

Abstract
Tominersen is an intrathecally administered antisense oligonucleotide targeting huntingtin mRNA which leads to a dose‐dependent, reversible lowering of cerebrospinal fluid (CSF) mutant huntingtin protein concentration in individuals with Huntington's disease. Nonlinear mixed‐effect population pharmacokinetic (PopPK) modeling was conducted to characterize the CSF and plasma pharmacokinetics (PK) of tominersen, and to identify and quantify the covariates that affect tominersen PKs. A total of 750 participants from five clinical studies with a dose range from 10 to 120 mg contributed CSF (n = 6302) and plasma (n = 5454) PK samples. CSF PK was adequately described by a three‐compartment model with first‐order transfer from CSF to plasma. Plasma PK was adequately described by a three‐compartment model with first‐order elimination from plasma. Baseline total CSF protein, age, and antidrug antibodies (ADAs) were the significant covariates for CSF clearance. Body weight was a significant covariate for clearances and volumes in plasma. ADAs and sex were significant covariates for plasma clearance. The developed PopPK model was able to describe tominersen PK in plasma and CSF after intrathecal administration across a range of dose levels, and relevant covariate relationships were identified. This model has been applied to guide dose selection for future clinical trials of tominersen in patients with Huntington's disease.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Tominersen is an intrathecally administered chimeric 2′‐O‐methoxyethyl (2‐MOE)‐modified antisense oligonucleotide (ASO) targeting huntingtin protein (HTT), under investigation in individuals with early manifest and manifest Huntington's disease. After administration of tominersen, dose‐dependent, reversible lowering of cerebrospinal fluid (CSF) mutant HTT was observed.
WHAT QUESTION DID THIS STUDY ADDRESS?
This analysis characterized the CSF and plasma pharmacokinetics (PK) of tominersen and identified the covariates affecting tominersen PKs, using plasma and CSF concentration–time profiles following intrathecal administration, at doses ranging from 10 to 120 mg.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This study provides a population PK (PopPK) model that describes both CSF and plasma PKs of tominersen. Furthermore, it provides an insight into drug distribution and variabilities in CSF PKs for ASOs after intrathecal administration.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
This PopPK model can be used to predict tominersen exposure in CSF and plasma after administration of various dosing regimens, to guide decision making during the clinical development program.
INTRODUCTION
Huntington's disease (HD) is a rare, genetic, neurodegenerative disease characterized by a triad of cognitive, behavioral, and motor symptoms. 1 , 2 , 3 It has a devastating impact on patients and entire families across generations, resulting in increasing disability, functional decline, and loss of independence, ultimately leading to death. 1 , 2 Onset normally occurs between 30 and 50 years of age and the median survival period is ~15 years after the onset of motor symptoms. 4
HD is believed to be caused by a cytosine‐adenine‐guanine (CAG) trinucleotide repeat expansion in the huntingtin gene on the fourth chromosome. 5 The expansion of this CAG repeat results in the production of a toxic mutant huntingtin protein (mHTT), which aggregates in and damages neurons in the central nervous system (CNS). 1 , 6 , 7 Given the monogenic nature of HD, huntingtin protein (HTT)‐lowering approaches are believed to alleviate HD pathogenesis. 8 There are currently no approved therapies that slow or stop disease progression.
Tominersen is an investigational chimeric 2′‐O‐methoxyethyl (2‐MOE)‐modified antisense oligonucleotide (ASO) targeting huntingtin gene mRNA. 8 It is non‐allele specific and therefore expected to reduce the production of all forms of HTT, including mHTT. Tominersen has been investigated in individuals with early manifest HD and manifest HD in five clinical studies (phase I/IIa study, NCT02519036; open‐label extension of the phase I/IIa study, NCT03342053; GENERATION HD1, NCT03761849; GEN‐PEAK, NCT04000594; and GEN‐EXTEND, NCT03842969) at doses ranging from 10 mg to 120 mg, for up to 25 months, and is administered as an intrathecal injection directly into the cerebrospinal fluid (CSF) to achieve efficient distribution to the CNS. In the phase I/IIa study, dose‐dependent, reversible lowering of mHTT protein in the CSF of individuals with early manifest HD was observed. 9 GENERATION HD1 is the largest phase III clinical trial of an HTT‐lowering therapy in HD to date and the data generated are providing insights helping us to understand the pharmacokinetics (PK) behavior and mechanism of action of tominersen, as a basis for a potential treatment approach.
