

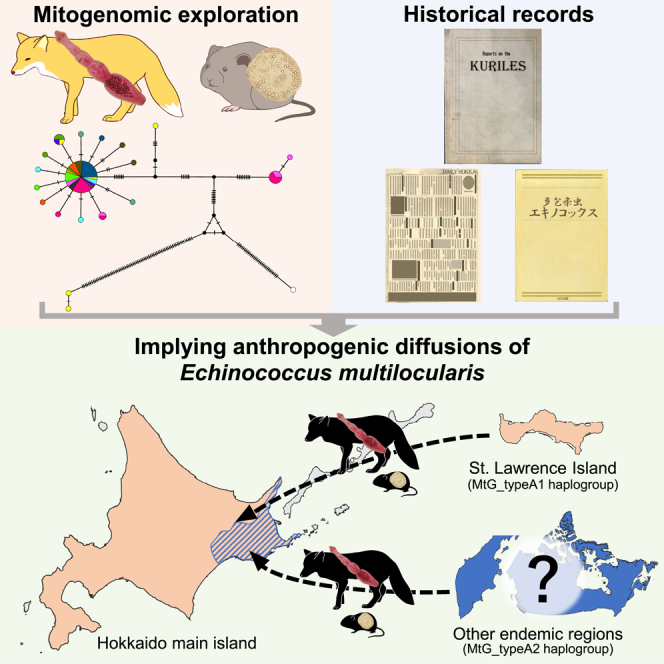
Summary
Animal movement across regions owing to human activity can lead to the introduction of pathogens, resulting in disease epidemics with medical and socioeconomic significance. Here, we validated the hypothesis that human activity, such as the transportation of infected animals, has played a significant role in introducing the zoonotic parasite Echinococcus multilocularis into Hokkaido, Japan, by synthesizing and evaluating parasite genetic data in light of historical records. Our analysis indicates that a major genetic group in Hokkaido originated from St. Lawrence Island, USA, which is in accordance with the route suggested by historical descriptions. Moreover, we identified a minor genetic group closely related to parasites found in Sichuan, China. This fact implies that parasite invasion in Japan may result from complex and inadvertent animal translocations. These findings emphasize the anthropogenic impacts on zoonotic parasite spread and provide a crucial perspective for preventing future potential epidemics.
Subject areas: Parasitology, Natural sciences, Biological sciences
Graphical abstract
Highlights
-
•
Past introduction routes of Echinococcus multilocularis were genetically investigated
-
•
Two distinct genetic groups were identified in the Hokkaido parasite population
-
•
Parasite invasion may result from complex and inadvertent animal translocations
-
•
Controls on animal translocations are required to prevent future disease epidemics
Parasitology; Natural sciences; Biological sciences
Introduction
Animal movement, whether anthropogenic or natural, heavily influences the spread of pathogens.1 Translocation of infected animals into non-endemic regions can cause medical and socioeconomic burdens.2,3 One example of such a pathogen is the small fox tapeworm Echinococcus multilocularis, which is distributed within a wide range of the northern hemisphere. The life cycle of E. multilocularis includes carnivores as definitive hosts and small rodents as intermediate hosts.4 Humans serve as dead-end hosts and are infected with a severe zoonotic disease, alveolar echinococcosis (AE), following oral ingestion of the parasite’s eggs, which are excreted in the feces of definitive hosts.5 Pet travel, international trade, and host animal migration are considered to have contributed to global increases in parasite distribution, raising public health concerns.6
Hokkaido, the northernmost prefecture of Japan, is an endemic area for AE.7 Red foxes, Vulpes vulpes, are the primary definitive hosts, and gray red-backed voles, Myodes rufocanus bedfordiae, are the most important intermediate hosts in this region.8 Two epidemics were recorded in the area, which was previously free of E. multilocularis.6 The first epidemic occurred on Rebun Island, located off the northwest coast of the main island of Hokkaido, and was presumably triggered by the introduction of E. multilocularis-infected foxes from the Kurile Islands during 1924–1926.8 This epidemic was identified due to the discovery of an AE patient from Rebun Island in 1937 and finally resulted in over 100 infected patients on the small island. However, the parasite appeared to have been mostly eliminated by the late 1950s, presumably due to the earlier eradication of definitive hosts on the island.7 In 1965, the second epidemic occurred in Eastern Hokkaido, near the western end of the Kurile Islands, and was not controlled, resulting in the spread of the parasite throughout the Hokkaido main island. The current prevalence of E. multilocularis in red foxes in Hokkaido is as high as 30%–40%, and approximately 20 human AE cases are identified annually.9 This equates to an incidence rate of around 0.42 per 100,000 inhabitants per year (between the years of 2010 and 2020).
Several lines of evidence indicate that human-mediated translocation of infected animals has played a significant role in the E. multilocularis invasions into Hokkaido. The most probable transmission routes described in previous studies10,11,12 are summarized in Figure 1. Briefly, infected voles were transferred from St. Lawrence Island, located in the Bering Strait, to Bering Island off the Kamchatka Peninsula in the late 19th century for fox feeding.11,13 This introduction resulted in a prevalence of E. multilocularis in intermediate hosts, Myodes rutilus, as high as approximately 50% on the island.14 In 1916–1917, 15 pairs of blue foxes, Vulpes lagopus, from Bering Island were introduced to the Middle Kurile Islands for the fox fur industry,15 followed by translocation of the infected foxes to other Kurile Islands. The foxes from the Kurile Islands are considered a parasite source on Rebun Island and Hokkaido main island. Although these routes have been inferred from some descriptions of human activities and epidemiological records, minimal data have been obtained to directly support this inference from a genetic perspective.16,17
Figure 1.

Map showing Hokkaido, Japan, and other islands relevant to this study
The possible introduction routes of Echinococcus multilocularis are indicated by black arrows with estimated year of the movements according to the literature. The inset illustrates the area enclosed by the red square.
Genetic information on endemic areas can provide a better understanding of disease dynamics. Previous genetic analyses have successfully shed light on the transmission dynamics of pathogens, including the origins and diffusion routes associated with human activity.18,19,20,21 A proper understanding of such past dynamics can help predict future diffusion and thus contribute to strategies for preventing further epidemics. Additionally, genetic characterization is also beneficial for assessing public health risks as parasites with distinct genetic backgrounds may vary in their zoonotic potential and severity.22,23
Here, we performed genetic analyses with complete mitochondrial genomes (mitogenomes) from 66 parasite specimens, consisting of 60 from Hokkaido main island, 5 from St. Lawrence Island in the USA, and 1 from Europe. The obtained data were further compared with those previously recorded in other endemic areas. The analyses provided genetic evidence that the parasite population in Hokkaido has two distinct ancestral locations of more than 3,000 km across the ocean, which indicates anthropogenic impacts on its diffusion process.
Results
Mitogenome construction
We analyzed 72 mitogenome sequences of E. multilocularis, including 66 from Hokkaido, 5 from St. Lawrence Island, and 1 from Europe. During the analysis, six of the Hokkaido mitogenome sequences (Sample IDs of HU_Em_018, 030, 031, 041, 044, and 049) were found to have nucleotide deletions of one to five bases in their coding sequences (CDSs) (Table 1), resulting in internal stop codons, and were excluded from subsequent molecular analysis. These deletions were confirmed through PCR and Sanger sequencing. As a result, we obtained a total of 66 complete mitogenomes, 60 from Hokkaido, 5 from St. Lawrence Island, and 1 from Europe (Table 1). These mitogenomes had a size range of 13,738–13,740 bp (Table 1) and consisted of 12 CDSs, 22 tRNA genes, and 2 ribosomal RNA genes, all of which were perfectly preserved. The accession numbers of the complete mitogenomes are listed in Table 1.
Table 1.
