

Abstract
The secretion of insulin from β-cells in the islet of Langerhans is governed by a series of metabolic and electrical events, which can fail during the progression of type2 diabetes (T2D). β-cells are electrically coupled via Cx36 gap junction channels, which coordinates the pulsatile dynamics of [Ca2+] and insulin release across the islet. Factors such as pro-inflammatory cytokines and free fatty acids disrupt gap junction coupling under invitro conditions. Here we test whether gap junction coupling and coordinated [Ca2+] dynamics are disrupted in T2D, and whether recovery of gap junction coupling can recover islet function. We examine islets from donors with T2D, from db/db mice, and islets treated with proinflammatory cytokines (TNF-α, IL-1β, IFN-ɣ) or free fatty acids (palmitate). We modulate gap junction coupling using Cx36 over-expression or pharmacological activation via modafinil. We also develop a peptide mimetic (S293) of the c-terminal regulatory site of Cx36 designed to compete against its phosphorylation. Cx36 gap junction permeability and [Ca2+] dynamics were disrupted in islets from both human donors with T2D and db/db mice, and in islets treated with proinflammatory cytokines or palmitate. Cx36 over-expression, modafinil treatment and S293 peptide all enhanced Cx36 gap junction coupling and protected against declines in coordinated [Ca2+] dynamics. Cx36 over-expression and S293 peptide also reduced apoptosis induced by proinflammatory cytokines. Critically S293 peptide rescued gap junction coupling and [Ca2+] dynamics in islets from both db/db mice and a sub-set of T2D donors. Thus, recovering or enhancing Cx36 gap junction coupling can improve islet function in diabetes.
Keywords: Diabetes, islet of Langerhans, gap junctions, Ca2+, peptide mimetic
Graphical Abstract
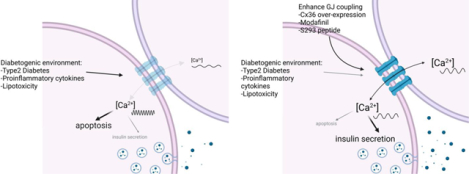
Left: Diabetogenic environments disrupt Cx36 gap junction coupling, Ca2+ activity/coordination and insulin secretion while increasing apoptosis. Right: Elevating Cx36 via genetic means, Modafinil or S293 peptide recovers Ca2+ activity/coordination and insulin secretion while reducing apoptosis.
INTRODUCTION
Type2 diabetes (T2D) results from β-cell failure in the context of obesity-related insulin resistance, such that insulin production does not meet physiological demands. Many patients with T2D require β-cell secretagogues or exogenous insulin therapy (Turner et al., 1999; van Raalte & Diamant, 2011) following the loss of first phase insulin secretion and reduced second phase insulin dynamics (Meier et al., 2005; Menge et al., 2011). While the etiology of T2D is multifactorial, it is becoming increasingly clear that T2D develops in patients after the onset of β-cell dysfunction (Pfeifer et al., 1981; Turner et al., 1999; Prentki & Nolan, 2006; Ray et al., 2009; van Raalte & Diamant, 2011). Both reduced β-cell mass as well as intrinsic β-cell dysfunction (de Koning et al., 2008; Bonner-Weir & O’Brien, 2008) have been shown to exist prior to clinical onset of disease (Xu et al., 2003; Abdulreda & Berggren, 2013). Therefore, therapeutics that maintain β-cell mass and function during T2D progression have the potential to prevent disease development. While inflammation and lipotoxicity are implicated in T2D, the mechanisms that determine the advancement of islet dysfunction in pre-T2D are poorly understood.
Pancreatic islets of Langerhans maintain blood glucose homeostasis by producing and secreting insulin. Insulin secretion is triggered by glucose-stimulated increases in intracellular free-calcium ([Ca2+]) (Prentki et al., 1997; Schuit et al., 2001; Benninger & Piston, 2014). Glucose-stimulated [Ca2+] increases are in the form of oscillations that generate pulses of insulin release, which are important for proper insulin action and glucose utilization (Meier et al., 2005; Bertram et al., 2007). Connexin-36 (Cx36) forms gap junctions (GJs) that electrically couple β-cells within islets (Ravier et al., 2005; Benninger et al., 2008). Loss-of-function studies have demonstrated that Cx36 GJs are critical for controlling oscillatory [Ca2+] across the islet to generate pulsatile insulin secretion under elevated glucose (Ravier et al., 2005; Benninger et al., 2008; Head et al., 2012), as well as to dampen spontaneous β-cell electrical activity and inhibit insulin secretion under low glucose (Speier et al., 2007; Benninger et al., 2011). In the absence of Cx36, there is a complete absence of gap junction electrical coupling, and disorganized and unsynchronized [Ca2+] oscillations lead to a complete loss of first phase insulin secretion, diminished second phase pulses and glucose intolerance (Ravier et al., 2005; Head et al., 2012). Furthermore, proper GJ coupling has been shown to be important for protecting against β-cell apoptosis and maintaining β-cell mass (Klee et al., 2011b, 2011a). While a direct role for GJ coupling in T2D progression remains unclear, the physiological consequences of islet uncoupling by a genetic disruption of Cx36 is analogous to pathological observations of patients T2D (Pfeifer et al., 1981; Menge et al., 2011), including glucose intolerance and altered insulin secretion dynamics suggesting a role for islet GJ uncoupling in the disease.
Chronic inflammation and gluco-lipotoxicity as a result of insulin resistance and metabolic stress in T2D has detrimental effects on β-cell function, including dysregulated [Ca2+] dynamics, reduced insulin synthesis and secretion, and increased β-cell apoptosis (Lee et al., 1994; McGarry, 2002; El-Assaad et al., 2003; Boden, 2005, 2008; Poitout et al., 2006; Hatanaka et al., 2014). For example, in obese individuals, excessive adipose mass releases increasing amounts of FFAs and pro-inflammatory cytokines into the bloodstream (Xu et al., 2003; Donath et al., 2008; Imai et al., 2016). These pro-inflammatory cytokines include tumor necrosis factor alpha (TNF-α) (Hotamisligil et al., 1993), interleukin (IL)-6 (Mahdi et al., 2012), IL-1β (Maedler et al., 2017), among others (Tataranni & Ortega, 2005; Marchetti, 2016). Pro-inflammatory cytokines can induce GJ uncoupling within mouse and human islets (Farnsworth et al., 2016a). Chronic FFA exposure can also induce GJ uncoupling within mouse and human islets (Hodson et al., 2013). In animal models of obesity, dysregulated [Ca2+] dynamics have also been observed (Ravier et al., 2002; Corezola do Amaral et al., 2020). Taken together, this suggests a role for GJ uncoupling in the development of islet dysfunction in T2D. However, little is known about the impact of chronic FFA and inflammation on Cx36 GJ uncoupling and [Ca2+] in prior to T2D and whether in fact this uncoupling significantly impacts the progression of islet dysfunction.
Cx36 gap junction coupling protects β-cells from the susceptibility of apoptosis (Klee et al., 2011a). Together with the role for GJ coupling in regulating insulin secretion, maintenance of Cx36 function in diabetic islets may have a therapeutic benefit. Modulation of GJ function with structural mimetic peptides has been demonstrated in treatment of cardiac arrhythmias (Eloff et al., 2003), myocardial infarction and ischemia (Hawat et al., 2012). The C-terminus of Cx36 contains many sites that regulate GJ function, including phosphorylation by PKA or CaMKII (Urschel et al., 2006; Kothmann et al., 2007; Alev et al., 2008), or interactions with microtubules (Brown et al., 2019) and tight junctions (Flores et al., 2008). Both PKA and PKCδ has been demonstrated to regulate Cx36 GJ coupling within the islet (Hodson et al., 2013; Farnsworth et al., 2016a). Thus, we hypothesized that maintaining Cx36 GJ coupling during inflammatory and lipotoxic events in T2D may provide a novel mechanism by which insulin secretion and β-cell mass can be maintained during pre-diabetes, and thus delay or prevent disease onset.
In this study, we sought to determine the pro-inflammatory and FFA environmental factors that influence GJ and Cx36 function, and to identify a mechanism by which GJ uncoupling could be prevented in the face of diabetogenic factors. We show that GJ coupling, insulin secretion, and cell viability are all negatively affected by diabetogenic environments, including in human T2D, animal models of diabetes and by pro-inflammatory cytokines and FFAs. We further demonstrate that reducing or preventing the decline in Cx36 gap junction coupling by either pharmacological activators or by a novel mimetic inhibitory peptide can preclude this dysfunction.
METHODS
Ethical Approval:
All experimental procedures were performed at the University of Colorado Anschutz Medical Campus, and were approved by the Institutional Care and Use Committee (IACUC) protocol #000024 and B-95817(05)1D. Work with human islets was considered ‘not human research’ COMIRB # 13–1895.
Animals:
Commercially-available wildtype female C57BL/6NHsd (Envigo stock #044) were used aged 12–16 weeks. Diabetes prone male and female C57BKS.Cg-Leprdb (“db/db”) mice were used aged 4–6 weeks (Jackson Laboratories stock #000642) with appropriate age- and sex-matched strain C57BLKS control mice (Jackson Laboratories stock #000662).
Islet isolation and culture:
For mouse islet isolation, animals were injected IP with ketamine/xylazine at 83/17 mg/kg to induce general anesthesia, with depth of anesthesia confirmed via absence of pedal withdrawal. Mice were euthanized via exsanguination or bilateral thoracotomy with cervical dislocation as a secondary method. Islets were isolated by a method previously described elsewhere following collagenase infusion, pancreas dissection and digestion, and hand picking of islets (Koster et al., 2002; Farnsworth et al., 2016a). Isolated islets were cultured overnight in RPMI medium 1640 (Life Technologies) supplemented with 10% FBS, 11mM glucose, 100 units/ml of penicillin, and 100 μg/ml of streptomycin at 37 °C under humidified 5% CO2.
Human islets were received from the Integrated Islet Distribution Program (https://iidp.coh.org/), obtained from either healthy donors or donors with type2 diabetes (Table 1) and were cultured overnight in CMRL medium 1066 (Fisher Scientific) prior to initiating experiments.
Table 1:
List of human donors from which islets were examined in this study, with the experiments performed and treatments applied and whether they responded to treatment.
| UNOS ID | Diabetes status | Experiments performed: | Treatments: | |||
|---|---|---|---|---|---|---|
| GSIS | Ca2+ | GJ FRAP | S293 pep | Modafinil | ||
| AA2482 | T2D | x | x | x | ||
| ABDG032 | T2D | x | x | x | ||
| ABFG183 | T2D | x | x | x | ||
| ACJV399 | T2D | x | x | x | x (resp.) | x |
| ADCD391 | T2D | x | x | x | x (resp.) | x |
| ADCI176 | T2D | x | x | x | x (non.resp.) | x |
| ADCY327 | T2D | x | x | x | x (resp.) | x |
| ADGD159 | T2D | x | x (non.resp.) | |||
| AECB422 | T2D | x | x | x | x (resp.) | x |
| AEC2011 | T2D | x | x | x | x (non.resp.) | x |
| AEDZ307 | T2D | x | x | x | x (non.resp.) | x |
| AAIQ245 | ND | x | x | |||
| ABD1375 | ND | x | x | |||
| ABEI419 | ND | x | x | |||
| ABFG183 | ND | x | ||||
| ABFE184B | ND | x | x | |||
| ABFV041 | ND | x | x | |||
| ABGK387A | ND | x | x | x | ||
| ABG2361 | ND | x | x | |||
| ABHC283 | ND | x | x | |||
| ABJR111 | ND | x | ||||
| ABJ5204 | ND | x | x | |||
| ACBO251 | ND | x | x | |||
| ACCR206A | ND | x | x | |||
| ACCZ181-A | ND | x | ||||
| ACDH493 | ND | x | x | |||
| ACEY057A | ND | x | x | |||
| ACFO053-AA | ND | x | ||||
| ACGK380 | ND | x | ||||
| ACH4140 | ND | x | x | |||
| ACIK323A | ND | x | ||||
| ACLE269 | ND | x | x | |||
| ADAM295 | ND | x | ||||
| ADAU462B | ND | x | x | |||
| ADBQ038 | ND | x | x | |||
| ADDQ143 | ND | x | ||||
| ADEJ409 | ND | x | ||||
Islet treatments:
For experiments involving treatment with pro-inflammatory cytokines, isolated mouse and human islets were treated for 1 hour in imaging buffer (125mM NaCl, 5.7mM KCl, 2.5mM CaCl2, 1.2mM MgCl2, 10mM HEPES, 0.1% bovine serum albumin (BSA), and 2mM glucose) or for 24 hours in either RPMI (mouse islets) or CMRL (human islets) based media. 1 hour treatments were used for imaging based measurements (FRAP for gap junction permeability, [Ca2+]), and 24 hour treatments used for viability and glucose-stimulated insulin secretion (GSIS) measurements. A “type-1” like cytokine cocktail consisted of 10 ng/ml of recombinant mouse tumor necrosis factor-α (TNF-α, R&D Systems, Minneapolis, MN), 5 ng/ml of recombinant mouse interleukin-1β (IL-1β, R&D Systems), and 100 ng/ml of recombinant mouse interferon-γ (IFN-γ, R&D Systems) (Farnsworth et al., 2016a). Where noted, a “type 2” like cytokine cocktail consisted of either IL1β+IL6 (1 ng/ml and 10 pg/ml respectively) or TNF-α+IL-1β (0.5 ng/ml and 1 ng/ml), which were used to mimic a proinflammatory cytokine environment in pre-T2 diabetes. For experiments involving treatment with free-fatty acid isolated mouse and human islets were incubated in 200 μM palmitate (bound to 0.17mM BSA) in either RPMI (mouse islets) or CMRL (human islets) for 24 hours or 48 hours. For experiments with an “untreated” condition, islets were maintained in RPMI/CMRL culture media prior to experiments, with no exchange of media.
