

Abstract
Sepsis is a complex disease due to dysregulated host response to infection. Oxidative stress and mitochondrial dysfunction leading to metabolic dysregulation are among the hallmarks of sepsis. The transcription factor NRF2 (Nuclear Factor E2-related faetor2) is a master regulator of the oxidative stress response, and the NRF2 mediated antioxidant response is negatively regulated by BTB and CNC homology 1 (BACH1) protein. This study tested whether Bach1 deletion improves organ function and survival following polymicrobial sepsis induced by cecal ligation and puncture (CLP). We observed enhanced post-CLP survival in Bach1−/− mice with a concomitantly increased liver HO-1 expression, reduced liver injury and oxidative stress, and attenuated systemic and tissue inflammation. After sepsis induction, the liver mitochondrial function was better preserved in Bach1−/− mice. Furthermore, BACH1 deficiency improved liver and lung blood flow in septic mice, as measured by SPECT/CT. RNA-seq analysis identified 44 genes significantly altered in Bach1−/− mice after sepsis, including HMOX1 and several genes in lipid metabolism. Inhibiting HO-1 activity by Zinc Protoporphyrin-9 worsened organ function in Bach1−/− mice following sepsis. We demonstrate that mitochondrial bioenergetics, organ function, and survival following experimental sepsis were improved in Bach1−/− mice through the HO-1-dependent mechanism and conclude that BACH1 is a therapeutic target in sepsis.
Keywords: Mitochondria, Cellular energetics, Sepsis, Oxidative stress, Antioxidant, NRF2, BACH1
1. Introduction
Sepsis affects over 48 million people worldwide, resulting in the death of about 11 million people annually [1]. Sepsis is a complex disease due to dysregulated host response to infection characterized by an initial hyperinflammatory phase followed by a sustained immunosuppressive phase, referred to as immunoparalysis, resulting in multi-organ dysfunction and death [2,3]. A decline in mitochondrial function and enhanced oxidative stress are also hallmarks of sepsis. Oxidative stress, immune cell function, and cellular energetics are closely interlinked in the metabolic derangement, and organ dysfunction observed in sepsis [4,5].
The transcription factor NRF2 (Nuclear Factor E2-related factor 2) plays a significant role as a master regulator of the oxidative stress response. Mitochondria and NADPH oxidases are the primary sources of reactive oxygen species (ROS) [6]. Several studies have demonstrated a protective role for NRF2 in endotoxemia and CLP-induced sepsis, leading to immune response modulation and improved survival [7–9]. NRF2 was also a critical regulator of innate memory in sepsis [4]. During basal conditions, NRF2 is associated with KEAP1, a substrate adaptor for the Cullin 3 ubiquitin ligase [10]. KEAP1 association leads to ubiquitination and proteasomal degradation of NRF2, thereby controlling its stability and cytoplasmic accumulation [11]. Disruption of Keap1 in myeloid leukocytes was protective in an experimental model of sepsis [12]. When cells are exposed to electrophilic or oxidative stress, ubiquitination of NRF2 is blocked, resulting in increased cytoplasmic levels of NRF2 and its translocation to the nucleus [10]. NRF2 associates with small MAF proteins in the nucleus, binds antioxidant response elements (ARE) on cytoprotective genes, and promotes transcription activity.
The transcription factor BTB and CNC homology 1 (BACH1) is a repressor of NRF2 function and has been shown to negatively regulate NRF2 binding to ARE sequences. BACH1 heterodimerizes with small MAF proteins and competes with NRF2 binding to overlapping domains of MARE and ARE, which are now collectively referred to as CNC-sMAF binding element [13–16]. Therefore, the ARE gene transcription is determined by the balance between BACH1 and NRF2 competition for the regulatory gene segment [17]. Bach1 gene deletion upregulates the expression of heme oxygenase −1 (HMOX-1, protein: HO-1), an NRF2 target gene, in several tissues [13]. There are two isoforms of heme oxygenase, HO-1 (inducible) and HO-2 (constitutive). They catalyze the conversion of pro-oxidant heme to biliverdin, carbon monoxide, and ferrous iron, which are antioxidants, anti-inflammatory, and anti-apoptotic [18]. The induction of HO-1 is a cytoprotective mechanism in response to cell stress, and HO-1 deficiency promotes the progression -of sepsis [18,19]. Furthermore, mice deficient in BACH1 were protected against left ventricular hypertrophy, atherosclerotic plaque, TNBS-induced colitis, LPS-induced liver injury, and oxidative stress in pancreatic beta cells [20–25]. In this study, we sought to determine the role of BACH1 in the outcome following sepsis and demonstrate that Bach1 deficiency profoundly affects cellular energetics and survival in a cecal ligation and puncture (CLP) model of sepsis in an HO-1 dependent manner.
2. Materials and methods
2.1. Animals
Male C57BL/6J mice (10–14 weeks old) were purchased from the Jackson Laboratory, USA. Heterozygous Bach1 knockout (Bach1+/−) mice were as described previously [26]. Bach1−/− male mice (10–14 weeks old) on the C57BL/6J background (backcrossed for more than 12 generations) were bred in the Augusta University animal facility. All experiments were approved by the Augusta University Institutional Animal Care and Use Committee (IACUC).
2.2. Sepsis model
The sepsis model in the mouse was performed using a well-established CLP procedure [27,28]. Following anesthesia, the mice were given a subcutaneous injection of 2 mg/kg carprofen sodium, the abdomen was disinfected, and a midline incision was made. The cecum was exposed, ligated below the ileocecal valve, and punctured once across the cecum using a 22-gauge needle. A small amount of fecal matter was gently squeezed out through the puncture sites. The cecum was placed back into the peritoneal cavity, and the abdominal wall was then closed. In the sham group, the mice underwent laparotomy and manipulation of the bowel, but ligation and perforation of the cecum were not performed. Following the procedures, the mice were resuscitated using normal saline (1 ml via subcutaneous injection). The mice were administered another dose of carprofen 24 h after the surgery. A subset of mice was euthanized at 24 h following CLP or sham surgery for molecular studies. SPECT imaging for tissue perfusion testing was done on day three, and the animals were euthanized immediately after the study. The animals in the survival study were monitored for up to 10 days, daily weight noted, and euthanized on day ten following the surgery. Zinc protoporphyrin-IX Treatment: Zinc protoporphyrin-IX (ZnPP, HO-1 inhibitor) (Sigma-Aldrich, St. Louis, MO) was dissolved in dimethyl sulfoxide (DMSO) and diluted with saline to a concentration of 50 mg/ml before injection. Bach1−/− mice were randomly divided into two groups: vehicle-treated CLP and ZnPP-treated CLP. Mice were intraperitoneally (i.p.) administered with vehicle or ZnPP (15 mg/kg) 1 h before the surgery [29,30]. Liver and plasma were collected 24 h after the CLP.
2.3. Mitochondria respiration
The oxygen consumption rate (OCR) of isolated liver mitochondria was measured using a Seahorse Extracellular Flux analyzer (Agilent, Boston, MA) [31,32]. Mitochondria were isolated using Mitochondria Isolation Buffer (Biovision) [33]. For OCR measurements of isolated liver mitochondria, the mitochondria were diluted to 4μg/25 μl in cold MAS buffer + substrate (pyruvate/malate) and plated in 25 μl per well. Wells without mitochondria were used for background correction. The plate was centrifuged to attach the mitochondria. After centrifugation, an additional 155 μl of MAS buffer + substrate was added to the wells. Mitochondria were checked under the microscope to ensure a homogeneous mitochondrial monolayer in each well and incubated at 37 °C for 10 min. OCR was measured as per manufacturer instructions. Three baseline measurements were obtained before any injection and three response measurements after each injection of ADP (4 mM), oligomycin (3 μM), FCCP (4 μM), and antimycin A (4 μM). OCR was calculated by the Seahorse Wave desktop software package (Agilent, Boston, MA). TMRE staining: Isolated mitochondria were stained with 200 nM TMRE (Sigma) for 15 min along with a baseline control pretreated with 10 μM FCCP for 10 min. Fluorescence intensity was read at Ex/Em = 549/575, and TMRE fluorescence was normalized to the control.