Geographic origins and haplotypes of the Echinococcus multilocularis parasites in this study
| Sample ID | Accession number | Sequence length (bp) | Collection city/site | Sub-prefecture | Collection year | Mitogenome haplotype | CYTB/ND2/COI haplotype |
|---|---|---|---|---|---|---|---|
| HU_Em_001 | LC720726 | 13,738 | Nemuro | Nemuro | 2019 | MtG_typeA1 | A4 |
| HU_Em_002 | LC720727 | 13,738 | Nemuro | Nemuro | 2019 | MtG_typeA7 | A4 |
| HU_Em_003 | LC720728 | 13,738 | Nemuro | Nemuro | 2019 | MtG_typeA1 | A4 |
| HU_Em_004 | LC720729 | 13,738 | Nemuro | Nemuro | 2019 | MtG_typeA2 | A27 |
| HU_Em_005 | LC720730 | 13,738 | Nemuro | Nemuro | 2020 | MtG_typeA1 | A4 |
| HU_Em_006 | LC720731 | 13,738 | Nemuro | Nemuro | 2020 | MtG_typeA1 | A4 |
| HU_Em_007 | LC720732 | 13,738 | Nemuro | Nemuro | 2020 | MtG_typeA2 | A27 |
| HU_Em_008 | LC720733 | 13,738 | Nemuro | Nemuro | 2020 | MtG_typeA5 | A4 |
| HU_Em_009 | LC720734 | 13,738 | Nemuro | Nemuro | 2020 | MtG_typeA1 | A4 |
| HU_Em_010 | LC720735 | 13,738 | Abashiri | Okhotsk | 2020 | MtG_typeA1 | A4 |
| HU_Em_011 | LC720736 | 13,738 | Abashiri | Okhotsk | 2020 | MtG_typeA1 | A4 |
| HU_Em_012 | LC720737 | 13,738 | Kiyosato | Okhotsk | 2019 | MtG_typeA1 | A4 |
| HU_Em_013 | LC720738 | 13,738 | Ozora | Okhotsk | 2020 | MtG_typeA1 | A4 |
| HU_Em_014 | LC720739 | 13,738 | Shari | Okhotsk | 2019 | MtG_typeA1 | A4 |
| HU_Em_015 | LC720740 | 13,738 | Shari | Okhotsk | 2019 | MtG_typeA1 | A4 |
| HU_Em_016 | LC720741 | 13,738 | Shari | Okhotsk | 2019 | MtG_typeA1 | A4 |
| HU_Em_017 | LC720742 | 13,738 | Koshimizu | Okhotsk | 2020 | MtG_typeA1 | A4 |
| HU_Em_018 | 13,738 | Iwamizawa | Sorachi | 2019 | Nucleotide deletion on ND3 | ||
| HU_Em_019 | LC720743 | 13,738 | Iwamizawa | Sorachi | 2020 | MtG_typeA1 | A4 |
| HU_Em_020 | LC720744 | 13,738 | Iwamizawa | Sorachi | 2020 | MtG_typeA1 | A4 |
| HU_Em_021 | LC720745 | 13,738 | Tsukigata | Sorachi | 2020 | MtG_typeA1 | A4 |
| HU_Em_022 | LC720746 | 13,738 | Tsukigata | Sorachi | 2020 | MtG_typeA9 | A4 |
| HU_Em_023 | LC720747 | 13,738 | Kushiro | Kushiro | 2019 | MtG_typeA1 | A4 |
| HU_Em_024 | LC720748 | 13,738 | Kushiro | Kushiro | 2019 | MtG_typeA3 | A27 |
| HU_Em_025 | LC720749 | 13,738 | Akkeshi | Kushiro | 2019 | MtG_typeA2 | A27 |
| HU_Em_026 | LC720750 | 13,738 | Hamanaka | Kushiro | 2020 | MtG_typeA5 | A4 |
| HU_Em_027 | LC720751 | 13,738 | Hamanaka | Kushiro | 2020 | MtG_typeA1 | A4 |
| HU_Em_028 | LC720752 | 13,738 | Asahikawa | Kamikawa | 2019 | MtG_typeA1 | A4 |
| HU_Em_029 | LC720753 | 13,738 | Asahikawa | Kamikawa | 2019 | MtG_typeA1 | A4 |
| HU_Em_030 | 13,738 | Asahikawa | Kamikawa | 2019 | Nucleotide deletion on ND1 | ||
| HU_Em_031 | 13,738 | Eniwa | Ishikari | 2020 | Nucleotide deletion on ND3 | ||
| HU_Em_032 | LC720754 | 13,738 | Eniwa | Ishikari | 2020 | MtG_typeA1 | A4 |
| HU_Em_033 | LC720755 | 13,738 | Eniwa | Ishikari | 2020 | MtG_typeA1 | A4 |
| HU_Em_034 | LC720756 | 13,738 | Bekkai | Nemuro | 2020 | MtG_typeA1 | A4 |
| HU_Em_035 | LC720757 | 13,738 | Bekkai | Nemuro | 2020 | MtG_typeA1 | A4 |
| HU_Em_036 | LC720758 | 13,738 | Bekkai | Nemuro | 2020 | MtG_typeA2 | A27 |
| HU_Em_037 | LC720759 | 13,738 | Bekkai | Nemuro | 2020 | MtG_typeA2 | A27 |
| HU_Em_038 | LC720760 | 13,738 | Esashi | Sōya | 2020 | MtG_typeA12 | A4 |
| HU_Em_039 | LC720761 | 13,738 | Esashi | Sōya | 2020 | MtG_typeA12 | A4 |
| HU_Em_040 | LC720762 | 13,738 | Tohma | Kamikawa | 2019 | MtG_typeA1 | A4 |
| HU_Em_041 | 13,738 | Higashikagura | Kamikawa | 2020 | Nucleotide deletion on ND3 | ||
| HU_Em_042 | LC720763 | 13,738 | Wakkanai | Sōya | 2020 | MtG_typeA11 | A30 |
| HU_Em_043 | LC720764 | 13,738 | Wakkanai | Sōya | 2019 | MtG_typeA1 | A4 |
| HU_Em_044 | 13,738 | Muroran | Iburi | 2019 | Nucleotide deletion on ND3 | ||
| HU_Em_045 | LC720765 | 13,738 | Muroran | Iburi | 2019 | MtG_typeA1 | A4 |
| HU_Em_046 | LC720766 | 13,738 | Embetsu | Rumoi | 2020 | MtG_typeA1 | A4 |
| HU_Em_047 | LC720767 | 13,738 | Teshio | Rumoi | 2020 | MtG_typeA12 | A4 |
| HU_Em_048 | LC720768 | 13,738 | Shinhidaka | Hidaka | 2020 | MtG_typeA13 | A4 |
| HU_Em_049 | 13,738 | Hidaka | Hidaka | 2019 | Nucleotide deletion on COI | ||
| HU_Em_050 | LC720769 | 13,738 | Sunagawa | Sorachi | 2020 | MtG_typeA1 | A4 |
| HU_Em_052 | LC720770 | 13,738 | Teshikaga | Kushiro | 2020 | MtG_typeA2 | A27 |
| HU_Em_053 | LC720771 | 13,738 | Shiraoi | Iburi | 2020 | MtG_typeA1 | A4 |
| HU_Em_054 | LC720772 | 13,738 | Samani | Hidaka | 2019 | MtG_typeA4 | A28 |
| HU_Em_055 | LC720773 | 13,738 | Yakumo | Oshima | 2020 | MtG_typeA10 | A4 |
| HU_Em_056 | LC720774 | 13,738 | Nakasatsunai | Tokachi | 2020 | MtG_typeA1 | A4 |
| HU_Em_057 | LC720775 | 13,738 | Ebetsu | Ishikari | 2020 | MtG_typeA1 | A4 |
| HU_Em_058 | LC720776 | 13,738 | Kamifurano | Kamikawa | 2020 | MtG_typeA8 | A4 |
| HU_Em_059 | LC720777 | 13,738 | Setana | Hiyama | 2019 | MtG_typeA1 | A4 |
| HU_Em_060 | LC720778 | 13,738 | Hokuto | Oshima | 2019 | MtG_typeA6 | A29 |
| HU_Em_061 | LC720779 | 13,738 | Nemuro | Nemuro | 1987 | MtG_typeA1 | A4 |
| HU_Em_062 | LC720780 | 13,738 | Koshimizu | Okhotsk | 1980s | MtG_typeA14 | A31 |
| HU_Em_063 | LC720781 | 13,738 | Tobetsu | Ishikari | 1980s | MtG_typeA15 | A3 |
| HU_Em_064 | LC720782 | 13,738 | Tobetsu | Ishikari | 2014 | MtG_typeA16 | A4 |
| HU_Em_065 | LC720783 | 13,738 | Kamiiso | Oshima | 1980s | MtG_typeA1 | A4 |
| HU_Em_066 | LC720784 | 13,738 | Kitami | Okhotsk | 1980s | MtG_typeA1 | A4 |
| HU_Em_067 | LC720785 | 13,737 | Yakumo | Oshima | 1980s | MtG_typeA1 | A4 |
| HU_Em_068 | LC720786 | 13,738 | St. Lawrence Island | 1989 | MtG_typeA1 | A4 | |
| HU_Em_069 | LC720787 | 13,739 | St. Lawrence Island | 1989 | MtG_typeN1 | N1 | |
| HU_Em_071 | LC720788 | 13,738 | St. Lawrence Island | 1989 | MtG_typeA17 | A2 | |
| HU_Em_073 | LC720789 | 13,740 | St. Lawrence Island | 1989 | MtG_typeN2 | N1 | |
| HU_Em_074 | LC720790 | 13,738 | St. Lawrence Island | 1951 | MtG_typeA12 | A4 | |
| HU_Em_075 | LC720791 | 13,738 | Europe | 1989 | MtG_typeE1 | E1 |
Characterization of mitogenome haplotypes in Hokkaido
The 60 E. multilocularis parasites from Hokkaido were classified into 16 haplotypes based on the CDSs of their mitogenome, with a haplotype diversity of 0.613 and nucleotide diversity of 0.00046. The reference sequence (AB018440) previously reported in Hokkaido24 did not match any of the 16 haplotypes. We designated these 16 mitogenome haplotypes as MtG_typeA1 to MtG_typeA16 (Asian haplotypes) following the analysis of partial sequences of selected genes, as explained below. The most common haplotype, the MtG_typeA1, was found in 61.7% (37 out of 60) of the parasites collected from 13 sub-prefectures in Hokkaido (Table 1; Figure 2). Our analysis showed that NADH dehydrogenase subunit (ND) 4, ND2, and ND3 had the highest number of parsimony-informative sites and that cytochrome b (CYTB) and ND4 had the highest haplotype diversity (Tables 2 and S1). The number of haplotypes distinguished by one mitochondrial CDS ranged from one to five.
