

ABSTRACT
HIV-infected macrophages are long-lived cells that represent a barrier to functional cure. Additionally, low-level viral expression by central nervous system (CNS) macrophages contributes to neurocognitive deficits that develop despite antiretroviral therapy (ART). We recently identified H3K9me3 as an atypical epigenetic mark associated with chronic HIV infection in macrophages. Thus, strategies are needed to suppress HIV-1 expression in macrophages, but the unique myeloid environment and the responsible macrophage/CNS-tropic strains require cell/strain-specific approaches. Here, we generated an HIV-1 reporter virus from a CNS-derived strain with intact auxiliary genes expressing destabilized luciferase. We employed this reporter virus in polyclonal infection of primary human monocyte-derived macrophages (MDM) for a high-throughput screen (HTS) to identify compounds that suppress virus expression from established macrophage infection. Screening ~6,000 known drugs and compounds yielded 214 hits. A secondary screen with 10-dose titration identified 24 meeting criteria for HIV-selective activity. Using three replication-competent CNS-derived macrophage-tropic HIV-1 isolates and viral gene expression readout in MDM, we confirmed the effect of three purine analogs, nelarabine, fludarabine, and entecavir, showing the suppression of HIV-1 expression from established macrophage infection. Nelarabine inhibited the formation of H3K9me3 on HIV genomes in macrophages. Thus, this novel HTS assay can identify suppressors of HIV-1 transcription in established macrophage infection, such as nucleoside analogs and HDAC inhibitors, which may be linked to H3K9me3 modification. This screen may be useful to identify new metabolic and epigenetic agents that ameliorate HIV-driven neuroinflammation in people on ART or prevent viral recrudescence from macrophage reservoirs in strategies to achieve ART-free remission.
IMPORTANCE
Macrophages infected by HIV-1 are a long-lived reservoir and a barrier in current efforts to achieve HIV cure and also contribute to neurocognitive complications in people despite antiretroviral therapy (ART). Silencing HIV expression in these cells would be of great value, but the regulation of HIV-1 in macrophages differs from T cells. We developed a novel high-throughput screen for compounds that can silence established infection of primary macrophages, and identified agents that downregulate virus expression and alter provirus epigenetic profiles. The significance of this assay is the potential to identify new drugs that act in the unique macrophage environment on relevant viral strains, which may contribute to adjunctive treatment for HIV-associated neurocognitive disorders and/or prevent viral rebound in efforts to achieve ART-free remission or cure.
KEYWORDS: HIV, macrophage, epigenetics, silencing, high-throughput screen
INTRODUCTION
Combination antiretroviral therapy (ART) is effective at blocking new rounds of HIV-1 infection, which suppresses viral load, prevents CD4+ T-cell loss, and prevents forward transmission. However, ART does not affect already-infected cells, which can persist and harbor virus for the life of the individual. The primary targets of HIV-1 are CD4+ T cells and macrophages. CD4+ T cells are the main infected cell type in lymphoid tissues and blood, whereas macrophage lineage cells are the principal host cell in the central nervous system (CNS). Activated, productively infected CD4+ T cells are generally killed by infection, whereas long-lived resting CD4+ T cells are typically latent, with little or no virus expression. In contrast, infected macrophages typically are not killed by infection and can produce virus for prolonged periods without achieving complete latency (1, 2).
While ART is highly effective under ideal circumstances, it requires life-long adherence to treatment. Only ~65% of people living with HIV in the US are durably suppressed, largely due to social and structural factors that limit sustained engagement in care (3). Viremia inevitably rebounds if ART is stopped, ignited by long-lived reservoir cells, resulting in both CD4+ T-cell loss and the ability to transmit to others. Thus, strategies to achieve a long-term aviremic state in the absence of continuous ART are a high priority. Much attention has been directed to eradication of the CD4+ T-cell reservoir or establishment of a state resistant to viral reactivation, but myeloid cells within the CNS may also serve to re-ignite systemic viremia in the absence of ART. Strategies to inhibit virus expression from the CNS macrophage reservoir are therefore needed.
Additionally, many people living with HIV develop HIV-associated neurocognitive disorders (HANDs) even with viral suppression. While HAND that develops on ART is milder than that in viremic individuals, it is nevertheless a common and important comorbidity that impairs function and quality of life (4 - 6). ART prevents new rounds of infection but does not prevent virus expression by persistently infected cells, and an important driver of HAND on ART is persistent or intermittent virus gene and protein expression by infected myeloid cells in brain that leads to bystander injury and neuroinflammation (7, 8). Thus, suppressing residual virus expression by these long-lived infected macrophages would be a valuable adjunct to prevent neurocognitive disease in these individuals.
