

Abstract
There are a number of mechanisms by which alkylmercury compounds cause toxic action in the body. Collectively, published studies reveal that there are some similarities between the mechanisms of the toxic action of the mono-alkyl mercury compounds methylmercury (MeHg) and ethylmercury (EtHg). This paper represents a summary of some of the studies regarding these mechanisms of action in order to facilitate the understanding of the many varied effects of alkylmercurials in the human body. The similarities in mechanisms of toxicity for MeHg and EtHg are presented and compared. The difference in manifested toxicity of MeHg and EtHg are likely the result of the differences in exposure, metabolism, and elimination from the body, rather than differences in mechanisms of action between the two.
Keywords: Arachidonic acid, Calcium homeostasis, Cell cycle/division, Ethylmercury, Glial cells, Glutamate, Glutamine, Glutathione (GSH), Leukotriene synthesis mechanism of toxicity, Membrane permeability/integrity, Methylmercury, Mitochondria, Neurotransmitter release nitric oxide, Oxidative stress, Reactive oxygen species, Receptor binding, ROS, Thimerosal
1. Preface
The alkyl mercury compounds methylmercury (MeHg) and ethylmercury (EtHg) have been shown to be toxic to humans and non-human animals as well (Driscoll et al. 2013). Both of those compounds have similar chemical properties, and both have been shown to disrupt the normal function of the CNS in a variety of animal species.
In mass poisonings from the consumption of MeHg-contaminated seafood in Japan (Kutsuna 1968) and MeHg-treated grain in Iraq (Bakir et al. 1973), frank developmental/birth effects and/or severe brain damage were observed in the children of some mothers who consumed large quantities of mercury-contaminated fish, bread, or grain products during pregnancy. In addition, a variety of neurologic effects were reported in both the 1968 and 1973 papers. Among these effects were deficits in cognitive and motor function. Some of the many specific effects observed were distal paresthesias, ataxia, unsteady gait, muscle weakness, impairment of the senses, irritability, memory loss, insomnia, and confusion. Similar neurologic disorders were seen in Iraq in 1956 and 1960, when food containing flour made from grain treated with EtHg p-toluene sulfonanilide was consumed (Bakir et al. 1973; Jalili and Abbasi 1961). In another reported incident (Zhang 1984), 41 individuals were poisoned by eating rice that had been treated with a grain disinfectant containing 2–2.5% EtHgCl2. Many of the symptoms and clinical signs were similar to those experienced in the ethyl- and methylmercury poisonings in Japan and Iraq. And while both of the alkyl compounds primarily effect the central nervous system, the rapid metabolism of ethylmercury to inorganic mercury may lead to kidney damage (Clarkson and Magos 2006), especially at higher amounts and with longer periods of exposure.
1.1. How Alkyl Mercury Compounds Get into the Food Web
Mercury enters the atmosphere from both natural sources (e.g., volcanos) and anthropogenic activities (e.g., burning of fossil fuels and industrial processes). As it gradually settles onto land and bodies of water, it is methylated by bacteria and other microorganisms to form MeHg. In aquatic and marine environments, the MeHg is biomagnified as it passes to larger and larger predatory organisms in the ecosystem. Top predatory fish typically have the greatest concentrations of MeHg in their muscle tissue.
The neurotoxic effects of alkyl mercury compounds appear to be similar across the vertebrate species studied. A primary source of exposure for humans and large marine animals is fish and shellfish. Many species of birds are piscivorous, so it logically follows that a number of bird species have been found to be negatively impacted by MeHg (Seewagen 2010). The effects reported in such species are primarily of neurologic origin and include ataxia, lethargy, reduced appetite and egg production, poor hatching success, and aberrant parental care. Non-avian species that feed on fish are also exposed to MeHg, and would be expected to manifest symptoms and clinical signs of MeHg effect, just as with humans and birds. Elevated MeHg levels have been reported in aquatic mammals that feed largely on fish, including pilot whales (Weihe et al. 1996), elephant seals (Cossaboon et al. 2015) and mink and river otter (Yates et al. 2005). But since there is currently a relative paucity of literature on the effects of alkyl mercury compounds and their respective mechanisms of toxic action compared with the plethora of such studies regarding humans, the information presented in this paper focuses primarily on mechanisms of action on human cells and tissue cultures.
2. Introduction
The affinity of mercury for sulfur has been known since the days of alchemy (Hughes 1957), and the strong affinity of sulfhydryl (-SH) groups for mercury has often been considered to be involved in the mechanism of toxic action of mercury and its compounds. Clarkson et al. (2007) reported that the high mobility of methylmercury (MeHg) in the body is due to the formation of small molecular weight thiol complexes that are readily transported across cell membranes.
There are a number of mechanisms by which alkylmercury compounds cause toxic action in the body (Faustman et al. 2002). This paper represents a summary of some of the studies regarding these mechanisms of action in order to facilitate the understanding of the many varied effects of alkylmercurials in the human body. The data reveal that there are some similarities between the mechanisms of the two mono-alkyl mercury compounds: methylmercury (MeHg) and ethylmercury (EtHg).
Mercury in any form will bind with sulfhydryl (-SH) groups in thiols or proteins. Sulfur is present in the amino acids cystine, cysteine, and methionine, as well as the cysteine-derivative taurine, which is plentiful in the bile. Since receptors in/on cell membranes are all constructed from proteins, those receptors are susceptible to attack by various forms of mercury. The majority of protein transport systems in higher eukaryotes contain cysteine, and the presence of cysteine residues exposed towards the extracellular environment make them potential sites of interaction with mercurials (Pochini et al. 2013). When mercury binds to one of the amino acid residues in receptor proteins, it blocks or attenuates that protein molecule’s range of availability for normal metabolic function. When the affected protein is normally used to transport a substance, such as calcium ion (Ca++), across the membrane of a cell or sub-cellular organelle, the selective permeability of that membrane and the proper physiologic function of that cell or organelle may be compromised.
Overall, the data reveal that there are some remarkable similarities between the toxic mechanisms of the mono-alkyl mercury compounds MeHg and EtHg. However, due to differences in metabolic rates and the subsequent elimination half-lives, the methyl and ethyl forms of organic mercury are presented separately in this paper. The effects of MeHg are provided in Table 1. The effects of EtHg/thimerosal are provided in Table 2. Because of the controversy between the preservative thimerosal (CDC 2015), which is metabolized to ethyl mercury and thiosalicylate, a comparatively benign compound, there is much more published data regarding thimerosal than EtHg by itself. Consequently, most of the studies regarding the toxicity of EtHg consist of experiments using the vaccine preservative thimerosal, in which the effective portion is EtHg.
Table 1.
Some mechanisms of toxic action proposed for MeHg
| Mechanism | Results | Reference(s) |
|---|---|---|
| Altered membrane permeability | MeHg attaches to cystine, enabling it to enter cells via a neutral amino acid transporter that also transports methionine into cells; also increases mitochondrial membrane permeability | Clarkson (1995); Clarkson et al. (2007); Pochini et al. (2013); Simmons-Willis et al. (2002) |
| Altered intracellular calcium homeostasis | Blocks cellular membrane Ca++ channels; causes release of mitochondrial Ca++ into the cytosol | Hare et al. (1993); Kang et al. (2006) Limke et al. (2004b); Marty and Atchison (1997); Minnema et al. (1987); Peng et al. (2002); Sirois and Atchison (2000); Szalai et al. (1999) |
| Oxidative stress/reactive oxygen species (ROS) | MeHg causes increase in ROS and subsequent depolarization of the mitochondrial membrane; decreases aconitase activity | Dreiem and Seegal (2007); Garg and Chang (2006); Myhre et al. (2003); Yin et al. (2007) |
| Effects on receptor binding/neurotransmitter release | Blocks GABA receptor sites; binds to NMDARs, inducing their expression; causes up-regulation of muscarinic ACh receptors | Basu et al. (2008); Coccini et al. (2000); Cooper et al. (2003); Fonfria et al. (2001); Ndountse and Chan (2008); Yuan and Atchison (2003) |
| Generation of arachidonic acid | Promotes the release of arachidonic acid, resulting in neurotoxicity | Shanker et al. (2002); Verity et al. (1994) |
| Effects on cycle/cell division | Causes arrest in the GM2/M-phase of the cell cycle through microtubule disruption | Burke et al. (2006); Castoldi et al. (2000); Gribble et al. (2005); Kim et al. (2007); Ou et al. (1999b); Rodier et al. (1984) |
| Effects on glutathione activity | Decreases glutathione activity, particularly in the cerebellum and mitochondria, thus providing less protection from oxidative stress due to MeHg | Carocci et al. (2014); Choi et al. (1996); Franco et al. (2006); Mori et al. (2007); Ndountse and Chan (2008) |
| Involvement with nitric oxide synthetase | MeHg causes an increase in nNOS protein, causing the overproduction of NO | Chuu et al. (2001); Shinyashiki et al. (1998) |
| Glutamate—glutamine uptake and homeostasis in glial cells | Disrupts glutamate homeostasis by disrupting glutamine/glutamate cycling; causes glutamate release from pre-synaptic neurons | Farina et al. (2003a, b); Manfroi et al. (2004); Yin et al. (2007) |
| Activation of calpain cascade leading to apoptosis | Cascade begins with increased intracellular Ca++ leading to microtubule degradation and cell death | Sakaue et al. (2005) |
Table 2.
Some mechanisms of toxic action proposed for EtHg/thimerosal
| Mechanism | Results | Reference(s) |
|---|---|---|
| Altered intracellular calcium homeostasis | Mobilization of intracellular Ca++; causes release of mitochondrial Ca++ into the cytosol; attenuates any increase in internal calcium ion concentration | Elferink (1999); Machaty et al. (1999); Sayers et al. (1993); Tornquist et al. (1999); Zarini et al. (2006) |
| Oxidative stress/reactive oxygen species (ROS) | EtHg causes increase in ROS and depolarization of the mitochondrial membrane; inhibits mitochondrial respiration; increased formation of superoxide and hydrogen peroxide | Sharpe et al. (2012) |
| Generation of arachidonic acid (AA) | Promotes release of AA; prevents reacylation of AA | Chen et al. (2003); Stuning et al. (1988) |
| Effects on cell division | Thimerosal destroyed cell spindle, arresting embryonic development | Machaty et al. (1999) |
| Effects on glutathione activity | Inhibits glutathione-S-transferase T1 (GST T1); depletes GSH | Muller et al. (2001); Wu et al. (2008) |
| Involvement with nitric oxide synthetase | Causes an increase in NOS | Chen et al. (2003) |
| Effects glutamate transport | Disrupts glutamate homeostasis by disrupting glutamine/glutamate cycling; decreases GLT-1 levels; significantly inhibition of GLAST activity | Mutkus et al. (2005) |
3. Methylmercury: Mechanisms of Toxic Action
Since Aschner and Aschner (1990) published that MeHg associates with thiol-containing amino acids due to the high affinity of the methylmercury cation (MeHg+) for sulfhydryl groups (-SH), many studies have further explored the mechanism(s) by which MeHg exerts its toxic effects on mammalian cells.
When mercury binds to protein receptors, the effects that occur following, or as a result of, that binding are key to understanding the ultimate mechanism or mechanisms of toxic action of mercury. Since these effects occur at both the cellular and sub-cellular levels, in vitro and in situ studies are necessary to examine the chemical mechanisms occurring at these levels. Together, in vitro and in situ studies have provided the most valuable evidence and explanations of exactly why and how mercury and its organic compounds cause damage to nervous tissue.
A number of recent studies have examined the sub-cellular mechanism of the neurotoxicity of MeHg. Impaired calcium homeostasis (Dreiem and Seegal 2007; Sirois and Atchison 2000), oxidative stress (Garg and Chang 2006; Ou et al. 1999a; Yin et al. 2007), and the alteration of glutamate homeostasis (Farina et al. 2003a, b; Ou et al. 1999a; Yin et al. 2007) have all been suggested as possible mechanisms contributing to neurotoxicity. This paper will discuss these proposed mechanisms, in addition to a number of other mechanisms of action. Kang et al. (2006) reported that cell damage caused by MeHg may occur through more than one mechanism, the effects of which may be additive or synergistic. Ndountse and Chan (2008) also indicated the likelihood of more than one mechanism contributing to the expressionof MeHg neurotoxicity. After reviewing the literature, we believe that there are indeed multiple mechanisms of action, which individually and collectively contribute to the adverse effects of MeHg on human health.
3.1. Altered Membrane Permeability
In a review of the mechanisms of mercury distribution in the body, Clarkson et al. (2007) described how the high mobility of MeHg in the body is due to the formation of small molecular weight thiol complexes that are readily transported across cell membranes. When MeHg attaches to the amino acid cysteine, the resulting structure mimics that of methionine, a large neutral amino acid (Clarkson 1992, 1995). This enables the MeHg-cysteine complex to enter cells on neutral amino acid transporters that also transport methionine into the cells. This is supported by the fact that the uptake of MeHg into the brain is inhibited by the presence of large, neutral amino acids such as leucine, methionine, and phenylalanine, and others (Clarkson 1995). Evidence presented by Clarkson et al. (2007) is also supported by the work of Simmons-Willis et al. (2002), who showed experimentally that the MeHg-l-cysteine complex is a substrate for human L-type large neutral amino acid transporters (LAT) LAT1 and LAT2.
Most MeHg in tissues is normally complexed with water-soluble sulfhydryl-containing molecules, primarily l-cysteine, GSH, hemoglobin, albumin, and other cysteine-containing polypeptides (Simmons-Willis et al. 2002). One possible explanation for the wide distribution of MeHg in the body is that an endogenously formed MeHg complex acts as a substrate for transporters that are also widely distributed throughout the body. Simmons-Willis et al. (2002) investigated the possibility that MeHg-l-cysteine, which is structurally similar to methionine, is a substrate for LAT. LAT1 and LAT2 are expressed in many tissues, including brain, and mediate the uptake of large neutral amino acids (Palacin et al. 1998; Verrey et al. 2000).
Using oocytes from the African clawed toad (Xenopus laevis), which expresses two of the L-type carriers expressed in humans [LAT1–4F2 heavy chain (LAT1–4F2hc) and LAT2–4F2hc], Simmons-Willis et al. (2002) injected oocytes with 50 nL of 110 mM stocks of amino acids. This resulted in intracellular concentrations of ~10 mM just prior to taking uptake measurements. In another part of the study, healthy oocytes were loaded with either [3H]methionine, and a 10:1 molar ratio of L-cysteine and [14C]MeHg giving approximate intracellular concentrations of 1 and 0.1 mM (100 μM), respectively.
Control oocytes were able to accumulate methionine from the medium containing 100 μM tritiated methionine, due to the presence of endogenous amino acid transporters. But when oocytes were injected with cRNA for LAT1–4F2hc or LAT2–4F2hc, methionine uptake was greatly enhanced. Further, LAT1- and LAT2 expressing oocytes also showed higher uptake of [14C] MeHg when applied as 100 μM MeHg-l-cysteine. Simmons-Willis et al. (2002) reported that the LAT1- and LAT2-stimulated uptake of MeHg from 100 μM MeHg-l-cysteine was significantly (p < 0.05) higher than from 100 μM methionine, indicating that the MeHg-l-cysteine complex may make an excellent substrate for these amino acid transporters. Those authors concluded that the LAT1 and LAT2 proteins may provide the route of MeHg entry into brain and other tissues and may account for the rapid and widespread tissue distribution of MeHg.
Pochini et al. (2013) provided supporting data by using proteoliposomes to investigate the effects of MeHg on the carnitine (OCTN2) transporter activity. Proteoliposomes have been shown to be an appropriate experimental model for investigating the interaction of transporters with chemical compounds, including MeHg (Oppedisano et al. 2010, 2011). Carnitine is an acyl carrier with respect to the mitochondrial membrane and stimulates fatty acid oxidation and synthesis in the mitochondria.
In their study, Pochini et al. (2013) added 10 μM [3H]carnitine to proteoliposomes. The subsequent addition of 8 μM MeHg was found to strongly inhibit carnitine transport. Fifteen minutes after the application of the MeHg, the transport was inhibited by more than 50%.
To test whether the cysteine (Cys) sulfhydryl groups could be involved in the observed inhibition of the carnitine transporter, Pochini et al. (2013) added 2 mM dithioerythritol (DTE) to the proteoliposomes after they were incubated with MeHg. DTE is a sulfur-containing sugar, known to be an excellent reducing agent. The addition of the DTE resulted in recovery of the transport. In contrast, DTE had virtually no effect when added to control proteoliposomes. These results indicated that the thiol groups of the protein were involved in the binding with MeHg. From their studies, Pochini et al. (2013) concluded that the interaction of mercury with transport systems essential in cell homeostasis seems to be a general mechanism of toxicity.
Garg and Chang (2006) examined the effects of MeHg concentrations from 0.5 to 40 μM on mitochondrial membrane potential in microglia. In this experiment, they observed significant depolarization of the mitochondrial membrane with increasing MeHg concentrations, but statistically significant (p < 0.001) only at concentrations of 20 μM or higher.
3.2. Altered Intracellular Calcium Homeostasis
In a review of possible mechanisms for the cytotoxic action of MeHg, Limke et al. (2004b) noted that intracellular calcium [Ca++]i undergoes cyclic changes in concentration during normal neurologic function, and that a large concentration gradient of calcium ion concentration typically exists across the neuronal membrane. As a result, the sustained elevation of [Ca++]i can be deleterious in two major ways: (1) a depletion of energy reserves resulting from excessive employment of ATP-dependent Ca++ pumps to restore resting membrane potential; and (2) the activation of catabolic functions, both of which can contribute to Ca++-mediated cell death (Limke et al. 2004a, b).
