

Abstract
In cancer, complex genome rearrangements and other structural alterations, including the amplification of oncogenes on circular extrachromosomal DNA elements (ecDNA), drive the formation and progression of tumors. EcDNA is a particularly challenging structural alteration. By untethering oncogenes from chromosomal constraints, ecDNA elevates oncogene copy number, drives intratumoral genetic heterogeneity, promotes rapid tumor evolution and results in treatment resistance. The profound changes in DNA shape and nuclear architecture generated by ecDNA, alter the transcriptional landscape of tumors, by catalyzing new types of regulatory interactions that do not occur on chromosomes. The current suite of tools for interrogating cancer genomes is well-suited for deciphering sequence, but has limited ability to resolve the complex changes in DNA structure and dynamics that ecDNA generates. Here, we review the challenges of resolving ecDNA form and function and discuss the emerging tool kit for deciphering ecDNA architecture and spatial organization, including what has been learned to date, about how this dramatic change in shape alters tumor development, progression, and drug resistance.
Keywords: ecDNA, cancer, homogeneously staining regions, HSRs, ecDNA evolution, ecDNA hubs, enhancer-hijacking
1. Introduction, discovery and nomenclature
Eukaryotic circular chromosomal structures have been part of the vernacular since the early days. Back in 1932, Barbara McClintock [68] discovered the presence of ring shaped chromosomes in maize, and observed that “ …a deletion in the short arm of the bmn chromosome comparable in extent to the most frequently observed size of the ring chromosome was also present…The ring chromosome did not synapse with the homologous region in the normal chromosome..” The notion of chromosomal deletion, circularization, acentricity, and independent segregation–hallmarks of extrachromosomal circular DNA formation– are all implicit in these prescient remarks. Nevertheless, it was another three decades before circular DNA structures were predicted in plants (wheat nuclei) and animal (boars) based on their biochemical similarity to E. coli [35]. In the same year, larger, extrachromosomal DNA were definitively documented through direct observation of cells in two publications. First, ‘minute chromatin bodies’ were observed in neuroblastoma cells during metaphase [19], where they often appeared paired as ‘double minutes’; second, ‘aberrant sized deleted chromosomes,’ were observed, distinct from the rod-shaped, condensed chromosomes typically observed in metaphase [58]. Subsequent research often used the terms double minutes, but also DMs, ‘double fragments of chromosomes,’ ‘double bodies,’ and ‘accessory chromatin’ to describe these extrachromosomal structures.
We use the term extrachromosomal circular DNA (eccDNA) to encompass all acentric (not containing a centromere), circular DNA structures in Eukaryotic cells. We contrast eccDNA with other large, chromosomal fragments that have circularized to form ring or neo-chromosomes and are often seen in highly rearranged tumor genomes [28]. Specifically, ring chromosomes have centromeres [67], including ectopic centromeres [117, 116], while eccDNA are acentric. EccDNA can be further classified based on size. Smaller elements (< 104 bp) appear to be abundant in all cells [35, 48, 118], sometimes express non-coding RNA capable of affecting gene expression [48, 84, 83] and have recently been shown to be immunostimulatory [118]. We refer to them as ‘micro-DNA’. Ring chromosomes and micro-DNA are not discussed further in this review. Instead, we focus on larger (104-107bp), acentric, circular molecules which are typically visible in an optical microscopic image. While there is some debate about terminology, we refer to them as ecDNA to emphasize that they are found not only as pairs (double-minutes) but as singletons, and also aggregated hubs (more on that later [37]); that they are largely derived from chromosomal DNA, but outside of it and the acentricity and extrachromosomality governs much of their (dys)-function.
2. Foundational experiments
The initial discoveries of ecDNA were quickly recapitulated in many other contexts, and usually in cancer cells [47, 99, 72, 65, 91]. Despite repeated observations of ecDNA, very basic questions regarding how the ecDNA replicated and segregated into daughter cells upon mitosis remained technically difficult to resolve, at least partly because ecDNA could only reliably be observed during metaphase when the chromosomes are compacted and ecDNA can be directly observed as distinct chromatin bodies. The discovery of cultured mouse cells, where the ecDNA were large and distinctive enough to be observed during anaphase, resolved some of these issues. Levan and Levan [54] concluded, based on orientation and positioning of chromosomes and ecDNA, that ecDNA did not orient with the rest of the chromosomes. Instead of being directly pulled by spindle fibers, they were either tethered to chromosomal ends, or clustered in peripheral, nucleolar regions. They also observed anaphase bridge formation with beads of ecDNA strung out along the bridge. Together, these results established the acentricity of ecDNA. Despite the lack of spindle attachments, ecDNA transmission into daughter cells was efficient; ecDNA tethered to chromosomes so they remained inside the nucleus upon cell division, and were not lost into the cytosol and degraded. These events were subsequently demonstrated and confirmed in live cells [40]. Soon after, Barker et al and others showed using BrdU labeling experiments that ecDNA were capable of replicating independently and at the same rate as chromosomal DNA [106,7]. Independently, and much later, the segregation into daughter cells was shown to be random and approximately Gaussian (as a limiting process of the Binomial distribution) [60, 52]. Another line of research suggested circularity, or at least non-linearity, of ecDNA in murine cell lines [33], and this was subsequently and beautifully confirmed with high resolution scanning electron microscopy images overlaid on DAPI stained metaphase images of cancer cells [119].
A series of remarkable papers coming out of the Schimke laboratory provided a foundation for ecDNA exploration. In studying a murine cancer cell line that had achieved resistance to Methotrexate (MTX), Alt et al. found a huge amplification of the Dihydrofolate reductase gene (DHFR) [4]. The DHFR copy number and resistance were both lost upon drug removal. The unstable amplification was due to ecDNA formation. However, a different cell-line showed a stable resistance phenotype and consistent but lower DHFR amplification, which was subsequently attributed to DHFR aggregation and integration in a chromosomal location to form a homogeneously staining region or HSR [41]. HSRs had previously been observed along with ecDNA in neuroblastoma cell-lines, where it was suggested that the HSRs were excised to form ecDNA [6]. However, observations of a colorectal cancer cell line (COLO320) across many passages reversed this conclusion. COLO320 cells originally carried ecDNA containing the oncogene c-Myc, but across many passages, a majority of the cells showed c-Myc to be integrated into a chromosomal location as HSRs [3], suggesting that ecDNA arose before HSR. The ecDNA and HSR mechanisms of amplification were directly correlated in an elegant experiment where continuous growth of the cell line over many passages under MTX treatment led to the DHFR gene being incorporated stably into the chromosome [42, 8].
While eccDNA were observed in ‘normal’ or non-stressed cells in other contexts [121], including animals and plants, they appeared to be mostly micro-DNA, not ecDNA [48]. Micro-DNA are indeed abundant in human and other cells [74]. There was also a suggestion early on that smaller, episomal structures could recombine into larger ecDNA [13], but even the episomes (size 105 bp) were still larger than typical micro-DNA. Moreover, other experiments suggested that ecDNA formation was a rare event and not adequately explained by recombination from smaller circular micro-DNA [32]. Until further evidence is revealed in support of a common origin, we will assume that micro-DNA and ecDNA are unrelated phenomena.
In fact, most of the early ecDNA observations were obtained in cancer cells. The ecDNA were large enough to carry genes, and many were found to amplify the copy number and activity of newly discovered oncogenes, including c-Myc [3] and n-Myc [45] that could provide a proliferative advantage to the cells. Other observations of ecDNA came from cells that were stressed in a manner that provided a fitness advantage to ecDNA carriers [41, 42, 8]. Notably, ecDNA carrying cells often lost them efficiently in the absence of selective pressure from drug treatment [4, 10], or simply in transference from mouse xenografts to cell cultures [42]. These observations remain relevant today. With technological improvements allowing for the testing of ecDNA presence in thousands of samples, ecDNA have rarely or never been observed in normal tissue from cancer patients even when the tumor cells of those patients have abundant ecDNA [108, 43]. Large ecDNA have been observed in plants (weeds) but, once again, as a pesticide resistance mechanism [73, 46].
Thus, while not apparent at the time, a retrospective reading of these papers as well as new results seem to confirm that unlike micro-DNA, ecDNA formation is a rare event, and its continued maintenance is unique to those cells in which ecDNA provide a fitness advantage.
3. Towards a model of ecDNA evolution
The results from the foundational experiments can be distilled to support a working model of ecDNA evolution, which we propose here as a strawman.
EcDNA are formed in a stochastic event that involves double strand breaks in the linear chromosome, followed by ligation to circularize the unprotected ends (See section 5).
ecDNA replicate independently at rates similar to linear chromosomes [7].
The replicated ecDNA segregate at random into the daughter cells [60, 52].
The cellular microenvironment imposes a selection bias for ecDNA carrying cells with direct implication on continued presence of ecDNA or their elimination from the population [4, 41, 42, 52].
These observations were recently put into a mathematical model and investigated systematically by Lange et al [52] (Also see (Figure 1)). Consider a scenario where a single ecDNA is generated and denote that time as t = 0. Each subsequent mitosis doubles the number of cells. Let Nk(t) describe the number of cells with exactly k ecDNA at time t of a population of cells. Consider one of the Ni(t) cells carrying i copies of ecDNA. The ecDNA self-replicate to form 2i ecDNA copies prior to mitosis. Random segregation leads to the two daughter cells obtaining k and 2i − k ecDNA copies according to the Binomial distribution for all 0 ≤ k ≤ 2i. evolving values of Nk(t) (distribution of ecDNA counts among cells) are governed by the following system of coupled differential equations:
| 1. |
| 2. |
Here, s denotes a selection coefficient so that ecDNA carrying cells are s-fold more likely to divide compared to non-carriers. Carriers have a fitness advantage when s > 1, disadvantage when s < 1, and are neutral for s = 1. While the coupled nature of the equations makes it difficult to solve them analytically, simulations allow us to model many aspects of ecDNA.
Figure 1. A model of ecDNA evolution.