The PKs of ASOs in humans following intravenous, subcutaneous, and, recently, intrathecal administration have been reported previously. 10 , 11 , 12 Following peripheral administration, ASOs are distributed broadly into most tissues; the major tissues that ASOs are distributed into are the liver, kidneys, bone marrow, adipocytes, and lymph nodes. 13 , 14 Oligonucleotides and their metabolites are known to be primarily excreted in urine and biliary elimination is considered to be a minor pathway. 14
Peripherally administered ASOs do not distribute into the CNS because the highly charged ASOs do not cross the intact blood–brain barrier efficiently. 15 Intrathecal administration is therefore an alternative route of administration (RoA) for ASOs targeting the CNS. Although drug kinetics after intrathecal administration remain poorly understood, intrathecally administered ASOs have been reported to distribute rapidly from the CSF in rats and nonhuman primates, likely due to the combination of uptake into CNS tissues and transfer to systemic circulation. 16 , 17 The terminal half‐life of ASOs in CSF is relatively long due to the slow elimination from CNS tissues. Recently, the use of intrathecal administration has been successfully implemented for nusinersen, which is indicated for the treatment of spinal muscular atrophy, 18 and was confirmed to be distributed throughout the spinal cord and brain in treated infants. 19
CSF concentrations are often considered the best available surrogate for brain target site concentrations in humans. Only very limited examples of CSF PK data in humans following intrathecal administration of ASOs have been reported, including tofersen and nusinersen. 11 , 12 Mathematical models describing PKs were previously developed, and a physiologically based PK model 17 and semimechanistic PK model 20 describing the PKs of nusinersen following intrathecal administration have been published. These models were developed using the PK data from nonhuman primates followed by scaling to humans. In addition, one population PK (PopPK) model developed based on the plasma and CSF PK data generated from pediatric patients with spinal muscular atrophy has been proposed. 21 The number of subjects and PK data included in the analysis were limited due to the nature of pediatric studies.
In this work, we describe a PopPK model to simultaneously characterize the CSF and plasma PKs of tominersen in patients with HD following intrathecal administration, and to identify the covariates that affect tominersen PK using both plasma and CSF concentration–time profiles from a total of 750 individuals with HD. The comprehensive understanding of PK characterization, including identification and quantification of the impact of covariates on tominersen PK, is essential for the tominersen clinical development program.
METHODS
Subject and study design
Plasma and CSF PK data collected in individuals with early manifest and manifest HD from five clinical studies were pooled. 9 , 22 , 23 , 24 , 25 For GENERATION HD1, GEN‐PEAK, and GEN‐EXTEND, only partial and preliminary data were available and included in the present work. The studies were conducted in accordance with the principles of the Declaration of Helsinki and were approved by the appropriate institutional review boards, local ethics committees, and regulatory agencies. All participants provided written informed consent.
A summary of the studies, including patient numbers, administered doses and dosing frequency, and CSF and plasma PK sampling times is provided in Table 1, in addition to a high‐level description of each study is provided in Appendix S1.
TABLE 1.
Summary of study designs and PK data included in the analysis.
| Study | Dose regimen | N of IT injection/length of the study | IT injection route | CSF PK assessments | Plasma PK assessments | N of subjects | N of observations a | |
|---|---|---|---|---|---|---|---|---|
| Plasma | CSF | |||||||
| Phase I/IIa study (NCT02519036) | 10 mg Q4W | 4 doses | LP | Before each dose | Before each dose; at 0.5, 1, 2, 3, 4, 5, 6, 8, 12, and 24 h after the first dose; and at 0.5, 1, 2, 3, 4, 5, and 24 h after the last dose | 3 | 50 | 0 |
| 30 mg Q4W | 6 | 102 | 17 | |||||
| 60 mg Q4W | 6 | 102 | 17 | |||||
| 90 mg Q4W | 9 | 154 | 28 | |||||
| 120 mg Q4W | 10 | 172 | 36 | |||||
| OLE of the phase I/IIa study (NCT03342053) | 120 mg Q4W | 15 months | LP | Before each dose | Before each dose; at 0.5, 1, 2, 3, 4, 5, 6, 8, 12 and 24 h after the first dose; and at 0.5, 1, 2, 3, 4, and 5 h after the dose on day 197 | 23 | 689 | 313 |
| 120 mg Q8W | 23 | 695 | 185 | |||||
| GEN‐PEAK c (NCT04000594) | 30 mg Q4W | 2 doses | 1st dose: catheter, 2nd dose: LP | Pre‐dose; at 2, 4, 8, 12, 16, 24, 36, 48, 60, and 72 h after the first dose; before the second dose; on days 43, 71, and 127; and at 6 months after last dose | Before each dose; at 1, 2, 4, 6, 8, 12, 16, 24, 36, 48, 60, 72, and 96 h; and 27 days after the first dose; at 1, 2, and 4 h after the second dose; and on days 30, 43, 71, and 127, and at the follow‐up visit | 4 | 67 | 45 |
| 60 mg Q4W | 4 | 73 | 40 | |||||
| 120 mg Q4W | 4 | 76 | 50 | |||||
| GENERATION HD1 c (NCT03761849) | 120 mg Q4W | 97 weeks | LP | Predose Q4W after protocol amendment, predose at weeks 1, 5, and thereafter Q8W | Predose at weeks 1, 13, 21, and thereafter Q16W and at 1, 2, and 4 h after the dose at week 37 | 35 | 91 | 209 |
| 120 mg Q8W | 295 | 1782 | 2300 | |||||
| 120 mg Q16W | 259 | 1032 | 2008 | |||||
| GEN‐EXTEND b , c (NCT03842969) | 120 mg Q4W | Up to 6 years | LP | One sample at inclusion and before each dose, and at end of study | One sample at inclusion Q16W for 48 weeks, every 32 weeks thereafter, and at end of study | 14 | 54 | 97 |
| 120 mg Q8W | 80 | 208 | 568 | |||||
| 120 mg Q8W (no loading) | 52 | 79 | 233 | |||||
| 120 mg Q16W | 19 | 22 | 58 | |||||
| 120 mg Q16W (no loading) | 51 | 6 | 98 | |||||
Abbreviations: CSF, cerebrospinal fluid; IT, intrathecal; LLOQ, lower limit of quantification; LP, lumbar puncture; N, number; PK, pharmacokinetic; Q4W, every 4 weeks; Q8W, every 8 weeks; Q16W, every 16 weeks.
