

RESUMEN
El síndrome QT largo (SQTL) congénito representa un grupo de enfermedades cardiacas de origen genético, caracterizado por la prolongación del intervalo QT y una onda T anormal en el electrocardiograma (ECG). Pueden tener una expresión dominante o recesiva, esta última asociada con sordera neurosensorial. En ambos casos su presentación clínica está asociada con síncopes recurrentes y muerte súbita como consecuencia de una taquicardia ventricular, específicamente a torsades de pointes. Actualmente se clasifican en función del defecto genético específico, pudiendo comprometer alrededor de 16 genes y casi 2000 mutaciones. Debe ser sospechada en individuos con la clínica relacionada, hallazgos electrocardiográficos y antecedentes familiares. El manejo está basado en la disminución o eliminación de los síntomas y, concomitantemente, la prevención de la muerte súbita (MS), en aquellos niños con sordera congénita el manejo requiere la aplicación de medidas propias del otorrinolaringólogo. Desde el punto de vista cardiovascular, el manejo implica la modificación de estilos de vida, principalmente la prohibición de deportes competitivos, entre ellos la natación, evitar la exposición a sonidos intensos o factores desencadenantes. La medicación usada abarca a los betabloqueadores y, más raramente, flecainida, ranozalina y verapamilo; el manejo invasivo consiste el implante de un cardiodesfibrilador o, incluso, la denervación simpática izquierda, cada una de ellas con sus propios riesgos y beneficios. En cualquiera de los casos debemos evitar las circunstancias que incrementen el intervalo QT, así como realizar el adecuado análisis de los beneficios y riesgos de cada posible medida invasiva.
Palabras clave: Síndrome de QT prolongado, Muerte Súbita, Pediatria, Torsades de Pointes
ABSTRACT
Congenital long QT syndrome (LQTS) represents a group of heart diseases of genetic origin characterized by prolongation of the QT interval and an abnormal T wave on the electrocardiogram (ECG). They can have a dominant or recessive expression, the latter associated with sensorineural deafness. In both cases, its clinical presentation is associated with recurrent syncope and sudden death as a consequence of ventricular tachycardia, specifically Torsades de Pointes. Currently they are classified according to the specific genetic defect, being able to compromise around 16 genes and almost 2000 mutations. It should be suspected in individuals with related symptoms, electrocardiographic findings, and family history. Management is based on the reduction or elimination of symptoms, and concomitantly the prevention of sudden death (SD), in those children with congenital deafness, the management requires the application of the otolaryngologist specialist’s own measures. The cardiovascular management implies the modification of lifestyles, mainly the prohibition of competitive sports, including swimming, avoiding exposure to loud sounds or triggers. The medications used include beta-blockers, and more rarely flecainide, ranozaline, and verapamil; invasive management consists of the implantation of a cardioverter defibrillator or even left sympathetic denervation, each with its own risks and benefits. In any of the cases, we must avoid the circumstances that increase the QT interval, as well as carry out the appropriate analysis of the benefits and risks of each possible invasive measure.
Keywords: Long QT Syndrome; Death, Sudden; Pediatrics; Torsades de Pointes
Las atenciones en cardiología pediátrica son múltiples y variadas, abarcan patologías estructurales con mínima repercusión en el estado general, hasta procesos con compromiso severo, son frecuentes los casos totalmente asintomáticos que debutan con eventos de muerte súbita (MS). Las arritmias cardiacas son alteraciones eléctricas del ritmo cardiaco, la generación de esta actividad puede ser errática, lenta o rápida (1, estas pueden no requerir tratamiento, autolimitándose; mientras que otras requerirán algún tipo de tratamiento, generalmente cuando son casos sintomáticos o cuando impliquen algún riesgo de muerte súbita, definida como «Aquella que ocurre de manera inesperada dentro de la primera hora desde el inicio de los síntomas o si se produce en ausencia de testigos cuando el fallecido ha sido visto en buenas condiciones menos de 24 h antes de hallarlo muerto» 2. Cuando el origen es una patología cardiaca se cataloga como muerte súbita cardiaca (MSC) (3.
En pediatría, la MSC tiene una incidencia de 0,7 - 6,4 cada 100 000 personas/año, afecta principalmente a pacientes de sexo masculino, abarcando de 60 a 75% del total de casos. Múltiples estudios sugieren que los defectos genéticos pueden ser una de las causas de estas muertes, pudiendo ocasionar anomalías cardiacas hereditarias que afectan los canales iónicos conocidas como canalopatías 4. Los canales iónicos son estructuras formadas por proteínas transmembrana cuya función es el paso de iones a través de la membrana, existen muchas alteraciones genéticas que pueden alterar la estructura genómica, por ende, alteran la función de estos canales, y no suelen acompañarse de malformaciones anatómicas, debutando con la MSC. Se han descrito muchas canalopatías, las principales son: el síndrome de Brugada (SBr), el síndrome de QT largo (SQTL), el síndrome de QT corto (SQTC) y la taquicardia ventricular polimórfica catecolaminérgica (TVPC) 5,6.
Prevalencia del SQTL
La prevalencia del SQTL es de 1:2000 a 1:7000 nacidos vivos, y la mortalidad anual es variable, dependiendo de cada mutación. La presentación clínica puede ser variada, desde pacientes asintomáticos con electrocardiograma (ECG) patológico, hasta pacientes que presentan síncopes recurrentes, muchas veces confundidos con convulsiones, fibrilación ventricular e, incluso, MSC, especialmente en jóvenes. Dado que la tasa de identificación y diagnóstico mediante ECG es elevada, se han realizado varios estudios preventivos en los cuales se realizan ECG a poblaciones concretas, como niños o deportistas. El SQTL es la mayor causa de MSC en población joven y varios estudios relacionan la MS en el niño y el SQTL 7,8.
Mecanismos fisiopatológicos y genéticos
La prolongación del intervalo QT puede producirse por una disminución en las corrientes repolarizadoras de potasio, que afecta a los canales de potasio (Iks, Ikr, Iki), o por una demora en el cierre del canal de sodio en los miocitos. Existen varios tipos diferentes de SQTL, cada uno caracterizado por mutaciones en genes que codifican para canales iónicos o algunas proteínas asociadas. Hasta el momento se han identificado alrededor de 16 genes y más de 2000 mutaciones responsables, los cuales representan a casi el 85% de todos los casos de SQTL 9,10.
Tiene una expresión homocigota, conocido como síndrome de Jervell y Lange-Nielsen, asociada con sordera neurosensorial y de presentación infrecuente; también tiene la forma heterocigota, llamada síndrome de Romano Ward (sin sordera). En la actualidad, se clasifica en función de las alteraciones genéticas encontradas, cerca del 80% de casos se explica por tres genes (KCNQ1, KCNH2 y SCN5A), mientras que las últimas alteraciones genéticas explican poco más del 5% de todos los casos, dependiendo de la población estudiada, ya que puede variar según edad, sexo o raza 11.
