

Abstract
Esophageal adenocarcinoma (EAC) patients have poor clinical outcomes, with an overall 5-year survival rate of 20%. Smoking is a significant risk factor for EAC. The role of WEE1, a nuclear kinase that negatively regulates the cell cycle in normal conditions, in EAC tumorigenesis and drug resistance is not fully understood. Immunohistochemistry staining shows significant WEE1 overexpression in human EAC tissues. Nicotine, nicotine-derived nitrosamine ketone, or 2% cigarette smoke extract treatment induces WEE1 protein expression in EAC, detected by western blot and immunofluorescence staining. qRT-PCR and reporter assay indicates that smoking induces WEE1 expression through miR-195-5p downregulation in EAC. ATP-Glo cell viability and clonogenic assay confirmed that WEE1 inhibition sensitizes EAC cells to docetaxel treatment in vitro. A TE-10 smoking machine with EAC patient-derived xenograft mouse model demonstrated that smoking induces WEE1 protein expression and resistance to docetaxel in vivo. MK-1775 and docetaxel combined treatment improves EAC patient-derived xenograft mouse survival in vivo. Our findings demonstrate, for the first time, that smoking-induced WEE1 overexpression through miRNA dysregulation in EAC plays an essential role in EAC drug resistance. WEE1 inhibition is a promising therapeutic method to overcome drug resistance and target treatment refractory cancer cells.
Keywords: esophageal adenocarcinoma, WEE1, smoking, miRNA, patient-derived xenograft, docetaxel
Graphical abstract
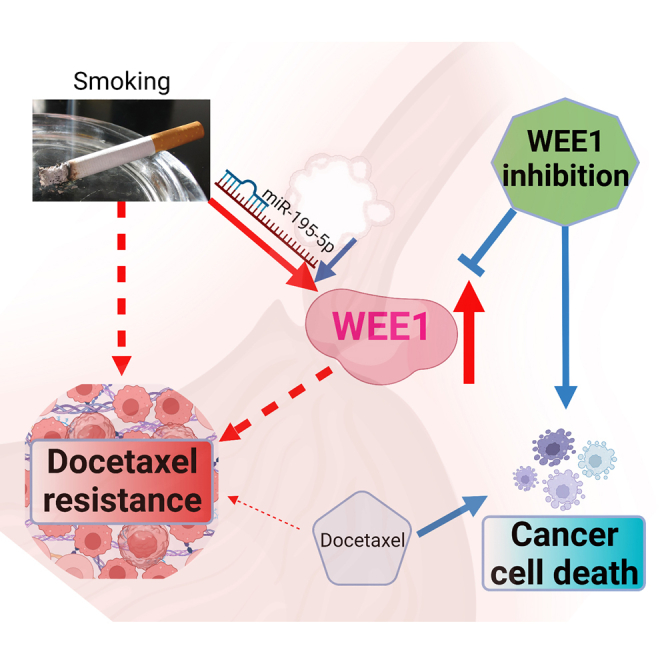
Chen and colleagues reveal a novel connection between smoking-induced WEE1 overexpression and drug resistance in esophageal adenocarcinoma (EAC). Their findings demonstrate that WEE1 inhibition, achieved through targeting miRNA dysregulation, holds promise as a therapeutic approach to overcome drug resistance and treat refractory cancer cells in EAC patients.
Introduction
Esophageal cancer is the eighth most diagnosed cancer and is the cause of the sixth highest number of cancer-related deaths worldwide.1 Globally, more than 604,000 new cases and more than 544,000 deaths were estimated from esophageal cancer in 2020.2 Over the past four decades, a sharp increase in esophageal adenocarcinoma (EAC) has been reported in the United States and Western countries.3 The estimated number of new cases in 2022 is over 20,000, and the estimated deaths are over 16,000 in the United States.4 The current seven standard types of esophageal cancer treatment include surgery, radiation therapy, chemotherapy, chemoradiation therapy, laser therapy, electrocoagulation, and immunotherapy.5 EAC is poorly responsive to treatment and has an unfavorable outcome, with an estimated 5-year survival rate of around 20% in the United States.4 Identification of new treatment approaches is urgently needed for this deadly disease.
According to the American Cancer Society, tobacco products, including cigarettes, cigars, pipes, and chewing tobacco, are a major risk factor for esophageal cancer.6 A recent study from Cook et al. used data from 12 studies in Barrett’s esophagus (BE) and esophageal adenocarcinoma (EAC) consortium (BEACON), containing 4,214 cases of EAC and 13,750 control subjects. In the pooled analyses of ever-cigarette smoking, the study reported statistically significant associations between EAC and esophagogastric junctional adenocarcinoma.7 Another study used primary data from 10 population-based case-control studies.8 Two cohort studies from the BEACON showed that current tobacco smoking was significantly associated with an increased risk of progression compared with never smoking. Moreover, their findings also indicated that tobacco smoking is the most potent risk factor for progression from BE to EAC.8
MicroRNAs (miRNAs) are short non-coding RNAs that negatively regulate protein expression. miRNA dysregulation has been shown to be a master regulator, playing a vital role in tumor initiation, progression, and metastasis.9,10,11 Our previous studies demonstrated unique miRNA expression signatures in human EAC and gastric cancer.12,13 We used human gastric tissue samples and the mouse model to show that miRNA dysregulation is conserved between human and mouse gastric cancers, promoting cancer survival and progression.14
WEE1, a nuclear kinase, phosphorylates and inactivates CDC2, arresting cancer cells in the G2-M checkpoint for DNA repair.15 Cancer cells with TP53 or RB pathway mutations evade their G1-S checkpoint and rely on the G2-M for DNA damage repair. Recent studies have shown that inhibition of WEE1 kinase is also effective in TP53 wild-type contexts and seems independent of TP53 status.16
In this study, we report that smoking induces WEE1 overexpression through miRNA dysregulation, promoting EAC cell drug resistance. WEE1 inhibition attenuates smoking-induced docetaxel resistance in EAC in vitro and in vivo models. Combining WEE1 inhibitor MK-1775 with docetaxel is a promising therapeutic option for EAC.
Results
WEE1 is overexpressed in human EAC patient samples, predicting poor survival
We explored WEE1 mRNA expression levels in different human cancers using the TNMplot online tool.17 Our analysis demonstrated that WEE1 mRNA was significantly upregulated in human gastrointestinal malignancies such as esophageal, stomach, and colon (Figure S1A). This result agrees with earlier reports showing overexpression of WEE1 in several human malignancies.18,19,20 WEE1 mRNA expression levels in 294 non-cancer normal and 375 esophageal cancer human tissue samples showed that WEE1 mRNA was significantly higher in EAC tissues compared with non-cancer normal esophageal samples (Figure 1A, p < 0.001). We tested 10 normal and 96 EAC de-identified human tissue samples to validate this finding. Immunohistochemistry staining of WEE1 in these samples indicated that the WEE1 protein is significantly overexpressed in human EAC compared with the normal esophagus (Figures 1B–1D, p < 0.01). Furthermore, analysis of 126 EAC patients' survival data from KMplot21 indicated that WEE1 high-expressing esophageal cancer patients had significantly worse survival than WEE1 low-expressing patients (Figure 1E, p < 0.05). To examine the WEE1 protein expression levels in the esophageal cell lines, we tested EPC2 (human normal esophageal epithelial cell line), CPA (non-dysplastic metaplasia, BE cell line), CPB (dysplastic BE epithelial cells), BART (non-neoplastic Barrett’s epithelial cells), and EAC cell lines (FLO-1, OE33, SK-GT4, OE19, and ESO26). Western blot data showed that WEE1 was relatively high in FLO-1, OE33, OE19, and ESO26 EAC cells compared with non-cancer cells (Figure S1B). These findings indicate that WEE1 is frequently overexpressed in human EACs, predicting poor patient survival.
Figure 1.

WEE1 is overexpressed in human esophageal adenocarcinoma
(A) WEE1 mRNA expression in the normal human non-cancer esophagus (Normal) and esophageal adenocarcinoma (Cancer) tissue samples. Data analyzed by TNMplot.com. (B) Immunohistochemistry (IHC) staining of WEE1 in human normal esophageal tissue samples. (C) IHC staining of WEE1 in human esophageal adenocarcinoma tissue samples. (D) Quantification data of (C and D). (E) Esophageal adenocarcinoma patient survival data analyzed according to different WEE1 expression levels, analyzed from KMplot.com ∗∗p<0.01.