Further, Peng et al. (2002) provided evidence that MeHg disrupts normal Ca++ channel functions, as seen in studies measuring the influx of radiolabeled calcium in synaptosomes (pinched-off nerve endings), ligand binding, and studies employing electrophysiological techniques.
One of the functions of the smooth endoplasmic reticulum (SER) is to serve as a storage site for [Ca++]i. As such, Ca++ in the cytosol must be actively transported against its concentration gradient into the SER, a process requiring ATP. Bearss et al. (2001) reported that the application of thapsigargin, an inhibitor of smooth endoplasmic reticulum Ca-ATPase activity, reduced the amplitude of MeHg-induced increase in [Ca++]i by 30% in rat cerebellar granule cells in primary culture.
Hare et al. (1993) used single NG108–15 cells preloaded with the fluorescent dye fura-2 to evaluate whether MeHg caused an increase in [Ca++]i. Whereas 0.5 μM MeHg had no effect, both 2 and 5 μM MeHg produced a biphasic increase in fluorescence. The initial increase in fluorescence was sustained, with the time to onset being concentration-dependent. The maximum increase, however, was not found to be dependent on the MeHg concentration. The initial phase was considered to likely be the result of an increase in Ca++ from both intra- and extra-cellular sources, since removal of Ca++ from the extracellular medium reduced, but did not eliminate, the increase. The time to the onset of the second phase was also concentration dependent. Thus, MeHg was found to alter the measured fura-2 fluorescence in the cells in both a concentration- and time-dependent manner. Hare et al. (1993) also concluded that the initial effect involved alterations in intracellular cation buffering, as well as an increase in the permeability of the plasma membrane to Ca++.
Sirois and Atchison (2000) used whole-cell patch clamp techniques to investigate the ability of MeHg to block Ca++ channel currents in cultures of neonatal cerebellar granule cells taken from 7-day-old rat pups of either gender. This cell type was chosen for this study due to their diversity of Ca++ channels, as initially reported by Randall and Tsien (1995). To determine whether MeHg is specific for one or more of the known sub-types of Ca++ channel receptor, Sirois and Atchison (2000) used a number of putative Ca++ channel antagonists (ω-conotoxin GVIA, ω-conotoxin MVIIC, ω-agatoxin IVA, calcicludine, and nimodipine), each with specific blocking characteristics.
To eliminate the possibility of mixing the release of intracellular calcium with the influx of extracellular calcium through calcium channels, Sirois and Atchison (2000) used the radiolabeled divalent barium ion (Ba++) instead of Ca++ in the bathing medium. While Ba++ is not normally a constituent of extracellular fluid bathing the neurons, the gated calcium channels are nonetheless highly permeable to Ba++, making Ba++ a logical surrogate for extracellular Ca++. In this study, the authors found that acute exposure to sub-micromolar concentrations of MeHg can block Ba++ currents carried through multiple Ca++ channel subtypes. They further reported that the channel-blocking effect of MeHg was seen well within concentrations seen during episodes of MeHg intoxication. And while the role that MeHg-induced calcium block plays in MeHg neurotoxicity remains to be determined fully, the low concentration at which these effects were seen in this study makes it likely that the effects on Ca++ channels at least contribute to the pathological damage observed in MeHg poisoning (Sirois and Atchison 2000). These authors further postulated that the blockage of Ca++ channels could possibly contribute to ultimate neuronal death in the cerebellum through the disruption of neurotransmitter release, cell growth and differentiation, protein synthesis, maintenance of the membrane potential, and regulation of [Ca++]i.
Additional support for the ability of MeHg to block Ca++ channels comes from work in that same lab which found that MeHg blocked Ca++ currents, although not completely, in human embryonic kidney cells in culture (Peng et al. 2002).
Altering intracellular calcium levels in brain capillary endothelial cells has a direct effect on blood-brain barrier permeability and transport (Paemeleire et al. 1999). Thus, substances such as organic mercury compounds, which cause the release of intracellularly bound Ca++, might not only have a direct effect on neuronal function, but may also increase the availability of mercury, and possibly other neurotoxicants, in the CNS.
Minnema et al. (1987) also found evidence of MeHg-induced release of mitochondrial calcium from rat brain synaptosomes. In a multi-faceted experiment, these researchers found that MeHg produced large effluxes of calcium from isolated mitochondria preloaded with radio-labeled calcium, but not from synaptosomes preloaded with radio-labeled calcium. They also found that 0.5–5.0 μm MeHg caused a concentration dependent increase in the spontaneous release of tritiated dopamine from striatal synaptosomes, gamma-amino butyric acid (GABA) from cerebellar cortical synaptosomes, and acetylcholine from hippocampal synaptosomes. These releases were determined to not be attributable to a MeHg-increase in calcium permeability of the synaptosomal membrane, since these increases persisted in the absence of extrasynaptosomal calcium (Minnema et al. 1987).
Some of the effects of MeHg intoxication involve the cerebellum, and some of those cerebellar effects are due to the preferential targeting of MeHg. At low concentrations, MeHg has been shown to disrupt the ability of rat cerebellar granule cells in primary culture to maintain the intracellular calcium concentration ([Ca++]i). The mitochondria in cerebellar granule cells can store and release large amounts of Ca++ (Budd and Nicholls 1996). One avenue of Ca++ release is through the mitochondrial permeability transition pore (MTP) (Bernardi et al. 1992; Bernardi and Petronilli 1996). Szalai et al. (1999) reported that, at the beginning of the apoptotic process, apoptotic stimuli induce a switch in mitochondrial calcium signaling by facilitation of a Ca++-induced opening of the MTP. Thus, signals evoked by large Ca++ pulses or IP3-mediated spikes in cytosolic Ca++ trigger mitochondrial permeability. Limke and Atchison (2002) demonstrated that MTP appears to play a significant role in the cellular effects following acute exposure of cerebellar granule neurons to MeHg.
Marty and Atchison (1997) had previously demonstrated that low (0.2–2.0 μM) concentrations of MeHg disrupt the ability of rat cerebellar granule cells to maintain [Ca++]i, resulting in a biphasic increase in [Ca++]i. Further, the initial phase involved release of intracellular Ca++ into the cytosol, followed by a secondary influx of extracellular Ca++. It had also been previously demonstrated that the opening of the MTP could be inhibited by the immunosuppressant cyclosporine A (CsA) (Bernardi et al. 1993). Limke and Atchison (2002) found that exposure to CsA for 10 min prior to the addition of MeHg delayed the time-to-onset of both the first- and second-phase elevations of intracellular Ca++ in a CsA concentration-dependent manner. The delay in both phases suggested to the authors that the two phases of increased intracellular Ca++ were interrelated, with the first phase causing or contributing in some way to the second phase. Limke and Atchison (2002) thus felt that the induction of the MTP may link the first and second [Ca++]i phases.
Kang et al. (2006) used both canine kidney and human neuroblastoma cells to further investigate the mechanism by which MeHg exerts its cytotoxicity. These researchers found that MeHg-induced toxicity was linked to the elevation of [Ca++]i through the activation of phosphatidylcholine-specific phospholipase C (PC-PLC),
To first study the change in [Ca++]i, Kang et al. (2006) loaded the calcium binding dye fura-2 into Marbin-Darby Canine Kidney (MDCK) cells. MeHg was found to increase [Ca++]I in a bimodal manner. Whereas the first phase sharply peaked by ~4-fold at approximately 80 s, the second phase was only gradually increased [Ca++]I by ~2-fold at 400 s. In order to determine whether this increase came from extracellular calcium or intracellular calcium stores, [Ca++]i was measured in the absence of extracellular Ca++. To accomplish this, the calcium binding agent ethylene glycol tetraacetic acid (EGTA) was added to the extracellular medium. In the presence of EGTA, the MeHg-induced increase in [Ca++]i was decreased by ~65% in the first phase and ~30% in the second phase, suggesting that the MeHg-induced increase in [Ca++]i could come from both intracellular and extracellular Ca++ sources. One source is the mobilization of Ca++ from [Ca++]i pools, and the other is through an increase in the permeability of the plasma membrane, as previously suggested by Atchison and Hare (1994).
Kang et al. (2006) also found that the antiviral, anti-tumoural xanthate D609, a competitive inhibitor of PC-PLC, almost completely abolished blocked MeHg-induced cytotoxicity in SH5YSY human neuroblastoma cells in a dose-dependent manner in both of the biphasic phases of [Ca++]i increase, providing strong evidence of the role of PC-PLC as a critical pathway in mediating the MeHg-induced toxicity.
To investigate whether mitochondrial dysfunction is involved in MeHg-induced neurotoxicity, Yin et al. (2007) exposed astrocytes to concentrations of 1, 5, or 10 μM MeHg for 1 or 6 h. Mitochondrial membrane potential was measured by quantification of TMRE fluorescence. The mitochondrial membrane potential was significantly (p < 0.001) reduced both at 1 h and 6 h measurements at all three MeHg concentrations.
Garg and Chang examined the effects of MeHg concentrations from 0.5 to 40 μM on mitochondrial membrane potential in microglia. In this experiment, they observed depolarization of the mitochondrial membrane with increasing MeHg concentrations, but the effect was significant (p < 0.001) only at concentrations of 20 μM or higher.
3.3. Oxidative Stress/Reactive Oxygen Species (ROS)
One demonstrated mechanism of MeHg toxicity is the generation of reactive oxygen species (ROS), which can lead to neuronal cell death. An excessive presence of ROS can trigger cytotoxicity through a mechanism involving the endoplasmic reticulum (ER) (Makino et al. 2014). One function of the ER is to synthesize, fold, and process secretory and transmembrane proteins (Oslowski and Urano 2011). Proteins enter the ER as unfolded polypeptide chains, which fold and mature in the lumen of the ER (Ron and Walter 2007). When unfolded proteins accumulate in the lumen of the ER, a condition called ER stress can occur. This ER stress is created due to an imbalance between the load of unfolded proteins entering the ER and the capacity of the cellular machinery to handle this load (Ron and Walter 2007).
Makino et al. (2014) investigated the means by which methylmercury interacts with an important ER enzyme, protein disulfide isomerase [PDI]. PDI is responsible for catalyzing the formation of disulfide bonds of maturing proteins in the ER. Any agent that interferes with the action of PDI can cause the accumulation of newly synthesized unfolded proteins in the ER, leading to ER stress. Makino and his fellow investigators hypothesized that methylmercury would affect PDI in the ER by covalently bonding with the enzyme and reducing its ability to act. They incubated human neuroblastoma SH-SY5Y cells with various concentrations of MeHg. The effect of MeHg on the cells was measured by an MTT assay [3-(4,5-dimethylthiazol-2-thiazolyl)-2,5diphenyl-tetrazolium bromide (MTT) assay]. The MTT is a tetrazolium reduction coloration assay which examines the ability of the treated cells to metabolize MTT and is used as a measure of their viability. Through this assay, Makino et al. (2014) found a dose-dependent effect of MeHg on the viability of the cells, especially at MeHg concentrations >4 μM MeHg.
To test whether the decrease in viability was tied to ER stress, levels of two indicators of ER stress, CHOP (transcription factor C/EBP homologous protein) and BiP (immunoglobulin binding protein) were measured. These two ER stress indicators were elevated by the presence of MeHg. Matrix-assisted laser desorption/ionization—time-of-flight (MALDI-TOF)/MS assays were also performed to specifically show that MeHg modifies PDI covalently at Cys 383 or 386 via bonding with thiol regions. They also looked at the effects of MeHg on the activity of PDI and found that it causes decreases in this enzyme’s activity. Thus, Makino et al. (2014) proposed that one possible mechanism for neuronal cell death caused by MeHg is the binding of Cys residues of the endoplasmic reticulum enzyme, PDI. This MeHg interference with the action of PDI could lead to ER stress (specifically through a build-up of unfolded protein residues), which if not corrected by the machinery of the cell’s unfolded protein response (UPR) could lead to cell death.
Limke et al. (2004a) examined the role of muscarinic receptors in MeHg-induced dysregulation in rat cerebellar granule cells in vitro through the use of fura-2 single-cell microfluorimetry. When atropine, a non-specific muscarine receptor antagonist, was added to the preparation, the onset of MeHg-induced Ca++ release into the cytosol was significantly delayed. In addition, depletion of smooth endoplasmic reticulum (SER) Ca++ with thapsigargin or down-regulation of muscarinic receptors and inositol-1,3,4-triphosphate (IP3) receptors with bethanechol caused similar reductions in the amplitude of the MeHg-induced Ca++ increase. Collectively, these suggest that MeHg interacts with muscarinic receptors to cause Ca++ release from the SER through activation of IP3 receptors.
Limke et al. (2004a) concluded that the results of their experiments suggested that interactions of MeHg with muscarinic receptors and resultant perturbation of Ca++ regulation within the SER may contribute to the selective vulnerability of cerebellar granule cells. Further, the importance of the SER as a MeHg target may lie in the effect of the released Ca++ on nearby mitochondria, and the localization of specific muscarinic receptor subtypes may contribute to the regional selectivity of MeHg toxicity within the CNS. These conclusions are supported by previous work in the same laboratory (Limke et al. 2003), which suggested that the SER contributes Ca++ to the observed mitochondrial dysregulation and subsequent neuronal death via an MTP-dependent pathway.
Active transport processes essential to the maintenance of the neuronal resting membrane potential require a great amount of the energy-carrying molecule adenosine triphosphate (ATP), which is produced in the mitochondria within the neurons through the oxidative metabolism of glucose. The effect of MeHg on mitochondrial metabolic function was examined by Dreiem and Seegal (2007) using rat striatal synaptosomes. Specifically, the study was designed to determine whether MeHg-induced elevations in reactive oxygen species (ROS) or alterations in intracellular calcium were the cause of compromised mitochondrial function. The formation of ROS was assessed through the use of dichloro-dihydro-fluorescein diacetate (DCFH-DA), a non-fluorescent, cell-permeable compound to form 2′7′-dichlorolfluorescein (DCF) (Myhre et al. 2003). Mitochondrial metabolic function was assessed by the ability to convert the dye methyl-thiazoletetrazolium to formazan. The authors reported that MeHg increased ROS levels, decreased mitochondrial function, and caused an increase in both cytosolic and mitochondrial calcium levels in the synaptosomes. When the synaptosomes-MeHg preparation was co-incubated with Trolox, an antioxidant, the MeHg-induced ROS level was significantly reduced; however, the Trolox failed to restore mitochondrial function. The elevation in calcium levels was found to be independent of extra-synaptosomal calcium. The authors concluded that the MeHg-induced mitochondrial dysfunction is not the result of increased ROS levels, but instead the ROS increase is a secondary event in MeHg toxicity. Further, they suggested that MeHg-induced elevations in mitochondrial calcium are responsible for the mitochondrial damage caused by MeHg (Dreiem and Seegal 2007).
Garg and Chang (2006) examined the effects of MeHg at various concentrations on the murine N9 microglial cell line. They treated microglial cells with various concentrations of MeHg for 30 or 60 min and found that ROS increased in a time-and dose-dependent manner. At concentrations of 20 μM or above, the increase in ROS was statistically significant (p < 0.001). It had previously been shown that the enzyme aconitase is a sensitive intracellular target for oxidative stress (Gardner et al. 1994). Aconitase is an enzyme catalyzing the conversion of citrate to isocitrate in the tricarboxylic acid (or citric acid) cycle, and is thus crucial to the production of the energy-carrying molecule adenosine triphosphate (ATP) from glucose. Garg and Chang (2006) thus performed experiments to study the effects of MeHg on aconitase at concentrations of 0, 5, 10, or 20 μM MeHg. Aconitase activity was found to be impaired by MeHg, with activity dropping by 14% at 5 μM, 53% at 10 μM, and 90% at a MeHg concentration of 20 μM. All decreases were statistically significant (p < 0.01 at 5 μM and p < 0.001 at 10 μM and 20 μM concentrations).
It had been previously reported (Eskes et al. 2002) that MeHg also decreased the production of the cytokine interleukin-6 (IL-6) in microglia. Garg and Chang also conducted experiments to study whether IL-6 production was affected in cultured N9 microglia. These cells were treated with various concentrations of MeHg for 8 h before measuring the production of IL-6 using ELISA. At concentrations of 10 μM, MeHg caused a significant increase in IL-6 production, despite the fact that this concentration greatly inhibited protein synthesis. The authors noted, however, that the role(s) of IL-6 in neurotoxicity is still unknown. Overall, Garg and Chang (2006) concluded that their study demonstrated that exposure to MeHg caused microglial cellular oxidative stress as determined by ROS generation, in addition to changes in mitochondrial membrane potential and aconitase activity.
Yin et al. (2007) studied the ability of MeHg to induce oxidative stress in primary astrocytes by measuring the levels of F2-isoprostanes (F2-Iso-Ps), a lipid peroxidation biomarker of oxidative injury. They found that 1 or 6 h of exposure of primary astrocytes to concentrations of 5 or 10 μM MeHg resulted in significant (p < 0.05) increases in F2-Iso-Ps levels. The highest increases were observed at a concentration of 5 μM for 6 h, suggesting a maximal effect at this concentration. A MeHg concentration of 1 μM did not cause a significant biomarker increase, with F2-Iso-Ps levels indistinguishable from controls.
3.4. Effects on Receptor Binding/Neurotransmitter Release
The muscarinic ACh (mACh) receptor has five different isotopes that regulate a diverse number of motor, sensory, and cognitive neurobehaviors (Basu et al. 2008). And while it is recognized that MeHg can affect the general population of mACh receptors and alter receptor protein levels, relatively little is known about the interaction of MeHg with specific isoforms of that receptor.