2.4. Liver hematoxylin and eosin (H&E) staining
Liver tissue was fixed in 4% paraformaldehyde and embedded in paraffin before sections were prepared. The sections were stained with H&E using standard methods.
2.5. Liver injury markers
To evaluate the liver injury in the mice after CLP or sham procedure, the levels of ALT and AST in the plasma were measured using the respective assay kits according to the manufacturer’s protocols (Abcam, Waltham, MA).
2.6. Inflammatory cytokine levels
The IL-1α, IL-6, MCP-1, IFN-γ, IL-17α, TNFα levels were measured in the mouse plasma 24 h after CLP by multiplex cytometric bead array (BD Biosciences, Franklin Lakes, NJ) according to the manufacturer’s instructions.
2.7. Measurement of plasma MDA and H2O2
Malondialdehyde (MDA) levels were determined in the plasma using TBARS (TCA Method) Assay Kit (Cayman Chemical, Ann Arbor, MI), and H2O2 levels in the plasma were measured using the Amplex Red hydrogen peroxide/peroxidase assay kit (Invitrogen, Carlsbad, CA) as per the manufacturer’s instructions.
2.8. Determination of myeloperoxidase (MPO) activity in liver tissue
According to the manufacturer’s instructions, MPO activity in the liver tissues of the mice was measured using an MPO Assay kit (Biovision, Milpitas, CA).
2.9. Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
RT-qPCR for TNF-α, IL-6, and MCP-1 in the liver tissues was performed as previously reported [34]. RNA was isolated by TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc., CA) and reverse-transcribed using PrimeScriptTM RT Master Mix (Takara Bio, Inc.). The sequences of the primers used for qPCR were as follows: TNF-α forward, 5′-TGC TGG GAA GCC TAA AAG G-3′ and reverse, 5′-CGA ATT TTG AGA AGA TGA TCC TG-3’; IL-6 forward, 5′-CCGGAGAGGAGACTTCACAG-3′ and reverse, 5′- TTCTGCAAGTGCATCATCGT-3’; MCP-1 forward,5′- CCC ACT CAC CTG CTG CTA CT -3′ and reverse, 5′- TCT GGA CCC ATT CCT TCT TG-3′, and β - actin forward, 5′ CGCCACCACTTCGCCATGGA 3′ and reverse, 5′ TACAGCCCGGGGAGCATCG 3’. Expression levels were normalized to β-actin. qPCR was performed using a Real-Time PCR instrument (AriaMx, Agilent Technologies) with SYBR Advantage qPCR mix. The thermocycling conditions were as follows: Initial denaturation for 30 s at 95 °C, followed by 40 cycles of 10 s at 95 °C, and 30 s at 60 °C. Relative mRNA expression was calculated using the 2-ΔΔCq method.
2.10. Western blotting
The tissues were homogenized in cold RIPA lysis buffer (Fisher Scientific, CA) with protease inhibitor (PIA32959; Fisher). Supernatants were collected, and protein concentrations determined using the DC Protein Assay kit (Biorad, Hercules, CA). The total lysates (30 μg protein/lane) were resolved in sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and the separated proteins were transferred to polyvinylidene difluoride (PVDF) membranes. The PVDF membranes were blocked with 5% nonfat milk before incubation with primary antibodies overnight at 4 °C. The membranes were washed three times, 5 min each, then incubated with appropriate HRP-conjugated secondary antibodies (Abcam, Waltham, MA) at room temperature for 1 h. Protein bands were detected with an ECL Western Blot Detection Kit (Thermo Fisher Scientific, Waltham, MA).
2.11. RNA-seq and data analysis
The RNA library preparation and RNA sequencing were performed in the laboratories of Novogene (San Diego, CA). The raw gene read count was used for differential expression analysis. A negative binomial model utilizing the DESeq2 package in R was used to compare expression differences between the control and knockout groups. The p-values were adjusted with the Benjamini-Hochberg method, and the significance level of α = 0.05 was used for the significance threshold. The enrichment analysis on gene ontology terms associated with significant differentially expressed genes was performed using the limma R package.
2.12. SPECT/CT imaging
Single-photon emission computed tomography (SPECT) images were acquired on a NanoScan™ (Mediso, Budapest, Hungary) using technetium-99m (Tc-99m) tagged red blood cells (RBC). Mice (n = 3 per group) were tested three days after surgery at the small animal imaging core at the Georgia Cancer Center at Augusta University. Tc-99m with radiochemical purity and specific activity greater than 99% and 55 GBq/μmol, respectively, was used. A pyrophosphate kit (Cardinal Health) was used to label RBC using in vivo labeling technique as per the manufacturer protocol. In short, one vial of pyrophosphate was dissolved in 10 ml of normal saline. Five minutes after dissolving pyrophosphate, 100 μl of the solution was IV administered to the mouse through the tail vein. After 20 min, 0.5 mCi of Tc-99m was administered to the mice through tail vein to tag RBC with Tc-99m. Thirty minutes after tagging RBC with Tc-99m, animals underwent SPECT-CT scanning. After the mice were placed in a prone position on the bed of an animal SPECT/CT scanner (NanoScan™, Mediso, Budapest, Hungary) under anesthesia with 2% isoflurane, whole body CT images were acquired for anatomical demarcation. Then SPECT images were acquired using frame mode in 60 projections (30 s/projection) using a four-head scanner fitted with high-resolution mouse collimators. The energy window was set at 140 keV ±15%.
Multiplanar SPECT-CT images were reconstructed for display using manufacturer-supplied software (Nucline, Mediso). For semi-quantitative analysis, we used ImageJ (NIH, USA). All SPECT images were formatted in analyze format and multiplanar reconstruction followed by summed images containing liver and lungs were created separately. The accumulated radioactivity of [99mTc] in tissues (different organs) was extracted from the images by drawing regions of interest (ROIs) then normalized to the whole body activity per volume (%ID/cm3). Whole-body activity was considered injected dose (ID).
2.13. Statistical analysis
Data were analyzed using GraphPad Prism 9 software (GraphPad Software, Inc.). Results were expressed as mean ± standard error of the mean (SEM). Statistical analysis was performed by one-way analysis of variance (ANOVA) with Tukey’s post hoc test. Between-group significance was tested by t-test. Differences between groups were considered statistically significant when p < 0.05. The statistical significance of lethality was analyzed using the Kaplan-Meier method and log-rank test.
3. Results
3.1. Bach1 deficiency reduces mortality following sepsis
We first tested whether BACH1 deficiency modulates survival in cecal ligation and puncture (CLP) model of sepsis. As shown in Fig. 1A, Bach1−/− mice demonstrated significantly improved survival after CLP surgery, with 80% mortality among wild-type (WT) mice within ten days after the CLP procedure, and 10% mortality in Bach1−/− mice. The body weight of Bach1−/− animals began to improve on the 5th day after surgery and was significantly higher from day six onwards (Fig. S1 (supplement)). The markers of oxidative stress, plasma H2O2 and MDA levels, and the liver MPO activity were significantly increased in WT mice following sepsis induction; their levels in Bach1−/− mice remained low, demonstrating a protective effect with Bach1 deficiency (Fig. 1B–D). The liver injury markers, plasma ALT and AST, were significantly elevated in WT mice subjected to CLP, but less in Bach1−/− mice (Fig. 1E and F). There was noticeable immune cell infiltration of the liver in WT mice after CLP. However, the infiltration was attenuated in Bach1−/− mice (Fig. 1G).
Fig. 1. BACH1 deficiency reduces mortality after CLP-induced sepsis.

