

Abstract
Chemogenetics refers to experimental systems that dynamically regulate the activity of a recombinant protein by providing or withholding the protein’s specific biochemical stimulus. Chemogenetic tools permit precise dynamic control of specific signaling molecules to delineate the roles of those molecules in physiology and disease. Yeast d-amino acid oxidase (DAAO) enables chemogenetic manipulation of intracellular redox balance by generating hydrogen peroxide only in the presence of d-amino acids. Advances in biosensors have allowed the precise quantitation of these signaling molecules. The combination of chemogenetic approaches with biosensor methodologies has opened up new lines of investigation, allowing the analysis of intracellular redox pathways that modulate physiological and pathological cell responses. We anticipate that newly developed transgenic chemogenetic models will permit dynamic modulation of cellular redox balance in diverse cells and tissues and will facilitate the identification and validation of novel therapeutic targets involved in both physiological redox pathways and pathological oxidative stress.
Keywords: oxidative stress, redox signaling, endothelial cells, cardiac myocytes, heart failure, hydrogen peroxide
THE EVOLVING ROLES OF REACTIVE OXYGEN SPECIES IN CARDIOVASCULAR TISSUES
For decades, reactive oxygen species (ROS) were viewed principally as pathological molecules that undermine normal cellular pathways. From heart failure to hypertension to atherosclerosis, it is difficult to identify a disease whose pathogenesis is not associated with oxidative stress (1–3). Several of the most widely prescribed drugs with cardiovascular targets, such as angiotensin-II receptor blockers, exert protective effects in part by attenuating endogenous production of ROS (4). However, a more nuanced role for ROS has recently emerged, with the stable ROS hydrogen peroxide (H2O2) identified as a critical inter- and intracellular messenger molecule with a role not only in pathology and toxicology but also in normal physiological responses in the cardiovascular system.
Understanding of the many biological roles of ROS has evolved significantly over the past century. ROS encompass a diverse set of molecules in which oxygen carries an unpaired electron. One of the simplest principles engrained in the minds of high-school chemistry students is that an atom with a free electron will avidly interact with other molecules—whether a lipid, protein, or DNA—to fill that empty orbital. The realization that ROS have the potential to irreversibly modify cellular proteins led to the vilification of cellular oxidants as being deleterious to cellular function. The image of delicate intracellular proteins rusting (as happens when iron is oxidized) does seem intuitively pathologic. Supporting this model of ROS as purely damaging molecules, innumerable diseases were found to have oxidative damage associated with their pathogenesis (1, 2, 5).
In the cardiovascular system, a unifying feature of the various animal models of heart failure used for drug development is an association with increased oxidative stress (see Table 1). For example, atherosclerotic models of heart disease such as the apolipoprotein E knockout mouse consistently show increased lipid oxidation (6), and increased mitochondrial ROS appears to be a hallmark of models of myocardial infarction (7). Similarly, several forms of heart failure are correlated with oxidative stress (8, 9). The cardiotoxic side-effects of multiple therapeutics have also been discovered to arise from excessive oxidative stress. For example, the cardiotoxic effects of several chemotherapeutic agents, including anthracycline, are mediated by oxidative stress (10).
Table 1.
Summary of animal models for human cardiovascular disease states that are associated with oxidative stress
| Human cardiovascular disease state | Animal models | Key review |
|---|---|---|
| Atherosclerosis | ApoE KO, others | 6 |
| Hypertension | Renal artery ligation, high-salt diet, Ang-II infusion | 125 |
| Heart failure (both HFrEF and HFpEF) | TAC, aortocaval fistula, genetic models, HFD and L-NAME | 8 |
| Diabetic cardiomyopathy | HFD, STZ, ob/ob mouse, db/db mouse | 126 |
| Myocardial infarction | LAD ligation, ischemia/reperfusion | 7 |
This table lists the principal animal models for various human cardiovascular states, with the unifying feature that all these diseases have been observed to be associated with excess reactive oxygen species. Abbreviations: Ang-II, angiotensin-II; HFD, high-fat diet; HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction; KO, knockout; L-NAME, N-ωnitro-l-arginine methyl ester; LAD, left anterior descending artery; STZ, streptozocin; TAC, transverse aortic constriction.
These observations led to the hypothesis that attenuating oxidative damage with exogenous antioxidants could fend off the many degenerative diseases associated with toxic levels of ROS. However, systemic antioxidant therapy has shown little success, with nearly every well-designed randomized controlled trial yielding negative results (11). Casting further doubt on the initial model of ROS as playing only deleterious roles, meta-analyses of antioxidant trials have even demonstrated potential harm associated with two of the most commonly used supplements (vitamins C and E) (12). These findings have led to a reassessment of the dogma that ROS are purely deleterious to cell function. Indeed, not all ROS are created equal (13). As shown in Figure 1a, the sequential addition of electrons to molecular oxygen (O2) leads to the formation of molecular species of varying reactivity and diffusibility, eventually leading to the formation of water (H2O). The addition of a single electron to O2 results in the formation of a superoxide anion (O2•−), which has a short half-life in cells, and its reactivity is harnessed to play a key role in antimicrobial defense within neutrophils. Superoxide can also be enzymatically or nonenzymatically reduced to form H2O2, which is not a free radical as it does not have an unpaired electron. H2O2 is relatively stable, and represents the most abundant cellular ROS, with critical roles in a broad range of physiological responses in cells (13). Addition of another electron to H2O2 forms the highly unstable hydroxyl radical, whose short half-life and high chemical reactivity limit it from dynamic regulation in biological systems. This oxidant therefore plays a limited role in cellular physiology. Adding a fourth and final electron yields water, completing the reduction of O2. This review focuses on the physiological and pathophysiological roles of H2O2, with an emphasis on recent studies using chemogenetic approaches that exploit the yeast enzyme d-amino acid oxidase (DAAO) to generate H2O2 and also to detect H2O2 using the biosensor HyPer, as shown in Figure 1b and discussed in detail below.
Figure 1.
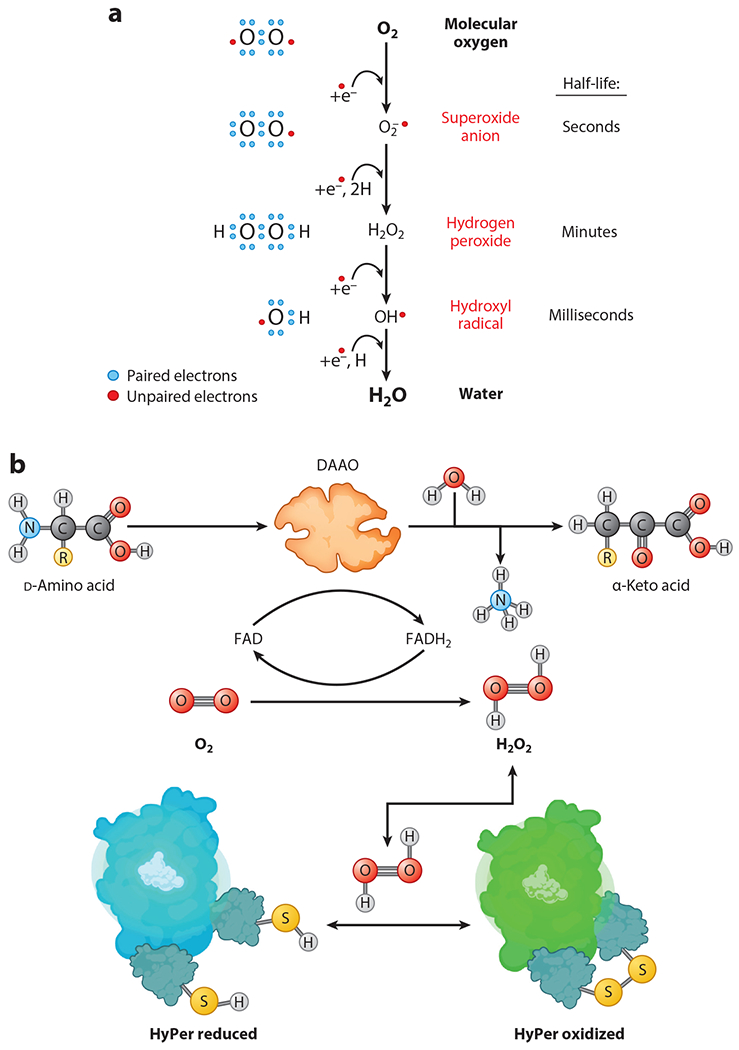
Intracellular redox reactions modulated by d-amino acid oxidase (DAAO). (a) Successive one-electron reduction of molecular oxygen (O2) to superoxide radical anion (O2•−), to hydrogen peroxide (H2O2), to the unstable hydroxyl radical (OH•), and finally to water (H2O). (b) Oxidation of d-amino acids by DAAO to form H2O2. DAAO converts d-amino acids into their corresponding α-keto acids along with equimolar H2O2 and ammonia (NH3), with O2 serving as a cosubstrate and flavin adenine dinucleotide (FAD) as the cofactor needed for catalysis of the enzymatic reaction. Panel b adapted from images created with BioRender.com.
Over the past several decades, new roles for H2O2 in physiological processes have emerged, ranging from receptor-mediated signal transduction (14) to hypoxic pulmonary vasoconstriction (15) and the activation of regulatory T cells (16). In the cardiovascular system, physiological roles for H2O2 have been established in the regulation of the key signaling protein endothelial nitric oxide synthase (eNOS, also known as NOS3) in endothelial cells (17) and cardiac myocytes (18, 19). In turn, eNOS generates the free radical gas nitric oxide (NO), which—akin to H2O2—can have diverse effects in target tissues: Low NOconcentrations play critical physiological roles in the modulation of blood pressure (20) and cardiac contractility (21), whereas higher concentrations can lead to organ dysfunction and disease (22). NO’s ability to rapidly control smooth muscle tone has led to the development of multiple drugs that control its downstream signaling processes, from the long-used NO donors nitroglycerin and sodium nitroprusside to the phosphodiesterase-5 inhibitor sildenafil and the recently developed direct soluble guanyl cyclase stimulator riociguat. In addition to its effects on NO metabolism, H2O2 has been implicated in insulin-modulated responses in the heart (23) and other tissues (24). Thus, a natural question with an elusive answer has emerged: How can the same signaling molecules apparently drive physiological processes in some situations yet induce pathology in others?