Estas mutaciones genéticas originan las canalopatías. Los canales iónicos son estructuras macromoleculares conformados por una proteína principal formadora del poro del canal y de proteínas reguladoras adyacentes que modifican la función del poro. La proteína principal, conocida como alfa, genera los tres subtipos más comunes de SQTL: SQTL tipo 1 (afección en el canal de potasio Iks); SQTL tipo 2 (afección en el canal de potasio IKr), y SQTL tipo 3 (afección en el canal de sodio). El síndrome de Jervell y Lange-Nielsen corresponde a las variedades de SQTL 1 y 5, mientras que el síndrome de Romano Ward abarca las variedades SQTL 1 hasta la 10 12,13.
El SQTL tipo 1 (SQTL1) es el subtipo más frecuente y explica cerca del 35 a 40% de casos, el ECG muestra una onda T de base ancha (Figura 1), se caracteriza porque los episodios arrítmicos se asocian al ejercicio, y es la natación una actividad gatillante para el inicio de una arritmia ventricular, llega a tener una mortalidad anual de 0,3%. El gen afectado es el KCNQ1 (KvLQT1), localizado en el cromosoma 11 (11p15,5), cuya principal función es la codificación de la subunidad alfa del canal de potasio Iks, por lo tanto, la corriente saliente de potasio disminuye, ocasionando la prolongación del potencial de acción 14-16.
Figura 1. Correlacion genotipo-fenotipo, y mortalidad anual en los sindromes QT largo mas frecuentes.
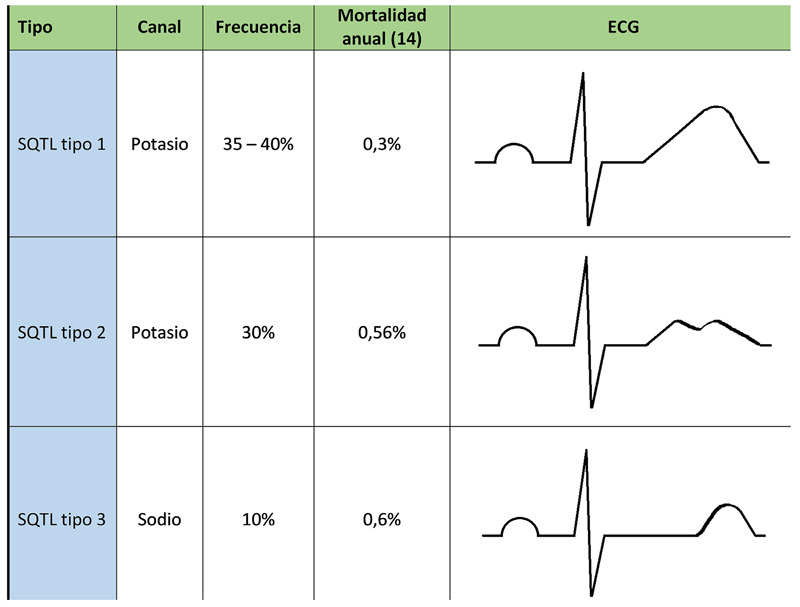
El SQTL tipo 2 explica hasta 30% de casos y puede mostrar una onda T de baja amplitud, con muesca o bífida (Figura 1); sin embargo, este patrón puede no presentarse en más del 20% de pacientes ya que la penetrancia es del 79%, y su mortalidad anual es de 0,6%. Se asocian con arritmias ventriculares en respuesta a estímulos auditivos intensos, estrés emocional y, con menor frecuencia, a la actividad física. El gen afectado es el KCNH2 (HERG), localizado en el cromosoma 7, encargado de la formación de la subunidad alfa del canal de potasio (IKr), cuya disfunción disminuye la corriente saliente de potasio durante la fase 3 del potencial de acción, prolongando su duración 14,17.
El SQTL tipo 3 produce cerca del 10% de casos y suele expresarse con una onda T acuminada, de aparición tardía haciendo evidente el alargamiento del segmento ST (Figura 1), que se puede apreciar en casi el 90% de casos por su alta penetrancia, y tiene una mortalidad anual de 0,56%. El riesgo de arritmias ventriculares es mayor durante el sueño, ya que es el periodo donde se produce mayor bradicardia, siendo más letales, pero menos frecuentes que en el SQTL tipo 1 y SQTL tipo 2. El gen afectado es el SCN5A, que codifica la subunidad alfa del canal de sodio, está localizado en el cromosoma 3 (3p21-24), cuya función defectuosa permite la entrada sostenida de sodio durante la fase 2 del potencial de acción, prolongando su duración 14,18.
El SQTL tipo 4 explica cerca del 1 % de los casos y el ECG muchas veces muestra un intervalo QT limítrofe, tiene un espectro de arritmias muy variable que incluye a la taquicardia ventricular polimorfa catecolaminérgica, fibrilación auricular y disfunción sinusal con bradicardia. El gen afectado es el ANKB localizado en el cromosoma 4 (4q25-27), que codifica la anquirina beta, cuya principal función es la de interrelacionar las proteínas de membrana de las células cardiacas, como la bomba de sodio, la bomba de potasio, el intercambiador sodio-calcio y el receptor inositol trifosfato, de esta manera, si existe disfunción de la anquirina beta se incrementa la concentración de calcio intracelular dando lugar a posdespolarizaciones precoces y tardías, y posteriormente a arritmias ventriculares 19,20
El SQTL tipo 5 explica entre 1 y 2% de casos. El gen alterado es el KCNE1, localizado en el cromosoma 21 (21q22.1-p22), este codifica la síntesis de la subunidad beta del canal Iks (subunidad minK). Tiene una penetrancia apenas superior al 20% para casos heterocigotos, y de 15% para homocigotos 20,21.
EL SQTL tipo 6 afecta a menos del 1%. El gen afectado es el KCNE2 localizado en el cromosoma 21 (21q22.1), este codifica la subunidad beta del canal de potasio (subunidad MiRP1) 19,20.
El SQTL tipo 7 (síndrome de Andersen-Tawil) representa menos del 1% de casos y corresponde a una alteración autosómica dominante; el electrocardiograma muestra un intervalo QTc ligeramente prolongado o, incluso, dentro de valores normales, pero con una onda U prominente. Se expresa clínicamente con extrasistolia ventricular, taquicardia ventricular polimórfica no sostenida, taquicardia bidireccional y torsades de pointes, además de las malformaciones corporales asociadas. El gen alterado es el KCNJ2 en el 70% de casos, el cual está ubicado en el cromosoma 17(17q23), codifica la proteína que conforma el canal rectificador de potasio Kir 2.1, y da origen al síndrome Andersen-Tawil tipo 1, mientras que los pacientes que no tienen esta mutación se les conoce como tipo non KCNJ2 22,23.