Number of observations greater than the lower limit of quantification.
Initial dose regimen.
Partial and preliminary data.
Bioanalytical methods
In the phase I/IIa study, plasma and CSF tominersen concentrations were determined using a hybridization cutting enzyme‐linked immunosorbent assay method. The lower limit of quantification (LLOQ) was 1 ng/mL in plasma and CSF. In all other studies, plasma and CSF tominersen concentrations were determined using hybridization electrochemiluminescence. The LLOQ was 0.05 ng/mL in plasma and 0.1 ng/mL in CSF.
PopPK model development
The PopPK models were developed in a stepwise manner. Because tominersen was administered intrathecally, the plasma PK distribution is considered to be affected by the drug distribution into the CNS. The structural model describing the PKs in CSF was developed first followed by a covariate analysis. Thereafter, the structural model for plasma was developed, again, followed by a covariate analysis. The parameters of the CSF model were kept fixed during the development of the plasma model to reduce runtimes and increase model stability. The analysis was performed on log‐transformed PK data. Samples below the LLOQ were excluded in the analysis but were taken into account in the visual predictive checks (VPCs).
Structural and stochastic model development
Disposition models for tominersen in CSF and plasma
According to the observed CSF and plasma PK profiles, two‐ and three‐compartment disposition models with first‐order elimination from the central compartments were evaluated for CSF and plasma PKs, respectively. The elimination from CSF was used as input to the central plasma compartment. The bioavailability to CSF compartment (F1) and the bioavailability from CSF to plasma (F2) were fixed to 1, implying that apparent clearances (CLs) and volumes (Vs) were estimated in plasma.
Interindividual variability for tominersen in CSF and plasma
Interindividual variability (IIV) was evaluated on all PK parameters based on the available PK data and were added in an exponential form (Equation 1):
| (1) |
where TVP is the typical value of the parameter P, P i is the individual value of the parameter, and η i is a normally distributed random variable with mean 0 and standard deviation ω.
Residual error for tominersen in CSF and plasma
Additive residual error models were used on the log‐transformed tominersen concentrations in CSF and plasma.
Covariate analysis
Covariates that had a strong rationale were evaluated as a part of the structural model development before the full covariate analysis. The following covariates were evaluated as a part of the structural model: body weight (BW) on CLs and on Vs for CSF and plasma, as well as the effects of RoA, lumbar site of administration on CSF clearance (CLCSF), and CSF volume withdrawn before the dose on volume of distribution in the central CSF compartment (V1,CSF), and the effect of antidrug antibodies (ADAs) on plasma clearance (CLplasma) and CLCSF. Covariates identified as statistically significant were kept in the model.
The stepwise covariate model building procedure
The covariate model building was performed using the stepwise covariate model building procedure (SCM) 26 in Perl‐speaks‐NONMEM. 27 , 28 Additional covariates were evaluated both on the CSF and plasma models. Age, sex, CAG, CAG age‐product score, caudate volume, ventricle volume, whole‐brain volume, total protein in CSF (TPCSF), and height were explored in the CSF model. Age, sex, alanine aminotransferase, total protein in plasma, and creatinine clearance (CrCL) were explored in the plasma model.
P values of 0.01 and 0.001 were used in the forward and backwards steps, respectively. In this analysis, adaptive scope reduction was added to the default SCM algorithm 29 to make the covariate search more efficient.
Continuous covariate relationships were coded as exponential models and categorical covariate relationships were coded as a fractional difference to the most common category. Equation (2a), (2b) illustrates this parameterization.
| (2a) |
| (2b) |
where Covref is a reference covariate value for cov m , to which the covariate model is normalized with the median.
Covariates that were identified as statistically significant in the SCM, but where the effect of the covariate was considered not clinically relevant (i.e., a difference in the parameter between two categories of <10%, or a <10% difference in the parameter between the 5th and 95th percentiles of the analysis population) were removed from the model.
Model discrimination
The performance of a model and selection between competing models was based on statistical and graphical assessments, including the inspection of graphical goodness‐of‐fit and changes in the objective function value provided by NONMEM. The differences in objective function values are nominally χ 2 distributed and a difference of 3.84 corresponds to an approximate p value less than 0.05 for 1 degree of freedom, considering that the difference is statistically significant.