Smoking induces WEE1 protein expression and activation in EAC cells
As smoking is a crucial risk factor during the progression from BE to EAC and our data showed that WEE1 mRNA is highly expressed in EAC (Figure 1A), we tested if smoking induces WEE1 expression in EAC. Western blot data indicated that low level of nicotine (100 ng/mL) treatment for 1 or 3 h induced WEE1 and downstream phosphorylation of CDC2 (p-CDC2) protein expression in both FLO-1 and OE33 cells (Figure 2A). Nicotine-derived nitrosamine ketone (NNK) is a vital tobacco-specific nitrosamine derived from nicotine, playing an essential role in carcinogenesis.22,23 Our data from western blot analysis showed that NNK treatment (10 μM) induced WEE1 and p-CDC2 protein expression levels from 3 to 48 h in a time-dependent manner (Figure 2B). To best simulate the smoking effect in vitro, we prepared cigarette smoking extract (CSE), as recently reported.24,25 Western blot results demonstrated that 2% CSE treatment induced WEE1 and p-CDC2 protein expression from 3 to 24 h in FLO-1, OE33, and CPB cells (Figures 2C–2E). Using immunofluorescence staining, our data indicated that 2% CSE and NNK treatment significantly increased WEE1 protein expression in FLO-1 cells (Figures 2F and 2G). To investigate how smoking induces WEE1 in EAC, we tested the mRNA expression of WEE1 after smoking treatment. qRT-PCR results demonstrated that NNK or 2% CSE treatment had a limited effect on regulating WEE1 mRNA in FLO-1 and OE33 cells (Figures S1B and S1C). In the meantime, we also tested the WEE1 protein half-life with or without CSE or NNK treatment. Surprisingly, western blot data indicated that CSE or NNK treatment did not increase the WEE1 half-life (Figures S2A–S2C). We analyzed the WEE1 mRNA expression in EAC from the TCGA database with smoking status. Although WEE1 mRNA was higher in EAC patients compared with adjacent normal esophagi as expected, there is no significant difference in WEE1 mRNA expression between the smoking or non-smoking EAC samples (Figures S2D and S2E). Although WEE1 mRNA levels are high in EACs, our data suggest that smoking increases WEE1 protein levels via mechanisms other than transcription or protein stability.
Figure 2.

Smoking induces WEE1 protein expression in EAC cells
(A) Western blot analysis of WEE1, p-CDC2 (Y15), and β-actin in FLO-1 and OE33 cells treated with nicotine for 1 or 3 h. (B) Bblot analysis of WEE1, p-CDC2 (Y15), and β-actin in FLO-1 and OE33 cells treated with NNK for different time points. (C–E) Western blot analysis of WEE1, p-CDC2 (Y15), and β-actin in FLO-1, OE33, and CPB cells treated with 2% cigarette smoking extract (CSE). (F) Immunofluorescence staining of WEE1 (green) in FLO-1 cells treated with 2% CSE or NNK. Scale bar, 50 μm. (G) Quantification of (F) ∗p<0.05, ∗∗p<0.01.
Smoking induces WEE1 protein expression through miRNA dysregulation
To investigate how smoking increases the WEE1 protein level in EAC, we tested if smoking regulates WEE1 through miRNAs. miRNAs can regulate gene expression by binding to complementary sequences in the 3′ untranslated region (3′ UTR) of mRNA molecules, leading to degradation or repression of translation. To find out the candidate miRNAs that can target WEE1, we analyzed three online databases, including miRTAR base, Target Scan, and miRDB. Our analysis indicates that miR-195-5p and miR-129-3p were among the top common miRNAs in all three databases (Figure 3A). A list of all the 33 miRNAs can be found in Table S2. To validate these miRNAs in regulating WEE1 expression, we overexpressed miR-195-5p or miR-129-3p in FLO-1 and OE33 cells. Western blot analysis indicated that miR-195-5p or miR-129-3p overexpression decreased WEE1 protein levels in EAC cells (Figure 3B). We treated FLO-1 or OE33 cells with NNK or 2% CSE to further test if smoking regulates these miRNAs. qRT-PCR results demonstrated that, in both cell lines, NNK or CSE treatment significantly decreased miR-195-5p expression levels (Figures 3C and 3D) but with limited effect on miR-129-3p expression (data not shown). Next, we investigated the possible binding of miR-195-5p on the WEE1 3′ UTR using the luciferase reporter containing the full length of WEE1 3′ UTR. qRT-PCR results confirmed that the reconstitution of miR-195-5p in FLO-1 and OE33 cells was successful (Figure 3E). Luciferase reporter assay results showed that reconstitution of miR-195-5p significantly decreased the reporter activity, indicating binding of the miR-195-5p to the 3′ UTR of WEE1 (Figure 3F, p < 0.001). To further confirm our findings, we mutated WEE1 3′ UTR by deleting the two miR-195-5p binding sites (Figure 3G). As expected, miR-195-5p reconstitution had little effect on the luciferase activity of the mutated reporter (Figure 3H). We performed CSE treatment with or without miR-195-5p reconstitution to further validate our findings. Western blot results showed that miR-195-5p reconstitution decreased not only the WEE1 level but also abrogated CSE-induced WEE1 protein level in FLO-1 and OE33 cells (Figure 3I). These data suggested that smoking increases the WEE1 protein level through miR-195-5p in EAC.
Figure 3.

Smoking induces WEE1 expression through miR-195-5p downregulation
(A) miRNAs targeting WEE1 were selected based on the common predicted binding in three online databases. (B) miR-129-3p or miR-195-5p reconstitution decreased WEE1 protein expression in FLO-1 and OE33 cells detected by western blot. WEE1 and β-actin protein expression levels were tested. (C) miR-195-5p expression was decreased by NNK or CSE treatment in FLO-1 cells examined by qRT-PCR. (D) Similar results as (C) were found in OE33 cells. (E) qRT-PCR validation of miR-195-5p reconstitution in FLO-1 and OE33 cells. (F) miR-195-5p reconstitution decreased WEE1 3′ UTR reporter activity in FLO-1 and OE33 cells. (G) Site mutations on the WEE1 3′ UTR reporter. Two miR-195-5p binding sites were deleted on the mutant reporter. (H) miR-195-5p reconstitution had little effect on the mutant WEE1 3′ UTR reporter lacking miR-195-5p binding sites. (I) Western blot analysis of WEE1, p-CDC2 (Y15), and β-actin in FLO-1, OE33 treated with CSE with or without miR-195-5p reconstitution. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
WEE1 mediates docetaxel resistance in EAC
Therapeutic resistance is a significant cause of EAC’s poor clinical outcome and recurrence. Single agents’ therapies often fail, and combination treatments are more likely to achieve clinical response. We, therefore, performed RNA sequencing analysis on FLO-1 and OE33 control or WEE1 siRNA knockdown cells to determine the WEE1 inhibition signature. The L1000 Fireworks Display analysis followed the RNA sequencing data to find the best drug combinations by comparing WEE1 inhibition-induced differential gene expression with different drug-induced transcriptomic signatures.26 Figure 4A showed significantly differentially expressed genes after WEE1 siRNA knockdown in OE33 cells. L1000FWD analysis using the WEE1 inhibition gene expression signature found that docetaxel was one of the top drugs with the lowest similarity scores with FLO-1 and OE33 WEE1 inhibition sequencing data, predicting the best potential synergistic effect (Figures 4B and 4C). Taxanes, including docetaxel, are included in several standard therapeutic options for EAC.27 Our data demonstrated that MK-1775, a specific WEE1 kinase inhibitor,16 significantly synergized with docetaxel in both FLO-1 and OE33 cells measured by ATP-Glo cell viability assay (Figures 4D and 4E, p < 0.001). Similar results were found in SK-GT4 and OE19 cells (Figures S3A and S3B). Western blot data confirmed that the WEE1 inhibitor decreased CDC2 phosphorylation as expected (Figure 4F, left panel). In the meantime, MK-1775 and docetaxel induced cleaved PARP, while the combination caused a dramatically higher level of cleaved PARP (Figure 4F, left panel). Similar results were found in FLO-1 cells treated with docetaxel combined with WEE1 inhibition, which was done by siRNA knockdown. The WEE1 inhibition and docetaxel treatment induced remarkably more cleaved PARP than single treatment or knockdown (Figure 4F, right panel). We acknowledge the noted difference in p-CDC2 levels between the WEE1 siRNA and MK-1775 treatment groups. The variance can be attributed to distinct treatment durations: 4 days for siRNA due to transfection needs and 2 days for MK-1775. This difference may lead to a more pronounced induction of WEE1 following docetaxel treatment in the siRNA group. While p-CDC2 is a known target of WEE1, its regulation in EAC cells could be influenced by other factors, such as PKMYT1.28 We performed Annexin V/PI staining in EAC cells treated with MK-1775, docetaxel, or a combination to confirm our findings further. Flow cytometry analysis indicated that MK-1775 and docetaxel treatment induced apoptosis measured by Annexin V in both cell lines; and the combination treatment induced significantly more cell apoptosis than single treatments (Figures 4G and 4H, p < 0.0001). Spheroids have diffusional limits to the mass transport of drugs, nutrients, and other factors, similar to in vivo tissues. Due to their mimicry of the physiological barriers to drug delivery in vivo, spheroids can be an improved assay format for testing efficacy.29 To test the effectiveness of MK-1775 and the docetaxel combination in vitro, our team performed EAC spheroids derived from OE33 cells. Our data showed that, although a single docetaxel treatment was not significantly effective in inhibiting spheroid growth, the combination treatment significantly decreased the diameter of spheroids (Figures 4I and 4J). Our data suggested that WEE1 inhibition synergized with docetaxel in EAC cells.