Figure 2.

Geographical mapping of detected mitogenome haplotypes of Echinococcus multilocularis in Hokkaido
The numbers of examined parasite specimens and frequency of each haplotype on 13 sub-prefectures in Hokkaido are shown with parentheses and pie charts, respectively. The colors of each pie chart correspond to the haplotypes listed on the right.
Table 2.
Comparison of mitochondrial gene variation from 61 Echinococcus multilocularis parasites
| Gene name | Gene product | Nucleotide length | Number of haplotypes | GC content (%) | Polymorphic sites (%) | Parsimony-informative sites (%)∗ | Haplotype diversity ±S.D.∗ | Nucleotide diversity ±S.D. |
|---|---|---|---|---|---|---|---|---|
| ND3 | NADH dehydrogenase subunit 5 | 1575 | 4 | 30.1 | 0.38 (6/1575) | 0.25 (4/1575) | 0.264 ± 0.069 | 0.00057 ± 0.00016 |
| COIII | Cytochrome c oxidase subunit III | 648 | 3 | 33.0 | 0.31 (2/648) | 0.15 (1/648) | 0.236 ± 0.066 | 0.00037 ± 0.00011 |
| CYTB | Cytochrome b | 1068 | 5 | 31.9 | 0.37 (4/1068) | 0.19 (2/1068) | 0.319 ± 0.073 | 0.00032 ± 0.00008 |
| ND4L | NADH dehydrogenase subunit 4L | 261 | 1 | 28.4 | 0.00 (0/261) | 0 (0/261) | 0.000 | 0.000 |
| ND4 | NADH dehydrogenase subunit 4 | 1215 | 5 | 31.3 | 0.41 (5/1215) | 0.32 (4/1215) | 0.370 ± 0.075 | 0.0005 ± 0.00011 |
| ATP6 | ATP synthase subunit 6 | 516 | 4 | 30.3 | 0.58 (3/516) | 0.19 (1/516) | 0.264 ± 0.069 | 0.00053 ± 0.00014 |
| ND2 | NADH dehydrogenase subunit 2 | 882 | 2 | 29.1 | 0.57 (5/882) | 0.57 (5/882) | 0.207 ± 0.063 | 0.00117 ± 0.00036 |
| ND1 | NADH dehydrogenase subunit 1 | 894 | 2 | 31.3 | 0.11 (1/894) | 0.11 (1/894) | 0.207 ± 0.063 | 0.00023 ± 0.00007 |
| ND3 | NADH dehydrogenase subunit 3 | 348 | 3 | 27.9 | 1.15 (4/348) | 0.86 (3/348) | 0.236 ± 0.066 | 0.00187 ± 0.00055 |
| COI | Cytochrome c oxidase subunit I | 1608 | 4 | 32.3 | 0.19 (3/1608) | 0.06 (1/1608) | 0.264 ± 0.069 | 0.00017 ± 0.00005 |
| 16S rDNA | 16S ribosomal RNA | 983 | 2 | 31.6 | 0.20 (2/983) | 0.20 (2/983) | 0.207 ± 0.063 | 0.00042 ± 0.00013 |
| 12S rDNA | 12S ribosomal RNA | 704 | 3 | 31.8 | 0.28 (2/704) | 0.14 (1/704) | 0.236 ± 0.066 | 0.00034 ± 0.00010 |
| COII | Cytochrome c oxidase subunit II | 582 | 4 | 34.9 | 0.52 (3/582) | 0 (0/582) | 0.097 ± 0.052 | 0.00017 ± 0.00009 |
| ND6 | NADH dehydrogenase subunit 6 | 456 | 1 | 30.9 | 0.00 (0/456) | 0 (0/456) | 0.000 | 0.000 |
| 3 protein-coding sequences (CYTB/ND2/COI) | 3558 | 7 | 31.1 | 0.34 (12/3558) | 0.22 (8/3558) | 0.372 ± 0.075 | 0.00046 ± 0.00012 | |
| 12 protein-coding sequences | 10053 | 17 | 31.1 | 0.36 (36/10053) | 0.22 (22/10053) | 0.626 ± 0.071 | 0.00046 ± 0.00012 | |
The analysis includes the reference sequence (AB018440) previously reported in Hokkaido
The three genes showing the highest value are shown in bold.
Mitogenome haplotypes of samples from St. Lawrence Island and Europe
We obtained five haplotypes from St. Lawrence Island based on mitochondrial 12 CDSs (Table 1). Two of these haplotypes (MtG_typeA1 and MtG_typeA12) were identical to those detected in Hokkaido (Table 1). The remaining three haplotypes were designated MtG_typeA17 and MtG_typeN1-N2 (North American haplotypes) (Table 1). The single haplotype from Europe was named MtG_typeE1 (European haplotype).
Haplotypes based on CYTB/ND2/COI sequences
We generated concatenated sequences of three complete mitochondrial genes (CYTB, ND2, and cytochrome c oxidase subunit I [COI]) from each mitogenome for comparative analysis with previous studies.25,26,27,28,29,30 Seven haplotypes were identified in Hokkaido based on these concatenated sequences. The haplotype A4, previously reported in Hokkaido,30 was assigned to 48 parasites, corresponding to 80% of the total number of the parasites. This haplotype was found in all sub-prefectures surveyed (Figure S1). The remaining haplotypes in Hokkaido were assigned new IDs of A27–A31, following the study by Nakao et al.30 for Asian isolates (Table 1). The haplotypes found on St. Lawrence Island and Europe had identical sequences to previously reported haplotypes A2 and N1, and E1, respectively (Table 1).25,30
Genetic analysis on mitogenome haplotypes
The median-joining network analysis of the mitogenome haplotypes recovered three distinct clades of E. multilocularis: the Asian, North American, and European clades (Figure 3A). MtG_typeA1 and the other 13 haplotypes (MtG_typeA4–MtG_typeA16), with one or two nucleotide mismatches from MtG_typeA1, constituted a star-like shape, which represented a major haplogroup in Hokkaido. In contrast, MtG_typeA2 and MtG_typeA3 formed a minor group with 19 or 20 nucleotide mismatches from MtG_typeA1, indicating that E. multilocularis in Hokkaido could be divided into two distinct haplogroups (Figure 3A). All seven parasites obtained from Hokkaido from the 1980s to 2014 were MtG_typeA1 or its satellite haplotypes (Table 1). The minor group was detected only in the eastern Hokkaido, Nemuro, and Kushiro sub-prefectures (Figure 2).
Figure 3.

Visualization of mitochondrial haplotype networks of Echinococcus multilocularis using multiple protein-coding sequences
Median-joining haplotype networks were constructed from mitochondrial (A) 12 protein-coding sequences (CDSs) and (B) three CDSs (CYTB/ND2/COI) of E. multilocularis. The circles represent mitochondrial DNA haplotypes. The color and size of each circle correspond to the sub-prefecture/location where the haplotype was detected and the haplotype number. For the haplotype network of three CDSs, concatenated sequences of haplotypes from Europe (E1–E5, EmPL1–EmPL15, Est1, and Est2), Asia (A1–A25), North America (N1–N2, EAB, ESK, ECA, and E4), and Inner Mongolia (O1) were included.