Model evaluation
In addition to the model selection criteria described above, simulation‐based diagnostics, such as prediction‐corrected VPCs (pcVPCs) were used to evaluate the predictive performance of the final model. 30
CSF and plasma PK simulation
The final PK model was used to simulate typical CSF and plasma PK profiles associated with various dosing regimens, to assess the influence of covariates and to support dose selection in future clinical studies. The test scenarios included tominersen intrathecal administration for 1.5 years, with doses ranging from 30 to 120 mg every 16 weeks. The impact of the covariates was assessed at 120 mg every 16 weeks.
Software
The analyses were performed using NONMEM version 7.4.4. 31 NONMEM runs were performed using the gfortran compiler, version 4.4.6. Data management and a graphical analysis were performed using R version 3.5.3. 32 SCM and VPCs were performed using PsN version 4.9.0. 27 , 28
RESULTS
Analysis dataset
A total of 5454 plasma samples and 6302 CSF samples from 750 individuals with HD were included in the analysis. A summary of the data included in the PK analysis is shown in Table 1. The percentage of PK data below the LLOQ was 4% in CSF PK data and 19% in plasma PK data, respectively. Baseline covariate data from the PopPK analysis set are summarized in Table S1; covariates associated with each intrathecal administration, such as RoA, lumbar site of administration, and volume of CSF withdrawn before intrathecal dosing are summarized in Table S2 and time‐varying ADA data are summarized in Table S3A,B.
Cerebrospinal fluid pharmacokinetics
CSF PK data are shown in Figure 1. CSF trough concentrations for the 120 mg dose from all studies versus time by dosing frequency are shown in Figure 1a. Accumulation of the drug in CSF was observed. For every‐4‐week dosing, steady‐state (SS) concentration was achieved at around week 37 at the latest, whereas for the every‐8‐week and every‐16‐week dosing regimens, SS concentration was reached after the loading dose and no further accumulation was observed at later timepoints. CSF PK profiles up to 72 h after the first dose and up to around 100 days after the latest dose in GEN‐PEAK at the dose range of 30–120 mg are shown in Figure 1b,c. These rich sampled data indicate a multiphase PK disposition in CSF and show tominersen to have a long terminal half‐life. The dose‐normalized CSF PK profiles in Figure 1c indicate that there was no obvious dose‐dependent trend in the PK profiles, suggesting that CSF PK is linear.
FIGURE 1.
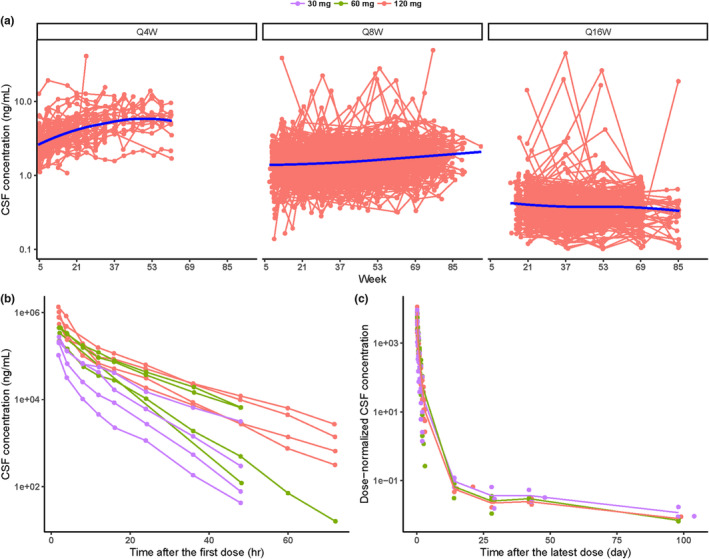
CSF concentration–time profiles for (a) individual trough concentrations at 120 mg Q4W, Q8W, and Q16W (red: individual observation; blue line: smooth), (b) individual concentrations up to 72 h after the first dose at the dose range of 30–120 mg in GEN‐PEAK (purple: 30 mg; green: 60 mg; red: 120 mg), (c) mean concentrations up to ~100 days after the latest dose at the dose range of 30–120 mg in GEN‐PEAK (purple: 30 mg; green: 60 mg; red: 120 mg). CSF, cerebrospinal fluid; Q4W, every 4 weeks; Q8W, every 8 weeks; Q16W, every 16 weeks.
Plasma pharmacokinetics
Plasma PK data are shown in Figure 2. Plasma trough concentrations for the 120 mg dose from all studies versus time by dosing frequency are shown in Figure 2a. SS concentration was achieved later in plasma compared with CSF. Mean plasma PK profiles up to 24 h in the phase I/IIa study (day 1 and day 85) and up to month 1 in the phase I/IIa study and GEN‐PEAK at the dose range of 10–120 mg are shown in Figure 2b,c. The plasma PK profiles supported a multiphase PK disposition. The dose‐normalized plasma PK profiles in Figure 2c showed that there was no obvious trend in the PK profiles among doses, suggesting that plasma PK is linear.
FIGURE 2.