Figure 4.

WEE1 mediates docetaxel resistance in EAC
(A) Differentially expressed genes in OE33 WEE1 siRNA knockdown cells compared with control siRNA transfected cells from RNA sequencing data. Red dots indicate significantly upregulated genes. Blue dots indicate significantly downregulated genes. (B) L1000FWD analysis predicted that docetaxel was one of the drugs with the lowest similarity score to WEE1 siRNA knockdown. (C) Drug similarity scores predicted by L1000FWD based on WEE1 siRNA RNA sequencing data from FLO-1 and OE33 cells. (D and E) SynergyFinder analysis results of FLO-1 and OE33 cells treated with MK-1775 (0–2 μM) and docetaxel (0–5 nM). (F) Left panel: western blot analysis of WEE1, PARP, cleaved PARP, p-CDC2 (Y15), and β-actin in FLO-1 cells treated with MK-1775 1 μM alone, docetaxel 2 nM alone, or a combination. Right panel: similar results in FLO-1 cells treated with docetaxel with or without WEE1 siRNA knockdown. (G and H) Left panels: flow cytometry analysis of Annexin V and PI staining in FLO-1 or OE33 cells treated with MK-1775 1 μM alone, docetaxel 2 nM alone, or a combination. Right panels: quantification of apoptotic cells in the left panels. (I) OE33 spheroids cells treated with MK-1775 alone, docetaxel alone, or a combination. (J) Quantifying data in (F). ∗∗p < 0.01, ∗∗∗∗p < 0.0001.
WEE1 inhibitor MK1775 overcomes smoking-induced docetaxel resistance in vitro
Since WEE1 inhibition and docetaxel treatment induced different gene expression patterns and were synergistic, we hypothesized that: (1) smoking-induced WEE1 could also generate a gene expression signature causing docetaxel resistance (Figure 5A) and (2) WEE1 inhibition eliminates smoking-induced docetaxel resistance. To test our hypothesis, we investigated if smoking causes docetaxel resistance through upregulating WEE1 expression in EAC. Firstly, using four EAC cell lines, FLO-1, OE33, SK-GT4, and OE19, we determined the IC50 values of WEE1 inhibitor MK-1775 (from 78 nM to 390 nm, Figure 5B) and docetaxel (from 0.15 to 0.65 nM, Figure 5C) for 5 days treatment. To better understand the effect of smoking on drug resistance, we cultured FLO-1 and OE33 cells with 2% CSE for 14 days to generate the CSE long-term (CSE-LT) cells. We treated FLO-1 CSE-LT, OE33 CSE-LT, and parental cells with docetaxel or MK-1775. ATP-Glo cell viability assay demonstrated that CSE-treated EAC cells had at least double dose higher IC50 values for docetaxel (Figures 5D and 5E). At the same time, the CSE-LT cells maintained a similar sensitivity to MK-1775 treatment (Figure S3C). Western blot analysis showed that overnight docetaxel treatment induced cleaved PARP and cleaved caspase-3 in FLO-1 and OE33 cells (Figure 5F, left three lanes of both panels). And CSE-LT cells demonstrated higher WEE1 protein levels and less cleaved PARP and cleaved caspase-3 treated by docetaxel (Figure 5F, right three lanes of both panels). Furthermore, FLO-1 CSE-LT cells were treated with a combination of MK-1775 and docetaxel. Our ATP-Glo cell viability data indicated that MK-1775 significantly synergized with docetaxel in FLO-1 CSE LT cells (Figure 5G). To test this synergy effect in a relatively long-term situation, we treated OE33 CSE-LT cells with docetaxel with or without MK-1775 treatment. Clonogenic assay results indicated that combination treatment significantly decreased clone size and number in OE33 CSE-LT cells (Figures 5H and 5I). These data suggested that smoking induces WEE1 expression and docetaxel resistance in EAC cells.
Figure 5.

WEE1 inhibitor MK1775 overcomes smoking-induced docetaxel resistance in vitro
(A) Graphic summary of the hypothesis that smoking induces WEE1 expression promoting docetaxel drug resistance in EAC cells. (B and C) ATP-Glo cell viability assay analysis of IC50 values of MK-1775, or docetaxel (DTX) in EAC cells. (D and E) ATP-Glo cell viability assay analysis of docetaxel IC50 values in FLO-1 or OE33 control or CSE long-term (CSE-LT)-treated cells. (F) Western blot results of WEE1, PARP, cleaved PARP, caspase-3, cleaved caspase-3, and β-actin in FLO-1 and OE33 control or CSE-LT cells treated with docetaxel overnight. (G) SynergyFinder analysis showed a significant synergy effect between WEE1 inhibitor MK-1775 and docetaxel in FLO-1 CSE-LT cells. (H) Clonogenic assay in OE33 cells treated with docetaxel or combined with MK-1775. (I) Quantification of (H). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
WEE1 inhibitor MK1775 overcomes smoking-induced docetaxel resistance in vivo
To test our findings in vitro, we generated an EAC patient-derived xenograft (PDX) mouse model with the TE-10 smoking machine for mice. As described in materials and methods, after 3 months of smoking, mice were implanted for EAC PDX followed by control, docetaxel alone, MK-1775 alone, or combination treatment in different groups (Figure 6A). Our data showed that, without smoking, MK-1775 or docetaxel treatment alone significantly decreased tumor volume compared with the control (Figures 6B, left panel, and 6C). In contrast, combination treatment delivered better tumor growth inhibition (Figures 6B, left panel, and 6C). While it is interesting to find out that smoking did not increase tumor growth, it did make xenograft tumors more resistant to MK-1775 or single docetaxel treatment, as there was no significant difference in tumor size in single treatment groups (Figures 6B, right panel, and 6C). Surprisingly, xenograft tumors in the smoking group were more sensitive to the combination treatment, showing a significant decrease in the tumor size compared with all other groups (Figures 6B, right panel, and 6C). When the tumor size reaches three times larger than the initial size as a potential survival index, our data showed that MK-1775/docetaxel single treatment or combination improved survival in non-smoking groups (Figure 6D). In contrast, only combination treatment in the smoking group affected survival. There was no survival difference for the single-drug treatments in the smoking groups (Figure 6E). Our western blot data from the xenograft tumor samples showed that smoking induced remarkably higher WEE1 and BCL-2 protein expression levels in mouse tumors. And both protein levels were decreased by the docetaxel alone or through combination treatment (Figure 6F). The combination treatment induced remarkably more cleaved PARP in the smoking group (Figure 6F). To investigate the mechanisms in which WEE1 was induced by smoking in vivo, we examined miR-195-5p expression levels in xenograft tumor samples. qRT-PCR results showed that, similar to in vitro, miR-195-5p expression was significantly lower in the smoking tumor samples with or without combination treatment (Figure 6G). To further investigate the mechanisms underlying the synergistic effect behind MK-1775 and docetaxel, we performed immunofluorescence staining of cleaved caspase-3 and detyrosinated tubulin in the four groups of the xenograft tumor samples. Our data suggested that MK-1775 single treatment and combination induced significantly higher cleaved caspase-3 than the control group in smoking and non-smoking tumors (Figures 7A and 7B, p < 0.001). On the other hand, docetaxel and combination treatment significantly decreased detyrosinated tubulin in smoking and non-smoking tumors compared with control samples (Figures 7C and 7D, p < 0.05). These data confirmed our in vitro findings that smoking induces WEE1 expression, promoting docetaxel drug resistance in EAC through miR-195-5P dysregulation.
Figure 6.