El SQTL tipo 8 (síndrome de Timothy) afecta a menos del 0,5% de casos, clínicamente se expresa con alteraciones dismórficas como fusiones interdigitales, malformaciones cardiacas, hipoglicemia y alteraciones cognitivas. Se produce por mutaciones en el gen CACNA1 que codifica el canal de calcio tipo L CaV 1.2, localizado en el cromosoma 12 (12p13.3) 24,25.
El SQTL tipo 9 explica menos del 1% de los casos. Se produce por mutaciones del gen CAV3, localizado en el cromosoma 3 (3p25), que codifica la caveolina 3, componente principal de la caveola, cuya principal función es la homeostasis de lípidos y la transducción de señales. Algunos canales iónicos como el canal de sodio Nav1.5 se localizan en la caveola 20,26.
El SQTL tipo 10 representa menos del 0,5% de casos, y su expresión clínica es grave, la mayoría de veces con intervalos QTC muy largos, por encima de los 600 ms, asociado con bradicardia fetal y bloqueo auriculoventricular. El gen alterado es el SCN4B, localizado en el cromosoma 11 (11q23) que codifica para la subunidad beta 4 de canal de sodio. El SQTL tipo 11 es un desorden ocasionado por una mutación en el gen AKAP9, que codifica la proteína Akinasa 9. Las sintrofinas son proteínas de las submembranas citoplasmáticas que forman parte de los complejos proteicos asociados a la distrofina, la sintrofina tipo alfa 1 (SNTA1) interactúa con el Nav1.5, esta mutación genera el SQTL tipo 12. Ambas representan menos del 0,5% de los casos. El SQTL tipo 13 se produce por alteración del gen KCNJ5, el ECG muestra un retraso desde el pico de la onda hasta el final de esta. Se han identificado otras mutaciones, por ejemplo, el receptor cardiaco de rianodina es codificado por el gen RyR2, y puede expresarse de manera heterocigota en pacientes con un intervalo QTC más largo de lo normal 20,27,28.
Diagnóstico
El ECG es la piedra angular para el diagnóstico del SQTL, la medición del intervalo QT y reconocimiento de ECG con repolarización alterada son elementos básicos para el diagnóstico y la determinación del pronóstico, la presencia de ondas T alteradas en algunos casos de SQTL también puede ayudar al diagnóstico. Existe una prolongación del intervalo QT (QT corregido > 460 ms en mujeres, > 450 ms en hombres y > 440 ms en niños) por una repolarización ventricular más prolongada, pudiendo originar un evento de torsades de pointes, su complicación más temida 13,29.
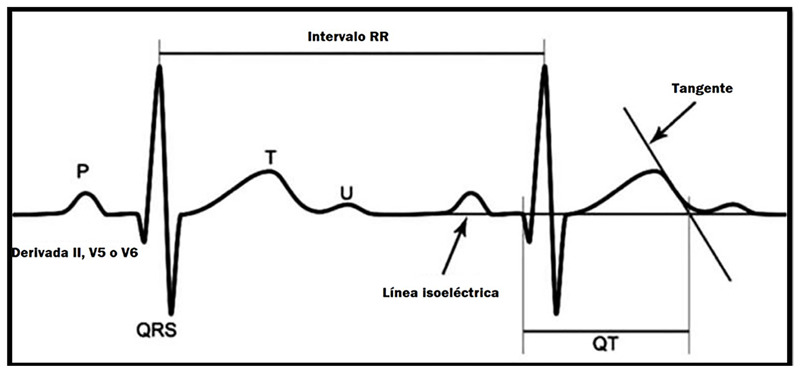
Un aspecto técnico que no debemos olvidar es que la medición del intervalo QT debe realizarse en las derivaciones II y V5 - V6, a una velocidad de 25 mm/s y una amplitud de 10 mm/mv, desde el inicio del QRS hasta el final de la onda T (la intersección de la rama descendente de la onda T y la línea isoeléctrica) sin incluir a la onda U, calculando el promedio de tres a cinco latidos (diez latidos en fibrilación auricular) y aplicando la corrección de Bazzet (Figura 2); cabe recordar que con frecuencias cardiacas por encima de cien latidos por minuto es menos exacta, por lo que se sugiere utilizar la fórmula de Friderica o de Hodges. Este detalle es importante en los neonatos, ya que manejan frecuencias cardiacas altas, haciendo imperativo que los equipos de cardiología pediátrica con conocimientos de arritmias sean los que se hagan cargo de estos pacientes 13,30-33.
Figura 2. Correcta medicion del intervalo QT.
El Holter de 24 h puede mostrar la variación circadiana del intervalo QT, sin embargo, su uso seriado no es rentable ya que los hallazgos son similares a los del ECG. En los casos donde los síntomas son infrecuentes se puede optar por el implante de un Holter subcutáneo, generalmente de 3 años de duración 34-36.
Respecto a la prueba de esfuerzo, se debe realizar con precaución, está indicado en pacientes con una puntuación de Schwartz de 3, es decir, de probabilidad intermedia, familiares asintomáticos con QTc en reposo <440 ms, también a pacientes sin un fenotipo o genotipo definido para adecuación terapéutica, y pacientes con síntomas inespecíficos al esfuerzo. Un QTc > 445 ms a los 4 min de recuperación, es un buen punto de corte para distinguir portadores de alguna mutación de no portadores; por otro lado, se pueden realizar pruebas de epinefrina provocadoras para diagnóstico y evaluación fenotípica 37,38.
El estudio genético, de disponibilidad limitada y de muy alto costo, contribuye al diagnóstico y, en algunos casos, al tratamiento y pronóstico. Pacientes con probabilidad intermedia diagnóstico (puntuación de Schwartz de 3) puede tener la enfermedad confirmada si tienen una mutación que causa SQTL. Por otro lado, la ausencia de mutación no excluye diagnóstico, ya que la posibilidad de que una prueba genética sea positiva es aproximadamente el 60%, y es en estos casos donde los familiares deberían ser estudiados para descartar que sean portadores, pues el 35% de estos son portadores silenciosos y pueden presentar un intervalo QTc normal y debutar con algún evento arrítmico si no se aplica una guía de comportamiento adecuada 10,14,19,37.
Diagnóstico según score de Schwartz (Tabla 1) 37
SQTL se define como:
• La puntuación de Schwartz ≥ 3,5, en ausencia de causas prolongaciones secundarias del intervalo QT y/o
• Presencia de mutación probada que causa SQTL o
• Intervalo QTc ≥ 500 ms en ECG seriados, en ausencia causas secundarias, incluso en pacientes asintomáticos.
