: 10.1007/978-3-540-77349-8_1 [DOI] [PubMed] [Google Scholar]
- 33. Murphy E, Yu D, Grimwood J, Schmutz J, Dickson M, Jarvis MA, Hahn G, Nelson JA, Myers RM, Shenk TE. 2003. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc Natl Acad Sci U S A 100:14976–14981. doi: 10.1073/pnas.2136652100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Umashankar M, Petrucelli A, Cicchini L, Caposio P, Kreklywich CN, Rak M, Bughio F, Goldman DC, Hamlin KL, Nelson JA, Fleming WH, Streblow DN, Goodrum F. 2011. A novel human cytomegalovirus locus modulates cell type-specific outcomes of infection. PLoS Pathog 7:e1002444. doi: 10.1371/journal.ppat.1002444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dutta N, Lashmit P, Yuan J, Meier J, Stinski MF. 2015. The human cytomegalovirus UL133-138 gene locus attenuates the lytic viral cycle in fibroblasts. PLoS One 10:e0120946. doi: 10.1371/journal.pone.0120946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang W, Taylor SL, Leisenfelder SA, Morton R, Moffat JF, Smirnov S, Zhu H. 2005. Human cytomegalovirus genes in the 15-kilobase region are required for viral replication in implanted human tissues in SCID mice. J Virol 79:2115–2123. doi: 10.1128/JVI.79.4.2115-2123.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kempová V, Lenhartová S, Benko M, Nemčovič M, Kúdelová M, Nemčovičová I. 2020. The power of human cytomegalovirus (HCMV) hijacked UL/B' functions lost in vitro. Acta Virol 64:117–130. doi: 10.4149/av_2020_202 [DOI] [PubMed] [Google Scholar]
- 38. Mlera L, Moy M, Maness K, Tran LN, Goodrum FD. 2020. The role of the human cytomegalovirus UL133-UL138 gene locus in latency and reactivation. Viruses 12:714. doi: 10.3390/v12070714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Petrucelli A, Rak M, Grainger L, Goodrum F. 2009. Characterization of a novel Golgi apparatus-localized latency determinant encoded by human cytomegalovirus. J Virol 83:5615–5629. doi: 10.1128/JVI.01989-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Umashankar M, Rak M, Bughio F, Zagallo P, Caviness K, Goodrum FD. 2014. Antagonistic determinants controlling replicative and latent states of human cytomegalovirus infection. J Virol 88:5987–6002. doi: 10.1128/JVI.03506-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li G, Rak M, Nguyen CC, Umashankar M, Goodrum FD, Kamil JP. 2014. An Epistatic relationship between the viral protein kinase UL97 and the UL133-UL138 latency locus during the human cytomegalovirus lytic cycle. J Virol 88:6047–6060. doi: 10.1128/JVI.00447-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang D, Li G, Schauflinger M, Nguyen CC, Hall ED, Yurochko AD, von Einem J, Kamil JP. 2013. The ULb' region of the human cytomegalovirus genome confers an increased requirement for the viral protein kinase UL97. J Virol 87:6359–6376. doi: 10.1128/JVI.03477-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Caviness K, Cicchini L, Rak M, Umashankar M, Goodrum F. 2014. Complex expression of the UL136 gene of human cytomegalovirus results in multiple protein isoforms with unique roles in replication. J Virol 88:14412–14425. doi: 10.1128/JVI.02711-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Caviness K, Bughio F, Crawford LB, Streblow DN, Nelson JA, Caposio P, Goodrum F. 2016. Complex interplay of the UL136 isoforms balances cytomegalovirus replication and latency. mBio 7:e01986. doi: 10.1128/mBio.01986-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mlera L, Zeltzer S, Collins-McMillen D, Buehler JC, Moy M, Caviness K, Cicchini L, Goodrum F. 2022. LXR-inducible host E3 Ligase IDOL targets a human cytomegalovirus reactivation determinant to promote latency. bioRxiv. doi: 10.1101/2022.11.15.516687:2022.11.15.516687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McSharry BP, Tomasec P, Neale ML, Wilkinson GWG. 2003. The most abundantly transcribed human cytomegalovirus gene (beta 2.7) is non-essential for growth in vitro. J Gen Virol 84:2511–2516. doi: 10.1099/vir.0.19298-0 [DOI] [PubMed] [Google Scholar]