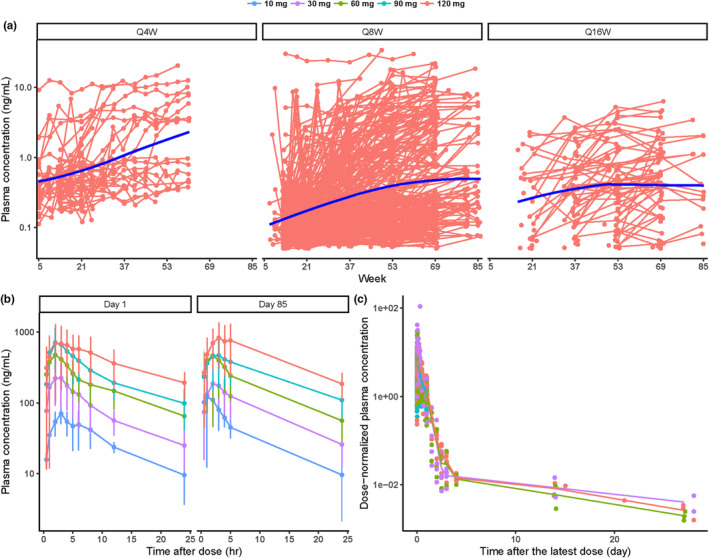
Plasma concentration–time profiles for (a) individual trough concentrations at 120 mg Q4W, Q8W, and Q16W (red: individual observation; blue line: smooth), (b) mean (± standard deviation) concentrations up to 24 h after the first (day 1) and the fourth dose (day 85) at the dose range of 10–120 mg in the phase I/IIa study (dark blue: 10 mg; purple: 30 mg; green: 60 mg; light blue: 90 mg; red: 120 mg), (c) mean concentrations up to 1 month after the latest dose at the dose range of 10–120 mg in the phase I/IIa study and GEN‐PEAK (dark blue: 10 mg; purple: 30 mg; green: 60 mg; light blue: 90 mg; red: 120 mg). Q4W, every 4 weeks; Q8W, every 8 weeks; Q16W, every 16 weeks.
PopPK model
A PopPK model was developed that described the plasma and CSF tominersen PK well (Figure 3). CSF and plasma were each described with a three‐compartment model with the elimination from the CSF central compartment providing the input to the plasma central compartment. The goodness‐of‐fit plots showed adequate agreement between predicted and observed plasma and CSF concentrations, without any significant trends (Figure S1). The pcVPCs for CSF concentrations versus time over the first 72 h postdose and for the trough concentrations are shown in Figure 4a,b, respectively. The pcVPCs for plasma concentrations versus time over the first 24 h postdose and for the trough concentrations are shown in Figure 4c,d, respectively. The pcVPC plots showed good predictive performance of the final model to capture the median of data both for plasma and CSF concentrations.
FIGURE 3.
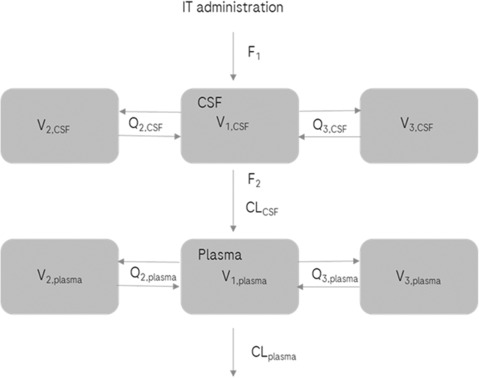
Schematic representation of the final PopPK model. CL, clearance; CLCSF, CL from the central CSF compartment; CLplasma, CL from the central plasma compartment; CSF, cerebrospinal fluid; F1, bioavailability to CSF; F2, bioavailability from CSF to plasma; IT, intrathecal; PopPK, population pharmacokinetic; Q, intercompartmental clearance; Q2,CSF, Q between the central CSF compartment and the first peripheral CSF compartment; Q3,CSF, Q between the central CSF compartment and the second peripheral CSF compartment; Q2,plasma, Q between the central plasma compartment and the first peripheral plasma compartment; Q3,plasma, Q between the central plasma compartment and the second peripheral plasma compartment; V, volume of distribution; V1,CSF, V in the central CSF compartment; V2,CSF, V in the first peripheral CSF compartment; V3,CSF, V in the second peripheral CSF compartment; V1,plasma, V in the central plasma compartment; V2,plasma, V in the first peripheral plasma compartment; V3,plasma, V in the second peripheral plasma compartment.
FIGURE 4.