Several mechanisms have been suggested for these seemingly opposite effects of H2O2. One intuitive explanation is that the effects of ROS are concentration dependent, with lower levels being required for physiological processes but higher levels eliciting toxic and pathologic responses. This view exemplifies the phenomenon of hormesis (see Figure 2), which can be defined as a biphasic dose response in which a chemical subserves a physiological role at lower concentrations (oxidative eustress) yet has a pathological effect at higher concentrations (oxidative stress) (25). Another explanation for the multiple roles of H2O2 in biology is that the subcellular location of ROS production determines the effects of signaling molecules (Figure 3). After all, the intracellular synthesis of H2O2 involves numerous enzymes and small molecules that are differentially localized within the cell (26). Adding an additional layer of regulation, the enzymes that catabolize H2O2 and other ROS, including peroxiredoxins, thioredoxins, catalase, and the glutathione system, have their own unique spatial localizations and are subject to dynamic regulation (27, 28).
Figure 2.
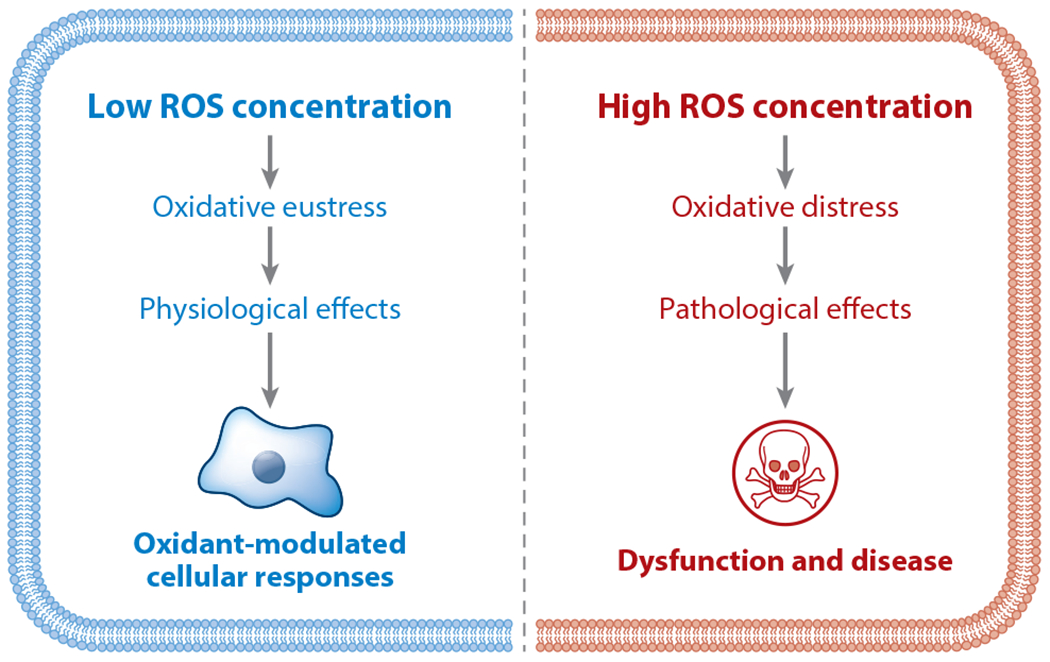
Reactive oxygen species (ROS) hormesis showing biphasic cellular responses to ROS. Hormesis can be defined as a biphasic dose response in which a specific chemical subserves a physiological role at lower concentrations yet has a pathological effect at higher concentrations. This is a fundamental feature of ROS in cells and tissues, with lower ROS concentrations causing oxidative eustress to mediate physiological cellular responses, while higher concentrations of ROS lead to oxidative distress and to the development of cellular dysfunction and disease (25). Figure adapted from images created with BioRender.com.
Figure 3.
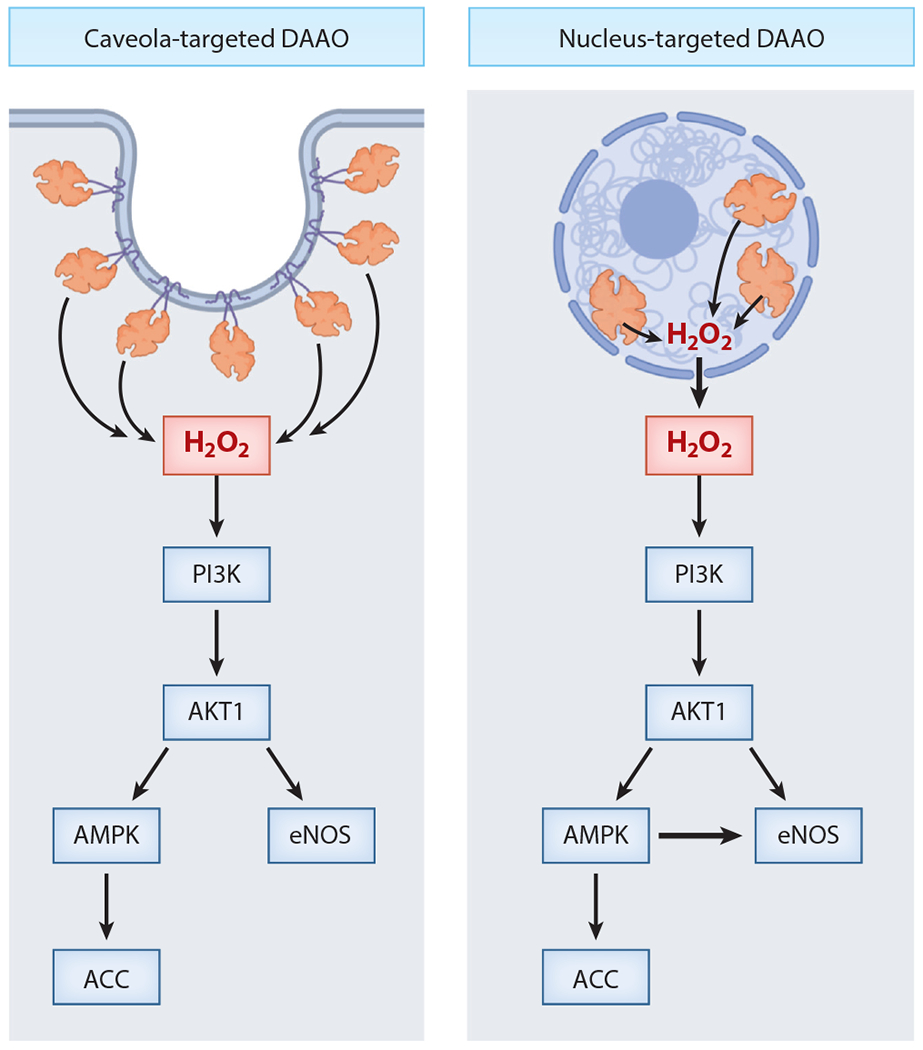
Schematic model for the effects of differentially targeted hydrogen peroxide (H2O2) on endothelial nitric oxide synthase (eNOS) phosphorylation pathways. This figure presents a model summarizing studies of endothelial cells expressing recombinant differentially targeted d-amino acid oxidase (DAAO). These experiments probed the effects of H2O2 generated in distinct subcellular compartments on oxidant-modulated phosphorylation pathways in vascular endothelial cells. DAAO is a soluble protein but can be targeted to different subcellular locales using N-terminal signal sequences. Following addition of the DAAO substrate d-alanine, the phosphorylation of key phosphoenzymes was analyzed. H2O2 synthesized by DAAO constructs targeted either to endothelial cell nucleus (right) or to caveolae (left) led to the activation of protein kinase pathways that promote the phosphorylation of eNOS. H2O2 generated in either the nucleus or the cytosol leads to phosphorylation of the kinases PI3K, Akt1, and AMP-activated protein kinase (AMPK) and to the phosphorylation of acetyl CoA carboxylase (ACC) and eNOS. But only the H2O2 generated in the endothelial cell nucleus, not in the caveolae, leads to eNOS phosphorylation via AMPK: Caveola-derived H2O2 does not promote eNOS phosphorylation via AMPK, suggesting that different cellular pools of AMPK may be differentially regulated by oxidants. This figure exemplifies the importance of the subcellular localization of oxidants in the modulation of cell signaling pathways. Figure adapted from Reference 32 using BioRender.com.
While specific pathologies and drug toxicities have been associated with increased absolute levels of H2O2 or unique spatial profiles of ROS production (e.g., excess mitochondrial ROS), the lack of tools to precisely control intracellular H2O2 production has made it difficult to tease apart correlation from causation. This review discusses emerging methods for the simultaneous manipulation and measurement of intracellular H2O2 aimed at delineating the transition from physiology to pathology and identifying therapeutic and toxic mechanisms of cardiovascular drugs.
INTRACELLULAR H2O2 METABOLISM AND TRANSPORT
Nature has devised multiple sources and sinks of ROS, allowing for a number of potential intracellular pathways and locales for the regulation of redox signaling. The intracellular enzymatic sources of ROS are localized to specific cellular organelles (26, 29), with the principal ROS-generating enzymes targeted to membrane structures (e.g., endoplasmic reticulum, plasma membrane). Enzymes with the exclusive role of generating ROS include the family of nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (30), which are expressed in most tissues and are necessary for mediating receptor-based signaling in the heart (23) and vasculature (31, 32). NADPH oxidases have distinctive subcellular localization profiles, with H2O2 derived from NADPH oxidase isoform 2 at the plasma membrane capable of diffusing back into the cell through aquaporins (33). Other oxidases, for example, monoamine oxidases and xanthine oxidases, generate ROS as a byproduct of catabolism of specific metabolites (34–36). The electron transport chain contributes the largest portion of basal oxidant production in cells and can be dynamically regulated in physiology and pathology (37–39).
The enzymatic systems responsible for ROS catabolism are no less complicated than those responsible for their generation. Glutathione, which exists in millimolar concentrations in the cell cytosol, can undergo oxidation of its cysteine sulfhydryl to form sulfenic acid and can then be recycled back to a reduced state by the actions of glutathione peroxidases and reductases (40). Glutathione’s importance in buffering oxidant stresses is highlighted by the fact that acetaminophen toxicity is mediated in part by a depletion in glutathione stores from the reduction of the toxic metabolite NAPQI (41). Peroxiredoxins, another key antioxidant buffering system, have different isoforms that localize to distinct cellular compartments and can not only buffer oxidants but also transfer oxidizing equivalents to cysteine-containing proteins, thereby regulating their function (42–44). Thioredoxins can reverse protein oxidation events by reducing disulfide bonds, and they can then be recycled through thioredoxin reductases (45, 46).