(A) 10-day survival following CLP surgery-induced sepsis represented by Kaplan-Meier curve (n = 10/group) show reduced mortality rate in Bach1−/− mice compare to WT mice. (B) plasma H2O2 levels. (C) liver MPO activity, (D) plasma MDA levels, (E-F), plasma ALT and AST levels. Data scatter plots expressed as mean ± SEM (n = 5–9). (G) Representative histopathological changes of hepatic parenchyma in WT and Bach1−/− mice. KO=Bach1−/−. Statistical significance was defined as p < 0.05. **** = p < 0.0001; *** = p < 0.001; ** = p < 0.01; * = p < 0.05.
3.2. BACH1 deficiency attenuates sepsis-induced inflammation
Sepsis in the mouse and the man is characterized by dysregulated systemic and tissue inflammation. To test the role of BACH1 in sepsis-induced inflammation, we first assayed the level of cytokines in the plasma of CLP and sham-operated WT and Bach1−/− mice at 24 h after the surgery using a multiplex cytometric bead array. IL-1α, IL-6, MCP-1, IFN-γ, IL17-α and TNF-α were all increased in the plasma of WT mice after the CLP procedure. The mean levels of the markers of inflammation in Bach1−/− mice after the induction of CLP were significantly less compared to the WT mice (Fig. 2A). Next, we investigated the effect of BACH1 deficiency on tissue inflammation by determining the expression of inflammatory genes in the liver. We tested the gene expression of TNF-α, IL-6, and MCP-1 and their levels were significantly increased in the liver of WT-CLP mice compared to Bach1−/−-CLP mice (Fig. 2B).
Fig. 2. BACH1 deficiency attenuated sepsis-induced cytokine levels.

(A) Plasma cytokines, IL-1α, IL-6, MCP-1, IL-17A, IFN-γ, and TNF-α, were measured by multiplex cytometric bead array using plasma harvested at 24 h after CLP or sham operation. n = 8–9 mice per group.; IL, interleukin; LPS lipopolysaccharide; MCP-1, Monocyte Chemoattractant Protein-1; IFN, Interferon; TNF, tumor necrosis factor. (B) Mice were subjected to sham or CLP surgery and liver cytokine expression was measured by real time PCR at 24 h. Mean values are shown as fold induction relative to β-actin and normalized to sham (n = 6–9 mice/group). Data expressed as mean ± SEM; Statistical significance was defined as p < 0.05. **** = p < 0.0001; *** = p < 0.001; ** = p < 0.01; * = p < 0.05. (C) Representative immunoblotting of BACH1, NRF2 and HO-1 in the liver tissues of WT and Bach1−/− mice after sham or CLP surgery. Loading control: GAPDH. Replicates = 2; (D) The Western blot images in panel D were quantified and presented. KO=Bach1−/−. Data expressed as mean ± SEM; Statistical significance was defined as *p < 0.05.
To determine the effector of BACH1-deficiency mediated improvement in inflammation and survival, we tested the change in protein expression of HO-1, a protein negatively regulated by BACH1 and important in anti-oxidant response. Interestingly Bach1−/− mice demonstrated a high basal level of HO-1 expression in the liver of sham-operated mice with a further increase following the CLP procedure (Fig. 2C and D). To further determine whether BACH1 deficiency modulates the NRF2 level, we tested the NRF2 protein level in the liver samples. The total NRF2 levels in the liver showed a significant increase in KO mice after CLP, consistent with the effect of BACH1 deficiency (Fig. 2C and D). However, further studies are required to ascertain whether the total levels of NRF2 observed parallels the amount of NRF2 bound to the native ARE sequences in WT and Bach1−/− mice following sepsis.
3.3. Mitochondrial respiration following sepsis improved in Bach1−/− mice
The trajectory of sepsis progression is marked by an energetic imbalance [35,36]. Sepsis is known to reduce mitochondrial function in various tissues. To assess the effect of BACH1 deficiency in energy homeostasis, we tested whether the mitochondrial function was improved in the liver of Bach1−/− mice following CLP. Mitochondria were isolated from the liver at 24 h after the CLP procedure, and OCR was measured in the presence of sequentially added ADP, oligomycin, FCCP, and anti-mycin A in a Seahorse analyzer. Although the basal respiration was not significantly different between the groups, there was a significant improvement in state 3 respiration following sepsis in Bach1−/− compared to WT mice (Fig. 3A and B). The elevated state 3u respiration further demonstrated the improved substrate oxidation with BACH1 deficiency. State 3 respiration is ADP-stimulated and determined by ATP production and substrate oxidation. A functional decline of processes associated with ATP production or substrate oxidation will reduce state 3 rates. However, when treated with oligomycin (ATP synthase inhibitor), the mitochondrial respiration (state 4o) will be primarily due to proton leak. Whereas, the state of respiration observed after treating with FCCP, an uncoupler, (state 3u) reflects only substrate oxidation. Therefore, a decrease in the respiratory component ratio (RCR; state3/state4o) or uncoupling control ratio (UCR; state 3u/state4o) demonstrates dysfunction in any of the components involved in mitochondrial function and are indicators of mitochondrial dysfunction [37, 38]. The RCR and UCR were significantly improved in mice deficient in BACH1, compared to WT mice,after CLP (Fig. 3C). Consistent with the improved mitochondrial function, loss of BACH1 also resulted in better-preserved mitochondrial membrane potential in the liver (Fig. 3D). However, these functional changes in mitochondria did not induce quantitative protein changes in the OXPHOS complexes as the protein level of various OXPHOS enzymes in the liver measured by Western Blot remained unchanged after CLP, irrespective of the level of BACH1 expression (Fig. S2).
Fig. 3. Effect of BACH1 deficiency on mitochondrial OCR.

(A–B) Representative respiration profile of mitochondria isolated from the liver at 24 h after surgery in WT and Bach1−/− mice. ADP, oligomycin, FCCP and antimycin A (AA) were sequentially added and OCR measured. Replicates = 3. (C) Respiratory control ratios (RCR: state 3/state 4°, and UCR: state 3u/state 4°). (D) TMRE fluorescence intensity (liver mitochondrial membrane potential) without vs with FCCP (see methods). KO=Bach1−/− Data presented as mean ± SEM; Statistical significance was defined as p < 0.05. **** = p < 0.0001; *** = p < 0.001; ** = p < 0.01; * = p < 0.05.
3.4. BACH1 deficiency improves microcirculation following sepsis
Sepsis adversely affects microcirculation resulting in reduced tissue oxygenation [39,40]. To assess sepsis-induced alteration in blood flow, we performed SPECT analysis in WT and Bach1−/− mice three days after the CLP procedure using Tc-99m tagged RBC. The procedure involved intravenous injection of Tc-99m and scanning of the whole body biodistribution by SPECT imaging. As shown in Fig. 4, there was significantly improved blood flow in the liver and lung of Bach1−/− mice compared to WT mice, after sepsis induction. SPECT scans were overlaid on CT images for anatomical localization.
Fig. 4. [99mTc]-RBC bio-distribution following sepsis in WT and Bach1−/− mice indicating tissue perfusion.
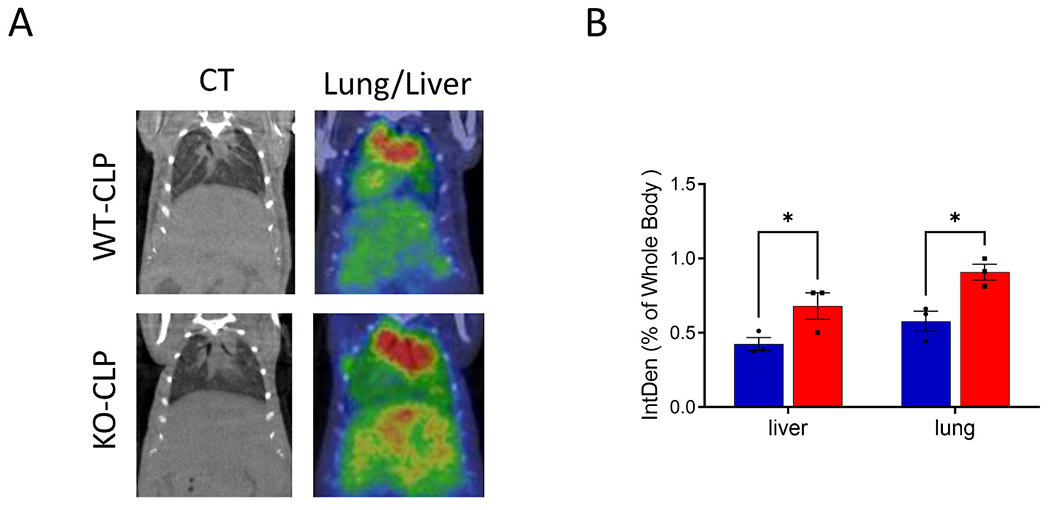
(A) Representative [99mTc]-RBC distribution in the liver and lung of the Bach1−/− and WT mice after CLP. The left panels are overlay CT images. (B) The mean integrated density (IntDen) in the liver and lung of the Bach1−/− (red bar) and WT (blue bar) mice versus the whole body. Data presented as mean ± SEM. KO=Bach1−/− Statistical significance was defined as *p < 0.05. 3 mice were used per group. See methods for details.