Combining MK-1775 and docetaxel improves EAC patient-derived xenograft mouse model survival with smoking in vivo
(A) Graphic summary of the EAC patient-derived xenograft experimental design. (B) Tumor growth curves in eight experimental groups with or without smoking. (C) Tumor size in eight experimental groups with or without smoking at the end of the experiment. (D and E) Probability of survival in non-smoking and smoking groups with single or combined drug treatment. (F) Western blot analysis of WEE1, BCL-2, PARP, cleaved PARP (Cl-PARP), and β-actin in mouse xenografts. (G) qRT-PCR analysis of miR-195-5p expression in mouse xenografts. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001.
Figure 7.

WEE1 inhibition induces apoptosis, and docetaxel promotes tubulin destabilization in EAC PDXs
(A) Immunofluorescence staining of cleaved caspase-3 (red), DAPI (blue) in EAC PDXs described in Figure 6. Scale bar, 50 μm. (B) Quantification data of (A). (C) Immunofluorescence staining of detyrosinated tubulin (green, stable tubulin) and DAPI (blue) in EAC PDXs described in Figure 6. Scale bar, 50 μm. (D) Quantification of (C) ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Discussion
Esophageal cancer is a deadly disease with limited treatment options, highlighting the need for new therapeutic targets.1 Although BE is a significant risk factor for EAC,30 smoking is an important risk factor for progressing from BE to EAC.7 In this study, we investigated the role of smoking and WEE1 in EAC and its potential as a therapeutic target. Our findings revealed that WEE1 is overexpressed in human EAC samples, and smoking induces high levels of WEE1 protein through miRNA dysregulation, leading to docetaxel resistance.
The overexpression of WEE1 in EAC is consistent with previous studies showing its upregulation in various human malignancies. In the meantime, targeting WEE1 has been identified as a potential therapeutic strategy for several cancers, including lung, ovarian, and gastric cancer.18,31,32 Our results suggest that WEE1 could also be a viable therapeutic target for EAC.
In our investigation, an unexpected observation was made regarding the subcellular localization of WEE1 in EAC cells. Traditionally, WEE1 has been documented to localize predominantly in the nucleoli.33 However, in our study, WEE1 appeared to localize in the cytoplasm, diverging from its typical nucleolar presence. This shift in localization might imply that WEE1 assumes diverse roles depending on its cellular location. Such unconventional cytoplasmic localization raises questions regarding potential alternative functional implications for WEE1 in the context of EAC cells. While this is an intriguing observation, our primary focus in this study is centered on elucidating the connection between smoking, WEE1 overexpression, and drug resistance. The unexpected cytoplasmic localization of WEE1 suggests novel functional implications that warrant deeper investigation. We are currently exploring these findings.
Smoking is a significant risk factor for the progression of BE to EAC, and our study demonstrates that smoking induces WEE1 protein expression in EAC cells. Interestingly, the smoking-induced WEE1 upregulation is not due to mRNA transcription or protein stability but rather miRNA dysregulation. It was indicated earlier that miR-424, miR-381, and miR-219-5p decreased WEE1 expression in renal cancer and Balb/C mice.34,35,36 In particular, we found that miR-195-5p directly binds to WEE1 3′ UTR and is most significantly associated with changes in WEE1 levels. Recent studies showed that miR-195 is downregulated in human esophageal cancer and plays a role as a tumor suppressor.37,38,39 Our results suggest that regulating WEE1 by different miRNAs could be cellular and context dependent. Despite the known role of miRNAs in modulating mRNA stability and translation, WEE1 mRNA levels remained relatively unchanged even as protein levels shifted after smoking exposure. This suggests that the presence of compensatory mechanisms, possibly a negative feedback loop, might stabilize WEE1 mRNA under smoking conditions. The detailed dynamics of this regulatory process merits further exploration. We recognize the significance of understanding the link between smoking status and WEE1 protein expression in human EAC. However, our tissue microarray data do not specify patients' smoking status, and further searches yielded no additional data on this relationship. Although smoking induces WEE1 in EAC, the abnormal WEE1 mRNA overexpression independent of smoking in EAC samples needs further investigation. WEE1 is an attractive therapeutic target for several malignancies, including lung, ovarian, and gastric cancer.18,20,32,40 According to recent publications, smoking promotes drug resistance in multiple human cancers, including the lung, prostate, and pancreas.41,42,43 Since WEE1 was induced by smoking in EAC, we tested if smoking causes drug resistance and whether it is WEE1 dependent. Our RNA sequencing data using WEE1 knockdown cells suggested that WEE1 inhibition and docetaxel treatment induce significantly different gene expression signatures. The F1000FWD method we used to predict the synergistic effect based on differential gene expression response after drug treatment has been tested in multiple human cancers.44,45,46,47,48 We found that smoking-induced docetaxel resistance was dependent on WEE1. We have examined the baseline WEE1 expression in EAC cancer cell lines and concurrently assessed the IC50 values of docetaxel. Our findings suggest that docetaxel sensitivity, gauged by IC50 values, showed no substantial differences among the EAC cells (data not shown). Utilizing the SynergyFinder tool, we observed that our synergy scores significantly exceeded the threshold suggested by the tool’s user manual for denoting synergism. Therefore, based on these scores, we infer that the MK-1775 WEE1 inhibitor synergized with docetaxel treatment in smoking-treated cells and non-smoking EAC cells, suggesting that combining these two drugs could be practical for EAC patients regardless of their smoking status. We have conducted cell-cycle analyses after single and combination drug treatments. While MK-1775 typically reduced cells in the G2/M phase and DTX induced G2/M arrest, the combination treatment presented a complex cell-cycle profile (data not shown). Such complexities in flow cytometry analysis, where typical cell-cycle signatures are obscured, have been reported in other drug treatment scenarios, as evidenced by Hoose et al..49 Given this intricate profile, consistently observed across repeated experiments, we have chosen not to derive specific conclusions for the combination treatment based on these analyses. Since WEE1 mRNA induction was not found in smoking-treated EAC cells, and the synergistic effect of WEE1 inhibition and docetaxel occurs in EAC cells even without smoking, we assume that smoking might not be the only reason for WEE1 activation in EAC.
To further investigate the smoking effect on WEE1 induction and examine the efficacy of WEE1 inhibition combined with docetaxel treatment in vivo, we used a mouse smoking machine joint with EAC PDX and drug treatment. Mice were exposed to smoking 3 months before tumor implantation to build up a host smoking background, as suggested by recent reports.50,51,52 As expected, WEE1 inhibition synergized with docetaxel treatment. Smoking decreased the survival of mice treated with WEE1 inhibitor or docetaxel alone. Interestingly, the combination treatment elicited a significant response in the smoking group, which initially took us by surprise. A plausible explanation could be that the xenograft tumor cells in the non-smoking group, which have a relatively lower WEE1 expression, may have limited response to WEE1 inhibition, and survived after combination treatment. In contrast, in the smoking group, WEE1 may have been induced in tumor cells with inherently lower endogenous WEE1 expression, making these cells more reliant on WEE1 for DNA repair and survival during chemotherapy. Hence, when the smoking group was exposed to the combination treatment, the high-WEE1 cells responded better to WEE1 inhibition than the WEE1 low cells in the non-smoking group. The staining of apoptosis maker and stabilized tubulin results indicated that docetaxel destroys tubulin while WEE1 inhibition induces apoptosis. The combination treatment showed both effects of WEE1 inhibition and the docetaxel treatment. The differences in inducing cell apoptosis and eliminating tubulin by WEE1 inhibition or docetaxel treatment are consistent with our hypothesis that there are different gene expression responses underlying the two treatments. These data validate our RNA sequencing data that WEE1 inhibition and docetaxel treatment target various aspects of cancer cell survival and suggest that combining drugs based on the discordance of responding gene patterns works in pre-clinical settings.
We observed elevated WEE1 expression in the smoking group, yet their tumor response to docetaxel was limited compared with the non-smoking group. Interestingly, the combination treatment was more effective in the smoking group. This might be because the smoking group’s tumors, with higher WEE1, became more dependent on it for DNA repair during chemotherapy, hence responding better to its inhibition. In contrast, the non-smoking group, with inherently lower WEE1 levels, may be less affected by its inhibition. We recognize our study’s constraint of using a single EAC PDX model, emphasizing the need for future research on predictive biomarkers and therapeutic response mechanisms. We acknowledge the importance of understanding smoking’s effect on docetaxel response in human EAC patients. While our in vivo study utilized a smoking machine to mimic patient conditions, the scarcity of related human data is a limitation. We hope our findings encourage more in-depth human studies in this domain. We recognize that our analysis did not show significant variations in cleaved caspase-3 or tubulin expression. The measurements taken at the end of a 4-week treatment might have affected these results. Although our data suggest a less responsive smoking group, earlier time point measurements could have provided a different perspective.