Infection of CD4+ T cells and macrophages differ in several important respects. CD4+ T-cell infection occurs mainly in the context of activated, proliferating cells (although some infection may occur in resting cells) (9, 10). In contrast, monocytes are generally resistant to infection but become permissive once they mature into non-dividing terminally differentiated macrophages (2, 11). Thus, macrophage infection occurs in the context of a distinct nuclear environment. We showed recently that HIV-1 exhibits a unique epigenetic landscape in primary macrophages characterized by an atypical epigenetic profile that differed from both acutely infected CD4+ T cells and latently infect JLAT T cells (12). Furthermore, all HIV-1 strains have the capacity to infect CD4+ T cells, but only a subset of virus isolates have the ability to infect and replicate in macrophages. While macrophage tropism is determined largely at entry by the ability to use the entry co-receptor CCR5 (and occasionally CXCR4) in conjunction with exceedingly low levels of CD4 expressed by macrophages (13), viral sequences obtained directly from brain often display unique features within the long terminal repeat (LTR) control region (14, 15). We recently found that HIV-infected macrophages were enriched in histone H3K9me3, an epigenetic mark typically associated with repetitive DNA and heterochromatin (12). Thus, the epigenetic features and nuclear environment that regulate viral expression likely differ in macrophages from that in CD4+ T cells, indicating the need for models that recapitulate these interactions.
We sought to develop a high-throughput screen (HTS) suitable to identify compounds that suppress HIV expression from established infection in macrophages. Because macrophage infection occurs in the context of a terminally differentiated non-dividing cell that may impact epigenetic regulatory control, whereas cell lines are continuously replicating, we employed differentiated post-mitotic primary human monocyte-derived macrophages (MDM). Because brain-derived macrophage-tropic HIV-1 isolates may have unique regulatory regions, we employed a primary virus cloned directly from brain of an infected individual. Finally, because HIV-1 accessory proteins have both known and potentially unknown regulatory functions, we constructed a reporter virus with all accessory genes intact. We show that this system can be used for screening libraries for compounds that silence HIV from established provirus which can then be validated in orthogonal assays of HIV/MDM suppression.
RESULTS
A brain-derived reporter virus suitable for primary macrophage-based screening
To best reflect regulation of virus in the myeloid reservoir in vivo, we considered several features important for a screening assay: (i) Because the CNS is the primary myeloid reservoir in the body, we generated a reporter construct based on a CNS-derived primary isolate, YU2, which was cloned directly from the brain of a patient with encephalopathy (16). (ii) While many reporters used to study HIV-1 regulation rely on LTR-only or other sub-viral constructs, we wished to conserve all potential known or unknown regulatory components in the context of an intact virus. Therefore, we used a viral molecular clone that was fully intact (except for env) into which we placed the reporter gene upstream of nef with a P2A ribosomal skip site so that both would be expressed and the reporter would be under the same control as nef. (iii) Infection of macrophages occurs only when they mature to terminally differentiated post-mitotic cells, which may result in unique epigenetic features that differ from replicating cells or cell lines, and thus likely best represented by differentiated MDM. (iv) Because non-dividing macrophages do not undergo cytoplasmic dilution, proteins may be more stable than in dividing cells, so we wished to employ a destabilized reporter that would be maximally responsive to changes in gene expression. (v) Although models with clonal integration can provide greater homogeneity, effects on viral gene expression following polyclonal integration of natural infection would be best reflected in a similar infection approach.
The reporter construct YU2/Δenv/nLuc is shown in Fig. 1A. Key features are that it is an env-deleted but otherwise intact viral genome cloned directly from human brain of a patient with HIV infection, possesses all accessory genes intact and its native LTRs, and carries a destabilized reporter in frame with and under the same control as nef expression.
Fig 1.

Primary MDM silencing assay. (A) Schematic of brain-derived macrophage-tropic HIV-1 reporter construct YU2/Δenv/nLuc. The destabilized nano-luciferase (nLuc) reporter was placed upstream of a P2A ribosomal skip site and inserted at the 5’ end of the nef gene of HIV-1 YU2. A premature stop was created in env by introduction of a 2-bp insertion at codon 59 of the open reading frame. (B) Overview of primary cell assay. Monocytes were cultured for 6 d to mature into MDM and then infected with the VSVg-pseudotyped reporter virus. They were cultured for 7 d more to allow infection to proceed, then re-plated into 384 well plates. One day later, test compounds were added and maintained in the cultures for 5 d. Cells were then harvested and nLuc for virus expression and CellGlo for cell viability measured in cell lysates of replicate plates. (C) Timing the completion of integration after reporter virus infection. The integrase inhibitor raltegravir (RTG) was added 1 d prior to, 1 d after, or 4 d after MDM were infected with the VSVg-pseudotyped reporter virus. By 4 d after infection, infection was resistant to integration blocking.
Figure 1B shows a schematic of the screening protocol. Monocytes were differentiated for 6 d into MDM, then infected with YU2/Δenv/nLuc pseudotyped with VSVg. We first determined the optimal number of MDM per well and determined that 2.5 × 103 cells per well of a 384-well plate was within the linear range of the luciferase assay and would allow 10 plates to be created from a typical donor yield of 10 × 106 MDM. We next considered the length of time to wait before adding test compounds to allow integration to complete and an epigenetic complex to form, to reflect suppression of virus expression from an established macrophage infection. We therefore carried out time-of-addition experiments with the integrase inhibitor raltegravir. As shown in Fig. 1C, raltegravir treatment prior to infection blocked >99% of Luc output, as expected; treatment 1 d after infection had a modest effect on Luc output, while treatment 4 d after infection had no effect. Thus, integration is complete by 4 d after infection. Thus, we reasoned that the addition of compounds at day 8 would allow completion of integration by 4 d plus another 4 d for maturation of epigenetic features that regulate HIV-1 expression and might be targeted for suppression.
