

RESUMEN
La miocardiopatía hipertrófica es la enfermedad miocárdica más frecuente de carácter genético (60-70%) causada por mutaciones en uno de varios genes del sarcómero que codifican componentes del aparato contráctil del corazón. Se caracteriza por una hipertrofia miocárdica desproporcionada en ausencia de causa secundaria. La presentación clínica es variable, desde asintomáticos hasta insuficiencia cardíaca o muerte súbita cardíaca. La hipertrofia y la configuración ventricular anormal pueden dar como resultado una obstrucción dinámica del flujo de salida del ventrículo izquierdo en la mayoría de los casos. El objetivo de las intervenciones terapéuticas es en gran medida reducir la obstrucción dinámica, con diferentes opciones terapéuticas que abarca la estratificación del riesgo de muerte súbita, tamizaje genético, modificaciones del estilo de vida y fármacos. A continuación se discute un caso de miocardiopatía hipertrófica septal, una entidad bastante frecuente y subdiagnosticada.
Palabras clave: Cardiomiopatías, Hipertrofia ventricular izquierda, Genética
ABSTRACT
Hypertrophic cardiomyopathy is the more commonly (60 to 70 percent) genetically determined disease of the heart muscle caused by mutations in one of several sarcomere genes that encode components of the heart’s contractile apparatus. It is characterized by disproportionate hypertrophy in the absence of a secondary cause. The clinical presentation is variable, ranging from asymptomatic to heart failure or sudden cardiac death. Hypertrophy and abnormal ventricular configuration can result in dynamic left ventricular outflow obstruction in most cases. The goal of therapeutic interventions is largely to reduce dynamic obstruction, with different therapeutic options encompassing risk stratification for sudden death, genetic screening, lifestyle modifications, and drugs. A case of hypertrophic septal cardiomyopathy, a fairly frequent and under-diagnosed entity, is discussed below.
Keywords: Cardiomyopathies; Hypertrophy, left ventricular; Genetics
Introducción
La miocardiopatía hipertrófica (MCH) es la miocardiopatía hereditaria más común, que se manifiesta como hipertrofia en ausencia de una causa secundaria, con una prevalencia aproximada de uno en 500 personas 1. Después del primer paciente reportado en 1868, fue hasta la década de los 60 donde el Dr. Eugene Braunwald recopiló y describió observaciones, casos e información relevante acerca de esta patología 2. Aproximadamente el 60% de los casos es explicada por mutaciones en uno o varios genes del sarcómero que codifican el componente del aparato contráctil 3. El diagnóstico es un reto, dada la heterogeneidad fenotípica. La característica inicial es una hipertrofia del ventrículo izquierdo con una amplia gama de manifestaciones clínicas que pueden llevar a obstrucción del tracto de salida del ventrículo izquierdo, disfunción diastólica, isquemia miocárdica y/o regurgitación mitral 4. Términos como miocardiopatía hipertrófica obstructiva han resultado confusos, ya que aproximadamente un tercio de los pacientes no desarrollan obstrucción del tracto de salida del ventrículo izquierdo 5.
Se presenta el caso de una paciente con MCH de predominio septal, considerada previamente una enfermedad rara con un pronóstico incierto y opciones de tratamiento limitadas.
Reporte de caso
Paciente femenina de 48 años de edad, sin antecedentes de muerte súbita familiar, con hipertensión arterial primaria e infección por SARS-CoV-2 recuperada, consulta a una clínica de cuarto nivel de atención por dolor torácico de características anginosas de 2 meses de evolución, acompañado de deterioro de su clase funcional y disnea NYHA II. Al examen físico al ingreso afebril, signos vitales con presión arterial de 170/84 mmHg, frecuencia cardiaca 68 lpm, frecuencia respiratoria de 20 rpm y saturación al ambiente de 96%, sin ingurgitación yugular, prueba de Rondot negativa; a la auscultación cardiopulmonar no había hallazgos significativos, no soplos ni desdoblamientos, el punto de impulso máximo estaba conservado, no edema de miembros inferiores. Se realizó electrocardiograma en ritmo sinusal, con frecuencia cardiaca de 75 lpm con signos de hipertrofia del ventrículo izquierdo y trastorno de la repolarización secundario. Se tomaron biomarcadores cardíacos los cuales estaban elevados, sin delta de aumento o disminución, por lo que se determinó estratificación no invasiva.
