

Abstract
Aim:
To assess the relative efficacy of disease-modifying therapies (DMTs) for relapsing multiple sclerosis (RMS) including newer therapies (ozanimod, ponesimod, ublituximab) using network meta-analysis (NMA).
Materials & methods:
Bayesian NMAs for annualised relapse rate (ARR) and time to 3-month and 6-month confirmed disability progression (3mCDP and 6mCDP) were conducted.
Results:
For each outcome, the three most efficacious treatments versus placebo were monoclonal antibody (mAb) therapies: alemtuzumab, ofatumumab, and ublituximab for ARR; alemtuzumab, ocrelizumab, and ofatumumab for 3mCDP; and alemtuzumab, natalizumab, and either ocrelizumab or ofatumumab (depending on the CDP definition used for included ofatumumab trials) for 6mCDP.
Conclusion:
The most efficacious DMTs for RMS were mAb therapies. Of the newer therapies, only ublituximab ranked among the three most efficacious treatments (for ARR).
Keywords: disease-modifying therapy, indirect treatment comparison, network meta-analysis, relapsing multiple sclerosis, systematic literature review
Multiple sclerosis (MS), a chronic demyelinating disease of the central nervous system (CNS), affects approximately 2.8 million people worldwide, with a substantial societal economic impact and high burden for patients and their caregivers [1–3]. Four major MS disease phenotypes are traditionally recognised: clinically isolated syndrome (CIS), relapsing-remitting MS (RRMS), secondary progressive MS (SPMS), and primary progressive MS (PPMS) [4]. Most patients present with RRMS, which is characterised by at least partly reversible episodes of neurologic deficits [5]. The term relapsing MS (RMS) has been used to describe both RRMS and SPMS with superimposed relapses [6].
Disease-modifying therapies (DMTs) are an essential part of the MS treatment pathway. Despite advancements in treatment that have improved outcomes for patients with MS, the disease remains incurable [7]. Current treatment strategy focuses on alleviating CNS inflammation, slowing disease progression, and reducing the recurrence of relapses, as keys to improving long-term outcomes. Although many DMTs are available for patients with RMS, the increasing complexity of the treatment landscape represents a challenge when recommending therapeutic options for individual patients. Factors such as efficacy, safety/tolerability, route of administration, availability, and costs vary among DMT options, and should be considered by patients and their healthcare providers when deciding on an appropriate therapy. High-efficacy monoclonal antibody (mAb) DMTs are often considered as first-line MS therapies, but further research is needed to establish the best DMT sequencing strategies.
The relative efficacy of all DMTs for RMS can be estimated using network meta-analysis (NMA), an indirect comparison method. Relative estimates of treatment effects are essential for both clinicians and payers, who must choose between newer therapies and those traditionally used in clinical practice. Although multiple NMAs have been published to date comparing RMS therapies, none of these analyses have collectively included several newer therapies (i.e., ozanimod, ponesimod, and ublituximab). We previously conducted an NMA to compare DMTs for patients with RMS, for several key efficacy outcomes [8]. For the present study, we sought to update our previous NMA to evaluate the relative efficacy of DMTs including ozanimod, ponesimod, and ublituximab.
Materials & methods
Systematic literature review
A systematic literature review (SLR) was performed in accordance with guidelines recommended by the National Institute of Health and Care Excellence (NICE) [9] and reported in alignment with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement [10]. Details of this literature review have been described previously [8]. In brief, we searched biomedical databases, conference proceedings, and trial registries. Randomised controlled trials (RCTs) of adult (age ≥18 years) patients with RMS that were reported in English were included. The following DMTs approved by the US FDA and/or European Medicines Agency (EMA) for the treatment of MS were considered as both interventions and comparators (with placebo also included as a comparator): alemtuzumab, cladribine, dimethyl fumarate, diroximel fumarate, fingolimod, glatiramer acetate, interferon beta (IFN β)-1a, IFN β-1b, mitoxantrone, natalizumab, ocrelizumab, ofatumumab, ozanimod, pegylated IFN β-1a, ponesimod, siponimod, and teriflunomide. To include the most recent data available for MS therapies, unapproved therapies at the time of the search were also considered (e.g., ublituximab). Trials were excluded if they included patients with CIS or PPMS. Searches were initially conducted in December 2019, with updates in April 2021 and March 2022. Additional details of the SLR are provided in Supplementary Appendix A.
The SLR eligibility criteria were broader than was required for the analysis reported in this study. Thus, the RCTs identified via the SLR were included in the NMA evidence base only if they met the following criteria: population was ≥75% RMS; interventions and comparators included DMTs approved for RMS by the FDA and/or EMA as of June 2022 or undergoing FDA and/or EMA review at this time; outcomes included at least one of annualised relapse rate (ARR), time to 3-month (or 12-week) confirmed disability progression (3mCDP), or time to 6-month (or 24-week) confirmed disability progression (6mCDP); the duration of the initial randomised portion of the trial was ≥48 weeks; and a full-text pivotal publication was available for the trial. If a trial did not report ARR but it was possible to calculate this outcome using reported data, then the trial was not excluded because of this criterion. For approved DMTs, only the maintenance doses specified by the FDA and/or EMA labels were considered for the NMAs.
Data was extracted by two independent reviewers into a standardised extraction form in Microsoft Excel (Microsoft Corporation, WA, USA). Study design characteristics (e.g., author, year and journal), intervention details (e.g., treatment, dose, route and frequency), patient inclusion/exclusion criteria, baseline patient characteristics (e.g., age, gender, baseline Expanded Disability Status Scale [EDSS] score, and disease duration), outcome data (i.e., ARR, 3mCDP, and 6mCDP) and trial-specific outcome definitions were extracted for all included RCTs, where reported. Discrepancies in collected data were resolved by consensus or a third independent reviewer. A risk of bias assessment of each included trial was conducted (Supplementary Appendix A) following the principles recommended in the Centre for Reviews and Dissemination guidance for undertaking reviews in healthcare [11].
Feasibility assessment
The validity of NMA results is dependent on the evidence meeting the exchangeability assumption. Under this assumption, all interventions being studied could have been included as comparators in a common RCT (i.e., treatment effect modifiers are similar across the included trials). Failure to meet this assumption can result in biased estimates of comparative effect and bring about misleading conclusions [12]. In the present study, a comprehensive qualitative assessment of cross-trial heterogeneity was performed in accordance with recommendations on the assessment of NMA feasibility [13–15]. Relevant trials were compared on the following domains: study design characteristics, patient inclusion/exclusion criteria, baseline patient characteristics, placebo response (where available), and outcome definitions. Extracted data used to inform the feasibility assessment are provided in Supplementary Appendix B.
Statistical analysis
Bayesian NMAs using random effects (REs) models for the analysis for each outcome of interest (ARR, 3mCDP, 6mCDP) were performed in accordance with methods outlined by the NICE Decision Support Unit to ensure robustness and transparency [16–18]. We used RE models because they make less stringent assumptions about the consistency of effects than fixed effects (FE) models, and so are more appropriate in cases where there are cross-trial differences in study and patient characteristics. All analyses were conducted using R version 3.6.1 [19], Just Another Gibbs Sampler version 4.3.0, and WinBUGS version 1.4.3 [20] and were based on burn-in and sampling durations of 60,000 iterations each. Input data for each analysis are provided in Supplementary Appendix C.
The deviance information criterion (DIC) and posterior mean of the residual deviance (ResDev) were calculated to assess model fit for each analysis. To assess whether the models had adequate fit to the data, we compared the ResDev from each NMA to the corresponding number of unconstrained data points. The Brooks-Gelman-Rubin statistic was assessed to ensure that convergence was reached for each analysis [20]. The ranking metrics Surface Under the cumulative ranking curve (SUCRA) and probability of a treatment being the best (p-best) were calculated for each analysis [21].