Tabla 1. Puntuación de Schwartz para el diagnóstico de SQTL.
| Característica | Puntos |
|---|---|
| Hallazgos electrocardiográficos | |
| QTc | |
| > 480 ms | 3 |
| 460 ms - 470 ms | 2 |
| 450 ms | 1 |
| Torsade de pointes | 2 |
| Alternancia de la onda T | 1 |
| Muesca en la onda T en 3 derivaciones | 1 |
| Baja FC para la edad | 0,5 |
| Historia | |
| Síncope | |
| Con estrés | 2 |
| Sin estrés | 1 |
| Sordera congénita | 0,5 |
| Historia familiar | |
| Definitivamente de SQTL congénito | 1 |
| Muerte súbida inexplicada en familiares inmediatos < 30 años | 0,5 |
Estratificación del riesgo
Recientemente, Goldenberg et al. publicaron una guía práctica de riesgo para SQTL 39.
• Riesgo elevado
- Torsades de pointes espontánea.
- Muerte súbita recuperada.
• Riesgo moderado
- QTc >500 ms.
- Síncopes previos.
• Riesgo bajo
- QTc <500 m.
- No síncopes previos.
La prueba de Viskin
Se define como histéresis al retraso en la adaptación que se presenta cuando se incrementa la FC como consecuencia del ejercicio o del ortostatismo, diversos investigadores observaron que los pacientes con SQTL presentaban una histéresis prolongada y, en algunos casos, había alternancia de la onda T en la FC máxima durante el esfuerzo físico 40. Viskin, en 2010, introdujo el término de estiramiento del QT, haciendo referencia a la falta de acortamiento del QT cuando se incrementa la FC. Esta sencilla prueba resulta muy útil en pacientes con un QT limítrofe (entre 450 y 480 ms) en quienes un incremento mayor de 50 ms del QTc con la FC máxima o el máximo estiramiento del QT tiene una sensibilidad del 90% y una especificidad del 86% para el diagnóstico de SQTL en el grupo de población con QTc entre 390 y 480 ms basales 41; en pacientes con el diagnóstico establecido nos sirve para hacer seguimiento, ya que se ha demostrado que este fenómeno se reduce de manera significativa con el uso de betabloqueadores 42,43. Por otro lado, la prueba de mesa basculante puede arrojar datos que apoyan al diagnóstico, ya que se observa alternancia de la onda T 44.
Manejo y tratamiento
En aquellos pacientes con probabilidad alta o intermedia de padecer SQTL, se debe indicar la modificación del estilo de vida, evitando realizar actividad física intensa 45, asimismo, hacer hincapié en la contraindicación de actividades acuáticas en todos los tipos de SQTL. Los portadores genéticos también deben evitar el deporte profesional y, en algunos casos, deberían ser tratados con betabloqueadores. Es de suma importancia evitar fármacos que pueden alargar el intervalo QT (www.torsades.org, www.crediblemeds.org), por lo que todo paciente o apoderado debe conocer esta lista.
Los betabloqueadores representan la primera línea en el manejo, se deben iniciar en los pacientes con alta probabilidad de SQTL (score de Schwartz ≥ 3,5), es decir, aquel paciente asintomático con QTc > 470 ms, o con clínica sospechosa (síncope de origen cardiaco o taquicardia/fibrilación ventricular documentada). En pacientes con diagnóstico de SQTL y QTC < 470 ms, el uso de bloqueadores tiene indicación de clase IIa. Los de uso más frecuente son propranolol, metropolol y nadolol, estos medicamentos tienen la mayor cantidad de investigaciones que demuestran su eficacia para reducir eventos arrítmicos y disminuir la mortalidad, algunos estudios demuestran que el metropolol es más efectivo en pacientes con SQTL tipo 1 46,47; además. también reduce los eventos en los tipos 2 y 3 de SQTL, mientras que el nadolol tiene mayor efectividad en SQTL tipo 2. La dosis objetivo de propranolol es de 2 a 3 mg/kg y puede aumentarse según síntomas, mientras que el metropolol se usa de 0,5 a 2 mg/kg y el nadolol de 0,5 a 1 mg/kg 46,48,49. En el SQTL tipo 3 puede usarse flecainida y ranozalina 50-52, mientras que el verapamilo se puede usar en el SQTL tipo 5 53.
Dentro de las medidas invasivas, el implante del cardiodesfibrilador es de importancia capital, esta se puede usar como terapia de primera línea en pacientes con una muerte súbita recuperada, o en ausencia de una respuesta adecuada a los betabloqueadores a dosis máxima, es decir, a la presencia recurrente de síncopes, a pesar de la medicación óptima o, incluso cuando existe contraindicación para el uso de estos 54. El cardiodesfibrilador es muy efectivo en adultos y niños, sin embargo, en estos últimos la decisión del implante como prevención primaria es complicada debido a los efectos colaterales como las descargas inapropiadas y el impacto en la esfera emocional, además de las dificultades anatómicas propias de este grupo de pacientes 55,56.
Estudios basados en el Registro de Rochester 57 refuerzan el uso del cardiodesfibrilador como prevención primaria, se destaca la posibilidad de choques apropiados, los cuales, a la vez, dependen de la duración del QTc (predictor más importante) y de la presencia de síncopes durante el tratamiento con betabloqueadores (tanto en los primeros choques apropiados como en los recurrentes). La probabilidad de una primera descarga apropiada es del 23%, con una mortalidad nula, sin embargo, en pacientes con SQTL sin cardiodesfibrilador la mortalidad llega a 17%. Adicionalmente, el uso de betabloqueadores disminuye el riesgo de una primera descarga apropiada, y es mucho más efectivo en SQTL tipo 1, donde la medicación tiene mayores efectos benéficos. A pesar de los beneficios demostrados, no podemos dejar de mencionar que los choques inapropiados (72%) son mucho más frecuentes que los choques apropiados, esto principalmente por la sobredetección de la onda T o por la presencia de arritmias supraventriculares 58-60. La programación del cardiodesfibrilador tiene como objetivo reducir los choques inapropiados, ya que su presencia causa hiperactividad adrenérgica, concomitantemente se incrementa el riesgo de recurrencia de la arritmia, pudiendo terminar en una tormenta eléctrica 61-64. El costo del dispositivo es alto, sin embargo, a largo plazo existe un beneficio económico significativo 65.