3.5. HO-1 is the most significant differentially expressed gene in the liver of Bach1−/− mice after CLP
To further examine the molecular basis of improved mitochondrial function and survival observed in Bach1−/− mice subjected to CLP, using RNA-seq, we performed gene expression profiling in the liver of Bach1−/− and WT mice subjected to CLP (Fig. 5). Among the 19767 genes probed by the RNAseq, only 44 genes showed a significant difference between the two groups, 24 genes were upregulated, and 20 were downregulated in Bach1−/− mice compared to WT mice subjected to CLP-sepsis (Table 1). The differential expression of these genes is shown in the heatmap (Fig. 5A). These genes were predominantly associated with lipid metabolism and oxidoreductase activity as per the gene ontology descriptions by IPA analysis (Fig. 5B). The expression level of the top 10 genes that were significantly up or downregulated in Bach1−/−-CLP compared to WT-CLP mice are shown in Fig. 5C and D, respectively. The RNA-seq data show that HMOX1 (the gene corresponding to HO-1 protein) is among the most upregulated genes, with the highest adjusted p-value, in Bach1−/− mice subjected to CLP compared to the WT mice. The upregulation of HMOX1 is consistent with the HO-1 protein expression observed and suggests that HO-1 is important in the beneficial effect due to Bach1 deficiency (Fig. 2C and D). The next most significantly upregulated gene was Slc48a1 (Solute carrier family 48, member 1), a heme transporter known to be a functional target of NRF2 and regulated by BACH1 [41]. Ehhadh transcript that was upregulated almost fourfold in Bach1−/− mice following CLP is an enzyme in fatty acid metabolism and closely linked to ATP synthesis and transport, mutations of which cause mitochondrial pathologies [42]. Interestingly, the most upregulated gene was the leptin receptor (Lepr). Though NRF2 improves leptin and insulin resistance, and adipokines have been associated with the prognosis of sepsis, the specific role of leptin receptor upregulation following sepsis with BACH1 deficiency is unclear [43,44]. A search for BACH1 target genes established from ChIP-seq data extracted from the ENCODE Transcription Factor Targets datasets and available at the Harmonizome portal shows 23 of the significantly altered genes to be a potential target of BACH1 (Table 1).
Fig. 5. Differentially expressed genes in the liver of Bach1−/− WT and mice following sepsis.

RNA sequencing (RNA-Seq) analysis of the wild type and Bach1−/− mice subjected to CLP-induced sepsis was performed to determine the gene expression levels. (A) Heat map showing the expression level of 44 differentially expressed genes in Bach1−/−-CLP mice as compared to WT-CLP. Red color represents higher expression, and green color represents lower expression. (B) Gene ontology terms and KEGG pathways significantly associated with 44 differentially expressed genes. Boxplots showing expression levels of top 10 upregulated (C) and top 10 downregulated genes (D) in WT (blue) and Bach1−/− (red) CLP groups. KO=Bach1−/− = 4 in each group. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Table 1.
Differentially expressed genes in the liver of WT and Bach1−/− mice after CLP.
| Gene symbol | Gene description | Fold change | adj. p-value (FDR) |
|---|---|---|---|
| Upregulated Genes | |||
| Hmox1a | Heme oxygenase 1 | 5.08 | 2.44 × 10−13 |
| Slc48a1a | Solute carrier family 48, member 1 | 2.51 | 1.20 × 10−10 |
| Ehhadh | Peroxisomal bifunctional enzyme | 3.78 | 3.12 × 10−06 |
| Rab36a | RAB36, member RAS oncogene family | 2.44 | 3.56 × 10−05 |
| Mup12 | Major urinary protein 12 | 2.04 | 3.56 × 10−05 |
| Cyp26b1 | Cytochrome P450, family 26, subfamily b, polypeptide 1 | 3.89 | 6.98 × 10−05 |
| Cyp4a32 | Cytochrome P450, family 4, subfamily a, polypeptide 32 | 2.87 | 1.38 × 10−03 |
| Abcb4 | ATP-binding cassette, sub-family B, member 4 | 1.60 | 1.64 × 10−03 |
| Trim80a | Tripartite motif-containing 80 | 4.22 | 2.14 × 10−3 |
| Asla | Argininosuccinate lyase | 1.81 | 2.86 × 10−03 |
| Gm15611 | Predicted gene 15611 | 3.84 | 3.00 × 10−03 |
| Lepra | Leptin receptor | 11.45 | 8.28 × 10−03 |
| Acaa1ba | Acetyl-Coenzyme A acyltransferase 1B | 2.35 | 8.28 × 10−03 |
| Elovl5a | ELOVL family member 5, elongation of long chain fatty acids | 2.01 | 1.15 × 10−02 |
| Crata | Carnitine acetyltransferase | 1.75 | 1.49 × 10−02 |
| Cyp4a10 | Cytochrome P450, family 4, subfamily a, polypeptide 10 | 2.39 | 2.74 × 10−02 |
| Chrna2 | Cholinergic receptor, nicotinic, alpha polypeptide 2 | 2.46 | 2.75 × 10−02 |
| Taok3a | TAO kinase 3 | 1.58 | 2.75 × 10−02 |
| Cyp7b1a | Cytochrome P450, family 7, subfamily b, polypeptide 1 | 1.70 | 3.66 × 10−02 |
| Cyp4a31 | Cytochrome P450, family 4, subfamily a, polypeptide 31 | 2.80 | 3.69 × 10−02 |
| Nipal1a | NIPA-like domain containing 1 | 3.49 | 4.69 × 10−02 |
| Abcc3a | ATP-binding cassette, sub-family C, member 3 | 1.73 | 4.69 × 10−02 |
| Retsata | Retinol saturase | 1.68 | 4.69 × 10−02 |
| Hmgcs2 | 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 2 | 1.55 | 4.69 × 10−02 |
| Downregulated Genes | |||
| Btg2a | B cell translocation gene 2 | 0.35 | 3.62 × 10−08 |
| Slc25a25a | Solute carrier family 25, member 25 | 0.41 | 1.58 × 10−05 |
| Plk3a | Polo like kinase 3 | 0.45 | 5.40 × 10−04 |
| Fmo5 | Flavin containing monooxygenase 5 | 0.56 | 1.64 × 10−03 |
| Gas6a | Growth arrest specific 6 | 0.58 | 1.64 × 10−03 |
| Flnca | Filamin C, gamma | 0.01 | 2.01 × 10−03 |
| Slc41a2 | Solute carrier family 41, member 2 | 0.65 | 2.86 × 10−03 |
| Tgtp1 | T cell specific gtpase 1 | 0.23 | 5.57 × 10−03 |
| Mid1 | Midline 1 | 0.42 | 8.34 × 10−03 |
| Egr1a | Early growth response 1 | 0.35 | 1.63 × 10−02 |
| Lyz2 | Lysozyme 2 | 0.49 | 1.63 × 10−2 |
| Bach1 | Basic leucine zipper transcription factor 1 | 0.47 | 1.72 × 10−02 |
| Tceal8 | Transcription elongation factor A-like 8 | 0.44 | 2.39 × 10−02 |
| Arid5aa | AT rich interactive domain 5A | 0.37 | 2.74 × 10−02 |
| Gm29825 | Predicted gene, 29825 | 0.01 | 2.75 × 10−02 |
| Trim11 | Tripartite motif-containing 11 | 0.65 | 4.42 × 10−02 |
| Saa3 | Serum amyloid A 3 | 0.46 | 4.69 × 10−02 |
| Sema4ca | Semaphorin-4C | 0.30 | 4.75 × 10−02 |
| Zfp36a | Zinc finger protein 36 | 0.45 | 4.80 × 10−02 |
| Tmem267 | Transmembrane protein 267 | 0.28 | 4.91 × 10−02 |
Target genes of the BACH1 transcription factor in ChIP-seq data as extracted from the ENCODE Transcription Factor Targets dataset. Data source: Harmonizome.