For ARR, a Poisson model was used with vague priors for treatment effects and between-trial variances. The ARR (mean), trial duration, and patient number were preferentially extracted for each treatment arm. Trial duration was extracted in weeks; where only the number of months or years was reported by a study, it was assumed that 1 year = 52 weeks and 12 months = 1 year. The total number of relapse events and total exposure (patient-years) were derived for the analysis using the extracted data. Several trials did not report ARR, but it was possible to calculate this outcome using other reported data (i.e., number of patients, number of relapses, and follow-up time). For the Boiko et al. trial, which was a non-inferiority trial, we pooled the two glatiramer acetate arms prior to analysis (the ARR was the same for both arms) [22].
In this study, CDP was assessed as a time-to-event outcome. For time to 3mCDPand time to 6mCDP, a continuous survival model on a log hazard scale was used with vague priors for treatment effects, and an informative prior distribution for between-trial variances (pharmaceutical vs pharmaceutical interventions; cause-specific mortality/major morbidity event: T2 ∼ log normal [-3.95, 1.792]) [23]. The use of an informative prior for between-trial variances reduces uncertainty in the analysis [24] and this approach aligns with previously published NMAs [8,25]. A vague prior was used for the ARR analysis for between-trial variances because an appropriate informative prior was not available for this outcome at the time of analysis. The mean hazard ratio (HR) for the time-to-event outcome and its 95% confidence interval (CI) were preferentially extracted for comparisons between treatment arms. Log-HR and its standard error (SE) were derived for the analysis by taking the natural log (Ln) of the mean HR and dividing the width of Ln of the CI limits by 1.96 × 2, respectively. If the time-to-event outcome was not reported but the proportion of patients with CDP was, then the log-HR and its SE were derived using formulae reported previously [26].
Because the CARE-MS I and CARE-MS II alemtuzumab trials did not publicly report time to 3mCDP results, we used the results of a meta-analysis by the Haute Autorité de Santé (HAS) for the three alemtuzumab trials (CAMMS223, CARE-MS I, and CARE-MS II) included in the evidence base [27]. The HAS meta-analysis reported a pooled time to 3mCDP estimate, which was used as the input in place of these three trials for the time to 3mCDP NMA model. This approach followed previously published NMAs [8,25].
To assess the impact of cross-trial differences for the time to CDP outcome definitions, patient-level progression data from the ASCLEPIOS I/II ofatumumab trials [28] were recalculated via post hoc analyses to align with CDP definitions used by other included trials. Two sets of ASCLEPIOS I/II data for time to 3mCDP and 6mCDP were used for separate NMAs: (1) CDP defined as per the ASCLEPIOS I/II protocol (‘predefined’) and (2) CDP reanalysed to align with multiple included trials (EVIDENCE, IFNB MS, OPERA I/II, TEMSO, TOWER, and ULTIMATE I/II) on the basis of the magnitude of increase in EDSS score required to be considered progression at different baseline scores but otherwise defined as per the ASCLEPIOS I/II protocol (‘EDSS-aligned’). For the EVIDENCE and IFNB MS trials, we only considered the EDSS score increases for the range of baseline scores specified in the trial inclusion/exclusion criteria when considering the alignment of their CDP definition with the other trials.
In ASCLEPIOS I/II (predefined CDP definition), progression was defined as an increase of ≥0.5-points in EDSS score from a baseline score of ≥5.5, an increase of ≥1-point from a baseline score of 1 to 5, or an increase of ≥1.5-points from a baseline score of 0. The baseline EDSS score was defined as the last EDSS assessment prior to the first dose of study medication. Disability progression had to be sustained for ≥90 days for CDP at three months (3mCDP) and ≥166 days for CDP at six months (6mCDP). Progression was confirmed at a scheduled visit in the absence of (confirmed or unconfirmed) relapse if, over the required time interval, all assessments met the progression criterion. In contrast, for the EDSS-aligned CDP definition, progression was defined as increase of ≥0.5-points in EDSS score from a baseline score of >5.5 or an increase of ≥1-point from a baseline score of 0 to 5.5. The definition otherwise aligned with the predefined CDP definition.
For our previous NMA, we included an analysis using ASCLEPIOS I/II CDP data reanalysed to align fully with the CDP definition reported for the OPERA I/II ocrelizumab trials, including not only the EDSS score increase requirements for progression but other definition components as well [8]. For the present study, we opted to include the EDSS-aligned analysis in place of the OPERA-aligned analysis to achieve consistency across a broader evidence base.
Inconsistency analysis
For each NMA, we evaluated inconsistency (i.e., agreement in treatment effects when both direct and indirect comparisons were available for pairwise contrasts) by comparing DIC values between the consistency model (i.e., main analysis) and the inconsistency model [18]. A difference of 3 points or greater was considered meaningful [29]. If there was a meaningful difference in DIC values, then we investigated sources of inconsistency by plotting the posterior mean deviance for the treatment arms of each trial in the inconsistency model versus in the consistency model in a scatterplot (as per NICE guidance) [18]. For each NMA, we also produced forest plots comparing treatment effect estimates from consistency and inconsistency models for pairwise treatment comparisons informed by at least two trials.
Sensitivity analyses
Sensitivity analyses were conducted to assess the impacts of the analytical methods and evidence base used for the NMAs. The following sensitivity analyses were conducted: (1) using FE models instead of RdE models, (2) including the ADVANCE trial, (3) including the INCOMIN trial, (4) excluding trials identified as potential contributors to inconsistency, and (5) excluding trials published prior to 2004.
The ADVANCE and INCOMIN trials [30,31] were only included in sensitivity analyses because they were identified as outliers within the NMA evidence base, and an analysis in which trials published prior to 2004 were excluded was of interest because reported ARR values for these trials were high compared with newer trials (see ‘Results’ for additional details).
Trials were identified as potential contributors to inconsistency if at least one of their treatment arms had a posterior mean deviance of >2 for the consistency model and/or inconsistency model. These trials were excluded one by one in iterative analyses that considered the magnitude of the difference between models for the posterior mean deviance. For example, the first analysis excluded the trial with the treatment arm displaying the greatest difference between consistency and inconsistency models, the second analysis excluded this trial as well as a second trial with a treatment arm displaying the next greatest difference between models, and so on. For each analysis, the DIC values were compared for the consistency and inconsistency models and trials continued to be iteratively excluded until the DIC values differed by no more than 3 (i.e., were not meaningfully different).
Results
Systematic literature review
After screening and full text appraisal, 728 records representing 93 RCTs were found to match the PICOS criteria for the SLR (Figure 1). During screening, 15,096 records were considered irrelevant, and the subsequent full text review resulted in the exclusion of 2966 records. Of the 93 RCTs identified via the SLR, 52 trials were excluded because they did not meet the eligibility criteria for the NMA, resulting in 41 trials being considered in the NMA feasibility assessment. Although data for the ULTIMATE I/II ublituximab trials were only available from a conference poster [32] at the time of the literature review, a full text publication became available during the NMA analysis [33] and values were confirmed to be the same between these two data sources. The risk of bias assessment using Centre for Reviews and Dissemination guidance [11] indicated that the overall risk of bias was generally low across the 41 RCTs included in the NMA evidence base (Supplementary Appendix A). There was some risk in terms of adequate concealment of treatment allocation and blinding, but the risk of bias was low for other assessed components (randomisation, prognostic factors, withdrawals/discontinuations, measured outcomes, and appropriate intention-to-treat analysis).
Figure 1. . PRISMA flow diagram of the study selection process.