In summary, our study offers valuable insights into the function of WEE1 in EAC and emphasizes its potential as a therapeutic target. We also reveal the synergistic impact of combining a WEE1 inhibitor with docetaxel treatment in EAC through in vitro experiments. Utilizing a specialized smoking machine for in vivo research, we discovered that the WEE1 inhibitor counteracts smoking-induced docetaxel resistance in EAC. These findings provide a scientific rationale for further exploration of WEE1 inhibition as a treatment strategy for EAC patients. Our study’s novelty lies in uncovering the role of smoking in inducing WEE1, promoting docetaxel resistance in EAC, and using a unique in vivo TE-10 smoking machine in mice to investigate these effects. These novel aspects contribute significantly to the field and warrant further research to improve treatment options for EAC patients.
Materials and methods
Cell culture and reagents
Immortalized non-neoplastic normal esophageal cell line EPC2, kindly provided by Dr. Anil Rustgi (Columbia University, NY), three human BE cell lines, BART (kindly supplied by Dr. Rhonda Souza, Baylor University Medical Center, Dallas, TX), CPA, and CPB (ATCC, Manassas, VA), were cultured with DMEM/F12 (Gibco, New York, NY) supplemented with 5% fetal bovine serum (FBS) (Gibco), 0.4 μg/mL hydrocortisone (Sigma-Aldrich, Saint Louis, MO), 1% penicillin/streptomycin (Gibco), 140 μg/mL bovine pituitary extract (Thermo Fisher Scientific, Waltham, MA), 20 mg/L adenine hydrochloride hydrate (Sigma-Aldrich), insulin-transferrin-sodium selenite medi supplement (Sigma-Aldrich), and 20 ng/mL recombinant epidermal growth factor (Sigma-Aldrich). EAC cell lines FLO-1 (ATCC), OE33 (kindly provided by Dr. David Beer), SK-GT4 (kindly supplied by Dr. Xiaochun Xu at MD Anderson), OE19, and ESO26 (purchased from MilliporeSigma, Burlington, MA) were cultured in DMEM, or RPMI 1640 medium (Gibco) supplemented with 10% FBS and 1% penicillin/streptomycin. All cell lines were grown at 37°C in 5% CO2. Cell lines were authenticated by Genetica DNA Laboratories (Burlington, NC) using short tandem repeat profiling. Cells from the stocks were authenticated using short tandem repeat markers and cultured for less than 6 months. Mycoplasma was tested periodically using the qRT-PCR method (SouthernBiotech, Birmingham, AL). WEE1 and p-CDC2 antibodies were obtained from Santa Cruz Biotechnology (Dallas, TX). PARP, cleaved PARP, caspase-3, cleaved caspase-3, and BCL-2 antibodies were purchased from Cell Signaling Technology (Beverly, MA). A detyrosinated tubulin antibody was obtained from Sigma. The β-actin antibody was obtained from Sigma-Aldrich. Mouse or rabbit secondary antibodies were purchased from Promega (Madison, WI). MK-1775 and docetaxel were supplied by Selleck Chemicals (Houston, TX).
Tissue microarray for EAC human samples
Tissue microarrays containing cores from paraffin-embedded, de-identified human (10 normal and 96 EAC cancer tissue samples) were obtained from Vanderbilt Tissue Pathology Core Resource. All tissue samples were coded and de-identified following institutional review board-approved protocols. The histology of all tissue samples was verified using H&E staining. A composite scoring system was developed for statistical analysis to integrate the immunohistochemistry signal intensity and the frequency of positive cells in the cytosol and nucleus. The intensity of staining is graded as 0 (negative), 1 (weak), 2 (moderate), and 3 (strong). The frequency is graded from 0 to 4 by the percentage of positive cells as follows: grade 0, <3%; grade 1, 3%–25%; grade 2, 25%–50%; grade 3, 50%–75%; grade 4, >75%. A composite expression score (CES) with a full range from 0 to 12 was used. Products = intensity of staining (0–3) × frequency (0–4). Index score 0 = products of 0, index score 1 = products of 1 and 2, index score 2 = products of 3 and 4, index score 3 = products of 6 through 12. CES was calculated as described previously.53 WEE1 gene expression in esophageal cancer and esophageal cancer patient survival data were analyzed through TNMplot.17
qRT-PCR and RNA sequencing
Total RNA was purified using the miRNeasy mini kit (QIAGEN). cDNA and miRNA cDNA was reverse transcribed as described before.13 The quantitative real-time PCR (qRT-PCR) was performed using a Bio-Rad CFX Connect Real-Time System with the threshold cycle number determined by Bio-Rad CFX manager software version 3.0. Primers were ordered from Integrated DNA Technologies (Coralville, IA). The sequences of qRT-PCR primers are given in Table S1. The results of target genes were normalized to the human HPRT1 gene. miRNA expressions were normalized to miR-191.12,54 Expression fold changes were calculated using the formula; 2(Rt – Et)/2(Rn – En) as described previously.55,56 Twelve RNA samples from FLO-1 and OE33 control siRNA (triplicates) or WEE1 siRNA (triplicates) cells were extracted. RNA sequencing was performed on a total of 1 μg RNA from each sample as described before.14 The Limma package in R performed differential gene expression analysis. The threshold for significance is a p value less than 0.05, with the log2 fold change larger than 1.5 or less than −1.5.
Western blot
Cells were harvested by 0.05% trypsin from the culture plate and centrifuged at 12,000 rpm 4°C for 2 min. Cell pellets were re-suspended in RIPA buffer containing protease inhibitor cocktail and phosphatase inhibitor sodium orthovanadate (Santa Cruz Biotechnology). The suspension was slowly shaken on ice for 30 min, with a vortex every 10 min. Protein concentrations were measured using a Bio-Rad Protein Assay (Bio-Rad, Hercules, CA). Proteins (10 μg) from each sample were subjected to SDS-PAGE and transferred onto nitrocellulose membranes (PerkinElmer, Waltham, MA). Membranes were blocked with 5% bovine serum albumin (Sigma-Aldrich). Membranes were probed with specific primary antibodies overnight at 4°C to detect target proteins. On the second day, membranes were washed for 10 min with TBS-T three times, followed by incubation with anti-rabbit or anti-mouse secondary antibodies for 2 h. Then membranes were washed 10 min × 3 times with TBS-T. Protein bands were detected using chemiluminescence reagents (Millipore, Billerica, MA) in GelDoc Go Imaging System (Bio-Rad).
Flow cytometry analysis of cell apoptosis and cell death
To quantitate drug-induced apoptosis, flow cytometry analysis of Annexin V and PI was performed using an FITC Annexin V apoptosis detection kit (BD Biosciences, San Jose, CA). FLO-1 and OE33 Cells were treated with 200 nM MK-1775, 2 nM docetaxel, or a combination for 72 h. Cells were then collected for FITC Annexin V and PI staining according to the manufacturer’s instruction and subjected to flow cytometry analysis at Flow Cytometry Shared Resource at Sylvester Comprehensive Cancer Center. Flow cytometry data were analyzed by FCS Express 7 software (https://denovosoftware.com/).
Cell viability ATP-Glo and clonogenic assay
FLO-1, OE19, SK-GT4, and OE33 cells were seeded at 1,500 cells per well in 96-well plates and treated with WEE1 inhibitor MK-1775 (range: 0 ng/mL to 20 μg/mL) or docetaxel (range: 0–20 nM) for 5 days. According to the manufacturer’s protocol, cell viability was measured using the CellTiter-Glo Cell Viability Assay (Promega, Madison, WI). Absorbance was obtained in a FluolarStar luminescence microplate reader (BMG Labtech). OE33 control, CSE long-term (CSE-LT) treated cells were seeded 1,000 cells/well in six-well plates treated with MK-1775 (range: 0.0–200 ng/mL) or docetaxel (range: 0–2 nM) or combination for 48 h. After treatments, cells were washed with PBS following incubation in a drug-free DMEM or RPMI medium for 10 to 12 days until exact clones were formed in control groups. Subsequently, the media were removed, and cells were fixed with 4% paraformaldehyde solution for 30 min at room temperature. The cells were then gently washed with PBS and stained overnight with crystal violet (0.05% crystal violet in 50% methanol). Following overnight staining, the excess dye was gently washed off with PBS. The plates were photographed. Colony formation and cell survival were evaluated by quantifying the dye signal in each well with ImageJ image analysis software (https://imagej.nih.gov/ij/).
