

Abstract
Human vertebral malformations (VMs) have an estimated incidence of 1/2000 and are associated with significant health problems including congenital scoliosis (CS) and recurrent organ system malformation syndromes such as VACTERL (vertebral anomalies; anal abnormalities; cardiac abnormalities; tracheo-esophageal fistula; renal anomalies; limb anomalies). The genetic cause for the vast majority of VMs are unknown. In a CS/VM patient cohort, three COL11A2 variants (R130W, R1407L and R1413H) were identified in two patients with cervical VM. A third patient with a T9 hemivertebra and the R130W variant was identified from a separate study. These substitutions are predicted to be damaging to protein function, and R130 and R1407 residues are conserved in zebrafish Col11a2. To determine the role for COL11A2 in vertebral development, CRISPR/Cas9 was used to create a nonsense mutation (col11a2L642*) as well as a full gene locus deletion (col11a2del) in zebrafish. Both col11a2L642*/L642* and col11a2del/del mutant zebrafish exhibit vertebral fusions in the caudal spine, which form due to mineralization across intervertebral segments. To determine the functional consequence of VM-associated variants, we assayed their ability to suppress col11a2del VM phenotypes following transgenic expression within the developing spine. While wildtype col11a2 expression suppresses fusions in col11a2del/+ and col11a2del/del backgrounds, patient missense variant-bearing col11a2 failed to rescue the loss-of-function phenotype in these animals. These results highlight an essential role for COL11A2 in vertebral development and support a pathogenic role for two missense variants in CS.
Introduction
With a collective incidence of ~1/2000, vertebral malformations (VMs) pose a significant health problem and form a major contributor to adolescent disability (1,2). Affected individuals may experience back and neck pain, disability or cosmetic disfigurement (3). VMs can occur in isolation or be associated with blastogenic syndromes including vertebral anomalies; anal abnormalities; cardiac abnormalities; tracheo-esophageal fistula; renal anomalies; limb anomalies (VACTERL), oculo-auriculo-vertebral spectrum, Klippel-Feil syndrome or congenital scoliosis. Other associated congenital malformations include anomalies of the spinal cord, kidney and/or heart (4).
In vertebrates, normal development and segmentation of the spine involves a complex, stepwise process involving multiple elements including connective and vascular tissues, cartilage, bone, muscle and the nervous system. In amniotes, vertebrae develop from transient embryonic structures known as somites (5,6). Somites are metameric blocks of paraxial mesoderm patterned along the axial midline of embryos. The process of somitogenesis along the embryonic axis occurs through a clock and wavefront model, which integrates Notch, Wnt and FGF signaling pathways. Since the positions of somite boundaries are essential for patterning the vertebrae and intervertebral discs, disruptions in somitogenesis are known to result in VMs such as hemivertebrae or fused vertebral segments (7,8). Experiments in mice suggest that environmental insults during embryogenesis, sometimes in combination with genetic mutations, can also contribute to the occurrence of VMs (9,10).
Advances in next generation sequencing technologies have led to the identification of genetic variants that are associated with structural birth defects and developmental disabilities. Pathogenic variants in TBX6 have recently been discovered to account for ~10% of the cases of congenital scoliosis (1,11), and sequence variants in PAX1 (12), DLL3 (13), WNT3A (14), POLR1D (15), T (Brachyury) (16), KIAA1217 (17), WBP11 (18), FBN1 (19) and FGFR1 (20) have also been identified in patients with VMs. However, the aetiology of the vast majority of VMs remains unknown. Improved understanding of the genetic cause of VMs can aid in clinical management, genetic counselling and possible treatment strategies.
Here, we performed whole exome sequencing on 37 patients with isolated non-syndromic VMs, including congenital scoliosis (vertebral malformations in the spine causing abnormal curvature). We identified three novel and likely deleterious variants in COL11A2: R130W, R1407L and R1413H. Using the zebrafish as a model, we demonstrated an essential role for COL11A2 in vertebral development, and provide evidence for deleterious consequences from two of these substitutions. We characterized col11a2 as haploinsufficient, and describe a severe vertebral fusion phenotype that results from mineralization defects in the absence of gross segmentation errors.
Results
Whole exome sequencing reveals VM-associated variants in COL11A2
We recruited a cohort of 81 patients with isolated VMs for whole exome sequencing (WES) or whole genome sequencing (WGS). Since VMs often present with both phenotypic and genetic heterogeneity, we selected 37 patients with isolated, non-syndromic VMs for WES/WGS analysis. We found two patients (Probands 1–2) with substitution variants in COL11A2 (Table 1 and Supplementary Material, Fig. S1). Proband 1 was a 45 year old adult female with C4-C5 fusion. Proband 2 was a 31 year old male adult with C3-C5 fusion. Parents for Probands 1 and 2 were not available for participation in the study. In a separate study, we identified a COL11A2 substitution in a 20 year old adult male from PUMCH with congenital scoliosis secondary to a T9 hemivertebra (Proband 3; Table 1). Proband 1 and 3 were found to carry the same substitution—R130W. Further segregation analysis showed that the variant in Proband 3 was inherited from their seemingly unaffected mother. Proband 2 was found to carry two COL11A2 variants, R1407L and R1413H, although we were not able to determine whether these variants were in cis- or trans- as parental DNA samples for each were not available. COL11A2 is not predicted to tolerate a loss-of-function mutation (gnomAD pLI = 0.7) (21), and all three of these sequence variants were predicted by PolyPhen to be damaging (Table 1) (22).
Table 1.
Whole exome sequencing—details of identified variants
| Subject | Vertebral phenotype | COL11A2 variant | gnomAD frequency | PolyPhen score | |
|---|---|---|---|---|---|
| Proband 1 | C4–C5 Fa | Exon 3 | c.C388T:p.R130W | 3.97e-5 | 0.996 |
| Proband 2 | C3–C5 F | Exon 60 | c.G4220T:p.R1407L | 0 | 0.996 |
| Exon 61 | c.G4238A:p.R1413H | 5.09e-5 | 0.871 | ||
| Proband 3 | T9 Hb | Exon 3 | c.C388T:p.R130W | 3.97e-5 | 0.996 |
Note: Proband 1–2 Marshfield Clinic Research Institute; Proband 3 Peking Union Medical College Hospital. Identified variants, their gnomAD frequency, and their PolyPhen scores.
aF = fusion
bH = hemivertebra
COL11A2 encodes for collagen α2(XI), a cartilaginous collagen that heterotrimerizes with collagen α1(XI) and α3(XI). COL11A2 functions to stabilize and maintain the collagenous matrix in cartilage. Highlighting the importance of type XI collagens in cartilaginous tissues, pathogenic variants in these genes are known to cause intervertebral disc disease (23) and osteochondrodysplasias like Stickler Syndrome (scoliosis), otospondylomegaepiphyseal dysplasia (VM), Weissenbacher–Zwëymuller syndrome (VM) and fibrochondrogenesis (24–30). Major features associated with these conditions include sensorineural hearing loss, shorter limbs, short stature and craniofacial defects. Fibrochondrogenesis patients also exhibit ‘pinched’ vertebrae (30), indicating that type XI collagens have a role in human vertebral development.
Consistent with observed phenotypes in humans, mouse Col11a1 loss-of-function homozygous mutants exhibit short limbs, craniofacial defects, shortened long bones and defective cartilage throughout the body leading to death at birth (31). Similarly, Col11a2 mutant mice exhibit a reduced body size and craniofacial defects, as well as disorganized collagen in the long bones (32). Zebrafish col11a2 mutant embryos have also been reported to exhibit structural jaw defects, as well as increased cartilage/bone stiffness and premature degradation of type II collagen at stages prior to spine formation (33). Although VM phenotypes have not been reported in these animal models, it is not clear that vertebral development was specifically examined. Given the external development and optical transparency of zebrafish embryos, which facilitate direct observation of developmental processes like vertebral formation, we employed zebrafish as a model to investigate the specific role for Col11a2 in vertebral development.
col11a2 is expressed in the zebrafish notochord
We reasoned that if col11a2 plays a role in vertebral development, it should be expressed in tissues that give rise to the spine. In amniotes, both the notochord and somites play important roles in patterning the spine, although the segmentated differentiation of somitic mesoderm has a greater contribution to spine development (34). Vertebrae and the outer part of the intervertebral disc (annulus fibrosis) are derived from somitic sclerotome, while notochordal cells form the inner part of the intervertebral disc (nucleus pulposus) (35). In zebrafish, however, vertebral bodies are formed predominantly from the notochord, while adjacent somites contribute to vertebral ribs and arches (36). Specifically, the zebrafish spine is patterned by segmentation of the notochord into alternating mineralized and cartilaginous domains that give rise to vertebrae and intervertebral discs, respectively (37).
To determine the expression pattern of col11a2, we performed whole mount RNA in situ hybridization on zebrafish embryos from 1 to 5 days post-fertilization (dpf). Notably, we observed staining in the notochord and craniofacial cartilage elements (Supplementary Material, Fig. S2), consistent with previous reports of col11a2 expression at 24–28 h post-fertilization (hpf) (38). Since the notochord is the precursor of the zebrafish spine, expression of col11a2 in this tissue supports a possible role in vertebral development.