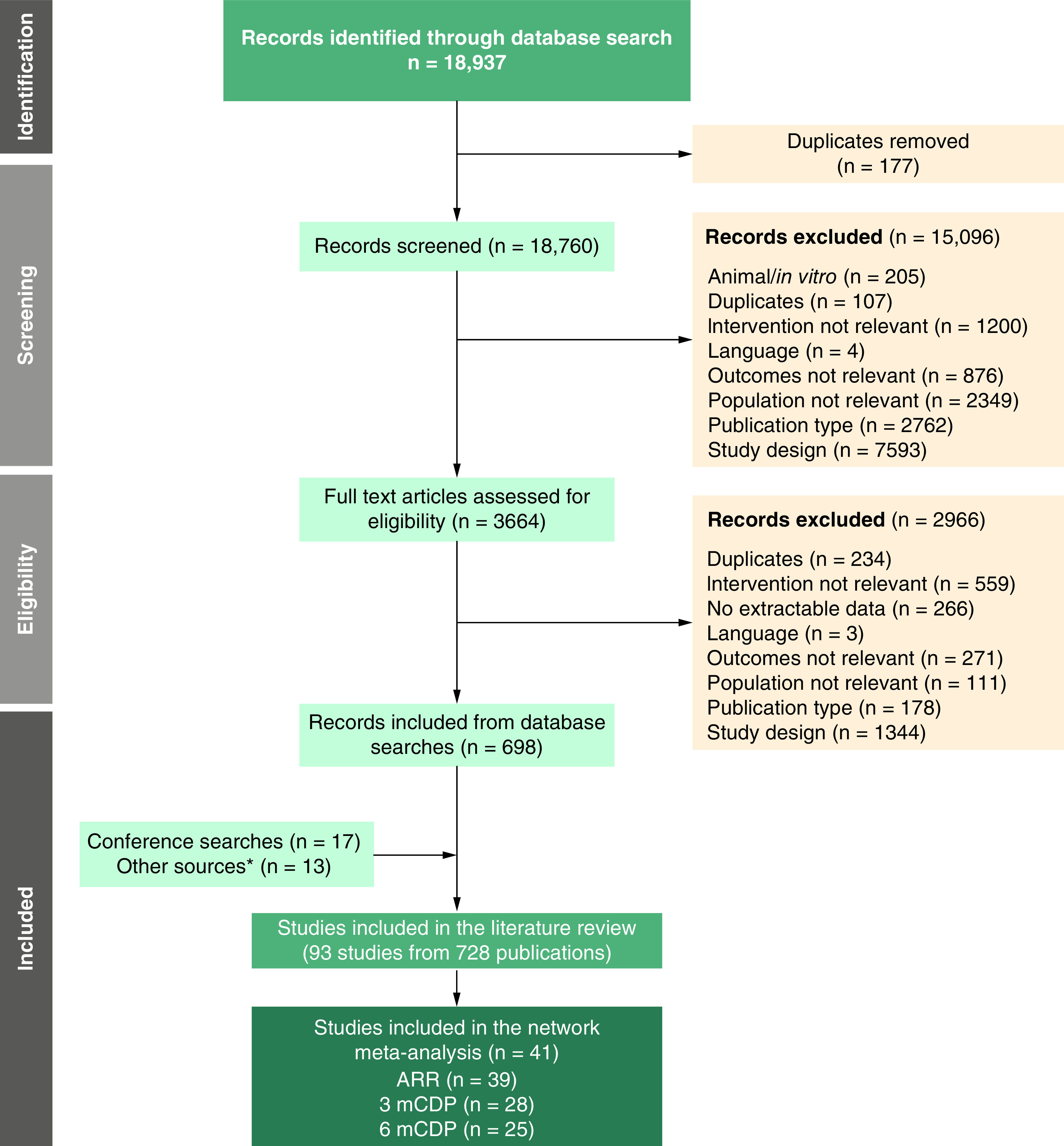
*Identified during bibliographic search of identified published systematic reviews.
3mCDP: 3-month confirmed disability progression; 6mCDP: 6-month confirmed disability progression; ARR: Annualised relapse rate.
Two of the 41 RCTs were excluded from the main analyses. We excluded the ADVANCE trial [30], which compared pegylated IFN β-1a with placebo, because it was previously identified by NICE as an outlier. In the technology appraisal guidance for ocrelizumab, the NICE committee reported that the inclusion of ADVANCE in a time to 6mCDP NMA resulted in pegylated IFN β-1a outperforming both other IFN β therapies and high-efficacy therapies (e.g., natalizumab) [34]. These results were deemed to be clinically implausible and so comparisons involving pegylated IFN β-1a were disregarded by the committee [34]. The INCOMIN trial [31], which compared subcutaneous IFN β-1b with intramuscular IFN β-1a, was also excluded because it was previously identified as an outlier. As noted in previous NMA publications [8,25] and a previous review of IFN β trials [35], the efficacy results for the two IFN β therapies were significantly different, which was contrary to clinical experience (i.e., other head-to-head IFN β trials). Separate sensitivity analyses including ADVANCE or INCOMIN in the evidence networks were conducted to explore the impact of these trials on NMA results. The number of RCTs included in each NMA by outcome is presented in Figure 1.
Feasibility assessment
Study and patient characteristics for the NMA evidence base are summarised in Supplementary Appendix B. The evidence base included RCTs published between 1987 and 2021. The design of these trials was similar, with most being phase III, double-blind, and having parallel allocation and a duration of ∼2 years. The three included alemtuzumab trials (CAMMS223, CARE-MS I, and CARE-MS II) were open-label trials. Across RCTs, patient inclusion/exclusion criteria were generally similar for age, baseline EDSS score, MS phenotype, and relapse and MRI lesion history (required previous relapses/lesions and absence of recent relapses/lesions). However, criteria related to disease duration and prior treatment experience were less similar between trials. Cross-trial differences were noted for several baseline patient characteristics, including time since first MS symptoms, number of gadolinium-enhancing (Gd+) lesions, volume of T2 lesions, and proportion with previous DMT experience. For outcome definitions, relapse and ARR were similarly defined for the purpose of comparison. As mentioned previously, several trials did not report ARR, but it was possible to calculate this outcome using other reported data. Some cross-trial differences were noted for the time to CDP outcome definitions. In particular, the magnitude of increase in EDSS score required to be considered progression at different baseline scores differed across trials. As described previously (see ‘Materials & methods’), to assess the impact of differences in the time to CDP definition between RCTs, we conducted a sensitivity analysis using ASCLEPIOS I/II ofatumumab data reanalysed to partially align with a CDP definition used by other included trials (thereby making these definitions more consistent across included trials). A final component of the NMA feasibility assessment was considering the placebo response for the 17 placebo-controlled RCTs in the evidence base as a proxy for differences between these trials. The proportion of patients with 3mCDP and 6mCDP in the placebo arms were generally similar across trials of a similar duration, but ARR values for placebo arms were comparatively high in trials published prior to 2004. As described previously (see ‘Materials & methods’), a sensitivity analysis excluding trials published prior to 2004 in the evidence network for ARR was conducted to assess the impact of these trials on NMA results.
NMA results
For each outcome, a network diagram visualising the evidence base used for the analysis is provided along with the results of the analysis as a forest plot comparing each treatment in the network with placebo. A league table and a table of SUCRA and p-best values for each analysis are provided in Supplementary Appendix D. Tables summarizing the direct evidence and NMA estimates for each treatment comparison by outcome are provided in Supplementary Appendix E.
ARR
The ARR network (Figure 2) was based on 39 RCTs and connected 19 treatments (including placebo). The mAb therapies (i.e., alemtuzumab, natalizumab, ocrelizumab, ofatumumab, and ublituximab) were the most efficacious treatments compared with placebo (rate ratio [RR]: 0.28 to 0.34) (Figure 3). The three most efficacious treatments versus placebo for reducing ARR were alemtuzumab, ofatumumab, and ublituximab (RR: 0.28 to 0.31) (Figure 3) The RR for ozanimod versus placebo was 0.43 and for ponesimod versus placebo was 0.47.