DNA/RNA transfection and WEE1 3′ UTR luciferase reporter
miR-195-5p, miR-129-3p, and control miRNA mimic were obtained from abmgood (Richmond, BC, Canada). miRNA and WEE1 siRNA (Thermo Fisher Scientific) transfection were performed with Lipojet transfection reagent (SignaGen Laboratories, Frederick, MD) following the manufacturer’s instructions. miRNA mimic or siRNA (60 pmol) were transfected into 1.5 × 105 FLO-1 or OE33 cells in one well of a six-well plate. Media were changed overnight. Cells were harvested for analysis 72 h after transfection. WEE1 3′ UTR reporter was purchased from Genecopoeia (Rockville, MD). One microgram of the reporter plasmid and 0.5 μg of β-galactosidase plasmid (Promega) were co-transfected into 1.5 × 105 FLO-1 or OE33 cells in one well of a six-well plate using Polyjet (SignaGen Laboratories). Media were changed overnight. Cells were harvested for analysis 72 h after transfection. WEE1 3′ UTR reporter mutation was generated using the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) following the manufacturer’s instructions.
Smoking treatments and PDXs
NNK solution and nicotine were obtained from Sigma-Aldrich. Methanol with the same volume of NNK or nicotine was added to the media as a control. CSE was prepared by bubbling smoke from two 1R6F research cigarettes into 20 mL of serum-free culture RPMI medium at one cigarette/min.57 After adjusting pH to 7.4, the medium was considered 100% CSE. Serum-free culture RPMI medium was used as a control for CSE treatment in vitro. We randomized 40 6-week-old NOD SCID mice (Charles River, Wilmington, MA) to an environment consisting of either filtered air (20 mice) or cigarette smoke (20 mice) for 3 months.58 Smoke exposure (6 h per day, 5 days per week) was standardized using a model TE-10 smoking machine51,59 and adjusted to maintain constant sidestream and mainstream smoke from 1R6F research cigarettes (Kentucky Tobacco Research and Development Center, Lexington, KY). EAC PDX was provided by Dr. Silvia Giordano.60 The PDX used in this study has KRAS amplification (approximately 30–50 copies) with TP53 mutation (p.Arg248Trp; p.Pro72Arg). For PDX implantation, one 24-well plate containing cold culture medium RPMI 1640 supplemented with antibiotics (Primocin, 100 μg/mL, InvivoGen, San Diego, CA), 1 mL of medium/well, was prepared. Tubes containing tumor samples were thawed and the tumor pieces placed in the 24-well plate containing the cold medium on ice. After 10 min, the tumor pieces were moved to a new well and, after a further 10 min, the tumor pieces were moved to a third well. Finally, after 10 min, the tumor pieces were moved to the last well, containing medium supplemented with antibiotics and 20% Matrigel (Fisher Scientific, Hampton, NH). With the animal anesthetized, shaved on the back, and disinfected superficially, the subcutaneous dorsal level of the loin skin was cut about 6–7 mm. One fragment (approximately 2–3 mm3 fragments) immersed in the medium supplemented with Matrigel was placed in the generated pocket. The incision was closed with a small amount of surgical glue. Forty NOD SCID mice with or without smoking were randomized into eight groups (1) control, (2) MK-1775 treatment, (3) docetaxel treatment, (4) MK-1775 + docetaxel treatment, (5) smoking, (6) smoking + MK-1775 treatment, (7) smoking + docetaxel treatment, and (8) smoking + MK-1775 + docetaxel treatment, when the average tumor volume reached around 150 mm3. Mouse weight was recorded, and tumor volumes were measured and calculated as volume = 0.5 × (long dimension) × (short dimension)2 twice weekly.14 Docetaxel (10 mg/kg) was intraperitoneally injected on day 1 per week for 4 weeks.61 The WEE1 inhibitor MK-1775 (Selleck Chemicals, Houston, TX) was delivered by oral gavage (30 mg/kg) on days 3 and 5 per week for 4 weeks in DMSO in 0.5% methylcellulose (Sigma-Aldrich).62 Mice were under the same schedule of smoking during the whole experiment as tumor implantation before. Tumor growth rate/regressions were calculated on day 28 after the initial treatment. The animal work is approved by the Institutional Animal Care and Use Committee Office (IACUC) of the University of Miami.
The assigned approval number of this study is 21-099 (Zheng Chen).
Immunofluorescence and immunohistochemistry
Immunofluorescence and immunohistochemistry were performed as described previously.63 Immunofluorescence assay was performed in paraffin-embedded sections of EAC PDX mouse tumor samples described above. Paraffin-embedded slides were deparaffinized after three incubations (3 min each time) in Histo-Clear (National Diagnostics, Atlanta, GA), followed by two incubations of 2 min in 100% ethanol, 95% ethanol, 70% ethanol, 50% ethanol, and double deionized water. In addition, slides were incubated in TE buffer (pH 8.0) for 10 min at 100°C for antigen unmasking. Slides were cooled to room temperature after antigen unmasking and blocked with 10% goat serum (Thermo Fisher Scientific, Waltham, MA) for 1 h. Slides were incubated with cleaved caspase-3 or detyrosinated tubulin antibody 1:250 overnight at 4°C. After washing in PBS 3 times, the slides were incubated with 1:500 goat anti-rabbit Alexa Fluor 488 or goat anti-rabbit Alexa Fluor 568 (Thermo Fisher Scientific, Weston, FL) secondary antibodies for 2 h in the dark. Then slides were covered with a mounting medium with 40,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA). Images were taken using an FV-1000 confocal microscope (Olympus America, Miami, FL). ImageJ software was used for cleaved caspase-3 or detyrosinated tubulin data quantification.
Acknowledgments
This study was supported by the US National Institutes of Health (1K22CA255424-01) and pilot funding from the Sylvester Comprehensive Cancer Center. The use of shared resources was supported by the Sylvester Comprehensive Cancer Center support grant (P30CA240139). This work’s content is solely the responsibility of the authors. It does not necessarily represent the official views of the National Institutes of Health or the University of Miami. We appreciate Dr. Ashok Saluja, the University of Miami, for technical support on the TE-10 smoking machine.
Author contributions
Conceptualization, Z.C., K.T., and M.O.I.; formal analysis, M.O.I., K.T., H.L., Z.C., Y.B., and S.S.; funding acquisition, Z.C.; investigation, Z.C., M.O.I., K.T., D.P., and M.S.; methodology, S.G., X.C., and D.B.; resources, W.E.-R., A.V.V., P.J.H., and Z.C.; supervision, Z.C.; validation, M.O.I. and K.T.; visualization, M.O.I., K.T., Z.C., Y.B., and X.C.; writing – original draft, M.O.I. and Z.C.; writing – review & editing, Z.C., K.T., H.L., D.P., A.V.V., and W.E.-R.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2023.08.012.
Supplemental information
Data and code availability
The data supporting this study’s findings are available in TNMplot at TNMplot.com and kmplot at kmplot.com. Source data are provided in this paper. All other data are available upon reasonable request.