Screening chemical libraries for silencing agents
We screened four chemical libraries (Table S1) encompassing ~6,000 compounds. The SelleckChem library (850 compounds), Spectrum library (2,500 compounds), and MedChem Anticancer library (2,500 compounds) were screened at a single concentration (10 nM), while the EPGN library (80 compounds) was screened at two concentrations (1 nM and 10 nM). Compounds were tested in two replicate plates. One plate was assayed for nLuc as a measure of virus expression from the integrated provirus (in the nef reading frame), and one plate was assayed for CellTiter-Glo as a measure of cellular viability and metabolic activity (based on ATP content). The overall HTS assay performed with a Z′ =0.52 indicating that is sufficiently robust for large-scale screening (Fig. S1). Our criteria for a “hit” were >80% inhibition of nanoLuc (surrogate for HIV transcription activity) and <25% inhibition of CellTiter-Glo (surrogate for cell toxicity). The first round screen (~6,000 compounds) identified 214 hits for HIV-1 macrophage infection silencing. A summary waterfall plot showing the top hits is shown in Fig. 2. Among the top hits for suppression of HIV transcription were nucleoside analogs (e.g., floxuridine, vidarabine, cytarabine, nelarabine), as well as napabucasin (STAT inhibitor), enasidenib (IDH2 inhibitor), and plx51107 (BET inhibitor). Interestingly, we also identified several activators of HIV transcription in MDM, such as cilnidipine, nimsulidipine, and nicardipine, which all fall into the class of calcium channel blockers.
Fig 2.

HTS of compounds. Waterfall plot of the HTS primary screen of 4,800 compounds from the SelleckChem library. Compounds were added at 10 μM, and duplicate plates were analyzed in parallel for nLuc expression from integrated provirus in MDM, and for CellTiter-Glo as an indication of cell viability. Compounds were ranked by percent inhibition of nanoLuc (Y-axis) and filtered for <25% change in CellTiter-Glo.
Dose titration analysis
The 214 hits from the primary screen were then assayed in a 10-point dose titration experiment (three-fold dilutions from 10 μM through 0.3 nM) using the identical protocol. A representative set of graphs is shown in Fig. 3. We found that nelarabine and fludarabine had among the best therapeutic windows, with nanoLuc IC50 values of ~10 nM and ~200 nM, respectively, and cell toxicity CC50 (CellTiter-Glo assay) >5 µM. Entecavir (nucleoside analog), pyroxamide (HDAC1 inhibitor), and M341 (HDAC inhibitor) also showed favorable therapeutic windows (Fig. 3).
Fig 3.

Titration screening of compounds. Compounds were tested at 10 concentrations from 0.1 nM to 10 mM. Representative compounds are shown that displayed dose-dependent separation between nLuc and CellGlo inhibition. Out of 214 compounds subject to titration screening, 24 showed dose-dependent separation between virus inhibition and cell toxicity.
Orthogonal validation using infectious virus
We next wished to test compounds in the context of live virus infection. First, to recapitulate the YU-2-based screen with a live virus, we used infectious virus derived from the YU-2 infectious molecular clone (IMC). To augment infectivity of this strain for maximal readout, the IMC was co-transfected with VSVg to generate mixed pseudotypes, yielding highly efficient infection. Monocytes were differentiated for 6 d into MDM, infected, and maintained in culture for an additional 5 d. The reverse transcriptase inhibitor efavirenz was then added to block new rounds of infection, and 3 d later test compounds were added. This timing was designed to allow integration to complete and epigenetic marks to assemble prior to compound addition. Effects on virus expression were assayed 5 d after that.
We first tested the effects of viral p24 Gag antigen both in supernatant and in cell lysates. Unexpectedly, high concentrations of the transcriptional inhibitor actinomycin D had minimal effect on p24 protein (data not shown). This result is likely due to high stability of the viral protein combined with the fact that virus budding in macrophages is largely into intracellular vesicles (17, 18), which together may make p24 very insensitive to changes in transcription.
Therefore, we assessed the effects of viral gene expression, focusing on two early gene transcripts, nef and tat. As shown in Fig. 4A, nelarabine, fludarabine, and to a lesser extent entecavir had dose-dependent selective suppression of HIV-1 gene expression from established macrophage infection.
Fig 4.

Testing of compounds in MDM infection with different HIV-1 strains. Six-day-old MDM were infected with three replication-competent CNS-derived HIV-1 strains. Five days after infection, efavirenz was added to prevent further rounds of infection. Three days later, compounds were added at four concentrations (0.3, 1, 3, and 10 μM) in the continued presence of efavirenz. Cells were harvested 5 d later and subject to qRT-PCR for expression of the viral tat (blue) and nef (red) transcripts, and cellular GUSB (green). Gene expression is shown as percent inhibition relative to untreated control infections, calculated by the ΔCt method. Infections were carried out using duplicate wells of MDM, and data reflect means ± SEM of three independent infectious using different MDM donors. (A) HIV-1 YU-2 derived from the IMC, carrying the VSVg envelope in mixed pseudotypes to boost infectivity; (B) HIV-1 JRFL; and (C) HIV-1 Jago.