Unfortunately, the tools to study this intricate network of oxidizing and reducing enzymes have been frustratingly limited and are confounded by low sensitivity or lack of specificity of both biochemical and immunochemical reagents. Biochemical readouts of cells and organs were among the first assays to reveal the correlation between increased oxidant forces in cells and pathology; these included direct measurement of protein sulfenation (47) and the ratio of reduced to oxidized NADPH and/or glutathione (48, 49). Small-molecule fluorescent redox indicators enabled the real-time measurement of cellular redox state (50). But even the most popular probe, dichlorofluorescein (DCF), is limited by a lack of specificity for oxidant species: NO, peroxynitrite, superoxide, and H2O2 can all oxidize the DCF probe (51). Moreover, DCF can itself generate superoxide and has a propensity to concentrate in certain cellular compartments (52).
To address these technical limitations, genetically encoded fluorescent biosensors have been engineered that have specificity for specific oxidants and can be targeted to a cellular compartment of interest (53, 54). These biosensors have clarified spatiotemporal changes in redox state in response to a variety of stimuli. The genetically encoded H2O2-specific HyPer biosensors have proved particularly useful for understanding the dynamics of H2O2 (55, 56). Many groups have utilized variants of HyPer biosensors to dissect redox signaling pathways in diverse model systems ever since Belousov et al. (57) first developed HyPer in 2006. The original HyPer biosensor was constructed as a circularly permuted yellow fluorescent protein (YFP)-based chimera in which YFP was flanked by cysteine-containing domains from the H2O2-sensitive Escherichia coli transcription factor OxyR. In a concentration-dependent response to H2O2, the sensor element OxyR can be reversibly oxidized to form an intramolecular disulfide bond that leads to a conformational rearrangement, causing a spectral change that yields a ratiometric signal and permits quantification of H2O2. Oxidation of HyPer in cells can be reversed by redox-active enzymes, including thioredoxin reductase or glutathione reductase (58). Different cellular compartments contain different combinations of reductants and oxidants that may modulate H2O2 metabolism and thereby affect the HyPer oxidation state. It is important to note that the YFP fluorophore in HyPer has a fluorescence response that can be affected by alterations in intracellular pH. This general limitation of many biosensors often requires performing control experiments in parallel. The SypHer biosensor, a H2O2-insensitive variant of HyPer, is a good control to confirm the validity of H2O2 levels acquired by HyPer (59). Changes in HyPer fluorescence that are duplicated by SypHer more likely result from pH than from H2O2 concentration changes. A new generation of HyPer biosensors for H2O2—dubbed HyPer7—was recently developed. HyPer7 is more sensitive than earlier HyPer variants for H2O2, shows faster responses, and is less sensitive to pH, as it is based on the less pH-sensitive fluorophore green fluorescent protein (GFP) (60).
The multiple generations of H2O2-sensitive fluorescent biosensors have provided ever-increasing granularity for monitoring redox changes in the cardiovascular system. In cardiac myocytes, HyPer has been used to document intracellular changes in H2O2 in response to angiotensin-II (19), and variants targeted to the mitochondria and caveolae helped to describe the subcellular pattern of redox changes with insulin stimulation (61). Subcellularly targeted variants of HyPer have also been applied to endothelial cells, where they have demonstrated primarily caveolar and cytosolic increases in H2O2 in response to the activation of purinergic receptors (17). While HyPer is the most widely used H2O2-specific fluorescent biosensor, several other families of probes have been developed that measure the overall redox state of a specific compartment rather than a specific oxidant species. The roGFP family of probes, which are ratiometric, calibratable, and relatively pH insensitive, has helped describe redox changes in other cardiovascular tissues (62–64). Another strategy to assess the redox state of a compartment with a real-time genetically encoded fluorescent probe has been to measure NAD/NADH ratios (65, 66). These many strategies to assess redox state have strengths and weaknesses. Probes that directly measure H2O2 enable experimenters to study a key signaling molecule as close to the enzymatic source as possible. By contrast, probes such as roGFP allow one to measure the net effect of oxidant molecules on the redox-sensitive machinery in the cell, producing a more functional read-out.
The realization offered by redox-sensitive biosensors that the redox states of subcellular compartments can independently change in response to a variety of stimuli has fueled the idea that the location of oxidant production is just as crucial as the amount of oxidant in determining the biological effects of these biochemical changes (67–69). However, the correlation of these changes with downstream biological effects does not imply a causal relationship. The task of demonstrating causation has proved to be more difficult, owing to the lack of a comprehensive toolbox for directly manipulating oxidant production in cells, let alone in whole organisms. Much of the current understanding of the causal role of oxidant signaling in biological processes comes from studies in which the source of ROS is ablated, either by small-molecule inhibitors (70) or by genetic knockout and knockdown of ROS-producing enzymes (71, 72). Another approach has been to mop up ROS after they have been produced, either by small-molecule antioxidants or by enzymes such as catalase or superoxide dismutase, depending on the oxidant species of interest (73). Finally, the application of exogenous oxidants (namely H2O2) has been helpful in demonstrating that ROS have the ability to independently activate certain signaling processes. However, the relevance of exogenous oxidants that must diffuse into the cell, particularly for studying processes where the oxidant signal is thought to originate from a specific compartment, is questionable. A somewhat more biological approach is to generate a steady lower level of H2O2 by incubating cultured cells with the H2O2-generating enzyme glucose oxidase along with its substrate (74)—but the ability of this approach to recapitulate endogenous signaling processes is unclear. This diverse set of tools, from inhibition of ROS production to metabolism of oxidants and application of exogenous oxidants, has helped reinforce the role of H2O2 in disease, but none have been able to define the effect of intracellular H2O2 production independent of a particular stimulus known to activate ROS-generating enzymes. The redox experimentalist’s toolbox has been missing a method that can dynamically produce H2O2 in a specific location on demand in order to isolate ROS as a unitary causal factor. The application of chemogenetic tools has filled this niche, and the combination of chemogenetic approaches with biosensors for ROS has enabled a better understanding of redox signaling in the cardiovascular system and beyond (Figure 4).
Figure 4.
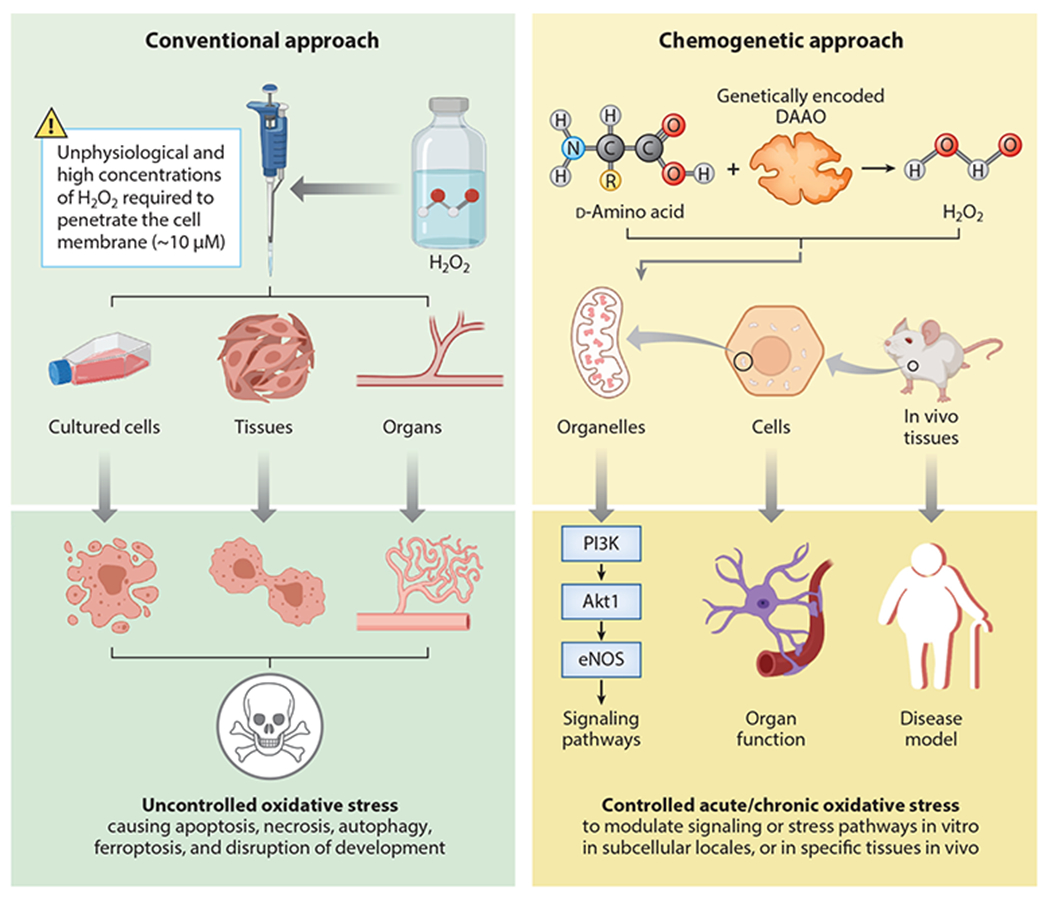
Comparison of conventional versus chemogenetic approaches to perturb cellular redox balance. (Left) Conventional approach to perturb intracellular redox balance by adding exogenous hydrogen peroxide (H2O2) or redox-active small molecules to cells or tissues. The direct addition of H2O2 to cells has highly variable effects on intracellular pathways due to the variable permeability of cell membranes to H2O2 and the differential distribution of H2O2 between intracellular organelles. Moreover, the concentrations of exogenous H2O2 needed to activate physiological pathways can also produce toxic effects on cells. Redox-active small molecules can also be added to cells, with variable (and often nonspecific) effects on intracellular oxidants. (Right) A chemogenetic approach that permits dynamic regulation of H2O2 generated by recombinant yeast d-amino acid oxidase (DAAO) expressed in mammalian cells. H2O2 can be reversibly generated in specific intracellular organelles by providing (or withholding) d-amino acids. DAAO can be cloned as a fusion protein along with the H2O2 biosensor HyPer to document H2O2 formation within specific subcellular compartments. Alternatively, the DAAO and HyPer can be differentially targeted to distinct subcellular organelles to study intracellular H2O2 diffusion from the site of H2O2 synthesis by DAAO to the organelle in which H2O2 is detected by HyPer. Chemogenetic approaches have been used both in cultured cells and in animals in vivo to dissect both physiological and pathophysiological oxidant-modulated pathways. Chemogenetic methods have enabled experimenters to isolate redox state as an independent variable of both in vitro and in vivo systems. Moreover, biosensors can be targeted to compartments distinct from the organelle expressing DAAO, enabling the experimenter to study the effects of oxidants generated in different subcellular locales. Figure adapted from images created with BioRender.com.