Fluorescence Recovery after Photobleaching:
To assess β-cell gap junction permeability in mouse and human islets, fluorescence recovery after photobleaching (FRAP) was used as previously established and described (Farnsworth et al., 2014). Briefly, islets were immobilized on MatTek dishes (MatTek Corp., Ashland, MA) that were coated with CellTak (BD Biosciences, San Jose, CA) and stained with 12.5μm Rhodamine123 (Sigma) for 30 min at 37 °C. Islets were imaged either on a Zeiss LSM 510 Meta confocal microscope with a ×40 1.2NA water immersion objective, or a Zeiss LSM 800 with a x40 1.2NA water immersion objective. Rhodamine123 was excited at 488 nm and images were collected with a 488-nm narrow band-pass dichroic beam splitter, 490-nm long pass dichroic beam splitter, and a 505-nm long pass emission filter. Procedurally, half of the islet area was selected and photobleached for 235.5 s at 316.05 milliwatt/cm2 and the fluorescence recovery was measured in the bleached area. Recovery rates were calculated from the inverse exponential fluorescence recovery curve for the entire bleached area (Farnsworth et al., 2014). Previous work from our lab has demonstrated that this method measures GJ permeability in β-cells only, as GJs do not couple α-cells.
FRAP data was either presented as raw recovery rates calculated from the recovery curve, or as the recovery rate normalized to paired untreated islets from experiments performed from on the same day (where indicated).
Intracellular Ca2+ Imaging and Analysis:
Intra-cellular fee-calcium ([Ca2+]) dynamics were assessed in islets stained with 4μm Fluo-4 AM for 1 hour at 35 ± 1°C in imaging buffer (125mM NaCl, 5.7mM KCl, 2.5mM CaCl2, 1.2mM MgCl2, 10mM HEPES, 0.1% bovine serum albumin (BSA), and 2 mM glucose). Islets were imaged on MatTek dishes and were maintained at 37 °C with an Eclipse-Ti wide field microscope (Nikon) and a ×20 0.75 NA Plan Apo objective. Images were acquired at 1 frame per second five minutes after stimulation with 11 mm glucose, for 300 seconds. Fluo-4 was imaged using a 490/40-nm band-pass excitation filter and a 525/36-nm band-pass emission filter (Chroma).
The proportion of the islet that showed synchronized oscillations was determined over the 300s intervals, as previously described (Hraha et al., 2014; Westacott et al., 2017). For mouse islets, a reference time-course was taken averaged over the whole islet or sub-region of the islet. A cross-correlation coefficient was then defined between this reference and the time-course corresponding to each pixel within the image that showed significant Fluo4 staining over background. A region was defined as synchronized if the cross-correlation coefficient was >0.75. The area of the islet showing synchronized oscillations was normalized to the total are of the islet. For human islets, elevations in [Ca2+] were identified for time-course corresponding to each pixel within the image that showed significant Fluo4 staining over background. Contagious pixels that showed coincident elevations in [Ca2+] for at least 50% of the oscillations were defined as synchronized regions. The area of the largest synchronized region was normalized to the total are of the islet.
[Ca2+] data was either presented as % synchronized area, or as the synchronized area normalized to paired untreated islets from experiments performed from on the same day (where indicated).
Islet Viability:
Following 24-h treatment in indicated condition, islets were incubated with a 1:1000 dilution of LIVE/DEAD™ Fixable Near-IR Dead Cell Stain (Life Technologies) diluted in HBSS for 30 min at room temperature. Islets were imaged on a Zeiss LSM 800 Meta confocal microscope with a ×40 1.2NA water immersion objective. The dye was excited with a 633-nm He-Ne laser with a UV/488/543/633-nm dichroic beam splitter, and images for live/dead cells were collected with a 650–710nm band-pass emission filter. Viability was calculated from manual counts of live and dead cells averaged over 3 images of increasing depth in each islet.
Glucose stimulated insulin secretion assay (GSIS):
Glucose stimulated insulin secretion (GSIS) was determined in static assays, as described previously (Farnsworth et al., 2016a; Westacott et al., 2017). Following 24-hour of experimental treatments, duplicates of ~5 IEQs per tube (mouse islets) and ~10 IEQs (human islets) were incubated in 500μl of Krebs-Ringer buffer (128.8mM NaCl, 5mM KCl, 1.2mM KH2PO4, 2.5mM CaCl2, 1.2mM MgSO4, 1mM HEPES, 0.1% BSA, pH 7.4) with 2mM glucose for 1 hour, followed by 1 hour stimulation with either 2mM or 20mM glucose. Islets were lysed by 2% Triton X-100 in deionized water followed by a freeze-thaw cycle. Both supernatant and islet lysate were collected for analysis of insulin secretion and content, respectively. Secreted insulin concentrations from both mouse and human GSIS assays were measured with a mouse ultrasensitive insulin ELISA kit (ALPCO; cross-reactive for human insulin) per the manufacturer’s instructions. GSIS data are reported as % insulin secretion (supernatant) normalized to insulin content (lysate), or insulin secretion at 20mM glucose normalized to insulin secretion at 2mM glucose (where indicated).
Cx36 Mimetic Peptide Design:
An eleven amino acid sequence corresponding to residues 288–298, flanking residue 293 (serine) of murine connexin-36 (QAKRKSVYEIR), was generated to mimic this portion of the c-terminus (“S293”). A 16-amino acid antennapedia domain (RQIKIWFQNRRMKWKK) was included on the N-terminus, immediately preceding the connexin-36 11 amino acid sequence to facilitate cellular internalization (Avignolo et al., 2008). A biotin tag was included on the N-terminus, and amide domain on the c-terminus, as shown in Figure 4A. This peptide was synthesized by the University of Colorado Peptide and Protein Chemistry Core Facility (shown in Figure 4A). A scrambled peptide (“Scr-S293”) was synthesized as a negative control (sequence: KYQREKVARIS). The sequence was manually designed by the CU-PPCC faculty. Scrambled peptide control experiments were performed alongside all S293 experiments to ensure peptide specificity and to account for off-target effects of non-Cx36 peptide sequences.
Statistical Analysis:
All data presented represent the average over all mice for each measurement. Unless indicated, data is presented as mean ± S.D. (with symbols representing individual datapoints). For human islets, data is presented as individual points per donor, together with the mean ± S.D. Statistical significance was determined using either a Student’s t-test or an analysis of variance (ANOVA) with Tukey’s post hoc analysis. An α of 0.05 was pre-determined prior to experiments to be statistically significant, and statistical significance is indicated where appropriate.
RESULTS
Reduced GJ function and [Ca2+] coordination in human islets from T2D donors:
To determine the role of β-cell gap junction (GJ) coupling in the pathogenesis of type2 diabetes (T2D), we first assessed GJ permeability and [Ca2+] dynamics in human islets from confirmed T2D donors and compared these with islets from age- and BMI-matched non-diabetic donors. Islets from T2D human donors exhibited a significant 28% reduction in recovery rate (reflecting GJ permeability, (Farnsworth et al., 2014)) compared to islets from non-diabetic human donors (Figure 1A, p=0.0012). There was no correlation between GJ permeability and HbA1c among the T2D group (p=0.59), indicating the decrease in coupling was unlikely to result from glucose toxicity but is caused by some other factor related to T2D progression.
Figure 1: Reduced GJ function and [Ca2+] coordination in human islets from T2D donors.
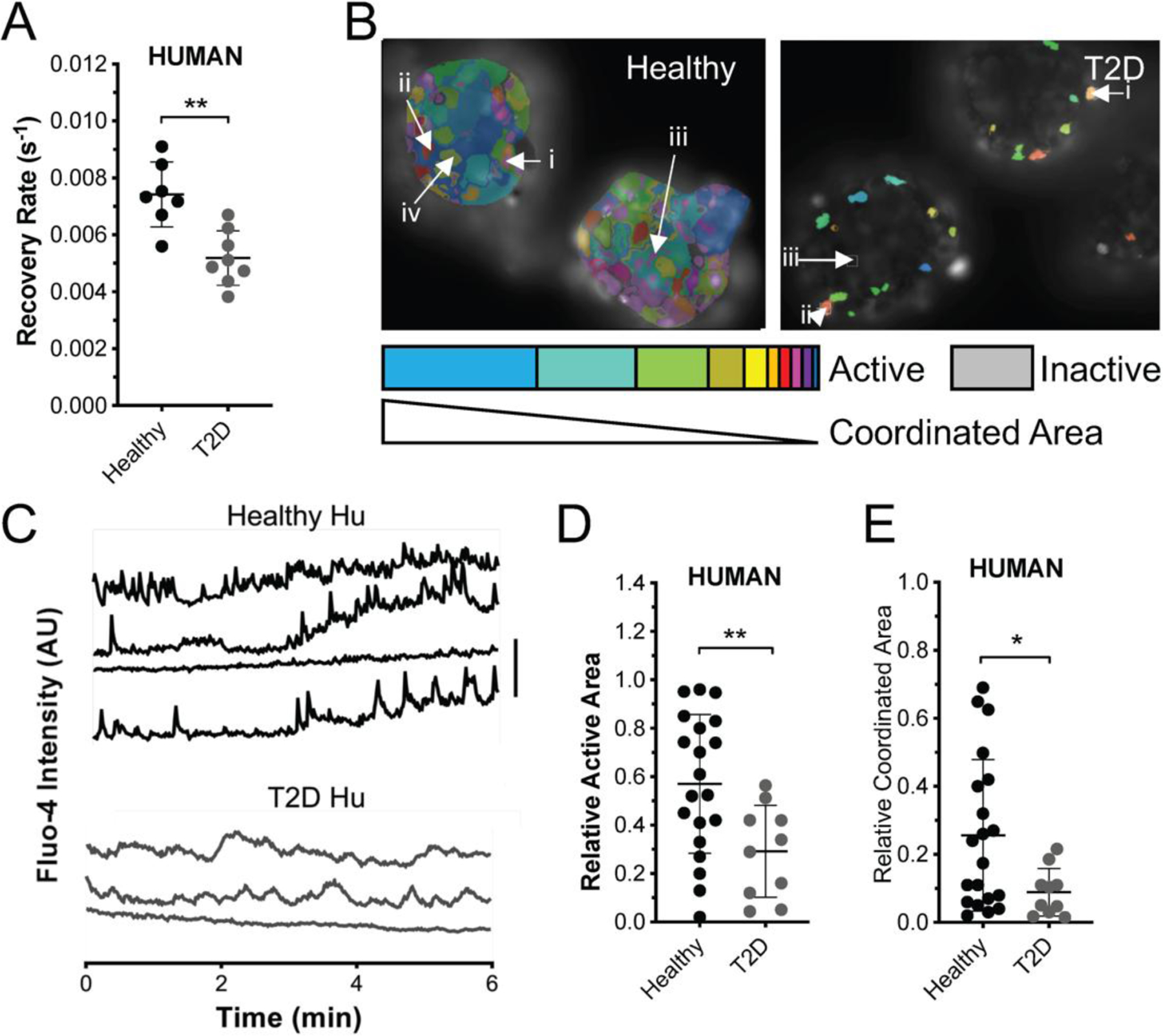
A) Gap junction permeability, as measured by FRAP recovery rate in islets from T2D donors and from age and BMI matched healthy donors. B) Representative false color map of [Ca2+] activity and coordination in human islet from a healthy donor (top) and from a T2D donor (bottom). [Ca2+] activity is represented by presence of false color, with each color representing a separate region of [Ca2+] coordination, as indicated in legend below. C) Time courses from individual cells (i–iv or i-iii) indicated in each of these islets. D) Area of [Ca2+] activity normalized to islet size (relative active area) averaged over human islets from each healthy and T2D donor. E) Largest area of coordinated [Ca2+] activity normalized to islet size (relative coordinated area) averaged over human islets from each healthy and T2D donor. Data in A representative of n=7 healthy donors and n=8 T2D donors. Data in D,E representative of n=20 healthy donors and n=10 T2D donors. In A ** represents p=0.0012, in D ** represents p=0.0099, in E * represents p=0.029, comparing groups indicated (all unpaired Student’s t-test). Data in A,D,E presented as one point per donor, with error bars representing mean±S.D.