Therefore, Basu et al. (2008) investigated the effects of MeHg on muscarinic cholinergic receptor subtypes M1 and M2 using a combination of in vitro competitive binding assays and examination of tissues from MeHg exposed mink. In this study, juvenile male mink (9 per treatment group) were fed diets containing MeHg concentrations of 0, 0.1, 0.5, 1, or 2 μg/g (ppm) diet for 89 days. Animals were then sacrificed and the entire brain was extracted from each skull, after which the occipital cortex and brain stem were dissected from the right hemisphere. Pirenzepine, a selective anti-muscarinic agent, was used in tritiated form to block M1 muscarinic ACh receptors, and [3H]-AFDX-384 was used as an M2 blocker.
In the in vitro tests, MeHg inhibited the binding of radioligands to the M1 and M2 receptors in the occipital cortex and brain stem of the mink. Whereas MeHg inhibition of [3H]-pirenzepine binding to M1 receptors was observed in both the cortex and brain stem, the effect was more pronounced in the cortex. Similarly, [3 H]-pirenzepine in vitro binding results showed that MeHg was more potent at inhibiting ligand binding in the occipital cortex than in the brain stem (Basu et al. 2008).
Conversely, in vivo exposure of the mink to dietary MeHg resulted in greater binding of radioligands to both M1 and M2 receptors (Basu et al. 2008). While an exposure-dependent increase in [3H]-pirenzepine binding was measured in both the occipital cortex and brain stem, only the increase in the cortex was statistically significant (p < 0.01, vs. p = 0.2 in the brain stem). In the case of [3H]-AFDX-384, a MeHg-dependent increase in binding was observed in the occipital cortex of mink exposed to concentrations of 0.5, 1, and 2 ppm MeHg (p < 0.01), but the increase in binding in the brain stem was non-significant (p = 0.1). The authors explained this difference in direction of binding change between in vitro and in vivo studies as being logical. They pointed out that previous neuropharmacologic studies have established that exposure of animals to the muscarinic agonist atropine results in the up-regulation of receptor protein (Wall et al. 1992). The relevance to the Basu et al. (2008) study is straight-forward, as the receptor binding inhibition outside of a whole-body situation could not result in a compensatory increase in protein necessary to make new receptors. However, the response to blockage of receptors in vivo over a period of time would be the compensatory up-regulation of receptors.
In another study, adult female Sprague-Dawley rats were provided MeHg in drinking water at nominal concentrations of 0, 2.5, or 10 μg/L for 16 consecutive days (Coccini et al. 2000). Water consumption and body weights were obtained daily and used to determine daily intakes of 0.5 or 2.0 mg MeHg/kg/day for the 2.5 and 10 μg/L concentrations, respectively. The rats were decapitated either immediately following the dosing period or 14 days after the cessation of MeHg administration. The cerebral cortex, cerebellum, and hippocampus were dissected out and examined for muscarinic receptors radiolabeled with [3H]quinuclidinyl benzilate. The authors found evidence of up-regulation of muscarinic ACh receptors. The density of these receptors was increased in only the cerebellum and hippocampus, but receptor affinity remained unaltered in all three brain areas. While an increase in muscarinic ACh receptors was not initially seen in the cerebral tissue, it was demonstrable in the cerebrum of animals examined two weeks after the termination of treatment.
Coccini et al. (2000) concluded that prolonged ingestion of low doses of MeHg by rats causes subtle molecular changes with adaptive imbalance of muscarinic ACh receptors in the hippocampus and cerebellum. These authors also noted that because cholinergic systems play an important role in learning and memory, as reported by Mash et al. (1985), the increased ACh receptor density caused by MeHg may be a deleterious process.
Yuan and Atchison (2003) conducted experiments using whole-cell recording techniques to examine why cerebellar granule cells are much more sensitive to MeHg than are their neighboring cerebellar Purkinje cells. A specific area of interest was whether the expression of phenotypically different GABAA receptor alpha subunits plays a role in this differential sensitivity. The premise was that if so, the responses of GABAA receptor-mediated inhibitory postsynaptic currents (IPSCs) in Purkinje and granule cells should differ in their response to MeHg. Despite the fact that a range of MeHg bath concentrations was used, the pattern of response was similar. Biphasic changes were observed in frequency and amplitude of both spontaneous IPSCs and miniature IPSCs recorded from Purkinje and granule cells in a concentration and time-dependent fashion. However, the magnitude of the changes in frequency or amplitude of postsynaptic current was independent of the concentration of MeHg, suggesting that either a fairly constant series of events is initiated once an effective MeHg concentration is achieved, or that the concentrations tested were all at the high end of the concentration-response relationship. To use lower MeHg concentrations, however, the authors felt would compromise the high quality of the continuous whole-cell recordings due to the concentration-dependent latent period preceding the time of onset of the response.
While thus unable to prove their hypothesis, Yuan and Atchison (2003) did establish that MeHg acts at both pre- and post-synaptic sites to alter GABAA receptor-mediated inhibitory synaptic transmission. And while the general patterns of effects on the two cell types were similar, GABAA receptors in granule cells did appear to be more sensitive to blockage by MeHg than are those in Purkinje cells, as spontaneous GABAergic currents in granule cells were blocked at an earlier time than were those in Purkinje cells.
To determine whether MeHg interacts specifically with the GABAA receptor, Fonfria et al. (2001) used intact mouse cerebellar granule cells as an in vitro model of neuronal selectivity of that organomercurial. In this study, the authors investigated whether MeHg had an effect on granular cells pretreated for 30 min. with the radioligand [3H]flunitrazepam, and found that binding was increased in a dose-dependent fashion. They further found that this increase was completely blocked by bicuculline and picrotoxinin, both GABAA receptor antagonists, and by the organochlorine pesticide gamma-endosulfan as well. It was also found that the increase in [3H]flunitrazepam binding in the presence of MeHg was independent of intracellular events, such as [Ca++]i, kinase activation/inactivation, or antioxidant conditions. Fonfria et al. (2001) concluded that MeHg interacts with the GABAA receptor by the way of alkylation of SH groups of cysteinyl residues found in GABAA receptor subunit sequences.
The effects of 40 μM, 400 μM, or 4 mM MeHg on the dopaminergic system of the rat striatum in conscious, freely moving animals were studied by Faro et al. (2000). All doses increased dopamine (DA) release (907 ± 31%, 2324 ± 156%, and 9032 ± 70%, for low, middle, and high dose concentrations, respectively). High-dose exposure also caused significant decreases in extracellular levels of the DA metabolites dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (AV). The authors attributed these effects to the MeHg-stimulated DA release, decreased DA intra-neuronal degradation, or both.
Gao et al. (2008) investigated the effects of MeHg on postnatal neurobehavioral development in mice. Mice from the MeHg treatment groups were given intraperitoneal injections of 0.1, 1, or 3 mg/kg MeHg chloride from postnatal days 15 through 17, while control mice were injected with sterile saline. On postnatal day 45, the mice were tested using the Morris water maze to evaluate spatial learning and memory. Significant differences were seen between 1 mg/kg and 3 mg/kg groups and controls with regard to two measured parameters. Twenty-four hours after the final tests, three mice from each group were sacrificed and the hippocampus removed. Increases in NR1, NR2A, and NR2B subunits of the N-methyl-D-aspartate (NMDA) dopamine receptor were expressed in the hippocampus, relative to controls. Expressed as a percentage of control values, subunit NR1 (NMDAR1) was increased by 10% in the 0.1 mg/kg group, 378% in the 1 mg/kg group, and 314% in the 3 mg/kg group mice. NR2A expression was increased by 21% in 0.1 mg/kg mice, 256% in 1 mg/kg mice, and 670% in 3 mg/kg mice, while NR2B expression was enhanced by 59%, 168%, and 252% in the 0.1 mg/kg, 1 mg/kg, and 3 mg/kg groups, respectively. Gao et al. (2008) concluded that the MeHg-induced subtle, but persistent, learning deficits and neurobehavioral abnormalities seen in the mice might be ascribed to alteration of the gene expression of specific NMDA receptor subunits in the hippocampus.
To investigate the role of the dopamine transporter in MeHg-induced dopamine release, Faro et al. (2002) administered adult female Sprague-Dawley rats MeHg dissolved in perfusion fluid and applied locally into the striatum via a dialysis probe. Intrastriatal infusion of 400 μM MeHg increased the extracellular dopamine levels to 1941% of baseline levels (± 199%). The MeHg-induced release of dopamine was not attenuated in the total absence of calcium in the bathing Ringer’s solution, nor was it attenuated after intraperitoneal (i.p.) pre-treatment with reserpine (a depletor of catecholamine stores in brain tissue) or the sodium channel blocker tetrodotoxin (TTX), suggesting that the dopamine release was independent of calcium and vesicular stores, as well as not affected by the blockade of voltage-sensitive sodium channels. Following infusion of KCl (75 mM) through the dialysis probe, Faro et al. (2002) found that MeHg caused a decrease in the KCl-evoked release of dopamine. The authors concluded that collectively, their experiments suggest that MeHg induces the release of dopamine via a transport-dependent mechanism. The MeHg-induced release of dopamine was also independent of calcium and vesicular stores.
It has been known for some time that NMDA receptors (NMDARs) are widely distributed in the mammalian brain and spinal cord, with particularly high densities in the hippocampus and cerebral cortex. Over-activation or long term stimulation can damage and eventually kill target neurons via a process known as excitotoxicity (Cooper et al. 2003). Based on this knowledge, Ndountse and Chan (2008) hypothesized that MeHg binds to NMDARs and induces their expression, ultimately leading to cell death. They tested this hypothesis by investigating the effects of MeHg on the NMDA component of the excitatory neurotransmitter system. Specifically, they investigated the functional roles of NMDARs on the induction of cell death in the SH-SY 5Y neuroblastoma cell line after exposure to MeHg. The stated objective of their study was to investigate the effects of MeHg on the concentration of NMDARs in the membrane of neuronal cells and to explore the mechanism of toxicity involved.
In the Ndountse and Chan (2008) study, binding to the NMDAR was measured using the tritiated antagonist [3H]-MK801. To measure the role of NMDA on the cultured neuroblastoma cells following MeHg application, the NMDAR antagonists MK801 (a.k.a. Dizocipine) and Memantine were used. Specific binding to the NMDARs was defined as the difference in [3H]MK801 bound in the presence and absence of unlabeled MK801 (100 μm). Cytotoxicity was determined using the level of lactate dehydrogenase (LDH) in the supernatant released from the cells. MeHg treatment levels ranged from 0.1 to 5 μM.
Ndountse and Chan found that the cytotoxic effects of MeHg in human SH-SY 5Y neuroblastoma cultured cells were both time- and concentration-dependent. LDH increases of 20%, 100%, and 170% were observed after 4 h of incubation with 1, 2.5, and 5 μM MeHg, respectively. After 24 h of treatment with 0.25, 0.5, 1, or 2.5 μM MeHg, LDH release was increased by 50%, 120%, 150%, and 190%, respectively, compared to control values.
Both of the NMDAR antagonists (100 μM) were found to significantly (p < 0.05) inhibit MeHg-induced neurotoxicity (72% with MK801 and 70% with Memantine). This finding was reported to suggest that the toxicity caused by MeHg was NMDA-dependent. To test for the involvement of Ca++ in the MeHg-induced neurotoxicity, Ndountse and Chan (2008) used the intracellular Ca++ chelator BAPTA-AM. They found that the inhibitory effect of BAPTA-AM on cytotoxicity was around 30%, but not complete. The conclusion of Ndountse and Chan (2008) was that their study demonstrates that MeHg binds and increases NMDARs and induces apoptosis and necrotic cell death on human SH-AY 5Y neuroblastoma cells.
3.5. Effects Involving Arachidonic Acid
Arachidonic acid (AA) is an important component of the phospholipid component of mammalian cell membranes (Stuning et al. 1988). Cultured cerebellar granule cells (Verity et al. 1994) and astrocytes (Shanker et al. 2002) have been used to demonstrate the release of AA in the presence of MeHg. Verity et al. (1994) pre-labeled primary cerebellar granule cultures with tritiated ([3H]) AA and exposed them to MeHg concentrations of 10–20 μM. The result was that MeHg induced [3H]AA release in both a concentration-dependent and time-dependent fashion. Verity et al. (1994) also looked at the role of phospholipase A2 (PLA2) in MeHg-induced cytotoxicity, Cytosolic PLA2 (cPLA2) is important in the maintenance of membrane phospholipids, catalyzing the production of arachidonic acid and affecting its release. They found that PLA2 activation preceded cytotoxicity in the cerebellar granule cells at lower, but not high, MeHg concentrations. Further, when the anti-malarial enzyme inhibitor mepacrine (100 μM) was present in the culture to inhibit MeHg-induced activation of PLA2, the cytotoxicity was not altered. This could mean that either PLA2 activation was not necessary for MeHg-induced cytotoxicity or that mepacrine failed to inhibit MeHg-induced PLA2 activation.
Shanker et al. (2002) conducted a study designed to determine the effects of MeHg on the regulatory expression and activity of astrocytic cPLA2 and AA release and look for a unifying target for MeHg-induced neurotoxicity. The astrocytes were obtained from cerebral cortices from 1-day-old Sprague-Dawley rat pups. The effect of MeHg on the activation of cPLA2 was measured by the lease of [3H]AA from astrocytes over a period of 120 min. Shanker et al. (2002) found a sustained increase in the rate of [3H]AA release in astrocytes treated with 2.5 or 5.0 μM MeHg. This release was statistically significant (p < 0.05) at 10, 30, 60, and 120 min in astrocytes treated with 5.0 μM MeHg, but only at 120 min in cultured astrocytes treated with 2.5 μM MeHg. This was consistent with the time- and concentration-dependent finding of Verity et al. (1994). To test for the involvement of cPLA2 in the observed [3H]AA release following MeHg treatment, Shanker et al. (2002) used the specific cPLA2 inhibitor AACOCF3. They found that pre-treatment of astrocytes for 3 h with 10 μM AACOCF3 reduced the release of [3H]AA following 5 μM MeHg to control levels (“statistically indistinguishable from control astrocytes”). The authors concluded that their results support the notion that cPLA2 stimulated hydrolysis and release of AA play a critical role in MeHg-induced neurotoxicity.
3.6. Effects on Cell Cycle/Cell Division
MeHg has long been known to interfere with the production of new neurons by adversely impacting the biological process of cell division (Rodier et al. 1984). This has been substantiated by a number of studies, including those reviewed next.
Castoldi et al. (2000) exposed in vitro cultures of rat cerebellar granule cells to MeHg concentrations of 0.5–1 μM or 5–10 μM. One hour of exposure to the higher concentration resulted in impairment of mitochondrial function and plasma membrane lysis, resulting in cell death. While the lower (0.5–1 μM) concentrations did not compromise cell viability or mitochondrial function at early time points, neuronal network fragmentation and depolymerization of microtubules were observed within 1.5 h. This damage continued to progress over time, and complete dissolution of microtubules and neuronal processes was seen after 18 h. The authors postulated that cytoskeletal breakdown and deprivation of neurotrophic support may play a role in delayed toxicity following MeHg exposure. Similarly, Miura et al. (1999) studied the relationship between changes in the cell cycle and the induction of apoptosis caused by MeHg in cultured mammalian cells, and reported that GM2/M-phase arrest through the disruption of microtubules is an important event in the development of apoptosis by MeHg. Consistent with altered mitotic division, Bahia et al. (1999) observed a reduction in the frequency of mitotic divisions following in vitro exposure of human lymphoblastoid (TK6) cells to MeHg.
While investigating the possibility that intracellular glutathione (GSH) synthesis may determine sensitivity to MeHg exposure, Ou et al. (1999a) found that while oxidative stress may mediate aspects of MeHg toxicity, disruption of GSH homeostasis alone is not responsible for the sensitivity of embryonic CNS cells to MeHg. In a separate study, Ou et al. (1999b) reported that the activation of cell cycle regulatory genes may be one mechanism by which MeHg interferes with the cell cycle in both adult and developing organisms.
Kim et al. (2007) used SH-SY5Y neuroblastoma cells to investigate whether or not MeHg alters the activity of regulatory proteins involved in the cell cycle. All-trans-retinoic acid (ATRA) was used in this study to induce differentiation of the neuroblastoma cells. The existence of retinoid receptors and related cytoplasmic binding proteins has previously been demonstrated in mammalian models (Maden et al. 1990; Ruberte et al. 1993); and it has been suggested that retinoids have a fundamental morphological action in the mammalian nervous system (Perez-Castro et al. 1989; Maden and Holder 1991). ATRA has been associated with several fundamental aspects of CNS development, including axonal growth (through modulation of nerve growth factor, or NGF), migration of elements of the neural crest (Perez-Castro et al. 1989; Maden and Holder 1991, 1992), and specifying the rostrocaudal position of the forebrain, midbrain, hindbrain, and spinal cord in the developing CNS (Maden and Holder 1992). Two days after ATRA-stimulated differentiation of neuroblastoma cells, Kim et al. (2007) performed cell cycle analysis using flow cytometry. Those researchers reported that MeHg treatment caused a significant change (p < 0.05) in the SH-SY5Y cell cycle. The G1/G0 portion of interphase was reduced in duration, and arrest of the S phase was reported.
Gribble et al. (2005) examined the role of p53, a major regulator of G1 and G2 stages of the cell cycle, in MeHg toxicity using isolated gestation day 14 (GD 14) mouse fibroblasts. In this study, mice heterozygous for the wild-type and null p53 allele were impregnated. At GD14, the pregnant mice were euthanized and gravid uteri were removed. Single cell fibroblast suspensions were subsequently isolated for testing. The fibroblasts were then exposed to sterile water solutions containing 0.25, 1.0, 2.5, or 4.0 μM MeHg. Gribble et al. (2005) found that MeHg caused a non-permanent delay in the G2 phase stage in p53+ cells at MeHg concentration of 1.0 μM and above. There was also an inhibition of G0/G1 cells to progress to G2. At 24 h following treatment, an average of 28% of untreated control cells had progressed to the G2 stage, compared with no progression to G2 in 1.0 and 2.5 μm MeHg treated cells. In addition, it was found that1.0 μM MeHg caused an approximate 3-fold larger percentage of p53+ cells to undergo apoptosis than p53 cells at 24 and 48 h.