We next tested two uncloned virus isolates. JRFL is a prototype CNS-derived macrophage-tropic strain that was isolated from brain tissue of a patient with HIV encephalopathy (19). Jago is a CNS-derived macrophage-tropic strain primary isolate derived from cerebrospinal fluid of a patient with HIV encephalopathy (20). Like YU-2, evidence for dose-dependent selective suppression of viral gene expression was seen with both CNS-derived isolates (Fig. 4B and C). Thus, features identified in our HTS were validated for some compounds in an authentic model of established HIV-1 macrophage infection.
Nelarabine decreases viral-associated H3K9me3 histone modification
To further investigate the mechanism of action of the nucleoside analogs on HIV activity in macrophages, we assayed several additional viral transcripts in MDM infected with YU-2 (Fig. 5). In particular, we wished to assay 5′ nascent transcripts that might not be dependent on Tat anti-termination activity and might best reflect transcriptional activity [TAR-short and TAR-long (21)]. Cells treated with nelarabine or fludarabine showed a significant reduction in TAR region RNA, as well as gag, tat, and nef RNA, supporting an effect on transcription. Since nelarabine had the better therapeutic window (less cytotoxicity in MDM), we focused on this drug for analysis of the HIV epigenome by ChIP assay.
Fig 5.
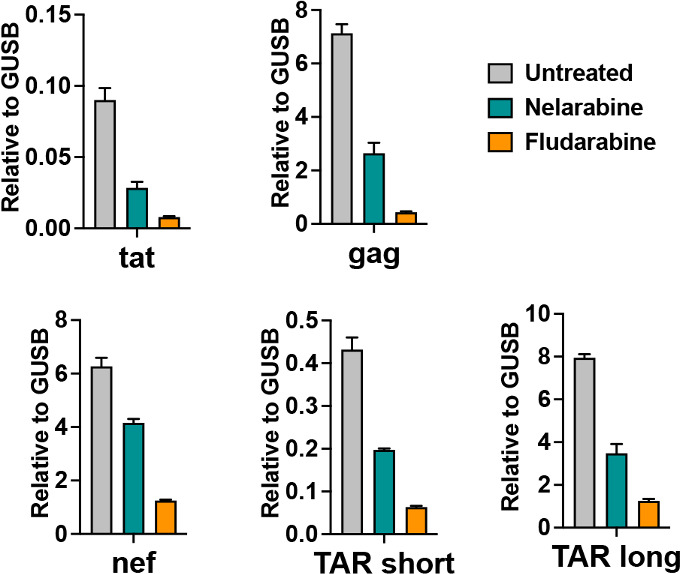
Nucleoside analogs nelarabine and fludarabine reduce HIV transcripts. MDM were infected with YU-2/VSVg and efavirenz added 5 d later to prevent new rounds of infection. Four days later, they were treated with or without nelarabine (3 µM) or fludarabine (3 μM) . RNA was isolated 7 d later and assayed by RT-qPCR with primers targeting HIV tat, gag, nef and two different regions of the 5′ nascent transcript (TAR-short and TAR-long). Transcription is shown as fold over cellular GUSB levels, calculated by the ΔΔCt method.
Previously, we showed that histone H3K9me3 was highly enriched on HIV genomes in actively infected MDM (12). We, therefore, assayed the effect of nelarabine treatment on H3K9me3 at various positions across the HIV genome (Fig. 6). We also examined cellular loci known to have high levels of H3K9me3 (e.g., subtelomeric sites 10q_CTCF and 10q_TERRA) and positions known to be depleted of H3K9me3 (e.g., transcription start sites for actin and GAPDH genes). As controls, we assayed total histone H3 and IgG control antibody. ChIP analysis showed that nelarabine treatment reduced H3K9me3 levels at most positions across the HIV genome, as well as at cellular subtelomeres. In contrast, nelarabine had no effect on total H3 or IgG levels. These results indicate that nucleoside analogs reduce transcription from the HIV provirus and that nelarabine treatment decreases H3K9me3 throughout the HIV genome and at cellular subtelomeres as well.
Fig 6.

Nelarabine reduces H3K9me3 on the HIV proviral genome and cellular subtelomeres. MDM were infected with YU-2/VSVg. Seven days later, efavirenz was added to block further rounds of reinfection, and 3 d later treated with or without nelarabine (3 µM). Six days later, crosslinking was carried out and nuclei isolated. Cells were assayed by ChIP for association of H3K9me3, total histone H3, or IgG control using primers positioned throughout the HIV genome, or at control loci in the human genome (actin, GAPDH, subtelomere 10q_CTCF, and 10q_TERRA). (A) HIV-1 genome regions targeted by PCR primers; (B) H3K9me3; (C) Total histone H3; and (D) IgG control. Data are presented as percent of input DNA for each primer pair and presented as means ± SEM for n = 3 technical replicates.
DISCUSSION
HIV-infected macrophages are found mainly in the CNS. If current cure-focused efforts to eradicate or silence HIV in the T-cell reservoir were to succeed, CNS macrophages could serve as an alternative long-lived reservoir and source of recrudescent viremia if ART were to be stopped. In addition, even during ART that effectively blocks new rounds of infection, intermittent or persistent low-level virus expression from these long-lived cells contributes to ongoing neuroinflammation and neurocognitive dysfunction that has emerged as an important comorbidity in virologically suppressed individuals. For both of those reasons, strategies to silence HIV in macrophages would be highly desirable. However, the unique features of macrophage infection and brain-derived macrophage-tropic virus variants suggest that focused efforts to target this interaction will be needed.