Ligand- and Substrate-Based Chemogenetic Approaches to Identifying Pharmacological Targets and Toxicological Mechanisms in the Cardiovascular System
Chemogenetics refers to experimental systems in which the activity of recombinant proteins in cells can be dynamically regulated by the addition or removal of specific biochemicals (75). There are many similarities between chemogenetic approaches and the various experimental systems known collectively as optogenetics: In optogenetics, the recombinant protein is dynamically modulated by light instead of by a chemical ligand or substrate (76) (Figure 5). In chemogenetics, the recombinant protein and/or its ligand or substrate is designed such that the protein is regulated by the addition or removal of its cognate small-molecule target. For example, a chemically engineered ligand can be added to cells expressing a genetically engineered recombinant receptor that is designed to respond exclusively to the novel ligand. These so-called designer receptors exclusively activated by designer drugs (DREADD) methods were initially exploited in neural cells and tissues (77), but applications have been extended to analyze receptor-modulated responses in adipose and hepatic tissues to identify G protein–coupled receptor (GPCR)-based targets for antidiabetic drugs (78).
Figure 5.
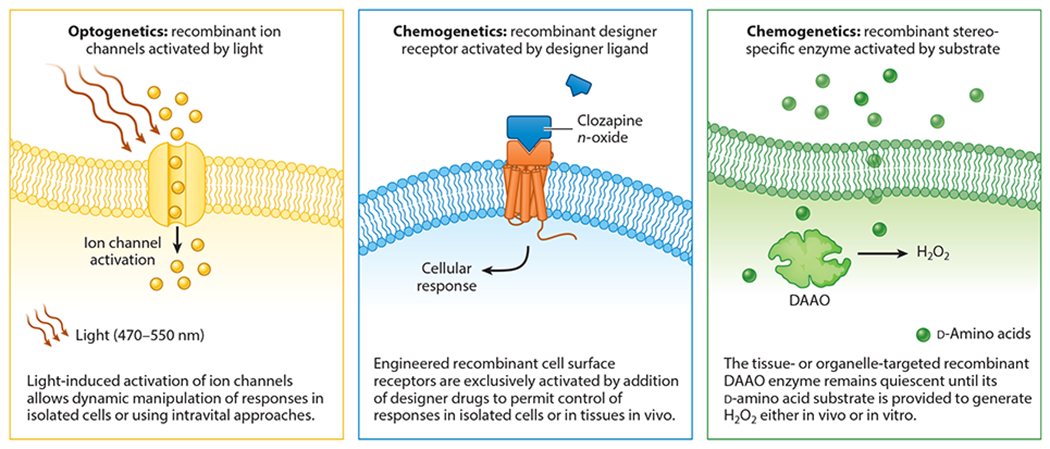
Comparison of optogenetics with ligand-activated versus substrate-dependent chemogenetic manipulation of cellular responses. The figure contrasts the basic features of dynamic cellular control by optogenetic or chemogenetic approaches. In experiments exploiting optogenetics (left), the recombinant protein (typically an ion channel) is dynamically modulated by light at a specific wavelength, leading to dynamic activation or deactivation of the channel either in vitro or using intravital approaches (76) to control channel activity. The middle and right panels contrast two different chemogenetic approaches: ligand-based cell activation [designer receptors exclusively activated by designer drugs (DREADD), middle] or substrate activated [d-amino acid oxidase (DAAO), right]. DREADD methods employ genetically engineered cell surface receptors—typically G protein–coupled receptors—that are designed to be activated only by highly specific ligands, which are designer drugs that are chemically modified to interact exclusively with their target receptor but otherwise elicit no other cellular response. Thus, recombinant DREADD receptors can be targeted to specific cells or tissues and remain quiescent until their cognate ligand is provided and activates the DREADD receptor, leading to the stimulation of receptor-mediated responses. In contrast to ligand-based DREADD chemogenetics, DAAO represents the archetype for substrate-based chemogenetic approaches, in which recombinant (yeast) DAAO is targeted to specific organelles within mammalian cells or tissues. The recombinant DAAO enzyme remains quiescent until its d-amino acid substrate is provided to the animal or cell, leading to the generation of hydrogen peroxide (H2O2) in the subcellular organelle to which the DAAO is targeted by means of N-terminal signal sequences. Figure adapted from images created with BioRender.com.
Akin to DREADD-based methods, chemogenetic approaches to modulate intracellular redox balance have involved the application of ROS-generating enzymes whose substrate is not present in the biological system of interest until the substrate is supplied to the cell or tissue. The archetypal chemogenetic approach in redox biology involves yeast DAAO, a family of enzymes that catalyze the stereoselective oxidation of d-amino acids to yield the corresponding keto acid, with the concomitant generation of equimolar ammonia and H2O2 (Figure 1b). The stereoselectivity of DAAOs is fundamental to their function in chemogenetics: Since mammalian cells and tissues principally contain primarily l-amino acids, a recombinant DAAO remains inactive until exogenous d-amino acids are provided, thus allowing an experimenter to effectively turn on and off H2O2 production wherever the DAAO is expressed.
Whether DREADD or DAAO-based, chemogenetic systems require the presence of several features in the biologic system in which they will be applied. First, the artificial receptor or enzyme must be easily expressed in the organ of interest. This can be accomplished using a number of molecular biological techniques, for example, the generation of transgenic animals or viral vectors driven by a tissue-specific promoter. Adeno-associated viruses (AAVs) have been used to express DREADDs in the brain (79) and DAAO in the heart (80). Due to their modular nature, with customizable tissue-specific promoters and capsid proteins with unique tissue tropisms, AAVs enable rapid prototyping and deployment of chemogenetic proteins. Another required feature, if one is to study long-term activation of a chemogenetic enzyme, is adequate oral bioavailability of the experimental ligand. Intravenous administration of a ligand can acutely activate an enzyme, and constant intraperitoneal infusion from an osmotically driven pump can supply a ligand for several days, but longer-term activation of a chemogenetic enzyme is most easily achieved by oral administration of a ligand. In the case of DAAO, intestinal transporters of amino acids appear to be relatively nonstereoselective (81, 82), which allows a d-ala substrate to be introduced in the drinking water of the animal. Similar to the inert optical stimulus used in optogenetics, the chemical stimulus should have minimal effect on the biological system on its own. While DREADD- and DAAO-based methods appear to have minimal off-target effects, there is some suggestion that clozapine N-oxide (CNO) has the ability to be metabolized to clozapine, a clinically used antipsychotic medication that can have its own unique neurobiological effects (83). Similarly, there is some suggestion that d-ala may have an independent diuretic effect when used at high doses in vivo (84).
Applications of DREADD-based chemogenetic approaches in the cardiovascular system.
DREADD methods employ genetically engineered receptors—typically GPCRs—that are designed to be activated only by highly specific ligands, which are designer drugs that are chemically modified to interact solely with their target receptor but otherwise elicit no other response. Thus, recombinant DREADD receptors can be targeted to specific cells or tissues and remain quiescent until their cognate ligand is provided and activates the DREADD receptor, leading to the stimulation of receptor-mediated responses. The most widely used DREADD receptor is the muscarinic cholinergic receptor together with the specific designer ligand CNO; other tissue-specific receptors and ligands are also being developed (85). Expressing the DREADD receptors under control of a tissue-specific promoter permits the dynamic control of receptor-mediated responses in specific cells in vivo.
Kaiser et al. (86) developed the first in vivo model to study the effects of Gq-coupled GPCRs on cardiac function. By expressing an M3Dq-based DREADD under a muscle creatine kinase promoter, they were able to target their Gq-based designer receptor to striated muscle, including the heart. They observed that dose-dependent activation of the receptor with CNO induced multiple cardiac arrythmias, including sinus and atrioventricular block, as well as ventricular tachycardia. The mechanism of this toxicity appeared to be driven by activation of the canonical phospholipase C signaling pathway, ultimately leading to the phosphorylation of connexin CX43 by protein kinase C. Multiple cardiovascular drugs target Gq-coupled GPCRs, including the vasoactive peptides angiotensin-II and vasopressin, as well as catecholamines with an affinity for the alpha adrenergic receptor such as phenylephrine, dopamine, norepinephrine, and epinephrine. By isolating the effect of a specific GPCR in the heart, these studies identified new therapeutic mechanisms of drugs such as angiotensin-II receptor blockers as well as arrhythmogenic side effects of vasopressor medications, independent of their effects on the vasculature (87).
DAAO-based chemogenetic approaches to identify therapeutic redox targets and toxicity of reactive oxygen species in the cardiovascular system.
While DREADDs have been applied to study pharmacological targets in vivo for over a decade, the application of DAAO as a chemogenetic tool to study oxidative stress and redox therapeutic targets in vitro is a more recent invention, and the only in vivo applications to date have been in the heart. DAAOs were described in the 1930s by Hans Krebs (88) when he observed that natural l-amino acids could be deaminated and oxidized by an enzymatic system distinct from that which can metabolize nonnatural d-amino acids. Since their discovery in liver and kidney preparations almost a century ago, DAAOs have been studied primarily in single-cell organisms such as bacteria and yeast. Mammalian DAAOs exist, with most of their function appearing to be in the central nervous system, where d-amino acids, including d-serine and N-methyl-d-aspartate (NMDA), are neurotransmitters. DAAOs in astrocytes are thought to facilitate the degradation of d-serine, which is a coagonist at the NMDA glutamate receptor (89). Genome-wide association studies (GWAS) recently linked gain of function mutations in the human DAAO gene with the pathogenesis of schizophrenia (90). Interestingly, the atypical antipsychotic medication risperidone can inhibit DAAO in vitro (91), raising the possibility that in addition to dopamine receptor antagonism, the drug’s efficacy arises from increasing the concentration of d-amino acid neurotransmitters. Outside of the nervous system, mammalian DAAOs have been discovered in the hepatobiliary and gastrointestinal systems, where they have been proposed to act as bacterial defense systems (92). More recently, human DAAO activity has been suggested to promote cellular senescence in vitro (93). The existence of mammalian DAAOs with still-emerging roles necessitates the use of recombinant DAAOs with faster kinetics in order to minimize the potential confounding effects of activating the endogenous form of the enzyme.