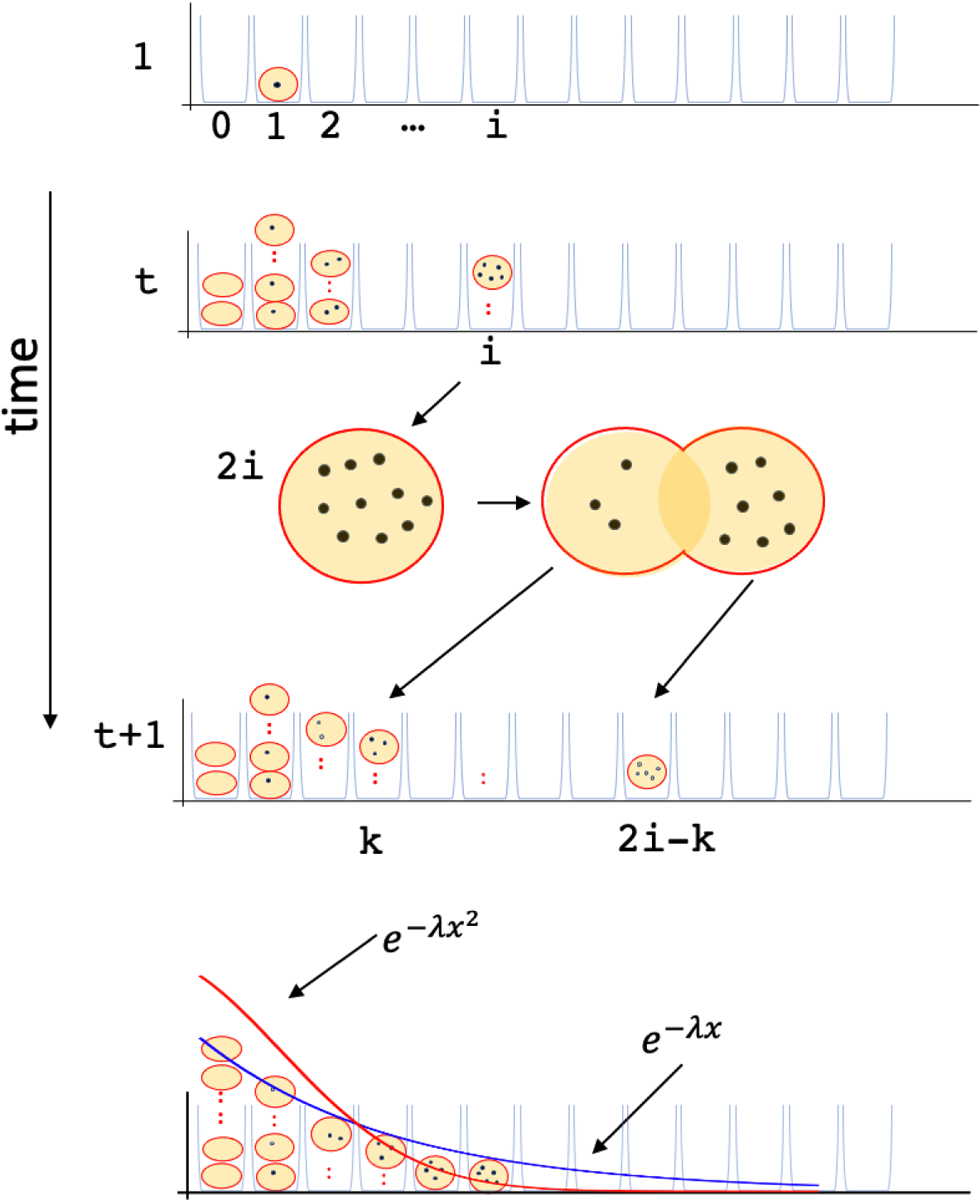
Bin i maintains the count Ni(t) of the number of cells with exactly i ≥ 0 ecDNA copies. At t = 1, N1(1) = 1 and all other bins are 0. An ecDNA-positive cell is s times more likely to be picked relative to an ecDNA-negative cell, where s denotes the selection coefficient. The chosen cell with i ecDNA copies replicates and divides into two daughter cells containing k and 2i − k ecDNA, chosen according to a Binomial distribution. Simulations suggest that in the absence of selection (s ≤ 1), the proportion of ecDNA-negative cells in the population increases to 1, but for s > 1, it rapidly diminishes. In the limit, the tail distribution of ecDNA counts follows a power-law with an exponential cut-off (dark black line), which is wider than the Normal distribution.
Simulations of the model showed in the absence of selection (s = 1)or with negative selection (s < 1), the fraction of cells carrying ecDNA (1 −N0(t)/N(t)) rapidly decays, completely consistent with ecDNA not being observed in ‘normal’ cells [52]. On the flip-side, N0(t)/N(t) approaches 0 in simulations with positive selection, even for small selective advantage. These complementary results suggest that ecDNA presence should coincide with the presence of a functional element that confers a functional advantage. We will explore this facet in Section 6.
The model also makes a direct prediction of the distribution of ecDNA in cells, with the tail probabilities suggesting a wider distribution than the Normal distribution, but somewhat narrower than an exponentially decaying distribution (e−λx2 ≤ Pr(Nk(t)/N(t) = x) ≤ e−λx for large x). The model predictions were nicely validated by comparing against experimentally observed ecDNA distributions in multiple cell lines, and choosing s to best fit the experimental data [52]. The results showed a good fit to the simulation based predictions of ecDNA count distribution, and indicated clearly that ecDNA were under positive selection. They also explain the relatively high heterogeneity of ecDNA copy number. Finally, with modest positive selection, the typical number of ecDNA per cell still remain in the hundreds.
Nevertheless, the model is simplistic in its treatment of selection, in that the fitness advantage depends solely on the presence/absence of ecDNA, and not on their abundance. It also does not account for the loss of ecDNA. Other models have worked with more complex selection regimes where the fitness is reduced with increasing ecDNA due to additional metabolic load [108]. With improved experimentation methods, the models could be refined to provide precise, quantitative predictions of the fitness advantage of ecDNA formation, and provide a foundation for exploring ecDNA evolution.
Even in its current, simple form, the model provides a useful framework for studying ecDNA. The remainder of this review will attempt to explain the experimental data using the model based framework, and we will also refine the model as we go along. Specifically, we discuss how ecDNA are formed, how prevalent they are in cancer or normal cells, the exact mechanisms of random segregation, what functional properties lead to positive selection, how they correlate with increase pathology of cancer, how these functional aspects could be co-opted for intervention and therapy, and the mechanisms by which ecDNA are lost under negative selection, or simply the absence of positive selection. Before we address these points, however, we consider a technical detour aimed at reliable detection of ecDNA.
4. Identifying ecDNA and elucidating their primary structure
4.1. Cytogenetic methods for detecting ecDNA
During metaphase, the chromosomes are compacted and aligned on the metaphase plate, making it easier to identify ecDNA as distinct structures after staining DNA with 4–6-diamidino-2-phenylindole (DAPI). Metaphase DAPI continues to be one of the most reliable method of ecDNA discovery, but until recently was a low throughput method. Turner et al [108] used automated computer-vision based methods to count the number of ecDNA per cell across a multitude of cells with high specificity (few false ecDNA calls), but somewhat lower sensitivity. Recent advances in deep neural networks have led to the development of fully convolutional neural networks (CNNs) for the problem of image segmentation [57], where the objective is to reliably assign a category or label to each pixel of the image. In an application of these ideas to metaphase images, CNN with the U-net architecture was trained to assign each pixel of the DAPI image to one of the following categories: ecDNA, cytoplasm, chromosome, and intact nucleus. The trained networks could identify ecDNA pixels with close to 85% accuracy, even when the ecDNA contours were proximal to chromosomes and not easily distinguished with computer vision methods [86]. The utilization of these methods helped identify ecDNA at scale in thousands of images and providing reliable estimates of the prevalence of ecDNA.
Recall from the model (Equations 1,2) that the high heterogeneity and wide tail of ecDNA distribution demands the sampling of a larger number of cells (20–200) for accurate estimates. The classification of a sample as ecDNA positive is determined by somewhat arbitrary but reasonable cut-offs of ≥ 2 ecDNA per cell [108] or a more aggressive measure of 1 ecDNA for every 2 cells on the average, with at least 20 cells sampled [43]. The estimation of ecDNA distribution has allowed for a precise calculation of the impact (selection strength) of drug treatment by measuring the change in ecDNA distribution [119, 86, 52].
While highly accurate, DAPI staining is not sufficient to resolve the different ecDNA forms that a cell may present. DAPI signals cannot be detected for micro-DNA, and very small ecDNA (~ 105 bp) may also be missed. The use of Fluorescent in situ hybridization (FISH) using specific DNA probes allows for the positioning of specific genomic regions inside or outside of chromosomes, and have revealed cells with small (< 100kbp) ecDNA [22]. It has also resolved the counts and copy numbers of multiple distinct ecDNA structures, each amplifying a different gene [37]. DNA FISH requires prior knowledge of the probes, and is typically used in conjunction with a DNA copy number analysis method to identify probes in genomic regions with focal copy number amplification.
Metaphase analysis requires growing cells, which are often not available from primary cancer tissues. DNA FISH and other hybridization methods have been used to validate the genomic content of ecDNA and also study amplification of specific DNA regions directly from interphase cells. The reliable identification of ecDNA in interphase cells remains an unsolved problem.
4.2. Next generation sequencing for ecDNA identification and structure
The genomics revolution, starting with the sequencing and assembly of the human genome [51, 111] and followed by the development of massively parallel next generation paired-end sequencing (NGS) and subsequent paired-end mapping of query DNA from donor samples allowed for the fine-mapping of both small nucleotide and larger, structural variants in the donor genome [89, 109, 123]. While revolutionary, “The Human Genome Project in the nineties sucked a lot of the oxygen” (Wahl, 2020) out of ecDNA research [20]. The early genomics projects focused more on detection of smaller single nucleotide variation, or ‘simpler’ structural variation in the form of measuring copy number changes and translocation events that were easiest to discover using genomic short reads. While aneuploidies and copy number amplifications were identified and treated as events with consequences, particularly in cancer, their spatial relationships and mechanistic aspects were set aside, or forgotten.
Methods for paired-end NGS based mapping [55] and structural variant identification began to be used for identifying the structural features of ecDNA (Figure 2a) and HSRs from cancer cell lines [104, 105, 90, 49] and xenografted tumor cells [112]. These methods have recently been extended and refined to automatically elucidate the genomic structure, or architecture, of ecDNA from whole genome sequencing data [108, 22, 120]. The computational problem is related to de novo genome assembly, but is distinct. First, unlike chromosomes which are typically diploid, ecDNA appear in multiple copies per cell and may have significant heterogeneity of structure. Second, in de novo genome assembly, there is no prior knowledge of the primary sequence, and the assembled sequence has to be generated by stitching overlapping fragments together. On the other hand, as the ecDNA segments are all derived from chromosomal segments from a known genomic reference, the fragments can be mapped to the reference first to identify distinct genomic regions mapping ecDNA (Figure 2b). Moreover, paired-end or split reads with ends mapping to distinct genomic segments describe breakpoints that help stitch those segments together in the correct orientation to elucidate the genomic structure of ecDNA. A number of methods have therefore adapted this method of directed assembly to determine the structure of ecDNA and other focal amplicons [90, 22, 120, 93]. Other graph and string based explorations of complex genomic rearrangements that include, but are not focused on ecDNA, are also becoming an essential part of the genomic toolkit [31, 12].
Figure 2. Directed assembly for ecDNA structure and detection.