Genetic analysis of CYTB/ND2/COI
The network analysis based on CYTB/ND2/COI haplotypes showed a genetic structure similar to that of mitogenome haplotypes, that is, two distinct haplogroups in Hokkaido. The major haplogroup included a haplotype from St. Lawrence Island (A4), whereas the minor haplogroup, detected only in the eastern part of Hokkaido, was clustered with haplotypes found in Sichuan, China (Figure 3B). The haplotypes neighboring A4 (A3, A28–A31) were unique samples collected in Hokkaido. The pairwise fixation index (Fst) values between Hokkaido and other endemic regions were high and significantly different among all combinations (Table 3), indicating a high degree of genetic differentiation in E. multilocularis population in Hokkaido.
Table 3.
Pairwise fixation index (Fst) among Echinococcus multilocularis subpopulations calculated using three mitochondrial genes
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|---|
| 1. Hokkaido, Japan | |||||||||
| 2. St. Lawrence Island | 0.833∗ | ||||||||
| 3. France | 0.914∗ | 0.762∗ | |||||||
| 4. Slovakia | 0.924∗ | 0.781∗ | 0.870∗ | ||||||
| 5. Kazakhstan | 0.597∗ | 0.611∗ | 0.948∗ | 0.992∗ | |||||
| 6. Kyrgyzstan | 0.626∗ | 0.843∗ | 0.918∗ | 0.928∗ | −0.0463 | ||||
| 7. Sichuan, China | 0.763∗ | 0.779∗ | 0.950∗ | 0.969∗ | 0.800∗ | 0.700∗ | |||
| 8. Indiana, USA | 0.949∗ | 0.713∗ | 0.979∗ | 1.00∗ | 0.992∗ | 0.954∗ | 0.978∗ | ||
| 9. Poland | 0.836∗ | 0.805∗ | 0.263∗ | 0.475∗ | 0.764∗ | 0.827∗ | 0.805∗ | 0.885∗ | |
| 10. Canada | 0.952∗ | 0.932∗ | 0.776∗ | 0.944∗ | 0.982∗ | 0.951∗ | 0.977∗ | 0.992∗ | 0.535∗ |
Significant values (p < 0.05) are indicated by an asterisk.
To investigate the diffusion process of E. multilocularis, we reconstructed the genealogy of three mitochondrial CDSs, including 64 haplotypes from 17 countries/regions. The generated Maximum clade credibility (MCC) tree showed that haplotypes in Hokkaido (A3–A4, A27–A31) fell into two clades, which is consistent with the results of the haplotype network. The major haplogroup was inferred to have originated from St. Lawrence Island, while the minor haplogroup was inferred to have originated from Sichuan (Figure 4A), with probabilities over 0.9 supporting these inferences. The Bayesian stochastic search variable selection (BSSVS) analysis identified a total of 19 diffusion routes of E. multilocularis, with four having a Bayes factor (BF) > 100, including two unidirectional routes to Hokkaido (Figure 4B; Table S2).
Figure 4.

Reconstruction of ancestral locations and diffusion process of Echinococcus multilocularis
(A) Maximum clade credibility tree generated with complete three protein-coding sequences (CYTB/ND2/COI). Branches are colored according to the most probable ancestral locations of the population. The probability of ancestral location is indicated via pie charts on nodes when the highest probability is less than 0.95. The color code is the same as that in Figure 3B, and tip codes indicate haplotypes and their sampling locations.
(B) Diffusion routes inferred by Bayesian stochastic search variable selection (BSSVS) analysis. Black arrows show routes with significant supports (100 > Bayes factor (BF) > 10), whereas red arrows show routes with decisive supports (BF > 100). The map was created by QGIS v3.16 (QGIS Association, 2022) using Natural Earth vector map data from https://www.naturalearthdata.com/downloads/.
Discussion
The dearth of research on the population genetics of E. multilocularis in Hokkaido has been a major obstacle to estimating the introduction process of this invasive parasite, in which anthropogenic activities are believed to be significantly involved. Understanding past events can provide precepts to prevent further diffusion and emergences of AE which are likely to occur in the context of global animal translocation. In this study, we synthesized complete mitogenomic data of E. multilocularis parasites to shed light on the human impacts on its diffusion.
Few studies have investigated the genetic characteristics of E. multilocularis in Hokkaido. One study by Okamoto et al. used a 391 bp fragment of COI sequences and detected no sequence variation in parasites from China, Hokkaido, and Alaska.16 Another study by Nakao et al. employed only six parasites to analyze CYTB/ND2/COI concatenated sequences, five of which were adult worms recovered from a single fox and probably derived from one cyst.30 These short sequence lengths and small sample sizes added ambiguity to the origin(s) of E. multilocularis population in Hokkaido. The current study analyzed mitogenomes from 66 parasites and found two haplotypes (MtG_typeA1 and MtG_typeA12) in both Hokkaido and St. Lawrence Island. Furthermore, we found that MtG_typeA1 was the dominant haplotype, comprising over 60% of the Hokkaido population, with a typical star-like-shaped haplotype network. This shape is typically observed when populations expand following introduction into new areas.31,32,33,34 These genetic structures, together with the link of decisive support (Figure 4), strongly suggest that St. Lawrence Island, located more than 3,000 km across the ocean, is one of the origins of E. multilocularis in Hokkaido, which supports the transmission routes indicated in previous studies (Figure 1).10,11,12
A striking feature of our study was that the parasite population in Hokkaido was divided into two haplogroups: MtG_typeA1 and MtG_typeA2 (Figure 3). The former includes haplotypes from St. Lawrence Island, whereas the latter is closely related to haplogroups from Sichuan, China (Figures 3B and 4A). Our analysis using BSSVS also strongly supported the two separated introduction routes to Hokkaido (Figure 4B). These results suggest that the parasite population in Hokkaido may have multiple origins. Indeed, it was documented that foxes were introduced into Kurile Islands for breeding from Canada and Sakhalin,35 which have endemic areas.23,36 Regarding intermediate hosts, previous reports have pointed out the possible transfer of voles on Bering Island from Kamchatka.14,37 Moreover, lemmings, which can serve as intermediate hosts,38,39 were reportedly brought into the Kurile Islands from overseas to provide fox feed, although the species and origin(s) have not been documented.40 While the accurate distribution of the MtG_TypeA2 (A27) haplogroup in the Northern Hemisphere remains unknown, these complex and inadvertent animal translocation events may explain the current genetic diversity of E. multilocularis in Hokkaido.
In the present study, we analyzed seven samples collected previously in Hokkaido (1980s–2014, Table 1) and demonstrated that they all belonged to the MtG_typeA1 haplogroup, which is specifically widely distributed throughout Hokkaido among samples collected in 2019–2020. Conversely, the MtG_typeA2 haplogroup comprised only samples obtained in 2019–2020 with a limited geographic distribution in the eastern part of Hokkaido, where the first human AE case was found. Although the absence of MtG_typeA2 haplogroup in 1980s–2014 might be attributed to the sample scarcity, these findings imply that multiple invasions of E. multilocularis have occurred in eastern Hokkaido. The parasite invasion into Rebun Island was most likely caused by anthropogenic introduction of infected foxes.12,41 However, the invasion route(s) into Hokkaido remain unclear as there has been no formal communication between the Hokkaido main island and the Kurile Islands for a long time.17 One possibility is that infected foxes naturally migrated to the area on drifting sea ice. From late February to early March, sea ice from the Okhotsk Sea drifts into the Nemuro Strait and the Goyoumai pass and sometimes covers these straits (Figure 1).42,43 The red fox (V. vulpes), the main definitive host in Hokkaido, has been observed to migrate on sea ice in other oceans in the Northern Hemisphere, such as the Beaufort Sea and Labrador Sea.44,45 Hasegawa reported a live fox on drifting ice in straits.10 Based on these observations, it can be speculated that E. multilocularis was brought into Hokkaido by spontaneous or accidental migration of foxes from the adjacent Kurile Islands. Indeed, the neighboring Kunashiri Island has a high prevalence of this parasite.17 Comparing the genetic compositions of V. vulpes populations in Hokkaido main island and Kurile Islands may further support this hypothesis. In fact, previous studies have successfully used genetic data to gain insights into introduction/migration events of foxes across the sea.46,47
A previous survey estimated the annual translocation of up to 30 infected dogs from Hokkaido to Honshu Island.48 This was in line with the study by Morishima et al., which found that some infected dogs were transported from Hokkaido.49 These reports imply that current inadvertent animal translocation can facilitate the further diffusion of the parasite and AE emergences. In fact, dogs infected with E. multilocularis have been reported in two prefectures of non-endemic areas on Honshu Island, Japan (see Figure 1).50,51 Microtus montebelli, a potential intermediate host, and a subspecies of the red fox, Vulpes vulpes japonica, are present in Honshu, implying that further epidemics are likely to occur in the future. Despite these risks, Japan has no regulations or quarantines on animal transportation. Similarly, Europe and North America, which have regions at risk of E. multilocularis invasions, have either no or insufficient controls, although infected pets have been reported.27,52,53 Applying pet mobility restrictions and appropriate veterinary controls, including diagnosis and deworming, is necessary to prevent further diffusion of the parasite.