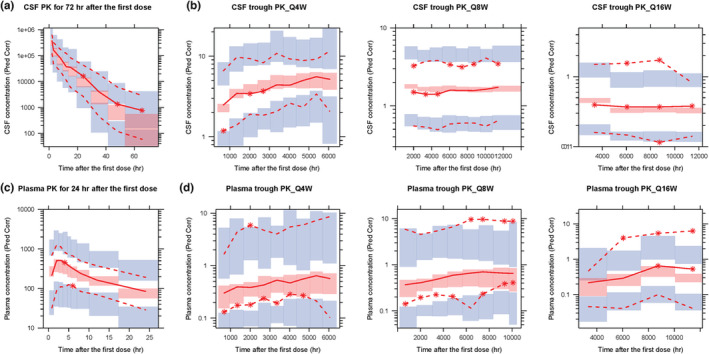
Prediction‐corrected visual predictive check of the final PopPK model for (a) CSF concentrations for the first 72 h, (b) CSF trough concentrations for the regimen of Q4W, Q8W, and Q16W, respectively, (c) plasma concentrations for the first 24 h and (d) plasma trough concentrations for the regimen of Q4W, Q8W, and Q16W, respectively. The red lines represent the 5th, 50th, and 95th percentiles of the observed data. The shaded areas represent the 90% confidence interval of the 5th, 50th, and 95th percentiles predicted by the model. CSF, cerebrospinal fluid; pred corr, prediction corrected; PK, pharmacokinetic; PopPK, population PK; Q4W, every 4 weeks; Q8W, every 8 weeks; Q16W, every 16 weeks.
The parameter estimates of the final model are shown in Table 2. In general, parameters were estimated with high precision across both the CSF and plasma models. Relative standard error (RSE) of the parameters of the CSF model was less than 16% in all parameters apart from V1,CSF, intercompartmental clearance (Q) between the central CSF compartment and the first peripheral CSF compartment (Q2,CSF), and the corresponding IIV parameters. For the plasma model, the RSE was less than 13% for all parameters. IIV was supported by the data and included on CLCSF, V1,CSF, and Q2,CSF of the CSF model and on CLplasma, Q between the central plasma compartment and the first peripheral plasma compartment (Q2,plasma), V in the first peripheral plasma compartment (V2,plasma), and V in the second peripheral plasma compartment (V3,plasma) of the plasma model. The shrinkage was relatively large for V1,CSF and Q2,CSF (88.4% and 88.7%, respectively) probably because individuals with sparse sampling did not contribute information on those parameters. The RSE was also higher for those parameters compared with other IIV parameters; however, they were still kept in the model because they were important for the model fit for the data in GEN‐PEAK, where rich CSF profiles were collected. The final PopPK model code is provided in Appendix S1.
TABLE 2.
Final PopPK model parameter estimates.
| Final model | Unit | Typical value | RSE (%) | SHR (%) |
|---|---|---|---|---|
| F1 | 1 FIXED | |||
| CLCSF | L/h | 0.0177 | 7.96 | |
| V1,CSF | L | 0.0436 | 33.1 | |
| Q2,CSF | L/h | 0.00982 | 27.3 | |
| V2,CSF | L | 0.0517 | 11.8 | |
| Q3,CSF | L/h | 0.0000121 | 15.7 | |
| V3,CSF | L | 0.0118 | 15.8 | |
| Total protein_CLCSF | −0.540 | 11.2 | ||
| ADA_CLCSF | −0.101 | 6.89 | ||
| Age_CLCSF | −0.00550 | 12.8 | ||
| F2 | 1 FIXED | |||
| CLplasma | L/h | 10.7 | 3.30 | |
| V1,plasma | L | 41.0 | 2.88 | |
| Q2,plasma | L/h | 13.4 | 7.98 | |
| V2,plasma | L | 283 | 6.15 | |
| Q3,plasma | L/h | 0.267 | 4.84 | |
| V3,plasma | L | 613 | 6.35 | |
| BW_CLs | 0.687 | 2.60 | ||
| BW_Vs | 0.866 | 1.54 | ||
| ADA_CLplasma | −0.673 | 0.639 | ||
| Sex_CLplasma | −0.186 | 12.1 | ||
| IIV a | ||||
| IIV on CLCSF | % | 16.5 | 3.22 | 6.65 |
| IIV on V1,CSF | % | 55.9 | 41.6 | 88.4 |
| IIV on Q2,CSF | % | 44.6 | 51.2 | 88.7 |
| IIV RUVCSF | % | 46.3 | 2.84 | 10.1 |
| IIV on CLplasma | % | 28.8 | 2.84 | 21.9 |
| IIV on Q2,plasma | % | 239 | 6.35 | 35.0 |
| IIV on V2,plasma | % | 64.5 | 9.33 | 52.0 |
| IIV on V3,plasma | % | 60.6 | 8.39 | 44.3 |
| IIV RUVplasma | 29.0 | 3.67 | 14.6 | |
| RUV a | ||||
| RUVCSF | % | 31.2 | 2.38 | 1.61 |
| RUVplasma | % | 53.6 | 1.94 | 7.97 |
Abbreviations: ADA, anti‐drug antibody; BW, body weight; CL, clearance; CLCSF, CL from the central CSF compartment; CLplasma, CL from the central plasma compartment; CLs, CLplasma, Q2,plasma and Q3,plasma; CSF, cerebrospinal fluid; F1, bioavailability to CSF; F2, bioavailability from CSF to plasma; IIV, interindividual variability; PopPK, population pharmacokinetic; Q, intercompartmental clearance; Q2,CSF, Q between the central CSF compartment and the first peripheral CSF compartment; Q3,CSF, Q between the central CSF compartment and the second peripheral CSF compartment; Q2,plasma, Q between the central plasma compartment and the first peripheral plasma compartment; Q3,plasma, Q between the central plasma compartment and the second peripheral plasma compartment; RSE, relative standard error; RUV, residual unexplained variability; SHR, shrinkage; V, volume of distribution; V1,CSF, V in the central CSF compartment; V2,CSF, V in the first peripheral CSF compartment; V3,CSF, V in the second peripheral CSF compartment; V1,plasma, V in the central plasma compartment; V2,plasma, V in the first peripheral plasma compartment; V3,plasma, V in the second peripheral plasma compartment; Vs, V1,plasma, V2,plasma, and V3,plasma.