Col11a2 is necessary for zebrafish vertebral development
To determine the function for Col11a2 in spine development, we employed established CRISPR/Cas9 gene targeting approaches to generate zebrafish col11a2 loss-of-function (LOF) alleles. We targeted exon 27, early in the triple helix domain of the protein (Fig. 1A) and established a stable 4 bp deletion that introduces a frameshift, multiple premature termination codons and is predicted to prematurely truncate the Col11a2 protein. We termed this allele col11a2L642*. Adult animals homozygous for the col11a2L642* allele exhibited a mild craniofacial defect that has been previously reported (33), and is not further characterized in this study. Strikingly, 60% of col11a2L642*/L642* mutant adults exhibited vertebral fusions in the caudal vertebrae of the spine, as determined by alizarin red staining or X-ray microcomputed tomography (μCT) (Fig. 1C and D). In comparison, the penetrance of vertebral fusion defects observed in wildtype animals was only 22% (Fig 2). The majority of col11a2L642*/L642* VMs appeared to be single fusion events, involving only two vertebral segments, that did not otherwise disrupt the curvature or gross appearance of the spine (Figs 1 and 2 and Supplementary Material, Fig. S3).
Figure 1.

Loss of function mutations in col11a2 cause vertebral fusion defects. (A) Schematic of the wildtype Col11a2 protein, showing its domain structure and the position at which gRNAs were targeted to generate the col11a2L642* mutation. This allele encodes a truncated protein, illustrated on the right. (B) Strategy used to create the col11a2del allele. Two gRNAs were targeted to the 5′ and 3′ ends of the col11a2 gene, to delete the intervening 45 kb genomic locus. The resulting col11a2del allele encodes a 13 amino acid peptide. (C) Alizarin red staining of adult wildtype and col11a2L642*/L642* zebrafish, showing vertebral fusion in the posterior spine of col11a2L642*/L642*mutants. Schematic to the right of each image highlights the structure of normal versus fused vertebrae. (D) In vivo staining with alizarin red at 16dpf and calcein at 21dpf labels the developing vertebrae. Fusions in col11a2L642*/L642* and col11a2del/del animals first form along the ventral edge of the spine (arrowheads) and proceed dorsally (asterisks). μCT scans at 8 wpf (weeks post fertilization) demonstrating mature fused vertebrae in each animal (asterisks).
Figure 2.

Vertebral fusion in all col11a2 genotypes is biased to the caudal end of the spine. Heatmaps indicating the position of vertebral fusions along the length of the spine, across col11a2 genotypes. Each row of the heatmap represents an individual animal at 21dpf, with colored blocks indicating the positions of fused vertebrae. Graph depicts fusion penetrance between all genotypes, and demonstrates that col11a2del/+ fish have significantly more fusions than wildtype, indicating haploinsufficiency. Significance was calculated by chi-square analysis and error bars represent 95% confidence interval. wt: wild type. ns—not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
In zebrafish, nonsense mutations can invoke nonsense mediated decay-associated transcriptional adaptation mechanisms that mask true loss-of-function phenotypes (39,40). Using CRISPR/Cas9 genome editing approaches, we therefore deleted the entire 45 kb col11a2 open reading frame to generate a ‘transcriptless’ col11a2del allele that is not expected to induce transcriptional compensation (Fig. 1B). Remarkably, the penetrance of vertebral fusions in homozygous col11a2del animals is increased to 90%, and the majority of col11a2del/del fish exhibit multiple (2–5) fusion events in the caudal vertebrae of the spine (Figs 1 and 2). Furthermore, 25% of these fusions encompass 3–4 vertebral segments. Altogether, these data demonstrate an essential role for col11a2 in vertebral development.
col11a2 is haploinsufficient
Having determined that both col11a2L642*/L642* and col11a2del/del mutant fish exhibit caudal vertebral fusions, we sought to assess the phenotype of heterozygote mutant animals. We found that col11a2L642*/+ fish develop vertebral fusions with 26% penetrance, which is not significantly different than the 22% penetrance of VMs observed in wildtype animals (Fig. 2). However, 51% of col11a2del/+ animals develop fusions, which is a significant increase over the wildtype level. Using RT-qPCR, we quantified col11a2 expression in col11a2del/+ heterozygotes to be 56% of wildtype expression levels (Supplementary Material, Fig. S4A). Together, these data indicate that col11a2 is haploinsufficient. Interestingly, vertebral fusions among col11a2del/+ fish are much milder than among col11a2del/del mutants, with most animals exhibiting only 1–2 fusions. This represents an intermediate phenotype between wildtype and col11a2del/del animals, suggesting that vertebral development is sensitive to the dosage of Col11a2. Haploinsufficiency of col11a2 supports our hypothesis that damaging, heterozygous variants from our patient cohort may have a causative role in the formation of vertebral malformations.
col11a2 mutants exhibit disorganized collagen fibrils
Despite strong col11a2 expression in the embryonic notochord, col11a2 mutants do not exhibit gross morphological defects at embryonic stages (Supplementary Material, Fig. S3). To more closely examine the effects of Col11a2 loss on the structure of the notochord extracellular matrix (ECM), we performed transmission electron microscopy (TEM) on transverse sections of the posterior trunk of 2 dpf wildtype and col11a2del/del embryos. Wildtype embryos exhibit a smooth, thick layer of collagen fibrils oriented parallel to the cross-section (Fig. 3). These fibrils, which compose the middle layer of the notochord ECM, run around the circumference of the notochord (41). In col11a2del/del embryos, these collagen fibrils appeared disorganized and exhibited bends and wave-like orientations that traversed outside of the plane of sectioning. Furthermore, the overall thickness of this layer is reduced (Supplementary Material, Fig. S5), which is consistent with the known role of COL11A2 in stabilizing other collagens (42,43).
Figure 3.

col11a2del/del mutants exhibit disorganized collagen fibrils. Representative TEM images of wildtype (left) and col11a2del/del mutant (right) zebrafish, depicting transverse sections through the notochord of 2dpf embryos (top) and sagittal sections through the spine of 14dpf juvenile fish (bottom). In 2dpf cross sections, brackets labelled ‘ECM’ denote the middle layer of the notochord ECM, which is composed of a thick layer of banded collagen in wildtype embryos. The fibrils of collagen are visible running parallel to the circumference of the notochord. In 2dpf col11a2del/del mutant embryos, the ECM layer is much thinner and exhibits more kinking and bending than wildtype controls. In 14dpf sagittal sections, the edge of the developing vertebrae (brackets, arrowheads) and the intervertebral disc boundary are indicated. In wildtype fish, the vertebra has a clear dense layer of mineral present, with a defined boundary between the vertebral and intervertebral disc region (asterisks), where mineral deposition stops. The intervertebral disc region shows a thick layer of evenly spaced collagen fibrils which are oriented perpendicular to the section. In col11a2del/del mutant animals, the mineral layer of the vertebra appears less dense, and the boundary between mineral and collagen is less defined. The collagen fibrils in these mutants appear denser, with very little space visible between them, and are not oriented perpendicular to the section suggesting a disorganized structure.
Having demonstrated a collagen defect in the ECM of col11a2del/del embryos, we next sought to examine the molecular phenotype of col11a2del/del juveniles at a later stage of spine development. We performed TEM on sagittal sections of the posterior trunk of 14 dpf wildtype and col11a2del/del animals, focusing on the boundary between the developing vertebral centra and intervertebral segments. Wildtype vertebrae exhibit a dense layer of mineral deposited upon a bed of collagen fibrils outside of the notochord (arrowheads, Fig. 3). The IVDs in these animals are composed of evenly spaced collagen fibrils that are oriented perpendicular to the sagittal plane of sectioning (asterisks, Fig. 3). The boundary between mineral and IVD is clear, with obvious delineation between these tissues. In contrast, col11a2del/del animals exhibited sparser and less dense mineral in the developing vertebrae. The collagen fibrils that compose the layer beneath the mineral as well as the IVD are more densely packed in col11a2del/del mutants, with little space evident between them (asterisk, Fig. 3). These fibrils also appeared misoriented and are not perpendicular to the plane of sectioning. Finally, the boundary between the vertebrae and IVDs was not clearly defined in this genotype—mineral appears more dispersed towards the edge of vertebrae but does not have a clearly defined edge as in wildtype.
VMs develop from fusion across intervertebral segments
To determine the functional consequences of the molecular collagen defect observed by TEM, we assessed vertebral development in col11a2L642*/L642* and col11a2del/del juveniles using alizarin red and calcein vital dyes, which label mineralized tissue (44,45). Together with the optical clarity of juvenile zebrafish, sequential staining using these vital dyes permits in vivo visualization of vertebrae formation over time. Zebrafish developmental stages were determined by standard length (SL) (46), which refers to the length from anterior most point of the snout to the base of the caudal fin. This measurement was found to explain more developmental variance than chronological age, since zebrafish developmental progression can be strongly affected by environmental factors such as temperature, rearing density and nutritional availability.
To begin, 5.5 mm SL (~16 dpf) wildtype, col11a2L642*/L642* and col11a2del/del fish were stained with alizarin red (Fig. 1D). Wildtype animals at this stage exhibit evenly spaced and regularly patterned centra. However, in col11a2L642*/L642* and col11a2del/del fish we observed developing centra with mineralization that extended across the intervertebral space along the ventral edge of the notochord. In col11a2del/del juveniles, these mineralization defects extended between multiple vertebral bodies in a row, whereas col11a2L642*/L642* juveniles exhibited single, isolated fusion events.