In a population of cells allowed to progress to the G2 stage, treatment with a MeHg concentration of 2.5 μM resulted in a time-dependent inability of G2 cells to progress in the cell cycle (Gribble et al. 2005). At 24 h, there was a 17.3% increase in the number of p53+ cells over the untreated cells; and at 48 h, 77.0% of the treated cells remained at the G2 stage, compared with an average of only 7.3% of the untreated G2 cells.
Gribble et al. (2005) concluded that low-dose MeHg causes cell cycle inhibition in the absence of cytotoxicity. Their study results suggest a mechanism involving p53 signaling that may have important implications for neurodevelopmental toxicity associated with MeHg.
Burke et al. (2006) further investigated the phenomenon of cell cycle interference by conducting both in vivo and in vitro studies using Sprague-Dawley rats. In the in vivo study, rats were mated, and the litter birth date was considered to be post-natal day 0 (P0). At day P7, rats were injected subcutaneously with vehicle (controls) or 0.1–30 μg/g MeHg. Rats were sacrificed at 7 h, 24 h, or 2 weeks, and the hippocampus and cerebellum were removed from whole brains. Tissues for cell cultures were obtained at embryonic day 14.5 for cortical precursors or day P7 pups for granule cells. Acute changes in DNA synthesis were evaluated at 7 h post-dosing, using [3H]-thymidine as a marker of DNA synthesis.
Burke et al. (2006) found that the incorporation of [3H]-thymidine decreased as the concentration of MeHg increased above 0.1 μg/g in the hippocampus. However, DNA synthesis in the cerebellum was unchanged across the entire dosage range of MeHg. The authors suggested that this indicated a region-specific response to MeHg, as total Hg levels were very similar in both areas of the brain. They also pointed out that since brain development relies on the production of new neurons, the acute decrease in the synthesis of DNA in the hippocampus might have permanent effects. This is supported by the observation that MeHg exposure on P7 resulted in a decrease in the cell number at P21 by 17%. In contrast, parallel studies of the cerebellum showed no significant change in cell number at 2 weeks post-exposure.
To determine whether the difference in the effects in the two brain regions was the result of differences in cellular characteristics, as opposed to regional differences, Burke et al. (2006) then conducted in vitro experiments. In isolated granule cells from the line that had shown resistance to MeHg in the in vivo studies, [3H] incorporation was decreased as the duration of exposure increased. Whereas 0.3 μM MeHg caused no change at 6 h, [3H]-thymidine incorporation was decreased by 17% at 24 h. Exposure of the granule cells to 3 μM MeHg resulted in a 43% decrease at 6 h and 92% at 24 h, indicating that granule cells are, in fact, responsive to MeHg, and they are not simply resistant to MeHg exposure.
Using bromodeoxyuridine (BrdU) labeling to assess the number of cells in S-phase during the last 2 h of MeHg incubation, Burke et al. (2006) found that in 6 h cerebellar granule cultures, MeHg induced a 73% reduction in [3H]-thymidine incorporation into DNA and a 43% decrease in BrdU labeling. The authors noted that this is consistent with the hypothesis that MeHg inhibits the transition from the G1 to the S phase of cell division. Based on the decrease in BrdU labeling, they also assessed the effects of MeHg on proteins involved in the transition of the G1 to S-phase of the cell cycle. One of these regulatory proteins is cyclin E. Burke et al. (2006) found that cyclin E is specifically targeted by MeHg; thus the effects of MeHg they observed on DNA synthesis may be the result of the MeHg-induced reduction in the availability of cyclin E.
3.7. Effects on Glutathione (GSH) Activity
The brain is particularly sensitive to oxidative injury, in large part due to its high rate of oxidative metabolism (Halliwell 1992). In the cellular cytosol, GSH serves as the major antioxidant, helping to scavenge ROS.
In a study described earlier in this paper, Ndountse and Chan (2008) used GSH levels as an indicator of cytotoxicity, measuring GSH levels in cultured human neuroblastoma cells incubated at MeHg concentrations of 0.1–5 μM. They found that GSH was decreased, even at non-toxic concentrations of MeHg, noting that GSH depletion is an indicator of oxidative stress. They further noted that they had found a good correlation between the observed decrease in GSH and cell death.
In a review of the neurodegenerative effects of mercury and its compounds, Carocci et al. (2014) note that the most important mechanism by which mercury causes toxicity is by mitochondrial damage via depletion of GSH, coupled with thiol binding which in turn generates free radicals. Mori et al. (2007) point out that the function of mitochondrial GSH would be expected to be essentially the same as cytosolic GSH with respect to MeHg toxicity. In this study, both whole tissue GSH and mitochondrial GSH were lower in the rat brains than in the livers, and the same was true of two other antioxidant enzymes, superoxide dismutase (SOD) and glutathione peroxidase (GPX). This occurred despite the higher oxygen consumption and subsequent ROS production in the brain. The authors suggested that the brain mitochondria might therefore function with marginal stability against oxidative stress. Further, the mitochondria of the cerebellum might be particularly sensitive to this, because of their higher oxygen consumption and ROS production than the cerebrum. In fact, the GSH levels measured in mitochondria in the cerebellum of intact rats was as low as one-third that of cerebral mitochondria. Mori et al. (2007) noted that the higher susceptibility to MeHg-induced oxidative damage in brain mitochondria (particularly in the cerebellum) compared with the liver mitochondria might account, at least in part, for the selective toxicity of MeHg. Mori et al. (2007) concluded that their data suggest that the high MeHg-induced activity in mitochondrial ROS generation and low activity in the brain’s defense system, particularly in the cerebellum, would easily cause a critical imbalance in ROS production and at least partially account for the selective neurotoxicity of MeHg.
Franco et al. (2006) examined the exclusive contribution of MeHg exposure through maternal milk on biochemical parameters related to the thiol status in the cerebellums of suckling mice. The thiol status was determined by GSH levels and GPX and glutathione reductase (GR) activity. In this study, 14 Swiss albino mice dams were randomly assigned to one of two groups of seven females each (one treatment and one control group). Pups (eight per litter) were maintained with their mothers, half of which were immediately exposed to MeHg (10 mg/L) in drinking water (treatment group) or to MeHg-free tap water ad libitum (control group). The exclusive route of MeHg exposure for the treated offspring was maternal milk.
Franco et al. (2006) found that the GSH level in the cerebellum was significantly higher (p < 0.05) in the MeHg-exposed dams than in the control rats. In the exposed pups, however, the response was completely the opposite, with MeHg exposure through breast milk resulting in a significant decrease in GSH levels (p < 0.05). Cerebellar GR activity was also significantly higher (p < 0.05) in MeHg exposed dams than in controls, but this increase was not seen in exposed pups. A two-tailed Pearson correlation test revealed a significant positive correlation between cerebellar GR activity and cerebellar GSH in mothers (p < 0.001). MeHg exposure did not affect GPX. Franco et al. (2006) concluded that decreased motor performance in the MeHg-exposed pups may be related to alterations in the cerebellar thiol status. They suggest that the increases in GSH levels and GR activity in the cerebellums of MeHg-exposed dams could represent a compensatory response to the oxidative effects of MeHg toward endogenous GSH. Franco et al. (2006) also suggest that the inability of the pups to perform this compensatory response is probably due to the immaturity of the CNS in the pups, making them more susceptible to the oxidative effects of MeHg on cerebellar thiol status.
The effects of MeHg on microglia were examined using the murine N9 microglial cell line (Garg and Chang 2006). Microglia are macrophage-like cells that make up ~15% of the cell population within the CNS. As such, they are responsible for removal of invading pathogens and are critical to the survival and maintenance of CNS neurons. They also secrete a number of proteins, such as IL-6, which can be either beneficial or detrimental to neuronal cells, depending on the internal conditions. To test for cytotoxicity, the cells were treated with various concentrations of MeHg for one day, and the viability of each treatment subsequently determined. Cytotoxicity was found to appear in a rapid and irreversible manner. Under the conditions of this experiment, MeHg at concentrations of 4, 8, 12, or 16 μM reduced cell viability by 12%, 59%, 85%, and 95%, respectively.
Targets of developmental MeHg exposure include neural cell adhesion molecules (NCAMs), which are sialoglyco conjugates whose proper temporal and spatial expression is important at all stages of neurodevelopment, especially during the formation of synapses. Dey et al. (1999) dosed rat pups subcutaneously with 7.0 mg/kg on every other day from postnatal days 3–13, and investigated the effects of MeHg on the temporal expression of NCAM during development. Postmortem examination of whole-cerebellum homogenates, cerebellar synaptosomes, and isolated cerebellar growth cones collected at postpartum days 15, 30, and 60 was conducted. Golgi sialytransferase activity analysis revealed significant reductions in samples collected at postnatal day 15; however, no such changes were found at postnatal days 30 or 60. In vitro studies revealed decreasing MeHg sensitivity of cerebellar sialytransferases with increasing developmental age. The authors concluded that MeHg-induced perturbation of the developmentally regulated expression of polysialylated NCAM during brain formation may disturb the stereotypic formation of neuronal contacts and contribute to the behavioral and morphologic disturbances seen following MeHg poisoning (Dey et al. 1999).
In a study of the in vivo degenerative effects of methylmercuric chloride on rat brain and cranial nerves, Kinoshita et al. (1999) demonstrated a disturbance in the integrity of microtubules and neurofilaments in the rat nervous system, particularly in the optic nerves. Specifically, electron microscopic examination revealed a marked decrease in microtubules and a moderate decrease of neurofilaments in the myelinated fibers of optic nerves in treated animals.
Faro et al. (2005) investigated the effect of pretreatment with GSH, cysteine, or methionine on MeHg-induced dopamine release from the rat striatum (Faro et al. 2005). Adult female Sprague-Dawley rats were administered a perfusion fluid containing either MeHg, MeHg following GSH pretreatment, MeHg following cysteine pretreatment, or MeHg following methionine pretreatment via a dialysis probe inserted directly into the striatum. The individual concentration of each of the four substances in the perfusion fluid was 400 μM. Infusion of 400 μM MeHg alone resulted in a 1941% (± 199%) increase in extracellular dopamine levels (vs. basal levels). Infusion of 400 μM following GSH pretreatment resulted in an increase in extracellular dopamine of only 465% (± 104%), or 76% less increase than that caused by MeHg alone. MeHg infusion following pretreatment with cysteine resulted in only an increase of 740% (± 149%), or only 62% lower than that induced by MeHg alone. Treatment with MeHg following methionine pretreatment resulted in no significant difference from MeHg alone.
In a study previously described in the current paper, Faro et al. (2005) pointed out that MeHg promotes a decrease in intracellular GSH levels, as previously demonstrated by Choi et al. (1996). Thus, pretreatment with GSH would to some extent ameliorate the impact of MeHg on existing cellular GSH, as found in this study. They also noted that the amino acid-cysteine has a free -SH group, whereas the amino acid methionine has a sulfur atom without the capacity to form -SH groups. Therefore, it is not surprising that the -SH group available in the cysteine pretreated group would bind with MeHg and serve to decrease dopamine release. But since the sulfur atom in methionine was unavailable for binding to MeHg, pretreatment with that amino acid had no effect on MeHg-induced dopamine release (Faro et al. 2005).
3.8. Involvement with Nitric Oxide Synthetase
Shinyashiki et al. (1998) examined the effects of MeHg on cerebral and cerebellar neuronal nitric oxide synthase (nNOS) isoforms in rat brain in vivo and in vitro. Eight week-old male Wistar rats were given subcutaneous doses of 10 mg MeHg/kg/day for 8 days. In vivo manifestation of neurotoxicity was considered to be hind limb crossing, which was evaluated as an indication of paralysis. Five animals were sacrificed each at 1, 2, 5, and 8 days after the first injection and when paralysis was achieved, and the cerebrum and cerebellum of each was removed and examined for MeHg levels. Effects on enzyme activity, interaction of NOS with mercuric compounds via thiols, and involvement of SH groups in MeHg-induced enzyme loss were examined in vitro.
Total Hg concentrations in blood, cerebrum, and cerebellum increased progressively through 8 days of exposure (Shinyashiki et al. 1998). The first hind limb crossing was seen 14.5 days after the first injection. The activity of nNOS increased in both the cerebrum and cerebellum, but in a different manner. In the cerebrum, nNOS activity increased significantly with time and peaked 5 days following the first injection (p < 0.01); it then declined slightly, but statistically significantly, at 8 days (p < 0.01). NOS activity in the cerebellum, however, was significantly increased only after 8 days (p < 0.05). In contrast to the in vivo results, in vitro tests showed a decrease in cerebellar nNOS activity in a concentration-dependent fashion following MeHg-induced covalent modification of thiol groups. Shinyashiki et al. (1998) suggested MeHg causes not only an increase in intracellular calcium, but also in nNOS protein, bringing about overproduction of NO.
Chuu et al. (2001) administered male mice oral gavage MeHg doses of 0.2 or 2.0 mg/kg/day for seven consecutive days. The animals were sacrificed by pentobarbital injection at 0, 5, and 11 weeks following cessation of treatment. Brainstem tissue was assayed for Na+/K+-ATPase activity immediately following the cessation of treatment and from animals sacrificed at 5 and 11 weeks post-treatment. Brainstem tissue and whole blood were also assayed using a nitric oxide (NO)/ozone chemiluminescence assay method. Their tests revealed MeHg-related inhibition of Na+/K+-ATPase activity in the brainstems of mice treated with 0.2 and 2.0 mg/kg MeHg, but this was observable only in those animals sacrificed immediately following treatment cessation; no such effect was seen in animals sacrificed at 5 or 11 weeks post-treatment. A significant (p < 0.05) and irreversible increase in brainstem NO level was seen at all times during the experimental course (0, 5, or 11 weeks post-treatment) at 2.0 mg/kg/day, compared to vehicle controls. No discernible change in nitric oxide level was seen at any post-treatment analysis period in animals receiving the 0.2 mg/kg dose. The authors concluded that high-dose MeHg intoxication is associated with a decrease in functional Na+/K+-ATPase activity in the brainstem of affected animals, secondary to excessive production of NO, leading to oto-toxicity and hearing loss (Chuu et al. 2001).
3.9. Effects on Glial Cells and Glutamate-Glutamine Uptake and Homeostasis
Of the non-neuronal support cells in the CNS, astrocytes are the most abundant. They participate in neurotransmitter synthesis, help to maintain K+ balance, assist in neuronal migration during development, play a prominent role in glutamate-glutamine homeostasis, and are a structural component of the blood-brain barrier.
To identify the mechanisms involved in astrocyte damage due to MeHg exposure, Yin et al. (2007) examined the effects of MeHg on glutamine uptake and expression of glutamine transporters in primary astrocyte cultures. Pre-treatment with 1 μM, 5 μM, or 10 μM MeHg for 30 min resulted in significant (p < 0.001) inhibition of glutamine uptake at both 1 and 5 min measurements at all three concentrations, compared to controls. The degree of inhibition was concentration, but not time, dependent. Differences in glutamate uptake with 10 μM MeHg was also significantly higher (p < 0.001) than uptake with 1 μM (p < 0.001) and 5 μM (p < 0.05) at both 1 and 5 min.
Yin et al. (2007) also investigated the molecular mechanisms associated with the effects of MeHg on glutamine uptake in cultured astrocytes by measuring the astrocytic amino acid transporter mRNAs by RT-PCR. Glutamine uptake is dependent upon several sodium-dependent transporters, including SNAT1, SNAT3, and ASCT2. MeHg treatment at 10 μM MeHg (but not at 1 μM or 5 μM) significantly (p < 0.05) reduced the mRNA expression of SNAT3 and ASCT2, but not SNAT1, when compared to controls. Yin et al. (2007) concluded that their data, when taken collectively, demonstrate an association between MeHg exposure and increased mitochondrial membrane permeability, alterations in glutamine/glutamate cycling, and oxidative injury resulting from increased ROS formation. They further suggested that the ultimate effect of MeHg is the initiation of multiple additive or synergistic mechanisms of a disruptive nature that lead to cellular dysfunction and cell death (Yin et al. 2007).
Farina et al. (2003a) examined the effects of MeHg on glutamate uptake by brain cortical slices. Two-month-old male Swiss Albino mice were given MeHg chloride equivalent to doses of 0, 10, or 40 mg/kg MeHg chloride in drinking water for 15 days and then sacrificed and the cerebral cortex removed for experimentation. The 40 mg/kg equivalent dosage, but not the 10 mg/kg dose, significantly (p = 0.013) decreased glutamate uptake in the cortical slices.
To evaluate the effects of in vivo MeHg exposure on glutamate release from synaptosomes and glutamate uptake by brain cortical slices, Farina et al. (2003b) divided 48 suckling Wistar rat pups into two groups of equal size. The pups received daily subcutaneous (s.c.) injections of MeHg dissolved in a 25 μM NaHCO3 solution beginning on post-natal day 3 (PND 3). The 24 MeHg-treated rat pups were divided in three groups of eight pups each that were sacrificed on PNDs 10, 17, and 24 for preparation of synaptosomal and cortical preparations. The 2 mg/kg dosage was based on a prior study by Miyamoto et al. (2001), in which the vulnerability of developing cortical neurons to MeHg was examined. Control rats (n=24) received daily s.c. injections of a 25 μM NaHCO3 solution.