Figure 2. . Network for annualised relapse rate outcome.

ALE: Alemtuzumab 12 mg; CLA: Cladribine 3.5 mg/kg; DMF: Dimethyl fumarate 240 mg twice a day; FIN: Fingolimod 0.5 mg; GA 20: Glatiramer acetate 20 mg; GA 40: Glatiramer acetate 40 mg; IFNB-1a IM: Interferon beta-1a 30 μg intramuscular; IFNB-1a SC 22: Interferon beta-1a 22 μg subcutaneous; IFNB-1a SC 44: Interferon beta-1a 44 μg subcutaneous; IFNB-1b SC: Interferon beta-1b 250 μg subcutaneous; NAT: Natalizumab 300 mg; OCR: Ocrelizumab 600 mg; OMB: Ofatumumab 20 mg; OZA: Ozanimod 1.0 mg; PBO: Placebo; PON: Ponesimod 20 mg; TER 7: Teriflunomide 7 mg; TER 14: Teriflunomide 14 mg; UTX: Ublituximab 450 mg.
Figure 3. . Forest plot for treatments compared with placebo for the annualised relapse rate analysis.

ALE: Alemtuzumab 12 mg; CLA: Cladribine 3.5 mg/kg; DMF: Dimethyl fumarate 240 mg twice a day; FIN: Fingolimod 0.5 mg; GA 20: Glatiramer acetate 20 mg; GA 40: Glatiramer acetate 40 mg; IFNB-1a IM: Interferon beta-1a 30 μg intramuscular; IFNB-1a SC 22: Interferon beta-1a 22 μg subcutaneous; IFNB-1a SC 44: Interferon beta-1a 44 μg subcutaneous; IFNB-1b SC: Interferon beta-1b 250 μg subcutaneous; NAT: Natalizumab 300 mg; OCR: Ocrelizumab 600 mg; OMB: Ofatumumab 20 mg; OZA: Ozanimod 1.0 mg; PBO: Placebo; PON: Ponesimod 20 mg; TER 7: Teriflunomide 7 mg; TER 14: Teriflunomide 14 mg; UTX: Ublituximab 450 mg.
Time to 3mCDP
The time to 3mCDP network (Figure 4) was based on 28 RCTs (3 of which were included via a meta-analysis by HAS; see ‘Materials & methods’ for details) and connected 18 treatments (including placebo). The mAb therapies (i.e., alemtuzumab, natalizumab, ocrelizumab, ofatumumab, and ublituximab) and ponesimod were the most efficacious treatments compared with placebo (hazard ratio [HR]: 0.39 to 0.59 for the predefined analysis, and 0.40 to 0.58 for the EDSS-aligned analysis) (Figure 5a and b). The three most efficacious treatments versus placebo for delaying 3mCDP were consistent between the predefined and EDSS-aligned analyses, though the rank-order did change: ocrelizumab, alemtuzumab, and ofatumumab (hazard ratio [HR]: 0.39 to 0.46) using the predefined definition (Figure 5a); and ocrelizumab, ofatumumab, and alemtuzumab (HR: 0.40 to 0.44) using the EDSS-aligned definition (Figure 5b). The HR for ozanimod versus placebo was 0.77 (both analyses).
Figure 4. . Network for 3-month confirmed disability progression outcome.

ALE: Alemtuzumab 12 mg; CLA: Cladribine 3.5 mg/kg; DMF: Dimethyl fumarate 240 mg twice a day; FIN: Fingolimod 0.5 mg; GA 20: Glatiramer acetate 20 mg; IFNB-1a IM: Interferon beta-1a 30 μg intramuscular; IFNB-1a SC 22: Interferon beta-1a 22 μg subcutaneous; IFNB-1a SC 44: Interferon beta-1a 44 μg subcutaneous; IFNB-1b SC: Interferon beta-1b 250 μg subcutaneous; NAT: Natalizumab 300 mg; OCR: Ocrelizumab 600 mg; OMB: Ofatumumab 20 mg; OZA: Ozanimod 1.0 mg; PBO: Placebo; PON: Ponesimod 20 mg; TER 7: Teriflunomide 7 mg; TER 14: Teriflunomide 14 mg; UTX: Ublituximab 450 mg.
Figure 5. . Forest plot for treatments compared with placebo for the (A) predefined and (B) Expanded Disability Status Scale-aligned 3-month confirmed disability progression analyses.
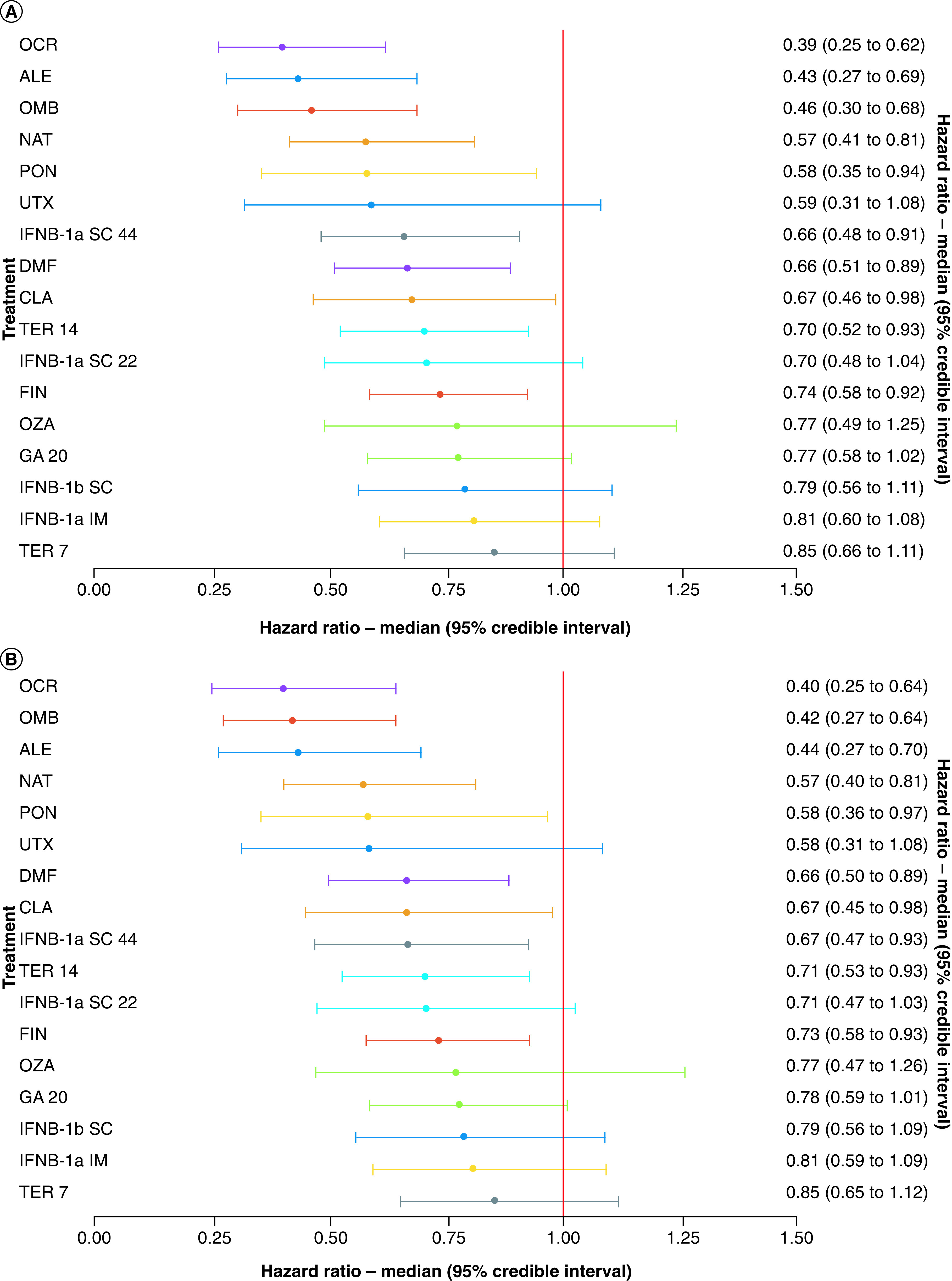
ALE: Alemtuzumab 12 mg; CLA: Cladribine 3.5 mg/kg; DMF: Dimethyl fumarate 240 mg twice a day; FIN: Fingolimod 0.5 mg; GA 20: glatiramer acetate 20 mg; IFNB-1a IM: Interferon beta-1a 30 μg intramuscular; IFNB-1a SC 22: Interferon beta-1a 22 μg subcutaneous; IFNB-1a SC 44: Interferon beta-1a 44 μg subcutaneous; IFNB-1b SC: Interferon beta-1b 250 μg subcutaneous; NAT: Natalizumab 300 mg; OCR: Ocrelizumab 600 mg; OMB: Ofatumumab 20 mg; OZA: Ozanimod 1.0 mg; PBO: Placebo; PON: Ponesimod 20 mg; TER 7: Teriflunomide 7 mg; TER 14: Teriflunomide 14 mg; UTX: Ublituximab 450 mg.