- 47. Collins-McMillen D, Rak M, Buehler JC, Igarashi-Hayes S, Kamil JP, Moorman NJ, Goodrum F. 2019. Alternative promoters drive human cytomegalovirus reactivation from latency. Proc Natl Acad Sci U S A 116:17492–17497. doi: 10.1073/pnas.1900783116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fehr AR, Yu D. 2011. Human cytomegalovirus early protein pUL21A promotes efficientviral DNA synthesis and the late accumulation of immediate-early transcripts. J Virol 85:663–674. doi: 10.1128/JVI.01599-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Camus S, Menéndez S, Cheok CF, Stevenson LF, Laín S, Lane DP. 2007. Ubiquitin-independent degradation of p53 mediated by high-risk human papillomavirus protein E6. Oncogene 26:4059–4070. doi: 10.1038/sj.onc.1210188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Erales J, Coffino P. 2014. Ubiquitin-independent proteasomal degradation. Biochim Biophys Acta 1843:216–221. doi: 10.1016/j.bbamcr.2013.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hwang J, Kalejta RF. 2007. Proteasome-dependent, ubiquitin-independent degradation of Daxx by the viral pp71 protein in human cytomegalovirus-infected cells. Virology 367:334–338. doi: 10.1016/j.virol.2007.05.037 [DOI] [PubMed] [Google Scholar]
- 52. Kalejta RF, Shenk T. 2003. Proteasome-dependent, ubiquitin-independent degradation of the Rb family of tumor suppressors by the human cytomegalovirus pp71 protein. Proc Natl Acad Sci U S A 100:3263–3268. doi: 10.1073/pnas.0538058100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Winkler LL, Hwang J, Kalejta RF. 2013. Ubiquitin-independent proteasomal degradation of tumor suppressors by human cytomegalovirus pp71 requires the 19S regulatory particle. J Virol 87:4665–4671. doi: 10.1128/JVI.03301-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Peppenelli M, Buehler J, Goodrum F. 2021. Human hematopoietic long-term culture (hLTC) for human cytomegalovirus latency and reactivation. Methods Mol Biol 2244:83–101. doi: 10.1007/978-1-0716-1111-1_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rak MA, Buehler J, Zeltzer S, Reitsma J, Molina B, Terhune S, Goodrum F. 2018. Human cytomegalovirus UL135 interacts with host adaptor proteins to regulate epidermal growth factor receptor and reactivation from latency. J Virol 92:e00919-18. doi: 10.1128/JVI.00919-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stern-Ginossar N, Weisburd B, Michalski A, Le VTK, Hein MY, Huang S-X, Ma M, Shen B, Qian S-B, Hengel H, Mann M, Ingolia NT, Weissman JS. 2012. Decoding human cytomegalovirus. Science 338:1088–1093. doi: 10.1126/science.1227919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Grainger L, Cicchini L, Rak M, Petrucelli A, Fitzgerald KD, Semler BL, Goodrum F. 2010. Stress-inducible alternative translation initiation of human cytomegalovirus latency protein pUL138. J Virol 84:9472–9486. doi: 10.1128/JVI.00855-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schommartz T, Tang J, Brost R, Brune W. 2017. Differential requirement of human cytomegalovirus UL112-113 protein isoforms for viral replication. J Virol 91:e00254-17. doi: 10.1128/JVI.00254-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Isomura H, Stinski MF, Kudoh A, Murata T, Nakayama S, Sato Y, Iwahori S, Tsurumi T. 2008. Noncanonical TATA sequence in the UL44 late promoter of human cytomegalovirus is required for the accumulation of late viral transcripts. J Virol 82:1638–1646. doi: 10.1128/JVI.01917-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kerry JA, Priddy MA, Kohler CP, Staley TL, Weber D, Jones TR, Stenberg RM. 1997. Translational regulation of the human cytomegalovirus pp28 (UL99) late gene. J Virol 71:981–987. doi: 10.1128/JVI.71.2.981-987.