In another paper, Farina et al. (2003b) found that glutamate release increased with age in both control and treated animals. The control pups sacrificed on PND 24 had significantly (p < 0.05) elevated glutamate release levels as compared to the 10- and 17-day sacrificed animals. The 24-day-old MeHg-treated pups had glutamate release levels significantly higher than the 24-day controls (p < 0.05) and the 10- and 17-PND-sacrificed MeHg-treated pups (p < 0.05). In contrast with the increase in glutamate with age and MeHg treatment, glutamate uptake by cortical neurons was found to decrease with age in both controls and MeHg-treated animals. In cortical slices from pups sacrificed on PND 24, there was a significant (p < 0.05) increase (56%) compared with age-matched controls. These authors concluded that their data suggest that the effect of MeHg on glutamate release from presynaptic nerve terminals could be involved in its neurotoxicity in suckling rats, and that the observed increase in glutamate uptake could correspond to a pathologic response to MeHg. Farina et al. (2003b) went on to suggest that it is possible that the neurologic deficits seen in MeHg-exposed children might be related, at least in part, to a MeHg-induced disturbance of glutamate homeostasis.
Due to the effects of MeHg on the brains of infants whose mothers had been exposed to MeHg during pregnancy, Manfroi et al. (2004) examined the effects of MeHg in weanling mice that had been exposed to MeHg only from the breast milk of their mothers. To do so, glutamate uptake by cerebellar slices from weanling mice was measured and compared with uptake from cerebellar slices from their mothers. While no differences found in glutamate uptake between exposed and control mothers, a significant (p< 0.05) decrease in glutamate uptake was observed in exposed weanlings, when compared to controls.
3.10. Other Mechanisms
Calpain is a Ca++-dependent cysteine peptidase (protease) that was first identified in rat brain by Guroff (1963). It has been reported that deregulation of calpain activity can result in tissue damage and is associated with events such as stroke and brain trauma (Goll et al. 2003). Calpain degrades many specific substrates, including microtubule-associated protein 2, α-fodrin, and p35.
Lee et al. (2000) reported that the calpain/cyclin-dependent kinase 5 cdk5/p35 cascade is important in neuronal cell apoptosis. The cdk5 activator, specifically expressed in neuronal cells, causes a cleavage of p35 to p25, which is followed by the redistribution and overactivation of cdk5 in the presence of p25. Caplain-induced conversion of p35 to p25 leads to the formation of a cdk5-p25 complex, which results in the hyperphosphorylation and degradation of many proteins, including tau and neurofilaments (Nath et al. 2000).
Sakaue et al. (2005) explored a possible mechanism by which MeHg leads to cell death in rat cerebellar granule cells. Their hypothesis was that MeHg activates the calpain-dependent cascade and leads to granular cell death in the cerebellum. To investigate this, they looked specifically at the calpain/cyclin dependent kinase (cdk5/p35) cascade which, when activated, leads to neuronal cell apoptosis. In their studies, cultured cerebellar granule neurons prepared from Wistar rats were treated with MeHg at concentrations ranging from 0 to 1 μM (1000 nM) for 24 or 48 h with or without calpain inhibitor II. Intracellular Ca++ concentrations were determined using the fluorescent Ca++ indicator, Fluo-3 (Fluo-3 acetoxymethyl ester), which specifically binds to calcium ions.
Sakaue and colleagues found that MeHg induced death in cerebellar granule cells in a dose dependent manner. In investigating the first stage of the calpain-activated cell death, which is an upsurge in intracellular calcium after MeHg treatment, the intracellular calcium increased in a dose-dependent manner. Even at the low dosage of 30 nM MeHg, significant (p < 0.05) cell death was observed. However, treatment with calpain inhibitor II restored the viability of cells treated with 30 nM or 100 nM MeHg from 76% to 89% and from 43% to 60%, respectively. Thus, the inhibitory effect of calpain inhibitor II was demonstrated to be statistically significant (p < 0.05), but incomplete.
Sakaue et al. (2005) also investigated the effect of MeHg on rat neuroblastoma cells for 48 h, and found that the sensitivity of the neuroblastoma cells to MeHg was less than the sensitivity of cerebellar granule cells. The viability of the B35 cells was found to be significantly (p < 0.05) decreased only at MeHg concentrations above 300 nM.
Sakaue et al (2005) also measured the cleavage of α-fodrin, a known calpain substrate, and found that it occurred in a dose-dependent manner with the addition of MeHg. They further noted that calpain inhibitor II did not completely stop the cascade that lead to calpain activated apoptosis, although it did reduce it. Those authors concluded that the calpain cascade may be one of the mechanisms that contribute to microtubule degradation that leads to cell death.
Although tau proteins are necessary for microtubule stabilization, Sakaue et al. reported that it was improbable that the phosphorylation of tau contributed to MeHg-induced cell death at very low concentrations. Rather, proteins containing cytoskeletons seemed to degrade and cause dysfunction via calpain-digestion in the neuronal cells treated with a very low dose of MeHg (Sakaue et al. 2005).
4. Ethylmercury: Mechanisms of Toxic Action
Ethylmercury is the other alkyl mercury compound discussed in this paper. While there has been considerable interest in ethylmercury in recent years due to its presence in the body as the result of the in vivo metabolism of the preservative thimerosal, almost all of the data regarding its metabolism have come from the studies of thimerosal and not ethylmercury itself.
Sodium ethylmercuric thiosalicylate (thimerosal; Merthiolate) is a mercury-containing preservative used in some multi-dose vials of vaccines and other products since the 1930s. It has the chemical name ethyl(2-mercaptobenzoato-(2-)-O,S) mercurate(1-) sodium and contains approximately 49% mercury by volume. Thimerosal is broken down by in vivo metabolic processes to ethylmercury (EtHg) and salicylic acid. Thimerosal is quickly metabolized in vivo, due to its reactions with protein and non-protein thiols (Wu et al. 2008).
4.1. Mobilization of Intracellular Ca++
Thimerosal has been shown to be a versatile sulfhydryl reagent, a mobilizer of intracellular calcium, and a modulator of cell function (Elferink 1999). It has been shown to induce the release of intracellular calcium stores in many cell types. This mobilization of calcium can in turn modulate a number of cell functions. Tornquist et al. (1999) found that thimerosal mobilized sequestered calcium and evoked modest store-dependent calcium entry in thyroid FRTL-5 cells. The mechanism of action was suggested to be mediated via activation of protein kinase C, as thimerosal potently stimulated the bonding of [3H]phorbol 12,13-dibutyrate and was without effect on store-operated calcium entry in cells treated with staurosporine or in cells with down-regulated protein kinase C. Whole-cell patch clamping experiments revealed that thimerosal did not depolarize the membrane potential. It was concluded that thimerosal attenuates any increase in internal calcium ion concentration, probably by activating a plasma membrane Ca++-ATPase (Tornquist et al. 1999).
Thimerosal has also been shown to be a potent activator of intracellular calcium release in pig oocytes. Such activation mimics the effects of sperm-induced release of intracellular calcium, as well as other activation events that occur in pig oocytes (Machaty et al. 1999). Wang et al. (1999) examined the temporal relationship between intracellular calcium transients, cortical granule exocytosis, and the zona reaction induced by thimerosal. These researchers found that thimerosal induced the same degree of exocytosis in oocytes that was caused by sperm penetration. Further, the zona block to sperm penetration in thimerosal-treated oocytes occurred within 35 min of cortical granule exocytosis and within 40 min of the first calcium transient. Machaty et al. (1999) found that the thimerosal-induced Ca++ release did not require the formation of IP3. In addition, thimerosal destroyed the meiotic spindle, preventing further development.
Calcium channels in skeletal muscle, cardiac muscle, and certain nerve fibers have a high affinity for the plant alkaloid ryanodine (Sitsapesan and Williams 2000). The receptors for which ryanodine has this particular affinity are known as ryanodine receptors (RyR). Eager and Dulhunty (1999) found that thimerosal reacts with specific cysteine residues on RyR, contributing to either activation or inhibition of the channel, depending on the domain and particular class of cysteine associated with that receptor.
Using whole-cell patch clamping to study the effects of thimerosal on tetrodotoxin (TTX) sensitive and TTX-resistant sodium channels in dorsal root ganglion neurons, Song et al. (2000) found that thimerosal blocked the two channel types in a dose-dependent fashion. The inhibition was considerably more pronounced in the TTX-resistant channels, but the effect was not reversed in either case with washing with thimerosal-free solution. The thimerosal-induced inactivation of both types of sodium channels would serve to diminish neuronal activity.
As previously noted in this paper, inositol 1,4,5-triphosphate (IP3) is involved in intracellular calcium homeostasis; and the binding of this compound to the inositol triphosphate receptor (IPR) is modulated by a number of compounds, including MeHg. IP3 plays its vital role in calcium homeostasis through its action as a second messenger responsible for the release of Ca++ from intracellular stores following receptor activation. Further, binding to the IPR is modulated by a number of compounds, including MeHg. In an earlier study, Sayers et al. (1993) investigated the mechanism by which thimerosal affects the biochemical properties of intracellular Ca++ pumps and channels using rat cerebellar microsomes and rabbit skeletal muscle and fluorescence monitoring techniques. The effect of up to 50 μM thimerosal on IP3 binding was assessed by the addition of thimerosal to cerebellar microsomes incubated with 40 nM [3H]IP3. In the rat cerebellar microsomes, a concentration-dependent inhibition of Ca++ uptake was observed, but 75 μM thimerosal was required for complete inhibition. In the rabbit skeletal muscle, the rate of Ca++ uptake by the sarcoplasmic reticulum was progressively decreased with increasing concentrations of thimerosal, with nearly complete inhibition occurring at thimerosal concentrations over 2 μM. Upon the addition of the Ca++ ionophore A23187 to enhance the permeability of the cell membrane to Ca++, Ca++-ATPase activity was completely inhibited by thimerosal, indicating that thimerosal had a direct, rather than non-specific, effect on the Ca++-ATPase. Thimerosal concentrations up to 50 μM had no effect on IP3 binding or on IP3 metabolism in the cerebellar microsomal preparation. Sayers et al. (1993) concluded that the opening of the IP3-sensitive Ca++ channel is a complex event, and that new models to explain IP3-induced Ca++ release were necessary.
Since the report of Sayers et al. (1993), other researchers have investigated the effects of thimerosal on IP3-mediated Ca++ release. Vanlingen et al. (1999) reported that the binding of IP3 to its membrane receptors can be differentially modulated by thimerosal. Using a preparation of cerebellar microsomes, these researchers found that thimerosal had a stimulatory effect on the binding of IP3 to IP3R1 receptors. Green et al. (1999) reported that the sensitivity of intracellular calcium stores to IP3 increases the affinity of the IP3 receptor in rat hepatocytes; and thimerosal was further shown to enhance agonist-specific differences in the oscillation of intracellular calcium in rat hepatocytes. Mason and Mahaut-Smith (2001) also reported the voltage-dependent Ca++ release in rat megakaryocytes following sensitization of IP3 receptors with thimerosal.
Bultynck et al. (2004) compared the functional and molecular effects of thimerosal on IP3R1 and IP3R3 receptors. Using a culture of A7r5 embryonic rat aorta cells, which express primarily the IP3R1 isoform, they found that thimerosal produced a modulated biphasic effect on the IP3induced Ca++ release and IP3-binding activity of IP3R1 receptors. Thimerosal (1 μM) was found to strongly potentiate the IP3-induced Ca++ release, and this was additive to the potentiation by the Ca++ itself. The authors stated that the additive effect of thimerosal and Ca++ may indicate that both agents cooperate to induce a conformational change to the IP3R1 receptor that is much more sensitive to activation by IP3. Bultynck et al. (2004) also studied the effects of thimerosal on R23–11 cells in culture. R23–11 cells are IP3R knockout cells derived from DT40 chicken B lymphoma cells by homologous recombination. These latter experiments showed that thimerosal potentiated IP3-induced Ca++ release and the IP3 binding activity of IP3R1 in triple IP3R knockout R23–11 cells, but not in the IP3R3 isoform, which lacks the N-terminal suppressor domain. Bultynck et al. (2004) concluded that, collectively, their data revealed a thimerosal-dependent intramolecular interaction within the N-terminal domain of IP3R1, and that this may be at least a part of the conformation changes occurring during the desensitization of IP3R1 by thimerosal. Further, IP3 may induce opening of the channel pore by modifying the interaction of the C-terminal channel domain with the N-terminal IP3-binding domain.
4.2. Effects on Mitochondria (Membrane and ROS)
Much of the increase in intracellular Ca++ is attributable to its release from the mitochondria (Sharpe et al. 2012). Sharpe et al. (2012) investigated the effects of ethylmercury (from thimerosal) on the mitochondria of normal human astrocytes, paying particular attention to mitochondrial function and the generation of specific antioxidants. These investigators pointed out that the types and levels of antioxidant enzymes of human astrocytes are somewhat different from most other cell types.
In their experiments, Sharpe and his co-investigators incubated astrocytic cells for 1 h, before performing fixation of the cells. Thimerosal solutions were prepared to a maximum concentration of 360 μM, and 10 μL were added to a 240 μL astrocytic volume. Additional 10 μL aliquots were added at 10-min intervals. Measurements of DNA, oxidized DNA bases, and blunt-ended breaks were also performed. Concentration dependence was determined by adding Thimerosal concentrations of 0–14.4 μM to the cell medium at t = 0, 10, 20, 30, 40, and 50 min, before fixation at 60 min. ROS generation was measured by dichlorohydrofluorescein (DCF) fluorescence in isolated mitochondria.
Treatment with 14.4 μM thimerosal caused ~50% decrease in mitochondrial membrane potential at 1 h, along with a 200% increase in mitochondrially generated oxidants. Time course evaluation of ethylmercury-induced changes revealed that the generation of ROS is an early event in the course of events leading to apoptosis and occurred prior to changes in the mitochondrial membrane potential. Further, thimerosal-treated astrocytic mitochondria were observed to generate four times the amount of ROS as control mitochondria. In comparison, steady state generation of ROS in areas with no mitochondria was unchanged.
Sharpe et al. (2012) reported a 5-fold increase in levels of oxidant-damaged mitochondrial DNA and increases in levels of mito-DNA nicks and blunt-ended breaks. Mitochondrial DNA was found to be far more vulnerable to ethylmercury-induced damage, compared with nuclear DNA. These researchers observed a 240 % increase in the levels of mitochondrial DNA breaks, a 300 % increase in 3′OH DNA nicks, and a 460 % increase in the levels of oxidized bases (apurinic or apyrimidinic sites). Since mitochondrial DNA is localized within the matrix, it was concluded that this is the main site of ROS generation.
Sharpe et al. (2012) also noted that EtHg not only inhibits mitochondrial respiration, leading to a drop in the steady state membrane potential, but also causes concurrent increases in the formation of superoxide, hydrogen peroxide, and Fenton/Haver-Weiss generated hydroxyl radical.
4.3. Effects on Arachidonic Acid, Leukotriene Synthesis, and Membrane Integrity
The levels of free AA in cells are known to be maintained by a balance between hydrolysis from membrane phospholipids by phospholipase A2 and reincorporation into phospholipids catalyzed by the L-type neutral amino acid carrier transport (LAT) system (Chilton et al. 1996). Thimerosal has been shown to be involved in the liberation and prolonged availability of arachidonic acid (AA) and AA metabolites in various mammalian tissues and cells (Chen et al. 2003; Hatzelmann et al. 1990; Kaever et al. 1988; Stuning et al. 1988; Zarini et al. 2006). Kaever et al. (1988) stated that the availability of arachidonic acid represents the fundamental prerequisite of cellular eicosanoid synthesis.
To examine the effects of thimerosal on μμLAT in human neutrophils, Zarini et al. (2006) isolated neutrophils from whole human blood, and incubated neutrophil microsomes and a mixture of standard lysophospholipids with AA Co-A in the presence or absence of 50 μM thimerosal. In the absence of thimerosal, the corresponding diacylphospholipids were formed. However, when the thimerosal was added to the incubated microsomes 2 min prior to the addition of the lysophospholipids, an almost complete inhibition of the formation of phospholipids was observed. Subsequent experiments in which the thimerosal was removed prior to the addition of the lysophosphate mixture revealed that the thimerosal-induced inhibition was irreversible.
Zarini et al. (2006) concluded that irreversible covalent binding of thimerosal to LAT inhibits activity of the LAT enzyme and the reacylation of AA into membrane phospholipids.
Arachidonic acid (AA) is an important constituent of the phospholipid component of mammalian cell membranes, and is present in esterified form in phospholipids and triglycerides in the membranes (Stuning et al. 1988). In vivo, 5-lipoxygenase in the cytosol translocates to the nuclear membrane and associates with 5-lipoxygenase activating protein (FLAP), which is believed to act as an AA transfer protein that presents the substrate to the 5-lipoxygenase (Hardin and Limbird 2001).
In the lab, AA can also be released from membranes upon cell activation by the Ca++-ionophore A23187. After release, AA may undergo oxidative metabolism through the enzyme cyclooxygenase, resulting in the formation of prostaglandins; through the cytochrome P450 system to produce a variety of metabolites including 19- or 20-hydroxy arachidonate and eicosatrienoic acids; or through lipoxygenases, including 5-lipoxygenase. The 5-lipoxygenase is one of the most important lipoxygenases, since it leads to the synthesis of leukotrienes (Hardin and Limbird, 2001). (See Fig. 1).
Fig. 1.