Interindividual variability and residual unexplained variability are expressed as coefficient of variation and in percentage of the parameter estimate.
Covariate analysis in CSF
TPCSF, age, and ADAs were identified as statistically significant covariates on CLCSF. No other covariates were identified on the CSF model parameters. CLCSF was decreased by ~19%, 18%, and 10%, respectively, when TPCSF was increased from 0.19 to 0.54 g/L (5th and 95th percentiles of the analysis population), when age was increased from 31 to 64 years (5th and 95th percentiles of the analysis population), and when the participants developed ADAs.
Covariate analysis in plasma
BW was a significant covariate for CLs and Vs in plasma, and sex and ADAs were significant covariates on CLplasma. The CLplasma was decreased by ~40%, 19%, and 67%, respectively, when BW was decreased from 95.5 to 52 kg (95th and 5th percentiles of the analysis population), when the participants were women, or developed ADAs. No other covariates were identified on the plasma model parameters.
Simulation of the tominersen PK profiles in CSF and plasma
The simulated plasma and CSF PK profiles following different every‐16‐week doses or covariate settings are presented in Figure 5. The median (5th–95th percentiles) of the CSF trough concentration at SS was ~0.074 (0.041–0.14), 0.15 (0.082–0.27), 0.22 (0.12–0.41), and 0.30 (0.16–0.54) ng/mL for doses of 30, 60, 90, and 120 mg, respectively, and the median (5th–95th percentiles) of the plasma trough concentration was 0.016 (0.0039–0.53), 0.033 (0.0078–0.11), 0.049 (0.012–0.16), and 0.065 (0.016–0.21) ng/mL at 30, 60, 90, and 120 mg, respectively. The variability in plasma PK data was much larger than that in CSF PK data. The CSF trough concentrations were increased by 24%, 46%, and 44% at SS concentration when the participant developed ADAs compared with an ADA‐negative participant, when TPCSF was increased from 0.19 to 0.54 g/L (5th and 95th percentiles of the analysis population), and when the age of a participant increased from 31 to 64 years (5th and 95th percentiles of the analysis population), respectively.
FIGURE 5.
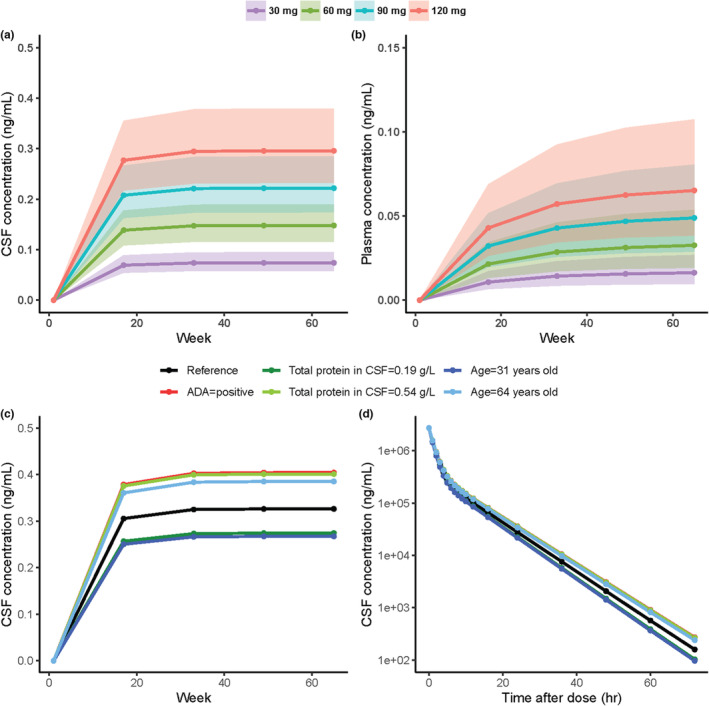
Model prediction for the doses of 30, 60, 90, and 120 mg Q16W with the final PopPK model for CSF trough concentrations (a) and plasma trough concentrations (b). The shaded areas indicate the 50% prediction interval for each dose. Impact of identified covariates on the predicted concentration following 120 mg Q16W for CSF trough concentrations (c) and CSF concentrations for the first 72 h (d). The black lines represent typical profiles at a condition of ADA = negative, sex = males, body weight = 75 kg, total protein in CSF = 0.35 g/L and age = 49. Each simulation condition is stratified by color and indicates which covariate differs from the conditions for the typical profiles. ADA, anti‐drug antibody; CSF, cerebrospinal fluid; PopPK, population pharmacokinetics; Q16W, every 16 weeks.