Experimental fish were allowed to develop until 8 mm SL (~21 dpf) and then stained with calcein. Alizarin red staining from 5.5 mm stages was still visible, while calcein stained new mineral deposited after that time. In wildtype animals, calcein highlighted growth of vertebral centra on both anterior and posterior surfaces, with even spacing and patterning along the length of the spine (Fig. 1D). In col11a2 mutant fish, vertebrae that had first exhibited ectopic alizarin red staining along the ventral notochord surface at 5.5 mm SL could still be detected. The outside surfaces of these affected centra appear to have developed normally, exhibiting new calcein-labelled vertebral growth. However, intervertebral segments between affected centra were now lost, and calcein stain highlighted ectopic mineralization across the entire dorsal-ventral axis, resulting in two completely fused vertebrae (Fig. 1D).
VMs are associated with ectopic mineralization, not defects in segmentation
To examine the mechanism of vertebral fusion in col11a2del/del animals, we utilized the chondrocytic reporter tg(col9a2::GFPCAAX) (47), the mineralized tissue reporter tgBAC(entpd5a::pKRed) (37), and the osteoblast reporter tg(osx::mCherryNTR) (48). The activity of these reporters has been previously characterized in wildtype zebrafish to define early segmentation of the notochord into cartilaginous and mineralized segments, and to show osteoblast recruitment to the mineralized segments (37). We aimed to examine their activity in col11a2del/del animals to determine whether col11a2del/del mutants exhibit an underlying patterning defect in the notochord.
Prior to 5.5 mm SL, both wildtype and col11a2del/del fish exhibit evenly spaced and alternating segments that are either entpd5a or col9a2 positive, corresponding to mineralized and cartilaginous segments, respectively (Fig. 4). This pattern is consistent with previous work investigating the segmentation of the notochord in wildtype animals (37). By 6 mm SL, wildtype fish exhibit the same pattern of alternating mineralized and cartilaginous domains, but col11a2del/del animals now show some ectopic entpd5a expression within the intervertebral space. These cells are also weakly positive for the col9a2 reporter, which likely represents perdurance of the GFP protein in these cells, as reported previously (37), since all notochord cells initially exhibit a col9a2 positive chondrocytic fate. This result may suggest that cells in chondrocytic intervertebral segments are changing to a mineralized fate.
Figure 4.

Vertebral fusions occur in the absence of gross notochord patterning and segmentation defects. Alternating domains of entpd5a and col9a2 positive cells are present and appear normal in both WT (left) and col11a2del/del mutant (right) embryos at 5.5 mm SL, prior to the onset of fusions (top). At the onset of fusions (6 mm SL; bottom), ectopic entpd5a positive cells are present in the previously cartilaginous domains of col11a2del/del mutant animals (arrowheads). These cells are also weakly col9a2 positive.
Since entpd5a positive cells signal to recruit osteoblasts (37), we reasoned that ectopic entpd5a positive cells between fusing vertebrae may result in abnormal recruitment of osteoblasts to those regions, producing mineral across developing intervertebral segments. We performed live imaging of tg(osx::mCherryNTR) with calcein staining, to visualize the localization of osteoblasts and the mineralizing vertebral centra (Fig. 5), respectively. At 5.5 mm SL, prior to fusion defects, we observed larger and more widespread clusters of osx::mCherryNTR positive cells in col11a2del/del fish than in wildtype. At 6 mm SL, when ectopic mineralization has begun along the ventral edge of the notochord, osteoblasts are present between fusing vertebral centra in col11a2del/del mutants (Fig. 5). These cells are concentrated immediately dorsal to calcein-positive deposits, following the ventral-to-dorsal pattern of vertebral fusion previously observed (Fig. 1). Altogether, our data supports a model where defects in collagen organization in co11a2 mutant zebrafish directs ectopic entpd5a positive sheath cell differentiation within intervertebral segments resulting in abnormal osteoblast recruitment, ectopic mineralization and vertebral fusion.
Figure 5.

col11a2del/del juveniles exhibit ectopic osteoblast recruitment to developing vertebrae, prior to fusion events. Larger and more widespread clusters of osteoblasts, labelled by osx::mCherryNTR (arrowheads), are present in col11a2del/del juveniles compared with wildtype controls prior to the onset of vertebral fusion (top). At later stages when fusions are forming (bottom), osteoblasts can be observed localizing dorsal to progressing fusions in col11a2del/del mutant fish (white arrows).
VM-associated COL11A2 variants are functionally damaging
To investigate the functional consequence of identified COL11A2 VM-associated variants, we assayed the ability of wildtype and mutant transgenic constructs to suppress col11a2del mutant phenotypes. R130 and R1407 residues are conserved in the zebrafish Col11a2, where R130 is homologous to the zebrafish R133 and R1407 is homologous to zebrafish R1542 (Supplementary Material, Fig. S6). Since the third identified VM-associated variant, R1413H, is not conserved in zebrafish (Supplementary Material, Fig. S6), it was not addressed in this study.
To begin, we cloned wildtype zebrafish col11a2 cDNA, and introduced substitutions corresponding to the patient variants to create col11a2 R133W and col11a2 R1542L variants. We then constructed transgenes with each of these col11a2WT, col11a2R133W and col11a2R1542L alleles, in which col11a2 gene expression was driven by (1) an aCNE7 promoter element (49) that drives embryonic notochord-specific expression up to 7 dpf (Fig. 6) or (2) a col2a1a promoter element (50) that drives expression in the notochord throughout embryonic stages, and within intervertebral segments of juvenile animals (Fig. 7). We generated multiple, stable lines for each transgenic element; crossed these transgenes into the col11a2del/del genetic background; and tested the ability of each transgene to suppress the penetrance and severity of vertebral fusions in col11a2del mutants. Fusion penetrance was measured as the fraction of animals sampled that exhibited any fusion defects, and fusion severity was measured as the number of fusions per animal, only among those exhibiting fusion defects.
Figure 6.

Expression of wildtype col11a2 under the aCNE7 promoter reduces the penetrance of fusion defects in col11a2del/+ fish, while patient mutation-bearing transgenes fail to significantly reduce fusion penetrance. (A) The aCNE7 enhancer element drives gene expression within the embryonic notochord, as demonstrated by an aCNE7::eGFP transgenic reporter. (B-D) Graphs reporting vertebral fusion penetrance (proportion of affected animals) and severity (number of fusions per affected animal) observed in col11a2del/+ fish carrying aCNE7::col11a2WT (B), aCNE7::col11a2R133W (C) and aCNE7::col11a2R1542L (D) transgenic lines. For penetrance graphs, error bars represent 95% confidence interval, and significance calculated using chi-squared analysis. For severity graphs, significance calculated using Student’s t-test. tg neg—transgene negative, tg pos—transgene positive, ns—not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Figure 7.
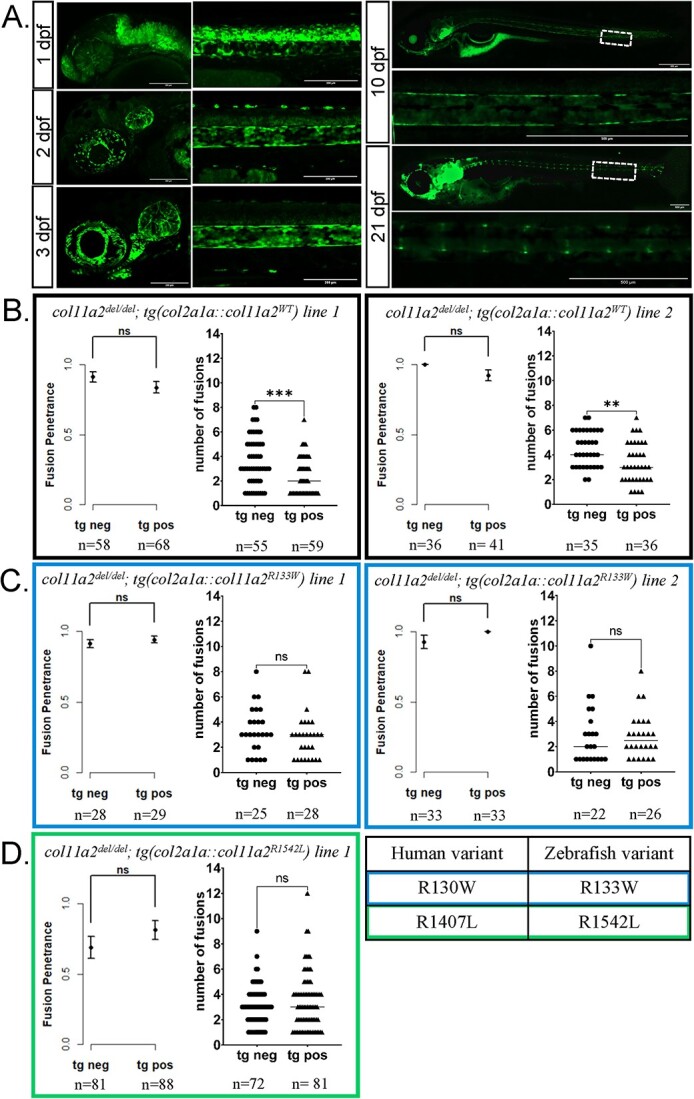
Expression of wildtype col11a2 under the col2a1a promoter reduces the severity, but not penetrance, of fusion defects in col11a2del/del fish, while patient mutation-bearing transgenes fail to significantly reduce fusion severity. (A) The col2a1a promoter drives gene expression expression in the embryonic notochord and later in the juvenile IVDs, as demonstrated by a col2a1a::eGFP transgenic reporter imaged from 1 to 21 dpf. (B-D) Graphs reporting vertebral fusion penetrance and severity observed in col11a2del/del fish carrying col2a1a::col11a2WT (B), col2a1a::col11a2R133W (C), and col2a1a::col11a2R1542L (D) transgenic lines. For penetrance graphs, error bars represent 95% confidence interval, and significance calculated using chi-squared analysis. For severity graphs, significance calculated using Student’s t-test. tg neg—transgene negative, tg pos—transgene positive, ns—not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Notably, two independent aCNE7::col11a2WT transgenic lines could significantly suppress the penetrance of vertebral fusions in col11a2del/+animals, and one tg(aCNE7::col11a2WT) transgene (line 1) also reduced the severity of VMs observed in col11a2del/+ fish (Fig. 6). Similarly, we found that two independent tg(col2a1a::col11a2WT) transgenic lines could significantly reduce vertebral fusion severity in col11a2del/del mutant animals (Fig. 7). Although the spatial pattern and duration of col11a2WT expression differ between aCNE7 and col2a1a transgenic elements (which likely influences their ability to suppress col11a2del/+ vs. col11a2del/del mutant phenotypes), these results indicate that transgenic suppressor assays can provide a quantitative means of assessing protein function.