3.6. HO-1 is a critical mediator in improving liver function following sepsis in Bach1−/− mice
BACH1 deficiency resulted in a significantly elevated HO-1 in the liver and a further increase following sepsis. To test whether the increased expression of HO-1 in Bach1−/− mice is causative in the beneficial effect following CLP surgery, Bach1−/− mice were pretreated with ZnPP, a potent inhibitor of HO-1 [29,30]. As shown in Fig. 6A and B, ZnPP treatment significantly reduced State 3 and State 3u respiration in isolated mitochondria from the liver, after CLP. The liver mitochondrial membrane potential was also less in Bach1−/−-CLP mice that received ZnPP (Fig. 6C). The increased plasma ALT, AST, and liver MPO activity also demonstrated exacerbated liver injury when HO-1 was inhibited by ZnPP in Bach1−/− mice (Fig. 6D–F). Overall, ALT, AST, MPO, and state 3 respiration measures in Bach1 KO-CLP mice treated with ZnPP were all trending towards the levels in untreated WT-CLP (Figs. 1 and 6).
Fig. 6. The beneficial effect due to BACH1 deficiency in CLP is dependent on HO-1.

(A and B) Representative Seahorse XF trace of mitochondrial respiration performed with liver mitochondria isolated from sepsis-induced Bach1−/− mice after intraperitoneal administration of ZnPP (15 mg/kg) or DMSO at 1 h before CLP. ADP, oligomycin, FCCP and antimycin A (AA) were sequentially added and OCR measured. (C) TMRE fluorescence intensity (liver mitochondrial membrane potential) in the absence or presence of FCCP. Data presented as mean ± SEM; Statistical significance was defined as *p < 0.05. (D-F) Hepatic function was assessed by testing alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels. Myeloperoxidase (MPO) activity in the liver is an indicator of oxidative stress. KO=Bach1−/−. Mean ± SEM (n = 4); Statistical significance was defined as *p < 0.05. **** = p < 0.0001; *** = p < 0.001; ** = p < 0.01; * = p < 0.05.
4. Discussion
The antioxidant response is a homeostatic response to oxidative stress. NRF2 is a master regulator of antioxidant response controlling the expression of many genes, including HMOX 1, that mediates the resolution of oxidative stress. Studies using activators of NRF2 and mice deficient in Keap1 have previously demonstrated a protective role against oxidative stress and inflammation in experimental models of sepsis [4,7,9,12]. Therefore, factors that promote the expression, stability, or activity of NRF2 can be expected to improve survival in sepsis. In this study, we used a well-established experimental model of sepsis, the CLP model, to test whether modulation of BACH1, a competitor of NRF2, can reduce oxidative stress and inflammation, improve microcirculation, and promote survival following sepsis.
While heterodimers of NRF2 and MAF promote the transcription of HMOX-1, the BACH1-MAF heterodimer blocks this interaction resulting in negative regulation of NRF2-mediated ARE gene transcription [45]. Our results show that the mice deficient in BACH1 have a significantly higher survival rate than the WT mice following CLP-induced sepsis. About 80% of WT mice died within ten days in our experiment, with most mortality observed after 48 h. However, the mortality rate in Bach1−/− mice was significantly low (10%), demonstrating that deficiency of BACH1 can protect the mice from CLP-induced sepsis.
To study the mechanistic basis of Bach1 deletion on sepsis, we focused our investigations mainly on the liver as it is an organ severely affected during sepsis, and the onset of liver injury following CLP procedure occurs within a few hours [3,46–48]. The mortality rates among sepsis patients with liver dysfunction are also higher than those of sepsis patients with respiratory system dysfunction or failure [3]. Furthermore, in our model, liver injury following CLP surgery was evident from the plasma AST and ALT elevation in the WT mice. The elevation of these injury markers was significantly less in the Bach1−/− mice indicating tissue protection accorded by the deficiency of the Bach1 gene. Furthermore, extensive immune cell infiltration into the liver parenchyma was seen in the histology of WT mice subjected to CLP, but not in the Bach1−/− mice. BACH1 deficiency also led to reduced oxidative stress as observed by lower levels of MPO and lipid peroxidation product MDA in the Bach1−/− mice subjected to CLP.
TNF-α is one of the pro-inflammatory cytokines that is elevated in the blood early during the course of sepsis. Within 24 h after the CLP, TNF-α was significantly elevated in the plasma; its gene expression in the liver was also increased considerably. BACH1 deficiency resulted in reduced TNF-α levels in the plasma along with declined gene expression in the liver. Similarly, plasma IL-1α, IL-6, IL-17a, IFN-γ and MCP-1 were reduced in Bach1−/− mice compared to WT mice. These results show that BACH1 modulates both systemic and tissue inflammation in experimental sepsis, and BACH1 deficiency provides an ‘anti-inflammatory’ protection.
Though a clear cause-effect relationship between mitochondrial functional alteration and organ dysfunction has not been established in sepsis, human and experimental models of sepsis demonstrate declined mitochondrial function [49–51]. Reduced activity of critical enzymes in OXPHOS, changes in membrane potential, and reduced mitochondrial respiration were previously observed in animal models of sepsis [49,50]. In this study, we tested oxygen consumption in mitochondria isolated from the liver of sepsis-induced mice. The experiments demonstrate that the absence of BACH1 reduced mitochondrial dysfunction after sepsis induction. The preservation of bioenergetics in Bach1−/− is also shown in the respiratory control ratio (RCR), the ratio of state 3 to state 4o, which is a good indicator of mitochondrial function as it reflects changes in oxidative phosphorylation [38]. Tighter coupling of oxidative phosphorylation was further ascertained in Bach1−/− mice as indicated by the increased uncoupling control ratio (UCR), which is the ratio of OCR in the presence of FCCP (state 3u) to the OCR in the presence of oligomycin (state 4°) [37,52]. The OCR assessments demonstrate better preservation of bioenergetics with sepsis in Bach1−/− animals, suggesting that inhibition of BACH1 can promote cellular energy availability and improve organ function.
The increased oxygen demand due to perfusion defects could be a significant cause of mitochondrial dysfunction in sepsis. The oxygen demand/supply ratio can be profoundly affected by disruption of microvascular flow. Microcirculatory dysfunction has been reported in patients with sepsis and experimental models of sepsis [39,40,53]. Furthermore, patients were at increased risk for death when microcirculation did not improve [54]. The laser speckle contrast imaging for measuring hepatic microcirculation in the rat showed that microcirculation defect is associated with liver injury and appears very early in sepsis [55]. In our study, the in vivo radioisotope-tagged RBC SPECT/CT imaging of WT and Bach1−/− mice three days after the CLP surgery showed a significant improvement in blood flow to the liver and lung.
The immunometabolic programming that rescue mice from sepsis, in the absence of BACH1, was evident from the results of RNA-seq studies. The gene expression analysis revealed a significantly altered expression of genes related to fatty acid oxidation, and oxidoreductase reactions. Consistent with this observation, other studies showed that a constitutive activation of NRF2 increased the expression of genes in the fatty acid oxidation pathway and deletion of the Bach1 gene in Apo E KO mice resulted in inhibition of atherosclerosis through upregulation HO-1 [24,56].