Se realizó un ecocardiograma transtorácico que reportó cardiopatía con fenotipo hipertrófico de predominio septal, dilatación auricular izquierda con una fracción de eyección del ventrículo izquierdo (FEVI) del 65%, además de disfunción diastólica del ventrículo izquierdo (VI) del tipo III (Figura 1). La perfusión miocárdica fue negativa para isquemia miocárdica inducida por estrés farmacológico. Dada la sospecha de hipertrofia de predominio septal, se realizó cardiorresonancia que corroboró la sospecha inicial, demostrándose ventrículo izquierdo con hipertrofia concéntrica severa de predominio septal (grosor de 29 mm) con fibrosis parcheada, alteraciones sugestivas de fenotipo de MCH. No se demostró movimiento anormal sistólico ni gradiente obstructivo (Figura 2). Debido a la ausencia de antecedentes familiares de muerte súbita cardíaca u otras condiciones y bajo riesgo de muerte súbita calculado por escala de SCD HCM de 3%, no se le consideró candidata a colocación de desfibrilador automático implantable. Se dio egreso con beta-bloqueador, cita por genética y seguimiento por cardiología con adecuada evolución ambulatoria.
Figura 1. Ecocardiograma transtorácico, vista cuatro cámaras, donde se observa hipertrofia de predominio septal con un máximo espesor de 20 mm en el segmento medio del septum inferior.

Figura 2. Cardiorresonancia, A: eje corto; B: corte sagital; C: cuatro cámaras, y D: axial que confirma presencia de hipertrofia septal máxima de 29 mm (flechas).

Discusión
La MCH se caracteriza por un grosor de la pared miocárdica mayor o igual a 15 milímetros por ecocardiografía, tomografía computarizada o resonancia magnética cardiaca, en ausencia de una causa cardiaca, sistémica o metabólica capaz de producir la magnitud de la hipertrofia evidente 6. Si el grosor de la pared es de 13-14 mm puede ser diagnosticado, si existe una historia familiar o una prueba genética positivo 7. En nuestro caso, la paciente tenía antecedente de hipertensión arterial, condición descrita hasta en la mitad de los casos, o incluso mayor según la cohorte reportada 8,9. El grosor de la pared mayor a 16 mm es raramente encontrado en pacientes con hipertensión 1.
En cuanto a la etiología y genética, la mayoría de casos de MCH se explican por mutaciones genéticas que involucran el sarcómero cardíaco, disco Z y/o las proteínas que controlan el calcio 10. Entre ellos, los más comunes son los que involucran la cadena pesada de beta miosina 7 (MYH7) y la proteína de unión a miosina C3 (MYBPC3), que afectan a aproximadamente el 70% de los pacientes con variante positiva 10.
La arquitectura del músculo cardíaco está muy definida por una red de proteínas de la matriz extracelular, una red que funciona como un andamio y contiene numerosas células, incluidos los fibroblastos cardíacos 11. El componente principal de la matriz es el colágeno, que consta de aproximadamente un 85% de colágeno de tipo I, que es importante para la resistencia, y un 11% de colágeno de tipo III que es importante para la elasticidad 11. En caso de fibrosis miocárdica, se altera el equilibrio entre la síntesis y la degradación de las proteínas de la matriz extracelular, lo que conduce a una formación excesiva de tejido conectivo fibroso 12. En la MCH se observa una fibrosis parcheada sola o en combinación con compromiso intersticial y principalmente de predominio basal, situación que puede ser sustrato para arritmias potencialmente fatales y remodelamiento del VI 11.