In contrast to col11a2WT transgene activity, neither tg(aCNE7:: col11a2R133W) nor tg(aCNE7::col11a2R1542L) lines could reduce vertebral fusion penetrance or severity in col11a2del/+animals (Fig. 6). Similarly, neither tg(col2a1a::col11a2R133W) or tg(col2a1a::col11a2R1542L) lines had any significant effects on vertebral fusion penetrance or severity in col11a2del/del fish (Fig. 7). To determine whether observed differences in phenotype suppression were due to insufficient levels of col11a2R133W and col11a2R1542L transgene expression, we quantified col11a2 expression levels from all established transgenic lines using RT-qPCR. No significant differences were observed between col11a2WT, col11a2R133W and col11a2R1542L transgene expression levels (Supplementary Material, Fig. S3B). Taken together, these results demonstrate that a short, early dose of wildtype col11a2 in the notochord is sufficient to suppress vertebral fusions in col11a2del mutant animals. However, since patient variants R133W and R1542L failed to suppress vertebral phenotypes, these substitutions are likely damaging to protein function and may therefore be contributing to the pathogenesis of VMs.
Discussion
In a CS/VM patient cohort, we identified two patients with COL11A2 variants. R130W and R1407L, R1413H variants were identified in two patients with cervical VM. A third patient with a T9 hemivertebra and the R130W variant was identified from a separate study. To demonstrate pathogenicity of these variants we have used a combination of live fluorescent reporters and in vivo staining to characterize two new zebrafish col11a2 mutant models and describe a highly penetrant and severe vertebral fusion phenotype. In creating both a premature termination codon as well as a deletion allele of col11a2, we highlighted the importance of utilizing transcriptless alleles to investigate gene function, as they can avoid nonsense mediated decay-associated transcriptional adaptation mechanisms that can mask true loss-of-function phenotypes (39,40), while revealing haploinsufficiency of the col11a2 locus. Furthermore, the haploinsufficiency observed in our mutant more accurately represents a clinically relevant model for vertebral fusion defects, since patients in this study as well as previous reports in the literature indicate heterozygous pathogenic variants in COL11A2 associate with skeletal dysplasias (24).
We demonstrated that although col11a2del/del mutant embryos do not exhibit any obvious morphological defects, collagen fibrils in the notochord ECM are disorganized at this stage. This subtle phenotype amplifies by juvenile stages into misoriented and denser collagen fibrils in the developing IVDs. Given that the collagenous matrix of the notochord is formed before mineralization begins, and that mineral is deposited into this pre-existing scaffold (51), it is likely that the thinner disorganized layer of collagen fibrils in col11a2del/del embryos may underlie ectopic mineralization defects observed at juvenile stages. Specifically, we demonstrated that vertebral fusion defects form in col11a2 mutant animals at ~5.5 mm SL. Prior to this stage, the segmentation of the developing notochord into alternating mineralized and cartilaginous segments appears to be normal as characterized by entpd5a and col9a2 reporters, respectively. At the onset of vertebral fusions, ectopic expression of entpd5a is evident in the intervertebral space. As fusions progress dorsally, the cartilaginous identity regresses towards the dorsal edge, while the mineralized region expands upwards. This dorsal-to-ventral pattern of fusion formation is consistent with the pattern of teleost vertebral formation, which begins with a basiventral origin (52). We have shown that fusion formation is correlated with osteoblast activity, measured by osx::mCherryNTR. Osteoblasts are precociously recruited to the mineralized notochord segments in col11a2del/del mutant animals, and these cells are likely responsible for the ectopic mineral that is produced in this genotype. This strong osteoblast recruitment, which precedes the formation of fusions, supports a mechanism where entpd5a positive segments recruit osteoblasts to the notochord and osteoblasts begin to produce mineralized matrix across the intervertebral disc region, fusing adjacent vertebrae together.
Our col11a2 mutant models highlight the involvement of the ECM in the formation and patterning of the spine, supporting previous work implicating other collagens in skeletal dysplasias. We have modelled two specific patient variants in the zebrafish, in both a homozygous and heterozygous col11a2 mutant background. Through the rescue experiments of col11a2del/+ fish, we have demonstrated that the patient VM-associated variants R130W and R1407L are likely damaging to protein function, and therefore are hypomorphic alleles of the gene. Interestingly, these experiments show that an early, short dose of functional wildtype col11a2 is sufficient to reduce fusion penetrance. This suggests that there may be a critical window early in development, prior to any mineralization, when functional Col11a2 is most important. Since collagens are very stable and have an extremely long half-life (53), it is also likely that the protein produced and secreted at early stages persists long enough to positively impact cartilage integrity during juvenile vertebral development. Heterozygous COL11A1 mouse mutants exhibit increasing levels of collagen II degradation over time (43), suggesting that collagen XI impacts stability of this major cartilaginous collagen. It is therefore possible that the early dose of Col11a2 prevents collagen II degradation in our zebrafish model. Although tg(aCNE7::col11a2WT) and tg(col2a1a::col11a2WT) expression resulted in significant reduction of VMs in col11a2 mutant animals, we were not able to suppress VMs to a wild type level. This is likely due to low levels of transgene expression, as measured by RT-qPCR, which was significantly less than col11a2 expression from endogenous alleles (Supplementary Material, Fig. S4). However, the fact that even this low dose of col11a2 suppressed vertebral fusions demonstrates the dose-sensitivity to this gene product, which is also consistent with haploinsufficiency producing an intermediate phenotype in col11a2del/+ animals.
Here, we utilized a transgene-based assay to test the functional consequences of patient variants, and showed using RT-qPCR that patient variant transgenes are expressed at levels comparable to the wildtype transgenes. This result supports that differential suppression of vertebral fusions by wildtype versus patient Col11a2 variants is likely due to differences in protein function, and not differences in gene expression levels. However, it is important to note that we did not test differential expression at the protein level, so it is possible that the abundance of patient variant and wildtype Col11a2 protein varies. For example, it is possible that patient variant Col11a2 is less stable than wildtype, and is degraded. Having identified the patient variants R130W and R1407L as hypomorphic pathogenic variants in this work, the question of how these substitutions affect protein function remains. The R130W substitution exists in the signal peptide domain of COL11A2. It is therefore possible that this substitution affects protein transport, potentially leading to a reduced amount of the protein being secreted to the ECM. Alternatively, this variant may affect post-translational modification of the molecule, and since collagens undergo extensive modifications, this could impact the ability of the protein to fold and crosslink successfully (54). Furthermore, this substitution encodes a change from a positively charged to a non-polar residue, which is likely to affect the peptide’s secondary structure. The substituted amino acid, tryptophan, is the least abundant of all amino acids in collagens, including Col11a2, and it has been suggested that this amino acid does not fit well into the helical collagen structure (54). It is worth noting the disparate phenotypes associated with the R130W variants characterized by cervical fusion anomalies in Proband 1 and thoracic hemivertebra in proband 3. The reason for this is unclear. It is possible that additional genetic and/or environmental factors may influence the position and nature of VMs in the presence of a COL11A2 mutant allele.
The other patient variant investigated here, R1407L, occurs in the C-terminus of the collagen triple helix domain, and so its pathogenic effects likely have a different mechanism than the earlier R130W substitution. The C-terminus of the triple helix domain nucleates collagen trimer assembly (55), which normally proceeds from the C- to the N-terminus, in a zipper-like pattern. As a result, pathogenic variants in the C-terminus that disrupt either nucleation or trimer polymerization are likely to disrupt collagen fibril structure more than a similar mutation in the N-terminus of the triple helix domain (54). This R1407L substitution results in a change to leucine, which is non-polar, from arginine, which is positively charged. It is possible that this change alters the ability of the protein to fold in that region, creating a kink or bend in the peptide that disrupts the collagen trimer. Other pathogenic variants in this region have been identified as causative for osteochondrodysplasias (24,26–29). The patients here, however, did not exhibit symptoms characteristic of these conditions, such as sensorineural hearing loss, shorter limbs, short stature and craniofacial defects. This may indicate that the phenotypic outcome of the individual is highly specific to the mutated residue, even within the same domain of the protein. Alternatively, genetic variation at other loci may influence the type and severity of morphological defects that result from similar pathogenic variants in COL11A2. The patient carrying this R1407L variant was also carrying R1413H, although it is not known whether these variants occurred in cis or trans to each other. Since R1413 is not conserved in zebrafish, it was not examined in this study. We showed evidence that R1407L is likely pathogenic, but if these substitutions are in trans, it is possible that R1413H is also damaging to protein function and the patient condition is the outcome of these two variants acting additively.