La simpatectomía izquierda o denervación simpática izquierda se usa en pacientes que tienen alguna contraindicación para el implante de un cardiodesfibrilador (o por elección del paciente), también se usa en pacientes con pobre respuesta, contraindicación o rechazo al uso de betabloqueadores. Los pacientes portadores de un cardiodesfibrilador y presencia de una tormenta eléctrica, a pesar del uso de betabloqueadores a dosis plena, también tienen indicación de clase I 66. Esta técnica implica el aislamiento de los primeros tres o cuatro ganglios torácicos, preservando la porción cefálica del ganglio estrellado, para evitar el síndrome de Horner. Las limitaciones son logísticas, ya que su aplicabilidad depende de la experiencia del grupo quirúrgico, cuyo acceso puede ser por toracotomía o por toracoscopía 67,68. En niños pequeños, el acceso es por el tercer espacio intercostal, donde hay una visualización adecuada del ganglio simpático y estrellado. El efecto benéfico se puede apreciar generalmente a partir del tercer mes de realizado, con reducción de hasta 39 ms, en muchos casos llegando a un intervalo QTc < 500 ms, casi en el 74% de casos se observa un efecto paradojal con incremento del intervalo QTc durante las primeras 24 h, con resolución posterior de hasta 3 meses de demora, tiempo que tarda en establecerse la remodelación eléctrica cardiaca 69-74. En casos seleccionados se puede realizar una simpatectomía bilateral 75.
A principios de este año se tuvo la primera experiencia con terapia génica para la corrección completa del SQTL tipo 1, el KCNQ1-SupRep provocó la supresión y sustitución de la estructura afectada en el gen KCNQ1, llegando a tener una función normal, esto abre una serie de nuevas estrategias de tratamiento dentro de nuestro arsenal terapéutico 76.
Pronóstico
Existe numerosa literatura acerca del SQTL, sin embargo, los datos validados científicamente son limitados. La mayoría de pacientes comprometidos llevan una vida normal, si aplicamos las recomendaciones dadas en las revisiones acerca del tema. Se debe educar a los padres de niños afectados, haciendo hincapié en el cumplimiento estricto del uso de betabloqueadores como base del esquema de tratamiento, así como evitar los medicamentos que prolongan el intervalo QT. Por otro lado, la existencia de técnicas más invasivas nos da un margen para poder influir un más en la calidad de vida de cada uno de nuestros pacientes. El rápido avance de esta área de estudio, abre cada vez nuevas esperanzas en su manejo y pronóstico.
Conclusiones
Los trastornos del ritmo cardiaco son frecuentes, sin embargo, las que obedecen a una causa genética son raros, como el SQTL. Su importancia radica en la posibilidad de debutar con la MSC. Para evitar estas situaciones, se recomienda realizar un ECG desde el nacimiento a todos los niños con antecedentes familiares; adicionalmente, se recomienda la realización de un ECG a todo niño que haga deporte de manera competitiva para descartar portadores asintomáticos de la enfermedad.
Los pilares para el manejo son la modificación del estilo de vida, el uso de betabloqueadores, el implante de un cardiodesfibrilador y la simpatectomía. En niños, el implante de un cardiodesfibrilador requiere de un prudente análisis ya que los efectos colaterales pueden causar gran impacto en la esfera física y emocional de nuestros pequeños pacientes.
Del mismo modo, la creciente cantidad de conocimiento acerca de los canales iónicos han mejorado nuestro conocimiento acerca de las causas y el tratamiento clínico e invasivo del SQTL; de esta manera, se identificarán nuevos agentes con menores efectos tóxicos; este nuevo conocimiento también nos ayuda a identificar a los familiares que son portadores de esta alteración, algunos de ellos sin afectación clínica, pero en riesgo de sufrir una MSC. Pese a estos grandes avances, todavía no se conocen todos los defectos genéticos causantes de estas arritmias malignas.
Footnotes
Financiamiento Autofinanciado.
Citar como: Melgar Quicaño L, Chipa Ccasani F. Síndrome de QT largo congénito. Arch Peru Cardiol Cir Cardiovasc. 2021;2(1):49-57. doi: 10.47487/apcyccv.v2i1.125
Referencias bibliográficas
- 1.Brugada J, Blom N, Sarquella-Brugada G, Blomstrom-Lundqvist C, Deanfield J, Janousek J, et al. Pharmacological and non-pharmacological therapy for arrhythmias in the pediatric population EHRA and AEPC-Arrhythmia Working Group joint consensus statement. EP Eur. 2013;15(9):1337–1382. doi: 10.1093/europace/eut082. [DOI] [PubMed] [Google Scholar]
- 2.Isbister J, Semsarian C. Sudden cardiac death an update. Intern Med J. 2019;49(7):826–833. doi: 10.1111/imj.14359. [DOI] [PubMed] [Google Scholar]
- 3.Jazayeri MA, Emert MP. Sudden Cardiac Death Who is at risk? Med Clin North Am. 2019;103(5):913–930. doi: 10.1016/j.mcna.2019.04.006. [DOI] [PubMed] [Google Scholar]
- 4.Sherwin ED, Berul CI. Sudden Cardiac Death in Children and Adolescents. Card Electrophysiol Clin. 2017;9(4):569–579. doi: 10.1016/j.ccep.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 5.Baltogiannis G, Conte G, Sieira J, De Ferrari GM, Brugada P. Editorial Sudden Cardiac Death and Channelopathies. Front Cardiovasc Med. 2020;7:605834–605834. doi: 10.3389/fcvm.2020.605834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sarquella-Brugada G, Campuzano O, Brugada R. Trastornos del ritmo cardiaco más frecuentes en pediatría síndrome de QT largo. Pediatr Integral. 2012;XVI(8):617–621. [Google Scholar]
- 7.Wong AR. Clinical and genetic analysis of long QT syndrome in two Malay children. Med J Malaysia. 2019;74(4):341–343. [PubMed] [Google Scholar]
- 8.Haverkamp W. Kongenitales Long-QT-Syndrom. Herz Kardiovaskuläre Erkrank. 2007;32(3):201–205. doi: 10.1007/s00059-007-2992-1. [DOI] [PubMed] [Google Scholar]