Bayesian phylogeographic analysis is an effective tool for reconstructing past diffusion routes using current genetic information. A previous attempt using this approach succeeded in identifying the global diffusion routes and origin of the closely related parasite Echinococcus granulosus.19,54 The present study supported 19 diffusion routes of E. multilocularis, including those into Hokkaido, some of which align with previous findings, such as invasions of European strains into North America.55,56 However, the results should be interpreted cautiously because there is a lack or shortage of datasets from regions where E. multilocularis is prevalent, such as Russia and several European countries, including Germany and Switzerland. Indeed, Knapp et al., 2009, who analyzed genetic polymorphisms of 571 worms using microsatellite markers, indicated that Southern Germany and Switzerland may serve as ancestral foci for peripheral areas,57 while our analysis did not reflect such diffusion routes (Figure 4B). This may have been attributed to the sampling bias: our dataset included only a single haplotype derived from one specimen in Germany in contrast to three haplotypes from ten specimens in France. Additionally, the absence of data from Russia, where the parasite population is composed of a mixture of Asian, European, North American, and Mongolian haplotypes,58 may have contributed to the direct connections between St. Lawrence Island and Sichuan, Kyrgyzstan, and Poland, which are likely more complex in reality. Bayesian phylogeographic analysis depends on sampling intensity and location, which is important for interpreting results.19 Future global studies with multiple gene markers, ideally the mitogenome, will improve our understanding of the global diffusion process of E. multilocularis.
Inadvertent animal translocation in the modern era is a likely cause of the invasion of E. multilocularis, which has led to the loss of many lives and continues to threaten Hokkaido. The tragic event of introducing parasites into previously unaffected regions is attributed to insufficient knowledge and a lack of awareness concerning the consequences of animal translocation. The present study, through parasite population genetics, highlights the impact of human activities on the spread of zoonotic diseases, and as such, is expected to promote the development of strategies to prevent further AE emergences. It is noteworthy that even today, anthropogenic activities such as transporting infected pet dogs to non-endemic areas without quarantine continue to pose a potential risk of further diffusion of E. multilocularis. Strategic planning and appropriate veterinary control of animal movement must be carried out with the inclusion of governments and experts, and the general public must be educated concerning these risks.
Limitations of the study
This study successfully identified 19 diffusion routes of E. multilocularis, using Bayesian phylogeographic analysis. However, these results may have been affected by a lack or insufficiency of data from regions where the parasite is prevalent. Further global investigations with multiple gene markers, ideally the mitogenome, would contribute to a more comprehensive understanding of the global diffusion process of E. multilocularis.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Oligonucleotides | ||
| Primers to amplify mitogenomes of E. multilocularis | This study. See Table S3. | N/A |
| Software and algorithms | ||
| Multiple Alignment using Fast Fourier Transform (MAFFT) | Katoh and Standley, 201362 | https://mafft.cbrc.jp/alignment/software/ |
| PopART v1.7 | University of Otago Popart | https://popart.maths.otago.ac.nz/ |
| DnaSP 6 | Rozas et al., 201764 | http://www.ub.edu/dnasp/ |
| Arlequin 3.5 | Excoffier and Lischer, 201065 | http://cmpg.unibe.ch/software/arlequin35/ |
| BEAST v1.10.4 | Suchard et al., 201867 | https://beast.community/ |
| BEAUti v1.10.4 | Suchard et al., 201867 | https://beast.community/ |
| Kakusan4 | Tanabe, 201168 | https://www.fifthdimension.jp/products/kakusan/ |
| LogCombiner v1.10.4 | Suchard et al., 201867 | https://beast.community/ |
| TreeAnnotator | Suchard et al., 201867 | https://beast.community/ |
| FigTree | Suchard et al., 201867 | https://beast.community/ |
| SpreaD3 v0.9.7 | Bielejec et al., 201670 | https://rega.kuleuven.be/cev/ecv/software/SpreaD3 |
| QGIS v3.16 | QGIS Association, 2022 | https://qgis.org/ja/site/index.html |
| Deposited data | ||
| Mitogenome sequences | This study. See Table 1. | N/A |
| Analysis settings | This study. | https://data.mendeley.com/datasets/pcpxkgfzfr/1 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ryo Nakao (ryo.nakao@vetmed.hokudai.ac.jp).
Material availability
No new reagents were created in this investigation.
Experimental model and subject details
Parasite DNA from Hokkaido
A total of 60 adult E. multilocularis were collected individually from the small intestines of 60 foxes captured in Hokkaido between April 2019 and March 2020 during an annual surveillance program conducted by the Hokkaido Institute of Public Health, Japan. The sample identifiers, collection cities/sites (36 municipalities), and 13 sub-prefectures of Hokkaido where the parasites were obtained are listed in Table 1. The parasites were fixed in 70% ethanol and inactivated by overnight heating at 60°C. Each inactivated parasite was rinsed three times with phosphate-buffered saline, and 30 μL of 50 mM NaOH was added. Then, samples were heated at 95°C for 10 min and neutralized using 5.3 μl of 1 M Tris-HCl, pH 7.0. The lysate was centrifuged at 20,000 × g for 5 min, and the supernatant was used as a DNA template for PCR. In addition, DNA from six parasites collected at different sites in Hokkaido between the 1980s and 2014 was used for PCR (HU_Em_61 to HU_Em_67 in Table 1). One of these parasites is E. multilocularis Nemuro strain (HU_Em_61), which was established from a male M. rufocanus bedfordiae captured in Nemuro city in 1987 and has been maintained using a cotton rat experimental infection cycle at the Hokkaido Institute of Public Health, Japan.59,60
Parasite DNA from St. Lawrence Island and Europe
DNA extracted from five larval cysts from St. Lawrence Island and a protoscolex of the European strain was included in the analysis. One cyst was obtained from Myodes rutilus in 1951, and the other four cysts were collected separately from four wild voles in the 1980s from St. Lawrence Island. The European strain was kindly provided by Dr. Auer at the University of Vienna, Austria, in 1989 and has been maintained at the Hokkaido Institute of Public Health, as mentioned above.61
Method details
Whole mitochondrial genome construction
The whole mitogenome sequences of E. multilocularis were amplified using two overlapping PCRs (Figure S2; Table S3). PCR primers for the short (EmMtG_S1F and EmMtG_S1R) and long fragments (EmMtG_L1F and EmMtG_L1R) were designed by aligning the complete mitogenome sequences of E. multilocularis (AB018440), E. shiquicus (AB208064), E. equinus (AB786665), and E. granulosus sensu stricto (AB786664) deposited in the database. PCR was performed in a 25 μl reaction mixture containing 5 μL of 5× PrimeSTAR GXL Buffer (Mg2+ plus) (TaKaRa Bio Inc., Shiga, Japan), 2.0 μl of dNTP mixture (2.5 mM each), 200 nM of each primer, 0.5 μl of PrimeSTAR GXL DNA Polymerase (TaKaRa Bio Inc.), and 1.0 μl of template DNA. The reaction conditions were 40–45 cycles of 98°C for 10 s, 60°C for 15 s, and 68°C for 7 min for the short fragment and 40–45 cycles of 98°C for 10 s, and 68°C for 10 min for the long fragment. For DNA samples that could not be amplified under these PCR conditions, we designed four primer sets for alternative overlapping PCRs (EmMtG_A1F and EmMtG_A1R, EmMtG_A2F and EmMtG_A2R, EmMtG_A3F and EmMtG_A3R, EmMtG_A4F and EmMtG_A4R) (Figure S2; Table S3). The reaction solution was prepared as described above, and the PCR conditions were 40–45 cycles at 98°C for 10 s, 60°C for 15 s, and 68°C for 4 min. The PCR products were analyzed by electrophoresis on a 1.0% agarose gel and purified with NucleoSpin® Gel and PCR Clean-up kit (Macherey-Nagel, Düren, Germany).