Unfortunately parents of probands 1 and 2 were not available for whole exome analysis. We plan to investigate other population cohorts for COL11A2 variants. Parent samples from Proband 3 were available and it was found that the COL11A2 variant identified in Proband 3 was inherited from an unaffected parent. While parental X-rays were not obtained, according to the medical record there was no clinically observable scoliosis in either parent. Variable penetrance may explain the occurrence of this phenomenon, which is commonly observed with other candidate genes for VM (16,17,56,57). Possible explanations for an incomplete penetrance finding include: (1) a mild phenotype that was unnoticed in an apparently clinically unaffected parent; (2) a mutant allele interacting with environmental factors; (3) an autosomal recessive phenotype in which the affected individual has a second coding or noncoding variant in COL11A2 that has not yet been identified or (4) an oligogenic phenotype where in which the affected individual has a second coding or noncoding variant in a different gene not yet recognized. Notably, incomplete VM penetrance was also observed in our zebrafish models, where 51% penetrance of vertebral fusions was obtained in col11a2del/+ animals compared with 90% penetrance in the homozygous col11a2del/del group.
Here, we identified patient variants in COL11A2 as pathogenic and likely causative for VMs. However, further work is necessary to clarify the role for this gene in VMs using larger patient cohorts. Although access to parental DNA samples was limited in this study, future work including parental DNA samples for each patient would inform our current understanding of the inheritance pattern for COL11A2-associated VMs. Here, we utilized the zebrafish as a model for human VMs. However, zebrafish vertebrae are not somite-derived, as in humans or mice, although somites do provide patterning cues for the zebrafish spine. It will therefore be interesting to validate results described here using amniote animal models, to determine how accurately this zebrafish model for VMs may recapitulate the developmental phenotype for humans.
Notably, we have also identified a strong positional bias for vertebral fusions in zebrafish, where VMs primarily affect the caudal vertebrae of the spine, even in wildtype fish. Given that fusions in wildtype animals exclusively affect the most posterior vertebrae of the spine, it is likely that the col11a2 pathogenic variants presented here exacerbate an existing susceptibility for fusion formation in that region. Previous work on zebrafish col27a1a and col27a1b gene knock-downs also showed a bias towards vertebral malformations in the distal posterior end of the spine (58), further supporting the idea that posterior vertebrae are inherently susceptible to these defects. Indeed, the caudal part of the zebrafish spine undergoes more stress than the rest of the spine in the process of normal locomotion. Previous work has shown that increased swimming exercise results in increased bone formation in the posterior caudal vertebrae of zebrafish as an adaptive response to this stress (59). The natural swimming pattern of the zebrafish involves a larger degree of tail flexion than any other part of the spine, raising the question of whether this inherent flexibility comes at a cost of increased fusion susceptibility. Interestingly, two of the three probands in this study had fusion defects in the cervical region, which is the most flexible part of the human spine. This underscores an intriguing parallel between our col11a2 zebrafish model and human patients, highlighting the impact of inherent structural characteristics in the formation of vertebral malformations and the utility of zebrafish models for understanding both genetic and biological mechanisms underlying human vertebral malformations.
Materials and Methods
Ethics statement
The collection and use of human DNA samples and data in this study was approved by Institutional Review Boards at the University of Wisconsin-Madison (2014-0520), the Marshfield Clinic Research Institute (GIA10504), the University of Illinois at Chicago (IRB of record, protocol 2020-1509), and the Ethics Committee of Peking Union Medical College Hospital (PUMCH). Written informed consent was required from all subjects at all institutions before being enrolled in study.
Zebrafish husbandry and experimental protocols were approved by the Hospital for Sick Children’s Animal Care Committee, and all protocols were performed in accordance with Canadian Council on Animal Care guidelines.
Recruitment
In order to identify candidate genes associated with VMs, an IRB approved human subjects protocol was implemented at participating study sites including Marshfield Clinic, Wisconsin, University of Wisconsin-Madison and the Hospital for Special Surgery NY. Subjects with VMs were recruited via the pediatric and adult population at the University of Illinois -Chicago. Clinical diagnoses of VM were reviewed and confirmed for each participant by radiologic imaging. Subjects with no identifiable cause for their VM were eligible for recruitment. Probands provided a blood and/or saliva specimen and completed a survey reviewing demographics and exposures during pregnancy in addition to a medical record review to identify possible associated birth defects. DNA was extracted from collected blood and/or saliva specimens from probands by established protocols. Whole exome sequencing and analysis were subsequently performed at Johns Hopkins University School of Medicine.
In a separate study, we consecutively enrolled 873 Chinese individuals (families) affected with congenital VM who underwent spinal surgery at Peking Union Medical College Hospital from November 2012 to November 2021 for correction of scoliosis or kyphosis, under the framework of Deciphering the disorders Involving Scoliosis and COmorbidities study. All individuals underwent a physical examination, spinal X-ray, spinal computed tomography, spinal magnetic resonance imaging, echocardiography and renal ultrasound. The diagnosis of VM was based on the spinal X-ray and was confirmed by both a radiologist and an orthopedic surgeon.
Whole exome sequencing
For samples sequenced at the Baylor-Hopkins Center for Mendelian Genomics (BHCMG), the Agilent SureSelect HumanAllExonV4_51MbKit_S03723314 was used for exome capture. Libraries were sequenced on the HiSeq2500 platform with onboard clustering using 125 bp paired-end runs and sequencing chemistry kits HiSeq PE Cluster Kit v4 and HiSeq SBS Kit v4. Intensity analysis and base calling were performed through the Illumina Real Time Analysis (RTA) software (version 1.17.20).
Variant filtering was done using the Variant Quality Score Recalibration method (60,61). For SNVs, the annotations of MQRankSum, QD, FS, SOR and ReadPosRankSum were used in the adaptive error model. Summary statistics on the multi-sample were calculated for each variant (PASS and FAIL) including counts and frequencies of alleles and genotypes, missing rates, overall quality scores, and mean depth. In addition, separate summary statistics files for just Coriell control samples (HapMap and 1000 Genomes subjects) and just BHCMG subjects for only PASS variants were generated. Variants were analyzed with the PhenoDB analysis tool (62) to filter for rare variants with a minor allele frequency < 0.01 in the 1000 Genome Project, Exome Variant Server, gnomAD, and in-house dataset.
For samples analyzed in China, Illumina paired-end libraries were prepared from DNA samples and subjected to exome capture using the VCRome SeqCap EZ Chice HGSC 96 Reactions capture reagent (Roche) or All Exon V6 + UTR r2 core design (Agilent), followed by sequencing on an Illumina HiSeq 4000 platform. Some samples were analyzed by genome sequencing; sequencing libraries were prepared using the KAPA Hyper Prep kit (KAPA Biosystems) according to the optimized manufacturer's protocol. Multiplex sequencing was performed using an Illumina HiSeq X-Ten or NovaSeq platform. The variant calling and annotation were performed by the in-house developed Peking Union Medical College Hospital Pipeline (63,64). In brief, single nucleotide variants and internal duplications and/or deletions (indels) were called using the HaplotypeCaller of the Genome Analysis Toolkit, version 3.4.0. Annotation for the de novo, compound heterozygotes, and homozygous inherited variants were calculated with Gemini (version 0.19.1) for in silico subtraction of parental variants from the proband’s variants, with accounting for read number information extracted from BAM files.
CRISPR/Cas9 mutagenesis
gRNAs were synthesized using Engen sgRNA synthesis kit, Streptococcus pyrogenes, and purified using Zymo RNA clean and concentrator kit, or ordered from IDT as Alt-R gRNAs. 1 nl of sgRNAs were injected at 100 ng/μl into one-cell staged zebrafish embryos. 1 nl of Alt-R gRNA was injected at 5 μm into one cell staged zebrafish embryos. A subset of injected embryos were lysed and PCR amplified around the site of cleavage to validate CRISPR efficiency by Sanger sequencing using TIDE analysis (65). To establish stable lines, F0 injected embryos were raised to adulthood and outcrossed to wildtype. F1 embryos were lysed and PCR amplified to test for transmission of insertion or deletion mutations.
For col11a2L642*, gRNA 5′- CCACCAGGACCTTCCTTCCC-3′, forward primer 5’-TGCTGACTTTCAGTTCATGTGAT-3′, reverse primer 5’-TTGAAAGTTATTTGTGATTGCTGA-3′. For col11a2del, gRNAs 5’-AACGAGGUCUCCAUGCACCA-3′ and 5’-TAATTGTWGCA CCTGTGGAC-3′, genotyping primers 5’-CCCGCCTTGTAGTCAAAC CA-3′, 5’-TCAGTGAAAGCACCGTCTTG-3′, and 5’-ACTGGACTGGTA AGTTGGCAT-3′.