GJ coupling coordinates oscillatory [Ca2+] dynamics across the islet. We quantified [Ca2+] dynamics in T2D human islets and compared them those dynamics measured from age- and BMI-matched non-diabetic controls (Figure 1B,C). Islets from T2D human donors had significantly reduced proportion of cells that showed elevated [Ca2+] (Figure 1D, ‘active area’: p=0.0099). Islets from T2D human donors also had a significantly reduced proportion of cells that showed synchronized [Ca2+] dynamics (Figure 1E, ‘coordinated area’: p=0.029). Again, there was no correlation between measures of [Ca2+] dynamics with HbA1c among the T2D donor islet group (active area p=0.75, coordinated area p=0.77). We also did not observe a correlation between fold-change in glucose-stimulated insulin secretion (GSIS) under static assay, with HbA1c (p=0.36). GSIS was similar between healthy and T2D groups (healthy 2.34±0.24, T2D 1.96±0.36, p=0.36). Taken together, these data indicate, for the first time, that islets from confirmed T2D humans have reduced gap junction function, which logically leads to disrupted coordinated [Ca2+] dynamics.
Diabetogenic environments disrupt GJ function and [Ca2+] coordination:
Given results from human T2D islets, we sought to understand the pathophysiological mechanisms for reduced GJ coupling. To this end, we performed similar experiments with islets isolated from the db/db mouse model of T2D: an animal model of obesity induced diabetes that results from leptin signaling deficiency. We examined db/db mice at 4 weeks of age, where animals were euglycemic, but glucose intolerant, and thus represent a “pre-diabetic” phase prior to marked β-cell decline. Compared to control animals (C57BLKS), islets from 4 week old db/db mice had significant reduction in GJ function as assessed with FRAP (Figure 2A; p=0.049). db/db islets also exhibited disorganized [Ca2+] elevations (Figure 2B), and reduced [Ca2+] synchronization compared to control islets (Figure 2C; p=0.002). Together with data from T2D human islets, FRAP and [Ca2+] data from db/db mouse islets indicates GJ uncoupling as a common pathophysiological feature in both human and mouse T2D.
Figure 2: Diabetogenic environments disrupt GJ function and [Ca2+] coordination.
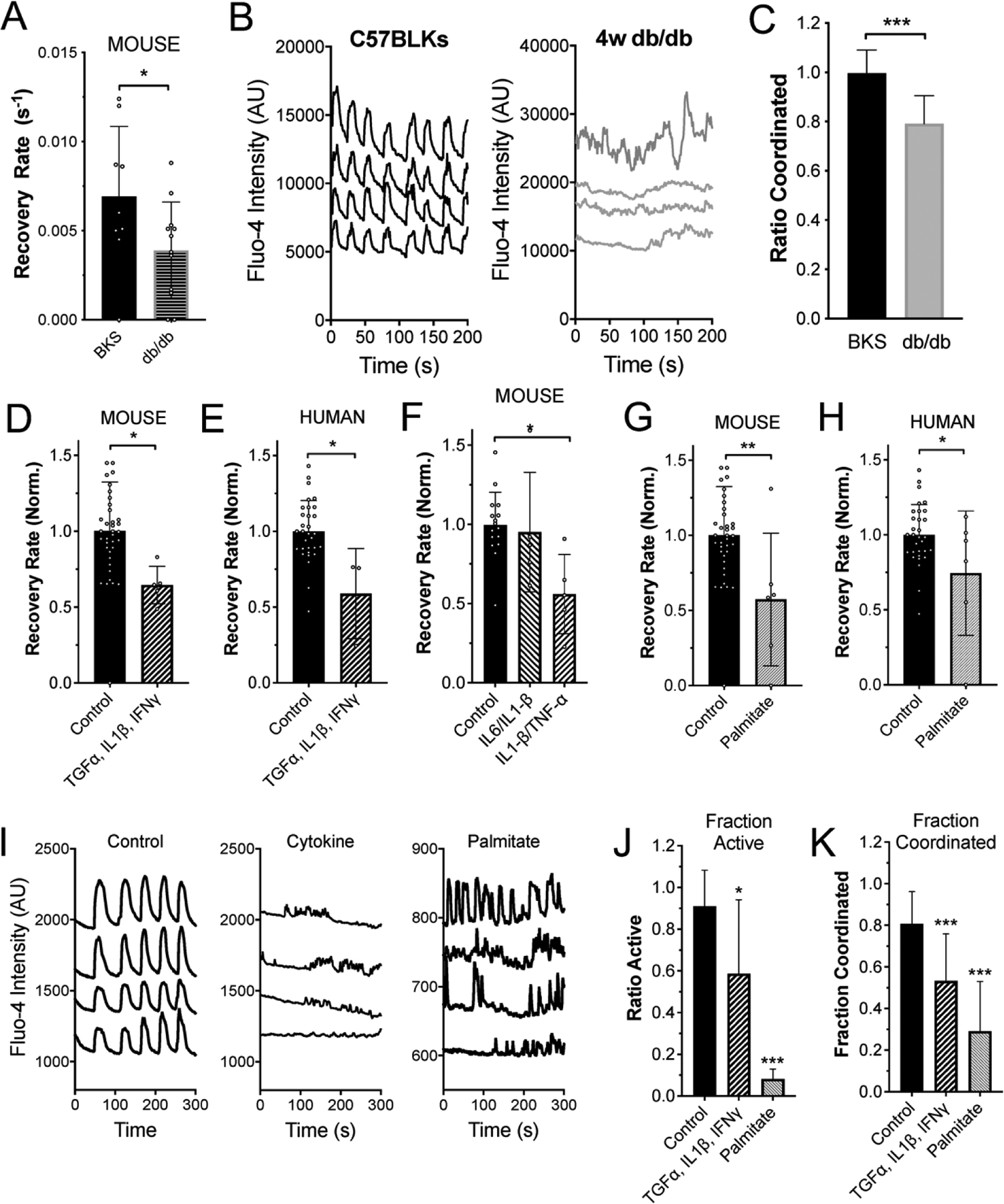
A) Mean gap junction permeability, as measured by FRAP recovery rate, in islets from 4w C57BLKS (BKS) controls and 4w db/db mice. B) Representative [Ca2+] time courses from four individual cells within an islet from a 4w C57BLKS control and 4w db/db mouse. C) Mean proportion of islets that show synchronized Ca2+ oscillations, normalized to the mean value observed in islets from BKS mice measured on the same day. D) Mean gap junction permeability, in untreated islets and islets treated with a cocktail of 1 ng/ml mouse recombinant TNF-α, 0.5 ng/ml IL-1β, 10 ng/ml IFN-γ. Data is normalized to the paired untreated islets. E) As in D for human islets treated with the same cytokine cocktail of TNF-α, IL-1β, IFN-γ. F) As in D for mouse islets treated with a cocktail of 1 ng/ml mouse recombinant TNF-α, 0.5 ng/ml IL-1β, or with a cocktail of 1 ng/ml IL1β, 10 pg/ml IL6. G) Mean gap junction permeability, in untreated mouse islets and islets treated with 200μM palmitate (bound to 0.17mM BSA). Data is normalized to the paired untreated islets. H) As in G for human islets treated with palmitate. I) Representative [Ca2+] time courses from four individual cells within an untreated islet (left), for an islet treated with TNF-α, IL-1β, IFN-γ (middle, as in D) or for an islet treated with palmitate (right, as in G). J) Area of [Ca2+] activity normalized to islet size (fraction active). K) Area of coordinated [Ca2+] normalized to islet size (fraction coordinated). Data in A representative of n=9 C57BLKS and n=12 db/db islets. Data in C representative of n=24 C57BLKS and n=5 db/db islets. Data in D representative of n=10 untreated and n=6 cytokine treated islets. Data in E representative of n=7 untreated and n=6 cytokine treated islets. Data in F representative of n=16 untreated and n=5 cytokine treated islets. Data in G representative of n=6 untreated and n=5 palmitate treated islets. Data in H representative of n=6 untreated and n=6 palmitate treated islets. In A * represents p=0.049; in C *** represents p=0.002; In D* represents p=0.020; in E * represents p=0.0066; in F * represents p=0.047; in G * represents p=0.007; in H * represents p=0.025; in J * represents p=0.0012, *** represents p<0.0001; in K *** represents p=0.0003 and p=<0.0001, comparing groups indicated (all unpaired Student’s t-test). Data in A, C-H, J,K presented as mean±S.D. with each data point representing an islet.
To delineate the diabetogenic mechanisms that lead to GJ uncoupling in T2D donor human and db/db mouse islets, we mimicked a diabetogenic environment by treating wildtype mouse islets and human islets from healthy donors with several cocktails of pro-inflammatory cytokines, as well as with palmitate, a long-chain free fatty acid. A cytokine cocktail of TNF-α, IL-1β, and IFN-γ reduced islet GJ uncoupling in both mouse and healthy human islets to a similar degree when normalized to untreated islets, as we have previously observed (Figure 2D, p=0.020; Figure 2E, p=0.0066). A cytokine cocktail of IL-1β, and TNF-α alone also reduced GJ coupling when normalized to untreated islets (Figure 2F, p=0.047). However, a cytokine cocktail of IL-1β and IL6 did not induce significant GJ uncoupling (Figure 2F). Finally, we assessed the ability of the long-chain free fatty acid palmitate to induce changes in GJ coupling. Treatment with 200 μM palmitate resulted in significantly reduced GJ coupling in both mouse islets (Figure 2G, 0.57±0.18, p=0.007) and human islets (Figure 2H, 0.74±0.17, p=0.025). These data clearly suggest that both IL-1β/TNF-α and/or free fatty acids have effects on mediating GJ uncoupling in the pre-T2D clinical milieu. These treatments that reduced GJ coupling also led to reductions in [Ca2+] activity and [Ca2+] coordination in mouse pancreatic islets treated in the same manner (Figure 2I–K).
Thus, convergent mechanisms may exist within the diabetogenic environment, such that both pro-inflammatory cytokine cocktails and FFAs (palmitate) induce GJ uncoupling and [Ca2+] desynchronization.
Increasing Cx36 gap junction coupling protects against islet decline and cell death:
Gap junctions in both mouse and human islets are formed by Cx36. To test whether elevating Cx36 gap junction coupling protects against islet decline under diabetogenic conditions we first over-expressed Cx36 in β-cells via a transgenic mouse, as previously characterized (Klee et al., 2011a). In islets from over-expression mice, Cx36 gap junction coupling was elevated compared to islets from age matched or littermate wild-type controls (Figure 3A). Following treatment with the cytokine cocktail of TNF-α, IL-1β, and IFN-γ Cx36 GJ coupling was elevated compared to controls (Figure 3A). Cell death was significantly less in islets from Rip-Cx36 mice compared to control mice, following treatment with the cytokine cocktail (Figure 3B), as previously observed (Klee et al., 2011a). While islets from Rip-Cx36 mice showed a trend to elevated GSIS compared to islets from non-transgenic controls, this increase was not statistically significant (Figure 3C).
Figure 3: Increasing Cx36 gap junction coupling protects against islet dysfunction and cell death.