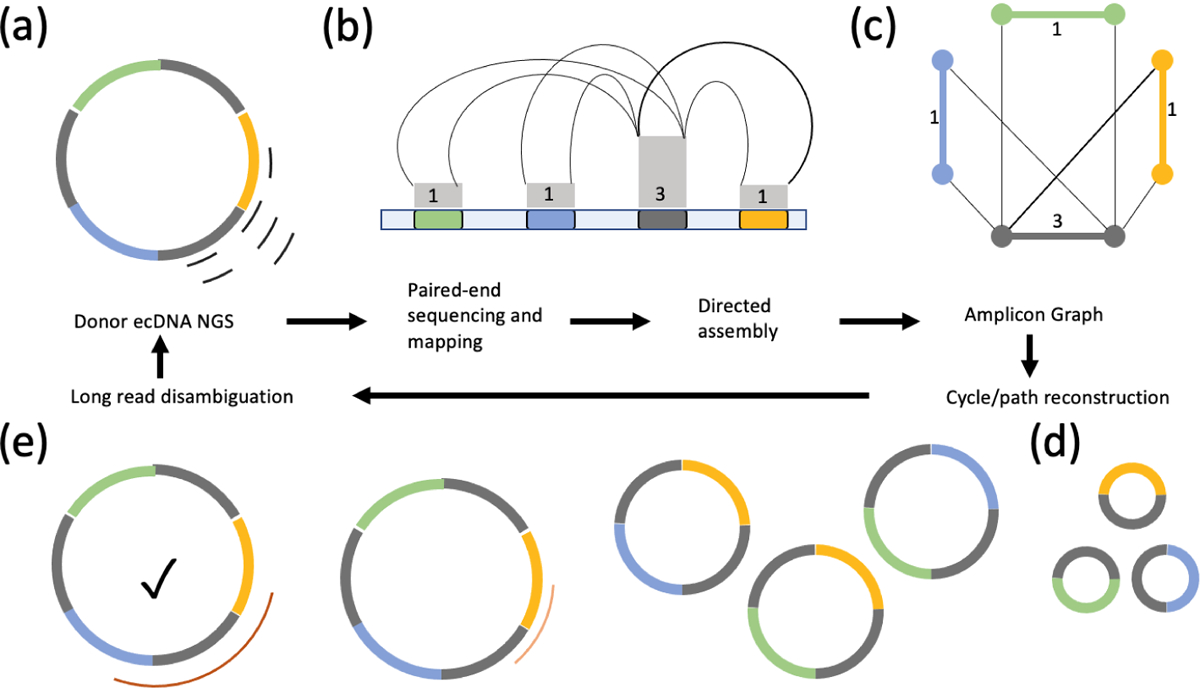
(a) Short-reads from whole genome sequencing also sample ecDNA structures. (b) Paired-end sequencing and mapping identifies breakpoints and copy number changes. Colored segments represent genomic intervals of an amplicon. Gray boxes represent estimates of the copy number and multiplicities of segments (numbers). Breakpoints connecting the segments are represented by thin black lines. (c) Directed assembly methods smooth out coverage and generate an amplicon graph representing all amplified segments and their multiplicities and breakpoints. (d,e) Paths and cycles in the amplicon graph help detect ecDNA (e.g. long, high-copy cycles) and their fine structure. Large segments with high multiplicities (gray segment), missing breakpoints, or heterogeneity of ecDNA all lead to ambiguity of reconstruction. All cycles in panels d and e are consistent with the breakpoint graph.(e) Long-reads help detect breakpoints that short-reads may have missed due to low complexity sequence. Long-reads that span high multiplicity regions resolve ambiguities in reconstruction. The checked circle represents the true reconstruction.
The Amplicon Architect method starts with identification of seed regions of focal amplification and explores breakpoints that connect these regions to other distinct regions. It next builds a graph where nodes correspond to the ends of segments. Edges represent either segments, or breakpoints connecting two distinct segments through deletion, inversion, or translocation. The resulting amplicon graph is a compact representation of all possible structures encoding the focal amplification (Figure 2c). Circular paths in the graph represent putative ecDNA structures, and in many cases a single (cyclic) path explains most of the copy number amplification of the genome, providing us with an unambiguous ecDNA structure. For complex, heterogeneous ecDNA structures, short reads may not suffice to provide a unique structure, especially when a large segment is repeated multiple times in the ecDNA. For instance, the amplicon graph constructed using short-reads sampled from the ecDNA in Figure 2a permits multiple ecDNA structures (Figure 2d).
Even in the absence of unique structure reconstruction, the amplicon graph provides a powerful abstraction of the amplicon structure, because it identifies the (possibly multi-chromosomal) genomic intervals that are part of the ecDNA (Figure 2b,c). Integration of functional information including chromatin accessibility, gene expression, chromatin conformation and DNA interactions onto the amplicon graphs can reveal important functional aspects of ecDNA (See section 6).
While genomic analysis is primarily used to understand the fine structure of ecDNA, it could also be utilized to predict the presence of ecDNA, based on the presence or absence of high copy cycles in the amplicon graph [22, 43, 120]. This argument needs some clarification because repeated tandem duplications could also mechanistically explain cycles in the amplicon graph. However, that explanation is contingent upon the re-use of identical breakpoints multiple times–an unlikely event. Kim et al [43] compared genomic and cytogenetic data to show that amplicon analysis could indeed predict ecDNA with high sensitivity and specificity (> 82% for both). In fact, because ecDNA are functionally different from chromosomal DNA (see Section 6), recent research has also utilized functional data, specifically, high-throughput sequencing of Transposase-Accessible Chromatin (ATAC-seq) to detect microDNA and ecDNA [75, 48], and future work is likely to incorporate other sources of functional data for reliable detection of ecDNA in bulk and single cell modes [119, 34, 16].
Despite these developments, ecDNA identification using short-reads has its challenges. EcDNA reintegrate back into chromosomes into non-native locations as HSRs. The integration often involves multiple copies of the ecDNA and preserve its fine structure, making it difficult to distinguish between ecDNA and HSRs. Koche et al [44] observed a palm-tree motif with the fronds indicative of chromosomal integration of ecDNA (HSR). The palm tree signal could be used to distinguish ecDNA from HSR but may not be visible when the integration happens at few (perhaps, one or two) locations. With short-read sequencing, it is hard to detect pure ecDNA from hybrid ecDNA and HSR structures. At the same time, much research [42, 105, 78, 37, 102] suggests that the movement between ecDNA and HSR is dynamic and that cell populations carrying both ecDNA and HSRs co-exist [38, 8], diminishing the need for a strict differentiation between these two states. A second challenge with short-read sequencing is that cyclic structures may be detected with multiple ‘fold-back’ discordant reads connecting duplicated segments head-to-head or tail-to-tail arising due to BFB cycles (See section 5). Conservatively, signatures of BFB [126] are used to preclude ecDNA calls prior to calling ecDNA [43].
4.3. Some long-reads are more equal than others
Breakpoints that lie in repetitive or low-complexity sequence at breakpoint junctions are often missed by short-reads, and this accounts for most of the false negatives in the short-read based ecDNA identification [43]. Additionally, the presence of repetitive sequence leads to ambiguous paths in the amplicon graph. It has long been a mantra that long-reads can help disambiguate complex assembly (or amplicon) graphs. Disambiguation can be achieved by reads that can span the entire repetitive region. For instance, the amplicon graph in Figure 2c supports many possible reconstructions obtained by traversing cycles (Figure 2d). However, the presence of a single long-read is sufficient to select the one true cycle as long as it spans the region of high multiplicity (Figure 2e). Long-read sequencing available via the SMRT and HiFi sequencing platforms of Pacific biosciences and Nanopore sequencing are large enough to theoretically identify all breakpoints of an amplicon, and they are being increasingly utilized for ecDNA analysis [22, 34]. However, current technologies typically have limited yield for DNA fragment sizes in excess of 40–50kbp. These lengths are large enough to span most common repeats in the human genome, where LINE elements are about 10 kbp, and therefore can identify most breakpoints. Single molecule sequencing also allows for the direct measurement of DNA methylation without the need for bisulfite conversion and have been used to elucidate ecDNA function [34]. However, current yields of very long-reads (> 200 kbp) are low. Therefore, large duplicated regions seen in ecDNA may not be spanned, making it difficult to resolve all ambiguities.
Optical mapping is emerging as another technology of interest for investigating structural variants in cancer genomes[39]. The technology is derived from one of the early innovations of genomics, where restriction fragment based physical mapping of clones allowed for identification of a clone and its overlapping partners without sequencing [23]. In its new incarnation, isolated DNA molecules up to 250kbp are at first stretched out in nano-channels. Next, specific motifs on the molecules are recognized and fluorescent imaged to provide optical restriction maps (OM) for each molecule [50]. The inter-marker distances can be used to detect overlapping fragments, and assemble very large molecules (exceeding 1Mbp) of restriction site locations. These OM contigs can be analyzed using specialized algorithms to identify structural variation [53, 59, 85] and have been used in conjunction with NGS to successfully disambiguate ecDNA structures (Figure 2e) and distinguish ecDNA from other amplicons [59, 16, 102]. They have been utilized to successfully reconstruct the architecture of large HSRs and BFB cycles, and to distinguish other forms of focal amplification from ecDNA [59, 102].
4.4. Purification before sequencing
In Circleseq, total genomic DNA, including ecDNA, is immobilized in a gel and digested with an exonuclease for digesting linear DNA and enriching for ecDNA [77]. The enrichment is followed by rolling circle amplification to amplify the circular DNA. Subsequent sequencing with short and long reads provide a clear guide to the ecDNA segments on the genome [34]. The technique has been shown to work even in single cells and promises to be a great tool in tracking ecDNA heterogeneity across populations, especially in conjunction with long-read sequencing [44]. It has been particularly successful for micro-DNA. Some challenges remain for the larger ecDNA molecules. Inefficient exonuclease digestion may lead to no-targeted sequence; multiple, distinct ecDNA segments can be identified but may be difficult to separate; the rolling circle amplification may not amplify the entire ecDNA region. Some of these issues were addressed using the ‘CRISPR-catch’ technology [36]. Strategically guided CRISPR based cuts linearize small ecDNA derived segments that can be separated from the larger chromosomal segments and from heterogeneous ecDNA using pulsed-field gel electrophoresis. Next generation sequencing of gel-bands followed by customized algorithmic analysis leads to simpler amplicon graphs with unique cyclic paths. Moreover, different bands and CRISPR guides provide a structural determination of ecDNA heterogeneity and can elucidate the structure of multiple distinct ecDNA in one sample.
Taken together, the tremendous growth of technologies for ecDNA detection have made it possible to estimate the prevalence of ecDNA in cancers and systematically explore its structure and function, which had long been a matter of some debate.
4.5. Estimating ecDNA prevalence
The prevalence of ecDNA has been a matter of some debate. A survey of 9,500 metaphases cells obtained from individuals with hereditary tumor, non-familial cancers and controls with no tumors identified ecDNA only in 15 images, and almost exclusively in patients with multiple endocrine neoplasia [91]. An estimate based on surveying the Mitleman database suggested that 1.5% of all cancer samples carried ecDNA [24]. In more recent work, Turner et al utilized automated image analysis to investigate over 2, 500 cancer and normal cell lines in metaphase stained with DAPI, representing multiple tumor subtypes. They found that nearly 40% of all samples carried ecDNA. Kim et al. [43] analyzed whole genome sequencing data from 3,212 tumors and 1,810 non-neoplastic samples, and found that 460 (14.3%) of tumor samples carried ecDNA, with no occurrence of ecDNA in normal samples. EcDNA have been independently shown to be abundant in other cancers including Glioma [21], Neuroblastoma [34], Medulloblastoma [16], and Oropharyngeal [82] cancers. Anecdotal evidence suggests that ecDNA arise early in tumor development and the frequency of ecDNA increases with progression of tumor, but these will need to be confirmed with systematic time course analyses of tumor progression. As technologies for ecDNA detection continue to improve, these estimates will be revised, but it is likely that ecDNA occurrence is a common, pan-cancer phenomenon.
5. Formation of ecDNA
5.1. Episome formation
The most direct model of ecDNA formation is the ‘episomal’ model that involves double strand breaks followed by re-ligation. Carroll et al investigated an experimental model of CHO cell line with an integrated CAD gene array, and showed the formation of ecDNA using Southern blotting methods [13]. They posited that the episomes are usually smaller (about 250kbp), but gradually enlarge to become ecDNA. They also showed that the ecDNA formation was concurrent with a deletion scar on the chromosome. Their results were generally consistent with independent observations of ecDNA formation [42, 8, 114].
Subsequently, Vogt et al used quantitative PCR and chromosome walking methods to elucidate the genetic content and ecDNA architecture in seven gliomas [113]. This structural analysis confirmed that the ecDNA was most likely formed by a circularization of a chromosome fragment. The DNA fragments overlapped the epidermal growth factor receptor (EGFR) gene [112], providing a selective advantage. However, they also observed that the corresponding chromosomal loci were not rearranged and the presence of ecDNA did not, in fact, coincide with a corresponding deletion in any of the glioma samples. Their results strongly suggested that a re-replicative or post-replicative event was responsible for the formation of each of these initial amplicons (Figure 3a). For example, in the case of 4 copies of the chromosome (two sister-chromatid pairs), the ecDNA could co-segregate with intact chromosomes during mitosis or sister chromatid repair could repair the chromosomal lesion. Alternatively, in a replication bubble model, an ecDNA might excise out of a replication fork and circularize. Meanwhile, fork regression and re-replication from flanking forks would lead to sister chromatids with no scars.