Dependiendo, en parte, del sitio y la extensión de la hipertrofia cardíaca, los pacientes con MCH pueden desarrollar obstrucción del tracto de salida del VI (75% de los casos), disfunción diastólica, isquemia miocárdica, regurgitación mitral, arritmias y disfunción autonómica 6. La disfunción diastólica puede estar explicada por una relajación ventricular prolongada y no uniforme, pérdida de succión ventricular, distensibilidad de la cámara disminuida y absorción anormal de calcio intracelular 6.
Por otra parte, en cuanto al curso clínico, la MCH puede aparecer en cualquier fase de la vida, desde la infancia hasta la séptima década de la vida. Se caracteriza por tener una hipertrofia ventricular izquierda desproporcionada que no es explicada por una enfermedad sistémica secundaria 6. La presentación clínica es variable, desde pacientes asintomáticos hasta pacientes que debutan con falla cardiaca, arritmias o con una muerte súbita (30-40% de los casos) causada por una obstrucción del tracto de salida del ventrículo izquierdo 6.
Para el diagnóstico y pronóstico, la historia clínica y el examen físico son fundamentales en el enfoque del paciente con sospecha de MCH. Se debe indagar por antecedentes familiares, presencia de síntomas, la detección incidental de un soplo predominantemente sistólico, un punto de impulso máximo desplazado o la presencia de un S4. Si el electrocardiograma muestra signos de hipertrofia del ventrículo izquierdo, con presencia de ondas Q patológicas en cara inferior, anormalidades auriculares, desviación del eje a la izquierda y ondas T profundamente invertidas (gigantes) de V2-V4, el siguiente paso es una imagen 6.
En general, la MCH es confirmada cuando el grosor de la pared del ventrículo izquierdo es mayor de 15 mm hasta 21-22 mm, en promedio, demostrada más comúnmente por ecocardiografía (1. Un engrosamiento de la pared más limitado (13-14 mm) también puede ser diagnosticado en aquellos pacientes con antecedente familiar en primer grado de consanguinidad 6. Si las imágenes por ecocardiografía no son concluyentes, está indicada la resonancia magnética cardiaca (RMC). La resonancia puede ser útil en la confirmación de la sospecha, además de evaluar la extensión y poder identificar diagnósticos diferenciales, como enfermedades infiltrativas o corazón de atleta 13.