References
- 1.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Morgan E., Soerjomataram I., Rumgay H., Coleman H.G., Thrift A.P., Vignat J., Laversanne M., Ferlay J., Arnold M. The Global Landscape of Esophageal Squamous Cell Carcinoma and Esophageal Adenocarcinoma Incidence and Mortality in 2020 and Projections to 2040: New Estimates From GLOBOCAN 2020. Gastroenterology. 2022;163:649–658.e2. doi: 10.1053/j.gastro.2022.05.054. [DOI] [PubMed] [Google Scholar]
- 3.Hur C., Miller M., Kong C.Y., Dowling E.C., Nattinger K.J., Dunn M., Feuer E.J. Trends in esophageal adenocarcinoma incidence and mortality. Cancer. 2013;119:1149–1158. doi: 10.1002/cncr.27834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Society A.C. Cancer Facts & Figures 2022. 2022. https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2022.html
- 5.Board P.A.T.E. Esophageal Cancer Treatment (Adult) (PDQ®)–Patient Version was originally published by the National Cancer Institute. 2021. https://www.cancer.gov/types/esophageal/patient/esophageal-treatment-pdq#_159
- 6.American Cancer Society Esophageal Cancer Risk Factors. 2017. https://www.cancer.org/cancer/esophagus-cancer/causes-risks-prevention/risk-factors.html
- 7.Cook M.B., Kamangar F., Whiteman D.C., Freedman N.D., Gammon M.D., Bernstein L., Brown L.M., Risch H.A., Ye W., Sharp L., et al. Cigarette smoking and adenocarcinomas of the esophagus and esophagogastric junction: a pooled analysis from the international BEACON consortium. J. Natl. Cancer Inst. 2010;102:1344–1353. doi: 10.1093/jnci/djq289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coleman H.G., Bhat S., Johnston B.T., McManus D., Gavin A.T., Murray L.J. Tobacco smoking increases the risk of high-grade dysplasia and cancer among patients with Barrett's esophagus. Gastroenterology. 2012;142:233–240. doi: 10.1053/j.gastro.2011.10.034. [DOI] [PubMed] [Google Scholar]
- 9.Shah M.Y., Ferrajoli A., Sood A.K., Lopez-Berestein G., Calin G.A. microRNA Therapeutics in Cancer - An Emerging Concept. EBioMedicine. 2016;12:34–42. doi: 10.1016/j.ebiom.2016.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rhim J., Baek W., Seo Y., Kim J.H. From Molecular Mechanisms to Therapeutics: Understanding MicroRNA-21 in Cancer. Cells. 2022;11 doi: 10.3390/cells11182791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gebrie A. Disease progression role as well as the diagnostic and prognostic value of microRNA-21 in patients with cervical cancer: A systematic review and meta-analysis. PLoS One. 2022;17 doi: 10.1371/journal.pone.0268480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saad R., Chen Z., Zhu S., Jia P., Zhao Z., Washington M.K., Belkhiri A., El-Rifai W. Deciphering the unique microRNA signature in human esophageal adenocarcinoma. PLoS One. 2013;8 doi: 10.1371/journal.pone.0064463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Z., Saad R., Jia P., Peng D., Zhu S., Washington M.K., Zhao Z., Xu Z., El-Rifai W. Gastric adenocarcinoma has a unique microRNA signature not present in esophageal adenocarcinoma. Cancer. 2013;119:1985–1993. doi: 10.1002/cncr.28002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Z., Li Z., Soutto M., Wang W., Piazuelo M.B., Zhu S., Guo Y., Maturana M.J., Corvalan A.H., Chen X., et al. Integrated Analysis of Mouse and Human Gastric Neoplasms Identifies Conserved microRNA Networks in Gastric Carcinogenesis. Gastroenterology. 2019;156:1127–1139.e8. doi: 10.1053/j.gastro.2018.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fang Y., McGrail D.J., Sun C., Labrie M., Chen X., Zhang D., Ju Z., Vellano C.P., Lu Y., Li Y., et al. Sequential Therapy with PARP and WEE1 Inhibitors Minimizes Toxicity while Maintaining Efficacy. Cancer Cell. 2019;35:851–867.e7. doi: 10.1016/j.ccell.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Linden A.A., Baturin D., Ford J.B., Fosmire S.P., Gardner L., Korch C., Reigan P., Porter C.C. Inhibition of Wee1 sensitizes cancer cells to antimetabolite chemotherapeutics in vitro and in vivo, independent of p53 functionality. Mol. Cancer Ther. 2013;12:2675–2684. doi: 10.1158/1535-7163.MCT-13-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bartha Á., Győrffy B. TNMplot.com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms22052622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao X., Kim I.K., Kallakury B., Chahine J.J., Iwama E., Pierobon M., Petricoin E., McCutcheon J.N., Zhang Y.W., Umemura S., et al. Acquired small cell lung cancer resistance to Chk1 inhibitors involves Wee1 up-regulation. Mol. Oncol. 2021;15:1130–1145. doi: 10.1002/1878-0261.12882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma L., Lin Y., Sun S.W., Xu J., Yu T., Chen W.L., Zhang L.H., Guo Y.C., Wang Y.W., Chen T., et al. KIAA1429 is a potential prognostic marker in colorectal cancer by promoting the proliferation via downregulating WEE1 expression in an m6A-independent manner. Oncogene. 2022;41:692–703. doi: 10.1038/s41388-021-02066-z. [DOI] [PubMed] [Google Scholar]
- 20.Cho J.G., Kim S.W., Lee A., Jeong H.N., Yun E., Choi J., Jeong S.J., Chang W., Oh S., Yoo K.H., et al. MicroRNA-dependent inhibition of WEE1 controls cancer stem-like characteristics and malignant behavior in ovarian cancer. Mol. Ther. Nucleic Acids. 2022;29:803–822. doi: 10.1016/j.omtn.2022.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lánczky A., Győrffy B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J. Med. Internet Res. 2021;23 doi: 10.2196/27633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo J., Kim D., Gao J., Kurtyka C., Chen H., Yu C., Wu D., Mittal A., Beg A.A., Chellappan S.P., et al. IKBKE is induced by STAT3 and tobacco carcinogen and determines chemosensitivity in non-small cell lung cancer. Oncogene. 2013;32:151–159. doi: 10.1038/onc.2012.39. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Yang S.H., Lee T.Y., Ho C.A., Yang C.Y., Huang W.Y., Lin Y.C., Nieh S., Lin Y.S., Chen S.F., Lin F.H. Exposure to nicotine-derived nitrosamine ketone and arecoline synergistically facilitates tumor aggressiveness via overexpression of epidermal growth factor receptor and its downstream signaling in head and neck squamous cell carcinoma. PLoS One. 2018;13 doi: 10.1371/journal.pone.0201267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang J., Chheda C., Lim A., Hauptschein D., Zayou L., Tang J., Pandol S.J., Edderkaoui M. HDAC4 Mediates Smoking-Induced Pancreatic Cancer Metastasis. Pancreas. 2022;51:190–195. doi: 10.1097/MPA.0000000000001998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sen T., Tong P., Diao L., Li L., Fan Y., Hoff J., Heymach J.V., Wang J., Byers L.A. Targeting AXL and mTOR Pathway Overcomes Primary and Acquired Resistance to WEE1 Inhibition in Small-Cell Lung Cancer. Clin. Cancer Res. 2017;23:6239–6253. doi: 10.1158/1078-0432.CCR-17-1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Z., Lachmann A., Keenan A.B., Ma'ayan A. L1000FWD: fireworks visualization of drug-induced transcriptomic signatures. Bioinformatics. 2018;34:2150–2152. doi: 10.1093/bioinformatics/bty060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ajani J.A., D'Amico T.A., Bentrem D.J., Chao J., Corvera C., Das P., Denlinger C.S., Enzinger P.C., Fanta P., Farjah F., et al. Esophageal and Esophagogastric Junction Cancers, Version 2.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2019;17:855–883. doi: 10.6004/jnccn.2019.0033. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt M., Rohe A., Platzer C., Najjar A., Erdmann F., Sippl W. Regulation of G2/M Transition by Inhibition of WEE1 and PKMYT1 Kinases. Molecules. 2017;22 doi: 10.3390/molecules22122045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mehta G., Hsiao A.Y., Ingram M., Luker G.D., Takayama S. Opportunities and challenges for use of tumor spheroids as models to test drug delivery and efficacy. J. Control Release. 2012;164:192–204. doi: 10.1016/j.jconrel.2012.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Institute N.C. Esophageal Cancer. 2023. https://www.cancer.gov/pediatric-adult-rare-tumor/rare-tumors/rare-digestive-system-tumors/esophageal
- 31.Li R., Chen J., Gao X., Jiang G. Transcription factor KLF2 enhances the sensitivity of breast cancer cells to cisplatin by suppressing kinase WEE1. Cancer Biol. Ther. 2021;22:465–477. doi: 10.1080/15384047.2021.1949228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vakili-Samiani S., Turki Jalil A., Abdelbasset W.K., Yumashev A.V., Karpisheh V., Jalali P., Adibfar S., Ahmadi M., Hosseinpour Feizi A.A., Jadidi-Niaragh F. Targeting Wee1 kinase as a therapeutic approach in Hematological Malignancies. DNA Repair (Amst) 2021;107 doi: 10.1016/j.dnarep.2021.103203. [DOI] [PubMed] [Google Scholar]