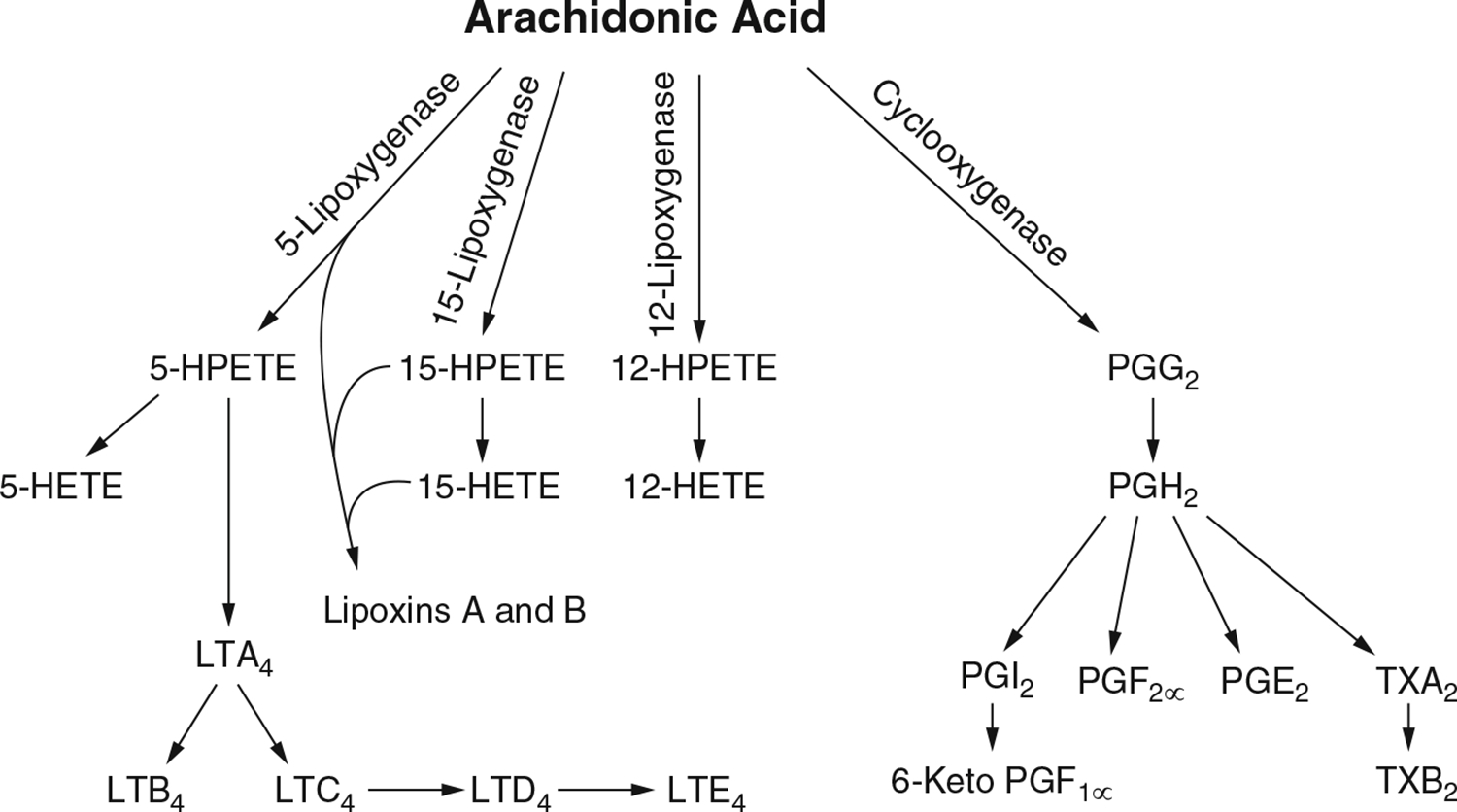
Pathways of leukotriene and prostaglandin synthesis from arachidonic acid (AA). Source: Collectively from Samuelsson (1982, 1983), and Samuelsson et al. (1987)
Because of its strong affinity of EtHg for SH groups, thimerosal is known to cause the inhibition of, among other enzymes, 5-lipoxygenase. In the presence of thimerosal, the metabolism of AA by human polymorphonuclear leucocytes resulted in the inhibition of 5-lipoxygenase, acetyltransferase, and the omega-oxidative system.
Stuning et al. (1988) investigated the effects of thimerosal on the transformation of AA via the 5-lipoxygenase pathway in human leukocytes stimulated with A23187. In their experiments, they collected blood from healthy human donors to isolate polymorphonuclear leukocytes (PMNs), which they incubated with Ca++-ionophore (usually 7.3 μM) in the presence of 2 mM Ca++. Thimerosal dissolved in phosphate buffered saline (PBS) was then added at times ranging from 0 to 0.5 min before stimulation, then allowing the reaction to proceed for 10 min. at 37 °C. In other experiments, 14C-labeled AA was given simultaneously with the stimulus. Separately, the metabolism of 3H-labeled LTB4 was analyzed using HPLC.
Stuning et al. (1988) found that the polymorphonuclear leukocytes (PMN) stimulated with the Ca-ionophore A23187 released a variety of arachidonic acid metabolites in a concentration dependent fashion. These metabolites included LTB4, LTC4, and several analogs of LBT4.
Thimerosal was found to exert a profound effect of the rate of formation of LTB4. Pre-incubation of the cell suspension with thimerosal 0–5 min prior to the stimulation with the Ca++-ionophore did not significantly alter the effect of thimerosal. Investigation of the effects of thimerosal on reacylation of AA into phospholipids and the formation of 5-hydroperoxyeicosatetraenoic acid (5-HPETE) and 5-hydroeicosatetraenoic acid (5-HETE) revealed that formation of labeled phospholipids, 5-HPETE, and 5-HETE was also decreased with increasing amounts of thimerosal. The concentration of the Ca++-ionophore stimulus did not significantly influence the effects of thimerosal.
The inhibition of the omega-oxidation system by thimerosal was further confirmed in experiments in which PMNs were incubated with (3H)LTB4. Pre-incubation with thimerosal at a concentration >31 μM, previously demonstrated to cause a marked decrease in the formation of omega-oxidation products, resulted in a 4.3 (± 0.3)-fold decrease in omega oxidation of (3H)LTB4 (Stuning et al. 1988). Stuning et al. (1988) further suggested that the interaction of environmental mercury with biological systems may have important implications regarding the efficacy of leukotriene synthesis.
4.4. Effects on Glutathione (GSH)
Muller et al. (2001) investigated the interaction of thimerosal with glutathione-S-transferases T1 (GST T1), an enzyme expressed solely in human erythrocytes and which displays genetic polymorphisms. Blood samples were collected from three (2 male, 1 female) healthy Caucasian adults previously identified as being either a non-conjugator of GST T1 (no enzyme activity), a normal conjugator (medium GST T1 activity), or super-conjugator (high GST T1 activity). Next, in vitro experiments were conducted using human erythrocytic and polymorphic GST T1 in the presence of a methyl chloride (MeCl) substrate and using an HPLC-fluorescence detection assay. In the case of the non-conjugator, thimerosal showed no effect due to the absence of GST T1. For the normal conjugator, a 2.77 mM thimerosal concentration (1.12 mg/L) was found to inhibit 50% of GST T1 activity; and 2.3 mM (0.93 mg/L) thimerosal resulted in a 50% inhibition in the blood of the super-conjugator. A 14.8 mM (6 mg/L) thimerosal concentration led to residual enzyme activities equal to those of the non-conjugator.
When Muller et al. (2001) used a 0.6 mM concentration of thimerosal and 10,000 ppm MeCl concentration in the incubation medium, a 30% inhibition of GST T1 was observed for both the normal and super-conjugator blood, compared with no effect for the non-conjugator blood. The authors concluded that the observed enzyme kinetics suggested a non-competitive inhibition of the human erythrocytic GST T1 enzyme and that sufficiently high concentrations of thimerosal may be able to change the phenotypic status of an individual by inhibition of the GST T1 enzyme, at least in vitro.
Wu et al. (2008) studied the importance of GSH in modulating the activity of thimerosal in experiments using human leukemia K562 cells and found that thimerosal undergoes an exchange reaction with cysteine, GSH, and human serum albumin and forms an EtHg adduct with single stranded DNA. In addition, the effect of thimerosal as a poisoning mechanism for topoisomerase H-alpha was also investigated as a mechanism of cytotoxicity in dividing cells. Topoisomerase is required to catalyze the double-stranded cleavage of DNA, allowing passage of a second DNA duplex through the break. The decatenation of DNA is known to be a topoisomerase H-specific reaction (Haldane and Sullivan 2001).
In their experiments, Wu et al. (2008) mixed 500 μL of 100 μM thimerosal with a similar quantity and concentration of cysteine in 50% methanol (v/v) at room temperature for 5 min, followed by the addition of 10 μL of 6% acetic acid (v/v). The same reaction of thimerosal with cysteine was carried out, with the exception of the addition of acetic acid. Wu et al. (2008) determined the ability of thimerosal to inhibit topoisomerase using a spectrofluorometric decantation assay as described in Hasinoff et al. (2006).
Wu et al. (2008) found that the reaction of thimerosal with cysteine produced a cysteine-EtHg adduct, and the reaction of thimerosal with GSH produced a GSH-thimerosal adduct, collectively confirming the ability of thimerosal to react with cysteine or GSH to produce a thiol-EtHg adduct and thiosalicylate. Given the high affinity of EtHg for thiols and the involvement of topoisomerase in the cleavage of double-stranded DNA, Wu et al. (2008) decided to investigate whether thimerosal could, in part, exert its cytotoxicity by inhibiting topoisomerase H-alpha through reacting with its free cysteine groups. In this part of their study, they found that thimerosal strongly inhibited the decatenation activity of purified topoisomerase H-alpha.
As the next step, Wu et al. (2008) decided to test whether GSH could protect isomerase H-alpha from the inhibitory effects of thimerosal. By adding various concentrations of GSH top topoisomerase H-alpha prior to thimerosal treatment, they found that an intracellular GSH level of 0.1–5 mM completely protected topoisomerase H-alpha decatenation activity from 5 to 20 μM thimerosal-induced inhibition.
In investigating the binding of thimerosal to DNA to induce DNA damage as a mechanism of cytotoxicity, Wu et al. (2008) found that treatment if K562 cells with thimerosal for 2 h resulted in a concentration-dependent reduction in remaining double-stranded DNA that achieved significance at all thimerosal concentrations of 50 of μM or higher, compared with cells not treated with thimerosal. From this, the authors concluded that thimerosal rapidly induces DNA damage in K562 cells. Wu et al. (2008) determined that these results suggest that the EtHg-thimerosal adduct did not significantly exchange with the free cysteines on topoisomerase H-alpha to inhibit it, and that the protective effect of GSH is consistent with thimerosal inhibiting topoisomerase H-alpha by binding to its free cysteines.
To assess the effects of lowering GSH levels on thimerosal-induced cytotoxicity, K562 cells in exponential growth were seeded (i.e., spread in a defined amount of cell suspension onto a plate) to yield a cell count of approximately 12,000 cells per well. At 24 or 48 h after seeding, cells were treated in the presence or absence of either 5 mM ()-2-oxo-4-thiazolidinecarboxylic acid (OTC) or 100 μM buthionine sulfoximine (BSO) and allowed to grow another 24 or 48 h, before treatment with vehicle or various concentrations of thimerosal, and allowed to grow another 72 h. (OTC is a non-thiol precursor of cysteine that is converted intracellularly to cysteine, whereas BSO is known to inhibit the rate-limiting enzyme for GSH synthesis.). The levels of OCT and BSO used in this experiment did not cause measurable cytotoxicity. However, BSO or OTC pretreatments followed by exposure to various concentrations of thimerosal for 72 h were remarkable. Cell growth was inhibited by thimerosal with an IC50 value of 2.0 for untreated cells. For the OTC-treated cells, this value was increased 1.3-fold. For the BSO-treated cells, the LC50 value was reduced 16.7-fold. A t-test of three IC50 determinations showed that the small OTC-dependent increase in IC50 was not significant (p = 0.22), whereas the BSO-dependent decrease in IC50 was highly significant (p < 0.0001), leading Wu et al. (2008) to conclude that GSH partly protected the thimerosal-treated cells from growth inhibition.
Overall, Wu et al. (2008) concluded that their studies showed that thimerosal reacts rapidly with cysteine, GSH, HSA, and single-stranded DNA to form EtHg adducts detectable by mass spectrometry. Further, they showed thimerosal to be a potent inhibitor of the decantation activity of topoisomerase H-alpha, and that depletion of GSH with BSO greatly increased the growth-inhibitory effects of thimerosal.
4.5. Effects on Glutamate Transport
Mutkus et al. (2005) studied the effects of thimerosal on the transport of the non-metabolizable glutamate analog [3H]-D-aspartate in Chinese hamster ovary (CHO) cells. In these experiments, the CHO cells were transfected with two glutamate transporter subtypes: GLAST (EAAT1) and GLT-1 (EAAT2) (EAAT=excitatory amino acid transport system). Mutkus et al. (2005) also report the results of studies to determine the effects of thimerosal on mRNA and protein levels of the glutamate transporters. The mutant CHO-K1 cell line DbB7 was chosen for their study since those cells possess essentially no endogenous Na+-dependent glutamate transport.
When GLAST- and GLT-1 transfected CHO cells were treated with 0, 10, or 20 μM thimerosal, none of the treatment levels caused a discernable change in GLAST mRNA expression. Under the same conditions, however, GLT-1 mRNA expression was significantly (p < 0.05) in 10 μM treated cells (126% of controls); but no such change was observed in 20 μM-treated cells.
In tests to examine glutamate transporter protein levels in thimerosal-treated CHO cells, Mutkus et al. (2005) found that 10 μM thimerosal caused a statistically significant (p < 0.001) increase in GLAST protein expression (198% of controls); however, 20 μM thimerosal GLAST levels were not significantly different from controls. In contrast, GLT-1 protein levels were reported to decrease in a concentration-dependent manner in 10 and 20 μM thimerosal-treated CHO cells. However, the 10 μM cells were not distinguishable from controls, whereas the 20 μM-treated cells showed significant (p < 0.001) reductions at 20 μM treatment levels.
In another experiment, Mutkus et al. (2005) examined [3H]aspartate uptake following 6 h of pretreatment with 0, 10, or 20 μM thimerosal, followed by measurement of [3H]aspartate uptake during the subsequent 1, 5, 15, and 30 min. These experiments demonstrated that thimerosal caused a significant decrease in the uptake of [3H]aspartate in transfected GLAST- and GLT-1 CHO cells. GLAST activity was significantly inhibited (p < 0.01) at the 1- and 5-min measurements with both 10 and 20 μM thimerosal. In the 10 μM treated cells, [3H]aspartate uptake had recovered, but the decrease remained significant (p < 0.01) in 20 μM-treated cells. At the 30-min measurement, both 10 and 20 μM cells were back to control levels. The effects of thimerosal on [3H]aspartate uptake in GLT-1 transfected cells was even more pronounced. The reduction in uptake was significant (p < 0.01) at all measurement times with both 10 and 20 μM treatment cells, with the sole exception of the 10 μM cells at the 30-min measurement.
The results of the Mutkus et al. (2005) experiments indicated that thimerosal treatment caused significant, selective changes in both glutamate transporter mRNA and protein expression in CHO cells. Thus, those authors concluded that their studies suggested that the application of thimerosal to the central nervous system might contribute to dysregulation of glutamate homeostasis.
4.6. Effects on Neurotransmitter Availability
Ida-Eto et al. (2011) administered thimerosal (1 mg/kg) intramuscularly to pregnant rats on gestational day 9, reported to be a susceptible time for the development of the fetal serotonergic system. Fetal serotonergic neurons were assessed at embryonic day 15, using anti-serotonin antibodies. A 1.9-fold increase in the number of serotonergic neurons in the lateral portion of the caudal raphe was observed in thimerosal group compared to control values (p < 0.01). The authors concluded that their results indicated that embryonic exposure to thimerosal affects early development of serotonergic neurons.
To put the results of Ida-Eto et al. (2011) in proper perspective, the amount of mercury in a single syringe of vaccine from a multi-dose vial containing thimerosal as a preservative is ~0.25 μg/injection. If this is divided by the weight of a female human (assumed to weigh 60 kg), the dose to a pregnant woman would be ~0.43 μg/kg bw, as opposed to the 1 mg/kg injected into pregnant rats by Ida-Ito et al. Thus, the dosage administered to the rats was ~2500× as high as the human dosage. This study shows that sufficiently high doses of thimerosal/EtHg injected at precisely the critical time in development can enhance overall serotonin (5-hydroxytryptamine, or 5-HT) availability; however, the specific relationship to amounts of thimerosal administered at any time in pregnancy is unclear. In addition, the unrealistically high mercury doses themselves limit the relevance of the Ida-Eto findings to potential mechanism, and not necessarily to the toxicity of mercury from thimerosal in vaccines.
In a follow-up study, Ida-Eto et al. (2013) administered thimerosal to pregnant rats at dosages of 1.0, 0.1, or 0.01 mg Hg/kg by intramuscular injection on embryonic day 9. At the 1 mg Hg/kg dosage, most of the pups were dead soon after birth. No major anomalies, growth retardation, or reduced number of delivered were seen at the other two dosages. On day 50 of life, male offspring were killed and serotonin (5-HT), dopamine (DA), and their metabolites 5-hydroxyindoleacetic acid (5-HIAA), 3,4-dihydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA) concentrations in brain tissue were measured. Females were not used, since brain serotonin levels are influenced by the estrus cycle. Concentrations of hippocampal 5-HT and striatal DA in the male brains were measured by HPLC. Hippocampal 5-HT was significantly increased in both the 0.01 mg Hg/kg (p < 0.05) and 0.1 mg Hg/kg (p < 0.01) treatment groups, compared with controls. Striatal DA levels were also significantly increased in the 0.01 mg Hg/kg (p < 0.05) and 0.1 mg Hg/kg (p < 0.01).treatment groups vs. controls.