These data demonstrate that BACH1 deficiency results in reduced systemic and tissue inflammation, reduced oxidative stress, improved mitochondrial function, better organ blood flow, and increased survival following CLP-induced sepsis. The mechanistic basis for the beneficial effect observed in Bach1−/− mice is likely due to the potentiation of antioxidant response by HO-1. NRF2 is known to be a direct regulator of HO-1. The two main checkpoints where NRF2 activity may be regulated are its point of association with KEAP1 (checkpoint 1) and the activation of ARE elements (checkpoint 2). Whereas ROS and electrophiles modify KEAP1 and modulate NRF2 stability at checkpoint 1, repressor factors such as BACH1 are expected to modulate NRF2 binding to ARE elements. The profound phenotypic change in the mice with BACH1 deficiency in terms of reduced susceptibility to sepsis would be expected to be contributed by the modulation of Nrf2 function, resulting in high expression levels of HO-1. The total NRF2 protein levels in the liver samples were significantly increased in Bach1−/− CLP mice compared to the controls. The NRF2 activation is reported to be associated with the nuclear export of BACH1 [57]. Additionally, ChIP or ChIP-seq studies may be necessary to further ascertain the level of NRF2 bound ARE-sequences on the native DNA in WT and Bach−/− mice following sepsis. Other mechanisms of HO-1 expression regulation by BACH1 also cannot be ruled out. ChIP analysis of the HMOX1 promoter clearly demonstrated both NRF2 and BACH1 binding sites [58]. Furthermore, HO-1 expression was significantly elevated in several tissues in the absence of both Bach1 and Nrf2 gene expression indicating that Nrf2 expression is not essential for HO-1 induction, and BACH1 was found to regulate enhancer availability of HMOX1 gene [26]. The regulation of HO-1 also involves a direct sensing of heme levels by BACH1 [59]. A functional genomic analysis of ventral midbrain samples from Bach1 KO and WT mice showed Bach1 ablation activates both NRF2 dependent ARE- and NRF2 independent non–ARE-mediated protective genes [60]. In ChIP assays, in the same study, both BACH1 and NRF2 were shown to bind to HO-1 enhancers, and both ARE and non-ARE-mediated HO-1 activation was observed [60].
Studies of HO-1-KO animal models and human cases of HO-1 deficiency show that HO-1 plays an important role in reducing oxidative stress and inflammation [61]. In a previous study, using HMOX-1−/−Bach1−/− mice, David Baltimore and colleagues demonstrated that in experimental autoimmune encephalomyelitis, the in vivo functional effect of BACH1 deficiency could be attributed to elevated expression of HO-1 [62]. Additionally, HO-1 expressed in innate immune cells showed anti-inflammatory effects and limited inflammatory tissue injury [63]. HO-1 deficient mice had high levels of circulating heme and reduced systemic levels of hemopexin, a heme sequestering protein, and were associated with increased lethality from sepsis [64]. Furthermore, exogenous administration of heme after low-grade infection in mice promoted tissue damage and severe sepsis. While these studies highlight the role of HO-1 regulation in reducing oxidative stress and disease pathology, the results collectively indicate a BACH1-HO-1 axis to be critical in sepsis pathogenesis. The HO-1 mediated catalytic reaction yields several breakdown products of the heme, including CO which is known to have therapeutic effects at lower doses [65]. To establish the role of HO-1 in protecting Bach1−/− mice from sepsis following CLP, we chemically inhibited HO-1 with ZnPP prior to the CLP procedure. ZnPP is an HO-1 inhibitor and abolishes the protective effects due to Nrf2 activation [57]. When Bach1−/− mice were pretreated with ZnPP, we observed enhanced CLP-induced liver injury, reduced liver mitochondrial respiration, and declined liver mitochondrial membrane potential confirming the role of HO-1 in the beneficial effect observed with BACH1 deficiency. One limitation of this experiment is that ZnPP may have off-target effects and may target other proteins such as HO-2 or other heme-dependent enzymes [66]. It also inhibits cyclin D1 in an HO-1 independent manner [67]. Future studies using small molecular inhibitors of BACH1 may further corroborate our findings. In other models, BACH1 inhibitors have been shown to induce HO-1 expression, antioxidant response and disease amelioration [60,68]. Nevertheless the results, collectively, demonstrate that the mechanism by which BACH1 deficiency imparts protection in sepsis involves upregulation of HO-1 and specific inhibitors of BACH1 may be a new therapeutic target in treating sepsis.
5. Conclusions
In summary, the findings reported in this study show that BACH1 deficiency enhances HO-1 levels and improves organ function and survival following sepsis. This preclinical study establishes BACH1 as a potential target in the treatment of sepsis and highlights the significance of putative inhibitors of BACH1 in improving organ function and survival following sepsis. The study further confirms that BACH1 is a viable alternative to NRF2 in targeting oxidative stress and systemic inflammation.
Supplementary Material
Acknowledgments
The work was partly supported by NIH grants R01 GM122059 (RPR) and R01 NS101967 (BT).
Abbreviations
- BACH1
BTB and CNC homology 1
- NRF2
Nuclear Factor E2-related factor 2
- ROS
Reactive Oxygen Species
- HMOX-1 or HO-1
Heme oxygenase-1
- RCR
Respiratory Control Ratio
- UCR
Uncoupled Respiration
- CLP
Cecal Ligation and Puncture
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.freeradbiomed.2022.06.005.
Declaration of competing interest
The authors have declared that no conflict of interest exists.
References
- [1].Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, Colombara DV, Ikuta KS, Kissoon N, Finfer S, Fleischmann-Struzek C, Machado FR, Reinhart KK, Rowan K, Seymour CW, Watson RS, West TE, Marinho F, Hay SI, Lozano R, Lopez AD, Angus DC, Murray CJL,Naghavi M, Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the Global Burden of Disease Study, Lancet 395 (10219) (2020) 200–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hotchkiss RS, Nicholson DW, Apoptosis and caspases regulate death and inflammation in sepsis, Nat. Rev. Immunol 6 (11) (2006) 813–822. [DOI] [PubMed] [Google Scholar]