Alizarin red/calcein staining and live imaging
For live imaging, juvenile zebrafish were stained in 0.02% alizarin red or 0.02% calcein in system water. Fish were stained in petri plates 5 h to overnight, and washed for 2 h to overnight in housing tanks of system water. For sequential imaging of the same animals, fish were anaesthetized in minimal tricane and imaged on an axiozoom, then washed in system water and returned to housing tanks. For terminal imaging, fish were euthanized in a tricane bath and imaged on an axiozoom. For genotyping after imaging, fish were lysed in 50 μl of buffer and proteinase K and 1 μl lysis was used for PCR analysis.
Fusion quantification
Vertebrae were counted manually based on full body calcein stained images taken on an axiozoom. Weberian vertebrae were not included in the count, the anteriormost vertebrae bearing a rib was annotated as ‘1’ for each sample. The position of fusions was recorded during this manual count.
In situ hybridization
Samples were dechorionated and fixed in 4% PFA in 1× PBS overnight. Samples were then rinsed in PBST 2 × 10 min, and dehydrated in methanol. Samples were stored in 100% methanol overnight at 4°C. Samples were washed in a series of 3:1, 1:1 and 1:3 of methanol:PBST for 5 min each. Samples were permeabilized in ProteinaseK (10 μg/ml)—5 minutes for 1 dpf, 45 min for 2 dpf, 1 h for 3 dpf, 1.25 h for 4 dpf and 1.5 h for 5 dpf. Embryos were quickly rinsed in PBST, and then washed again in PBST for 5 min before fixation with 4% PFA in PBS for 20 min at room temperature. Samples were then washed 2× for 5 min in PBST, and incubated in hyb solution at 65°C overnight. Probe was prepared in hyb solution at 1 μg/ml, and embryos were incubated with probe for 60 h at 65°C. Embryos were washed for 30 min in hyb solution at 65°C, followed by a series of 50% formamide, 50% 5× SSC:2× SSC in 3:1, 1:1, and then 1:3 ratios. Samples were then washed in 2× SSC, and then in 0.2× SSC 0.0.1% Tween-20, followed by a series of 3:1, 1:1 and 1:3 0.2× SSC:PBST. Samples were blocked with 1× blocking reagent +5% heat inactivated sheep serum for 3 h at room temperature. Samples were then incubated with Fab-AP antibody (Boehringer) at 1:2500 overnight at 4°C. For detection, samples were washed 8× 15 min each in maleate buffer, followed by three washes of 15 min each in staining buffer, followed by NBT/BCIP at 20 μl/ml in staining buffer. Reactions were developed at 28°C for 1 h in the dark.
Transmission electron microscopy
TEM was performed with assistance from the Nanoscale Biomedical Imaging Facility at the Hospital for Sick Children, Toronto. Zebrafish samples were fixed in glutaraldehyde for 1 h at room temperature followed by overnight at 4°C. Samples were sectioned with standard protocols and imaged on a Hitachi HT7800 transmission electron microscope. ECM thickness was measured using Fiji, and was measured as the distance from the most medial to most distal collagen fibril in the ECM layer.
Confocal microscopy
Zebrafish were treated with minimal tricane to anaesthetize and standard length was measured on a dissecting microscope to select individuals for imaging. Selected animals were euthanized by tricane overdose and mounted laterally on glass bottom dishes in 1% low melt agarose. Images were acquired on a LSM 710 confocal microscope (Zeiss). Maximum intensity projections were created using Fiji.
RT-qPCR
RT-qPCR was performed using the Luna Universal qPCR and RT-qPCR kit (NEB), with 10 ng of template RNA per well. Thermocycler conditions were 60°C annealing temperature, 30 sec/extension cycle, and 40 cycles were run on a Roche Light Cycler 96 HRM machine. Primers for col11a2 were 5’-GAGAAGCCTGGGTCCTTGTT-3′ and 5’-TTGGCAGTGGTAGGTGAGG-3′.
Transgene cloning/injection
RNA was extracted from 1 dpf zebrafish embryos and reverse transcribed to create a cDNA library using the SuperScript IV Reverse Transcriptase kit and protocol (Thermofisher). The col11a2 cDNA was cloned into pCS2+ in three separate amplicons using the following primer pairs:
5’-ATGGATATTCGGAAGAAGCGGA-3′ and 5’-GCCCTTTAGGCCCCAACAAAC-3′.
5’-GACCCCGTGGTTTGTTGGGG-3′ and 5’-CATCTCCCTTGGCACCAAACTG-3′.
5’-TCAACAGGGACAGTTTGGTGC-3′ and 5’-TTATCCCAGGAAACAGACAGGT-3’.
PCR amplicons were assembled together by overlap extension PCR and cloned into pDONR221 via established Gateway cloning protocols. The aCNE7 promoter element was a gift from Dr Ian Scott, and the col2a1a promoter was cloned as in Dale et al. 2011 (50). Transgenes were assembled via the Gateway LR cloning protocol, and injected at 25 pg/μl into single cell staged zebrafish embryos.
Supplementary Material
Acknowledgements
Authors gratefully acknowledge the SickKids’ Nanoscale Biomedical Imaging Facility for assistance with TEM sample preparation and imaging, the SickKids’ Zebrafish Facility technicians for excellent zebrafish care.
Conflicts of Interest statement. The authors declare no competing interests.
Contributor Information
Denise Rebello, Program in Developmental & Stem Cell Biology, The Hospital for Sick Children, Toronto, Ontario M5G 0A4, Canada; Department of Molecular Genetics, The University of Toronto, Toronto, Ontario M5S 1A8, Canada.
Elizabeth Wohler, McKusick-Nathans Department of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA.
Vida Erfani, Program in Developmental & Stem Cell Biology, The Hospital for Sick Children, Toronto, Ontario M5G 0A4, Canada; Department of Molecular Genetics, The University of Toronto, Toronto, Ontario M5S 1A8, Canada.
Guozhuang Li, Department of Orthopedic Surgery, Key Laboratory of Big Data for Spinal Deformities, Beijing Key Laboratory for Genetic Research of Skeletal Deformity, State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100730, China.
Alexya N Aguilera, Department of Pediatrics, University of Illinois-Chicago, Chicago, IL 60612, USA.
Alberto Santiago-Cornier, Genetic Section, San Jorge Children’s and Women’s Hospital, San Juan, Puerto Rico 00912, USA; Department of Public Health, Ponce Health Sciences University, Ponce, Puerto Rico 00912, USA.
Sen Zhao, Department of Orthopedic Surgery, Key Laboratory of Big Data for Spinal Deformities, Beijing Key Laboratory for Genetic Research of Skeletal Deformity, State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100730, China; Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA.
Steven W Hwang, Shriners Children’s-Philadelphia, Philadelphia, PA 19140, USA.
Robert D Steiner, Department of Pediatrics, University of Wisconsin, Madison, WI 54449, USA; Marshfield Clinic Health System, Marshfield, WI 54449, USA.
Terry Jianguo Zhang, Department of Orthopedic Surgery, Key Laboratory of Big Data for Spinal Deformities, Beijing Key Laboratory for Genetic Research of Skeletal Deformity, State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100730, China.
Christina A Gurnett, Department of Neurology, Washington University in St. Louis, St. Louis, MO 63110, USA.
Cathleen Raggio, Hospital for Special Surgery, New York, NY 10021, USA.
Nan Wu, Department of Orthopedic Surgery, Key Laboratory of Big Data for Spinal Deformities, Beijing Key Laboratory for Genetic Research of Skeletal Deformity, State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100730, China.
Nara Sobreira, McKusick-Nathans Department of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA.
Philip F Giampietro, Department of Pediatrics, University of Illinois-Chicago, Chicago, IL 60612, USA.
Brian Ciruna, Program in Developmental & Stem Cell Biology, The Hospital for Sick Children, Toronto, Ontario M5G 0A4, Canada; Department of Molecular Genetics, The University of Toronto, Toronto, Ontario M5S 1A8, Canada.
Data Availability
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
Funding
Canadian Institutes of Health Research (FDN-167285) and the Canada Research Chair program awarded to b.c.; National Institutes of Health funding (R03 HD099516-01A1) and Dr Asok K. Ray and Purnima Ray Professorship in Pediatrics awarded to P.F.G.; National High Level Hospital Clinical Research Funding (2022-PUMCH-D-007, 2022-PUMCH-C-033) and CAMS Innovation Fund for Medical Sciences (CIFMS) (2021-I2M-1-051) both to T.J.Z. and N.W.