To this end, DAAO from the yeast Rhodotorula gracilis has become the most widely applied isoform in chemogenetic systems, owing to its favorable kinetics compared to mammalian homologs. For the commonly used alanine substrate, the Km for yeast DAAO is two orders of magnitude lower than for porcine DAAO [2.8 nM versus 420 mM (94)]. Similarly, the yeast DAAO is over an order of magnitude more efficient than human DAAO (89, 95). These faster kinetic properties of yeast DAAO are essential to enable its use as an experimental tool. Because it can be activated by substrate concentrations far below the Km of endogenous mammalian DAAOs (96), off-target effects from the activation of endogenous DAAOs by exogenous d-amino acids can be minimized. In addition to chemogenetic applications aimed at better understanding the roles of H2O2 in cardiovascular biology and heart failure therapeutics, DAAOs have been investigated as targeted anticancer therapies. Relying on the known pathological effects of excess H2O2, DAAOs have been overexpressed in tumor cells, which can be killed by supplying a d-amino acid substrate that spares normal cells (97). These efforts have led to the development of novel engineered DAAOs with even more favorable kinetics than natural yeast-derived enzymes and may one day be deployed in chemogenetic studies as well. Different DAAO isoforms have different substrate selectivity, but most in vivo studies exploiting DAAO have utilized d-alanine (d-ala), which has the dual advantages of having both a low Km and low cost [d-ala is used in commercial applications and is available in bulk (98)]. Yeast DAAO shows activity on a wide variety of d-amino acid substrates (94), and amino acids other than alanine, including nonnatural amino acids, warrant future investigation. Of particular interest is the potential to supply d-cysteine as a substrate, which can generate hydrogen sulfide (H2S), a diffusible gas, which has been shown to play a central role in signaling in a diverse set of organ systems (99–103). Furthermore, the fact that DAAO is a genetically encoded enzyme enables it to be targeted to specific subcellular locations and permits it to be fused to a ROS biosensor such as the H2O2-sensitive HyPer, allowing for the simultaneous production of H2O2 (by DAAO) and the real-time quantitation of intracellular H2O2 levels using live-cell fluorescent imaging (58, 96).
Although an important addition to the redox pharmacologist’s tool box, DAAO-based chemogenetic systems possess several experimental inconveniences. In addition to the potentially confounding effects of activation of endogenous DAAOs discussed above, the reactions catalyzed by DAAO (Figure 1b) yield two important byproducts of H2O2 production: an alpha ketoacid derived from the substrate d-amino acid and equimolar ammonia. Oxidation of the d-ala substrate generates pyruvate, which is relatively ubiquitous in metabolically active tissues as an end product of glycolysis and a precursor for the tricarboxylic acid cycle (104). Ammonia, on the other hand, has been reported to activate some of the same signaling responses as H2O2, including the mechanistic target of rapamycin (mTOR) pathway (105). However, the cellular concentrations of ammonia and pyruvate are several orders of magnitude higher than that of H2O2 (106, 107). Ammonia also has its own buffering system through glutamine pathways (56). It is also important to note that pyruvate and ammonia are produced in equimolar quantities as H2O2 by DAAO. While it is difficult to definitively measure the amount of H2O2 generated by DAAO in cells, it has been estimated that full enzyme activation results in [H2O2]i of approximately 50 nM, about double the [H2O2] in most resting cells (108). Because intracellular concentrations of pyruvate and ammonia are orders of magnitude higher than the typical levels of H2O2, a nominal relative increase in these products is expected, whereas H2O2 levels may increase twofold or greater (depending on the level of recombinant protein expression and the duration of enzyme activation). While little plausibility exists for meaningful changes in pyruvate or ammonia concentrations following DAAO activation, definitive measurement of these metabolites is lacking and will be important for excluding their potential roles in responses elicited by DAAO activation. A similar caveat applies when d-cysteine is provided as a substrate for DAAO: H2S will be generated (109) as well as H2O2, and H2S could then modulate a range of downstream signaling responses (110, 111). However, the chemogenetic generation of H2S from DAAO/d-cysteine has not yet been explored in detail in cellular models.
In Vitro Applications of DAAO-Based Chemogenetics to Study Mechanisms of H2O2 Toxicity
Following the adaptation and optimization of DAAO from yeast to mammalian systems discussed above, some of its first applications explored the ability of intracellular H2O2 generation to induce a shift from physiology to pathology. Huang & Sikes (112) expressed DAAO along with the fluorescent biosensor HyPer in HeLa cells to investigate the parameters that drive apoptosis in response to oxidative stress. After applying a kinetic model to infer the intracellular H2O2 concentration from changes in HyPer fluorescence, they observed that not one but two thresholds must be crossed in order to reliably trigger cell death: First, the absolute intracellular H2O2 concentration must be more than ~40 nM, and second, the concentration integrated over time must exceed ~50 nM/h (113). Unless both of these conditions were met, toxicity did not reliably occur.
The next steps in the in vitro application of DAAO chemogenetics leveraged the ability to target the enzyme to specific subcellular compartments, thereby allowing for both spatial and temporal control of H2O2 production and enabling the study of local ROS in toxicity. Stein et al. (114) targeted DAAO to the mitochondrial matrix of HeLa cells and demonstrated the ability to progressively oxidize protein sulfhydryl groups in not only the mitochondrial matrix but also the cytosol in a dose-dependent manner. By measuring the dimerization of peroxiredoxin isoforms 2 (in the cytosol) and 3 (in the mitochondrial matrix), they demonstrated that supplying increasing amounts of d-ala substrate induced increasing oxidation of thiol groups. This progression to sulfenic residues (able to dimerize peroxiredoxins through disulfide bond formation) and then to the nonreactive/irreversible sulfinic moiety occurred primarily after mitochondrial generation of H2O2 at lower doses of d-ala but spread to the cytosol at higher doses. Similar to prior observations with cytosolic DAAO expression, a time-integrated exposure to d-ala correlated with collapse of the mitochondrial membrane potential and cellular toxicity.
While excessive amounts of ROS have long been known to be capable of inducing cell death (at least when supplied exogenously as H2O2), the diverse physiological roles of oxidants as signaling molecules have proved more difficult to understand. In order to better define the spatiotemporal kinetics of the diffusion of endogenous oxidants, Mishina et al. (115) targeted DAAO to the nucleus and measured diffusion of H2O2 by measuring changes in fluorescence of the H2O2-sensitive biosensor HyPer. They anchored HyPer to the cytoskeleton by fusing it to keratin, thereby limiting the ability of the probe to diffuse over short time periods. Upon addition of the DAAO substrate d-ala, the HyPer fluorescence revealed a gradient of oxidation in the cytosol that extended from the nucleus to the plasma membrane. The authors then used small-molecule inhibitors to study the reducing systems in the cells that controlled this gradient and limited H2O2 diffusion. Inhibition of catalase had no effect on the gradient from nucleus to plasma membrane, while inhibition of the thioredoxin system abolished the gradient. This newly discovered role of thioredoxin in maintaining spatial gradients of ROS suggests that it may serve as a therapeutic target for either attenuating ROS diffusion (through potentiation of the enzyme) or facilitating ROS spread (through its inhibition).
The realization that intracellular H2O2 production can establish redox gradients without necessarily inducing cell death allowed attention to turn to determining which physiological signaling processes regulate endogenous H2O2 generation and may in turn identify novel pharmacologic targets. Eroglu et al. (116) expressed recombinant DAAO in immortalized human endothelial cells and used multispectral imaging methods with NO and Ca2+ biosensors to document that H2O2-promoted eNOS phosphorylation takes place without eNOS activation (as measured by NO production) or Ca2+ mobilization. Saravi et al. (32) expressed DAAO in endothelial cells and targeted the enzyme to multiple compartments, including the cytosol, nucleus, and plasmalemmal caveolae, which are lipid-rich microdomains in the plasma membrane with a high density of signaling machinery. Generation of H2O2 in all three locations led to the downstream phosphorylation of eNOS, but the protein kinase pathways involved in eNOS phosphorylation were found to depend on the subcellular organelle in which the H2O2 was generated. Cytosolic, caveolar, and nuclear H2O2 all appeared to induce phosphorylation and activation of the kinase Akt, which then promotes the phosphorylation of the AMP-activated protein kinase (AMPK) and eNOS. Inhibition of AMPK prevented H2O2 generated in the nucleus from subsequently inducing the phosphorylation of eNOS, but AMPK inhibition had no effect on the ability of H2O2 generated in the cytosol or caveolae to induce eNOS phosphorylation (Figure 3). These observations suggest that generation of H2O2 in different subcellular compartments may differentially affect intracellular signaling responses. These results could not have been reached without the application of chemogenetic tools to control H2O2 production in specific subcellular compartments.
In vivo applications of DAAO-based chemogenetics.
The studies discussed thus far have expressed DAAO in cell lines, which serve as convenient and easily manipulated systems, but they are limited in their ability to predict changes in the physiology of primary cell types, let alone whole organisms. Given the association of deranged redox signaling in many human diseases (see Table 1), the transition from physiological to pathological redox signaling in human physiology has long garnered intense study and interest as a potential therapeutic target in heart disease. The first in vivo application of chemogenetic redox tools was in the heart. Steinhorn et al. (80) expressed a cytosolic variant of DAAO fused to the H2O2-sensitive fluorescent biosensor HyPer in the hearts of rats through a viral vector. The complementary DNA (cDNA) for HyPer, DAAO, and a nuclear exclusion sequence were fused and driven by a cardiac-specific cardiac troponin T (cTnT) promoter, all of which were packaged in an AAV. Serotype 9 was chosen, as it exhibits cardiac tropism and an altered heparin-binding domain that limits hepatic uptake, thereby increasing systemic bioavailability (117, 118). Rat pups were infected with a single intravenous injection of the viral vector carrying the cDNA for the HyPer-DAAO fusion protein. The adult rat hearts demonstrated strong expression of the construct, and ventricular myocytes isolated from these animals were capable of producing H2O2 when exposed to d-ala but not l-ala. By applying a fusion construct between DAAO and the HyPer biosensor, real-time production of H2O2 in response to DAAO activation could be measured by ratiometric cell imaging. In vitro exposure of the myocytes to d-ala induced both inflammatory and antioxidant transcriptional responses (80).