Figure 3. Mechanisms of ecDNA formation.
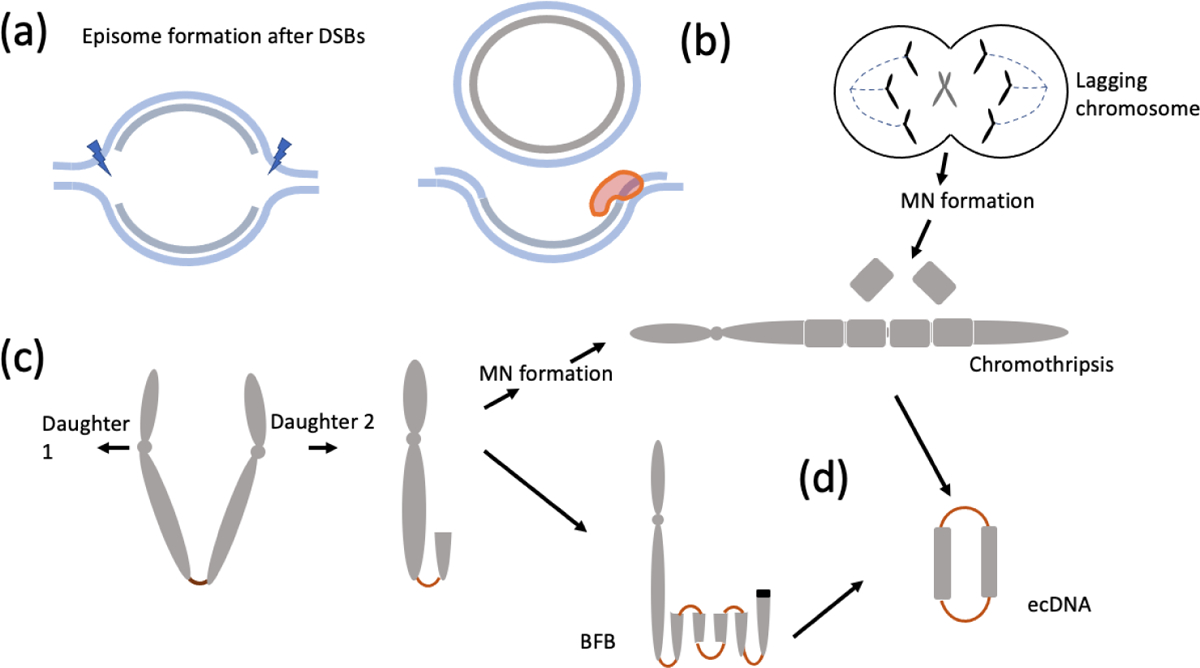
(a) episomes formed by replication fork stalling at a bubble, DNA breakage and subsequent re-ligation leads to ecDNA formation from the one strand while repair in the second strand allows for replication to proceed without a deletion ‘scar.’ (b) Mis-segregation errors may lead to a lagging chromosome followed by micronuclei formation. Shattering in subsequent mitoses and re-ligation generate a ‘chromothriptic’ chromosome. (c) Telomere loss and sister-chromatid bridging leads to broken ends or lagging chromosomes. Repeated breakage fusion bridge cycles may lead to a rearranged chromosome with an HSR-like signature. (d) EcDNA may form by fragments breaking off and circularizing during BFB cycles or chromothripsis.
Subsequent experiments analyzing the fine sequence of multiple ecDNA and HSR structures were consistent with the episome model as the basis of ecDNA formation [104, 105]. Those experiments also failed to identify a corresponding deletion scar in the chromosome that matched the initial excision, and therefore supported the post-replicative model. Storlazzi et al[105] also identified strong structural similarities between ecDNA and HSR structures, indicating a common origin between the two modes of amplification. The fusion of the two double-stranded breaks was found to be mediated by by non-homologous end-joining (NHEJ), a repair mechanism that is largely error free and can occur at all stages of the cell cycle [15, 71].
5.2. Chromothripsis
With the rapid advent of genomic methods including whole genome sequencing, ecDNA formation could be studied in a larger context of the genome formation. Stephens et al observed extensive rearrangements, largely within a single chromosome of a patient with chronic lymphocytic leukemia [103]. Moreover, the copy numbers on the chromosome followed a distinct oscillating patterns, suggesting a one time, catastrophic shattering event they called Chromothripsis. Subsequent work suggested that micronucleus formation was a critical event for chromothripsis occurrence [127]. Mitotic errors could lead to mis-segregation of chromosomes and their physical isolation into aberrant micronuclear structures. Subsequent mitoses resulted in catastrophic shattering and recombination, or chromothripsis. Additionally, the rearranged byproducts could include ecDNA, implicating chromothripsis as a source of ecDNA formation (Figure 3b) [62]. In an elegant confirmation of this idea, an experimental model of inducible y-chromosome centromere inactivation was used to generate a mis-segregation error leading to a lagging chromosome, micronucelus formation, and chromothripsis [63, 62]. Whole-genome sequencing of clonally propagated rearrangements suggested extensive rearrangements and translocations consistent with chromothriptic breakage and re-ligation, copy number changes, and also ecDNA formation [61].
5.3. Breakage Fusion Bridge
The breakage fusion bridge cycle was first described by Barbara McClintock as a genomic abnormality in maize [69, 70]. A telomeric break leads to the formation of a chromatin bridge between sister chromatids during replication (Figure 3c). When the sister chromatids are pulled apart during mitosis, an unequal cleavage might lead to a rearranged chromosome with a duplicated inversion at one end and a broken end that is rescued by bridge formation with a sister chromatid and a repeat of the BFB cycle (Figure 3d). It was suggested that BFB could result in genome instability and over-expression of oncogenes due to gene amplification [56]. BFB mediated rearrangements have often been described in explaining tumor genome arrangements, notably in the case of HER2 amplified breast cancers [96, 66]. Umbreit et al utilized multiple experimental methods to induce telomere loss, including low-dose topoisomerase II inhibition and CRISPR/Cas9 mediated telomere loss on chromosome 4, allowing them to observe the consequence of bridge formation at high resolution [110]. Telomere loss and bridge formation triggered a series of catastrophic sequence of events that could lead to extensive rearrangements and a chromothripsis like signature. Specifically, the bridge formation did not only occur in sister-chromatids but also among different chromosomes, resulting in multi-chromosomal rearrangement events. In parallel, the BFB cycle was shown to result in breaking off and circularization of the re arranged fragments to form ecDNA [97] combining chromothripsis, BFB cycles, and ecDNA as part of a common smorgasbord of DNA instability.
5.4. Oncoviral sequences complete the circle
HPV integration into the human genome in squamous cells of the cervix and oropharyngeal cavity is a major source of genome instability, and integration is associated with poor outcome for the patient [79]. Analysis of the whole genome data from HPV16/18 positive cell-lines revealed extensive rearrangements involving both viral and human sequences [2]. Similar analyses of head and neck cancer samples suggested the presence of hybrid episomal structures [80]. A reconstruction of the amplicon structures using reference HPV16/18 and human genome as references revealed the presence of hybrid ecDNA in nearly 50% of cervical cancer samples [22], and human-only or hybrid ecDNA in a third of head and neck cancer samples [82]. EcDNA formation could be associated with increased expression of viral and human oncogenes, leading to increased genomic instability.
Taken together, these results suggest that ecDNA formation is a complex event, a consequence of compromised DNA damage repair, and can occur through multiple, distinct but non-exclusive rearrangement mechanisms. The initial event leading to ecDNA formation is likely to be a stochastic, as analyses of breakpoints support non-homologous end joining (NHEJ) at junctions [105, 63]. Even in ecDNA amplifying the same oncogene, the exact breakpoints on either side of the oncogene are not conserved [43]. Nevertheless, it is not understood if the events are completely stochastic or if they have a tissue specific bias. More experimental work is needed to clarify these and other missing facets of ecDNA formation.
6. Functional characteristics of ecDNA
Selection is a key aspect of ecDNA maintenance in cells. Increased expression of oncogenes can increase the fitness of cancer cells in terms of growth and proliferation. Indeed, over 50% of the oncogenes that showed > 8× copy number amplification were found on ecDNA in a pan-cancer analysis [43]. Tissue specificity of ecDNA architecture has also been observed. An analysis of focal amplifications (and likely ecDNA) in the Cancer Genome Atlas data showed enrichment of MDM4 and EGFR in glioblastoma, while breast cancer amplifications were enriched in MYC and ERBB2 [22]. Thus, while ecDNA formation might be stochastic, tissue specificity of gene expression programs may lead to different selection pressure on an oncogene carrying ecDNA in different tissue types, resulting in a tissue specific architecture. The explanation is not entirely satisfactory, however, because it suggests high diversity of ecDNA at formation, followed by selection based pruning for specific ecDNA. One possible explanation is that the formation of ecDNA, with respect to the location of double-stranded breaks, is itself regulated, but the mechanisms for tissue specific ecDNA formation remain to be elucidated.
6.1. ecDNA chromatin is highly accessible
It was known that ecDNA carry accessible chromatin and genes on ecDNA are expressed, although not uniformly, providing a functional basis for selection [100]. Initially, the over-expression was entirely attributed to the increase in copy number of the gene carried by ecDNA. To test this, a systematic assessment of chromatin accessibility using the Assay for Transposase-Accessible Chromatin (ATAC-seq [11, 18]), and a related visual method (ATAC-see [17]) was conducted on multiple cancer cell-lines and primary tumor samples [119]. The analysis revealed that ecDNA contain among the most accessible chromatin in the cell, and lack the higher order conformation typical of heterochromatin allowing for significantly higher levels of transcriptional activity (Figure 4a). A large pan-cancer study of over 3, 000 samples showed tremendous increase in expression of oncogenes on ecDNA relative to their expression on chromosomes, even after correcting for the increased copy number [43]. Similarly, over expression of the NTF3 gene was observed in an ecDNA in a neuroblastoma sample and its extrachromosomal origin was confirmed using allele specific expression [44]. The results emphasized how the unique architecture and epigenetics of ecDNA provided carrier cells with increased fitness, resulting in proliferation and increased tumor pathology.
Figure 4. Functional characteristics of ecDNA.