The HIV Cure agenda includes efforts both to increase expression from the resting CD4+ T-cell latent reservoir to enable virus detection and targeting (kick-and-kill) and to prevent reactivation of HIV from latency in these cells (lock-and-block). The lock-and-block approach may have some advantages for CNS macrophage infection, however. First, kick-and-kill may be challenging in the CNS, where acutely increased virus expression and/or immune-mediated recognition and killing could have deleterious consequences (22). Second, long-lived infected macrophages exhibit intermittent or persistent low-level virus expression, in contrast to latent resting CD4+ T cells. Indeed, even transformed cell line models for human myeloid cell infection such as the human microglia-derived microglia/HIV line undergo progressive spontaneous virus reactivation in culture (23). Thus, lock-and-block might have utility even in the context of ART-suppressed patients by reducing virus expression and consequent neuroinflammation. Our HTS assay provides a unique primary cell and CNS virus model for identifying agents that may be useful in this strategy.
The mechanisms that control HIV expression in macrophages are not well understood, but evidence suggests that it differs from better-studied CD4+ T cells. Factors implicated in HIV regulation in microglial cells include the transcription factor C/EBP-alpha, histone demethylase LSD1, HIC1, HMGA1, COUP-TF, CTIP2, and HPI-alpha (24 - 27). We recently reported that HIV-1 proviruses in MDM have a unique atypical histone methylation pattern characterized by heterochromatin-associated H3K9me3 across the whole HIV genome, combined with H3K9ac and H3K27ac activation markers at the LTR (12). This pattern was distinct from both activated CD4+ T cells replicating virus and the JLAT cell model of latent T-cell infection. We also found enrichment of 5-hydroxymethylcytosine across the proviral genome in MDM, which was similar to activated infected CD4+ T cells and contrasted with 5-methylcytosine enrichment in latent JLATs. Thus, strategies that modulate macrophage viral gene expression will likely differ from what works in latent resting T cells. Here, we found that nelarabine reduced H3K9me3 at both HIV and cellular genomic loci, suggesting that nucleoside analogs may affect HIV through some epigenetic pathways. Future studies will be required to address this potential mechanism of action.
Nelarabine, fludarabine, and entecavir demonstrated silencing activity in our screen and showed evidence of activity in a whole virus infection assay with three CNS-derived virus strains. Nelarabine and fludarabine are purine nucleoside analogs that inhibit DNA synthesis and are used as anticancer agents. While these agents would be too toxic for therapeutic use in this context, they may point toward novel mechanisms or other compounds with lesser toxicity. Entecavir is a nucleoside reverse transcriptase inhibitor used to treat hepatitis B virus infection. While entecavir has some modest activity against HIV-1 reverse transcriptase (28), our model isolates late stages of infection after reverse transcription and integration, so it is likely working through alternative mechanisms. Given the distinct mechanisms of HIV-1 regulation in macrophages compared to T cells, future studies be of interest to determine the effects of the agents identified here on virus expression or reactivation from latency in CD4+ T cells. We also identified several calcium channel blockers that active HIV transcription. Although we did not further characterize these compounds, they may be of interest for the kick-and-kill strategy, as well as potential risks for the wide use of these drugs in the HIV population. Nevertheless, these results demonstrated the ability to identify novel lead compounds in this screen.
In conclusion, the use of brain-derived viruses and primary human myeloid cells will be important to model the unique virus/cell environment in efforts to silence HIV within this compartment. While less efficient than cell lines and partial constructs, our system offers a viable approach to HTS for such agents.
MATERIALS AND METHODS
Brain-derived macrophage-tropic primary isolate reporter virus
The IMC of HIV-1 YU2 (16) with intact accessory genes, kindly provided by B. Hahn (University of Pennsylvania), was used as the basis for the construct. Mutations were introduced by PCR and primers used are shown in Table S2. First, a 2-bp insertion was introduced into codon 59 of env, leading to a frameshift and stop codon. A Not1 site was then introduced between the termination codon of env and initiation codon of nef by overlap extension PCR. A reporter cassette was generated by sequential PCRs consisting of (a) a 5′ Not1 site; (b) a Kozak initiation sequence (5 bp); (c) the NanoLucP (nLuc) reporter gene (513 bp) containing luciferase fused to a PEST destabilization sequence (120 bp) (Promega pNL1.2) lacking its termination codon; (d) a P2A ribosomal skip sequence (66 bp) to enable expression of both NanoLucP and Nef; and (e) a 3’ Not1 site. The reporter cassette was introduced into the Δenv YU2 IMC at the Not1 site between env and nef, which placed nLuc and nef in-frame. This construct was designated YU2/Δenv/nLuc.
To generate pesudotyped reporter virions, pYU2/Δenv/nLuc and pVSVg (1 μg each) were co-transfected into 293T cells using Fugene (Promega). Cells were washed, re-fed, and supernatant was harvested 3 d later and treated with DNAse for 20 min (to avoid inadvertent transfection of target cells). Reporter virus was quantified by p24 antigen content by ELISA, and stored at −80°C until use.