Ida-Eto et al. (2013) also found that the 5-HT metabolite 5-HIAA was increased in the 0.01 mg Hg/kg treated group, but the increase was not significant when compared with controls. The increase in the 0.1 mg Hg/kg vs. controls was significant (p < 0.05), however. The concentrations of striatal DOPAC and HVA were not significantly statistically different from controls. Those authors concluded that embryonic exposures to thimerosal at the concentrations tested produced lasting impairment of the brain monoaminergic system.
5. Summary and Conclusions
There are many commonalities/similarities in the mechanisms of toxic action of methylmercury and ethylmercury (from thimerosal). Thimerosal is quickly metabolized in vivo, due to its reactions with protein and non-protein thiols (Wu et al. 2008), so the effects of thimerosal reported in numerous articles is very likely the result of exposure to the metabolite ethylmercury.
Evidence for the similarity of the various mechanisms of toxicity include the following:
Both MeHg and EtHg bind to the amino acid cysteine (Clarkson 1995; Wu et al. 2008).
Both MeHg and EtHg promote the release of arachidonic acid, resulting (on a potentially positive side) in a marked increase in leukotriene production. However, both MeHg and EtHg strongly inhibit the reacylation of arachidonic acid, thus inhibiting the reincorporation of this fatty acid into membrane phospholipids (Shanker et al. 2002; Verity et al. 1994; Zarini et al. 2006).
Both decrease glutathione activity, thus providing less protection from the oxidative stress caused by MeHg and EtHg (Carocci et al. 2014; Ndountse and Chan (2008); Choi et al. 1996; Franco et al. 2006; Mori et al. 2007; Muller et al. 2001; Ndountse and Chan 2008; Wu et al. 2008).
Both cause an increase in NOS, causing an overproduction of NO (Chen et al. 2003; Chuu et al. 2001; Shinyashiki et al. 1998).
Both disrupt glutamate homeostasis (Farina et al. 2003a, b; Manfroi et al. 2004; Mutkus et al. 2005; Yin et al. 2007).
Both cause oxidative stress/creation of ROS (Dreiem and Seegal 2007; Garg and Chang 2006; Myhre et al. 2003; Sharpe et al. 2012; Yin et al. 2007).
Both alter intracellular calcium homeostasis (Elferink 1999; Hare et al. 1993; Kang et al. 2006; Limke et al. 2004b; Machaty et al. 1999; Marty and Atchison 1997; Minnema et al. 1987; Peng et al. 2002; Sayers et al. 1993; Sirois and Atchison, 2000; Szalai et al. 1999; Tornquist et al. 1999; Zarini et al. 2006).
Both cause effects on cell division by damaging the spindle apparatus during mitosis (Burke et al. 2006; Castoldi et al. 2000; Gribble et al. 2005; Kim et al. 2007; Ou et al. 1999b; Machaty et al. 1999; Rodier et al. 1984).
Both cause effects on receptor binding/neurotransmitter release involving one or more transmitters (Basu et al. 2008; Coccini et al. 2000; Cooper et al. 2003; Fonfria et al. 2001; Ida-Eto et al. 2011; Ndountse and Chan 2008; Yuan and Atchison 2003).
Both cause DNA damage or impair DNA synthesis (Burke et al. 2006; Sharpe et al. 2012; Wu et al. 2008).
The difference in toxicity between MeHg and EtHg is likely the result of the more rapid metabolism and elimination of EtHg than MeHg and the amount of the mercurial form to which significant exposure is most likely to occur (i.e., MeHg exposure from frequent fish consumption vs. very small and widely spaced exposure to EtHg through thimerosal in multi-dose vials of vaccine).
Footnotes
Disclaimer The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Agency for Toxic Substances and Disease Registry.
References
- Aschner M, Aschner JL (1990) Mercury neurotoxicity: mechanisms of blood-brain barrier transport. Neurosci Biobehav Rev 14(2):169–176 [DOI] [PubMed] [Google Scholar]
- Atchison WD, Hare MF (1994) Mechanisms of methylmercury-induced neurotoxicity. FASEB J 8:622–629 [DOI] [PubMed] [Google Scholar]
- Bahia MO, De Amorim MI, Burbano RR, Vincent S, Dubeau H (1999) Genotoxic effects of mercury on in vitro cultures of human cells. An Acad Bras Cienc 71:437–443 [PubMed] [Google Scholar]
- Bakir F, Damluji SF, Amin-Zaki Murtadha M, Khalidi A, al-Rawi NY, Tikriti S, Dahahir HI, Clarkson TW, Smith JC, Doherty RA (1973) Methylmercury poisoning in Iraq. Science 181:230–241 [DOI] [PubMed] [Google Scholar]
- Basu N, Scheuhammer AM, Rouvinen-Watt K, Evans RD, Grochowina N, Chan LHM (2008) The effects of mercury on muscarinic cholinergic receptor subtypes (M1 and M2) in captive mink. Neurotoxicology 29:328–334 [DOI] [PubMed] [Google Scholar]
- Bearss JJ, Limke TL, Atchison WD (2001) Methylmercury (MeHg) causes calcium release from smooth endoplasmic reticulum (SER) inositol-1,4,5-triphosphate receptors in rat cerebellar granule neurons. Toxicology 60:184 [Google Scholar]
- Bernardi P, Petronilli V (1996) The permeability transition pore as a mitochondrial calcium release channel: a critical appraisal. J Bioenerg Biomembr 28:131–138 [DOI] [PubMed] [Google Scholar]
- Bernardi P, Veronese P, Petronilli V (1993) Modulation of the mitochondrial cyclosporine A-sensitive permeability transition pore. I. Evidence for two separate Me2+ binding sites with opposing effects on the pore opening probability. J Biol Chem 268:1005–1010 [PubMed] [Google Scholar]
- Bernardi P, Vassanelli S, Veronese P, Colonna R, Szabo I, Zorati M (1992) Modulation of the mitochondrial permeability transition pore. Effect of protons and divalent cations. J Biol Chem 267:2934–2939 [PubMed] [Google Scholar]
- Budd SL, Nicholls DG (1996) A reevaluation of the role of mitochondrial in neuronal Ca2+ homeostasis. J Neurochem 66:403–411 [DOI] [PubMed] [Google Scholar]
- Bultynck G, Szlufcik K, Nadif Kasri N, Assefa Z, Callewaert G, Missiaen L, Parys JB, De Smedt H (2004) Thimerosal stimulates Ca2+ flux through inositol 1,4,5-trisphosphate receptor type 1 but not type 3 via modulation of an isoform-specific Ca2+-dependent intramolecular interaction. Biochem J (published 11 Mar 2004 as ms BJ20040072):1–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke K, Cheng Y, Li B, Petrov A, Joshi P, Bermqn R, Reuhl K, DiCicco-Bloom E (2006) Methylmercury elicits rapid inhibition of cell proliferation in the developing brain and decreases cell cycle regulator, cyclin e. Neurotoxicology 27(6):970–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carocci A, Rovito N, Sinicropi MS, Gehchi G (2014) Mercury toxicity and neurodegenerative effects. Rev Environ Contam Toxicol 229:1–18 [DOI] [PubMed] [Google Scholar]
- Castoldi AF, Barni S, Turin I, Gandini C, Manzo L (2000) Early acute necrosis, delayed apoptosis and cytoskeletal breakdown in cultured cerebellar granule neurons exposed to methylmercury. J Neurosci Res 59:775–787 [DOI] [PubMed] [Google Scholar]
- CDC (2015) Centers for Disease Control and Prevention. Vaccines do not cause autism. http://www.cdc.gov/vaccinesafety/concerns/autism.html
- Chen YJ, Jiang H, Quilley J (2003) The nitric oxide- and prostaglandin-independent component of the renal vasodilator effect of thimerosal is mediated by epoxyeicosatrienoic acids. J Pharmacol Exp Ther 304(3):1292–1298 [DOI] [PubMed] [Google Scholar]
- Chilton FH, Fonteh AN, Surette ME, Triggiani M, Winklaer JD (1996) Control of arachidonate levels within inflammatory cells. Biochim Biophys Acta 1299:1–15 [DOI] [PubMed] [Google Scholar]
- Choi BH, Yee S, Robles M (1996) The effects of glutathione glycoside in methylmercury poisoning. Toxicol Appl Pharmacol 141(2):357–364 [DOI] [PubMed] [Google Scholar]
- Chuu J-J, Hsu C-J, Lin-Shiau S-Y (2001) Abnormal auditory brainstem responses for mice treated with mercurial compounds: involvement of excessive nitric oxide. Toxicology 162:11–22 [DOI] [PubMed] [Google Scholar]
- Clarkson TW (1995) Environmental contaminants in the food chain. Am J Clin Nutr 61 (3):682s–686s [DOI] [PubMed] [Google Scholar]
- Clarkson TW (1992) Mercury: major issues in environmental health. Environ Health Perspect 100:31–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson TW, Magos L (2006) The toxicology of mercury and its chemical compounds. Crit Rev Toxicol 36(8):609–662 [DOI] [PubMed] [Google Scholar]
- Clarkson TW, Vyas JB, Ballatori N (2007) Mechanisms of mercury disposition in the body. Am J Ind Med 50:757–764 (Review article) [DOI] [PubMed] [Google Scholar]
- Coccini T, Randine G, Candura SM, Nappi RE, Prockop LD, Manzo L (2000) Low-level exposure to methylmercury modifies muscarinic cholinergic receptor binding characteristics in rat brain and lymphocytes: physiologic implications and new opportunities in biologic monitoring. Environ Health Perspect 108(10):29–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JR, Bloom FE, Roth RH (eds) (2003) The biochemical basis of neuropharmacology, 8th edn. Oxford University Press, Oxford [Google Scholar]
- Cossaboon JM, Ganguli PM, Flegal AR (2015) Mercury offloaded in Northern elephant seal hair affects coastal seawater surrounding rookery. Proc Natl Acad Sci U S A 112(39):12058–12052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey PM, Gochfeld M, Reuhl KR (1999) Developmental methylmercury administration alters cerebellar PSA-NCAM expression and Golgi sialyltransferase activity. Brain Res 845:139–151 [DOI] [PubMed] [Google Scholar]
- Dreiem A, Seegal RF (2007) Methylmercury-induced changes in mitochondrial function in striatal synaptosomes are calcium-dependent and ROS-independent. Neurotoxicology 28:720–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll CT, Mason RP, Chan HM, Jacob DJ, Pironne N (2013) Mercury as a global pollutant: sources, pathways, and effects. Environ Sci Technol 47:4967–4983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eager KR, Dulhunty AF (1999) Cardiac ryanodine receptor activity is altered by oxidizing reagents in either luminal or cytoplasmic solution. J Membr Biol 167:205–214 [DOI] [PubMed] [Google Scholar]
- Elferink JGR (1999) Thimerosal: a versatile sulfhydryl reagent, calcium mobilize, and cell function-modulating agent. Gen Pharmacol 33:1–6 [DOI] [PubMed] [Google Scholar]
- Eskes C, Honegger P, Juillerat-Jeanneret L, Monett-Tschudi F (2002) Microglial reaction induced by noncytotoxic methylmercury treatment leads to neuroprotein via interactions with astrocytes and IL-6 release. Glia 37:43–52 [DOI] [PubMed] [Google Scholar]
- Farina M, Frizzo MES, Soares FAA, Schwalm FD, Detrich MO, Zeni G, Rocha JBT, Souza DO (2003a) Ebselen protects against methylmercury-induced inhibition of glutamate uptake by cortical slices from adult mice. Toxicol Lett 144:351–357 [DOI] [PubMed] [Google Scholar]
- Farina M, Dahm KCS, Schwalm FD, Brusque AM, Frizzo MES, Zeni G, Souza DO, Rocha JBT (2003b) Methylmercury increases glutamate release from brain synaptosomes and glutamate uptake by cortical slices from suckling rat pups: modulatory effect of ebselen. Toxicol Sci 73:135–140 [DOI] [PubMed] [Google Scholar]
- Faro LRF, do Nascimento JLM, Campos F, Vidal L, Alfonso M, Duran R (2005) Protective effects of glutathione and cysteine on the methylmercury-induced striatal dopamine release in vivo. Life Sci 77:444–451 [DOI] [PubMed] [Google Scholar]
- Faro LRF, do Nascimento JLM, Alfonso M, Duran R (2002) Mechanism of action of methylmercury on in vivo striatal dopamine release: possible involvement of dopamine transporter. Neurochem Int 40:455–465 [DOI] [PubMed] [Google Scholar]
- Faro LR, do Nascimento JL, San Jose JM, Alfonso M, Durán R (2000) Intrastriatal administration of methylmercury increases in vivo dopamine release. Neurochem Res 25:225–229 [DOI] [PubMed] [Google Scholar]
- Faustman EM, Ponce RA, Ou YC, Mendoza MAC, Lewandowski TL, Kavanagh T (2002) Investigations of methylmercury-induced alterations in neurogenesis. Environ Health Perspect 110(Suppl 5):859–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfria E, Rodrigues-Farre E, Sunol C (2001) Mercury interaction with the GABAA receptor modulates the benzodiazepine binding site in primary cultures of mouse cerebellar granule cells. Neuropharmacology 41:819–833 [DOI] [PubMed] [Google Scholar]
- Franco JL, Teixeira A, Meotti FC, Ribas CM, Stringari J, Pomblum SCG, Moro AM, Bohrer D, Bairos AV, Dafre AL, Santos ARS, Farina M (2006) Cerebellar thiol status and motor deficit after lactational exposure to methylmercury. Environ Res 102:22–28 [DOI] [PubMed] [Google Scholar]
- Gao Y, Ding W, Shi R, Tian Y (2008) Effects of methylmercury on postnatal neurobehavioral development in mice. Neurotoxicol Teratol 30(6):462–467 [DOI] [PubMed] [Google Scholar]
- Gardner PR, Nguyen DD, White CW (1994) Aconitase is a sensitive and critical target of oxygen poisoning in cultured mammalian cells and in rat lungs. Proc Natl Acad Sci U S A 91:12248–12252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg TK, Chang IY (2006) Methylmercury causes oxidative stress and cytotoxicity in microglia: attenuation by 15-deoxy-delat 12, 14-prostaglandin J2. J Neuroimmunol 171(1–2):17–28 [DOI] [PubMed] [Google Scholar]
- Goll DE, Thompson VF, Li H, Wei W, Cong J (2003) The caplain system. Physiol Rev 83(3):731–801 [DOI] [PubMed] [Google Scholar]
- Green AK, Cobbold PH, Dixon CJ (1999) Thimerosal enhances agonist-specific differences between [Ca2+]i oscillations induced by phenylephrine and ATP in single rat hepatocytes. Cell Calcium 25:173178. [DOI] [PubMed] [Google Scholar]
- Gribble EJ, Hong S-W, Faustman EM (2005) The magnitude of methylmercury-induce cytotoxicity and cell arrest is p53-dependent. Birth Def Res A Clin Mol Teratol 73(1):29–38 [DOI] [PubMed] [Google Scholar]
- Guroff G (1963) A neutral calcium-activated proteinase from the soluble fraction of rat brain. J Biol Chem 239:149–155 [PubMed] [Google Scholar]
- Haldane H, Sullivan DM (2001) DNA topoisomerase II-catalyzed DNA decantation. In: Osheroff N, Bjornsti M-A (eds) Methods of molecular biology 95: DNA topoisomerase protocols: enzymology and drugs. Humana Press Inc., Totowa, NJ [Google Scholar]
- Halliwell B (1992) Reactive oxygen species and the central nervous system. J Neurochem 59(5):16091623. [DOI] [PubMed] [Google Scholar]
- Hardin JG, Limbird LE (2001) In: Hardin JG, Limbird LE (eds) Goodman and Gilman’s the pharmacological basis of therapeutics, 10th edn. McGraw-Hill Medical Publishing Division, New York [Google Scholar]
- Hare MF, McGinnis KM, Atchison WD (1993) Methylmercury increases intracellular concentrations of Ca++ and heavy metals in NG108–15 cells. J Pharmacol Exp Ther 266(3):1626–1635 [PubMed] [Google Scholar]
- Hasinoff BB, Wu X, Yalowich JC, Goodfellow B, Laufer RS, Adedayo O, Dmitrienko GI (2006) Kinamycins A and C, bacterial metabolites that contain an unusual diazo group, as potential new anticancer agents: antiproliferative and cell effects. Anticancer Drugs 17:825–837 [DOI] [PubMed] [Google Scholar]
- Hatzelmann A, Haurand M, Ullrich V (1990) Involvement of calcium in the thimerosal-stimulated formation of leukotriene by fMLP in human polymorphonuclear leukocytes. Biochem Pharmacol 39(3):559–567 [DOI] [PubMed] [Google Scholar]
- Hughes WL (1957) A physiochemical rationale for the biologic activity of mercury and its compounds. Ann N Y Acad Sci 65:454–460 [DOI] [PubMed] [Google Scholar]
- Ida-Eto M, Oyabu A, Ohkawara T, Tashiro Y, Narita N, Narita M (2013) Prenatal exposure to organomercury, thimerosal, persistently impairs the serotonergic and dopaminergic systems in the rat brain: Implications for association with developmental disorders. Brain Dev 35:261–264 [DOI] [PubMed] [Google Scholar]
- Ida-Eto M, Oyabu A, Ohkawara T, Tashiro Y, Narita N, Narita M (2011) Embryonic exposure to thimerosal, an organomercury compound, causes abnormal early development of serotonergic neurons. Neurosci Lett 505:61–64 [DOI] [PubMed] [Google Scholar]