- 9.Kwok S, Liu AP, Chan CY, Lun K, Fung JL, Mak CC, et al. Clinical and genetic profile of congenital long QT syndrome in Hong Kong a 20-year experience in paediatrics. Hong Kong Med J. 2018;24(6):561–570. doi: 10.12809/hkmj187487. [DOI] [PubMed] [Google Scholar]
- 10.Zullo A, Frisso G, Detta N, Sarubbi B, Romeo E, Cordella A, et al. Allelic Complexity in Long QT Syndrome A Family-Case Study. Int J Mol Sci. 2017;18(8):1633–1633. doi: 10.3390/ijms18081633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lahrouchi N, Tadros R, Crotti L, Mizusawa Y, Postema PG, Beekman L, et al. Transethnic Genome-Wide Association Study Provides Insights in the Genetic Architecture and Heritability of Long QT Syndrome. Circulation. 2020;142(4):324–338. doi: 10.1161/CIRCULATIONAHA.120.045956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsuda S, Ohnuki Y, Okami M, Ochiai E, Yamada S, Takahashi K, et al. Jervell and Lange-Nielsen syndrome with novel KCNQ1 and additional gene mutations. Hum Genome Var. 2020;7(1):34–34. doi: 10.1038/s41439-020-00121-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma N, Cortez D, Disori K, Imundo JR, Beck M. A Review of Long QT Syndrome Everything a Hospitalist Should Know. Hosp Pediatr. 2020;10(4):369–375. doi: 10.1542/hpeds.2019-0139. [DOI] [PubMed] [Google Scholar]
- 14.Modell SM, Bradley DJ, Lehmann MH. Genetic testing for long QT syndrome and the category of cardiac ion channelopathies. PLoS Curr. 2012;4 doi: 10.1371/4f9995f69e6c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faridi R, Tona R, Brofferio A, Hoa M, Olszewski R, Schrauwen I, et al. Mutational and phenotypic spectra of KCNE1 deficiency in Jervell and Lange-Nielsen Syndrome and Romano-Ward Syndrome. Hum Mutat. 2019;40(2):162–176. doi: 10.1002/humu.23689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oertli A, Rinné S, Moss R, Kääb S, Seemann G, Beckmann BM, et al. Molecular Mechanism of Autosomal Recessive Long QT-Syndrome 1 without Deafness. Int J Mol Sci. 2021;22(3):1112–1112. doi: 10.3390/ijms22031112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ono M, Burgess DE, Schroder EA, Elayi CS, Anderson CL, January CT, et al. Long QT Syndrome Type 2 Emerging Strategies for Correcting Class 2 KCNH2 (hERG) Mutations and Identifying New Patients. Biomolecules. 2020;10(8):1144–1144. doi: 10.3390/biom10081144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pérez-Riera AR, Barbosa-Barros R, Daminello Raimundo R, da Costa de Rezende Barbosa MP, Esposito Sorpreso IC, de Abreu LC. The congenital long QT syndrome Type 3 An update. Indian Pacing Electrophysiol J. 2018;18(1):25–35. doi: 10.1016/j.ipej.2017.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wallace E, Howard L, Liu M, O'Brien T, Ward D, Shen S, et al. Long QT Syndrome Genetics and Future Perspective. Pediatr Cardiol. 2019;40(7):1419–1430. doi: 10.1007/s00246-019-02151-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Medeiros-Domingo A, Iturralde-Torres P, Ackerman MJ. Clinical and Genetic Characteristics of Long QT Syndrome. Rev Esp Cardiol Engl Ed. 2007;60(7):739–752. doi: 10.1016/S1885-5857(08)60010-9. [DOI] [PubMed] [Google Scholar]
- 21.Roberts JD, Asaki SY, Mazzanti A, Bos JM, Tuleta I, Muir AR, et al. An International Multicenter Evaluation of Type 5 Long QT Syndrome A Low Penetrant Primary Arrhythmic Condition. Circulation. 2020;141(6):429–439. doi: 10.1161/CIRCULATIONAHA.119.043114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Veerapandiyan A, Statland JM, Tawil R. In: GeneReviews(r) Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mirzaa G, et al., editors. Seattle(WA): University of Washington, Seattle; 1993. Andersen-Tawil Syndrome. [PubMed] [Google Scholar]
- 23.Kukla P, Biernacka E, Baranchuk A, Jastrzebski M, Jagodzinska M. Electrocardiogram in Andersen-Tawil Syndrome New Electrocardiographic Criteria for Diagnosis of Type-1 Andersen-Tawil Syndrome. Curr Cardiol Rev. 2014;10(3):222–228. doi: 10.2174/1573403x10666140514102528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mellor GJ, Panwar P, Lee AK, Steinberg C, Hathaway JA, Bartels K, et al. Type 8 long QT syndrome pathogenic variants in CACNA1C-encoded Cav1.2 cluster in STAC protein binding site. EP Eur. 2019;21(11):1725–1732. doi: 10.1093/europace/euz215. [DOI] [PubMed] [Google Scholar]
- 25.Sepp R, Hategan L, Bácsi A, Cseklye J, Környei L, Borbás J, et al. Timothy syndrome 1 genotype without syndactyly and major extracardiac manifestations. Am J Med Genet A. 2017;173(3):784–789. doi: 10.1002/ajmg.a.38084. [DOI] [PubMed] [Google Scholar]
- 26.Cutler MJ, Kaufman ES. To Be or Not to Be Long-QT Syndrome Type 9. Circ Cardiovasc Genet. 2013;6(5):439–440. doi: 10.1161/CIRCGENETICS.113.000345. [DOI] [PubMed] [Google Scholar]
- 27.Wang F, Liu J, Hong L, Liang B, Graff C, Yang Y, et al. The phenotype characteristics of type 13 long QT syndrome with mutation in KCNJ5 (Kir3 4-G387R) Heart Rhythm. 2013;10(10):1500–1506. doi: 10.1016/j.hrthm.2013.07.022. [DOI] [PubMed] [Google Scholar]
- 28.Waddell-Smith KE, Skinner JR. Update on the Diagnosis and Management of Familial Long QT Syndrome. Heart Lung Circ. 2016;25(8):769–776. doi: 10.1016/j.hlc.2016.01.020. [DOI] [PubMed] [Google Scholar]
- 29.Garson A, Dick M, Fournier A, Gillette PC, Hamilton R, Kugler JD, et al. The long QT syndrome in children An international study of 287 patients. Circulation. 1993;87(6):1866–1872. doi: 10.1161/01.cir.87.6.1866. [DOI] [PubMed] [Google Scholar]
- 30.Bazett HC. An Analysis of The Time-Relations of Electrocardiograms. Ann Noninvasive Electrocardiol. 1997;2(2):177–194. doi: 10.1111/j.1542-474X.1997.tb00325.x. [DOI] [Google Scholar]