References
- 1. Wu, N., Ming, X., Xiao, J., Wu, Z., Chen, X., Shinawi, M., Shen, Y., Yu, G., Liu, J., Xie, H. et al. (2015) TBX6 null variants and a common hypomorphic allele in congenital scoliosis. NEJM, 372, 341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Heiskanen, S., Syvänen, J., Helenius, I., Kemppainen, T., Löyttyniemi, E., Gissler, M. and Raitio, A. (2022) Increasing prevalence and high risk of associated anomalies in congenital vertebral defects: a population-based study. J. Pediatr. Orthop., 42, e538–e543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Giampietro, P.F., Dunwoodie, S.L., Kusumi, K., Pourquié, O., Tassy, O., Offiah, A.C., Cornier, A.S., Alman, B.A., Blank, R.D., Raggio, C.L., Glurich, I. and Turnpenny, P.D. (2009) Progress in the understanding of the genetic etiology of vertebral segmentation disorders in humans. Ann. N. Y. Acad. Sci., 1151, 38–67. [DOI] [PubMed] [Google Scholar]
- 4. Nan, W. (2023) Retrospective analysis of associated anomalies in 636 patients with operatively treated congenital scoliosis. J. Bone Jt. Surg.Am., 105, 537–548. [DOI] [PubMed] [Google Scholar]
- 5. Pourquie, O. (2001) Vertebrate somitogenesis. Annu. Rev. Cell Dev. Biol., 17, 311–350. [DOI] [PubMed] [Google Scholar]
- 6. Pourquié, O. (2011) Vertebrate segmentation: from cyclic gene networks to scoliosis. Cell, 145, 650–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eckalbar, W.L., Fisher, R.E., Rawls, A. and Kusumi, K. (2012) Scoliosis and segmentation defects of the vertebrae. Wiley Interdiscip. Rev. Dev. Biol., 1, 401–423. [DOI] [PubMed] [Google Scholar]
- 8. Erol, B., Kusumi, K., Lou, J. and Dormans, J.P. (2002) Etiology of congenital scoliosis. UPOJ, 15, 37–42. [Google Scholar]
- 9. Giampietro, P.F., Raggio, C.L., Blank, R.D., McCarty, C., Broeckel, U. and Pickart, M.A. (2013) Clinical, genetic and environmental factors associated with congenital vertebral malformations. Mol. Syndromol., 4, 94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sparrow, D.B., Chapman, G., Smith, A.J., Mattar, M.Z., Major, J.A., O’Reilly, V.C., Saga, Y., Zackai, E.H., Dormans, J.P., Alman, B.A. et al. (2012) A mechanism for gene-environment interaction in the etiology of congenital scoliosis. Cell, 149, 295–306. [DOI] [PubMed] [Google Scholar]
- 11. Liu, J., Wu, N., Yang, N., Takeda, K., Chen, W., Li, W., Du, R., Liu, S., Zhou, Y., Zhang, L. et al. (2019) TBX6-associated congenital scoliosis (TACS) as a clinically distinguishable subtype of congenital scoliosis: further evidence supporting the compound inheritance and TBX6 gene dosage model. Genet. Med., 21, 1548–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Giampietro, P.F., Raggio, C.L., Reynolds, C.E., Shukla, S.K., McPherson, E., Ghebranious, N., Jacobsen, F.S., Kumar, V., Faciszewski, T., Pauli, R.M. et al. (2005) An analysis of PAX1 in the development of vertebral malformations. Clin. Genet., 68, 448–453. [DOI] [PubMed] [Google Scholar]
- 13. Giampietro, P.F., Raggio, C.L., Reynolds, C., Ghebranious, N., Burmester, J.K., Glurich, I., Rasmussen, K., McPherson, E., Pauli, R.M., Shukla, S.K. et al. (2006) DLL3 as a candidate gene for vertebral malformations. Am. J. Med. Genet. A, 140A, 2447–2453. [DOI] [PubMed] [Google Scholar]
- 14. Ghebranious, N., Raggio, C.L., Blank, R.D., McPherson, E., Burmester, J.K., Ivacic, L., Rasmussen, K., Kislow, J., Glurich, I., Jacobsen, F.S. et al. (2007) Lack of evidence of WNT3A as a candidate gene for congenital vertebral malformations. Scoliosis, 2, 13. 10.1186/1748-7161-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Giampietro, P.F., Armstrong, L., Stoddard, A., Blank, R.D., Livingston, J., Raggio, C.L., Rasmussen, K., Pickart, M., Lorier, R., Turner, A. et al. (2015) Whole exome sequencing identifies a POLRID mutation segregating in a father and two daughters with findings of Klippel-Feil and Treacher Collins syndromes. Am. J. Med. Genet., 167A, 95–102. [DOI] [PubMed] [Google Scholar]
- 16. Ghebranious, N., Blank, R.D., Raggio, C.L., Staubli, J., McPherson, E., Ivacic, L., Rasmussen, K., Jacobsen, F.S., Faciszewski, T., Burmester, J.K. et al. (2008) A missense T (Brachyury) mutation contributes to vertebral malformations. J. Bone Miner. Res., 23, 1576–1583. [DOI] [PubMed] [Google Scholar]
- 17. Al Dhaheri, N., Wu, N., Zhao, S., Wu, Z., Blank, R.D., Zhang, J., Raggio, C., Halanski, M., Shen, J., Noonan, K. et al. (2020) KIAA1217: a novel candidate gene associated with isolated and syndromic vertebral malformations. Am. J. Med. Genet., 182, 1664–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Martin, E.M.M.A., Enriquez, A., Sparrow, D.B., Humphreys, D.T., McInerney-Leo, A.M., Leo, P.J., Duncan, E.L., Iyer, K.R., Greasby, J.A., Ip, E. et al. (2020) Heterozygous loss of WBP11 function causes multiple congenital defects in humans and mice. Hum. Mol. Genet., 29, 3662–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lin, M., Zhao, S., Liu, G., Huang, Y., Yu, C., Zhao, Y., Wang, L., Zhang, Y., Yan, Z., Wang, S. et al. (2020) Identification of novel FBN1 variations implicated in congenital scoliosis. J. Hum. Genet., 65, 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang, S., Chai, X., Yan, Z., Zhao, S., Yang, Y., Li, X., Niu, Y., Lin, G., Su, Z., Wu, Z., Zhang, T.J. and Wu, N. (2021) Novel FGFR1 variants are associated with congenital scoliosis. Genes., 12, 1126. 10.3390/genes12081126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lek, M., Karczewski, K.J., Minikel, E.V., Samocha, K.E., Banks, E., Fennell, T., O’Donnell-Luria, A.H., Ware, J.S., Hill, A.J., Cummings, B.B. et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Adzhubei, I.A., Schmidt, S., Peshkin, L., Ramensky, V.E., Gerasimova, A., Bork, P., Kondrashov, A.S. and Sunyaev, S.R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mio, F., Chiba, K., Hirose, Y., Kawaguchi, Y., Mikami, Y., Oya, T., Mori, M., Kamata, M., Matsumoto, M., Ozaki, K. et al. (2007) A functional polymorphism in COL11A1, which encodes the α1 chain of type XI collagen, is associated with susceptibility to lumbar disc herniation. Am. J. Med. Genet., 81, 1271–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vikkula, M., Madman, E.C.M., Lui, V.C.H., Zhidkova, N.I., Tiller, G.E., Goldring, M.B., van Beersum, S.E.C., de Waal Malefijt, M.C., van den Hoogen, F.H.J., Ropers, H.-H. et al. (1995) Autosomal dominant and recessive osteochondrodysplasias associated with the COL11A2 locus. Cell, 80, 431–437. [DOI] [PubMed] [Google Scholar]
- 25. Annunen, S., Körkkö, J., Czarny, M., Warman, M.L., Brunner, H.G., Kääriäinen, H., Mulliken, J.B., Tranebjærg, L., Brooks, D.G., Cox, G.F. et al. (1999) Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/stickler phenotypes. Am. J. Med. Genet., 65, 974–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Melkoniemi, M., Brunner, H.G., Manouvrier, S., Hennekam, R., Superti-Furga, A., Kääriäinen, H., Pauli, R.M., van Essen, T., Warman, M.L., Bonaventure, J., Miny, P. and Ala-Kokko, L. (2000) Autosomal recessive disorder otospondylomegaepiphyseal dysplasia is associated with loss-of-function mutations in the COL11A2 gene. Am. J. Med. Genet., 66, 368–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brunner, H.G., van Beersum, S.E., Warman, M.L., Olsen, B.R., Ropers, H.-H. and Mariman, E.C. (1994) A stickler syndrome gene is linked to chromosome 6 near the COL11A2 gene. Hum. Mol. Genet., 3, 1561–1564. [DOI] [PubMed] [Google Scholar]
- 28. Pihlajamaa, T., Prockop, D.J., Faber, J., Winterpacht, A., Zabel, B., Giedion, A., Wiesbauer, P., Spranger, J. and Ala-Kokko, L. (1998) Heterozygous glycine substitution in the COL11A2 gene in the original patient with the Weissenbacher-Zweymüller syndrome demonstrates its identity with heterozygous OSMED (nonocular stickler syndrome). Am. J. Med. Genet., 80, 115–120. [DOI] [PubMed] [Google Scholar]
- 29. Vuoristo, M.M., Pappas, J.G., Jansen, V. and Ala-Kokko, L. (2004) A stop codon mutation in COL11A2 induces exon skipping and leads to non-ocular stickler syndrome. Am. J. Med. Genet., 130A, 160–164. [DOI] [PubMed] [Google Scholar]
- 30. Tompson, S.W., Faqeih, E.A., Ala-Kokko, L., Hecht, J.T., Miki, R., Funari, T., Funari, V.A., Nevarez, L., Krakow, D. and Cohn, D.H. (2012) Dominant and recessive forms of fibrochondrogenesis resulting from mutations at a second locus, COL11A2. Am. J. Med. Genet., 158A, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li, Y., Lacerda, D.A., Warman, M.L., Beier, D.R., Yoshioka, H., Ninomiya, Y., Oxford, J.T., Morris, N.P., Andrikopoulos, K., Ramirez, F. et al. (1995) A fibrillar collagen gene, Col11a1, is essential for skeletal morphogenesis. Cell, 80, 423–430. [DOI] [PubMed] [Google Scholar]