Se debe brindar asesoría genética cuando los antecedentes familiares o la evaluación clínica hacen sospechar de otra afección genética que se sabe que causa hipertrofia del ventrículo izquierdo, por ejemplo, enfermedad de Fabry o enfermedades por almacenamiento lisosómico, y en miembros de la familia de primer grado de un paciente con MCH 10.
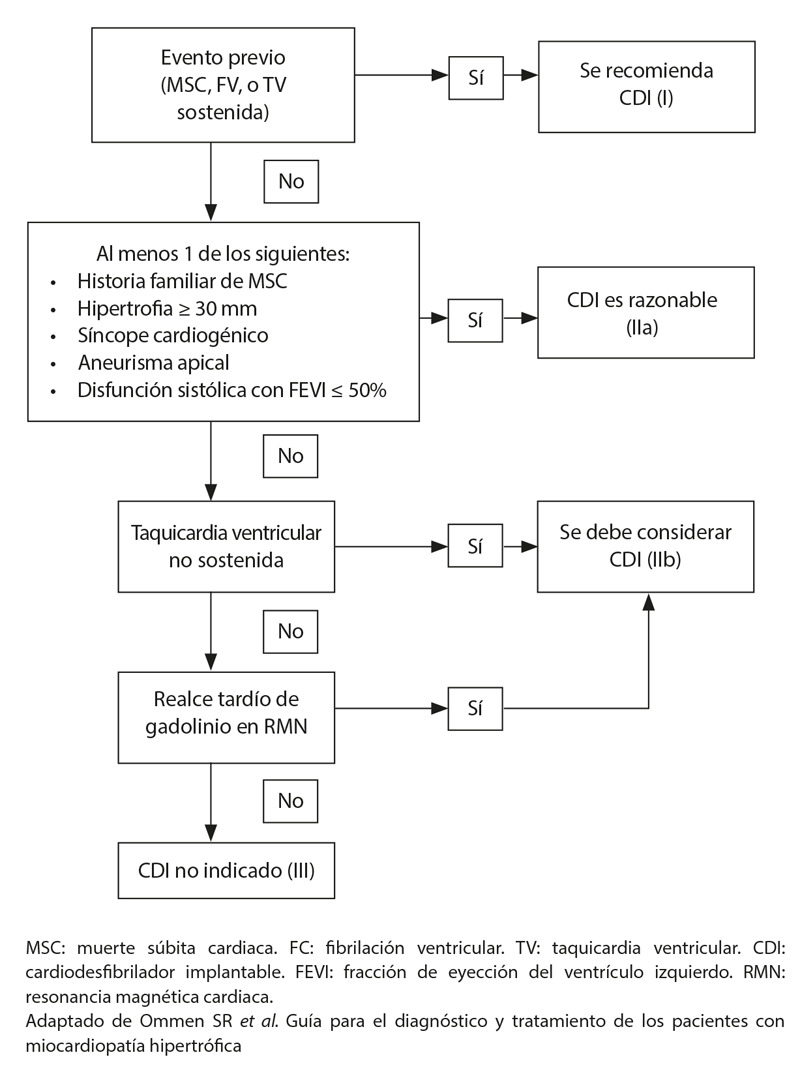
La estratificación del riesgo de muerte súbita para uso de desfibrilador cardioversor implantable (CDI), se puede realizar por medio de la calculadora HCM risk-SCD, a pesar de que algunos autores no lo recomiendan por su alta especificidad y baja sensibilidad 14. En esta se estima, que ante un riesgo >6% se debe considerar implantar un desfibrilador, entre 4-6% (puede ser considerado) y <4% no está generalmente indicado 6. En pacientes con historia de muerte súbita, grosor de la pared del ventrículo izquierdo mayor o igual a 30 mm, síncope cardiogénico secundario a una arritmia ventricular, aneurisma apical del ventrículo izquierdo y disfunción sistólica, se debe ofrecer el uso de CDI, independientemente del riesgo 6. En la Figura 3 se presenta un algoritmo donde se resumen las diferentes indicaciones del CDI y el nivel de evidencia.
Figura 3. Indicaciones de CDI en pacientes con miocardiopatía hipertrófica.
El manejo debe dirigirse hacia el control de los síntomas, control de las comorbilidades y prevención de eventos cardiovasculares mayores. Los betabloqueadores o calcioantagonistas no dihidropiridínicos podrían ser una opción terapéutica para disminuir el consumo de oxígeno, prevenir arritmias ventriculares y disminuir la frecuencia cardiaca para mejorar el llenado diastólico 4. Algunos reportes y series de casos han planteado la ablación por catéter y el manejo quirúrgico en pacientes que desarrollan arritmias ventriculares y tienen obstrucción del tracto de salida del ventrículo izquierdo, respectivamente 6.
A manera de conclusión, la MCH es una miocardiopatía genética infradiagnosticada causada por mutaciones del sarcómero cardíaco, que da como resultado una expresión fenotípica heterogénea con engrosamiento de la pared del ventrículo izquierdo. Tiene una presentación clínica muy variable, desde pacientes asintomáticos y otros con limitación significativa del estado funcional. El diagnóstico requiere una evaluación exhaustiva, incluida la estratificación del riesgo de muerte súbita cardiaca. El tratamiento depende del estado de los síntomas, la presencia de obstrucción del tracto de salida y la coexistencia de otras comorbilidades como fibrilación auricular, arritmias ventriculares y falla cardiaca.
Footnotes
Financiamiento: Autofinanciado.