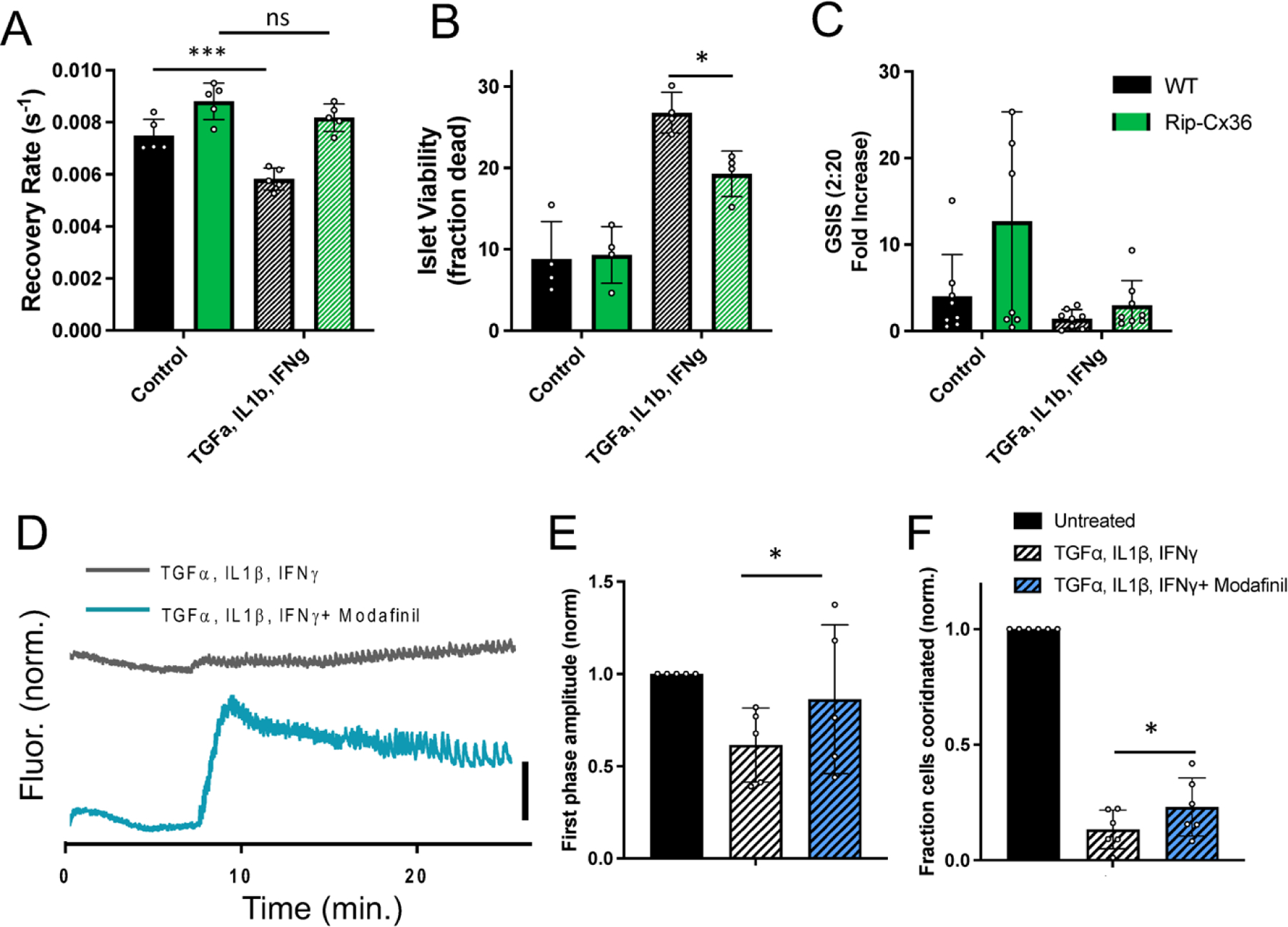
A) Mean gap junction permeability, as measured by FRAP recovery rate, in islets from wild-type and Rip-Cx36 overexpression mice, that are either untreated or treated with a cocktail of 1 ng/ml mouse recombinant TNF-α, 0.5 ng/ml IL-1β, 10 ng/ml IFN-γ. B) Islet viability, as indicated by the proportion of cells labelled as dead, in islets as in A. C) Glucose-stimulated Insulin secretion (GSIS) in islets as in A. D) Representative [Ca2+] time courses averaged over an islet treated with TNF-α, IL-1β, IFN-γ as in A-C (grey) or an islet treated with TNF-α, IL-1β, IFN-γ as in A-C plus 50 μM modafinil. E) Amplitude of [Ca2+] elevation ~10 minutes after glucose elevation, for untreated islets and treatments in D. Data is normalized to the elevation measured in untreated islets. F) Area of coordinated [Ca2+] normalized to islet size (fraction coordinated), for untreated islets and treatments in D. Data is normalized to the elevation measured in untreated islets. Data in A representative of n=5 wild-type and n=5 Rip-Cx36 mice; data in B representative of n=4 wild-type and n=4 Rip-Cx36 mice; data in C representative of n=8 wild-type and n=8 Rip-Cx36 mice; data in E representative of n=5 mice for each treatment, data in F representative of n=6 mice for each treatment. In A * represents p=0.0007, ns represents p=0.21; in B * represents p=0.02; in E * represents p=0.04; in B * represents p=0.05; comparing groups indicated (all unpaired Student’s t-test). Data in A-C, E-F presented as mean±S.D. with each data point representing an islet (A,B,E,F) or independent experiment (C).
Modafinil has been shown to elevate Cx36 GJ coupling in the islet (Farnsworth et al., 2014; Westacott et al., 2017), albeit with the mechanisms poorly understood (Urbano et al., 2007). Modafinil treatment significantly elevated both the first phase elevation in [Ca2+] (Figure 3D,E) and synchronization of Ca2+ oscillations (Figure 3D,F) following treatment with the cytokine cocktail. Therefore elevating Cx36 GJ coupling through genetic or pharmacological means provides protection against disruptions to [Ca2+] dynamics and blunts cell death induced by diabetogenic conditions.
Cx36-S293 is critical for GJ uncoupling in diabetogenic environments:
Cx36 GJ coupling can be regulated by phosphorylation of various residues on the protein, particularly on the intra-cellular loop and c-terminus (Urschel et al., 2006; Li et al., 2014; Ivanova et al., 2015; Zhang et al., 2015). Phosphorylation of Cx36 at serine-293 (S293) by PKA was shown to induce Cx36 uncoupling in AII Amacrine cells in the mouse retina, as determined by dye tracer experiments (Urschel et al., 2006; Kothmann et al., 2009). Furthermore, dynamic regulation of Cx35 coupling by phosphorylation and de-phosphorylation at serine-273 in the zebrafish (analogous to S293 on the mammalian Cx36) retina is regulated by PKA (Kothmann et al., 2007). Our lab has also demonstrated a role for PKCδ in regulating β-cell Cx36 GJ coupling (Farnsworth et al., 2016a). Given the role of PKA and PKCδ in regulating Cx36 (in the islet and elsewhere), we hypothesized that prevention of Cx36-S293 phosphorylation in the context of the diabetogenic environment described above could lead to preservation of GJ coupling and downstream factors such as [Ca2+] dynamics. To this end, we synthesized a novel mimetic inhibitory peptide targeted toward S293 (“S293”) on the Cx36 c-terminus (Figure 4A). This synthetic peptide included an antennapedia internalization domain (Avignolo et al., 2008); an 11-amino acid sequence corresponding to the Cx36 sequence flanking S293 (Cx36:288–298); and an N-terminal biotin tag. Effective intracellular internalization of the peptide occurred in dissociated β-cells from mouse islets (Figure 4B), as measured by fluorescence imaging of an Alexa-488 tagged streptavidin that bound to the biotin tag on the S293 peptide. We also synthesized a peptide with internalization domain and biotin tag, but with a scrambled sequence of Cx36:288–298 (“Scr-S293”).
Figure 4: Cx36-S293 is critical for GJ uncoupling in diabetogenic environments.
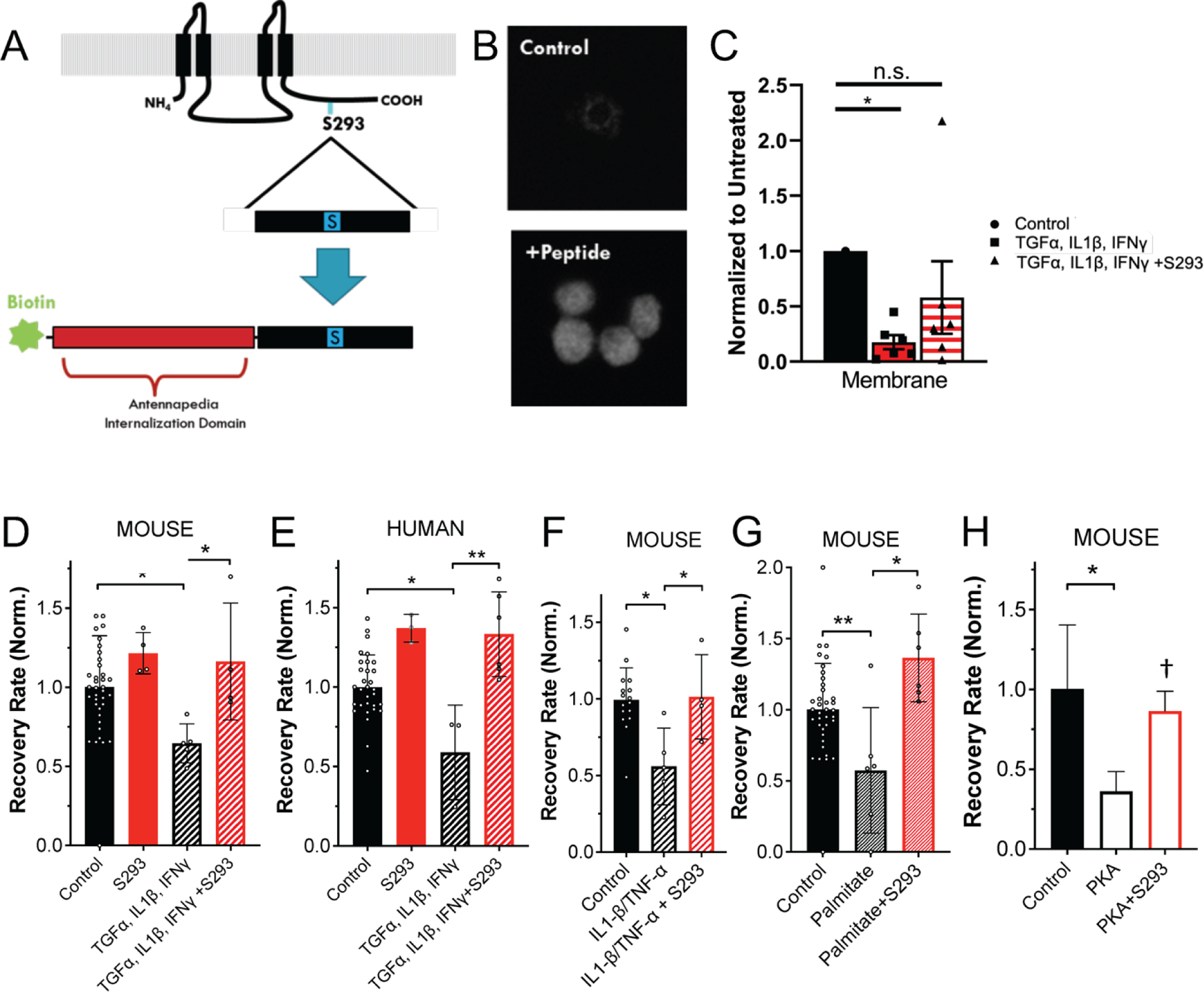
A) Schematic of S293 peptide mimetic of Cx36 c-terminus. B) Fluorescence image of biotin labelling in control, untreated cells and cells treated with S293 mimetic. C) Mean intensity of membrane-localized Cx36, quantified from western blot of membrane fraction of mouse islets treated with a cocktail of 1 ng/ml mouse recombinant TNF-α, 0.5 ng/ml IL-1β, 10 ng/ml IFN-γ and/or 1 μM S293 peptide. D) Mean gap junction permeability, as measured by FRAP recovery rate, in mouse islets treated with a cocktail of 1 ng/ml mouse recombinant TNF-α, 0.5 ng/ml IL-1β, 10 ng/ml IFN-γ and/or 1 μM S293 peptide. Data is normalized to the paired untreated islets. E) As in D for human islets. F) As in D for mouse islets treated with a cocktail of 1 ng/ml mouse recombinant TNF-α, 0.5 ng/ml IL-1β, and/or 1 μM S293 peptide. G) As in D for mouse islets treated with 200μM palmitate (bound to 0.17mM BSA) and/or 1 μM S293 peptide. H) As in D for mouse islets treated with 300uM 6-Bnz-cAMP (PKA) and/or 1 μM S293 peptide. Data in C representative of n=6 experiments; data in D representative of islets from n=5 mice; data in E representative of islets from n=6 donors; data in F representative of islets from n=4 mice; data in G representative of islets from n=7 mice; data in H representative of islets from n=2 mice. In C * represents p<0.01 D * represents p=0.021; in E ** represents p=0.007; in F * represents p=0.036; in G * represents p=0.004; in H * represents p=0.037, † represents p=0.056; comparing groups indicated (all unpaired Student’s t-test). Data in C-H presented as mean±S.D. with each data point representing an islet (D-H) or independent experiment (C).
We tested the function of the S293 peptide and whether it could block the GJ uncoupling induced by the cocktails of pro-inflammatory cytokines and palmitate (Figure 2), as well as the reduced membrane localization (Farnsworth et al., 2016b). Incubation of islets with the S293 peptide reduced the loss of membrane localized Cx36 (Figure 4C), and prevented the cytokine-mediated decreases in GJ permeability in response to TNF-α, IL-1β, and IFN-γ in mouse islets (Figure 4D, p= 0.021) and in human islets (Figure 4E, p=0.007). We observed a small increase in GJ permeability observed when human islets were treated with S293 peptide alone (Figure 4E, 37±4% increase, p=0.03), but not in the case of mouse islets (Figure 4D, 22±6% increase, p=0.33). The S293 inhibitory peptide was effective in preventing cytokine-mediated (TNF-α, IL-1β, and IFN-γ) decreases in GJ permeability, down to 30 nM concentration, highlighting its potency (not shown). Similarly, incubation of mouse islets with S293 peptide in the presence of TNF-α, IL-1β alone prevented decreases in GJ permeability (Figure 4F, p=0.036).