Monocyte-derived macrophages
Primary human monocytes were isolated from healthy donors by leukapheresis followed by negative selection (Rosette-Sep; StemCell Technologies) and provided by the Immunology Core of the Penn Center for AIDS Research under their IRB-approved protocols. Monocytes were cultured in Iscove’s modified Dulbecco’s medium containing 10% human AB serum with penicillin, streptomycin, and 1% glutamine (MDM media).
High-throughput screen
Monocytes were plated in 6-well plates (2 × 106 cells/well) and cultured for 6 d to allow differentiation into MDM. MDM were then infected with the reporter virus using 30 ng viral p24 antigen per well. Seven days later, the media was removed, cells were detached using ethylenediaminetetraacetic acid and gentle scraping, washed, and re-suspended in MDM media to which the protease inhibitor nelfinavir (2 μM) was added (as an additional precaution against infectious virus to be handled in the HTS facility).
Cells were transported to the HTS facility and plated at 2,500 cells/well in white, opaque, tissue culture-treated 384-well plates (Griener 781080) in 40 μL of MDM media containing nelfinavir using the MicroFlo bulk dispenser (Biotek). The next day, 40 nL of 1000X test compound in 100% dimethylsulfoxide (DMSO) was added using the Janus MDT Nanohead (PerkinElmer). As a positive control, actinomycin (5 μM final concentration) or bortezomib (10 μM) was added to some wells. Parallel assay plates were set up such that one could be used to measure NanoLuc reporter activity, while the other could be used for cell viability. Three days later, 20 μL of either NanoLuc reagent (Promega, N1120) or CellTiterGlo (Promega, G7573) was added using the MicroFlo bulk dispenser. After 15–30 min, the luminescence was measured using the Envision Multimode plate reader (PerkinElmer). Relative luminescence unit (RLU) values were converted to percent inhibition (luciferase expression) or percent toxicity (CellTiterGlo), where 0% was equal to the RLU values in the presence of DMSO, and 100% was equal to the RLU in the presence of 5 μM actinomycin for luciferase reporter activity, or 10 μM bortezomib for cell viability. Z′ values were calculated according to the standard formula of Z′ = (3*SDLowControl +3*SDHighControl)/(HighControl – LowControl). The screen established a Z ′=0.52 (Fig. S2). Hits were cherry picked and revalidated by 10-point titration curves, which was carried out using duplicate wells.
Live virus macrophage infections
An orthogonal live virus assay for viral suppression used replication-competent CNS-derived HIV-1 primary isolates JRFL (19) and Jago (20), as well as YU2 derived from the IMC. JRFL and Jago virus stocks were generated in peripheral blood mononuclear cells. To boost infectivity titers of YU2, the intact IMC was co-transfected with VSVg to generate mixed pseudotypes carrying both HIV-1 and VSV envelope glycoproteins and harvested 3 d later. Monocytes were plated at 7 × 105 cells/well in 48-well plates and infected 6 d later using 7 ng p24 antigen per well of each virus. One day after infection, the inoculum was removed and cells were re-fed. Four days later, the reverse transcriptase inhibitor efavirenz (2 μM) was added to prevent further rounds of infection, and test compounds (re-purchased as fresh stocks; ThermoFisher Scientific) were added 3 d later. This approach ensured that treatment would reflect effects on established infection rather than possibly inhibiting early steps of new rounds of infection. Cells were maintained in culture for another 5 d and then harvested. RNA was extracted (Qiagen RNeasy mini kit), reverse transcribed using random hexamers or decamers, and qPCR carried out for viral tat and nef sequences and the cellular GUSB housekeeping gene by SYBR Green. Expression of tat, nef and GUSB transcripts was calculated relative to untreated control infections using the ΔCt method. Experiments were done three times using cells from different donors. Additional analysis was carried out using primers to detect gag gene transcription along with two different primer sets targeting extremely nascent 5′ transcripts spanning the TAR region as described in reference (21) and modified to match YU2 sequence. Primers used for RT-qPCR are shown in Table S3.
ChIP assay
Chromatin immunoprecipitation (ChIP) assays were performed as described previously (12). MDM were infected with HIV-1 YU2 transcomplemented with VSVg, 7 d later treated with efavirenz (2 μM) to block new rounds of infection, and 3 d later treated with or without nelarabine (3 μM). Six days later, DNA/chromatin was crosslinked, sheared, and immunoprecipitated with antibodies to H3K9me3 (Diagenode; catalog no. C15410056), pan-histone H3 (MilliporeSigma; catalog no. 07–690), or control rabbit IgG (Cell Signaling; catalog no. 2729S). Precipitated DNA was quantified by qPCR using primers positioned throughout the HIV genome, or at loci in the human genome (actin, GAPDH, subtelomere 10q_CTCF, and 10q_TERRA). The percent of input DNA was calculated for each target subjected to immunoprecipitation. Details including qPCR primers are described in references (12, 29).
ACKNOWLEDGMENTS
This work was supported by NIH grant R61/33-AI133696.