Illumina sequencing libraries were constructed from the purified amplicons using the Nextera XT DNA Library Preparation Kit (Illumina, Hayward, CA, USA) and sequenced on an Illumina MiSeq platform using the MiSeq reagent kit v3 for 600 cycles. CLC Genomics Workbench v20.0.4 (Qiagen, Hilden, Germany) was used to map reads against the reference mitogenome (AB018440) to obtain complete mitogenome sequences of the samples. The 66 complete mitogenomes of E. multilocularis from Japan, St. Lawrence Island, and Europe were deposited in the DNA Data Bank of Japan (https://www.ddbj.nig.ac.jp/index.html) under accession numbers LC720726–LC720791 (Table 1).
Quantification and statistical analysis
Genetic data analyses
In total, 12 CDSs were extracted from each mitogenome sequence and concatenated. The concatenated sequences were aligned using MAFFT v7.450 software.62 To visualize the relationships between the haplotypes based on the 12 CDSs, a median-joining haplotype network analysis was performed using PopART v1.7.63 Genetic indices, including haplotype diversity (Hd) and nucleotide diversity () were calculated using the DnaSP v6.12.03.64
To compare our data with those previously reported from other endemic regions, we employed three mitochondrial CDSs (CYTB, ND2, and COI) in addition to the analysis of 12 CDSs. Following a concatenation of the selected CDSs, we added haplotypes from Asia (A1–25), North America (N1–N2, EAB, ESK, ECA, and E4), Inner Mongolia (O1), and Europe (E1–E5, EmPL1-15, Est1, and Est2) reported in previous studies for the analysis25,26,27,28,29,30 (Table S4). Sequence alignment and haplotype network analyses were performed as described above. Genetic distance between two sub-populations of the parasite was evaluated by pairwise Fst, which was calculated using Arlequin 3.5.65
A Bayesian discrete phylogeographic approach66 was performed to infer the diffusion process of E. multilocularis. This approach annotates tree nodes with the most probable ancestral locations and enables us to estimate the phylogeographic diffusion process using BSSVS. Specifically, we employed haplotypes based on the three selected CDSs (CYTB/ND2/COI) described above and assigned their sampling locations. Identical haplotypes from a single location were considered single haplotypes. BEAUti v1.10.4 was used to generate an XML file for BEAST v1.10.4.67 The best-fit model for nucleotide substitution was estimated by Kakusan4,68 and the Hasegawa-Kishino-Yano (HKY) model, with estimated base frequencies and gamma heterogeneity of four categories, was applied.69 The other priors were as follows: uncorrelated relaxed clock with a lognormal relaxed distribution for clock model, strict clock for discrete trait, asymmetric substitution model for discrete trait substitution model, constant-size tree model, and default. We carried out two independent runs of 2×108 generations and sampled every 20,000 generations. Tracer v1.6 was used to check the convergence and determine the number of samples to be removed as burn-in. We combined the remaining logs and trees with LogCombiner v1.10.4, discarded the first 10% of each run, and generated maximum clade credibility tree using TreeAnnotator. The MCC tree was displayed in FigTree. Finally, SpreaD3 v0.9.770 was used to calculate BFs and identify strongly supported diffusion routes (BF > 10).19
Acknowledgments
We are grateful to members of our laboratory for helpful discussions and support in the development of our research. We also extend our sincere appreciation to anonymous reviewers whose advice and comments improved this article. This research was funded by Japan Society for the Promotion of Science (JSPS) KAKENHI [Japan] Grant Numbers 20K06402 and 23H02369, by a research grant from JSPS for young scientists, by the World-leading Innovative and Smart Education (WISE) Program (1801) from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and by Research expenses from Hokkaido University Ambitious Doctoral Fellowship for sustainable development goals.
Author contributions
Conceptualization, N.H., R.N., G.K., and N.N.; Methodology, N.H., Y.O., W.M.A.M., M.A.M.M., and G.K.; Investigation, N.H., R.N., Y.O., T.I., H.K., E.C., and G.K.; Resources, T.I., H.K., M.O., and K.Y.; Data Curation, N.H. and R.N.; Writing – Original Draft, N.H.; Writing – Review & Editing, R.N., K.Y., and N.N.; Visualization, N.H.; Supervision, R.N. and N.N.; Funding Acquisition, N.H., R.N., K.Y., and N.N.
Declaration of interests
The authors declare no conflict of interest.
Published: August 26, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107741.
Supplemental information
Data and code availability
-
•
The sequences analyzed in this study are available under the accession numbers provided in Table S4.
-
•
Original codes, including software settings, have been deposited to Mendeley Data: https://data.mendeley.com/datasets/pcpxkgfzfr/1
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Fèvre E.M., Bronsvoort B.M.D.C., Hamilton K.A., Cleaveland S. Animal movements and the spread of infectious diseases. Trends Microbiol. 2006;14:125–131. doi: 10.1016/j.tim.2006.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marano N., Arguin P.M., Pappaioanou M., Marguerite P. Impact of globalization and animal trade on infectious disease ecology. Emerg. Infect. Dis. 2007;13:1807–1809. doi: 10.3201/eid1312.071276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karesh W.B., Cook R.A., Bennett E.L., Newcomb J., James N. Wildlife trade and global disease emergence. Emerg. Infect. Dis. 2005;11:1000–1002. doi: 10.3201/eid1107.050194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Torgerson P.R., Macpherson C.N.L. The socioeconomic burden of parasitic zoonoses: Global trends. Vet. Parasitol. 2011;182:79–95. doi: 10.1016/j.vetpar.2011.07.017. [DOI] [PubMed] [Google Scholar]
- 5.Romig T., Deplazes P., Jenkins D., Giraudoux P., Massolo A., Craig P.S., Wassermann M., Takahashi K., De La Rue M. Advances in Parasitology. Elsevier; 2017. Ecology and life cycle patterns of Echinococcus species; pp. 213–314. [DOI] [PubMed] [Google Scholar]
- 6.Davidson R.K., Romig T., Jenkins E., Tryland M., Robertson L.J. The impact of globalisation on the distribution of Echinococcus multilocularis. Trends Parasitol. 2012;28:239–247. doi: 10.1016/j.pt.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 7.Ito A., Romig T., Takahashi K. Perspective on control options for Echinococcus multilocularis with particular reference to Japan. Parasitology. 2003;127:S159–S172. doi: 10.1017/s0031182003003718. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi K., Uraguchi K., Kudo S. The epidemiological status of Echinococcus multilocularis in animals in Hokkaido, Japan. Mamm. Stud. 2005;30:101–105. [Google Scholar]
- 9.Yagi K. The control measures of alveolar echiococcosis conducted by Hokkaido local government. Rep. Hokkaido Inst. Public Health. 2017;67:1–7 (in Japanese). [Google Scholar]
- 10.Hasegawa M. (1969). On the role of foxes in Echinococcus multilocularis in Hokkaido. Rep. Hokkaido Inst. Public Health 19, 82–91 (in Japanese).
- 11.Yamashita J. Echinococcus and echinococcosis. Prog. Med. Parasitol. Jpn. Tokyo Meguro Parasitol. Mus. 1973:64–123. [Google Scholar]
- 12.Yamashita J. Studies on echinococcosis: Ⅱ. Echinococcosis in Japan. Jpn. J. Vet. Res. 1956;4:64–74. [Google Scholar]
- 13.Rausch R., Schiller E.L. Studies on the Helminth Fauna of Alaska. XXIV. Echinococcus sibiricensis n. sp., from St. Lawrence Island. J. Parasitol. 1954;40:659–662. doi: 10.2307/3273705. [DOI] [PubMed] [Google Scholar]
- 14.Barabash-Nikiforov I. Mammals of the Commander Islands and the surrounding sea. J. Mammal. 1938;19:423–429. [Google Scholar]
- 15.Ishino K. (1925). Fur industry in the Kuriles (Ⅳ). Chigakuzasshi Jpn. Geogr. J. 37, 351–358 (in Japanese).