Time to 6mCDP
The time to 6mCDP network (Figure 6) was based on 25 RCTs and connected 16 treatments (including placebo). The mAb therapies (i.e., alemtuzumab, natalizumab, ocrelizumab, ofatumumab, and ublituximab) and cladribine were the most efficacious treatments compared with placebo (hazard ratio [HR]: 0.41 to 0.54 for the predefined analysis, and 0.43 to 0.54 for the EDSS-aligned analysis) (Figure 7a and b). The three most efficacious treatments versus placebo for delaying 6mCDP varied between analyses: alemtuzumab, natalizumab, and ocrelizumab (HR: 0.41 to 0.46) using the predefined definition (Figure 7a); and alemtuzumab, natalizumab, and ofatumumab (HR: 0.43 to 0.48) using the EDSS-aligned definition (Figure 7b). The HR for ozanimod versus placebo was 1.01 (both analyses) and for ponesimod versus placebo was 0.66 (predefined) or 0.67 (EDSS-aligned).
Figure 6. . Network for 6-month confirmed disability progression outcome.

ALE: Alemtuzumab 12 mg; CLA: Cladribine 3.5 mg/kg; DMF: Dimethyl fumarate 240 mg twice a day; FIN: Fingolimod 0.5 mg; GA 20: Glatiramer acetate 20 mg; IFNB-1a IM: interferon beta-1a 30 μg Intramuscular; IFNB-1a SC 44: Interferon beta-1a 44 μg subcutaneous; NAT: Natalizumab 300 mg; OCR: Ocrelizumab 600 mg; OMB: Ofatumumab 20 mg; OZA: Ozanimod 1.0 mg; PBO: Placebo; PON: Ponesimod 20 mg; TER 7: Teriflunomide 7 mg; TER 14: Teriflunomide 14 mg; UTX: Ublituximab 450 mg.
Figure 7. . Forest plot for treatments compared with placebo for the (A) predefined and (B) Expanded Disability Status Scale-aligned 6-month confirmed disability progression analyses.

ALE: Alemtuzumab 12 mg; CLA: Cladribine 3.5 mg/kg; DMF: Dimethyl fumarate 240 mg twice a day; FIN: Fingolimod 0.5 mg; GA 20: glatiramer acetate 20 mg; IFNB-1a IM: Interferon beta-1a 30 μg intramuscular; IFNB-1a SC 44: Interferon beta-1a 44 μg subcutaneous; NAT: Natalizumab 300 mg; OCR: Ocrelizumab 600 mg; OMB: Ofatumumab 20 mg; OZA: Ozanimod 1.0 mg; PBO: Placebo; PON: Ponesimod 20 mg; TER 7: Teriflunomide 7 mg; TER 14: Teriflunomide 14 mg; UTX: Ublituximab 450 mg.
Inconsistency analysis
Results of the inconsistency analysis are provided in Supplementary Appendix F. Of the three outcomes analysed in this study, only the ARR analysis had a meaningful difference (i.e., 3 points or greater) in DIC values between consistency and inconsistency models. The residual deviance scatterplot revealed that both arms of the Bornstein et al. trial [36] had relatively large deviance values for both consistency and inconsistency models, and arms of the GOLDEN [37] and TENERE [38] trials were noticeably lower/higher for inconsistency versus consistency (i.e., these trials were potential contributors to inconsistency). After excluding the TENERE, GOLDEN, and Bornstein et al. trials in a sensitivity analysis for ARR, the difference in DIC values between the models was not meaningful. The NMA results of this sensitivity analysis did not differ appreciably from the main analysis.
Sensitivity analyses
Detailed results of the sensitivity analyses are provided in Supplementary Appendix G. Two key differences were observed for the results of sensitivity analyses compared with the main analyses. For the sensitivity analysis including the ADVANCE trial, pegylated IFN β-1a was included among the top treatments versus placebo for delaying 6mCDP. For the sensitivity analysis including the INCOMIN trial, IFN β-1b was the top treatment versus placebo for delaying 6mCDP. The results for each outcome were otherwise robust to sensitivity analyses.
Discussion
It is not practical to assess all available DMTs for patients with RMS in a single RCT, so robust statistical methods are required to simultaneously compare the efficacy of these therapies. In the present study, NMA was used to generate comparative efficacy data for DMTs, including several newer therapies (ozanimod, ponesimod, and ublituximab) that to our knowledge have not yet been collectively compared in a published NMA. This analysis can inform decisions by both clinicians and payers.
Our NMA results showed that treatment with mAb therapies was generally associated with the greatest reduction in ARR and delay of 3mCDP and 6mCDP compared with placebo, in patients with RMS. This finding for mAb therapies was shared by previous NMAs that broadly compared the efficacy of DMTs for RMS [8,25,39–46]. Current MS treatment strategy focuses on alleviating CNS inflammation, slowing disease progression, and reducing the recurrence of relapses. The ideal DMT for treating patients with RMS is multifaceted in its efficacy. Our results demonstrated that of the mAb therapies, only alemtuzumab and ofatumumab were among the three most efficacious therapies versus placebo for reducing ARR and delaying 3mCDP and 6mCDP. Ocrelizumab was among the three most efficacious therapies for both CDP outcomes but not ARR, and natalizumab and ublituximab only ranked among the three most efficacious therapies for one of ARR, 3mCDP, and 6mCDP.