- [3].Yan J, Li S, Li S, The role of the liver in sepsis, Int. Rev. Immunol 33 (6) (2014) 498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ohl K, Fragoulis A, Klemm P, Baumeister J, Klock W, Verjans E, Boll S, Mollmann J, Lehrke M, Costa I, Denecke B, Schippers A, Roth J, Wagner N, Wrack C, Tenbrock K, Nrf2 is a central regulator of metabolic reprogramming of myeloid-derived suppressor cells in steady state and sepsis, Front. Immunol 9 (2018) 1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Beury DW, Carter KA, Nelson C, Sinha P, Hanson E, Nyandjo M, Fitzgerald PJ, Majeed A, Wali N, Ostrand-Rosenberg S, Myeloid-Derived suppressor cell survival and function are regulated by the transcription factor Nrf2, J. Immunol 196 (8) (2016) 3470–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lee H, Jose PA, Coordinated contribution of NADPH oxidase- and mitochondria-derived reactive oxygen species in metabolic syndrome and its implication in renal dysfunction, Front. Pharmacol 12 (2021), 670076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gunne S, Heinicke U, Parnham MJ, Laux V, Zacharowski K, von Knethen A, Nrf2-A molecular target for sepsis patients in critical Care, Biomolecules 10 (12) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kong X, Thimmulappa R, Kombairaju P, Biswal S, NADPH oxidase-dependent reactive oxygen species mediate amplified TLR4 signaling and sepsis-induced mortality in Nrf2-deficient mice, J. Immunol 185 (1) (2010) 569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S, Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis, J. Clin. Invest 116 (4) (2006) 984–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhang DD, Chapman E, The role of natural products in revealing NRF2 function, Nat. Prod. Rep 37 (6) (2020) 797–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Baird L, Yamamoto M, The molecular mechanisms regulating the KEAP1-NRF2 pathway, Mol. Cell Biol 40 (13) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kong X, Thimmulappa R, Craciun F, Harvey C, Singh A, Kombairaju P, Reddy SP, Remick D, Biswal S, Enhancing Nrf2 pathway by disruption of Keap1 in myeloid leukocytes protects against sepsis, Am. J. Respir. Crit. Care Med 184 (8) (2011) 928–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yusoff FM, Maruhashi T, Kawano KI, Nakashima A, Chayama K, Tashiro S, Igarashi K, Higashi Y, Bach1 plays an important role in angiogenesis through regulation of oxidative stress, Microvasc. Res 134 (2021), 104126. [DOI] [PubMed] [Google Scholar]
- [14].Otsuki A, Yamamoto M, Cis-element architecture of Nrf2-sMaf heterodimer binding sites and its relation to diseases, Arch Pharm. Res. (Seoul) 43 (3) (2020) 275–285. [DOI] [PubMed] [Google Scholar]
- [15].Kurokawa H, Motohashi H, Sueno S, Kimura M, Takagawa H, Kanno Y, Yamamoto M, Tanaka T, Structural basis of alternative DNA recognition by Maf transcription factors, Mol. Cell Biol 29 (23) (2009) 6232–6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Igarashi K, Sun J, The heme-Bach1 pathway in the regulation of oxidative stress response and erythroid differentiation, Antioxidants Redox Signal. 8 (1–2) (2006) 107–118. [DOI] [PubMed] [Google Scholar]
- [17].Davudian S, Mansoori B, Shajari N, Mohammadi A, Baradaran B, BACH1, the master regulator gene: a novel candidate target for cancer therapy, Gene 588 (1) (2016) 30–37. [DOI] [PubMed] [Google Scholar]
- [18].Araujo JA, Zhang M, Yin F, Heme oxygenase-1, oxidation, inflammation, and atherosclerosis, Front. Pharmacol 3 (2012) 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Fujioka K, Kalish F, Zhao H, Wong RJ, Stevenson DK, Heme oxygenase-1 deficiency promotes severity of sepsis in a non-surgical preterm mouse model, Pediatr. Res 84 (1) (2018) 139–145. [DOI] [PubMed] [Google Scholar]
- [20].Kondo K, Ishigaki Y, Gao J, Yamada T, Imai J, Sawada S, Muto A, Oka Y, Igarashi K, Katagiri H, Bach1 deficiency protects pancreatic beta-cells from oxidative stress injury, Am. J. Physiol. Endocrinol. Metab 305 (5) (2013) E641–E648. [DOI] [PubMed] [Google Scholar]
- [21].Zhang X, Guo J, Wei X, Niu C, Jia M, Li Q, Meng D, Bach1: function, regulation, and involvement in disease, Oxid. Med. Cell. Longev 2018 (2018), 1347969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Iida A, Inagaki K, Miyazaki A, Yonemori F, Ito E, Igarashi K, Bach1 deficiency ameliorates hepatic injury in a mouse model, Tohoku J. Exp. Med 217 (3) (2009) 223–229. [DOI] [PubMed] [Google Scholar]
- [23].Harusato A, Naito Y, Takagi T, Uchiyama K, Mizushima K, Hirai Y, Higashimura Y, Katada K, Handa O, Ishikawa T, Yagi N, Kokura S, Ichikawa H, Muto A, Igarashi K, Yoshikawa T, BTB and CNC homolog 1 (Bach1) deficiency ameliorates TNBS colitis in mice: role of M2 macrophages and heme oxygenase-1, Inflamm. Bowel Dis 19 (4) (2013) 740–753. [DOI] [PubMed] [Google Scholar]
- [24].Watari Y, Yamamoto Y, Brydun A, Ishida T, Mito S, Yoshizumi M, Igarashi K, Chayama K, Ohshima T, Ozono R, Ablation of the bach1 gene leads to the suppression of atherosclerosis in bach1 and apolipoprotein E double knockout mice, Hypertens. Res 31 (4) (2008) 783–792. [DOI] [PubMed] [Google Scholar]
- [25].Mito S, Ozono R, Oshima T, Yano Y, Watari Y, Yamamoto Y, Brydun A, Igarashi K, Yoshizumi M, Myocardial protection against pressure overload in mice lacking Bach1, a transcriptional repressor of heme oxygenase-1, Hypertension 51 (6) (2008) 1570–1577. [DOI] [PubMed] [Google Scholar]
- [26].Sun J, Hoshino H, Takaku K, Nakajima O, Muto A, Suzuki H, Tashiro S, Takahashi S, Shibahara S, Alam J, Taketo MM, Yamamoto M, Igarashi K, Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene, EMBO J 21 (19) (2002) 5216–5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hubbard WJ, Choudhry M, Schwacha MG, Kerby JD, Rue LW 3rd, Bland KI, Chaudry IH, Cecal ligation and puncture, Shock 24 (Suppl 1) (2005) 52–57. [DOI] [PubMed] [Google Scholar]
- [28].Subramani K, Raju SP, Chu X, Warren M, Pandya CD, Hoda N, Fulzele S, Raju R, Effect of plasma-derived extracellular vesicles on erythrocyte deformability in polymicrobial sepsis, Int. Immunopharm 65 (2018) 244–247. [DOI] [PubMed] [Google Scholar]
- [29].Li HB, Zhang XZ, Sun Y, Zhou Q, Song JN, Hu ZF, Li Y, Wu JN, Guo Y, Zhang Y, Shi J, Yu JB, HO-1/PINK1 regulated mitochondrial fusion/fission to inhibit pyroptosis and attenuate septic acute kidney injury, BioMed Res. Int 2020 (2020), 2148706. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [30].Park JS, Choi HS, Yim SY, Lee SM, Heme oxygenase-1 protects the liver from septic injury by modulating TLR4-mediated mitochondrial quality control in mice, Shock 50 (2) (2018) 209–218. [DOI] [PubMed] [Google Scholar]
- [31].Warren M, Subramani K, Schwartz R, Raju R, Mitochondrial dysfunction in rat splenocytes following hemorrhagic shock, Biochim. Biophys. Acta (BBA) - Mol. Basis Dis 1863 (10 Pt B) (2017) 2526–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zheng H, Yu WM, Shen J, Kang S, Hambardzumyan D, Li JY, Shen Y, Kenney AM, Chen J, Qu CK, Mitochondrial oxidation of the carbohydrate fuel is required for neural precursor/stem cell function and postnatal cerebellar development, Sci. Adv 4 (10) (2018) eaat2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Subramani K, Lu S, Warren M, Chu X, Toque HA, Caldwell RW, Diamond MP, Raju R, Mitochondrial targeting by dichloroacetate improves outcome following hemorrhagic shock, Sci. Rep 7 (1) (2017) 2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Jian B, Wang D, Chen D, Voss J, Chaudry I, Raju R, Hypoxia-induced alteration of mitochondrial genes in cardiomyocytes: role of Bnip3 and Pdk1, Shock 34 (2) (2010) 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Carre JE, Singer M, Cellular energetic metabolism in sepsis: the need for a systems approach, Biochim. Biophys. Acta 1777 (7–8) (2008) 763–771. [DOI] [PubMed] [Google Scholar]
- [36].Vachharajani V, McCall CE, Epigenetic and metabolic programming of innate immunity in sepsis, Innate Immun. 25 (5) (2019) 267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Solsona-Vilarrasa E, Fucho R, Torres S, Nunez S, Nuno-Lambarri N, Enrich C, Garcia-Ruiz C, Fernandez-Checa JC, Cholesterol enrichment in liver mitochondria impairs oxidative phosphorylation and disrupts the assembly of respiratory supercomplexes, Redox Biol. 24 (2019), 101214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Brand MD, Nicholls DG, Assessing mitochondrial dysfunction in cells, Biochem. J 435 (2) (2011) 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tyagi A, Sethi AK, Girotra G, Mohta M, The microcirculation in sepsis, Indian J. Anaesth 53 (3) (2009) 281–293. [PMC free article] [PubMed] [Google Scholar]