We also tested whether the S293 peptide could prevent free-fatty acid (100μM palmitate) mediated decreases in GJ coupling. Decreases in GJ coupling were prevented in islets treated with palmitate and S293 peptide, when compared to islets treated with palmitate alone (Figure 4G, p=0.004). PKA activation has been suggested to mediate the decrease in GJ coupling induced by elevated free-fatty acids. Incubation with the PKA activator 6-Bnz-cAMP reduced GJ coupling. However, decreases in GJ coupling were prevented in islets treated with 6-Bnz-cAMP and S293 peptide (Figure 4H, p=0.056).
Taken together, these data demonstrate how the S293 peptide can restore declines in mouse and human islet GJ coupling and thus also suggest that Serine-293 on Cx36 may be a convergent target for both pro-inflammatory cytokines and the FFA palmitate to reduce GJ coupling.
Inhibition of Cx36 Serine-293 maintains islet function and cell viability:
We next tested whether using the S293 peptide to prevent GJ uncoupling in the context of a diabetogenic environment would preserve synchronized Ca+2 dynamics and β-cell viability, as occurs under genetic or pharmacological elevation in Cx36 GJ coupling (Figure 3). In addition to the effects on GJ coupling and [Ca2+], incubation of mouse islets with proinflammatory cytokines increases cell death and decreases GSIS (see Figure 5C,E). Incubation with the S293 peptide in the presence of cytokines restored [Ca2+] oscillatory dynamics (Figure 5A), and significantly increased the proportion of the islet that was active in response to glucose (Figure 5B, p=0.0009). In contrast, incubation with the scrambled peptide resulted in disorganized and unsynchronized [Ca2+] dynamics remaining (Figure 5A). Incubation with the S293 peptide in the presence of cytokines significantly reduced cytokine-induced β-cell death, by a factor of ~40% (Figure 5C, p=0.0003). The reduction in β-cell death by incubation with the S293 peptide was not observed in islets from Cx36 knockout mice (Figure 5D). This is consistent with a role for Cx36 in protecting against apoptosis (Figure 3A, (Klee et al., 2011a)). Finally, incubation of mouse islets with cytokines and S293 peptide restored GSIS at 20 mM glucose (Figure 5E, p= 0.0003). In islets from Cx36 knockout mice incubation with the S293 peptide in the presence of cytokines led to a small elevation in GSIS, although this increase was not statistically significant (Figure 5F, p=0.11).
Figure 5: Inhibition of Cx36 Serine-293 maintains islet function and cell viability.
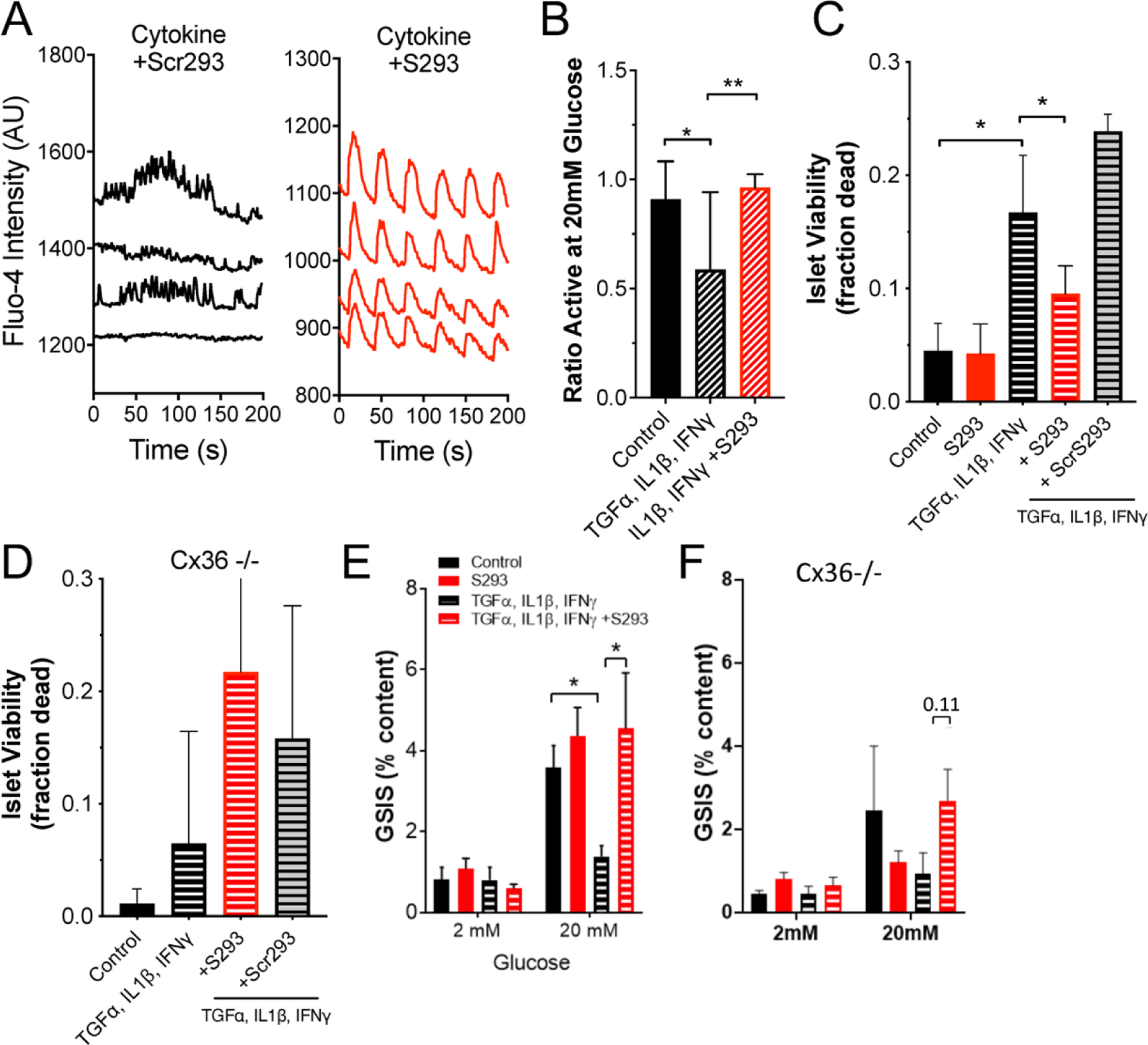
A) Representative [Ca2+] time courses from four individual cells within an islet treated with a cocktail of 1 ng/ml mouse recombinant TNF-α, 0.5 ng/ml IL-1β, 10 ng/ml IFN-γ plus 1 μM scrambled peptide (cytokine+Scr293, left) or within an islet treated with a cocktail of 1 ng/ml mouse recombinant TNF-α, 0.5 ng/ml IL-1β, 10 ng/ml IFN-γ plus 1 μM S293 peptide (cytokine+S293, right). B) Area of [Ca2+] activity normalized to islet size (fraction active) for islets treated with TNF-α, IL-1β, = IFN-γ plus = S293 peptide, as in A. C) Islet viability, as indicated by the proportion of cells labelled as dead, under a cocktail of 10 ng/ml mouse recombinant TNF-α, 5 ng/ml IL-1β, 100 ng/ml IFN-γ and/or 1 μM S293 peptide or 1 μM scrambled (Scr) peptide. D) As in C for islets from Cx36 knockout mice. E) Glucose-stimulated Insulin secretion (GSIS) in islets treated with a cocktail of 1 ng/ml mouse recombinant TNF-α, 0.5 ng/ml IL-1β, 10 ng/ml IFN-γ plus 1 μM S293 peptide. F) As in E for islets from Cx36 knockout mice. Data in B representative of islets from n=5 mice; data in C representative of islets from n=6 mice; data in D representative of islets from n=8 mice; data in E representative of islets from n=5 mice; data in F representative of islets from n=6 mice. In B ** represents p=0.021; in C ** represents p=0.0003; In E ** represents p=0.0003; comparing groups indicated (all unpaired Student’s t-test). Data in B-F presented as mean±SD.
Together, these data support that hypothesis that preventing GJ uncoupling precludes the disruption to coordinated [Ca2+] dynamics, GSIS and islet cell death that is observed when islets are exposed to a diabetogenic environment.
Inhibition of Cx36 Serine-293 maintains islet function in db/db mouse islets:
The pathophysiology of T2D is complex and multifactorial (DeFronzo et al., 2015), thus culture of islets with pro-inflammatory cytokines or FFAs alone have a limited ability to model the islet microenvironment during T2D. We therefore tested the ability of S293 peptide to restore GJ coupling and [Ca2+] dynamics in islets from the db/db animal model of T2D. In islets isolated from 4-week old db/db mice the S293 peptide restored regular oscillatory [Ca2+] dynamics compared to in untreated db/db islets (Figure 6A). However, these oscillations had increased frequency compared to untreated control islets. When quantified, S293 peptide treatment in db/db islets increased the total fraction of the islet that was coordinated compared to untreated db/db islets (Figure 6B, p=0.002). Consistent with these data, we observed that S293 protected against decreases in GJ coupling in db/db islets. Significant decreases in GJ coupling were observed in untreated or scrambled peptide-treated db/db islets compared to equivalently treated control BLKS islets (Figure 6C; p=0.05 for BKS vs. db/db, p=0.067 for db/db vs. db/db+S293). However no decrease was observed in scrambled peptide-treated db/db islets compared to equivalently treated control BLKS islets. Thus in pre-diabetic db/db mouse islets, S293 peptide mimetic can restore both GJ coupling and [Ca2+] dynamics.
Figure 6: Inhibition of Cx36 Serine-293 maintains islet function in db/db mouse islets.
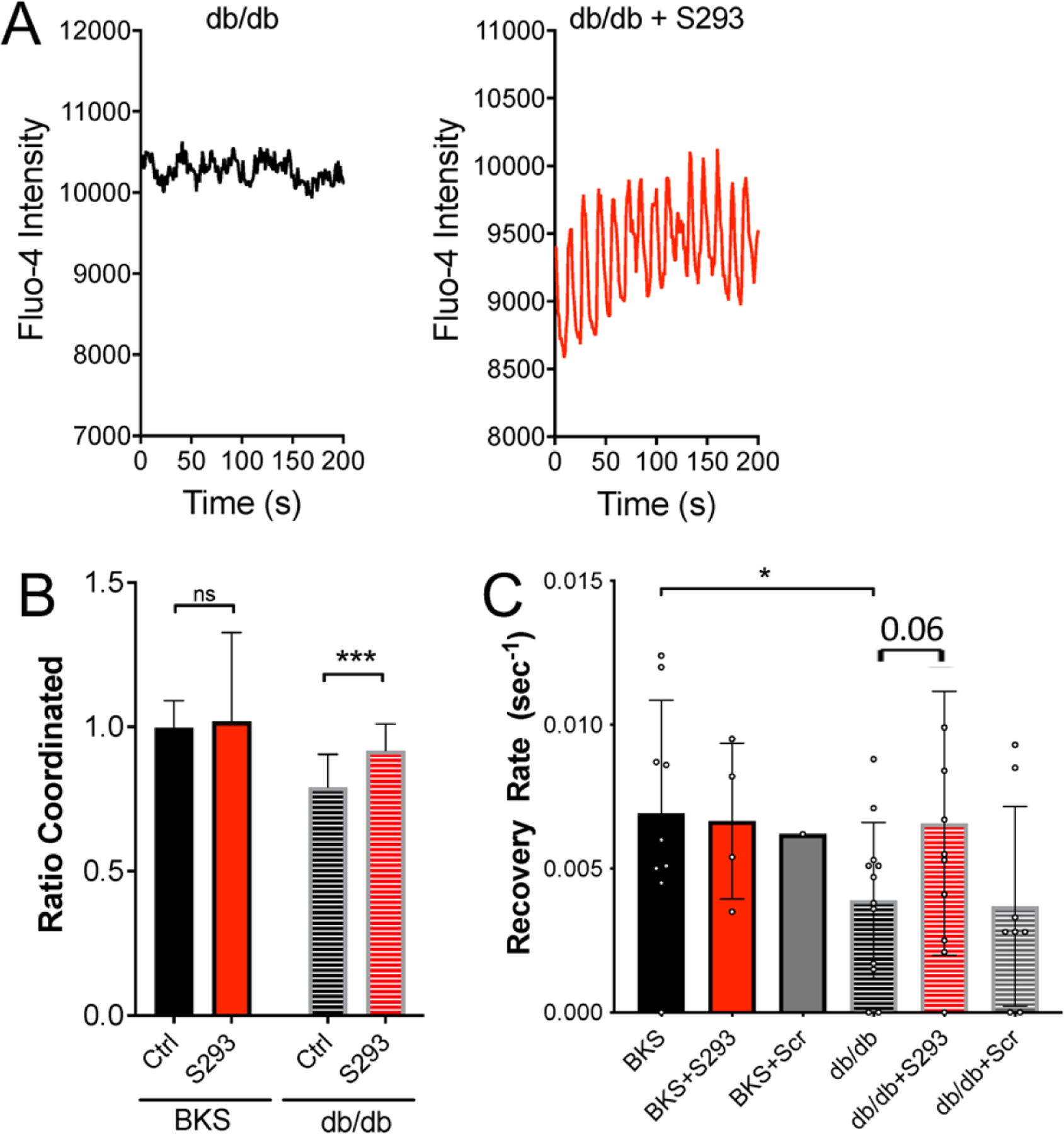
A) Representative [Ca2+] time courses averaged over an islet from a 4w db/db mouse, that is either untreated (left) or treated with S293 peptide (right). B) Area of coordinated [Ca2+] normalized to islet size (ratio coordinated) for islets from 4w C57BLKS (WT) or db/db mice that are either untreated (Ctrl) or treated with S293 peptide as in A. Data is normalized to paired untreated C57BLKS islets. C) Mean gap junction permeability, as measured by FRAP recovery rate, for islets isolated from 4w C57BLKS mice (BKS) that are untreated, treated with the S293 peptide (+S293), or treated with the scrambled peptide (+Scr); and for islets isolated from 4w db/db mice that are untreated, treated with the S293 peptide (+S293), or treated with the scrambled peptide (+Scr). Data in B representative of islets from n=5 mice; data in C representative of islets from n=5 mice. In B *** represents p=0.002; in C * represents p=0.05; comparing groups indicated (all unpaired Student’s t-test). Data in B,C presented as mean±S.D. with each data point representing an islet.