- 16.Okamoto M., Bessho Y., Kamiya M., Kurosawa T., Horii T. Phylogenetic relationships within Taenia taeniaeformis variants and other taeniid cestodes inferred from the nucleotide sequence of the cytochrome c oxidase subunit I gene. Parasitol. Res. 1995;81:451–458. doi: 10.1007/BF00931785. [DOI] [PubMed] [Google Scholar]
- 17.Satoh M., Nakaya K., Nakao M., Xiao N., Yamasaki H., Sako Y., Naitoh Y., Kondo S., Kobayashi M., Ohtaishi N., Ito A. Short report: Echinococcus multilocularis confirmed on Kunashiri Island, 15 kilometers from the eastern part of Hokkaido, Japan. Am. J. Trop. Med. Hyg. 2005;72:284–288. [PubMed] [Google Scholar]
- 18.Hansen H., Bachmann L., Bakke T.A. Mitochondrial DNA variation of Gyrodactylus spp. (Monogenea, Gyrodactylidae) populations infecting Atlantic salmon, grayling, and rainbow trout in Norway and Sweden. Int. J. Parasitol. 2003;33:1471–1478. doi: 10.1016/S0020-7519(03)00200-5. [DOI] [PubMed] [Google Scholar]
- 19.Kinkar L., Laurimäe T., Acosta-Jamett G., Andresiuk V., Balkaya I., Casulli A., Gasser R.B., van der Giessen J., González L.M., Haag K.L., et al. Global phylogeography and genetic diversity of the zoonotic tapeworm Echinococcus granulosus sensu stricto genotype G1. Int. J. Parasitol. 2018;48:729–742. doi: 10.1016/j.ijpara.2018.03.006. [DOI] [PubMed] [Google Scholar]
- 20.Knapp J., Umhang G., Wahlström H., Al-Sabi M.N.S., Ågren E.O., Enemark H.L. Genetic diversity of Echinococcus multilocularis in red foxes from two Scandinavian countries: Denmark and Sweden. Food Waterborne Parasitol. 2019;14:e00045. doi: 10.1016/j.fawpar.2019.e00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Susetya H., Sugiyama M., Inagaki A., Ito N., Mudiarto G., Minamoto N. Molecular epidemiology of rabies in Indonesia. Virus Res. 2008;135:144–149. doi: 10.1016/j.virusres.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 22.Bartel M.H., Seesee F.M., Worley D.E. Comparison of Montana and Alaska isolates of Echinococcus multilocularis in gerbils with observations on the cyst growth, hook characteristics, and host response. J. Parasitol. 1992;78:529–532. [PubMed] [Google Scholar]
- 23.Davidson R.K., Lavikainen A., Konyaev S., Schurer J., Miller A.L., Oksanen A., Skírnisson K., Jenkins E. Echinococcus across the north: Current knowledge, future challenges. Food Waterborne Parasitol. 2016;4:39–53. doi: 10.1016/j.fawpar.2016.08.001. [DOI] [Google Scholar]
- 24.Nakao M., Yokoyama N., Sako Y., Fukunaga M., Ito A. The complete mitochondrial DNA sequence of the cestode Echinococcus multilocularis (Cyclophyllidea: Taeniidae) Mitochondrion. 2002;1:497–509. doi: 10.1016/s1567-7249(02)00040-5. [DOI] [PubMed] [Google Scholar]
- 25.Alvarez Rojas C.A., Kronenberg P.A., Aitbaev S., Omorov R.A., Abdykerimov K.K., Paternoster G., Müllhaupt B., Torgerson P., Deplazes P. Genetic diversity of Echinococcus multilocularis and Echinococcus granulosus sensu lato in Kyrgyzstan: The A2 haplotype of E. multilocularis is the predominant variant infecting humans. PLoS Negl. Trop. Dis. 2020;14:e0008242. doi: 10.1371/journal.pntd.0008242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karamon J., Stojecki K., Samorek-Pieróg M., Bilska-Zajac E., Rózycki M., Chmurzynska E., Sroka J., Zdybel J., Cencek T. Genetic diversity of Echinococcus multilocularis in red foxes in Poland: The first report of a haplotype of probable Asian origin. Folia Parasitol. 2017;64:2017.007. doi: 10.14411/fp.2017.007. [DOI] [PubMed] [Google Scholar]
- 27.Kuroki K., Morishima Y., Neil J., Beerntsen B.T., Matsumoto J., Stich R.W. Intestinal echinococcosis in a dog from Missouri. J. Am. Vet. Med. Assoc. 2020;256:1041–1046. doi: 10.2460/javma.256.9.1041. [DOI] [PubMed] [Google Scholar]
- 28.Laurimaa L., Süld K., Moks E., Valdmann H., Umhang G., Knapp J., Saarma U. First report of the zoonotic tapeworm Echinococcus multilocularis in raccoon dogs in Estonia, and comparisons with other countries in Europe. Vet. Parasitol. 2015;212:200–205. doi: 10.1016/j.vetpar.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 29.Massolo A., Klein C., Kowalewska-Grochowska K., Belga S., MacDonald C., Vaughan S., Girgis S., Giunchi D., Bramer S.A., Santa M.A., et al. European Echinococcus multilocularis identified in patients in Canada. N. Engl. J. Med. 2019;381:384–385. doi: 10.1056/nejmc1814975. [DOI] [PubMed] [Google Scholar]
- 30.Nakao M., Xiao N., Okamoto M., Yanagida T., Sako Y., Ito A. Geographic pattern of genetic variation in the fox tapeworm Echinococcus multilocularis. Parasitol. Int. 2009;58:384–389. doi: 10.1016/j.parint.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 31.Gaubert P., Godoy J.A., Del Cerro I., Palomares F. Early phases of a successful invasion: Mitochondrial phylogeography of the common genet (Genetta genetta) within the Mediterranean Basin. Biol. Invasions. 2009;11:523–546. doi: 10.1007/s10530-008-9268-4. [DOI] [Google Scholar]
- 32.López M., Foronda P., Feliu C., Hernández M. Genetic characterization of black rat (Rattus rattus) of the Canary Islands: Origin and colonization. Biol. Invasions. 2013;15:2367–2372. doi: 10.1007/s10530-013-0466-3. [DOI] [Google Scholar]
- 33.Tollenaere C., Brouat C., Duplantier J.M., Rahalison L., Rahelinirina S., Pascal M., Moné H., Mouahid G., Leirs H., Cosson J.F. Phylogeography of the introduced species Rattus rattus in the western Indian Ocean, with special emphasis on the colonization history of Madagascar. J. Biogeogr. 2010;37:398–410. doi: 10.1111/j.1365-2699.2009.02228.x. [DOI] [Google Scholar]
- 34.Wang Y., Halbert S., Mohamed S., Delatte H., Reynaud B., Beattie G.A.C., Holford P., Lu J., Cen Y. Mitochondrial genomes reveal diverse lineages of Diaphorina citri Kuwayama (Hemiptera: Sternorrhyncha: Psyllidae) in Kenya and La Réunion. Biol. Invasions. 2021;23:3109–3117. doi: 10.1007/s10530-021-02560-1. [DOI] [Google Scholar]
- 35.Hokkaido Government Chishima Tyousajo . 1957. Chishima Tyousasyo. (in Japanese) [Google Scholar]
- 36.Torgerson P.R., Keller K., Magnotta M., Ragland N. The global burden of alveolar echinococcosis. PLoS Negl. Trop. Dis. 2010;4:e722. doi: 10.1371/journal.pntd.0000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stejneger L. Contributions to the history of the Commander Islands. No.1. Notes on the natural history, including descriptions of new cetaceans. Proc. U. S. Natl. Mus. 1883;6:58–89. [Google Scholar]
- 38.Holt D.W., Hanns C., O’hara T., Burek K., Frantz R. New distribution records of Echinococcus multilocularis in the brown lemming from Barrow, Alaska, USA. J. Wildl. Dis. 2005;41:257–259. doi: 10.7589/0090-3558-41.1.257. [DOI] [PubMed] [Google Scholar]
- 39.Rausch R.L., Jentoft V. Studies on the helminth fauna of Alaska. XXXI. Observations on the propagation of the larval Echinococcus multilocularis Leuckart, 1863, in vitro. J. Parasitol. 1957;43:1–8. [PubMed] [Google Scholar]
- 40.Yamada S. (2020). Fox introduction to the islands of Chishima (Kuril Islands), Karafuto, and Hokkaido during the 1910s-40s. Bull. Hokkaido Mus. 5, 265–282 (in Japanese).