- [40].Abraham E, Singer M, Mechanisms of sepsis-induced organ dysfunction, Crit. Care Med 35 (10) (2007) 2408–2416. [DOI] [PubMed] [Google Scholar]
- [41].Warnatz HJ, Schmidt D, Manke T, Piccini I, Sultan M, Borodina T, Balzereit D, Wruck W, Soldatov A, Vingron M, Lehrach H, Yaspo ML, The BTB and CNC homology 1 (BACH1) target genes are involved in the oxidative stress response and in control of the cell cycle, J. Biol. Chem 286 (26) (2011) 23521–23532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Forst AL, Reichold M, Kleta R, Warth R, Distinct mitochondrial pathologies caused by mutations of the proximal tubular enzymes EHHADH and GATM, Front. Physiol 12 (2021), 715485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yagishita Y, Uruno A, Fukutomi T, Saito R, Saigusa D, Pi J, Fukamizu A, Sugiyama F, Takahashi S, Yamamoto M, Nrf2 improves leptin and insulin resistance provoked by hypothalamic oxidative stress, Cell Rep. 18 (8) (2017) 2030–2044. [DOI] [PubMed] [Google Scholar]
- [44].Jacobsson S, Larsson P, Johansson G, Norberg M, Wadell G, Hallmans G, Winso O, Soderberg S, Leptin independently predicts development of sepsis and its outcome, J. Inflamm 14 (2017) 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Igarashi K, Kurosaki T, Roychoudhuri R, BACH transcription factors in innate and adaptive immunity, Nat. Rev. Immunol 17 (7) (2017) 437–450. [DOI] [PubMed] [Google Scholar]
- [46].Nesseler N, Launey Y, Aninat C, Morel F, Malledant Y, Seguin P, Clinical review: the liver in sepsis, Crit. Care 16 (5) (2012) 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Vandewalle J, Steeland S, Van Ryckeghem S, Eggermont M, Van Wonterghem E, Vandenbroucke RE, Libert C, A study of cecal ligation and puncture-induced sepsis in tissue-specific tumor necrosis factor receptor 1-deficient mice, Front. Immunol 10 (2019) 2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yu Q, Zou L, Yuan X, Fang F, Xu F, Dexmedetomidine protects against septic liver injury by enhancing autophagy through activation of the AMPK/SIRT1 signaling pathway, Front. Pharmacol 12 (2021), 658677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Arulkumaran N, Deutschman CS, Pinsky MR, Zuckerbraun B, Schumacker PT, Gomez H, Gomez A, Murray P, Kellum JA, Workgroup AX, Mitochondrial function in sepsis, Shock 45 (3) (2016) 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Singer M, The role of mitochondrial dysfunction in sepsis-induced multi-organ failure, Virulence 5 (1) (2014) 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ham PB 3rd, Raju R, Mitochondrial function in hypoxic ischemic injury and influence of aging, Prog. Neurobiol 157 (2017) 92–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kim SK, Belzer FO, Southard JH, Loss of mitochondrial respiratory function and its suppression during cold ischemic preservation of rat livers with University of Wisconsin solution, Hepatology 16 (3) (1992) 742–748. [DOI] [PubMed] [Google Scholar]
- [53].Caraballo C, Jaimes F, Organ dysfunction in sepsis: an ominous trajectory from infection to death, Yale J. Biol. Med 92 (4) (2019) 629–640. [PMC free article] [PubMed] [Google Scholar]
- [54].Charlton M, Sims M, Coats T, Thompson JP, The microcirculation and its measurement in sepsis, J Intensive Care Soc 18 (3) (2017) 221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wu Y, Ren J, Zhou B, Ding C, Chen J, Wang G, Gu G, Liu S, Li J, Laser speckle contrast imaging for measurement of hepatic microcirculation during the sepsis: a novel tool for early detection of microcirculation dysfunction, Microvasc. Res 97 (2015) 137–146. [DOI] [PubMed] [Google Scholar]
- [56].Ludtmann MH, Angelova PR, Zhang Y, Abramov AY, Dinkova-Kostova AT, Nrf2 affects the efficiency of mitochondrial fatty acid oxidation, Biochem. J 457 (3) (2014) 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ahuja M, Ammal Kaidery N, Yang L, Calingasan N, Smirnova N, Gaisin A, Gaisina IN, Gazaryan I, Hushpulian DM, Kaddour-Djebbar I, Bollag WB, Morgan JC, Ratan RR, Starkov AA, Beal MF, Thomas B. Distinct Nrf2 signaling mechanisms of fumaric acid esters and their role in neuroprotection against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced experimental Parkinson’s-like disease, J. Neurosci 36 (23) (2016) 6332–6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Reichard JF, Motz GT, Puga A, Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1, Nucleic Acids Res. 35 (21) (2007) 7074–7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kitamuro T, Takahashi K, Ogawa K, Udono-Fujimori R, Takeda K, Furuyama K, Nakayama M, Sun J, Fujita H, Hida W, Hattori T, Shirato K, K Igarashi S Shibahara, Bach1 functions as a hypoxia-inducible repressor for the heme oxygenase-1 gene in human cells, J. Biol. Chem 278 (11) (2003) 9125–9133. [DOI] [PubMed] [Google Scholar]
- [60].Ahuja M, Ammal Kaidery N, Attucks OC, McDade E, Hushpulian DM, Gaisin A, Gaisina I, Ahn YH, Nikulin S, Poloznikov A, Gazaryan I, Yamamoto M, Matsumoto M, Igarashi K, Sharma SM, Thomas B, Bach1 derepression is neuroprotective in a mouse model of Parkinson’s disease, Proc. Natl. Acad. Sci. U. S. A 118 (45) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Yachie A, Heme oxygenase-1 deficiency and oxidative stress: a review of 9 independent human cases and animal models, Int. J. Mol. Sci 22 (4) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].So AY, Garcia-Flores Y, Minisandram A, Martin A, Taganov K, Boldin M, Baltimore D. Regulation of APC development, immune response, and autoimmunity by Bach1/HO-1 pathway in mice, Blood 120 (12) (2012) 2428–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Gozzelino R, Jeney V, Soares MP, Mechanisms of cell protection by heme oxygenase-1, Annu. Rev. Pharmacol. Toxicol 50 (2010) 323–354. [DOI] [PubMed] [Google Scholar]
- [64].Larsen R, Gozzelino R, Jeney V, Tokaji L, Bozza FA, Japiassu AM, Bonaparte D, Cavalcante MM, Chora A, Ferreira A, Marguti I, Cardoso S, Sepulveda N, Smith A, Soares MP, A central role for free heme in the pathogenesis of severe sepsis, Sci. Transl. Med 2 (51) (2010) 51ra71. [DOI] [PubMed] [Google Scholar]
- [65].Ryter SW, Therapeutic potential of heme oxygenase 1 and carbon monoxide in acute organ injury, critical illness, and inflammatory disorders, Antioxidants 9 (11) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zeitler L, Fiore A, Meyer C, Russier M, Zanella G, Suppmann S, Gargaro M, Sidhu SS, Seshagiri S, Ohnmacht C, Kocher T, Fallarino F, Linkermann A, Murray PJ, Anti-ferroptotic mechanism of IL4i1-mediated amino acid metabolism, Elife 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].La P, Fernando AP, Wang Z, Salahudeen A, Yang G, Lin Q, Wright CJ, Dennery PA, Zinc protoporphyrin regulates cyclin D1 expression independent of heme oxygenase inhibition, J. Biol. Chem 284 (52) (2009) 36302–36311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Attucks OC, Jasmer KJ, Hannink M, Kassis J, Zhong Z, Gupta S, Victory SF, Guzel M, Polisetti DR, Andrews R, Mjalli AM, Kostura MJ, Induction of heme oxygenase I (HMOX1) by HPP-4382: a novel modulator of Bach1 activity, PLoS One 9 (7) (2014), e101044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.