After demonstrating that a viral vector could achieve expression and activation of DAAO in cardiac myocytes, the authors chronically activated the enzyme in vivo. d-ala was added to the water of control animals (expressing only HyPer) and of animals expressing the HyPer-DAAO construct (80). Given the more prominent role of l-amino acids in mammalian physiology, gastrointestinal transport ofd-amino acids has received far less study, but as discussed above, intestinal transporters responsible for amino acid uptake are relatively agnostic to amino acid chirality (81), an important feature facilitating in vivo chemogenetic approaches. Chronic (4-week) activation of DAAO in rat hearts induced systolic dysfunction and a dilated cardiomyopathy, demonstrating for the first time that increased oxidative stress is sufficient to cause heart failure. The systolic dysfunction was associated with a decrease in phospholamban phosphorylation at both the PKA and CamKII sites, reflecting decreased β-adrenergic flux (119–121). Consistent with a chronic shift toward an oxidative state, the availability of glutathione reducing power was diminished, while antioxidant transcriptional programs appeared to be activated.
After characterizing this new model of heart failure driven by the toxic effects of excess ROS production, Sorrentino et al. (84) investigated whether drugs used to treat heart failure could alter the deranged oxidant signaling and heart failure phenotype. Heart failure was induced in rats by intracardiac expression of DAAO and the addition of d-ala substrate in the drinking water, and then the rats were treated with either the combination drug sacubitril (a neprilysin inhibitor) (122) plus valsartan (an angiotensin receptor blocker) or valsartan alone. Both drugs reversed the systolic dysfunction caused by chronic DAAO activation. The authors also found that the degree of intracellular oxidation, as measured by 8-hydroxyguanosine (123), was reduced, suggesting that part of the mechanism of these drugs may be a restoration of redox state toward physiological levels. Additionally, chronic oxidative stress by activation of DAAO led to a shift in metabolic substrate preference to glucose, as measured by glucose uptake by 18F-FDG positron emission tomography, a change that was also reversed by drug treatment. These observations are similar to several other animal models of heart failure and are consistent with findings in cardiac tissues of human heart failure patients (124). The finding that chemogenetic heart failure is reversible following drug treatment may permit the identification of new therapeutic targets using this animal model. This new heart failure model helps to establish a proof of principle that chemogenetic approaches can be used to establish a pathological approach caused by oxidative stress and to then successfully reverse the phenotype with drug treatment. In so many disease states in which oxidative stress is implicated, association and causation are confounded by the complexity of disease pathogenesis. The application of chemogenetics permits the dynamic control of redox stress and can help to establish a causal role for ROS and facilitate the identification of new therapeutic targets.
CONCLUDING REMARKS
Chemogenetic approaches have provided important new insights into diverse pathways in normal cells and in disease states. DREADD-based chemogenetic approaches are now being expanded to study cardiovascular tissues and recently have identified new therapeutic targets of currently used drugs and have uncovered new pathways leading to arrhythmogenicity following activation of Gq-based GPCRs by a vasoactive drug. DAAO-based approaches show great promise both in permitting the dissection of intracellular oxidant pathways and in identifying the transition from physiological to toxic levels of ROS. In addition, the generation of informative new animal models of chemogenetic oxidative stress has the potential to aid in the identification of new therapeutic targets for heart failure. To this point, in vivo models based upon DAAO have been limited to virus (AAV)-based methods to target the recombinant enzyme to specific tissues. The availability of transgenic mouse lines expressing DAAO under the control of tissue-specific promoters should permit analyses of the diverse disease states in which oxidative stress has been implicated not only in the heart but also in the many other organ systems that have lacked tools to precisely manipulate subcellular redox states in vivo. These transgenic mouse models will not only facilitate the development and validation of novel therapeutic targets in diseases caused by pathological oxidative stress but also permit the study of oxidants that are involved in physiological redox eustress pathways.
SUMMARY POINTS.
Chemogenetic approaches based on d-amino acid oxidase (DAAO) have enabled the precise control of intracellular reactive oxygen species (ROS) levels and permit the analysis of both the toxic effects of excess ROS and physiological pathways that involve ROS-dependent signaling.
Chemogenetic generation of redox stress in the heart rapidly leads to cardiac dysfunction, which can be reversed by drug treatment. This new form of heart failure isolates oxidative stress as a single causative factor and can serve as a model for validating drugs that modulate redox pathways in heart failure and other disease states.
Subcellular targeting of DAAO enables the study of the role of subcellular ROS production in physiology and disease.
The DAAO-based model of oxidative stress can be expanded to other tissues through transgenic models with inducible expression of the DAAO enzyme, enabling the study of redox stress in the many other tissues where alterations in redox state have been associated with disease.
ACKNOWLEDGMENTS
T.M. acknowledges support from National Institutes of Health grants R21AG063073 and R01HL152173 and from the Brigham Research Institute Fund to Sustain Research Excellence. E.E. acknowledges support from the Scientific and Technological Research Council of Turkey (118C242) and Austrian Science Foundation grant FWF J4113.
DISCLOSURE STATEMENT
Some of the published research studies described in this review (e.g., Reference 84) were supported by an investigator-initiated grant from Novartis that was provided to the laboratory of T.M. The authors are not aware of any other affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Cui H, Kong Y, Zhang H. 2012. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal Transduct 2012:646354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leopold JA, Loscalzo J. 2005. Oxidative enzymopathies and vascular disease. Arterioscler. Thromb. Vasc. Biol 25:1332–40 [DOI] [PubMed] [Google Scholar]
- 3.Kattoor AJ, Pothineni NVK, Palagiri D, Mehta JL. 2017. Oxidative stress in atherosclerosis. Curr. Atheroscler. Rep 19(11):42. [DOI] [PubMed] [Google Scholar]
- 4.Forrester SJ, Booz GW, Sigmund CD, Coffman TM, Kawai T, et al. 2018. Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol. Rev 98(3):1627–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X, Wang W, Li L, Perry G, Lee H, Zhu X. 2014. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis 1842(8):1240–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Emini Veseli B, Perrotta P, De Meyer GRA, Roth L, Van der Donckt C, et al. 2017. Animal models of atherosclerosis. Eur. J. Pharmacol 816:3–13 [DOI] [PubMed] [Google Scholar]
- 7.Lindsey ML, Bolli R, Canty JM, Du X-J, Frangogiannis NG, et al. 2018. Guidelines for experimental models of myocardial ischemia and infarction. Am. J. Physiol. Circ. Physiol 314(4):H812–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Houser SR, Margulies KB, Murphy AM, Spinale FG, Francis GS, et al. 2012. Animal models of heart failure: a scientific statement from the American Heart Association. Circ. Res 111(1):131–50 [DOI] [PubMed] [Google Scholar]
- 9.Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, et al. 2019. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 568(7752):351–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cardinale D, Iacopo F, Cipolla CM. 2020. Cardiotoxicity of anthracyclines. Front. Cardiovasc. Med 7:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jenkins DJA, Kitts D, Giovannucci EL, Sahye-Pudaruth S, Paquette M, et al. 2020. Selenium, antioxidants, cardiovascular disease, and all-cause mortality: a systematic review and meta-analysis of randomized controlled trials. Am. J. Clin. Nutr 112(6):1642–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bjelakovic G, Nikolova D, Gluud C. 2013. Antioxidant supplements to prevent mortality. JAMA 310(11):1178–79 [DOI] [PubMed] [Google Scholar]
- 13.Sies H. 2017. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: oxidative eustress. Redox. Biol 11:613–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nathan C, Cunningham-Bussel A. 2013. Beyond oxidative stress: an immunologist’s. Nat. Rev. Immunol 13(5):349–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waypa GB, Marks JD, Guzy RD, Mungai PT, Schriewer JM, et al. 2013. Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. Am. J. Respir. Crit. Care Med 187(4):424–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, et al. 2019. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 565:495–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalwa H, Sartoretto JL, Martinelli R, Romero N, Steinhorn BS, et al. 2014. Central role for hydrogen peroxide in P2Y1 ADP receptor-mediated cellular responses in vascular endothelium. PNAS 111(9):3383–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sartoretto JL, Kalwa H, Shiroto T, Sartoretto SM, Pluth MD, et al. 2012. Role of Ca2+ in the control of H2O2-modulated phosphorylation pathways leading to eNOS activation in cardiac myocytes. PLOS ONE 7(9):e44627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sartoretto JL, Kalwa H, Pluth MD, Lippard SJ, Michel T, et al. 2011. Hydrogen peroxide differentially modulates cardiac myocyte nitric oxide synthesis. PNAS 108(38):15792–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsieh H-J, Liu C-A, Huang B, Tseng AH, Wang DL. 2014. Shear-induced endothelial mechanotransduction: the interplay between reactive oxygen species (ROS) and nitric oxide (NO) and the pathophysiological implications. J. Biomed. Sci 21:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang ZM, Gao E, Fonseca F, Hayashi H, Shang X, et al. 2013. Convergence of G protein-coupled receptor and nitric oxide pathways determines the outcome to cardiac ischemic injury. Sci. Signal 6(299):ra95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jian Z, Han H, Zhang T, Puglisi J, Izu LT, et al. 2014. Mechanochemotransduction during cardiomyocyte contraction is mediated by localized nitric oxide signaling. Sci. Signal 7(317):ra27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steinhorn B, Sartoretto JL, Sorrentino A, Romero N, Kalwa H, et al. 2017. Insulin-dependent metabolic and inotropic responses in the heart are modulated by hydrogen peroxide from NADPH-oxidase isoforms NOX2 and NOX4. Free Radic. Biol. Med 113:16–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldstein BJ, Mahadev K, Wu X, Zhu L, Motoshima H. 2005. Role of insulin-induced reactive oxygen species in the insulin signaling pathway. Antioxid. Redox Signal 7(7–8):1021–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sies H. 2014. Role of metabolic H2O2 generation: redox signaling and oxidative stress. J. Biol. Chem 289(13):8735–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ushio-Fukai M. 2009. Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxid. Redox Signal 11(6):1289–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Netto LES, Antunes F. 2016. The roles of peroxiredoxin and thioredoxin in hydrogen peroxide sensing and in signal transduction. Mol. Cells 39(1):65–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferguson GD, Bridge WJ. 2019. The glutathione system and the related thiol network in Caenorhabditis elegans. Redox Biol. 24:101171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maron BA, Michel T. 2012. Subcellular localization of oxidants and redox modulation of endothelial nitric oxide synthase. Circ. J 76(11):2497–512 [DOI] [PubMed] [Google Scholar]