- 41.Inukai T., Yamashita J., Mori H. Most probable route of introduction of Echinococcus into the island of Rebun. J. Fac. Agric. Hokkaido Univ. 1955;50:134–139. [Google Scholar]
- 42.Motoi T., Chan W.-L., Miyakawa T., Usui N. Sea ice, watermass and freshwater processes/Coastal lagoons Sea-ice flow from the Okhotsk Sea to the Pacific Ocean through the Nemuro Strait in 2008. PICES Sci. Rep. 2009;36:143–153. [Google Scholar]
- 43.Otsuki M., Akamatsu T., Nobetsu T., Mizuguchi D., Mitani Y. Diel changes in ribbon seal Histriophoca fasciata vocalizations during sea ice presence in the Nemuro Strait, Sea of Okhotsk. Polar Biol. 2018;41:451–456. doi: 10.1007/s00300-017-2203-3. [DOI] [Google Scholar]
- 44.Andriashek D., Kiliaan H.P., Taylor M.K. Observations on foxes, Alopex lagopus and Vulpes vulpes, and wolves, Canis lupus, on the off-shore sea ice of northern Labrador. Can. Field Nat. 1985;99:86–89. [Google Scholar]
- 45.Jung T.S., Suitor M.J., Barykuk S., Nuyaviak J., Gordon D.C., Gordon Jr D., Pokiak E. Red Fox (Vulpes vulpes) scavenging on the spring sea ice: Potential implications for Arctic food webs. Can. Field Nat. 2020;134:144–146. doi: 10.22621/CFN.V134I2.2375. [DOI] [Google Scholar]
- 46.Norén K., Carmichael L., Fuglei E., Eide N.E., Hersteinsson P., Angerbjörn A. Pulses of movement across the sea ice: population connectivity and temporal genetic structure in the arctic fox. Oecologia. 2011;166:973–984. doi: 10.1007/s00442-011-1939-7. [DOI] [PubMed] [Google Scholar]
- 47.Atterby H., Allnutt T.R., MacNicoll A.D., Jones E.P., Smith G.C. Population genetic structure of the red fox (Vulpes vulpes) in the UK. Mamm. Res. 2015;60:9–19. doi: 10.1007/s13364-014-0209-6. [DOI] [Google Scholar]
- 48.Doi R., Matsuda H., Uchida A., Kanda E., Kamiya H., Konno K., Tamashiro H., Nonaka N., Oku Y., Kamiya M. (2003). Possibility of invasion of Echinococcus multilocularis into Honshu with pet dogs from Hokkaido and overseas. Nihon Koshu Eisei Zasshi 50, 639–649 (in Japanese). [PubMed]
- 49.Morishima Y., Sugiyama H., Arakawa K., Kawanaka M. Echinococcus multilocularis in dogs, Japan. Emerg. Infect. Dis. 2006;12:1292–1294. doi: 10.3201/eid1208.051241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morishima Y., Tomaru Y., Fukumoto S.I., Sugiyama H., Yamasaki H., Hashimoto C., Harada K. Canine echinococcosis due to Echinococcus multilocularis: A second notifiable case from mainland Japan. Jpn. J. Infect. Dis. 2016;69:448–449. doi: 10.7883/yoken.JJID.2015.573. [DOI] [PubMed] [Google Scholar]
- 51.Yamamoto N., Morishima Y., Kon M., Yamaguchi M., Tanno S., Koyama M., Maeno N., Azuma H., Mizusawa H., Kimura H., et al. The first reported case of a dog Infected with Echinococcus multilocularis in Saitama Prefecture, Japan. Jpn. J. Infect. Dis. 2006;59:351–352. [PubMed] [Google Scholar]
- 52.Romig T., Thoma D., Weible A.-K. Echinococcus multilocularis – a zoonosis of anthropogenic environments? J. Helminthol. 2006;80:207–212. doi: 10.1079/joh2006347. [DOI] [PubMed] [Google Scholar]
- 53.Wahlström H., Enemark H.L., Davidson R.K., Oksanen A. Present status, actions taken and future considerations due to the findings of E. multilocularis in two Scandinavian countries. Vet. Parasitol. 2015;213:172–181. doi: 10.1016/j.vetpar.2015.07.037. [DOI] [PubMed] [Google Scholar]
- 54.Zhao Y., Gesang D., Wan L., Li J., Qiangba G., Danzeng W., Basang Z., Renzhen N., Yin J., Gongsang Q., et al. Echinococcus spp. and genotypes infecting humans in Tibet Autonomous Region of China: a molecular investigation with near-complete/complete mitochondrial sequences. Parasites Vectors. 2022;15:75. doi: 10.1186/s13071-022-05199-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gesy K., Hill J.E., Schwantje H., Liccioli S., Jenkins E.J. Establishment of a European-type strain of Echinococcus multilocularis in Canadian wildlife. Parasitology. 2013;140:1133–1137. doi: 10.1017/S0031182013000607. [DOI] [PubMed] [Google Scholar]
- 56.Massolo A., Liccioli S., Budke C., Klein C. Echinococcus multilocularis in North America: The great unknown. Parasite. 2014;21:73. doi: 10.1051/parasite/2014069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Knapp J., Bart J.M., Giraudoux P., Glowatzki M.L., Breyer I., Raoul F., Deplazes P., Duscher G., Martinek K., Dubinsky P., et al. Genetic diversity of the cestode Echinococcus multilocularis in red foxes at a continental scale in Europe. PLoS Negl. Trop. Dis. 2009;3:e452. doi: 10.1371/journal.pntd.0000452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Konyaev S.V., Yanagida T., Nakao M., Ingovatova G.M., Shoykhet Y.N., Bondarev A.Y., Odnokurtsev V.A., Loskutova K.S., Lukmanova G.I., Dokuchaev N.E., et al. Genetic diversity of Echinococcus spp. in Russia. Parasitology. 2013;140:1637–1647. doi: 10.1017/S0031182013001340. [DOI] [PubMed] [Google Scholar]
- 59.Imasato Y., Nakao R., Irie T., Kouguchi H., Yagi K., Nariaki N., Katakura K. Characterization of microRNAs expressed in the cystic legion of the liver of Mus musculus perorally infected with Echinococcus multilocularis Nemuro strain. Parasitol. Int. 2021;81:102247. doi: 10.1016/j.parint.2020.102247. [DOI] [PubMed] [Google Scholar]
- 60.Nakao R., Kameda Y., Kouguchi H., Matsumoto J., Dang Z., Simon A.Y., Torigoe D., Sasaki N., Oku Y., Sugimoto C., et al. Identification of genetic loci affecting the establishment and development of Echinococcus multilocularis larvae in mice. Int. J. Parasitol. 2011;41:1121–1128. doi: 10.1016/j.ijpara.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 61.Yagi K., Ito T., Kawase S., Takahashi K. (2001). Establishment of European isolate of Echinococcus multilocularis and its identification by PCR-RFLP. Rep. Hokkaido Inst. Public Health 51, 72–75 (in Japanese).
- 62.Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leigh J.W., Bryant D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015;6:1110–1116. doi: 10.1111/2041-210X.12410. [DOI] [Google Scholar]
- 64.Rozas J., Ferrer-Mata A., Sánchez-DelBarrio J.C., Guirao-Rico S., Librado P., Ramos-Onsins S.E., Sánchez-Gracia A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017;34:3299–3302. doi: 10.1093/molbev/msx248. [DOI] [PubMed] [Google Scholar]
- 65.Excoffier L., Lischer H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010;10:564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- 66.Lemey P., Rambaut A., Drummond A.J., Suchard M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009;5:e1000520. doi: 10.1371/journal.pcbi.1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Suchard M.A., Lemey P., Baele G., Ayres D.L., Drummond A.J., Rambaut A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4:vey016. doi: 10.1093/ve/vey016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tanabe A.S. Kakusan4 and Aminosan: two programs for comparing nonpartitioned, proportional and separate models for combined molecular phylogenetic analyses of multilocus sequence data. Mol. Ecol. Resour. 2011;11:914–921. doi: 10.1111/j.1755-0998.2011.03021.x. [DOI] [PubMed] [Google Scholar]
- 69.Hasegawa M., Kishino H., Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- 70.Bielejec F., Baele G., Vrancken B., Suchard M.A., Rambaut A., Lemey P. SpreaD3: Interactive visualization of spatiotemporal history and trait evolutionary processes. Mol. Biol. Evol. 2016;33:2167–2169. doi: 10.1093/molbev/msw082. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The sequences analyzed in this study are available under the accession numbers provided in Table S4.
-
•
Original codes, including software settings, have been deposited to Mendeley Data: https://data.mendeley.com/datasets/pcpxkgfzfr/1
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.