Citar como: Forero S, Moreno NL. Miocardiopatía hipertrófica septal, la gran simuladora. Arch Peru Cardiol Cir Cardiovasc. 2021;2(4):274-278. doi: 10.47487/apcyccv.v2i4.171
Referencias bibliográficas
- 1.Geske JB, Ommen SR, Gersh BJ. Hypertrophic Cardiomyopathy Clinical Update. JACC Heart Fail. 2018;6(5):364–375. doi: 10.1016/j.jchf.2018.02.010. [DOI] [PubMed] [Google Scholar]
- 2.Braunwald E. Hypertrophic cardiomyopathy The first century 1869-1969. Glob Cardiol Sci Pract. 2012;2012(1):5–5. doi: 10.5339/gcsp.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ingles J, Burns C, Barratt A, Semsarian C. Application of Genetic Testing in Hypertrophic Cardiomyopathy for Preclinical Disease Detection. Circ Cardiovasc Genet. 2015;8(6):852–859. doi: 10.1161/CIRCGENETICS.115.001093. [DOI] [PubMed] [Google Scholar]
- 4.Maron BJ. Clinical Course and Management of Hypertrophic Cardiomyopathy. N Engl J Med. 2018;379(7):655–668. doi: 10.1056/NEJMra1710575. [DOI] [PubMed] [Google Scholar]
- 5.Maron MS, Rowin EJ, Olivotto I, Casey SA, Arretini A, Tomberli B, et al. Contemporary Natural History and Management of Nonobstructive Hypertrophic Cardiomyopathy. J Am Coll Cardiol. 2016;67(12):1399–1409. doi: 10.1016/j.jacc.2016.01.023. [DOI] [PubMed] [Google Scholar]
- 6.Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P, et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy. J Am Coll Cardiol. 2020;76(25):e159–e240. doi: 10.1016/j.jacc.2020.08.045. [DOI] [PubMed] [Google Scholar]
- 7.Maron BJ, Ommen SR, Semsarian C, Spirito P, Olivotto I, Maron MS. Hypertrophic Cardiomyopathy Present and Future, With Translation Into Contemporary Cardiovascular Medicine. J Am Coll Cardiol. 2014;64(1):83–99. doi: 10.1016/j.jacc.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Geske JB, Ong KC, Siontis KC, Hebl VB, Ackerman MJ, Hodge DO, et al. Women with hypertrophic cardiomyopathy have worse survival. Eur Heart J. 2017;38(46):3434–3440. doi: 10.1093/eurheartj/ehx527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aslam F, Haque A, Foody J, Shirani J. The frequency and functional impact of overlapping hypertension on hypertrophic cardiomyopathy a single-center experience. J Clin Hypertens (Greenwich) 2010;12(4):240–245. doi: 10.1111/j.1751-7176.2009.00251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Konno T, Chang S, Seidman JG, Seidman CE. Genetics of Hypertrophic Cardiomyopathy. Curr Opin Cardiol. 2010;25(3):10–10. doi: 10.1097/HCO.0b013e3283375698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eijgenraam TR, Silljé HHW, de Boer RA. Current understanding of fibrosis in genetic cardiomyopathies. Trends Cardiovasc Med. 2020;30(6):353–361. doi: 10.1016/j.tcm.2019.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Piek A, de Boer RA, Silljé HHW. The fibrosis-cell death axis in heart failure. Heart Fail Rev. 2016;21(2):199–211. doi: 10.1007/s10741-016-9536-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maron MS, Rowin EJ, Maron BJ. How to Image Hypertrophic Cardiomyopathy. Circ Cardiovasc Imaging. 2017;10(7):e005372. doi: 10.1161/CIRCIMAGING.116.005372. [DOI] [PubMed] [Google Scholar]
- 14.Maron BJ, Rowin EJ, Maron MS. Evolution of risk stratification and sudden death prevention in hypertrophic cardiomyopathy Twenty years with the implantable cardioverter-defibrillator. Heart Rhythm. 2021;18(6):1012–1023. doi: 10.1016/j.hrthm.2021.01.019. [DOI] [PubMed] [Google Scholar]