- 30.Bedard K, Krause K-H. 2007. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev 87(1):245–313 [DOI] [PubMed] [Google Scholar]
- 31.Li J-M. 2002. Intracellular localization and preassembly of the NADPH oxidase complex in cultured endothelial cells. J. Biol. Chem 277(22):19952–60 [DOI] [PubMed] [Google Scholar]
- 32.Saravi SSS, Eroglu E, Waldeck-Weiermair M, Sorrentino A, Steinhorn B, et al. 2020. Differential endothelial signaling responses elicited by chemogenetic H2O2 synthesis. Redox Biol. 36:101605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Montiel V, Bella R, Michel LYM, Esfahani H, De Mulder D, et al. 2020. Inhibition of aquaporin-1 prevents myocardial remodeling by blocking the transmembrane transport of hydrogen peroxide. Sci. Transl. Med 12(564):eaay2176. [DOI] [PubMed] [Google Scholar]
- 34.Granger DN, Kvietys PR. 2015. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol. 6:524–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Furuhashi M. 2020. New insights into purine metabolism in metabolic diseases: role of xanthine oxidoreductase activity. Am. J. Physiol. Endocrinol. Metab 319(5):E827–34 [DOI] [PubMed] [Google Scholar]
- 36.Kaludercic N, Mialet-Perez J, Paolocci N, Parini A, Di Lisa F. 2014. Monoamine oxidases as sources of oxidants in the heart. J. Mol. Cell. Cardiol 73:34–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holmström KM, Finkel T. 2014. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol 15(6):411–21 [DOI] [PubMed] [Google Scholar]
- 38.Finkel T. 2011. Signal transduction by reactive oxygen species. J. Cell Biol 194(1):7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murphy MP. 2009. How mitochondria produce reactive oxygen species. Biochem. J 417(1):1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oestreicher J, Morgan B. 2019. Glutathione: subcellular distribution and membrane transport. Biochem. Cell Biol 97(3):270–89 [DOI] [PubMed] [Google Scholar]
- 41.Yoon E, Babar A, Choudhary M, Kutner M, Pyrsopoulos N. 2016. Acetaminophen-induced hepatotoxicity: a comprehensive update. J. Clin. Transl. Hepatol 4(2):131–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fomenko DE, Koc A, Agisheva N, Jacobsen M, Kaya A, et al. 2011. Thiol peroxidases mediate specific genome-wide regulation of gene expression in response to hydrogen peroxide. PNAS 108(7):2729–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kil IS, Lee SK, Ryu KW,Woo HA, Hu MC, et al. 2012. Feedback control of adrenal steroidogenesis via H2O2-dependent, reversible inactivation of peroxiredoxin III in mitochondria. Mol. Cell 46(5):584–94 [DOI] [PubMed] [Google Scholar]
- 44.Van Laer K, Dick TP. 2016. Utilizing natural and engineered peroxiredoxins as intracellular peroxide reporters. Mol. Cells 39(1):46–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang J, Duan D, Osama A, Fang J. 2021. Natural molecules targeting thioredoxin system and their therapeutic potentials. Antioxid. Redox Signal 34(14):1083–107 [DOI] [PubMed] [Google Scholar]
- 46.Fan Y, Makar M, Wang MX, Ai H-W. 2017. Monitoring thioredoxin redox with a genetically encoded red fluorescent biosensor. Nat. Chem. Biol 13(9):1045–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lo Conte M, Carroll KS. 2013. The redox biochemistry of protein sulfenylation and sulfinylation. J. Biol. Chem 288(37):26480–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qin F, Rounds NK, Mao W, Kawai K, Liang CS. 2001. Antioxidant vitamins prevent cardiomyocyte apoptosis produced by norepinephrine infusion in ferrets. Cardiovasc. Res 51(4):736–48 [DOI] [PubMed] [Google Scholar]
- 49.Zimmer HG. Regulation of and intervention into the oxidative pentose phosphate pathway and adenine nucleotide metabolism in the heart. Mol. Cell. Biochem 160–161:101–9 [DOI] [PubMed] [Google Scholar]
- 50.Ashoka AH, Ali F, Tiwari R, Kumari R, Pramanik SK, Das A. 2020. Recent advances in fluorescent probes for detection of HOCl and HNO. ACS Omega 5(4):1730–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang X, Wang L, Carroll SL, Chen J, Wang MC, Wang J. 2018. Challenges and opportunities for small-molecule fluorescent probes in redox biology applications. Antioxid. Redox Signal 29(6):518–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Winterbourn CC. 2014. The challenges of using fluorescent probes to detect and quantify specific reactive oxygen species in living cells. Biochim. Biophys. Acta 1840(2):730–38 [DOI] [PubMed] [Google Scholar]
- 53.Zhu H, Fan J, Du J, Peng X. 2016. Fluorescent probes for sensing and imaging within specific cellular organelles. Acc. Chem. Res 49(10):2115–26 [DOI] [PubMed] [Google Scholar]
- 54.Fujikawa Y, Roma LP, Sobotta MC, Rose AJ, Diaz MB, et al. 2016.Mouse redox histology using genetically encoded probes. Sci. Signal 9(419):rs1. [DOI] [PubMed] [Google Scholar]
- 55.Bilan DS, Belousov VV 2016. HyPer family probes: state of the art. Antioxid. Redox Signal 24(13):731–51 [DOI] [PubMed] [Google Scholar]
- 56.Braissant O, McLin VA, Cudalbu C. 2013. Ammonia toxicity to the brain. J. Inherit. Metab. Dis 36(4):595–612 [DOI] [PubMed] [Google Scholar]
- 57.Belousov VV, Fradkov AF, Lukyanov KA, Staroverov DB, Shakhbazov KS, et al. 2006. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat. Methods 3(4):281–86 [DOI] [PubMed] [Google Scholar]
- 58.Bilan DS, Belousov VV 2016. New tools for redox biology: from imaging to manipulation. Free Radic. Biol. Med 109:167–88 [DOI] [PubMed] [Google Scholar]
- 59.Matlashov ME, Bogdanova YA, Ermakova GV, Mishina NM, Ermakova YG, et al. 2015. Fluorescent ratiometric pH indicator SypHer2: applications in neuroscience and regenerative biology. Biochim. Biophys. Acta 1850(11):2318–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pak VV, Ezerina D, Lyublinskaya OG, Pedre B, Tyurin-Kuzmin PA, et al. 2020. Ultrasensitive genetically encoded indicator for hydrogen peroxide identifies roles for the oxidant in cell migration and mitochondrial function. Cell Metab. 31(3):642–53.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Steinhorn B, Sartoretto JL, Sorrentino A, Romero N, Kalwa H, et al. 2017. Insulin-dependent metabolic and inotropic responses in the heart are modulated by hydrogen peroxide from NADPH-oxidase isoforms NOX2 and NOX4. Free Radic. Biol. Med. 113(August):16–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Waypa GB, Marks JD, Guzy RD, Mungai PT, Schriewer JM, et al. 2013. Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. Am. J. Respir. Crit. Care Med 187(4):424–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Waypa GB, Marks JD, Guzy R, Mungai PT, Schriewer J, et al. 2010. Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ. Res 106(3):526–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ezerina D, Morgan B, Dick TP. 2014. Imaging dynamic redox processes with genetically encoded probes. J. Mol. Cell. Cardiol 73:43–49 [DOI] [PubMed] [Google Scholar]
- 65.Zou Y, Wang A, Shi M, Chen X, Liu R, et al. 2018. Analysis of redox landscapes and dynamics in living cells and in vivo using genetically encoded fluorescent sensors. Nat. Protoc 13(10):2362–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hung YP, Albeck JG, Tantama M, Yellen G. 2011. Imaging cytosolic NADH-NAD+ redox state with a genetically encoded fluorescent biosensor. Cell Metab. 14(4):545–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bubb KJ, Drummond GR, Figtree GA. 2020. New opportunities for targeting redox dysregulation in cardiovascular disease. Cardiovasc. Res 116(3):532–44 [DOI] [PubMed] [Google Scholar]
- 68.Waypa GB, Smith KA, Schumacker PT. 2016. O2 sensing, mitochondria and ROS signaling: The fog is lifting. Mol. Aspects Med 47–48:76–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Santolini J, Wootton SA, Jackson AA, Feelisch M. 2019. The redox architecture of physiological function. Curr. Opin. Physiol 9:34–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stefanska J, Pawliczak R. 2008. Apocynin: molecular aptitudes. Mediators Inflamm. 2008:106507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang M, Brewer AC, Schroder K, Santos CXC, Grieve DJ, et al. 2010. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. PNAS 107(42):18121–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang M, Prosser BL, Bamboye MA, Gondim ANS, Santos CX, et al. 2015. Contractile function during angiotensin-II activation: Increased Nox2 activity modulates cardiac calcium handling via phospholamban phosphorylation. J. Am. Coll. Cardiol 66(3):261–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rapti K, Diokmetzidou A, Kloukina I, Milner DJ, Varela A, et al. 2017. Opposite effects of catalase and MnSOD ectopic expression on stress induced defects and mortality in the desmin deficient cardiomyopathy model. Free Radic. Biol. Med 110(June):206–18 [DOI] [PubMed] [Google Scholar]
- 74.Leskovac V, Trivić S, Wohlfahrt G, Kandrac J, Pericin D. 2005. Glucose oxidase from Aspergillus niger: the mechanism of action with molecular oxygen, quinones, and one-electron acceptors. Int. J. Biochem. Cell Biol 37(4):731–50 [DOI] [PubMed] [Google Scholar]