Inhibition of Cx36 Serine-293 restores GJ coupling and GSIS in type2 diabetic human islets:
We finally tested the ability of S293 peptide to restore the function of islets isolated from human donors with confirmed T2D. As described before we observed a small elevation in GJ coupling in islets from all healthy human donors upon incubation with S293 peptide (Figure 7A). Incubation of T2D human islets with S293 peptide restored GJ coupling with varying degrees (Figure 7B). No increase in GJ coupling was observed upon incubation of T2D human islets with scrambled peptide (Figure 7C). There was no significant change in glucose-stimulated insulin secretion in islets from healthy donor following S293 treatment (Figure 7D). The change in GSIS was highly variable following S293 treatment among the islets from T2D donors. However, when we separately analyzed GSIS data from islets isolated from the donors that showed a recovery in GJ coupling (an increase in recovery rate by >1.1), 3 out of 4 “responders” showed an increase in GSIS (Figure 7E). In contrast none of the “non-responders” showed an increase in GSIS (Figure 7E). There was also no significant change in glucose-stimulated insulin secretion in islets from T2D following scrambled peptide treatment (Figure 7F).
Figure 7: Inhibition of Cx36 Serine-293 restores GJ coupling and GSIS in type2 diabetic human islets.
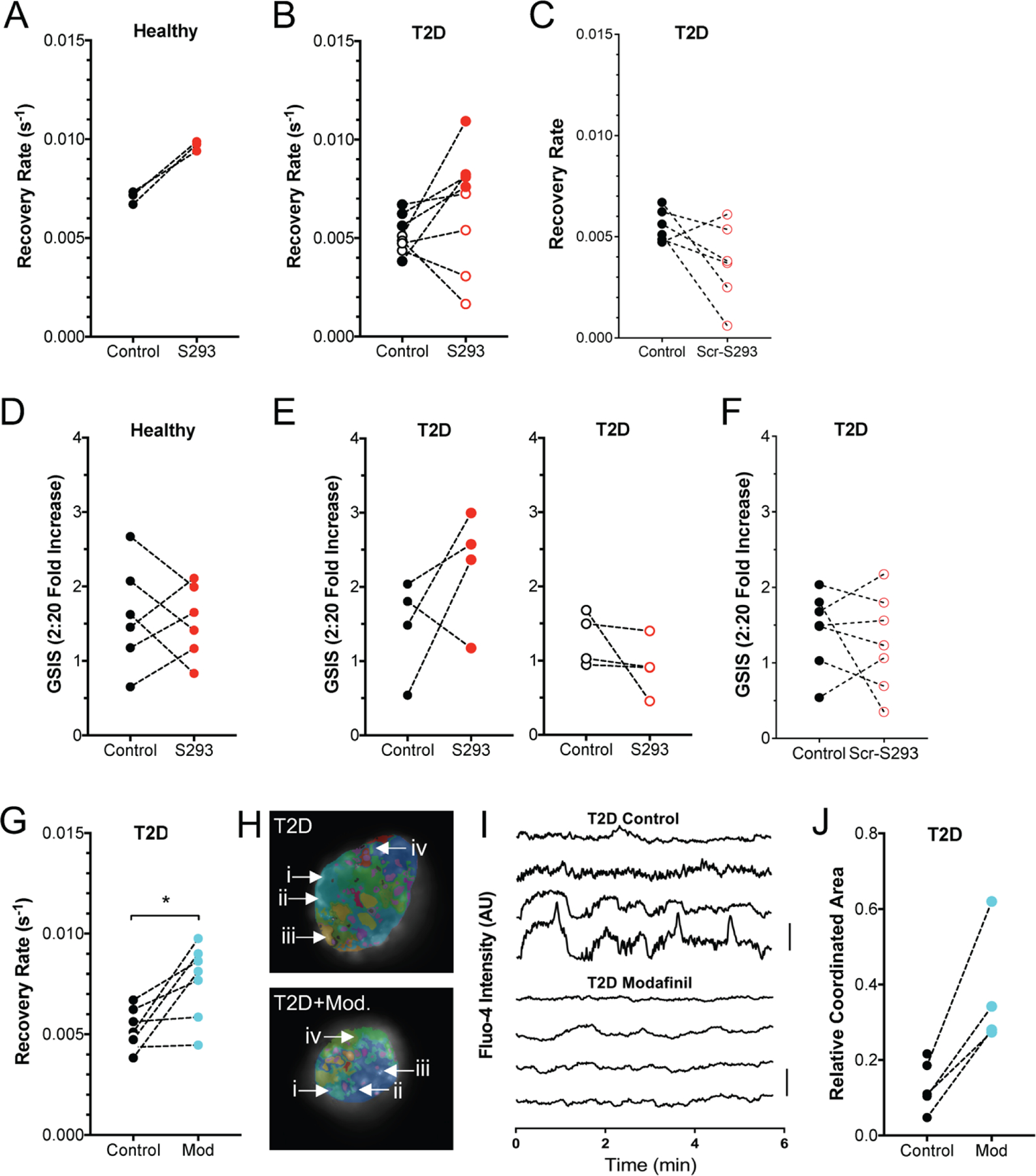
A) Gap junction permeability, as measured by FRAP recovery rate, for human islets isolated from healthy donors, that are either untreated (Control) or treated with S293 peptide. Data is presented as the mean recovery rate over islets from each donor, with dashed line connecting data for each donor. B) As in A for human islets isolated from donors with T2D, that are either untreated or treated with S293 peptide. C) As in A for human islets isolated from donors with T2D, that are either untreated or treated with scrambled peptide (Scr-S293). D) Glucose-stimulated Insulin secretion (GSIS) for human islets isolated from healthy donors, that are either untreated (Control) or treated with S293 peptide, as in A. E) GSIS as in D, for human islets isolated from T2D donors, that are either untreated (Control) or treated with S293 peptide. Data is presented as the mean recovery rate over islets from each donor, with dashed line connecting data for each donor. Left corresponds to those donors in B that showed a >10% elevation in GJ coupling by S293 peptide. Right corresponds to those donors in B that showed a decrease or <10% elevation in GJ coupling by S293 peptide. F) GSIS as in D, for human islets isolated from T2D donors, that are either untreated (Control) or treated with scrambled peptide (Scr-S293). G) Gap junction permeability for human islets isolated from donors with T2D that are untreated (control) or treated with 100μM modafinil (Mod). H) Representative false color map of [Ca2+] coordination in human islet from a T2D donor that is either untreated (T2D) or treated with 100μM modafinil (T2D+Mod). [Ca2+] activity is represented by presence of false color, with each color representing a separate region of [Ca2+] coordination, as in Figure 1 (see legend therein). I) Time courses from individual cells (i–iv) indicated in each of these islets. J) Largest area of coordinated [Ca2+] activity normalized to islet size (relative coordinated area) averaged over human islets from each T2D donor that are untreated (control) or treated with 100μM modafinil (Mod). Data in A representative of 3 donors; data in B representative of 8 donors; data in C representative of 6 donors; data in D representative of 6 donors; data in E representative of 4 donors each; data in F representative of 7 donors; data in G representative of 7 donors; data in J representative of 4 donors. In G * represents p=0.018; in J * represents p=0.04; comparing groups indicated (paired Student’s t-test). Data in A-G, J presented as one point per donor.
We also tested whether modafil would provide similar recoveries in islet function. We observed a highly variable increase in GJ coupling in islets from T2D donors following treatment with modafinil (Figure 7G). Following [Ca2+] imaging, we also observed a substantial increase in the fraction of the islet that was coordinated in islets from T2D donors (Figure 7H).
These data show that the S293 peptide mimetic, as well as pharmacological elevations in GJ coupling were able to restore [Ca2+] dynamics and glucose-stimulated insulin secretion in islets from human donors with T2D.
DISCUSSION
Type2 diabetes results from insufficient secretion of insulin in the context of insulin resistance, followed by subsequent further decline in β-cell mass. Dysfunction to β-cells within the islet of Langerhans is one main cause for this lack of sufficient insulin production. Cx36 gap junction (GJ)-mediated electrical coupling is critical to the regulation of pulsatile insulin secretion. GJ coupling is diminished by conditions associated with diabetes, including by inflammatory cytokines, lipotoxic conditions, in animal models of diabetes and in aged human islets (Hodson et al., 2013; Farnsworth et al., 2016a; Westacott et al., 2017; Corezola do Amaral et al., 2020). In this study we sought to further define how conditions associated with diabetes disrupt GJ coupling within the islet and determine how a disruption to GJ coupling impacts downstream [Ca2+], insulin secretion and cell viability under diabetogenic conditions. We found that transgenic over-expression of Cx36, pharmacological activation of GJ coupling, and a novel peptide mimetic that restores Cx36 GJ coupling all protect against islet dysfunction by retaining [Ca2+] dynamics and cell viability.
Importance of GJ coupling to islet function in health and in diabetes.
Gap junction channels are critical for coordinating and enhancing glucose-regulated electrical activity and pulsatile insulin secretion across the islet. Cx36 deficient mice lack any electrical coupling between β-cells in the islet, highlighting how Cx36 gap junctions are the driver of this electrical coupling. These mice are glucose intolerant as a result of both diminished first phase insulin secretion and disrupted pulsatile insulin secretion (Head et al., 2012). Thus, diminished Cx36 GJ coupling under diabetogenic conditions, and in subjects with T2D, would disrupt pulsatile insulin secretion and glucose tolerance. Indeed loss of first phase secretion is a hall mark of pre-T2D, and individuals with T2D show diminished pulsatile insulin as in Cx36 null animals (Menge et al., 2011). Thus, loss of Cx36 GJ coupling likely explains, in part, the reduced glucose tolerance in T2D,
Cx36 GJ coupling is also protective against cell death induced by cytotoxic conditions such as by proinflammatory cytokines (Klee et al., 2011a). Indeed, we demonstrated through multiple means that elevating Cx36 GJ coupling or protecting against its decline reduces apoptosis (Figure 3, 5). Thus, the loss of GJ coupling in diabetes likely exacerbates cell death and islet decline. Interestingly however, we did not find a strong role for Cx36 GJ coupling to be protective against declines in the regulation of insulin secretion induced by proinflammatory cytokines (Figures 3,5), but did see a role for Cx36 GJ coupling to be protective against disrupted insulin secretion in T2D subjects (Figure 7). Thus the loss of GJ coupling in diabetes may also influence the regulation of insulin release.
Through utilizing multiple approaches and models of diabetes we can have high confidence as to the protective role provided by Cx36 GJ coupling. We observed improvements in cell viability and GSIS using the Cx36:S293 mimetic, which were not observed in Cx36 deficient islets. Data from Cx36 deficient islets indicates that potential off-target effects of the Cx36:S293 mimetic are unlikely to contribute significantly to cell death, but may contribute slightly to GSIS. Qualitatively similar results in terms of cell death, GSIS and [Ca2+] following Cx36 over expression and following modafinil-based increases in GJ coupling (which may have off target effects) support Cx36 playing a key role.