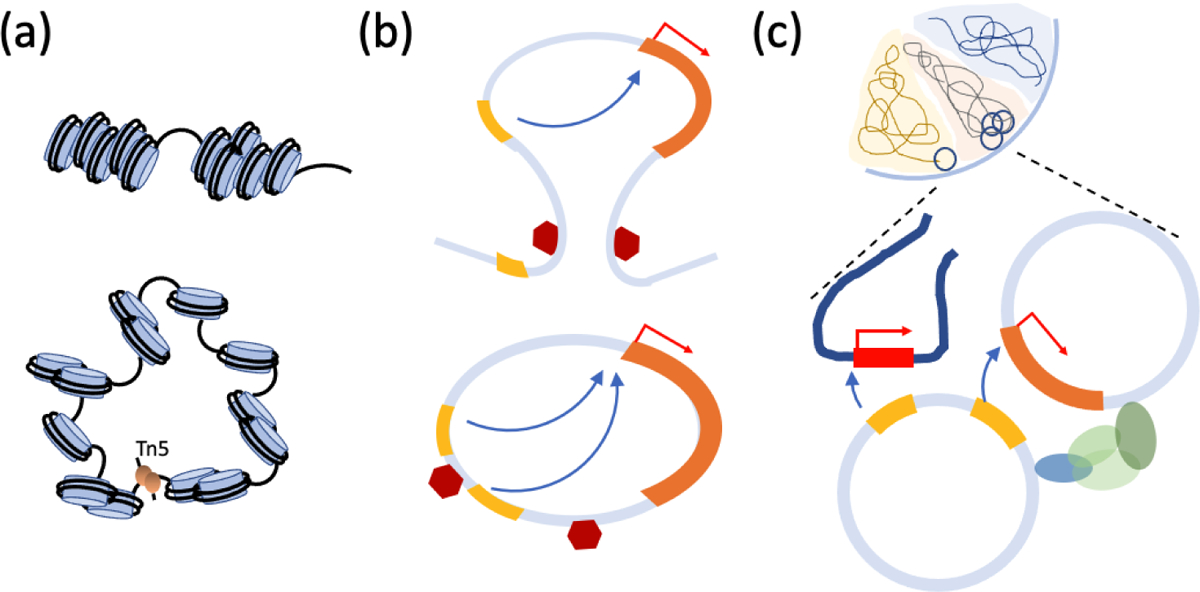
(a) In contrast with chromosomes (top-panel), ecDNA (bottom-panel) is highly accessible, resulting in over-expression of genes on ecDNA. The expression level remains high even after correcting for copy number. (b) Topology Associated domains (TADs) provide the regulatory elements that control gene regulation, and shield the gene body from outside enhancers (top-panel). The circular structure of ecDNA changes the regulatory circuitry through hijacking of enhancers outside the TAD. (c) ecDNA form hubs and interact with chromosomes, resulting in enhancer activity that regulates genes in other ecDNA, and even on chromosomes.
6.2. Enhancer hijacking
These results were rapidly extended in multiple directions, providing new insights into ecDNA biology. Chromatin capture technologies identify spatial proximity of linearly distal regions of the chromosome, suggesting loop formation and topological domains that serve as boundaries of gene regulation [64]. An independent 4C-seq chromatin capture experiment anchored on the EGFR promoter was used to interrogate Glioblastoma samples and revealed a remarkable pattern [75, 119]. In the sample where EGFR copy number was not amplified, the expected strong contacts against known upstream enhancers were observed and none were outside the 480kbp topological domain that contained EGFR (Figure 4b). However, in other samples, where EGFR was amplified on ecDNA, multiple new contacts were observed. The new contacts were outside the known topology associated domain, suggesting a rewiring of the regulatory circuitry [75].
In a similar experiment in neuroblastoma, a MYCN containing ecDNA was found to connect distal regions of chromosome 2 into a single amplicon [34]. Hi-C showed enhancer hijacking events connecting enhancers in the distal region to the n-Myc promoter. The contact maps confirmed a new chromatin domain (neo-TAD formation) where “genes, enhancers, and insulators from distal parts of the genome” formed a novel, spatially interacting neighborhood. Non-conventional chromatin interactions have also been reported in Medulloblastoma patients [16], and are likely to be a common feature of ecDNA gene regulation.
6.3. Regulatory trans-interactions
Remarkably, a ChIA-PET analysis on GBM-patient-derived neurosphere cell lines revealed that enhancer hijacking and regulatory rewiring is not limited to being within the ecDNA structure [128]. Two cell-lines, one carrying c-Myc and another carrying EGFR showed extensive contacts within ecDNA but also between regions of ecDNA and other chromosomal regions (Figure 4c). The contact sites on ecDNA and their chromosomal targets were mostly enriched at promoters. The ecDNA regions were enriched with marks of enhancer activity, including accessible chromatin and H3K27 acetylation, suggestive of regulatory function. Further, transfection of artificial ecDNA into ecDNA negative cell lines increased activation of many chromosomal genes. Together, their results are suggestive of an ‘ecDNA party bus’, that brings a mobile enhancer to change the expression of chromosomal genes [1]. The question remains if these interactions are stochastic or recur in different cell types.
Aggregation of ecDNA had been observed previously but was generally assumed to be an aberrant condition suggestive of DNA damage in ecDNA, untethering of ecDNA from the chromosomes and their eviction via micronuclei formation [54]. However, Hung et al [37] observed a strong local concentration of the ecDNA FISH signal within the nuclei of many ecDNA-positive cancer cells, despite arising from tens to hundreds of distinct ecDNA molecules. They suggested that hub formation was central to ecDNA biology. Live imaging showed that the hub formation was not static. During mitosis and in metaphase, the hubs separated into smaller particles and tethered to chromosomes. After mitosis, the hubs reformed in the G1 phase. Hung et al also identified a bromodomain and extraterminal (BET) protein, Brd4, that was critical to hub maintenance. A BET inhibitor, JQ1, strongly and specifically disrupted ecDNA hubs. In the SNU-16 cell-line, two distinct ecDNA molecules, one carrying FGFR2, and the other carrying Myc were found inthe hubs. Enhancer elements on both ecDNA cooperatively regulated Myc, thus establishing a novel instance of enhancer hijacking involving distinct ecDNA. EcDNA hub formation was independently observed using a newly developed ecTag analysis [124]. As the hubs dissociate in metaphase, hub-formation is not inconsistent with random segregation. Inefficient dissociation could lead to non-random segregation and demand a revision of Equations 1,2. Note, however, that with clumped ecDNA, the segregation is more likely to be unequal, which would only exacerbate ecDNA mediated pathology.
These results underscore the high accessibility of ecDNA chromatin, and the dramatic reorganization of primary structure that results in the formation of new topologically assisted domains and rewiring of the circuitry regulating genes on ecDNA. Moreover, enhancers on ecDNA can regulate distal genes on chromosomes or other ecDNA that are proximal due to hub formation. The tremendous dysregulation of oncogenes achieved by ecDNA leads to persistence of ecDNA due to a selective advantage, further leading to proliferation, increased pathology, and poor survival outcomes for patients whose tumor cells carry ecDNA [43].
7. Therapy, evolution, resistance and fate
The increased pathology of ecDNA positive tumors underscores the need for intervention, while the unique biology of ecDNA raises hope of vulnerabilities specific to ecDNA. Identifying new targets and therapeutic intervention strategies are considered a ‘grand challenge’ for cancer [25].
7.1. ecDNA evolve and adapt to varying selection pressure
The early ecDNA research already pointed to the central role of ecDNA in tumor evolution and resistance. Dihydrofolate reductase (DHFR) is an enzyme that reduces dihydrofolic acid to tetrahydrofolic acid which is an important component of multiple pathways of nucleotide synthesis. MTX inhibits DHFR with a high affinity and is used as an anti-cancer agent. High doses of MTX were compensated by the formation of large numbers of ecDNA carrying DHFR [10, 41, 42]. The analysis also revealed mutations on DHFR that appeared during the ecDNA formation and propagated with the increase in ecDNA copies [30, 29], although the functional characteristics of these mutations on ecDNA fitness has not been explored since those early results.
Somewhat surprisingly, continued treatment with MTX led to the integration of ecDNA into HSR as more stable structures with amplified copies of DHFR [42]. Independently, Amler and Schwab [5] identified multiple tandem arrays of DNA segments containing the N-myc oncogene in multiple neuroblastoma cell lines. They suggested a model involving spontaneous reintegration of (a single copy of) ecDNA followed by multiple tandem duplications. However, this model is less attractive because most observed ecDNA structures reveal conserved breakpoints down to the base-pair level [43]. The multiple tandem duplication model would require for each event to re-use the same breakpoints; however, despite considerable work in fragile site identification [26, 27, 76], a correlation between ecDNA breakpoints and fragile sites has not been reported. We conjecture that aggregation of ecDNA into hubs [37], followed by recombination into a larger structure, and a single integration event generates an HSR derived from ecDNA (Figure 5).
Figure 5. Plasticity of ecDNA and HSR.
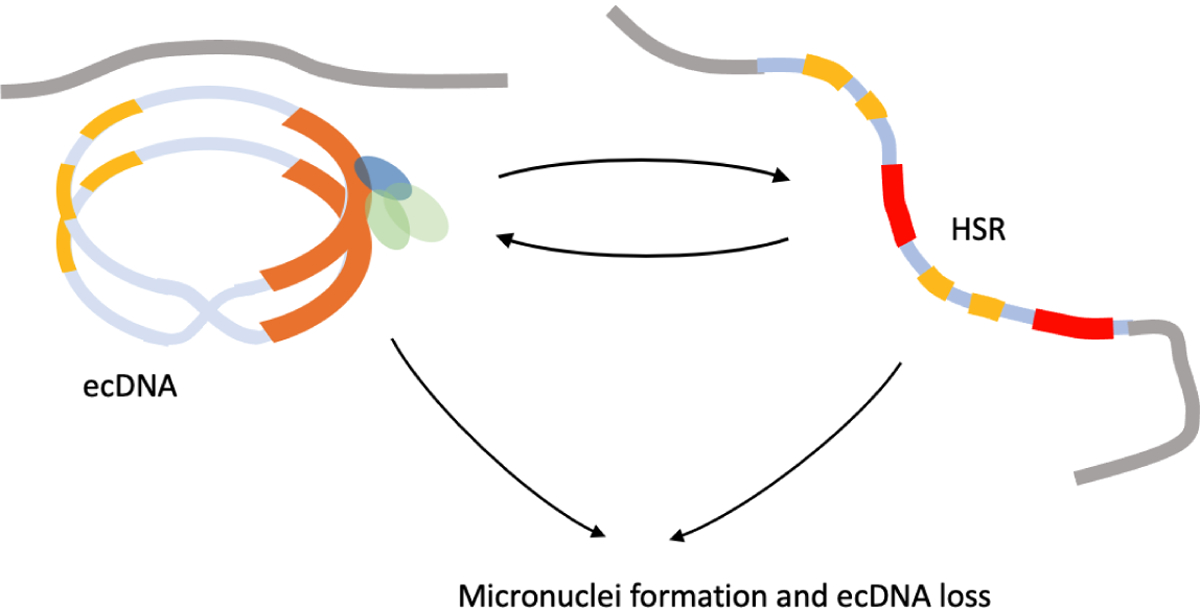
Aggregated ecDNA can re-combine into larger structures. Continued selection pressure that selects for amplification may lead to ecDNA formation, aggregation, and subsequent integration into a multi-copy HSR. HSRs also show plasticity and change length. Removal of selection or DNA damage leads to micronuclei formation and loss of ecDNA.
HSR formation also represents a mechanism of resistance. The SF295 cell-line was shown to be ecDNA positive with a G protein related to the family of ABC transporters, ABCG2 on ecDNA [87]. At higher levels of dosage of the drug mitoxantrone, the number of ecDNA reduced but were accompanied by a concomitant rise in HSRs, suggesting that the ecDNA had re-integrated into multiple, non-native chromosomal locations in response to treatment. Spectral karyotyping and FISH analysis showed evidence of integration of ABCG2 along with a marked increase in structural variation (deletions, insertions and translocations of genomic fragments) that were not apparent in the parental SF295 line prior to drug treatment. Nathanson et al [78] performed similar experiments in a Glioblastoma cell-line, and found something remarkable. First, the cell-line already carried EGFR-vIII— a mutant of the receptor tyrosine kinase EGFR—on ecDNA suggesting that ecDNA was a stable part of the maintenance of the cell-line. Upon drug-treatment with Erlotinib, which targets EGFR, the ecDNA reintegrated into the chromosome as HSR. The reintegration was concurrent with the arrival of a second ecDNA species containing FGFR2. Although HSRs were considered to be a stable form of amplification, removal of drug led to a re-emergence of EGFR carrying ecDNA. Sequence based analysis confirmed that the ecDNA in the naive cells and in the drug-removed cells had essentially identical structures, although with additional rearrangements in ecDNA in the drug-removed cells [78, 22]. Recently, Song et al [102] investigated the impact of BRAF and MEK inhibitors on a melanoma cell line carrying a mutant form of BRAF. The results provided another intriguing example of ecDNA mediated drug resistance. With increasing drug dosage, an ecDNA arose that dramatically increased BRAF copy number. Over continued drug treatment, the ecDNA reintegrated into chromosome 3 in an aggregated HSR-like structure. Passage of the drug-addicted single cell cloned cell line in low drug or the absence of drug treatment resulted in shortening or loss of the HSR. This result echoes some early observations on the plasticity of HSRs[9], but now demonstrating faster kinetics.