- 75.Vlasov K, Van Dort CJ, Solt K. 2018. Optogenetics and chemogenetics. In Chemical and Biochemical Approaches for the Study of Anesthetic Function Part B, ed. Eckenhoff RG, Dmochowski IJ. 603:181–96. Cambridge, MA: Academic; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim CK, Adhikari A, Deisseroth K. 2017. Integration of optogenetics with complementary methodologies in systems neuroscience. Nat. Rev. Neurosci 18(4):222–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Urban DJ, Roth BL. 2015. DREADDs (designer receptors exclusively activated by designer drugs): chemogenetic tools with therapeutic utility. Annu. Rev. Pharmacol. Toxicol 55:399–417 [DOI] [PubMed] [Google Scholar]
- 78.Wang L, Zhu L, Meister J, Bone DBJ, Pydi SP, et al. 2021. Use of DREADD technology to identify novel targets for antidiabetic drugs. Annu. Rev. Pharmacol. Toxicol 61:421–40 [DOI] [PubMed] [Google Scholar]
- 79.Dimidschstein J, Chen Q, Tremblay R, Rogers SL, Saldi G-A, et al. 2016. A viral strategy for targeting and manipulating interneurons across vertebrate species. Nat. Neurosci 19(12):1743–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Steinhorn B, Sorrentino A, Badole S, Bogdanova Y, Belousov V, Michel T. 2018. Chemogenetic generation of hydrogen peroxide in the heart induces severe cardiac dysfunction. Nat. Commun 9(1):4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sahoo S, Aurich MK, Jonsson JJ, Thiele I. 2014. Membrane transporters in a human genome-scale metabolic knowledgebase and their implications for disease. Front. Physiol 5:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sorrentino A, Michel T. 2020. Redox à la carte: novel chemogenetic models of heart failure. Br. J. Pharmacol 177(14):3162–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Manvich DF, Webster KA, Foster SL, Farrell MS, Ritchie JC, et al. 2018.The DREADD agonist clozapine N-oxide (CNO) is reverse-metabolized to clozapine and produces clozapine-like interoceptive stimulus effects in rats and mice. Sci. Rep 8(1):3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sorrentino A, Steinhorn B, Troncone L, Saravi SSS, Badole S, et al. 2019. Reversal of heart failure in a chemogenetic model of persistent cardiac redox stress. Am.J. Physiol. Heart Circ. Physiol 317(3):H617–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Magnus CJ, Lee PH, Bonaventura J, Zemla R, Gomez JL, et al. 2019. Ultrapotent chemogenetics for research and potential clinical applications. Science 364(6436):eaav5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kaiser E, Tian Q, Wagner M, Barth M, Xian W, et al. 2019. DREADD technology reveals major impact of Gq signalling on cardiac electrophysiology. Cardiovasc. Res 115(6):1052–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ferrantini C, Coppini R, Sacconi L. 2019. Cardiomyocyte-specific Gq signalling and arrhythmias: novel insights from DREADD technology. Cardiovasc. Res 115(6):992–94 [DOI] [PubMed] [Google Scholar]
- 88.Krebs HA. 1935. Metabolism of amino-acids: deamination of amino-acids. Biochem. J 29(7):1620–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pollegioni L, Sacchi S, Murtas G. 2018. Human D-amino acid oxidase: structure, function, and regulation. Front. Mol. Biosci 5(November):107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu Y-L, Wang S-C, Hwu H-G, Fann CS- J, Yang U-C, et al. 2016. Haplotypes of the D-amino acid oxidase gene are significantly associated with schizophrenia and its neurocognitive deficits. PLOS ONE 11(3):e0150435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Abou El-Magd RM, Park HK, Kawazoe T, Iwana S, Ono K, et al. 2010. The effect of risperidone on D-amino acid oxidase activity as a hypothesis for a novel mechanism of action in the treatment of schizophrenia. J. Psychopharmacol 24(7):1055–67 [DOI] [PubMed] [Google Scholar]
- 92.Sasabe J, Miyoshi Y, Rakoff-Nahoum S, Zhang T, Mita M, et al. 2016. Interplay between microbial d-amino acids and host d-amino acid oxidase modifies murine mucosal defence and gut microbiota. Nat. Microbiol 1(July):16125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nagano T, Yamao S, Terachi A, Yarimizu H, Itoh H, et al. 2019. d-amino acid oxidase promotes cellular senescence via the production of reactive oxygen species. Life Sci. Alliance 2(1):e201800045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pollegioni L, Langkau B, Tischer W, Ghisla S, Pilone MS. 1993. Kinetic mechanism of d-amino acid oxidases from Rhodotorula gracilis and Trigonopsis variabilis. J. Biol. Chem 268(19):13850–57 [PubMed] [Google Scholar]
- 95.Sacchi S, Lorenzi S, Molla G, Pilone MS, Rossetti C, Pollegioni L. 2002. Engineering the substrate specificity of d-amino-acid oxidase. J. Biol. Chem 277(30):27510–16 [DOI] [PubMed] [Google Scholar]
- 96.Matlashov ME, Belousov VV, Enikolopov G. 2014. How much H2O2 is produced by recombinant D-amino acid oxidase in mammalian cells? Antioxid. Redox Signal 20(7):1039–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rosini E, Pollegioni L, Ghisla S, Orru R, Molla G. 2009. Optimization of D-amino acid oxidase for low substrate concentrations—towards a cancer enzyme therapy. FEBS J 276(17):4921–32 [DOI] [PubMed] [Google Scholar]
- 98.Pollegioni L, Molla G. 2011. New biotech applications from evolved D-amino acid oxidases. Trends Biotechnol. 29(6):276–83 [DOI] [PubMed] [Google Scholar]
- 99.Chai Q, Lu T, Wang XL, Lee HC. 2014. Hydrogen sulfide impairs shear stress-induced vasodilation in mouse coronary arteries. Pflugers Arch. 467(2):329–40 [DOI] [PubMed] [Google Scholar]
- 100.Zhang H, Bai Z, Zhu L, Liang Y, Fan X, et al. 2020. Hydrogen sulfide donors: therapeutic potential in anti-atherosclerosis. Eur.J. Med. Chem 205:112665. [DOI] [PubMed] [Google Scholar]
- 101.Cheng Z, Kishore R. 2020. Potential role of hydrogen sulfide in diabetes-impaired angiogenesis and ischemic tissue repair. Redox Biol. 37:101704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ngowi EE, Sarfraz M, Afzal A, Khan NH, Khattak S, et al. 2020. Roles of hydrogen sulfide donors in common kidney diseases. Front. Pharmacol 11:1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ngowi EE, Afzal A, Sarfraz M, Khattak S, Zaman SU, et al. 2021. Role of hydrogen sulfide donors in cancer development and progression. Int. J. Biol. Sci 17:73–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.San Martín A, Ceballo S, Baeza-Lehnert F, Lerchundi R, Valdebenito R, et al. 2014. Imaging mitochondrial flux in single cells with a FRET sensor for pyruvate. PLOS ONE 9(1):e85780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Merhi A, Delree P, Marini AM. 2017. The metabolic waste ammonium regulates mTORC2 and mTORC1 signaling. Sci. Rep 7:44602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Skowrońska M, Albrecht J. 2013. Oxidative and nitrosative stress in ammonia neurotoxicity. Neurochem. Int 62(5):731–37 [DOI] [PubMed] [Google Scholar]
- 107.Adeva-Andany M, López-Ojén M, Funcasta-Calderón R, Ameneiros-Rodríguez E, Donapetry-García C, et al. 2014. Comprehensive review on lactate metabolism in human health. Mitochondrion 17:76–100 [DOI] [PubMed] [Google Scholar]
- 108.Huang BK, Stein KT, Sikes HD. 2016. Modulating and measuring intracellular H2O2 using genetically encoded tools to study its toxicity to human cells. ACS Synth. Biol 5(12):1389–95 [DOI] [PubMed] [Google Scholar]
- 109.Shibuya N, Kimura H.2013. Production of hydrogen sulfide from d-cysteine and its therapeutic potential. Front. Endocrinol 4:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kanagy NL, Szabo C, Papapetropoulos A. 2017. Vascular biology of hydrogen sulfide. Am. J. Physiol. Cell Physiol 312(5):C537–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bełtowski J 2019. Synthesis, metabolism, and signaling mechanisms of hydrogen sulfide: an overview. In Vascular Effects of Hydrogen Sulfide: Methods and Protocols, ed. Bełtowski J, pp. 1–8. New York: Humana; [DOI] [PubMed] [Google Scholar]
- 112.Huang BK, Sikes HD. 2014. Quantifying intracellular hydrogen peroxide perturbations in terms of concentration. Redox Biol. 2(1):955–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Huang BK, Stein KT, Sikes HD. 2016. Modulating and measuring intracellular H2O2 using genetically encoded tools to study its toxicity to human cells. ACS Synth. Biol 5(12):1389–95 [DOI] [PubMed] [Google Scholar]
- 114.Stein KT, Moon SJ, Sikes HD. 2018. Mitochondrial H2O2 generation using a tunable chemogenetic tool to perturb redox homeostasis in human cells and induce cell death. ACS Synth. Biol 7(9):2037–44 [DOI] [PubMed] [Google Scholar]
- 115.Mishina NM, Bogdanova YA, Ermakova YG, Panova AS, Kotova DA, et al. 2019. Which antioxidant system shapes intracellular H2O2 gradients? Antioxid. Redox Signal 31(9):664–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Eroglu E, Saravi SSS, Sorrentino A, Steinhorn B, Michel T. 2019. Discordance between eNOS phosphorylation and activation revealed by multispectral imaging and chemogenetic methods. PNAS 116(40):20210–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Samulski RJ, Muzyczka N. 2014. AAV-mediated gene therapy for research and therapeutic purposes. Annu. Rev. Virol 1:427–51 [DOI] [PubMed] [Google Scholar]
- 118.Grimm D, Lee JS, Wang L, Desai T, Akache B, et al. 2008.In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J. Virol 82(12):5887–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Grimm M, Brown JH. 2010. β-Adrenergic receptor signaling in the heart: role of CaMKII. J. Mol. Cell. Cardiol 48(2):322–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mattiazzi A, Mundina-Weilenmann C, Guoxiang C, Vittone L, Kranias E. 2005. Role of phospholamban phosphorylation on Thr17 in cardiac physiological and pathological conditions. Cardiovasc. Res 68(3):366–75 [DOI] [PubMed] [Google Scholar]
- 121.Sulakhe PV, Vo XT. 1995. Regulation of phospholamban and troponin-I phosphorylation in the intact rat cardiomyocytes by adrenergic and cholinergic stimuli: roles of cyclic nucleotides, calcium, protein kinases and phosphatases and depolarization. Mol. Cell. Biochem 149–150:103–26 [DOI] [PubMed] [Google Scholar]
- 122.McMurray JJV, Packer M, Desai AS, Gong J, Lefkowitz MP, et al. 2014. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med 371(11):993–1004 [DOI] [PubMed] [Google Scholar]
- 123.Kong Q, Lin CG. 2010. Oxidative damage to RNA: mechanisms, consequences, and diseases. Cell. Mol. Life Sci. 67(11):1817–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ziaeian B, Fonarow GC. 2016. Epidemiology and aetiology of heart failure. Nat. Rev. Cardiol 13(6):368–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lerman LO, Kurtz TW,Touyz RM, Ellison DH, Chade AR, et al. 2019. Animal models of hypertension: a scientific statement from the American Heart Association. Hypertension 73(6):e87–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bugger H, Abel ED. 2009. Rodent models of diabetic cardiomyopathy. Dis. Model. Mech 2(9–10):454–66 [DOI] [PubMed] [Google Scholar]