We thank B. Hahn for the YU2 IMC. We also acknowledge assistance from the Penn Center for AIDS Research (P30-AI045008) and the Wistar Molecular Screening Core supported by The Wistar Institute Cancer Center Core Grant (P30-CA010815).
Contributor Information
Paul M. Lieberman, Email: Lieberman@wistar.org.
Ronald G. Collman, Email: collmanr@pennmedicine.upenn.edu.
Frank Kirchhoff, Ulm University Medical Center, Ulm, Baden-Württemberg, Germany .
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/jvi.00653-23.
Fig. S1 and S2 and Tables S1 to S3.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Sattentau QJ, Stevenson M. 2016. Macrophages and HIV-1: an unhealthy constellation. Cell Host Microbe 19:304–310. doi: 10.1016/j.chom.2016.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Collman R, Hassan NF, Walker R, Godfrey B, Cutilli J, Hastings JC, Friedman H, Douglas SD, Nathanson N. 1989. Infection of monocyte-derived macrophages with human immunodeficiency virus type 1 (HIV-1). monocyte-tropic and lymphocyte-tropic strains of HIV-1 show distinctive patterns of replication in a panel of cell types. J Exp Med 170:1149–1163. doi: 10.1084/jem.170.4.1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harris NS, Johnson AS, Huang Y-LA, Kern D, Fulton P, Smith DK, Valleroy LA, Hall HI. 2019. Vital signs: status of human immunodeficiency virus testing, viral suppression, and HIV preexposure prophylaxis - United States, 2013-2018. MMWR Morb Mortal Wkly Rep 68:1117–1123. doi: 10.15585/mmwr.mm6848e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Métral M, Darling K, Locatelli I, Nadin I, Santos G, Brugger P, Kovari H, Cusini A, Gutbrod K, Tarr PE, Calmy A, Lecompte TD, Assal F, Monsch A, Kunze U, Stoeckle M, Schwind M, Schmid P, Pignatti R, Di Benedetto C, Du Pasquier R, Cavassini M, NAMACO study group, Swiss HIV Cohort Study . 2020. The Neurocognitive assessment in the metabolic and aging cohort (NAMACO) study: baseline participant profile. HIV Med 21:30–42. doi: 10.1111/hiv.12795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Portilla I, Reus S, León R, van-der Hofstadt C, Sánchez J, López N, Boix V, Merino E, Portilla J. 2019. Neurocognitive impairment in well-controlled HIV-infected patients: a cross-sectional study. AIDS Res Hum Retroviruses 35:634–641. doi: 10.1089/AID.2018.0279 [DOI] [PubMed] [Google Scholar]
- 6. Saylor D, Dickens AM, Sacktor N, Haughey N, Slusher B, Pletnikov M, Mankowski JL, Brown A, Volsky DJ, McArthur JC. 2016. HIV-associated Neurocognitive disorder - pathogenesis and prospects for treatment. Nat Rev Neurol 12:309. doi: 10.1038/nrneurol.2016.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Johnson TP, Nath A. 2022. Biotypes of HIV-associated neurocognitive disorders based on viral and immune pathogenesis. Curr Opin Infect Dis 35:223–230. doi: 10.1097/QCO.0000000000000825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kolson DL. 2022. Developments in neuroprotection for HIV-associated neurocognitive disorders (HAND). Curr HIV/AIDS Rep 19:344–357. doi: 10.1007/s11904-022-00612-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chavez L, Calvanese V, Verdin E. 2015. HIV latency is established directly and early in both resting and activated primary CD4 T cells. PLoS Pathog 11:e1004955. doi: 10.1371/journal.ppat.1004955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dai J, Agosto LM, Baytop C, Yu JJ, Pace MJ, Liszewski MK, O’Doherty U. 2009. Human immunodeficiency virus integrates directly into naive resting CD4+ T cells but enters naive cells less efficiently than memory cells. J Virol 83:4528–4537. doi: 10.1128/JVI.01910-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rich EA, Chen IS, Zack JA, Leonard ML, O’Brien WA. 1992. Increased susceptibility of differentiated mononuclear phagocytes to productive infection with human immunodeficiency virus-1 (HIV-1). J Clin Invest 89:176–183. doi: 10.1172/JCI115559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lu F, Zankharia U, Vladimirova O, Yi Y, Collman RG, Lieberman PM. 2022. Epigenetic landscape of HIV-1 infection in primary human macrophage. J Virol 96:e0016222. doi: 10.1128/jvi.00162-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gorry PR, Francella N, Lewin SR, Collman RG. 2014. HIV-1 envelope-receptor interactions required for macrophage infection and implications for current HIV-1 cure strategies. J Leukoc Biol 95:71–81. doi: 10.1189/jlb.0713368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ross HL, Gartner S, McArthur JC, Corboy JR, McAllister JJ, Millhouse S, Wigdahl B. 2001. HIV-1 LTR C/EBP binding site sequence configurations preferentially encountered in brain lead to enhanced C/EBP factor binding and increased LTR-specific activity. J Neurovirol 7:235–249. doi: 10.1080/13550280152403281 [DOI] [PubMed] [Google Scholar]