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Seo J-Y, Britt WJ. 2006. Sequence requirements for localization of human cytomegalovirus tegument protein pp28 to the virus assembly compartment and for assembly of infectious virus. J Virol 80:5611–5626. doi: 10.1128/JVI.02630-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wakeman BS, Izumiya Y, Speck SH. 2017. Identification of novel Kaposi's sarcoma-associated herpesvirus orf50 transcripts: discovery of new RTA Isoforms with variable transactivation potential. J Virol 91:e01434-16. doi: 10.1128/JVI.01434-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Reuven NB, Antoku S, Weller SK. 2004. The UL12.5 gene product of herpes simplex virus type 1 exhibits nuclease and strand exchange activities but does not localize to the nucleus. J Virol 78:4599–4608. doi: 10.1128/jvi.78.9.4599-4608.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bughio F, Umashankar M, Wilson J, Goodrum F. 2015. Human cytomegalovirus UL135 and UL136 genes are required for postentry tropism in endothelial cells. J Virol 89:6536–6550. doi: 10.1128/JVI.00284-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Michel D, Pavić I, Zimmermann A, Haupt E, Wunderlich K, Heuschmid M, Mertens T. 1996. The Ul97 gene product of human cytomegalovirus is an early-late protein with a nuclear localization but is not a nucleoside kinase. J Virol 70:6340–6346. doi: 10.1128/JVI.70.9.6340-6346.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Buehler J, Zeltzer S, Reitsma J, Petrucelli A, Umashankar M, Rak M, Zagallo P, Schroeder J, Terhune S, Goodrum F. 2016. Opposing regulation of the EGF receptor: a molecular switch controlling cytomegalovirus latency and replication. PLoS Pathog 12:e1005655. doi: 10.1371/journal.ppat.1005655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Buehler J, Carpenter E, Zeltzer S, Igarashi S, Rak M, Mikell I, Nelson JA, Goodrum F. 2019. Host signaling and EGR1 transcriptional control of human cytomegalovirus replication and latency. PLoS Pathog 15:e1008037. doi: 10.1371/journal.ppat.1008037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Smith NA, Chan GC, O’Connor CM. 2021. Modulation of host cell signaling during cytomegalovirus latency and reactivation. Virol J 18:207. doi: 10.1186/s12985-021-01674-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Stanton RJ, Prod’homme V, Purbhoo MA, Moore M, Aicheler RJ, Heinzmann M, Bailer SM, Haas J, Antrobus R, Weekes MP, Lehner PJ, Vojtesek B, Miners KL, Man S, Wilkie GS, Davison AJ, Wang ECY, Tomasec P, Wilkinson GWG. 2014. HCMV pUL135 remodels the actin cytoskeleton to impair immune recognition of infected cells. Cell Host Microbe 16:201–214. doi: 10.1016/j.chom.2014.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ferrell JE, Ha SH. 2014. Ultrasensitivity part III: cascades, bistable switches, and oscillators. Trends Biochem Sci 39:612–618. doi: 10.1016/j.tibs.2014.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sinzger C, Hahn G, Digel M, Katona R, Sampaio KL, Messerle M, Hengel H, Koszinowski U, Brune W, Adler B. 2008. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J Gen Virol 89:359–368. doi: 10.1099/vir.0.83286-0 [DOI] [PubMed] [Google Scholar]
- 72. Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33:e36. doi: 10.1093/nar/gni035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. 2000. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A 97:5978–5983. doi: 10.1073/pnas.100127597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hu Y, Smyth GK. 2009. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 347:70–78. doi: 10.1016/j.jim.2009.06.008 [DOI] [PubMed] [Google Scholar]
- 75. Smith MS, Goldman DC, Bailey AS, Pfaffle DL, Kreklywich CN, Spencer DB, Othieno FA, Streblow DN, Garcia JV, Fleming WH, Nelson JA. 2010. Granulocyte-colony stimulating factor reactivates human cytomegalovirus in a latently infected humanized mouse model. Cell Host Microbe 8:284–291. doi: 10.1016/j.chom.2010.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw viral genome sequences read have been deposited in the Sequence Read Archive (SRA) (BioProject ID: PRJNA926319).