These results are all consistent with the model equations 1,2 in a positive selection setting under the assumption that ecDNA and HSRs are interconvertible states, although not functionally equivalent. More research is needed to clarify the functional differences between HSR and ecDNA states. The model makes other simplifying assumptions: namely, cells do not die and ecDNA are never lost. Thus, the impact of positive or negative selection is governed by a single parameter s which changes the distribution of ecDNA among cells, making it wider or narrower. While it is likely that the fitness of the tumor cell also depends upon the number of ecDNA, not just their presence of absence, more experimental data is needed to refine the model.
7.2. DNA damage and ecDNA loss
The role of the DNA damage sensing and repair system in the genesis and progression of ecDNA remains incompletely understood. ecDNA arises from a diversity of potential mechanisms, implicating different defects in DNA damage sensing and repair pathways. Further, the DDR pathways engaged to promote ecDNA evolution, once ecDNAs are formed, may differ from those involved in its inception. Consequently, there is a great deal about the link between DNA damage and ecDNA that remains to be discovered. Snapka and Varshavsky [101] confirmed earlier findings that ecDNA are lost once the MTX selection pressure is removed, over 25–30 cell doublings. As new ecDNA are not being created, this effect could be explained simply due to random drift without requiring negative selection. Remarkably, they also observed that low, non-cytotoxic doses of hyroxyurea (HU) led to an acceleration of ecDNA loss with 90% of the cells losing ecDNA in 4–5 doublings. These findings were confirmed and extended to multiple cell-lines and cytotoxic drugs [115, 94]. Importantly, the loss of ecDNA was not mediated by inhibition of DNA synthesis; instead, the low doses of HU were accompanied by an increase in micronuclei formation (Figure 5). These results were bolstered by a patient study that showed a slowing of progression in patients with advanced Ovarian cancer upon low doses of HU treatment [88]. Similarly, low doses of radiation were associated with a loss of Myc carrying ecDNA in a breast cancer cell-line, primarily due to their entrapment in micronuclei [92].
The reduction in ecDNA upon cytotoxic drug treatment through micronuclei formation was recapitulated in other contexts [107]. COLO320-DM lines, which had earlier been shown to be ecDNA positive [3] also lost ecDNA through micronuclei formation upon HU treatment [95]. Yu et al [125] found gemcitabine to be similarly effective at greatly reduced doses for the loss of ecDNA from the ovarian cancer cell line UACC-1598. This was explained by proposing a mechanism in which the cytotoxic drugs induce damage to ecDNA, which break away from their chromosomal tethers. The lack of tethering leads to lagging of ecDNA aggregates during cell division, resulting in formation of micronuceli enriched with ecDNA segments, and eventual eviction of ecDNA. Recent results suggest, however, that ecDNA aggregation (or, hub formation) during interphase may be a natural part of ecDNA maintenance, and not a consequence of DNA damage [37]. The ecDNA hubs disaggregate during mitosis but are retained in the daughter nuclei by tethering to segregating chromosomes. Through reasons that are not fully understood, DNA damage disrupts this process, leading to lagging aggregates of ecDNA and subsequent micronuceli formation. The micronuclei may be lost in subsequent cell-divisions or re-integrate into the nucleus, where they form larger ecDNA, and/or integrate into HSRs [81], possibly at locations with double strand break lesions [97].
The Poly(ADP-ribose) polymerase 1 (PARP1) is a central enzyme involved in many facets of DNA repair including single-strand DNA break, nucleotide excision, and alternative nonhomologous end-joining pathways. PARP inhibition, possibly in conjunction with other cytotoxic therapy, can accelerate DNA damage in cancer cells [98]. It is particularly effective when homology directed repair has been compromised, for example, through BRCA mutations [14]. As sister chromatids are not available to ecDNA, it is possible that homology directed repair is compromised, pointing to the viability of PARP inhibition for treating ecDNA positive tumors. However, the integration of ecDNA into HSRs is also accelerated by PARP inhibitors, suggesting a possible mode of resistance [97], and due to the importance of NHEJ in ecDNA formation, DNA-PKcs inhibitors have been shown to reduce ecDNA formation [97]. Future research aimed at understanding the role of DNA damage repair pathways in ecDNA maintenance is likely to be of high interest for targeting ecDNA [122].
8. Conclusion
The 23 pairs of chromosomes describe the instructions that make life possible. The instructions are typically hidden and revealed only in tightly regulated settings to ensure that only the requisite type and amount of RNA–the precursor to the production of the molecular machinery–is expressed as needed for the cell to function in specific contexts. It is perhaps not surprising that much cellular machinery is dedicated to duplicating this information with high fidelity so that each daughter cell receives nearly identical copies of the book of the life, and to ensuring that the instructions are exposed at the right time and place.
We are reminded of the American architect Louis Sullivan, who coined the concept, “Form follows function” to describe the principle of how the shape of a building or an object reflects its function or purpose. The phrase also aptly describes ecDNA. The circular shape, and the lack of centromeres, directly contributes to the non-chromosomal inheritance. Thus, the daughter cells do not receive the same book. Second, the instructions are highly accessible and regulatory circuits are rewired, leading to a dysregulation of the expressed genes. It is likely that the dysregulation imposes negative fitness in general, leading to rapid elimination of ecDNA. Thus, ecDNA are rarely seen in normal cells. However, in a few unfortunate instances, the extra copies of the gene (and an excess of gene products) provide a fitness advantage to the cells which then proliferate at the expense of neighboring cells, leading to cancer. EcDNA positive cancers adapt faster through a rapid change in copy number of oncogenes, reveal novel resistance pathways, are less likely to mediate an immune response, and result in worse outcomes for patients. Finally, ecDNA are prevalent, with a fifth to a third of all cancer samples being ecDNA positive. New tools, that provide insight into the shape and spatial organization of the genome of ecDNA-containing cancers, are providing unique insight into their biology. In that light, although ecDNA were discovered nearly 60 years ago, their prevalence in cancer, and their central role in cancer pathogenicity, is only just beginning to be appreciated.
ACKNOWLEDGMENTS
The authors thank the anonymous referee for many suggestions and corrections. VB was supported in part by grants U24CA264379, and R01GM114362 from the NIH. P.S.M. was supported by a grant from The National Brain Tumour Society and NIH R01-CA238349.
Footnotes
DISCLOSURE STATEMENT
VB is a co-founder, consultant, SAB member and has equity interest in Boundless Bio, inc. and Abterra, Inc. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. P.S.M. is a co-founder of Boundless Bio, Inc. He chairs the Scientific Advisory Board, for which he is compensated.
LITERATURE CITED
- 1.Adelman K, Martin BJE. 2021. ecDNA party bus: Bringing the enhancer to you. Mol Cell 81(9):1866–1867 [DOI] [PubMed] [Google Scholar]
- 2.Akagi K, Li J, Broutian TR, Padilla-Nash H, Xiao W, et al. 2014. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res 24(2):185–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alitalo K, Schwab M, Lin CC, Varmus HE, Bishop JM. 1983. Homogeneously staining chromosomal regions contain amplified copies of an abundantly expressed cellular oncogene (c-myc) in malignant neuroendocrine cells from a human colon carcinoma. Proc Natl Acad Sci U S A 80(6):1707–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alt FW, Kellems RE, Bertino JR, Schimke RT. 1978. Selective multiplication of dihydrofolate reductase genes in methotrexate-resistant variants of cultured murine cells. Journal of Biological Chemistry 253(5):1357–1370 [PubMed] [Google Scholar]
- 5.Amler LC, Schwab M. 1989. Amplified N-myc in human neuroblastoma cells is often arranged as clustered tandem repeats of differently recombined DNA. Mol Cell Biol 9(11):4903–4913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balaban-Malenbaum G, Gilbert F. 1977. Double minute chromosomes and the homogeneously staining regions in chromosomes of a human neuroblastoma cell line. Science 198(4318):739–741 [DOI] [PubMed] [Google Scholar]
- 7.Barker PE, Drwinga HL, Hittelman WN, Maddox AM. 1980. Double minutes replicate once during S phase of the cell cycle. Exp Cell Res 130(2):353–360 [DOI] [PubMed] [Google Scholar]
- 8.Beverley SM, Coderre JA, Santi DV, Schimke RT. 1984. Unstable DNA amplifications in methotrexate-resistant Leishmania consist of extrachromosomal circles which relocalize during stabilization. Cell 38(2):431–439 [DOI] [PubMed] [Google Scholar]
- 9.Biedler JL, Melera PW, Spengler BA. 1980. Specifically altered metaphase chromosomes in antifolate-resistant Chinese hamster cells that overproduce dihydrofolate reductase. Cancer Genetics and Cytogenetics 2(1):47–60 [Google Scholar]
- 10.Brown PC, Beverley SM, Schimke RT. 1981. Relationship of amplified dihydrofolate reductase genes to double minute chromosomes in unstably resistant mouse fibroblast cell lines. Molecular and Cellular Biology 1(12):1077–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10(12):1213–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cameron DL, Baber J, Shale C, Valle-Inclan JE, Besselink N, et al. 2021. GRIDSS2: comprehensive characterisation of somatic structural variation using single breakend variants and structural variant phasing. Genome Biol 22(1):202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carroll SM, DeRose ML, Gaudray P, Moore CM, Needham-Vandevanter DR, et al. 1988. Double minute chromosomes can be produced from precursors derived from a chromosomal deletion. Molecular and Cellular Biology 8(4):1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang HY, Nuyten DSA, Sneddon JB, Hastie T, Tibshirani R, et al. 2005. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proceedings of the National Academy of Sciences 102(10):3738–3743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang HY, Pannunzio NR, Adachi N, Lieber MR. 2017. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 18(8):495–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chapman OS, Luebeck J, Wani S, Tiwari A, Pagadala M, et al. 2021. The landscape of extrachromosomal circular dna in medulloblastoma. bioRxiv doi: 10.1101/2021.10.18.464907. [DOI] [Google Scholar]
- 17.Chen X, Shen Y, Draper W, Buenrostro JD, Litzenburger U, et al. 2016. ATAC-see reveals the accessible genome by transposase-mediated imaging and sequencing. Nat Methods 13(12):1013–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, et al. 2017. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14(10):959–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cox D, Yuncken C, Spriggs AI. 1965. Minute Chromatin Bodies in Malignant Tumours of Childhood. The Lancet 286(7402):55–58 [DOI] [PubMed] [Google Scholar]
- 20.Cross R 2020. The curious DNA circles that make treating cancer so hard. Chemical and Engineering News 98(40) [Google Scholar]