Of the newer therapies included in this analysis (ozanimod, ponesimod, and ublituximab), none consistently ranked among the three most efficacious treatments relative to placebo for both ARR and CDP outcomes. The efficacy of ozanimod, a non-mAb therapy, was numerically worse than placebo with respect to time to 6mCDP (HR of 1.01); this was not seen for any other included DMT. This finding was not unexpected because the ozanimod trials, RADIANCE-B and SUNBEAM, demonstrated that time to 6mCDP was appreciably more delayed with IFN β-1a compared with ozanimod (HR of 1.41; i.e., ozanimod did not delay disease progression) [47,48]. These results may have contributed to recent negative decisions by health technology assessment agencies for ozanimod [49,50]. Specifically, NICE highlighted the lack of clarity regarding the effect of ozanimod on disability progression as a reason for not recommending it as a treatment option [50]. Ponesimod, another non-mAb therapy, was not among the most efficacious treatments for ARR or time to 6mCDP, although it was ranked fifth among the included treatments relative to placebo for time to 3mCDP (for both predefined and EDSS-aligned analyses). Notably, the OPTIMUM ponesimod trial found time to 3mCDP and time to 6mCDP did not differ significantly between ponesimod and teriflunomide 14 mg, another relatively low efficacy therapy [51]. Ublituximab, a mAb therapy, ranked among the three most efficacious mAb therapies relative to placebo in the NMA for ARR. However, ublituximab ranked sixth out of included treatments relative to placebo for time to 3mCDP, behind ponesimod (for both predefined and EDSS-aligned analyses), and fourth (predefined) or fifth (EDSS-aligned) relative to placebo for time to 6mCDP. Notably, unlike the other mAb therapies, the ublituximab versus placebo comparison for time to 3mCDP and time to 6mCDP (both predefined and EDSS-aligned analyses) was not statistically significant (i.e., the 95% credible interval crossed unity). In alignment with this result, the ULTIMATE I/II ublituximab trials found that time to 3mCDP was not statistically different between ublituximab and teriflunomide 14 mg [33].
The NMA results were robust to multiple sensitivity analyses conducted to assess the impacts of the analytical methods and evidence base used for the NMAs: using FE models instead of RE models, excluding trials identified as potential contributors to inconsistency, and excluding trials published prior to 2004. As expected, the results of the sensitivity analyses including specific trials (ADVANCE and INCOMIN) previously identified as outliers differed from the main analyses. For the sensitivity analysis including the ADVANCE trial, pegylated IFN β-1a was included among the most efficacious DMTs compared with placebo for delaying 6mCDP. For the sensitivity analysis including the INCOMIN trial, IFN β-1b was the most efficacious therapy versus placebo for delaying 6mCDP. These results are unlikely to be valid given the relatively low ranking of other IFN β therapies in the NMAs and the otherwise consistent ranking of mAb therapies as the most efficacious DMTs versus placebo for each analysed outcome. Further, health technology assessment agencies have noted in their recommendations for newer DMTs that IFN β therapies are not high-efficacy therapies [34,49].
Cross-trial differences were noted for baseline patient characteristics, CDP outcome definitions (i.e., EDSS score requirements for confirming disease progression), and the ARR placebo response (i.e., ARR results for placebo arms). Previously published NMAs of DMTs for patients with RMS used a similar evidence base as the present study and so were impacted by the same cross-trial differences we identified, and did not deem these differences substantial enough to prevent an analysis [8,25,39,41,43,44]. Further, results of the predefined and EDSS-aligned 3mCDP and 6mCDP analyses as well as our sensitivity analyses conducted to assess the impact of cross-trial differences on the NMA (i.e., excluded trials published prior to 2004) demonstrated that the main analyses were robust and consistent. Although meta-regression was considered as another approach to explore the impact of cross-trial differences, it requires that multiple RCTs inform each connection in the network so that the impact of differences in baseline characteristics can be teased apart from ‘true’ relative efficacy as measured in an indirect comparison. In the evidence networks in this study, most direct connections were informed by only one trial thereby limiting the validity of a meta-regression.
Overall, the magnitude and consistency of the NMA results indicate that mAb therapies, and specifically alemtuzumab and ofatumumab, represent the best treatment option for patients with RMS in terms of reducing relapse frequency and delaying disease progression. The NMA results also provide evidence on the relative efficacy of newer DMTs for patients with RMS and may be used to populate future cost-effectiveness analyses and support reimbursement decisions and/or guide clinical practice on the most efficient use of healthcare resources.
To our knowledge, this study is the most recent and comprehensive NMA comparing DMTs for patients with RMS. Given the recent advances in the treatment of RMS, this NMA specifically provides data on the comparative efficacy of newer therapies (ozanimod, ponesimod, and ublituximab) for key clinical end points commonly assessed in MS clinical trials. The analyses conducted for this study aligned with best practices for conducting and reporting NMAs [16–18] to ensure transparency and reproducibility. Further, an NMA feasibility assessment was undertaken to ensure the evidence base was sufficiently similar for NMA results to be valid as well as to identify potential sources of cross-trial heterogeneity for further investigation via sensitivity analyses. Despite our initial use of a conference poster for the ublituximab trials, we confirmed the results when the full text data for the trials became available (post-SLR) and found that the results from the poster were consistent with the full text publication that became available during our study [33].
This study is not without limitations. First, there were limited RCT data to inform the comparative efficacy of DMTs for RMS, with relatively few trials per treatment comparison in the NMA networks. Further, because many trials did not share a central common comparator (e.g., placebo) there were substantial distances between some treatments. These network characteristics increase the potential for biased treatment effect estimates and so limit the strength of the analysis. In addition, the limited number of trials informing each treatment comparison prevented the effective use of meta-regression to adjust for potential sources of cross-trial heterogeneity. However, the characteristics of the networks are an inherent limitation of NMAs evaluating the comparative efficacy of DMTs for RMS and highlight the need for additional head-to-head RCTs with active treatments in this space. Second, we used RE models for our main analysis, which produced estimates with relatively wide credible intervals compared with those generated using FE models. Nevertheless, we identified the use of RE models as the most suitable approach to account for the cross-trial heterogeneity from the broad evidence base for our analysis, in accordance with NICE guidance [16–18]. Our use of RE models also aligned with many other published NMAs comparing the efficacy of DMTs for RMS [8,25,39,41,43,44]. Third, only ARR, 3mCDP, and 6mCDP were considered in our analysis. Although these outcomes are among the most reported end points in RMS trials, they are typically based on relatively short follow-up periods and so may not reflect the chronic course of RMS. Discontinuation- and adverse event-related outcomes were not examined in this study because these outcomes typically suffer from many confounding factors. For example, RCTs often lack sufficient follow-up times and sample sizes to detect differences in discontinuation- and adverse event-related outcomes, making it difficult to interpret quantitative analyses of these outcomes [8]. Notably, these limitations are shared by the many other published NMAs comparing the efficacy of DMTs for RMS, which used a similar evidence base and NMA methodology as our study, as they are inherent to the treatment landscape and RCT availability for this disease [8,25,39,41,43,44]. Lastly, our analysis considered only data from clinical trials and did not incorporate real-world data. We focused on clinical trial data to minimize the extent of cross-study heterogeneity, which would be expected to substantially increase if real-world data were included. An assessment of disease-modifying therapy (DMT) effectiveness using real world data would be an interesting topic for future research but was beyond the scope of the present study. Although NMAs are useful for comparing interventions for which no direct evidence is available, they are ultimately but one source of data that can assist decision making by healthcare stakeholders. It is important to note that the relative additional benefit of highly effective DMTs presented herein comes from comparisons of their average ability to reduce relapses and delay progression. However, the average benefit of a DMT may not apply to an individual patient because response to a given DMT among patients may greatly vary.
We focused our analysis on DMT efficacy because the primary treatment goals of therapies for RMS are preventing relapses, reducing accumulation of neurologic impairment and disability over time, and reducing brain inflammation and injury. However, there are multiple factors beyond efficacy that influence choice of therapy. It is important to consider the balance of efficacy and risk, as well as patient values and preferences. The choice of DMT for RMS should consider individual patient views about therapy efficacy, safety, and tolerability (with a goal of minimizing risks and side effects); convenience and preferred method of administration (e.g., willingness to perform injections); readiness and urgency to begin treatment; and likelihood of adherence. A separate discussion is warranted for patients engaging in family planning. Lastly, the availability of some DMTs for patients may be limited due to geographic distribution, national regulations, cost, and insurance restrictions.