- 15. Ross HL, Nonnemacher MR, Hogan TH, Quiterio SJ, Henderson A, McAllister JJ, Krebs FC, Wigdahl B. 2001. Interaction between CCAAT/enhancer binding protein and cyclic AMP response element binding protein 1 regulates human immunodeficiency virus type 1 transcription in cells of the monocyte/macrophage lineage. J Virol 75:1842–1856. doi: 10.1128/JVI.75.4.1842-1856.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li Y, Kappes JC, Conway JA, Price RW, Shaw GM, Hahn BH. 1991. Molecular characterization of human immunodeficiency virus type 1 cloned directly from uncultured human brain tissue: identification of replication-competent and -defective viral genomes. J Virol 65:3973–3985. doi: 10.1128/JVI.65.8.3973-3985.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Raposo G, Moore M, Innes D, Leijendekker R, Leigh-Brown A, Benaroch P, Geuze H. 2002. Human macrophages accumulate HIV-1 particles in MHC II compartments. Traffic 3:718–729. doi: 10.1034/j.1600-0854.2002.31004.x [DOI] [PubMed] [Google Scholar]
- 18. Nkwe DO, Pelchen-Matthews A, Burden JJ, Collinson LM, Marsh M. 2016. The intracellular plasma membrane-connected compartment in the assembly of HIV-1 in human macrophages. BMC Biol 14:50. doi: 10.1186/s12915-016-0272-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Koyanagi Y, Miles S, Mitsuyasu RT, Merrill JE, Vinters HV, Chen IS. 1987. Dual infection of the central nervous system by AIDS viruses with distinct cellular tropisms. Science 236:819–822. doi: 10.1126/science.3646751 [DOI] [PubMed] [Google Scholar]
- 20. Chen W, Sulcove J, Frank I, Jaffer S, Ozdener H, Kolson DL. 2002. Development of a human neuronal cell model for human immunodeficiency virus (HIV)-Infected macrophage-induced neurotoxicity: apoptosis induced by HIV type 1 primary isolates and evidence for involvement of the Bcl-2/bcl-xL-sensitive intrinsic apoptosis pathway. J Virol 76:9407–9419. doi: 10.1128/jvi.76.18.9407-9419.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lassen KG, Bailey JR, Siliciano RF. 2004. Analysis of human immunodeficiency virus type 1 transcriptional elongation in resting CD4+ T cells in vivo. J Virol 78:9105–9114. doi: 10.1128/JVI.78.17.9105-9114.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nühn MM, Gumbs SBH, Buchholtz N, Jannink LM, Gharu L, de Witte LD, Wensing AMJ, Lewin SR, Nijhuis M, Symons J. 2022. Shock and kill within the CNS: a promising HIV eradication approach. J Leukoc Biol 112:1297–1315. doi: 10.1002/JLB.5VMR0122-046RRR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alvarez-Carbonell D, Ye F, Ramanath N, Dobrowolski C, Karn J. 2019. The glucocorticoid receptor is a critical regulator of HIV latency in human microglial cells. J Neuroimmune Pharmacol 14:94–109. doi: 10.1007/s11481-018-9798-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marban C, Redel L, Suzanne S, Van Lint C, Lecestre D, Chasserot-Golaz S, Leid M, Aunis D, Schaeffer E, Rohr O. 2005. COUP-TF interacting protein 2 represses the initial phase of HIV-1 gene transcription in human microglial cells. Nucleic Acids Res 33:2318–2331. doi: 10.1093/nar/gki529 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25. Cherrier T, Le Douce V, Eilebrecht S, Riclet R, Marban C, Dequiedt F, Goumon Y, Paillart J-C, Mericskay M, Parlakian A, Bausero P, Abbas W, Herbein G, Kurdistani SK, Grana X, Van Driessche B, Schwartz C, Candolfi E, Benecke AG, Van Lint C, Rohr O. 2013. CTIP2 is a negative regulator of P-TEFb. Proc Natl Acad Sci U S A 110:12655–12660. doi: 10.1073/pnas.1220136110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Le Douce V, Colin L, Redel L, Cherrier T, Herbein G, Aunis D, Rohr O, Van Lint C, Schwartz C. 2012. LSD1 cooperates with CTIP2 to promote HIV-1 transcriptional silencing. Nucleic Acids Res 40:1904–1915. doi: 10.1093/nar/gkr857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Le Douce V, Forouzanfar F, Eilebrecht S, Van Driessche B, Ait-Ammar A, Verdikt R, Kurashige Y, Marban C, Gautier V, Candolfi E, Benecke AG, Van Lint C, Rohr O, Schwartz C. 2016. HIC1 controls Cellular- and HIV-1- gene transcription via interactions with CTIP2 and HMGA1. Sci Rep 6:34920. doi: 10.1038/srep34920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McMahon MA, Jilek BL, Brennan TP, Shen L, Zhou Y, Wind-Rotolo M, Xing S, Bhat S, Hale B, Hegarty R, Chong CR, Liu JO, Siliciano RF, Thio CL. 2007. The HBV drug entecavir - effects on HIV-1 replication and resistance. N Engl J Med 356:2614–2621. doi: 10.1056/NEJMoa067710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chau CM, Lieberman PM. 2004. Dynamic chromatin boundaries delineate a latency control region of Epstein-Barr virus. J Virol 78:12308–12319. doi: 10.1128/JVI.78.22.12308-12319.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 and S2 and Tables S1 to S3.