It is important to note that we solely utilize in vitro based experiment’s in this study, combining mouse and human islet models. However, there are challenges to examining our questions invivo. Firstly, the Cx36 over-expression decreased slightly under inflammatory conditions and thus invivo protection against declines in GJ coupling may be limited. Peptides have low half-life in circulation and thus the S293 mimetic would likely provide limited action following systemic delivery. Modafinil would also have other actions invivo, as it can impacts CNS function. As such whether the protection against apoptosis and disruptions to GSIS is sufficient to impact diabetes is still to be determined. As a second point, while Cx36 is the predominant connexin in both mouse and human beta-cells (Miranda et al., 2022), the precise role for Cx36 GJ coupling in human islets is not fully determined. Other mechanism have also been suggested to contribute to synchronizing [Ca2+] dynamics in the human islet via paracrine communication (Rodriguez-Diaz et al., 2011; van der Meulen et al., 2015; Almaça et al., 2016; Menegaz et al., 2019). We have previously demonstrated that improving GJ coupling using pharmacological activators in aged human islets improves [Ca2+] regulation and first phase GSIS (Westacott et al., 2017). Nevertheless, we still observed significant variability among human donors, with batches responding effectively to the S293 mimetic and/or modafinil (responders) and batches not (non-responders). This response did not significantly dependent on the state of the islets as measured by GSIS or [Ca2+], but may depend on some other factor.
Overall, increasing Cx36 GJ coupling or protecting against its decline protects against disruptions to islet [Ca2+], GSIS and cell viability, across both mouse and human islets.
Regulation of Cx36 by S293 peptide mimetic:
We demonstrated that treatments that restore or elevate Cx36 GJ coupling are protective against declines in islet function ([Ca2+], GSIS in human T2D) and cell death. Key points worthy of discussion are how mechanistically our treatments elevate GJ coupling and how elevated GJ coupling provides such protection.
Robust protection is provided by the S293 mimetic peptide. This mimetic peptide was designed to provide a competitive substrate for kinases that would normally phosphorylate the native S293 site, as well as other regulators of Cx36 that would bind this region. This S293 site has been identified as a critical for the regulation of Cx36 GJ coupling in AII amacrine cells, where it is phosphorylated by PKA (Urschel et al., 2006). It is also identified as a phosphorylation site for PKCδ. PKA is activated within human islets during lipotoxic conditions and mediates the disruption to GJ coupling (Hodson et al., 2013). We show that the S293 peptide can block the decrease to GJ coupling induced by PKA activation (Figure 4). Similarly, PKCδ is activated within mouse and human islets during inflammatory conditions and mediates the disruption to GJ coupling (Farnsworth et al., 2016a). Therefore, the S293 peptide mimetic may be competing out factors that either bind or phosphorylate the key regulatory site(s) on the Cx36 cytoplasmic tail that regulates channel closure or trafficking. The mechanisms by which modafinil promotes electrical coupling are less clear. Reports in studying its action on Cx36 in the CNS suggest activation of CamKII and phosphorylation sites distinct from those phosphorylated by PKA/PKC. Thus, phosphorylation sites that promote Cx36 channel opening may be targeted. However, microtubule binding to this c-terminal region has been reported (Brown et al., 2019), and thus regulation of trafficking is also possible. Indeed, the S293 peptide blunts the decrease in membrane localized Cx36 induced by pro-inflammatory cytokines, which is consistent with improved trafficking of Cx36. Detailed phospho-proteomic measurements will be required in future studies to delineate whether the S293 site or other sites are phosphorylated or dephosphorylated by diabetogenic conditions (pro-inflammatory cytokines, FFA, db/db mice, T2D donor) and the interventions we make (Cx36:S293 peptide, modafinil). We also note that a Cx43 peptide mimetic (alphaCT1) can promote gap junction aggregation (Hunter et al., 2005); and we cannot exclude that the S293 peptide also directly regulates membrane-localized Cx36 organization. However the limited impact of the S293 peptide under baseline conditions (Figure 4) argues against this.
The other major question is how Cx36 increases providing protection against cell death. Retaining normal GJ coupling would maintain normal coordinated [Ca2+] dynamics, and this maintained [Ca2+] would contribute to improved GSIS. Cx36 GJ coupling also protects against apoptosis, as we demonstrate here and we and others have previously demonstrated (Klee et al., 2011a). The mechanism underlying this are poorly defined. However, published studies from our lab suggest the increased cytosolic [Ca2+] at lower glucose levels induced by loss of Cx36 increases the susceptibility to apoptosis (Farnsworth et al., 2022). This is consistent with well-established results in which elevated cytosolic [Ca2+] and calcineurin activation contributes to intrinsic apoptotic mechanisms (Wang et al., 1999).
Relevance to diabetes and greater context:
The decline in functional β-cell mass is important in the pathophysiology of diabetes, determining overt disease onset, progression and success of treatment. Therapies are needed to preserve β-cell mass and existing β-cell function: such therapies would prolong the duration until diabetes onset and thus lower the risk of diabetic complications, as well as lessening the financial and physical burden to the patient. Our results suggest that targeting Cx36 GJ coupling, and particularly targeting it to prevent its disruption in T2D can preserve normal GSIS dynamics and cell viability.
Targeting Cx36 is also strongly supported by its role in the pathogenesis in T2D. Several studies, including results presented here, demonstrate its disruption under diabetogenic conditions (Carvalho et al., 2012; Hodson et al., 2013; Farnsworth et al., 2016a; Westacott et al., 2017; Corezola do Amaral et al., 2020). We further demonstrate its disruption in human donors with T2D compared to age matched healthy donors (Figure 1). Our results also suggest this is linked to the development of T2D and not as a result of subsequent poor glucose control given lack of any correlation with HbA1c. Importantly the improvement in function after targeting Cx36 via the mimetic peptide or via modafinil further support an important role in human T2D. In addition to these results, gene variants in the GJD2 gene (encoding Cx36) are associated with T2D (Cigliola et al., 2016). The disease associated variant has reduced coupling, further supporting a role for Cx36 in T2D and thus targeting it for T2D prevention.
The most effective strategy we found to target Cx36 GJ coupling was the Cx36:S293 mimetic peptide. This showed effective protection under diabetogenic conditions in both mouse and human islets, including from T2D donors. However, we were unable to effectively test it invivo given the short circulation time of peptides before clearance. Delivering peptides for therapeutic use is a major challenge: once-weekly delivery systems such as for GLP1R agonists are complex. Implementing such a delivery system, designing a more stable mimetic peptide, or targeting the peptide directly to the islet microenvironment will be needed in future work. Modafinil which is an approved drug, also provided protection in a range of exvivo models and assessing its effectiveness in human patients at risk for T2D would be useful. Our results examining transgenic over-expression of Cx36 also suggest a potential challenge in using this method to assess the role of Cx36 in protecting against T2D progression invivo. While protection against apoptosis was provided by transgenic over-expression of Cx36, GJ coupling still did decrease significantly under diabetogenic conditions. We did not test whether this over-expression could still protect against islet decline in animal models of T2D. However, we have observed a minor protection against disease protection in animal models of T1D (Farnsworth et al., 2022). In this case autoimmunity is still progressing and other means to target β-cell death do not completely abrogate disease progression.
A general limitation in treating T2D is the fact that the disease is complex and multifactorial, with no single pathway dominant in regulating islet decline. Thus, targeting solely Cx36 GJ coupling is unlikely to be sufficient to fully abrogate disease. Furthermore, even if we can preserve beta cell function, the continuing metabolic stress caused by insulin resistance will likely mean that the preservation will be temporary. Nevertheless, targeting Cx36 GJ coupling could be effective as part of a broader strategy, such as targeting ER stress and stimulating GSIS.
Supplementary Material
KEY POINTS.
Cx36 gap junction permeability and associated coordination of [Ca2+] dynamics is diminished in human type 2 diabetes (T2D) and mouse models of T2D.
Enhancing Cx36 gap junction permeability protects against disruptions to the coordination of [Ca2+] dynamics.
A novel peptide mimetic of the Cx36 c-terminal regulatory region protects against declines in Cx36 gap junction permeability.
Pharmacological elevation in Cx36 or Cx36 peptide mimetic recovers [Ca2+] dynamics and GSIS in human T2D and mouse models of T2D.
ACKNOWLEDGEMENTS:
The authors acknowledge the guidance and expertise of Dr. Robert Hodges, Director of the University of Colorado Protein Peptide Synthesis core for his guidance in the design and synthesis of both S293 and scrambled mimetic peptide sequences.
FUNDING:
This work was supported by NIH grants R01 DK102950, R01 DK106412 (RKPB), F32 DK112525 (JRS), F31 DK107043 (MJW), F32 DK102276 (NLF), F31 DK126360 (JMD); and by Juvenile Diabetes Research Foundation Grants 5-CDA-2014-198-A-N (RKPB), 3-APF-2019-749-A-N (NLF) and 3-PDF-2019-741-A-N (VK). Microscopy experiments were performed in the Advanced Light Microscopy Core, supported in part by NIH grants P30 NS048154, P30 DK116073.
DATA AVAILABILITY:
All raw data will be made available upon request.
REFERENCES:
- Abdulreda MH & Berggren P-O (2013). Islet inflammation in plain sight. Diabetes Obes Metab 15, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alev C, Urschel S, Sonntag S, Zoidl G, Fort AG, Höher T, Matsubara M, Willecke K, Spray DC & Dermietzel R (2008). The neuronal connexin36 interacts with and is phosphorylated by CaMKII in a way similar to CaMKII interaction with glutamate receptors. Proc Natl Acad Sci U S A 105, 20964–20969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almaça J, Molina J, Menegaz D, Pronin AN, Tamayo A, Slepak V, Berggren P-O & Caicedo A (2016). Human beta cells produce and release serotonin to inhibit glucagon secretion from alpha cells. Cell Rep 17, 3281–3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avignolo C, Bagnasco L, Biasotti B, Melchiori A, Tomati V, Bauer I, Salis A, Chiossone L, Mingari MC, Orecchia P, Carnemolla B, Neri D, Zardi L & Parodi S (2008). Internalization via Antennapedia protein transduction domain of an scFv antibody toward c-Myc protein. FASEB J Off Publ Fed Am Soc Exp Biol 22, 1237–1245. [DOI] [PubMed] [Google Scholar]
- Benninger RKP, Head WS, Zhang M, Satin LS & Piston DW (2011). Gap junctions and other mechanisms of cell-cell communication regulate basal insulin secretion in the pancreatic islet. J Physiol 589, 5453–5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benninger RKP & Piston DW (2014). Cellular communication and heterogeneity in pancreatic islet insulin secretion dynamics. Trends Endocrinol Metab TEM 25, 399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benninger RKP, Zhang M, Head WS, Satin LS & Piston DW (2008). Gap junction coupling and calcium waves in the pancreatic islet. Biophys J 95, 5048–5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram R, Sherman A & Satin LS (2007). Metabolic and electrical oscillations: partners in controlling pulsatile insulin secretion. Am J Physiol Endocrinol Metab 293, E890–900. [DOI] [PubMed] [Google Scholar]
- Boden G (2005). Free fatty acids and insulin secretion in humans. Curr Diab Rep 5, 167–170. [DOI] [PubMed] [Google Scholar]
- Boden G (2008). Obesity and free fatty acids. Endocrinol Metab Clin North Am 37, 635–646, viii–ix. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner-Weir S & O’Brien TD (2008). Islets in Type 2 Diabetes: In Honor of Dr. Robert C. Turner. Diabetes 57, 2899–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CA, Del Corsso C, Zoidl C, Donaldson LW, Spray DC & Zoidl G (2019). Tubulin-Dependent Transport of Connexin-36 Potentiates the Size and Strength of Electrical Synapses. Cells; DOI: 10.3390/cells8101146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CPF, Oliveira RB, Britan A, Santos-Silva JC, Boschero AC, Meda P & Collares-Buzato CB (2012). Impaired β-cell-β-cell coupling mediated by Cx36 gap junctions in prediabetic mice. Am J Physiol Endocrinol Metab 303, E144–151. [DOI] [PubMed] [Google Scholar]