Conclusion
New treatment options continue to emerge for patients with RMS; hence, there is a need to disseminate clinical efficacy knowledge constantly and rapidly to help individuals make up-to-date and well-informed healthcare decisions. Here, we used NMA to synthesize the current treatment landscape for patients with RMS and incorporated recently introduced DMTs to gain a comprehensive and contemporary view of the comparative efficacy of DMTs. The mAb therapies were generally the most efficacious DMTs for all outcomes studied (ARR, 3mCDP, and 6mCDP), and this result was robust to sensitivity analyses. Of the included mAb therapies, only the immune cell-depleting mAbs alemtuzumab and ofatumumab ranked among the three most efficacious treatments for reducing ARR and delaying time to 3mCDP and 6mCDP. None of the newer treatment options (i.e., ozanimod, ponesimod, and ublituximab) consistently ranked among the three most efficacious treatments versus placebo for both ARR and CDP outcomes. These NMA results indicate that many therapies continue to offer substantial benefits for patients with RMS in terms of reducing ARR and delaying CDP despite the introduction of newer treatment options. This study provides the most recent comparative efficacy data and may help facilitate future treatment comparisons and health economic analyses for reimbursement decision makers and help guide clinical practice in disease area with limited head-to-head evidence.
Summary points.
Increasingly effective disease-modifying therapies (DMTs) have emerged for the treatment of patients with relapsing multiple sclerosis (RMS), but evidence regarding the comparative efficacy of newer therapies (ozanimod, ponesimod, ublituximab) is lacking.
The aim of this study was to assess the relative efficacy of DMTs for RMS using network meta-analysis (NMA).
For this systematic review and NMA, we searched biomedical databases, conference proceedings, and trial registries for randomised controlled trials (RCTs) published up to March 2022.
We included RCTs in the NMA if they met the following criteria: population was ≥75% RMS; interventions and comparators included DMTs approved for RMS in the United States and Europe as of June 2022 or undergoing review at this time; outcomes included annualised relapse rate (ARR) and/or time to 3-month or 6-month confirmed disability progression (3mCDP and 6mCDP); the randomised portion of the trial was ≥48 weeks; and a full-text pivotal trial publication was available.
Bayesian NMAs for ARR and time to 3mCDP and 6mCDP were conducted, with potential sources of uncertainty explored via sensitivity analyses.
For each outcome, the three most efficacious treatments versus placebo were monoclonal antibody (mAb) therapies: alemtuzumab, ofatumumab, and ublituximab for ARR; alemtuzumab, ocrelizumab, and ofatumumab for 3mCDP; and alemtuzumab, natalizumab, and either ocrelizumab or ofatumumab (depending on the CDP definition used for included ofatumumab trials) for 6mCDP.
The monoclonal antibody therapies were the most efficacious DMTs for RMS, with alemtuzumab and ofatumumab ranking among the three most efficacious treatments for reducing ARR and delaying time to 3mCDP and 6mCDP.
Of the newer therapies, only ublituximab ranked among the three most efficacious treatments, for the single outcome of ARR.
Supplementary Material
Footnotes
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: https://bpl-prod.literatumonline.com/doi/10.57264/cer-2023-0016
Author contributions
IA Samjoo, N Adlard and J Banhazi were responsible for study conception and design. All authors were responsible for the acquisition, analysis, and interpretation of data. IA Samjoo, C Drudge and S Walsh were responsible for drafting the manuscript, and all authors were responsible for critically revising/reviewing the manuscript and for providing final approval. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work, and have given their approval for this version to be published.
Financial & competing interests disclosure
This work was funded by Novartis Pharma AG. IA Samjoo, C Drudge, and S Walsh are employees of EVERSANA™. EVERSANA receives consultancy fees from pharmaceutical and device companies, including Novartis Pharma AG. S Tiwari, R Brennan, I Boer, DA Häring, N Adlard and J Banhazi are salaried employees of Novartis. L Klotz received compensation for serving on Scientific Advisory Boards for Alexion, Biogen, Bristol-Myers Squibb, Genzyme, Horizon, Janssen, Merck Serono, Novartis, Roche, and Viatris. L Klotz received speaker honoraria and travel support from Bayer, Biogen, Bristol-Myers Squibb, Genzyme, Grifols, Merck Serono, Novartis, Roche, Santhera, and Teva. L Klotz receives research support from the German Research Foundation, the IZKF Münster, IMF Münster, Biogen, Immunic AG, Novartis, and Merck Serono. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Data sharing statement
The data that support the findings of this work are publicly available.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit https://creativecommons.org/licenses/by-nc-nd/4.0/
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Compston A, Coles A. Multiple sclerosis. Lancet 372(9648), 1502–1517 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Filippi M, Bar-Or A, Piehl F et al. Multiple sclerosis. Nat. Rev. Dis. Primers 4(1), 43 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Kobelt G, Thompson A, Berg J, Gannedahl M, Eriksson J. New insights into the burden and costs of multiple sclerosis in Europe. Mult. Scler. 23(8), 1123–1136 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lublin FD, Reingold SC, Cohen JA et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology 83(3), 278–286 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.National Multiple Sclerosis Society. Relapsing-remitting MS (RRMS) (2022). https://www.nationalmssociety.org/What-is-MS/Types-of-MS/Relapsing-remitting-MS
- 6.European Medicines Agency. Clinical investigation of medicinal products for the treatment of multiple sclerosis (2015). ; • This publication provides a definition for relapsing multiple sclerosis.
- 7.Hauser SL, Cree BaC. Treatment of multiple sclerosis: a review. Am. J. Med. 133(12), 1380–1390.e1382 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; • This paper provides an overview of treatment approaches for multiple sclerosis.
- 8.Samjoo IA, Worthington E, Drudge C et al. Comparison of ofatumumab and other disease-modifying therapies for relapsing multiple sclerosis: a network meta-analysis. J. Comp. Eff. Res. 9(18), 1255–1274 (2020). [DOI] [PubMed] [Google Scholar]
- 9.National Institute for Health and Care Excellence. Single technology appraisal: user guide for company evidence submission template (2017).
- 10.Moher D, Liberati A, Tetzlaff J, Altman DG. Prisma Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Ann. Intern. Med. 151(4), 264–269 (2009). [DOI] [PubMed] [Google Scholar]; • This paper presents a commonly used protocol for conducting systematic literature reviews.
- 11.Centre for Reviews and Dissemination. Systematic Reviews: CRD's Guidance for Undertaking Reviews in Health Care. University of York, York, UK: (2009). [Google Scholar]
- 12.Salanti G. Indirect and mixed-treatment comparison, network, or multiple-treatments meta-analysis: many names, many benefits, many concerns for the next generation evidence synthesis tool. Res. Synth. Methods 3(2), 80–97 (2012). [DOI] [PubMed] [Google Scholar]; • This paper provides guidance for conducting network meta-analyses of randomised controlled trials.
- 13.Hoaglin DC, Hawkins N, Jansen JP et al. Conducting indirect-treatment-comparison and network-meta-analysis studies: report of the ISPOR Task Force on Indirect Treatment Comparisons Good Research Practices: part 2. Value Health 14(4), 429–437 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Jansen JP, Fleurence R, Devine B et al. Interpreting indirect treatment comparisons and network meta-analysis for health-care decision making: report of the ISPOR Task Force on Indirect Treatment Comparisons Good Research Practices: part 1. Value Health 14(4), 417–428 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Jansen JP, Naci H. Is network meta-analysis as valid as standard pairwise meta-analysis? It all depends on the distribution of effect modifiers. BMC Med 11(1), 159 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dias S, Sutton AJ, Ades A, Welton NJ. Evidence synthesis for decision making 2: a generalized linear modeling framework for pairwise and network meta-analysis of randomized controlled trials. Med. Decis. Making 33(5), 607–617 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This paper provides guidance for conducting network meta-analyses of randomised controlled trials.