- 33.Mueller S., Haas-Kogan D.A. WEE1 Kinase As a Target for Cancer Therapy. J. Clin. Oncol. 2015;33:3485–3487. doi: 10.1200/JCO.2015.62.2290. [DOI] [PubMed] [Google Scholar]
- 34.Chen B., Duan L., Yin G., Tan J., Jiang X. Simultaneously expressed miR-424 and miR-381 synergistically suppress the proliferation and survival of renal cancer cells---Cdc2 activity is up-regulated by targeting WEE1. Clinics (Sao Paulo) 2013;68:825–833. doi: 10.6061/clinics/2013(06)17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen B., Duan L., Yin G., Tan J., Jiang X. miR-381, a novel intrinsic WEE1 inhibitor, sensitizes renal cancer cells to 5-FU by up-regulation of Cdc2 activities in 786-O. J. Chemother. 2013;25:229–238. doi: 10.1179/1973947813Y.0000000092. [DOI] [PubMed] [Google Scholar]
- 36.Lou W., Zhang X., Hu X.Y., Hu A.R. MicroRNA-219-5p Inhibits Morphine-Induced Apoptosis by Targeting Key Cell Cycle Regulator WEE1. Med. Sci. Monit. 2016;22:1872–1879. doi: 10.12659/MSM.895439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu Q., Xu J.L., Chen W.Q., Xu W.X., Song Y.X., Tang W.J., Xu D., Jiang M.P., Tang J. Roles and mechanisms of miR-195-5p in human solid cancers. Biomed. Pharmacother. 2022;150 doi: 10.1016/j.biopha.2022.112885. [DOI] [PubMed] [Google Scholar]
- 38.Li Y., Wu D., Wang P., Li X., Shi G. miR-195 Regulates Proliferation and Apoptosis through Inhibiting the mTOR/p70s6k Signaling Pathway by Targeting HMGA2 in Esophageal Carcinoma Cells. Dis. Markers. 2017;2017 doi: 10.1155/2017/8317913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao X., Lu M., Xu W., Liu C., Wu J. miR-195 inhibits esophageal cancer cell proliferation and promotes apoptosis by downregulating YAP1. Int. J. Clin. Exp. Pathol. 2019;12:275–281. [PMC free article] [PubMed] [Google Scholar]
- 40.Jin M.H., Nam A.R., Bang J.H., Oh K.S., Seo H.R., Kim J.M., Yoon J., Kim T.Y., Oh D.Y. WEE1 inhibition reverses trastuzumab resistance in HER2-positive cancers. Gastric Cancer. 2021;24:1003–1020. doi: 10.1007/s10120-021-01176-7. [DOI] [PubMed] [Google Scholar]
- 41.Hamilton G., Rath B. Smoking, inflammation and small cell lung cancer: recent developments. Wien. Med. Wochenschr. 2015;165:379–386. doi: 10.1007/s10354-015-0381-6. [DOI] [PubMed] [Google Scholar]
- 42.Andersson U., Johansson S., Landstrom M., Bjermer L., Henriksson R. Smoking enhanced the expression of MDR-1 in rat prostatic carcinoma. Oncol. Rep. 1997;4:953–956. doi: 10.3892/or.4.5.953. [DOI] [PubMed] [Google Scholar]
- 43.Chen X., Zhang W., Liu R., Zhu Z., Gong M., Wang Q., Qian W., Wu Z., Ma Q., Wang Z. NNK from tobacco smoking enhances pancreatic cancer cell stemness and chemoresistance by creating a beta2AR-Akt feedback loop that activates autophagy. Mol. Oncol. 2022;16:2881–2895. doi: 10.1002/1878-0261.13230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Q., Lv L., Ma P., Zhang Y., Deng J., Zhang Y. Identification of an Autophagy-Related Pair Signature for Predicting Prognoses and Immune Activity in Pancreatic Adenocarcinoma. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.743938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qiu L., Tao A., Liu F., Ge X., Li C. Potential prognostic value of a eight ferroptosis-related lncRNAs model and the correlative immune activity in oral squamous cell carcinoma. BMC Genom. Data. 2022;23:80. doi: 10.1186/s12863-022-01097-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu T.P., Hsieh Y.Y., Chou C.J., Yang P.M. Systematic polypharmacology and drug repurposing via an integrated L1000-based Connectivity Map database mining. R. Soc. Open Sci. 2018;5 doi: 10.1098/rsos.181321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gu L., Liu Y., Jiang C., Sun L., Zhou H. Identification and clinical validation of metastasis-associated biomarkers based on large-scale samples in colon-adenocarcinoma. Pharmacol. Res. 2020;160 doi: 10.1016/j.phrs.2020.105087. [DOI] [PubMed] [Google Scholar]
- 48.Chen Z.A., Tian H., Yao D.M., Zhang Y., Feng Z.J., Yang C.J. Identification of a Ferroptosis-Related Signature Model Including mRNAs and lncRNAs for Predicting Prognosis and Immune Activity in Hepatocellular Carcinoma. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.738477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoose S.A., Duran C., Malik I., Eslamfam S., Shasserre S.C., Downing S.S., Hoover E.M., Dowd K.E., Smith R., 3rd, Polymenis M. Systematic analysis of cell cycle effects of common drugs leads to the discovery of a suppressive interaction between gemfibrozil and fluoxetine. PLoS One. 2012;7 doi: 10.1371/journal.pone.0036503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tommasi S., Zheng A., Besaratinia A. Exposure of mice to secondhand smoke elicits both transient and long-lasting transcriptional changes in cancer-related functional networks. Int. J. Cancer. 2015;136:2253–2263. doi: 10.1002/ijc.29284. [DOI] [PubMed] [Google Scholar]
- 51.Reed R.M., Borgan S.M., Eberlein M., Goldklang M., Lewis J., Miller M., Navab M., Kim B.S. Tobacco Smoke Exposure Reduces Paraoxonase Activity in a Murine Model. Int. J. Biomed. Sci. 2017;13:20–25. [PMC free article] [PubMed] [Google Scholar]
- 52.Liu H., Ding L., Zhang Y., Ni S. Circulating endothelial microparticles involved in lung function decline in a rat exposed in cigarette smoke maybe from apoptotic pulmonary capillary endothelial cells. J. Thorac. Dis. 2014;6:649–655. doi: 10.3978/j.issn.2072-1439.2014.06.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Katsha A., Soutto M., Sehdev V., Peng D., Washington M.K., Piazuelo M.B., Tantawy M.N., Manning H.C., Lu P., Shyr Y., et al. Aurora kinase A promotes inflammation and tumorigenesis in mice and human gastric neoplasia. Gastroenterology. 2013;145:1312–1322.e1. doi: 10.1053/j.gastro.2013.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peltier H.J., Latham G.J. Normalization of microRNA expression levels in quantitative RT-PCR assays: identification of suitable reference RNA targets in normal and cancerous human solid tissues. RNA. 2008;14:844–852. doi: 10.1261/rna.939908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.El-Rifai W., Moskaluk C.A., Abdrabbo M.K., Harper J., Yoshida C., Riggins G.J., Frierson H.F., Jr., Powell S.M. Gastric cancers overexpress S100A calcium-binding proteins. Cancer Res. 2002;62:6823–6826. [PubMed] [Google Scholar]
- 57.Wang V., Heffer A., Roztocil E., Feldon S.E., Libby R.T., Woeller C.F., Kuriyan A.E. TNF-alpha and NF-kappaB signaling play a critical role in cigarette smoke-induced epithelial-mesenchymal transition of retinal pigment epithelial cells in proliferative vitreoretinopathy. PLoS One. 2022;17 doi: 10.1371/journal.pone.0271950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sahin Ersoy G., Zhou Y., İnan H., Taner C.E., Cosar E., Taylor H.S. Cigarette Smoking Affects Uterine Receptivity Markers. Reprod. Sci. 2017;24:989–995. doi: 10.1177/1933719117697129. [DOI] [PubMed] [Google Scholar]
- 59.Wong H.P.S., Li Z.J., Shin V.Y., Tai E.K.K., Wu W.K.K., Yu L., Cho C.H. Effects of cigarette smoking and restraint stress on human colon tumor growth in mice. Digestion. 2009;80:209–214. doi: 10.1159/000231898. [DOI] [PubMed] [Google Scholar]
- 60.Corso S., Isella C., Bellomo S.E., Apicella M., Durando S., Migliore C., Ughetto S., D'Errico L., Menegon S., Moya-Rull D., et al. A Comprehensive PDX Gastric Cancer Collection Captures Cancer Cell-Intrinsic Transcriptional MSI Traits. Cancer Res. 2019;79:5884–5896. doi: 10.1158/0008-5472.CAN-19-1166. [DOI] [PubMed] [Google Scholar]
- 61.Sehdev V., Katsha A., Ecsedy J., Zaika A., Belkhiri A., El-Rifai W. The combination of alisertib, an investigational Aurora kinase A inhibitor, and docetaxel promotes cell death and reduces tumor growth in preclinical cell models of upper gastrointestinal adenocarcinomas. Cancer. 2013;119:904–914. doi: 10.1002/cncr.27801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moser R., Xu C., Kao M., Annis J., Lerma L.A., Schaupp C.M., Gurley K.E., Jang I.S., Biktasova A., Yarbrough W.G., et al. Functional kinomics identifies candidate therapeutic targets in head and neck cancer. Clin. Cancer Res. 2014;20:4274–4288. doi: 10.1158/1078-0432.CCR-13-2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu S., Belkhiri A., El-Rifai W. DARPP-32 increases interactions between epidermal growth factor receptor and ERBB3 to promote tumor resistance to gefitinib. Gastroenterology. 2011;141:1738–1748.e1-2. doi: 10.1053/j.gastro.2011.06.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting this study’s findings are available in TNMplot at TNMplot.com and kmplot at kmplot.com. Source data are provided in this paper. All other data are available upon reasonable request.