- Cigliola V, Populaire C, Pierri CL, Deutsch S, Haefliger J-A, Fadista J, Lyssenko V, Groop L, Rueedi R, Thorel F, Herrera PL & Meda P (2016). A Variant of GJD2, Encoding for Connexin 36, Alters the Function of Insulin Producing β-Cells. PloS One 11, e0150880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corezola do Amaral ME, Kravets V, Dwulet JM, Farnsworth NL, Piscopio R, Schleicher WE, Miranda JG & Benninger RKP (2020). Caloric restriction recovers impaired β-cell-β-cell gap junction coupling, calcium oscillation coordination, and insulin secretion in prediabetic mice. Am J Physiol Endocrinol Metab 319, E709–E720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFronzo RA, Ferrannini E, Groop L, Henry RR, Herman WH, Holst JJ, Hu FB, Kahn CR, Raz I, Shulman GI, Simonson DC, Testa MA & Weiss R (2015). Type 2 diabetes mellitus. Nat Rev Dis Primer 1, 15019. [DOI] [PubMed] [Google Scholar]
- Donath MY, Schumann DM, Faulenbach M, Ellingsgaard H, Perren A & Ehses JA (2008). Islet inflammation in type 2 diabetes: from metabolic stress to therapy. Diabetes Care 31 Suppl 2, S161–164. [DOI] [PubMed] [Google Scholar]
- El-Assaad W, Buteau J, Peyot M-L, Nolan C, Roduit R, Hardy S, Joly E, Dbaibo G, Rosenberg L & Prentki M (2003). Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology 144, 4154–4163. [DOI] [PubMed] [Google Scholar]
- Eloff BC, Gilat E, Wan X & Rosenbaum DS (2003). Pharmacological Modulation of Cardiac Gap Junctions to Enhance Cardiac Conduction Evidence Supporting a Novel Target for Antiarrhythmic Therapy. Circulation 108, 3157–3163. [DOI] [PubMed] [Google Scholar]
- Farnsworth NL, Hemmati A, Pozzoli M & Benninger RKP (2014). Fluorescence recovery after photobleaching reveals regulation and distribution of connexin36 gap junction coupling within mouse islets of Langerhans. J Physiol 592, 4431–4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnsworth NL, Piscopio RA, Schleicher WE, Ramirez DG, Miranda JG & Benninger RKP (2022). Modulation of Gap Junction Coupling Within the Islet of Langerhans During the Development of Type 1 Diabetes. Front Physiol 13, 913611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnsworth NL, Walter RL, Hemmati A, Westacott MJ & Benninger RKP (2016a). Low Level Pro-inflammatory Cytokines Decrease Connexin36 Gap Junction Coupling in Mouse and Human Islets through Nitric Oxide-mediated Protein Kinase Cδ. J Biol Chem 291, 3184–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnsworth NL, Walter RL, Hemmati A, Westacott MJ & Benninger RKP (2016b). Low Level Pro-inflammatory Cytokines Decrease Connexin36 Gap Junction Coupling in Mouse and Human Islets through Nitric Oxide-mediated Protein Kinase Cδ *. J Biol Chem 291, 3184–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores CE, Li X, Bennett MVL, Nagy JI & Pereda AE (2008). Interaction between connexin35 and zonula occludens-1 and its potential role in the regulation of electrical synapses. Proc Natl Acad Sci 105, 12545–12550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatanaka M, Maier B, Sims EK, Templin AT, Kulkarni RN, Evans-Molina C & Mirmira RG (2014). Palmitate induces mRNA translation and increases ER protein load in islet β-cells via activation of the mammalian target of rapamycin pathway. Diabetes 63, 3404–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawat G, Hélie P & Baroudi G (2012). Single intravenous low-dose injections of connexin 43 mimetic peptides protect ischemic heart in vivo against myocardial infarction. J Mol Cell Cardiol 53, 559–566. [DOI] [PubMed] [Google Scholar]
- Head WS, Orseth ML, Nunemaker CS, Satin LS, Piston DW & Benninger RKP (2012). Connexin-36 gap junctions regulate in vivo first- and second-phase insulin secretion dynamics and glucose tolerance in the conscious mouse. Diabetes 61, 1700–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodson DJ, Mitchell RK, Bellomo EA, Sun G, Vinet L, Meda P, Li D, Li W-H, Bugliani M, Marchetti P, Bosco D, Piemonti L, Johnson P, Hughes SJ & Rutter GA (2013). Lipotoxicity disrupts incretin-regulated human β cell connectivity. J Clin Invest 123, 4182–4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS & Spiegelman BM (1993). Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259, 87–91. [DOI] [PubMed] [Google Scholar]
- Hraha TH, Bernard AB, Nguyen LM, Anseth KS & Benninger RKP (2014). Dimensionality and size scaling of coordinated Ca(2+) dynamics in MIN6 β-cell clusters. Biophys J 106, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter AW, Barker RJ, Zhu C & Gourdie RG (2005). Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol Biol Cell 16, 5686–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Dobrian AD, Morris MA, Taylor-Fishwick DA & Nadler JL (2016). Lipids and immunoinflammatory pathways of beta cell destruction. Diabetologia 59, 673–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova E, Yee CW & Sagdullaev BT (2015). Increased phosphorylation of Cx36 gap junctions in the AII amacrine cells of RD retina. Front Cell Neurosci 9, 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klee P, Allagnat F, Pontes H, Cederroth M, Charollais A, Caille D, Britan A, Haefliger J-A & Meda P (2011a). Connexins protect mouse pancreatic β cells against apoptosis. J Clin Invest 121, 4870–4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klee P, Lamprianou S, Charollais A, Caille D, Sarro R, Cederroth M, Haefliger J-A & Meda P (2011b). Connexin implication in the control of the murine beta-cell mass. Pediatr Res 70, 142–147. [DOI] [PubMed] [Google Scholar]
- de Koning EJP, Bonner-Weir S & Rabelink TJ (2008). Preservation of β-cell function by targeting β-cell mass. Trends Pharmacol Sci 29, 218–227. [DOI] [PubMed] [Google Scholar]
- Koster JC, Remedi MS, Flagg TP, Johnson JD, Markova KP, Marshall BA & Nichols CG (2002). Hyperinsulinism induced by targeted suppression of beta cell KATP channels. Proc Natl Acad Sci U S A 99, 16992–16997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothmann WW, Li X, Burr GS & O’Brien J (2007). Connexin 35/36 is phosphorylated at regulatory sites in the retina. Vis Neurosci 24, 363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothmann WW, Massey SC & O’Brien J (2009). Dopamine-stimulated dephosphorylation of connexin 36 mediates AII amacrine cell uncoupling. J Neurosci Off J Soc Neurosci 29, 14903–14911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Hirose H, Ohneda M, Johnson JH, McGarry JD & Unger RH (1994). Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: impairment in adipocyte-beta-cell relationships. Proc Natl Acad Sci U S A 91, 10878–10882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Chuang AZ & O’Brien J (2014). Regulation of photoreceptor gap junction phosphorylation by adenosine in zebrafish retina. Vis Neurosci 31, 237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, Kaiser N, Halban PA & Donath MY (2017). Glucose-induced β cell production of IL-1β contributes to glucotoxicity in human pancreatic islets. J Clin Invest 127, 1589–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdi T et al. (2012). Secreted frizzled-related protein 4 reduces insulin secretion and is overexpressed in type 2 diabetes. Cell Metab 16, 625–633. [DOI] [PubMed] [Google Scholar]
- Marchetti P (2016). Islet inflammation in type 2 diabetes. Diabetologia 59, 668–672. [DOI] [PubMed] [Google Scholar]
- McGarry JD (2002). Banting lecture 2001: dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 51, 7–18. [DOI] [PubMed] [Google Scholar]
- Meier JJ, Veldhuis JD & Butler PC (2005). Pulsatile insulin secretion dictates systemic insulin delivery by regulating hepatic insulin extraction in humans. Diabetes 54, 1649–1656. [DOI] [PubMed] [Google Scholar]
- Menegaz D, Hagan DW, Almaça J, Cianciaruso C, Rodriguez-Diaz R, Molina J, Dolan RM, Becker MW, Schwalie PC, Nano R, Lebreton F, Kang C, Sah R, Gaisano HY, Berggren P-O, Baekkeskov S, Caicedo A & Phelps EA (2019). Mechanism and effects of pulsatile GABA secretion from cytosolic pools in the human beta cell. Nat Metab 1, 1110–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menge BA, Grüber L, Jørgensen SM, Deacon CF, Schmidt WE, Veldhuis JD, Holst JJ & Meier JJ (2011). Loss of inverse relationship between pulsatile insulin and glucagon secretion in patients with type 2 diabetes. Diabetes 60, 2160–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meulen T, Donaldson CJ, Cáceres E, Hunter AE, Cowing–Zitron C, Pound LD, Adams MW, Zembrzycki A, Grove KL & Huising MO (2015). Urocortin3 mediates somatostatin-dependent negative feedback control of insulin secretion. Nat Med 21, 769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda JG, Schleicher WE, Wells KL, Ramirez DG, Landgrave SP & Benninger RKP (2022). Dynamic changes in β-cell [Ca2+] regulate NFAT activation, gene transcription, and islet gap junction communication. Mol Metab 57, 101430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer MA, Halter JB & Porte D (1981). Insulin secretion in diabetes mellitus. Am J Med 70, 579–588. [DOI] [PubMed] [Google Scholar]
- Poitout V, Hagman D, Stein R, Artner I, Robertson RP & Harmon JS (2006). Regulation of the insulin gene by glucose and fatty acids. J Nutr 136, 873–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentki M & Nolan CJ (2006). Islet beta cell failure in type 2 diabetes. J Clin Invest 116, 1802–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentki M, Tornheim K & Corkey BE (1997). Signal transduction mechanisms in nutrient-induced insulin secretion. Diabetologia 40, S32–S41. [DOI] [PubMed] [Google Scholar]
- van Raalte DH & Diamant M (2011). Glucolipotoxicity and beta cells in type 2 diabetes mellitus: target for durable therapy? Diabetes Res Clin Pract 93 Suppl 1, S37–46. [DOI] [PubMed] [Google Scholar]
- Ravier MA, Güldenagel M, Charollais A, Gjinovci A, Caille D, Söhl G, Wollheim CB, Willecke K, Henquin J-C & Meda P (2005). Loss of connexin36 channels alters beta-cell coupling, islet synchronization of glucose-induced Ca2+ and insulin oscillations, and basal insulin release. Diabetes 54, 1798–1807. [DOI] [PubMed] [Google Scholar]
- Ravier MA, Sehlin J & Henquin JC (2002). Disorganization of cytoplasmic Ca(2+) oscillations and pulsatile insulin secretion in islets from ob/obmice. Diabetologia 45, 1154–1163. [DOI] [PubMed] [Google Scholar]
- Ray KK, Seshasai SRK, Wijesuriya S, Sivakumaran R, Nethercott S, Preiss D, Erqou S & Sattar N (2009). Effect of intensive control of glucose on cardiovascular outcomes and death in patients with diabetes mellitus: a meta-analysis of randomised controlled trials. The Lancet 373, 1765–1772. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Diaz R, Dando R, Jacques-Silva MC, Fachado A, Molina J, Abdulreda MH, Ricordi C, Roper SD, Berggren P-O & Caicedo A (2011). Alpha cells secrete acetylcholine as a non-neuronal paracrine signal priming beta cell function in humans. Nat Med 17, 888–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuit FC, Huypens P, Heimberg H & Pipeleers DG (2001). Glucose sensing in pancreatic beta-cells: a model for the study of other glucose-regulated cells in gut, pancreas, and hypothalamus. Diabetes 50, 1–11. [DOI] [PubMed] [Google Scholar]
- Speier S, Gjinovci A, Charollais A, Meda P & Rupnik M (2007). Cx36-mediated coupling reduces beta-cell heterogeneity, confines the stimulating glucose concentration range, and affects insulin release kinetics. Diabetes 56, 1078–1086. [DOI] [PubMed] [Google Scholar]
- Tataranni PA & Ortega E (2005). A burning question: does an adipokine-induced activation of the immune system mediate the effect of overnutrition on type 2 diabetes? Diabetes 54, 917–927. [DOI] [PubMed] [Google Scholar]
- Turner RC, Cull CA, Frighi V & Holman RR (1999). Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: progressive requirement for multiple therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS) Group. JAMA 281, 2005–2012. [DOI] [PubMed] [Google Scholar]
- Urbano FJ, Leznik E & Llinás RR (2007). Modafinil enhances thalamocortical activity by increasing neuronal electrotonic coupling. Proc Natl Acad Sci U S A 104, 12554–12559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urschel S, Höher T, Schubert T, Alev C, Söhl G, Wörsdörfer P, Asahara T, Dermietzel R, Weiler R & Willecke K (2006). Protein kinase A-mediated phosphorylation of connexin36 in mouse retina results in decreased gap junctional communication between AII amacrine cells. J Biol Chem 281, 33163–33171. [DOI] [PubMed] [Google Scholar]
- Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF & Reed JC (1999). Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science 284, 339–343. [DOI] [PubMed] [Google Scholar]
- Westacott MJ, Farnsworth NL, St Clair JR, Poffenberger G, Heintz A, Ludin NW, Hart NJ, Powers AC & Benninger RKP (2017). Age-Dependent Decline in the Coordinated [Ca2+] and Insulin Secretory Dynamics in Human Pancreatic Islets. Diabetes 66, 2436–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA & Chen H (2003). Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112, 1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Li H, Liu X, O’Brien J & Ribelayga CP (2015). Circadian clock control of connexin36 phosphorylation in retinal photoreceptors of the CBA/CaJ mouse strain. Vis Neurosci 32, E009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data will be made available upon request.