- 31.Cobos Gil MA, García Rubira JC. ¿Quién fue el autor de la fórmula de Bazett? Rev Esp Cardiol. 2008;61(8):896–897. doi: 10.1157/13125006. [DOI] [PubMed] [Google Scholar]
- 32.Fridericia LS. Die Systolendauer im Elektrokardiogramm bei normalen Menschen und bei Herzkranken. Acta Med Scand. 2009;53(1):469–486. doi: 10.1111/j.0954-6820.1921.tb15167.x. [DOI] [Google Scholar]
- 33.Hodges M. Rate Correction of the QT Interval. Cardiac Electrophysiol Rev. 1997;1:360–363. [Google Scholar]
- 34.Extramiana F, Maison-Blanche P, Badilini F. Circadian Modulation of QT Rate Dependence in Healthy Volunteers Gender and Age Differences. J Electrocardiology. 1999;32(1):33–43. doi: 10.1016/S0022-0736(99)90019-5. [DOI] [PubMed] [Google Scholar]
- 35.Mauriello DA, Johnson JN, Ackerman MJ. Holter Monitoring in the Evaluation of Congenital Long QT Syndrome. Pacing Clin Electrophysiol. 2011;34(9):1100–1104. doi: 10.1111/j.1540-8159.2011.03102.x. [DOI] [PubMed] [Google Scholar]
- 36.Vilcant V, Kousa O, Hai O. StatPearls. Treasure Island(FL): StatPearls Publishing; 2020. Implantable Loop Recorder. [PubMed] [Google Scholar]
- 37.Schwartz PJ, Crotti L. QTc Behavior During Exercise and Genetic Testing for the Long-QT Syndrome. Circulation. 2011;124(20):2181–2184. doi: 10.1161/CIRCULATIONAHA.111.062182. [DOI] [PubMed] [Google Scholar]
- 38.Churet M, Luttoo K, Hocini M, Haïssaguerre M, Sacher F, Duchateau J. Diagnostic reproducibility of epinephrine drug challenge interpretation in suspected long QT syndrome. J Cardiovasc Electrophysiol. 2019;30(6):896–901. doi: 10.1111/jce.13926. [DOI] [PubMed] [Google Scholar]
- 39.Goldenberg I, Moss AJ. Long QT Syndrome. J Am Coll Cardiol. 2008;51(24):2291–2300. doi: 10.1016/j.jacc.2008.02.068. [DOI] [PubMed] [Google Scholar]
- 40.Márquez MF. El síndrome de QT largo una breve revisión del diagnóstico electrocardiográfico incluyendo la prueba de Viskin. Arch Cardiol México. 2012;82(3):243–247. doi: 10.1016/j.acmx.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 41.Viskin S, Postema PG, Bhuiyan ZA, Rosso R, Kalman JM, Vohra JK, et al. The Response of the QT Interval to the Brief Tachycardia Provoked by Standing. J Am Coll Cardiol. 2010;55(18):1955–1961. doi: 10.1016/j.jacc.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krahn AD, Yee R, Chauhan V, Skanes AC, Wang J, Hegele RA, et al. Beta blockers normalize QT hysteresis in long QT syndrome. Am Heart J. 2002;143(3):528–534. doi: 10.1067/mhj.2002.120408. [DOI] [PubMed] [Google Scholar]
- 43.Muñoz-Esparza C, Zorio E, Domingo Valero D, Peñafiel-Verdú P, Sánchez-Muñoz JJ, García-Molina E, et al. Valor del "test de bipedestación" en el diagnóstico y la evaluación de la respuesta al tratamiento con bloqueadores beta en el síndrome de QT largo. Rev Esp Cardiol. 2017;70(11):907–914. doi: 10.1016/j.recesp.2016.12.028. [DOI] [PubMed] [Google Scholar]
- 44.Gu B, Liu T, Yang L, Zhang H, Xin Y, Wang J. Head-up tilt test induces T-wave alternans in long QT syndrome with KCNQ1 gene mutation. Medicine (Baltimore) 2020;99(20):e19818. doi: 10.1097/MD.0000000000019818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marrakchi S, Kammoun I, Bennour E, Laroussi L, Ben Miled M, Kachboura S. Inherited primary arrhythmia disorders cardiac channelopathies and sports activity. Herz. 2020;45(2):142–157. doi: 10.1007/s00059-018-4706-2. [DOI] [PubMed] [Google Scholar]
- 46.Abu-Zeitone A, Peterson DR, Polonsky B, McNitt S, Moss AJ. Efficacy of Different Beta-Blockers in the Treatment of Long QT Syndrome. J Am Coll Cardiol. 2014;64(13):1352–1358. doi: 10.1016/j.jacc.2014.05.068. [DOI] [PubMed] [Google Scholar]
- 47.Chockalingam P, Crotti L, Girardengo G, Johnson JN, Harris KM, van der Heijden JF, et al. Not All Beta-Blockers Are Equal in the Management of Long QT Syndrome Types 1 and 2. J Am Coll Cardiol. 2012;60(20):2092–2099. doi: 10.1016/j.jacc.2012.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Han L, Liu F, Li Q, Qing T, Zhai Z, Xia Z, et al. The Efficacy of Beta-Blockers in Patients With Long QT Syndrome 1-3 According to Individuals' Gender, Age, and QTc Intervals A Network Meta-analysis. Front Pharmacol. 2020;11:579525–579525. doi: 10.3389/fphar.2020.579525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Waddell-Smith KE, Li J, Smith W, Crawford J, Skinner JR. B-Blocker Adherence in Familial Long QT Syndrome Circ Arrhythm. Electrophysiol. 2016;9(8):e003591. doi: 10.1161/CIRCEP.115.003591. [DOI] [PubMed] [Google Scholar]
- 50.Chorin E, Hu D, Antzelevitch C, Belardinelli L, Zeltser D, Barajas-Martinez H, et al. Ranolazine for Congenital Long-QT Syndrome Type III Experimental and Long-Term Clinical Data. Circ Arrhythm Electrophysiol. 2016;9(10):e004370. doi: 10.1161/CIRCEP.116.004370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cano J, Zorio E, Mazzanti A, Arnau MA, Trenor B, Priori SG, et al. Ranolazine as an Alternative Therapy to Flecainide for SCN5A V411M Long QT Syndrome Type 3 Patients. Front Pharmacol. 2020;11:580481–580481. doi: 10.3389/fphar.2020.580481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chorin E, Taub R, Medina A, Flint N, Viskin S, Benhorin J. Long-term flecainide therapy in type 3 long QT syndrome. Europace. 2018;20(2):370–376. doi: 10.1093/europace/euw439. [DOI] [PubMed] [Google Scholar]
- 53.Sakata S, Kurata Y, Li P, Notsu T, Morikawa K, Miake J, et al. Instability of KCNE1-D85N that Causes Long QT Syndrome Stabilization by Verapamil: Instability Of KCNE1-D85N. Pacing Clin Electrophysiol. 2014;37(7):853–863. doi: 10.1111/pace.12360. [DOI] [PubMed] [Google Scholar]
- 54.Pick JM, Batra AS. Implantable cardioverter-defibrillator implantation for primary and secondary prevention indications and outcomes. Cardiol Young. 2017;27(S1):S126–S131. doi: 10.1017/S1047951116002365. [DOI] [PubMed] [Google Scholar]