- Jalili HA, Abbasi AH (1961) Poisoning by ethyl mercury toluene sulfonanilide. Br J Ind Med 18:303–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaever V, Goppelt-Strube M, Resch K (1988) Enhancement of eicosanoid synthesis in mouse peritoneal macrophages by the organic mercury compound thimerosal. Prostaglandins 35(6):885–902 [DOI] [PubMed] [Google Scholar]
- Kang MS, Jeong JY, Seo JH, Jeon HJ, Jung KM, Chin M-R, Moon C-K, Bonventre JV, Jung SY, Kim DK (2006) Methylmercury-induced toxicity is mediated by enhanced intracellular calcium through activation of phosphatidylcholine-specific phospholipase C. Toxicol Appl Pharmacol 216:206–215 [DOI] [PubMed] [Google Scholar]
- Kim Y-J, Kim Y-S, Kim M-S, Ryu J-C (2007) The inhibitory mechanism of methylmercury on differentiation of human neuroblastoma cells. Toxicology 234(1–2):1–9 [DOI] [PubMed] [Google Scholar]
- Kinoshita Y, Ohnishi A, Kohaki K, Yokota A (1999) Apparent diffusion coefficient on rat brain and nerves intoxicated with methylmercury. Environ Res 80:348–354 [DOI] [PubMed] [Google Scholar]
- Kutsuna M (ed) (1968) Minamata disease. Study group of Minamata disease. Kumamatio University, Japan, pp 1–4 [Google Scholar]
- Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH (2000) Neurotoxicity induces cleavage of p35 to p25 by caplain. Nature 405:360–364 [DOI] [PubMed] [Google Scholar]
- Limke TL, Heidemann SR, Atchison WD (2004a) Disruption of intraneuronal divalent cation regulation by methylmercury: Are specific targets involved in altered neuronal development and cytotoxicity in methylmercury poisoning? Neurotoxicology 25:741–760 [DOI] [PubMed] [Google Scholar]
- Limke TL, Bearss JJ, Atchison WD (2004b) Acute exposure to methylmercury causes Ca2+ dysregulation and neuronal death in rat cerebellar granule cells through an M3 muscarinic receptor linked pathway. Toxicol Sci 80:60–68 [DOI] [PubMed] [Google Scholar]
- Limke TL, Otero-Montanez JK, Atchison WD (2003) Evidence for interactions between intracellular calcium stores during methylmercury-induced intracellular calcium dysregulation in rat cerebellar granule neurons. J Pharmacol Exp Ther 304:949–958 [DOI] [PubMed] [Google Scholar]
- Limke TL, Atchison WD (2002) Acute exposure to methylmercury opens the mitochondrial permeability transition pore in rat cerebellar granule cells. Toxicol Appl Pharmacol 178:52–61 [DOI] [PubMed] [Google Scholar]
- Machaty Z, Wang WH, Day BN, Prather RS (1999) Calcium release and subsequent development induced by modification of sulfhydryl groups in porcine oocytes. Biol Reprod 61:1384–1391 [DOI] [PubMed] [Google Scholar]
- Maden M, Holder N (1992) Retinoic acid and development of central nervous system. Bioessays 14:431–438 [DOI] [PubMed] [Google Scholar]
- Maden M, Holder N (1991) The involvement of retinoic acid in the development of the vertebrate central nervous system. Development Suppl. 2:87–94 [PubMed] [Google Scholar]
- Maden M, Ong DE, Chytil F (1990) Retinoid-binding protein distribution in the developing mammalian nervous system. Development 109(1):75–80 [DOI] [PubMed] [Google Scholar]
- Makino K, Okuda K, Sugino E, Nishiya T, Toyama T, Iwawaki T, Fujimura M, Kumagai Y, Uchara T (2014) Correlation between attenuation of protein disulfide isomerase activity through S-mercuration and neurotoxicity induced by methylmercury. Neurotox Res 27:99–105. doi: 10.1007/s12640-014-9494-8 [DOI] [PubMed] [Google Scholar]
- Manfroi CB, Schwalm FD, Cereser V, Abreu F, Oliveira A, Bizarro L, Rocha JBT, Frizzo MES, Souza DO, Faroma M (2004) Maternal milk as methylmercury source for suckling mice: neurotoxic effects involved with the cerebellar glutamatergic system. Toxicol Sci 81:172–178 [DOI] [PubMed] [Google Scholar]
- Marty MS, Atchison WD (1997) Pathways mediating Ca2+ entry in rat cerebellar granule cells following in vitro exposure to methyl mercury. Toxicol Appl Pharmacol 147:319–330 [DOI] [PubMed] [Google Scholar]
- Mash DC, Flynn DD, Polter LT (1985) Loss of M2 muscarinic receptors in the cerebral cortex in Alzheimer’s disease and experimental cholinergic denervation. Science 228:1115–1117 [DOI] [PubMed] [Google Scholar]
- Mason MJ, Mahaut-Smith MP (2001) Voltage-dependent Ca 2+ release in rat megakaryocytes requires functional IP3 receptors. J Physiol 533:175–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnema DJ, Cooper GP, Greenland RD (1987) Effects of methylmercury on neuro-transmitter release from rat brain synaptosomes. Toxicol Appl Pharmacol 99(3):510–521 [DOI] [PubMed] [Google Scholar]
- Miura K, Koide N, Himeno S, Nakagawa I, Imura N (1999) The involvement of microtubular disruption in methylmercury-induced apoptosis in neuronal and nonneuronal cell lines. Toxicol Appl Pharmacol 160:279–288 [DOI] [PubMed] [Google Scholar]
- Miyamoto K, Nakanishi H, Moriguchi S, Fukuyama N, Eto K, Wakammiya J, Murao K, Arimura K, Osame M (2001) Involvement of enhanced sensitivity of N-methyl-D-aspartate receptors in vulnerability of developing cortical neurons to methylmercury neurotoxicity. Brain Res 901:252–258 [DOI] [PubMed] [Google Scholar]
- Mori N, Yasutake A, Hirayama K (2007) Comparative study of activities in reactive oxygen species production/defense system in mitochondria of rat brain and liver, and their susceptibility to methylmercury toxicity. Arch Toxicol 81(11):769–778 [DOI] [PubMed] [Google Scholar]
- Muller M, Westphal G, Vesper A, Bunger J, Hallier E (2001) Inhibition of the human erythrocytic glutathione-S-transferase T1 (GST T1) by thimerosal. Int J Hyg Environ Health 203 (5–6):479–481 [DOI] [PubMed] [Google Scholar]
- Mutkus L, Aschnr JL, Syversen T, Shanker G, Sonnewald U, Aschner M (2005) In vitro uptake of glutamate in GLAST- and GLT-1-transfected mutant CHO-K1 cells is inhibited by the ethylmercury-containing preservative thimerosal. Biol Trace Elem Res 105(1–3):71–86 [DOI] [PubMed] [Google Scholar]
- Myhre O, Andersen JM, Aarnes H, Fonnum F (2003) Evaluation of the probes 2′7′-dichlorofluoresein diacetate, luminal, and lucigenin as indicators of reactive species formation. Biochem Pharmacol 65(10):1575–1582 [DOI] [PubMed] [Google Scholar]
- Nath R, Davis M, Probert AW, Kupina NC, Ren X, Schielke GP, Wang KW (2000) Processing of cdk5 activator p35 to its truncated form (p25) by calpain in acutely injured neuronal cells. Biochem Biophys Res Commun 274:16–21 [DOI] [PubMed] [Google Scholar]
- Ndountse LT, Chan HM (2008) Methylmercury increases N-methyl-D-aspartate receptors on human SH-SY 5Y neuroblastoma cells leading to neurotoxicity. Toxicology 249:251–255 [DOI] [PubMed] [Google Scholar]
- Oppedisano F, Pochini L, Broer S, Indiveri C (2011) The B°AT1 amino acid transporter from rat kidney reconstituted in liposomes: kinetics and inactivation by methylmercury. Biochim Biophys Acta 1808:2551–2558 [DOI] [PubMed] [Google Scholar]
- Oppedisano F, Galluccio M, Indiveri C (2010) Inactivation by Hg2+ and methylmercury of the glutamine/amino acid transporter (ASCT2) reconstituted in liposomes: prediction of the involvement of a CXXC motif by homology modeling. Biochem Pharmacol 80:1266–1273 [DOI] [PubMed] [Google Scholar]
- Oslowski CM, Urano F (2011) Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol 490:71–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou YC, White CC, Krejsa CM, Ponce RA, Kavanagh TJ, Faustman EM (1999a) The role of intracellular glutathione in methylmercury-induced toxicity in embryonic neural cells. Neurotoxicology 20:793–804 [PubMed] [Google Scholar]
- Ou YC, Thompson SA, Ponce RA, Schroeder J, Kavanagh TJ, Faustman EM (1999b) Induction of the cell cycle regulatory gene p21 (WAF1, C1P1) following methylmercury exposure in vitro and in vivo. Toxicol Appl Pharmacol 157(3):203–212 [DOI] [PubMed] [Google Scholar]
- Palacin M, Estevez R, Bertran J, Zorzano A (1998) Molecular biology of mammalian plasma membrane amino acid transporters. Physiol Rev 78:969–1054 [DOI] [PubMed] [Google Scholar]
- Paemeleire K, de Hemptinne A, Leybaert L (1999) Chemically, mechanically, and hyperosmolarity induced calcium responses of rat cortical capillary endothelial cells in culture. Exp Brain Res 126:473–481 [DOI] [PubMed] [Google Scholar]
- Peng S, Hajela RK, Atchison WD (2002) Effects of methylmercury on human neuronal L-type calcium channels transiently expressed in human embryonic kidney cells (HEK-293). J Pharmacol Exp Ther 302(2):424–432 [DOI] [PubMed] [Google Scholar]
- Perez-Castro AV, Toth-Rogler LE, Wei L-N, Nguyen-Huu MC (1989) Spatial and temporal pattern of expression of the cellular retinol-binding protein during mouse embryogenesis. Proc Natl Acad Sci U S A 86(22):8813–8817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pochini L, Peta V, Indiveri C (2013) Inhibition of the OCTN2 carnitine transporter by HgCl(2) and methylmercury in the proteoliposome experimental model: insights in the mechanism of toxicity. Toxicol Mech Methods 23(2):68–76 [DOI] [PubMed] [Google Scholar]
- Randall A, Tsien RW (1995) Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J Neurosci 15:2995–3012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier PM, Aschner M, Sager PR (1984) Mitotic arrest in the developing CNS after prenatal exposure to methylmercury. Neurobehav Toxicol Teratol 6:379–385 [PubMed] [Google Scholar]
- Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8:519–529 [DOI] [PubMed] [Google Scholar]
- Ruberte E, Friederich V, Chambon P, Morriss-Kay G (1993) Retinoic acid receptors and cellular retinoid binding proteins. III. Their differential transcript distribution during mouse nervous system development. Development 118(1):267–282 [DOI] [PubMed] [Google Scholar]
- Sakaue M, Okazaki M, Hara S (2005) Very low levels of methylmercury induce cell death of cultured rat cerebellar neurons via calpain activation. Toxicology 213:97–106 [DOI] [PubMed] [Google Scholar]
- Samuelsson B (1982) From studies of biochemical mechanisms to novel biological mediators: prostaglandin endoperoxides, thromboxanes and leukotrienes. Nobel Lecture, December 8, 1982. Department of Physiological Chemistry, Karolinska Institutet, S-104 01 Stockholm, Sweden: [DOI] [PubMed] [Google Scholar]
- Samuelsson B (1983) Leukotrienes: mediators of immediate hypersensitivity reactions and inflammation. Science 220:568–575 [DOI] [PubMed] [Google Scholar]
- Samuelsson B, Dahlen S-E, Lindgren JA, Rouzer CA, Serhan CN (1987) Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science 237:1171–1176 [DOI] [PubMed] [Google Scholar]
- Sayers LG, Brown GR, Michell RH, Michelangeli F (1993) The effects of thimerosal on calcium uptake and inositol 1,4,5-trisphosphate-induced calcium release in cerebellar microsomes. Biochem J 289:883887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seewagen CL (2010) Threats of environmental mercury to birds: knowledge gaps and priorities for future research. Bird Conserv Int 20:112–123 [Google Scholar]
- Shanker G, Matkus LS, Walker SJ, Aschner M (2002) Methylmercury enhances arachidonic acid release and cytosolic phospholipase A2 expression in primary cultures of neonatal astrocytes. Mol Brain Res 106:1–11 [DOI] [PubMed] [Google Scholar]
- Sharpe MA, Livingston AD, Baskin DS (2012) Thimerosal-derived ethylmercury is a mitochondrial toxin in human astrocytes: possible role of Fenton chemistry in the oxidation and breakage of mtDNA. J Toxicol 2012: Article ID 373678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinyashiki M, Kumagai Y, Nakajima H, Nagafune J, Homma-Takeda S, Sagai M, Shimojo N (1998) Differential changes in rat brain nitric oxide synthase in vivo and in vitro by methylmercury. Brain Res 798:147–155 [DOI] [PubMed] [Google Scholar]
- Simmons-Willis TA, Koh AS, Clarkson TW, Ballatori N (2002) Transport of a neurotoxicant by molecular mimicry: the methylmercury-L-cysteine complex is a substrate for human L-type large neutral amino acid transporter (LAT) 1 and LAT2. Biochem J 367:239–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirois JE, Atchison WD (2000) Methylmercury affects multiple subtypes of calcium channels in rat cerebellar granule cells. Toxicol Appl Pharmacol 167:1–11 [DOI] [PubMed] [Google Scholar]
- Sitsapesan R, Williams AJ (2000) Do inactivation mechanisms rather than adaptation hold the key to understanding ryanodine receptor channel gating? J Gen Physiol 116:867–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Jang YY, Shin YK, Lee MY, Lee C-S (2000) Inhibitory action of thimerosal, a sulfhydryl oxidant, on sodium channels in rat sensory neurons. Brain Res 864(1):105–113 [DOI] [PubMed] [Google Scholar]
- Stuning M, Brom J, Konig W (1988) Multiple effects of ethylmercurithiosalicylate on the metabolization of arachidonic acid by human neutrophils. Prostaglandins Leukot Essent Fatty Acids 32:1–7 [DOI] [PubMed] [Google Scholar]
- Szalai G, Krishnamurthy R, Hajnoczky G (1999) Apoptosis driven by IP3-linked mitochondrial calcium signals. EMBO J 18(22):6349–6361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornquist K, Vainio P, Titievsky A (1999) Redox modulation of intracellular free calcium concentration in thyroid FRTL-5 cells; evidence for an enhanced extrusion of calcium. Biochem J 339:621–628 [PMC free article] [PubMed] [Google Scholar]
- Vanlingen S, Sipma H, Missianen L, De Smedt H, De Smet P, Casteels R, Parys JB (1999) Modulation of type 1, 2, and 3 inositol 1,4,5-trisphosphate receptors by cyclic ADP-ribose and thimerosal. Cell Calcium 25:107–114 [DOI] [PubMed] [Google Scholar]
- Verity MA, Sarafian T, Pacifici EHK, Sevanian A (1994) Phospholipase A2 stimulation by MeHg in neuron culture. J Neurochem 62(2):705–714 [DOI] [PubMed] [Google Scholar]
- Verrey F, Meier C, Rossier G, Kuhn LC (2000) Glycoprotein-associated amino acid exchangers: broadening the range of transport specificity. Plfugers Arch 440:503–512 [DOI] [PubMed] [Google Scholar]
- Wall SJ, Yasuda RP, Li M, Ciesla W, Wolf BB (1992) Differential regulation of subtypes M1-M5 of muscarinic receptors in forebrain by chronic atropine administration. J Pharmacol Exp Ther 262(2):584–588 [PubMed] [Google Scholar]
- Wang WH, Machaty Z, Abeydeera LF, Prather RS, Day BN (1999) Time course of cortical and zona reactions of pig oocytes upon intracellular calcium increase induced by thimerosal. Zygote 7:79–86 [DOI] [PubMed] [Google Scholar]
- Weihe P, Grandjean P, Debes F, White R (1996) Health implications for Faroe Islanders of heavy metals and PCBs from pilot whales. Sci Total Environ 186:141–148 [DOI] [PubMed] [Google Scholar]
- Wu X, Liang H, O’Hara KA, Yalowich JC, Hasinoff BB (2008) Thiol-modulated mechanisms of the cytotoxicity of thimerosal and inhibition of DNA topoisomerase H alpha. Chem Res Toxicol 21(2):483–493 [DOI] [PubMed] [Google Scholar]
- Yates DE, Mayack DT, Munney K, Evers DC, Major A, Kaur T, Taylor RJ (2005) Mercury levels in mink (Mustels vison) and river otter (Lontra canadensis) from northeastern North America. Ecotoxicology 14:263–274 [DOI] [PubMed] [Google Scholar]
- Yin Z, Milatovic D, Aschner JL, Syversen T, Rocha JBT, Souza DO, Sidoryk M, Albrecht J, Aschner M (2007) Methylmercury induces oxidative injury, alterations in permeability and glutamine transport in cultured astrocytes. Brain Res 1131(1):1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Atchison WD (2003) Methylmercury differentially affects GABAA receptor-mediated spontaneous IPSCs in Purkinje and granule cells of rat cerebellar slices. J Physiol 550 (1):191–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarini S, Gijon MA, Folco G, Murphy RC (2006) Effect of arachidonic acid reacylation on leukotriene biosynthesis in human neutrophils stimulated with granulocyte-macrophage colony stimulating factor and formyl-methionyl-leucyl-phenylalanine. J Biol Chem 281(15):10134–10142 [DOI] [PubMed] [Google Scholar]
- Zhang J (1984) Clinical observations in ethyl mercury chloride poisoning. Am J Ind Med 8:251–258 [DOI] [PubMed] [Google Scholar]