- 21.DeCarvalho AC, Kim H, Poisson LM, Winn ME, Mueller C, et al. 2018. Discordant inheritance of chromosomal and extrachromosomal DNA elements contributes to dynamic disease evolution in glioblastoma. Nature Genetics 2018 50:5 50(5):708–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deshpande V, Luebeck J, Nguyen NPD, Bakhtiari M, Turner KM, et al. 2019. Exploring the landscape of focal amplifications in cancer using AmpliconArchitect. Nature Communications 2019 10:1 10(1):1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evans G 1993. Physical mapping of complex genomes. United States Patent USOO529726A. [Google Scholar]
- 24.Fan Y, Mao R, Lv H, Xu J, Yan L, et al. 2011. Frequency of double minute chromosomes and combined cytogenetic abnormalities and their characteristics. J Appl Genet 52(1):53–59 [DOI] [PubMed] [Google Scholar]
- 25.Foulkes I, Sharpless NE. 2021. Cancer Grand Challenges: Embarking on a New Era of Discovery. Cancer Discov 11(1):23–27 [DOI] [PubMed] [Google Scholar]
- 26.Fungtammasan A, Walsh E, Chiaromonte F, Eckert KA, Makova KD. 2012. A genome-wide analysis of common fragile sites: what features determine chromosomal instability in the human genome? Genome Res 22(6):993–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fungtammasan A, Walsh E, Chiaromonte F, Eckert KA, Makova KD. 2016. Corrigendum: A genome-wide analysis of common fragile sites: What features determine chromosomal instability in the human genome? Genome Res 26(10):1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garsed D, Marshall O, Corbin V, Hsu A, Di Stefano L, et al. 2014. The Architecture and Evolution of Cancer Neochromosomes. Cancer Cell 26(5):653–667 [DOI] [PubMed] [Google Scholar]
- 29.Haber DA, Beverley SM, Kiely ML, Schimke RT. 1981. Properties of an altered dihydrofolate reductase encoded by amplified genes in cultured mouse fibroblasts. The Journal of biological chemistry 256(18):9501–10 [PubMed] [Google Scholar]
- 30.Haber DA, Schimke RT. 1981. Unstable amplification of an altered dihydrofolate reductase gene associated with double-minute chromosomes. Cell 26(3 Pt 1):355–62 [DOI] [PubMed] [Google Scholar]
- 31.Hadi K, Yao X, Behr JM, Deshpande A, Xanthopoulakis C, et al. 2020. Distinct Classes of Complex Structural Variation Uncovered across Thousands of Cancer Genome Graphs. Cell 183(1):197–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hahn PJ, Nevaldine B, Longo JA. 1992. Molecular structure and evolution of double-minute chromosomes in methotrexate-resistant cultured mouse cells. Molecular and Cellular Biology 12(7):2911–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamkalo BA, Farnham PJ, Johnston R, Schimke RT. 1985. Ultrastructural features of minute chromosomes in a methotrexate-resistant mouse 3T3 cell line. Proceedings of the National Academy of Sciences of the United States of America 82(4):1126–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Helmsauer K, Valieva ME, Ali S, Chamorro González R, Schöpflin R, et al. 2020. Enhancer hijacking determines extrachromosomal circular MYCN amplicon architecture in neuroblastoma. Nature Communications 2020 11:1 11(1):1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.HOTTA Y, BASSEL A. 1965. MOLECULAR SIZE AND CIRCULARITY OF DNA IN CELLS OF MAMMALS AND HIGHER PLANTS. Proc Natl Acad Sci U S A 53:356–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hung K, Luebeck J, Dehkordi S, Coruh C, Law J, et al. 2021. Targeted profiling of human extrachromosomal DNA by CRISPR-CATCH. bioRxiv doi: 10.1101/2021.11.28.470285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hung KL, Yost KE, Xie L, Shi Q, Helmsauer K, et al. 2021. ecDNA hubs drive cooperative intermolecular oncogene expression. Nature 600(7890):731–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inazawa J, Abe T, Inoue K, Nishigaki H, Horiike S, et al. 1989. Simultaneous existence of double minute chromosomes and a homogeneously staining region in a retinoblastoma cell line (Y79) and amplification of N-myc at HSR. Cancer Genetics and Cytogenetics 37(1):133–137 [DOI] [PubMed] [Google Scholar]
- 39.Jaratlerdsiri W, Chan EKF, Petersen DC, Yang C, Croucher PI, et al. 2017. Next generation mapping reveals novel large genomic rearrangements in prostate cancer. Oncotarget 8(14):23588–23602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kanda T, Sullivan KF, Wahl GM. 1998. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol 8(7):377–385 [DOI] [PubMed] [Google Scholar]
- 41.Kaufman RJ, Brown PC, Schimke RT. 1979. Amplified dihydrofolate reductase genes in unstably methotrexate-resistant cells are associated with double minute chromosomes. Proceedings of the National Academy of Sciences of the United States of America 76(11):5669–5673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaufman RJ, Brown PC, Schimke RT. 1981. Loss and stabilization of amplified dihydrofolate reductase genes in mouse sarcoma S-180 cell lines. Mol Cell Biol 1(12):1084–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim H, Nguyen NP, Turner K, Wu S, Gujar AD, et al. 2020. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nature Genetics 52(9):891–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koche RP, Rodriguez-Fos E, Helmsauer K, Burkert M, MacArthur IC, et al. 2020. Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma. Nat Genet 52(1):29–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kohl NE, Kanda N, Schreck RR, Bruns G, Latt SA, et al. 1983. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell 35(2 PART 1):359–367 [DOI] [PubMed] [Google Scholar]
- 46.Koo DH, Molin WT, Saski CA, Jiang J, Putta K, et al. 2018. Extrachromosomal circular DNA-based amplification and transmission of herbicide resistance in crop weed Amaranthus palmeri. Proceedings of the National Academy of Sciences of the United States of America 115(13):3332–3337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kucheria K 1968. Double minute chromatin bodies in a sub-ependymal glioma. Br J Cancer 22(4):696–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kumar P, Dillon LW, Shibata Y, Jazaeri AA, Jones DR, Dutta A. 2017. Normal and Cancerous Tissues Release Extrachromosomal Circular DNA (eccDNA) into the Circulation. Molecular Cancer Research 15(9):1197–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.L’Abbate A, Macchia G, D’Addabbo P, Lonoce A, Tolomeo D, et al. 2014. Genomic organization and evolution of double minutes/homogeneously staining regions with MYC amplification in human cancer. Nucleic Acids Res 42(14):9131–9145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lam ET, Hastie A, Lin C, Ehrlich D, Das SK, et al. 2012. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat Biotechnol 30(8):771–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lander ES, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409(6822):860–921 [DOI] [PubMed] [Google Scholar]
- 52.Lange JT, Chen CY, Pichugin Y, Xie L, Tang J, et al. 2021. Principles of ecdna random inheritance drive rapid genome change and therapy resistance in human cancers. bioRxiv doi: 10.1101/2021.06.11.447968. [DOI] [Google Scholar]
- 53.Leung AK, Kwok TP, Wan R, Xiao M, Kwok PY, et al. 2017. OMBlast: alignment tool for optical mapping using a seed-and-extend approach. Bioinformatics 33(3):311–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Levan A, Levan G. 1978. Have double minutes functioning centromeres? Hereditas 88(1):81–92 [DOI] [PubMed] [Google Scholar]
- 55.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lo AW, Sabatier L, Fouladi B, Pottier G, Ricoul M, Murnane JP. 2002. DNA amplification by breakage/fusion/bridge cycles initiated by spontaneous telomere loss in a human cancer cell line. Neoplasia 4(6):531–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Long J and Shelhamer E and Darrell T 2015. Fully convolutional networks for semantic segmentation. In 2015 IEEE Conference on Computer Vision and Pattern Recognition (CVPR) [DOI] [PubMed] [Google Scholar]
- 58.LUBS HA, SALMON JH. 1965. THE CHROMOSOMAL COMPLEMENT OF HUMAN SOLID TUMORS. II. KARYOTYPES OF GLIAL TUMORS. J Neurosurg 22:160–168 [DOI] [PubMed] [Google Scholar]
- 59.Luebeck J, Coruh C, Dehkordi SR, Lange JT, Turner KM, et al. 2020. AmpliconReconstructor integrates NGS and optical mapping to resolve the complex structures of focal amplifications. Nature Communications 2020 11:1 11(1):1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lundberg G, Rosengren AH, Håkanson U, Stewénius H, Jin Y, et al. 2008. Binomial Mitotic Segregation of MYCN-Carrying Double Minutes in Neuroblastoma Illustrates the Role of Randomness in Oncogene Amplification. PLOS ONE 3(8):e3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ly P, Brunner SF, Shoshani O, Kim DH, Lan W, et al. 2019. Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nature Genetics 51(4):705–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ly P, Cleveland DW. 2017. Rebuilding Chromosomes After Catastrophe: Emerging Mechanisms of Chromothripsis. Trends in Cell Biology 27(12):917–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ly P, Teitz LS, Kim DH, Shoshani O, Skaletsky H, et al. 2017. Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat Cell Biol 19(1):68–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maass PG, Barutcu AR, Rinn JL. 2019. Interchromosomal interactions: A genomic love story of kissing chromosomes. Journal of Cell Biology 218(1):27–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Madhavi R, Guntur M, Ghosh R, Ghosh PK. 1990. Double minute chromosomes in the leukocytes of a young girl with breast carcinoma. Cancer Genet Cytogenet 44(2):203–207 [DOI] [PubMed] [Google Scholar]
- 66.Marotta M, Onodera T, Johnson J, Budd GT, Watanabe T, et al. 2017. Palindromic amplification of the ERBB2 oncogene in primary HER2-positive breast tumors. Sci Rep 7:41921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marshall OJ, Chueh AC, Wong LH, Choo KH. 2008. Neocentromeres: new insights into centromere structure, disease development, and karyotype evolution. Am J Hum Genet 82(2):261–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McClintock B 1932. A correlation of ring-shaped chromosomes with variegation in Zea Mays. PNAS 18(12):672–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McClintock B 1939. The Behavior in Successive Nuclear Divisions of a Chromosome Broken at Meiosis. Proc Natl Acad Sci U S A 25(8):405–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McClintock B 1941. The Stability of Broken Ends of Chromosomes in Zea Mays. Genetics 26(2):234–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Meng X, Qi X, Guo H, Cai M, Li C, et al. 2015. Novel role for non-homologous end joining in the formation of double minutes in methotrexate-resistant colon cancer cells. Journal of Medical Genetics 52(2):135–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mitra AB, Murty VV, Luthra UK. 1983. Double-minute chromosomes in the leukocytes of a patient with a previous history of cervical carcinoma. Cancer Genet Cytogenet 8(2):117–122 [DOI] [PubMed] [Google Scholar]
- 73.Molin WT, Yaguchi A, Blenner M, Saski CA. 2020. The EccDNA replicon: A heritable, extranuclear vehicle that enables gene amplification and glyphosate resistance in amaranthus palmeri. Plant Cell 32(7):2132–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Møller HD, Lin L, Xiang X, Petersen TS, Huang J, et al. 2018. CRISPR-C: circularization of genes and chromosome by CRISPR in human cells. Nucleic Acids Res 46(22):e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Morton AR, Dogan-Artun N, Faber ZJ, MacLeod G, Bartels CF, et al. 2019. Functional Enhancers Shape Extrachromosomal Oncogene Amplifications. Cell 179(6):1330–1341.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mrasek K, Schoder C, Teichmann AC, Behr K, Franze B, et al. 2010. Global screening and extended nomenclature for 230 aphidicolin-inducible fragile sites, including 61 yet unreported ones. Int J Oncol 36(4):929–940 [DOI] [PubMed] [Google Scholar]