- 55.DeWitt ES, Abrams DJ. Implantable cardioverter-defibrillators in children. Arch Dis Child. 2015;100(3):265–270. doi: 10.1136/archdischild-2011-301536. [DOI] [PubMed] [Google Scholar]
- 56.González Corcia MC, Sieira J, Pappaert G, de Asmundis C, Chierchia GB, La Mier M, et al. Implantable Cardioverter-Defibrillators in Children and Adolescents With Brugada Syndrome. J Am Coll Cardiol. 2018;71(2):148–157. doi: 10.1016/j.jacc.2017.10.082. [DOI] [PubMed] [Google Scholar]
- 57.Clinical Cardiovascular Research Center . LQTS Registry. New Yprk: University of Rochester Medical Center; 2021. [Google Scholar]
- 58.Biton Y, Rosero S, Moss AJ, Goldenberg I, Kutyifa V, McNitt S, et al. Primary prevention with the implantable cardioverter-defibrillator in high-risk long-QT syndrome patients. Europace. 2019;21(2):339–346. doi: 10.1093/europace/euy149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kwiatkowska J, Budrejko S, Wasicionek M, Meyer-Szary FJ, Lubinski A, Kempa M. Long-term follow-up of implantable cardioverter-defibrillators in children Indications and outcomes. Adv Clin Exp Med. 2019;29(1):123–133. doi: 10.17219/acem/110313. [DOI] [PubMed] [Google Scholar]
- 60.Lewandowski M, Syska P, Kowalik I, Maciag A, Sterlinski M, Atenska-Pawlowska J, et al. Fifteen years' experience of implantable cardioverter defibrillator in children and young adults Mortality and complications study. Pediatr Int. 2018;60(10):923–930. doi: 10.1111/ped.13660. [DOI] [PubMed] [Google Scholar]
- 61.Asakai H, Shimizu A, Mitsuhashi T, Ueyama T, Yokoshiki H, Nishii N, et al. Current Trends in Implantable Cardioverter-Defibrillator Therapy in Children - Results From the JCDTR Database. Circ J. 2018;83(1):52–55. doi: 10.1253/circj.CJ-18-0712. [DOI] [PubMed] [Google Scholar]
- 62.Molina Lerma MJ, Álvarez M, Tercedor L. Desfibrilador implantable, ¿la solución definitiva. Cardiocore. 2016;51(3):104–107. doi: 10.1016/j.carcor.2016.05.003. [DOI] [Google Scholar]
- 63.Rhodes T, Weiss R. Device Therapy in the Setting of Long QT Syndrome. Card Electrophysiol Clin. 2015;7(3):479–486. doi: 10.1016/j.ccep.2015.05.018. [DOI] [PubMed] [Google Scholar]
- 64.Kriebel T, Müller MJ, Ruschewski W, Krause U, Paul T, Schneider H. Value of Regular Defibrillation Threshold Testing After Extracardiac Implantable Cardioverter Defibrillator Placement in Small Children During Mid-Term Follow-Up. JACC Clin Electrophysiol. 2018;4(7):936–943. doi: 10.1016/j.jacep.2018.03.012. [DOI] [PubMed] [Google Scholar]
- 65.Haag MB, Hersh AR, Toffey DE, Sargent JA, Stecker EC, Heitner SB, et al. Cost-Effectiveness of Implantable Cardioverter-Defibrillators in Children with Cardiac Conditions Associated with Risk for Sudden Cardiac Death. Pediatr Cardiol. 2020;41(7):1484–1491. doi: 10.1016/j.hrthm.2020.03.018. [DOI] [PubMed] [Google Scholar]
- 66.Dusi V, Pugliese L, Castelletti S, Dagradi F, Crotti L, Mori A, et al. Cardiac sympathetic denervation evolving technique, expanding indications. Eur Heart J. 2018;39(Suppl 1) doi: 10.1093/eurheartj/ehy564.P297. [DOI] [Google Scholar]
- 67.Lampridis S, Antonopoulos A, Kakos C, Mitsos S, Patrini D, Lawrence DR, et al. Video-thoracoscopic left cardiac sympathetic denervation for long-QT syndrome. Asian Cardiovasc Thorac Ann. 2021;29:186–190. doi: 10.1177/0218492320971492. [DOI] [PubMed] [Google Scholar]
- 68.Li K, Yang J, Guo W, Lv T, Guo J, Li J, et al. Video-Assisted Thoracoscopic Left Cardiac Sympathetic Denervation in Chinese Patients with Long QT Syndrome. Int Heart J. 2018;59(6):1346–1351. doi: 10.1536/ihj.17-727. [DOI] [PubMed] [Google Scholar]
- 69.Anand S, Jain V, Agarwala S, Sachdeva S, Kothari S. Thoracoscopic left cardiac sympathetic denervation in a child with refractory long QT syndrome. J Indian Assoc Pediatr Surg. 2019;24(4):297–297. doi: 10.4103/jiaps.JIAPS_144_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anderson HN, Bos JM, Rohatgi RK, Ackerman MJ. The Effect of Left Cardiac Sympathetic Denervation on Exercise in Patients with Long QT Syndrome. JACC Clin Electrophysiol. 2019;5(9):1084–1090. doi: 10.1016/j.jacep.2019.06.008. [DOI] [PubMed] [Google Scholar]
- 71.Dusi V, De Ferrari GM, Pugliese L, Schwartz PJ. Cardiac Sympathetic Denervation in Channelopathies. Front Cardiovasc Med. 2019;6:27–27. doi: 10.3389/fcvm.2019.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dusi V, Ajijola OA. Cardiopulmonary Performance After Left Cardiac Sympathetic Denervation for Long QT Syndromes. JACC Clin Electrophysiol. 2019;5(9):1091–1092. doi: 10.1016/j.jacep.2019.08.005. [DOI] [PubMed] [Google Scholar]
- 73.Han J, Ackerman MJ, Moir C, Cai C, Xiao PL, Zhang P, et al. Left cardiac sympathetic denervation reduces skin sympathetic nerve activity in patients with long QT syndrome. Heart Rhythm. 2020;17(10):1639–1645. doi: 10.1016/j.hrthm.2020.03.023. [DOI] [PubMed] [Google Scholar]
- 74.Mingyang X, Weijie C, Yuehui Y. Renal denervation for treating congenital long QT syndrome shortening the QT interval or modulating sympathetic tone?-Author's reply. Europace. 2019;21(11):1756–1757. doi: 10.1093/europace/euz266. [DOI] [PubMed] [Google Scholar]
- 75.Akkus M, Seyrek Y, Kafali HC, Ergül Y. Bilateral cardiac sympathetic denervation in children with long-QT syndrome and catecholaminergic polymorphic ventricular tachycardia. J Electrocardiol. 2020;61:32–36. doi: 10.1016/j.jelectrocard.2020.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dotzler M, Kim J, Gendron W, Zhou W, Ye D, Bos M, et al. Suppression-Replacement KCNQ1 Gene Therapy for Type 1 Long QT Syndrome. Circulation. 2021;143:1411–1425. doi: 10.1161/CIRCULATIONAHA.120.051836. [DOI] [PubMed] [Google Scholar]