- 32. Li, S.-W., Takanosu, M., Arita, M., Bao, Y., Ren, Z.-X., Maier, A., Prockop, D.J. and Mayne, R. (2001) Targeted disruption of Col11a2 produces a mild cartilage phenotype in transgenic mice: comparison with the human disorder otospondylomegaepiphyseal dysplasia (OSMED). Dev. Dyn., 222, 141–152. [DOI] [PubMed] [Google Scholar]
- 33. Lawrence, E.A., Kague, E., Aggleton, J.A., Harniman, R.L., Roddy, K.A. and Hammond, C.L. The mechanical impact of col11a2 loss on joints; col11a2 mutant zebrafish show changes to joint development and function, which leads to early-onset osteoarthritis. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci., 14, 20170335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ward, L., Pang, A.S.W., Evans, S.E. and Stern, C.D. (2018) The role of the notochord in amniote vertebral column segmentation. Dev. Biol., 439, 3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Choi, K.-S., Cohn, M.J. and Harfe, B.D. (2008) Identification of nucleus pulposus precursor cells and notochordal remnants in the mouse: implications for disk degeneration and chordoma formation. Dev. Dyn., 237, 3953–3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fleming, A., Keynes, R. and Tannahill, D. (2004) A central role for the notochord in vertebral patterning. Dev., 131, 873–880. [DOI] [PubMed] [Google Scholar]
- 37. Wopat, S., Bagwell, J., Sumigray, K.D., Dickson, A.L., Huitema, L.F.A., Poss, K.D., Schulte-Merker, S. and Bagnat, M. (2018) Spine patterning is guided by segmentation of the notochord sheath. Cell Rep., 22, 2026–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Baas, D., Malbouyres, M., Haftek-Terreau, Z., Le Guellec, D. and Ruggiero, F. (2009) Craniofacial cartilage morphogenesis requires zebrafish col11a1 activity. Matrix Biol., 28, 490–502. [DOI] [PubMed] [Google Scholar]
- 39. El-Brolosy, M.A., Kontarakis, Z., Rossi, A., Kuenne, C., Günther, S., Fukuda, N., Kikhi, K., Boezio, G.L.M., Takacs, C.M., Lai, S.-L. et al. (2019) Genetic compensation triggered by mutant mRNA degradation. Nature, 568, 193–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ma, Z., Zhu, P., Shi, H., Guo, L., Zhang, Q., Chen, Y., Chen, S., Zhang, Z., Peng, J. and Chen, J. (2019) PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature, 568, 259–263. [DOI] [PubMed] [Google Scholar]
- 41. Trapani, V., Bonaldo, P. and Corallo, D. (2017) Role of the ECM in notochord formation, function and disease. J. Cell Sci., 130, 3203–3211. [DOI] [PubMed] [Google Scholar]
- 42. Blaschke, U.K., Eikenberry, E.F., Hulmes, D.J.S., Galla, H.-J. and Bruckner, P. (2000) Collagen XI nucleates self-assembly and limits lateral growth of cartilage fibrils *. J. Biol. Chem., 275, 10370–10378. [DOI] [PubMed] [Google Scholar]
- 43. Rodriguez, R.R., Seegmiller, R.E., Stark, M.R. and Bridgewater, L.C. (2004) A type XI collagen mutation leads to increased degradation of type II collagen in articular cartilage11. Osteoarthr. Cartil., 12, 314–320. [DOI] [PubMed] [Google Scholar]
- 44. Bensimon-Brito, A., Cardeira, J., Dionísio, G., Huysseune, A., Cancela, M.L. and Witten, P.E. (2016) Revisiting in vivo staining with alizarin red S - a valuable approach to analyse zebrafish skeletal mineralization during development and regeneration. BMC Dev. Biol., 16, 2. 10.1186/s12861-016-0102-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Du, S.J., Frenkel, V., Kindschi, G. and Zohar, Y. (2001) Visualizing normal and defective bone development in zebrafish embryos using the fluorescent chromophore calcein. Dev. Biol., 238, 239–246. [DOI] [PubMed] [Google Scholar]
- 46. Parichy, D.M., Elizondo, M.R., Mills, M.G., Gordon, T.N. and Engeszer, R.E. (2009) Normal table of postembryonic zebrafish development: staging by externally visible anatomy of the living fish. Dev. Dyn., 238, 2975–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Garcia, J., Bagwell, J., Njaine, B., Norman, J., Levic, D.S., Wopat, S., Miller, S.E., Liu, X., Locasale, J.W., Stainier, D.Y.R. and Bagnat, M. (2017) Sheath cell invasion and trans-differentiation repair mechanical damage caused by loss of Caveolae in the zebrafish notochord. Curr. Biol., 27, 1982–1989.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Singh, S.P., Holdway, J.E. and Poss, K.D. (2012) Regeneration of amputated zebrafish fin rays from de novo osteoblasts. Dev. Cell, 22, 879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yuan, X., Song, M., Devine, P., Bruneau, B.G., Scott, I.C. and Wilson, M.D. (2018) Heart enhancers with deeply conserved regulatory activity are established early in zebrafish development. Nat. Commun., 9, 4977. 10.1038/s41467-018-07451-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dale, R.M. and Topczewski, J. (2011) Identification of an evolutionarily conserved regulatory element of the zebrafish col2a1a gene. Dev. Biol., 357, 518–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang, S., Kryvi, H., Grotmol, S., Wargelius, A., Krossøy, C., Epple, M., Neues, F., Furmanek, T. and Totland, G.K. (2013) Mineralization of the vertebral bodies in Atlantic salmon (Salmo salar L.) is initiated segmentally in the form of hydroxyapatite crystal accretions in the notochord sheath. J. Anat., 223, 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bensimon-Brito, A., Cancela, M.L., Huysseune, A. and Witten, P.E. (2012) Vestiges, rudiments and fusion events: the zebrafish caudal fin endoskeleton in an evo-devo perspective. Evol. Dev., 14, 116–127. [DOI] [PubMed] [Google Scholar]
- 53. Tiku, M.L. and Madhan, B. (2016) Preserving the longevity of long-lived type II collagen and its implication for cartilage therapeutics. Ageing Res. Rev., 28, 62–71. [DOI] [PubMed] [Google Scholar]
- 54. Myllyharju, J. and Kivirikko, K.I. (2001) Collagens and collagen-related diseases. Ann. Med., 33, 7–21. [DOI] [PubMed] [Google Scholar]
- 55. McLaughlin, S.H. and Bulleid, N.J. (1998) Molecular recognition in procollagen chain assembly. Matrix Biol., 16, 369–377. [DOI] [PubMed] [Google Scholar]
- 56. Edwards, Y.H., Putt, W., Lekoape, K.M., Stott, D., Fox, M., Hopkinson, D.A. and Sowden, J. (1996) The human homolog T of the mouse T (Brachyury) gene; gene structure, cDNA sequence, and assignment to chromosome 6q27. Genome Res., 6, 226–233. [DOI] [PubMed] [Google Scholar]
- 57. Papapetrou, C., Drummond, F., Reardon, W., Winter, R., Spitz, L. and Edwards, Y.H. (1999) A genetic study of the human T gene and its exclusion as a major candidate gene for sacral agenesis with anorectal atresia. J. Med. Genet., 36, 208–213. [PMC free article] [PubMed] [Google Scholar]
- 58. Christiansen, H.E., Lang, M.R., Pace, J.M. and Parichy, D.M. (2009) Critical early roles for col27a1a and col27a1b in zebrafish notochord morphogenesis, vertebral mineralization and post-embryonic axial growth. PLoS One, 4, e8481. 10.1371/journal.pone.0008481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Suniaga, S., Rolvien, T., vom Scheidt, A., Fiedler, I.A.K., Bale, H.A., Huysseune, A., Witten, P.E., Amling, M. and Busse, B. (2018) Increased mechanical loading through controlled swimming exercise induces bone formation and mineralization in adult zebrafish. Sci. Rep., 8, 3646. 10.1038/s41598-018-21776-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. DePristo, M.A., Banks, E., Poplin, R., Garimella, K.V., Maguire, J.R., Hartl, C., Philippakis, A.A., del Angel, G., Rivas, M.A., Hanna, M. et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet., 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Van der Auwera, G.A., Carneiro, M.O., Hartl, C., Poplin, R., del Angel, G., Levy-Moonshine, A., Jordan, T., Shakir, K., Roazen, D., Thibault, J. et al. (2013) From fastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinformatics, 43, 11.10.1-11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sobreira, N., Schiettecatte, F., Boehm, C., Valle, D. and Hamosh, A. (2015) New tools for Mendelian disease gene identification: PhenoDB variant analysis module; and GeneMatcher, a web-based tool for linking investigators with an interest in the same gene. Hum. Mutat., 36, 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang, K., Zhao, S., Liu, B., Zhang, Q., Li, Y., Liu, J., Shen, Y., Ding, X., Lin, J., Wu, Y. et al. (2018) Perturbations of BMP/TGF-β and VEGF/VEGFR signalling pathways in non-syndromic sporadic brain arteriovenous malformations (BAVM). J. Med. Genet., 55, 675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhao, S., Zhang, Y., Chen, W., Li, W., Wang, S., Wang, L., Zhao, Y., Lin, M., Ye, Y., Lin, J. et al. (2021) Diagnostic yield and clinical impact of exome sequencing in early-onset scoliosis (EOS). J. Med. Genet., 58, 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Brinkman, E.K., and van Steensel, B. (2019). Rapid Quantitative Evaluation of CRISPR Genome Editing by TIDE and TIDER. In: Luo, Y. (eds) CRISPR Gene Editing. Methods in Molecular Biology, vol 1961. Humana Press, New York, NY. 10.1007/978-1-4939-9170-9_3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.