- 77.Møller HD. 2020. Circle-Seq: Isolation and Sequencing of Chromosome-Derived Circular DNA Elements in Cells. Methods Mol Biol 2119:165–181 [DOI] [PubMed] [Google Scholar]
- 78.Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, et al. 2014. Targeted Therapy Resistance Mediated by Dynamic Regulation of Extrachromosomal Mutant EGFR DNA. Science (New York, N.Y.) 343(6166):72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nulton TJ, Kim NK, DiNardo LJ, Morgan IM, Windle B. 2018. Patients with integrated HPV16 in head and neck cancer show poor survival. Oral Oncol 80:52–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nulton TJ, Olex AL, Dozmorov M, Morgan IM, Windle B. 2017. Analysis of The Cancer Genome Atlas sequencing data reveals novel properties of the human papillomavirus 16 genome in head and neck squamous cell carcinoma. Oncotarget 8(11):17684–17699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Oobatake Y, Shimizu N. 2020. Double-strand breakage in the extrachromosomal double minutes triggers their aggregation in the nucleus, micronucleation, and morphological transformation. Genes Chromosomes Cancer 59(3):133–143 [DOI] [PubMed] [Google Scholar]
- 82.Pang J, Nguyen N, Luebeck J, Ball L, Finegersh A, et al. 2021. Extrachromosomal DNA in HPV-Mediated Oropharyngeal Cancer Drives Diverse Oncogene Transcription. Clin Cancer Res 27(24):6772–6786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Paulsen T, Malapati P, Shibata Y, Wilson B, Eki R, et al. 2021. MicroDNA levels are dependent on MMEJ, repressed by c-NHEJ pathway, and stimulated by DNA damage. Nucleic Acids Res 49(20):11787–11799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Paulsen T, Shibata Y, Kumar P, Dillon L, Dutta A. 2019. Small extrachromosomal circular DNAs, microDNA, produce short regulatory RNAs that suppress gene expression independent of canonical promoters. Nucleic Acids Res 47(9):4586–4596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Raeisi Dehkordi S, Luebeck J, Bafna V. 2021. FaNDOM: Fast nested distance-based seeding of optical maps. Patterns (N Y) 2(5):100248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rajkumar U, Turner K, Luebeck J, Deshpande V, Chandraker M, et al. 2019. EcSeg: Semantic Segmentation of Metaphase Images Containing Extrachromosomal DNA. iScience 21:428–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rao VK, Wangsa D, Robey RW, Huff L, Honjo Y, et al. 2005. Characterization of ABCG2 gene amplification manifesting as extrachromosomal DNA in mitoxantrone-selected SF295 human glioblastoma cells. Cancer Genetics and Cytogenetics 160(2):126–133 [DOI] [PubMed] [Google Scholar]
- 88.Raymond E, Faivre S, Weiss G, McGill J, Davidson K, et al. 2001. Effects of hydroxyurea on extrachromosomal DNA in patients with advanced ovarian carcinomas. Clinical Cancer Research 7(5):1171–1180 [PubMed] [Google Scholar]
- 89.Sachidanandam R, Weissman D, Schmidt SC, Kakol JM, Stein LD, et al. 2001. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409(6822):928–933 [DOI] [PubMed] [Google Scholar]
- 90.Sanborn JZ, Salama SR, Grifford M, Brennan CW, Mikkelsen T, et al. 2013. Double Minute Chromosomes in Glioblastoma Multiforme Are Revealed by Precise Reconstruction of Oncogenic Amplicons. Cancer Research 73(19):6036–6045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Scappaticci S, Fossati GS, Valenti L, Scabini M, Tateo S, et al. 1995. A search for double minute chromosomes in cultured lymphocytes from different types of tumors. Cancer Genet Cytogenet 82(1):50–53 [DOI] [PubMed] [Google Scholar]
- 92.Schoenlein PV, Barrett JT, Kulharya A, Dohn MR, Sanchez A, et al. 2003. Radiation therapy depletes extrachromosomally amplified drug resistance genes and oncogenes from tumor cells via micronuclear capture of episomes and double minute chromosomes. Int J Radiat Oncol Biol Phys 55(4):1051–1065 [DOI] [PubMed] [Google Scholar]
- 93.Shale C, Baber J, Cameron DL, Wong M, Cowley MJ, et al. 2020. Unscrambling cancer genomes via integrated analysis of structural variation and copy number. bioRxiv doi: 10.1101/2020.12.03.410860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shimizu N, Itoh N, Utiyama H, Wahl GM. 1998. Selective entrapment of extrachromosomally amplified DNA by nuclear budding and micronucleation during S phase. J Cell Biol 140(6):1307–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shimizu N, Misaka N, Utani K. 2007. Nonselective DNA damage induced by a replication inhibitor results in the selective elimination of extrachromosomal double minutes from human cancer cells. Genes Chromosomes Cancer 46(10):865–874 [DOI] [PubMed] [Google Scholar]
- 96.Shimizu N, Shingaki K, Kaneko-Sasaguri Y, Hashizume T, Kanda T. 2005. When, where and how the bridge breaks: Anaphase bridge breakage plays a crucial role in gene amplification and HSR generation. Experimental Cell Research 302(2):233–243 [DOI] [PubMed] [Google Scholar]
- 97.Shoshani O, Brunner SF, Yaeger R, Ly P, Nechemia-Arbely Y, et al. 2021. Chromothripsis drives the evolution of gene amplification in cancer. Nature 591(7848):137–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Slade D 2020. PARP and PARG inhibitors in cancer treatment. Genes Dev 34(5–6):360–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Smith CA, Vinograd J. 1972. Small polydisperse circular DNA of HeLa cells. J Mol Biol 69(2):163–178 [DOI] [PubMed] [Google Scholar]
- 100.Smith G, Taylor-Kashton C, Dushnicky L, Symons S, Wright J, Mai S. 2003. c-Myc-Induced Extrachromosomal Elements Carry Active Chromatin. Neoplasia (New York, N.Y.) 5(2):110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Snapka RM, Varshavsky A. 1983. Loss of unstably amplified dihydrofolate reductase genes from mouse cells is greatly accelerated by hydroxyurea. Proceedings of the National Academy of Sciences 80(24):7533–7537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Song K, Minami JK, Huang A, Dehkordi SR, Lomeli SH, et al. 2021. Plasticity of extrachromosomal and intrachromosomal BRAF amplifications in overcoming targeted therapy dosage challenges. Cancer Discovery [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, et al. 2011. Massive GenomicRearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 144(1):27–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Storlazzi CT, Fioretos T, Surace C, Lonoce A, Mastrorilli A, et al. 2006. MYC-containing double minutes in hematologic malignancies: evidence in favor of the episome model and exclusion of MYC as the target gene. Hum Mol Genet 15(6):933–942 [DOI] [PubMed] [Google Scholar]
- 105.Storlazzi CT, Lonoce A, Guastadisegni MC, Trombetta D, D’Addabbo P, et al. 2010. Gene amplification as double minutes or homogeneously staining regions in solid tumors: origin and structure. Genome Res 20(9):1198–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Takayama S, Uwaike Y. 1988. Analysis of the replication mode of double minutes using the PCC technique combined with BrdUrd labeling. Chromosoma 97(3):198–203 [DOI] [PubMed] [Google Scholar]
- 107.Tanaka T, Shimizu N. 2000. Induced detachment of acentric chromatin from mitotic chromosomes leads to their cytoplasmic localization at G(1) and the micronucleation by lamin reorganization at S phase. J Cell Sci 113 (Pt 4):697–707 [DOI] [PubMed] [Google Scholar]
- 108.Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, et al. 2017. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 2017 543:7643 543(7643):122–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, et al. 2005. Fine-scale structural variation of the human genome. Nat Genet 37(7):727–732 [DOI] [PubMed] [Google Scholar]
- 110.Umbreit NT, Zhang CZ, Lynch LD, Blaine LJ, Cheng AM, et al. 2020. Mechanisms generating cancer genome complexity from a single cell division error. Science 368(6488) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Venter JC, et al. 2001. The sequence of the human genome. Science 291(5507):1304–1351 [DOI] [PubMed] [Google Scholar]
- 112.Vogt N, Gibaud A, Lemoine F, de la Grange P, Debatisse M, Malfoy B. 2014. Amplicon rearrangements during the extrachromosomal and intrachromosomal amplification process in a glioma. Nucleic Acids Research 42(21):13194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vogt N, Lefèvre SH, Apiou F, Dutrillaux AM, Cör A, et al. 2004. Molecular structure of double-minute chromosomes bearing amplified copies of the epidermal growth factor receptor gene in gliomas. Proc Natl Acad Sci U S A 101(31):11368–11373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Von Hoff DD, Forseth B, Clare CN, Hansen KL, VanDevanter D. 1990. Double minutes arise from circular extrachromosomal DNA intermediates which integrate into chromosomal sites in human HL-60 leukemia cells. J Clin Invest 85(6):1887–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Von Hoff DD, Waddelow T, Forseth B, Davidson K, Scott J, Wahl G. 1991. Hydroxyurea Accelerates Loss of Extrachromosomally Amplified Genes from Tumor Cells. Cancer Research 51:6273–6279 [PubMed] [Google Scholar]
- 116.Voullaire L, Saffery R, Davies J, Earle E, Kalitsis P, et al. 1999. Trisomy 20p resulting from inverted duplication and neocentromere formation. Am J Med Genet 85(4):403–408 [PubMed] [Google Scholar]
- 117.Voullaire LE, Slater HR, Petrovic V, Choo KH. 1993. A functional marker centromere with no detectable alpha-satellite, satellite III, or CENP-B protein: activation of a latent centromere? Am J Hum Genet 52(6):1153–1163 [PMC free article] [PubMed] [Google Scholar]
- 118.Wang Y, Wang M, Djekidel MN, Chen H, Liu D, et al. 2021. eccDNAs are apoptotic products with high innate immunostimulatory activity. Nature 599(7884):308–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wu S, Turner KM, Nguyen N, Raviram R, Erb M, et al. 2019. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 2019 575:7784 575(7784):699–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Xu K, Ding L, Chang TC, Shao Y, Chiang J, et al. 2019. Structure and evolution of double minutes in diagnosis and relapse brain tumors. Acta Neuropathol 137(1):123–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Y Hota AB. 1965. Molecular size and circularity of DNA in cells of mammals and higher plants. Proc Natl Acad Sci U S A 53:356–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yan Y, Guo G, Huang J, Gao M, Zhu Q, et al. 2020. Current understanding of extrachromosomal circular DNA in cancer pathogenesis and therapeutic resistance. Journal of Hematology and Oncology 13(1):1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yang L, Luquette LJ, Gehlenborg N, Xi R, Haseley PS, et al. 2013. Diverse mechanisms of somatic structural variations in human cancer genomes. Cell 153(4):919–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yi E, Gujar AD, Guthrie M, Kim H, Zhao D, et al. 2021. Live-cell imaging shows uneven segregation of extrachromosomal DNA elements and transcriptionally active extrachromosomal DNA hubs in cancer. Cancer Discov [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yu L, Zhao Y, Quan C, Ji W, Zhu J, et al. 2013. Gemcitabine Eliminates Double Minute Chromosomes from Human Ovarian Cancer Cells. PLoS ONE 8(8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zakov S, Kinsella M, Bafna V. 2013. An algorithmic approach for breakage-fusion-bridge detection in tumor genomes. Proc Natl Acad Sci U S A 110(14):5546–5551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang CZ, Spektor A, Cornils H, Francis JM, Jackson EK, et al. 2015. Chromothripsis from DNA damage in micronuclei. Nature 2015 522:7555 522(7555):179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhu Y, Gujar AD, Wong CH, Tjong H, Ngan CY, et al. 2021. Oncogenic extrachromosomal DNA functions as mobile enhancers to globally amplify chromosomal transcription. Cancer Cell 39(5):694–707 [DOI] [PMC free article] [PubMed] [Google Scholar]