- 17.Dias S, Sutton AJ, Welton NJ, Ades A. Evidence synthesis for decision making 3: heterogeneity—subgroups, meta-regression, bias, and bias-adjustment. Med. Decis. Making 33(5), 618–640 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dias S, Welton NJ, Sutton AJ, Caldwell DM, Lu G, Ades A. Evidence synthesis for decision making 4: inconsistency in networks of evidence based on randomized controlled trials. Med Decis Making 33(5), 641–656 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria: (2019). [Google Scholar]
- 20.Spiegelhalter D, Thomas A, Best N. WinBUGS user manual. MRC Biostat Unit; (2004). [Google Scholar]
- 21.Salanti G, Ades A, Ioannidis JP. Graphical methods and numerical summaries for presenting results from multiple-treatment meta-analysis: an overview and tutorial. J. Clin. Epidemiol. 64(2), 163–171 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Boiko AN, Lashch NY, Sharanova SN et al. A comparative placebo-controlled clinical trial of the efficacy and safety of glatiramer acetate 20 mg in patients with remitting multiple sclerosis: first-year study results. Neurosci. Behav. Physiol. 48(3), 351–357 (2018). [Google Scholar]
- 23.Turner RM, Jackson D, Wei Y, Thompson SG, Higgins JPT. Predictive distributions for between-study heterogeneity and simple methods for their application in Bayesian meta-analysis. Stat. Med. 34(6), 984–998 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ren S, Oakley JE, Stevens JW. Incorporating genuine prior information about between-study heterogeneity in random effects pairwise and network meta-analyses. Med. Decis. Making 38(4), 531–542 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCool R, Wilson K, Arber M et al. Systematic review and network meta-analysis comparing ocrelizumab with other treatments for relapsing multiple sclerosis. Mult. Scler. Relat. Disord. 29, 55–61 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Watkins C, Bennett I. A simple method for combining binomial counts or proportions with hazard ratios for evidence synthesis of time-to-event data. Res. Synth. Methods 9(3), 352–360 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Haute Autorité De Santé. Commission de la Transparence: alemtuzumab (2016).
- 28.Hauser SL, Bar-Or A, Cohen JA et al. Ofatumumab versus teriflunomide in multiple sclerosis. N. Engl. J. Med. 383(6), 546–557 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Dias S, Ades AE, Welton NJ, Jansen JP, Sutton AJ. Network Meta-Analyses for Decision-Making. John Wiley & Sons, NJ, USA: (2018). [Google Scholar]; •• This book provides guidance for conducting network meta-analyses of randomised controlled trials.
- 30.Calabresi PA, Kieseier BC, Arnold DL et al. Pegylated interferon beta-1a for relapsing-remitting multiple sclerosis (ADVANCE): a randomised, phase 3, double-blind study. Lancet Neurol. 13(7), 657–665 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Durelli L, Verdun E, Barbero P et al. Every-other-day interferon beta-1b versus once-weekly interferon beta-1a for multiple sclerosis: results of a 2-year prospective randomised multicentre study (INCOMIN). Lancet 359(9316), 1453–1460 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Steinman L, Fox E, Hartung H-P et al. Efficacy and safety of ublituximab vs teriflunomide in patients with relapsing multiple sclerosis: results from two phase 3 studies ULTIMATE I & ULTIMATE II. Presented at: 2021 American Academy of Neurology Virtual Annual Meeting. (17–22 April 2021). [Google Scholar]
- 33.Steinman L, Fox E, Hartung H-P et al. Ublituximab versus teriflunomide in relapsing multiple sclerosis. N. Engl. J. Med. 387(8), 704–714 (2022). [DOI] [PubMed] [Google Scholar]
- 34.National Institute for Health and Care Excellence. Technology appraisal guidance TA533: ocrelizumab for treating relapsing-remitting multiple sclerosis (2018).
- 35.Vartanian T. An examination of the results of the EVIDENCE, INCOMIN, and phase III studies of interferon beta products in the treatment of multiple sclerosis. Clin. Ther. 25(1), 105–118 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Bornstein MB, Miller A, Slagle S et al. A pilot trial of Cop 1 in exacerbating-remitting multiple sclerosis. N. Engl. J. Med. 317(7), 408–414 (1987). [DOI] [PubMed] [Google Scholar]
- 37.Comi G, Patti F, Rocca MA et al. Efficacy of fingolimod and interferon beta-1b on cognitive, MRI, and clinical outcomes in relapsing-remitting multiple sclerosis: an 18-month, open-label, rater-blinded, randomised, multicentre study (the GOLDEN study). J. Neurol. 264(12), 2436–2449 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vermersch P, Czlonkowska A, Grimaldi LM et al. Teriflunomide versus subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis: a randomised, controlled phase 3 trial. Mult. Scler. 20(6), 705–716 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Chen C, Zhang E, Zhu C et al. Comparative efficacy and safety of disease-modifying therapies in patients with relapsing multiple sclerosis: a systematic review and network meta-analysis. J. Am. Pharm. Assoc. (2022) (Online ahead of print). [DOI] [PubMed] [Google Scholar]
- 40.Fogarty E, Schmitz S, Tubridy N, Walsh C, Barry M. Comparative efficacy of disease-modifying therapies for patients with relapsing remitting multiple sclerosis: systematic review and network meta-analysis. Mult. Scler. Relat. Disord. 9, 23–30 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Giovannoni G, Lang S, Wolff R et al. A systematic review and mixed treatment comparison of pharmaceutical interventions for multiple sclerosis. Neurol. Ther. 9(2), 359–374 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Institute for Clinical and Economic Review. Disease-modifying therapies for relapsing-remitting and primary-progressive multiple sclerosis: effectiveness and value (2017).
- 43.Li H, Hu F, Zhang Y, Li K. Comparative efficacy and acceptability of disease-modifying therapies in patients with relapsing-remitting multiple sclerosis: a systematic review and network meta-analysis. J. Neurol. 267, 3489–3498 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Liu Z, Liao Q, Wen H, Zhang Y. Disease modifying therapies in relapsing-remitting multiple sclerosis: a systematic review and network meta-analysis. Autoimmun. Rev. 20(6), 102826 (2021). [DOI] [PubMed] [Google Scholar]
- 45.Lucchetta RC, Tonin FS, Borba HHL et al. Disease-modifying therapies for relapsing-remitting multiple sclerosis: a network meta-analysis. CNS Drugs 32(9), 813–826 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Siddiqui MK, Khurana IS, Budhia S, Hettle R, Harty G, Wong SL. Systematic literature review and network meta-analysis of cladribine tablets versus alternative disease-modifying treatments for relapsing-remitting multiple sclerosis. Curr. Med. Res. Opin. 34(8), 1361–1371 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Cohen JA, Comi G, Selmaj KW et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (RADIANCE): a multicentre, randomised, 24-month, phase 3 trial. Lancet Neurol. 18(11), 1021–1033 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Comi G, Kappos L, Selmaj KW et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (SUNBEAM): a multicentre, randomised, minimum 12-month, phase 3 trial. Lancet Neurol. 18(11), 1009–1020 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Canadian Agency for Drugs and Technologies in Health. Reimbursement reviews: ozanimod (2021).
- 50.National Institute for Health and Care Excellence. Technology appraisal guidance TA706: ozanimod for treating relapsing-remitting multiple sclerosis (2021).
- 51.Kappos L, Fox RJ, Burcklen M et al. Ponesimod compared with teriflunomide in patients with relapsing multiple sclerosis in the active-comparator phase 3 OPTIMUM study: a randomized clinical trial. JAMA Neurol. 78(5), 558–567 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.