DISCUSSION
A PopPK model was developed based on pooled data from five clinical studies where tominersen was intrathecally administered at doses ranging from 10 to 120 mg at frequencies of every 4 weeks, every 8 weeks, and every 16 weeks. In total, 6302 CSF concentrations and 5454 plasma concentrations from 750 individuals with HD were included in the analysis. The developed PopPK model successfully described tominersen PKs in plasma and CSF simultaneously. TPCSF, age, and ADAs were identified as clinically relevant covariates on the tominersen PK in CSF, whereas BW, ADAs, and sex were identified as clinically relevant covariates on the tominersen PK in plasma.
The percentage of PK data below the LLOQ was 4% in CSF, and it was therefore considered appropriate to exclude the below limit of quantification (BLOQ) samples from the CSF PK model development. 33 For plasma, the percentage of PK data below the LLOQ was 19%, and for this reason, the M3 method was initially implemented. 34 However, the model did not converge when the M3 method was used, probably due to the high complexity of the model, and therefore, the BLOQ samples were excluded from the plasma PK model development.
The tominersen PK both in CSF and plasma showed multiphase PK kinetics and a long terminal half‐life. After intrathecal administration, ASOs are rapidly distributed to CNS tissues and the long terminal half‐life in CSF is considered to correspond to a slow rate of ASOs dissociating from the CNS tissue. 16 , 17 The terminal half‐life for CSF PKs is calculated to be ~1 month based on the estimated PK parameters. This calculated long half‐life supports the proposed infrequent administration for tominersen.
The plasma PK displayed dose‐proportional properties whereas assessment of the CSF PK linearity was challenging due to limitations associated with available data, such as a majority of sparse sampling at mainly one dose level (120 mg) and low sensitivity of the bioanalytical assay used in the phase I/IIa study (in which multiple dose levels were administered). Dose was tested as a covariate on the CSF PK parameters but was not statistically significant on any of the parameters. Therefore, a linear model was considered to be adequate to describe the CSF PK data.
The design of GEN‐PEAK, which included rich CSF sampling via an indwelling intrathecal catheter up to 72 h postdose, was invaluable to enable characterization of the full concentration–time profile in CSF, including identification of a three‐compartment disposition model. Due to the small sample size of this study (N = 12), some IIV parameters could not be estimated, and others displayed a large shrinkage and higher RSE, but they were nevertheless kept in the model as they were important for the description of those data. Notably, CL parameters were estimated with good precision (both population and IIV), which provides confidence on the model predictions at trough, including the impact of covariates. A three‐compartment model for plasma PKs was supported by the data and the parameters were well‐estimated (RSE < 13%). The variability in plasma concentration was larger than that observed in CSF, which is likely a result of being further from the point of administration. The VPC showed that the model captured the median of the data very well, although the data variability was not perfectly captured.
Oligonucleotides are metabolized by nucleases that are ubiquitously expressed by cells in most tissues. 14 Based on the available data it was not possible to identify the fraction of tominersen that was metabolized in the CSF space and it was assumed that tominersen was fully cleared into plasma. Additional data, for instance, following intravenous administration of tominersen, could have provided further insights.
Age, TPCSF, and ADAs were clinically relevant covariates for the CSF PKs. CSF concentrations increased with increased age, increased TPCSF, and the presence of ADAs in plasma. Reduction in CSF flow in elderly individuals with HD has been reported previously. 35 It is thought that this reduction can lead to a decrease in CLCSF, and therefore an increase in CSF concentrations. In addition, ASOs, including tominersen, are characterized by high plasma protein binding (>85%) primarily to albumin which in systemic circulation (and after systemic administration) prevents loss of the ASO through urinary excretion and promotes tissue uptake. 36 High protein binding in the CSF results in slower elimination of the CSF protein‐bound drug from the CSF compartment compared with free tominersen analogous to the systemic situation which is in line with the identified TPCSF covariate effect. 14 Importantly, the validated bioanalytical assay used to measure tominersen in CSF measures total tominersen (both the protein–tominersen conjugate and free tominersen) concentrations. The impact of the increased CSF concentrations of tominersen due to binding to CSF proteins or ADAs on the efficacy and safety profiles needs to be investigated further to clarify if this is likely to impact the pharmacologic activity of tominersen.
ADAs, BW, and sex were clinically relevant covariates on the plasma PKs; however, covariates corresponding to kidney and liver function were not identified as statistically significant. The identification of ADAs as a significant covariate is consistent with known effects of ADAs on plasma PKs of ASOs where presence of ADAs results in an increase in plasma trough concentrations after repeated administration. 37 Urine is considered to be a primary elimination pathway of ASOs and their metabolites; however, the results suggest that renal function did not have an impact on tominersen plasma PKs with the range of CrCL that was included in the analysis (CrCL: 51–150 mL/min). Hepatic insufficiencies are expected to have less effect on the clearance of oligonucleotides, 14 and the analysis result is consistent with previous knowledge.
The physicochemical properties of 2′‐MOE ASOs are reported to be very similar across sequences and species, and PK similarity in plasma has been reported, 10 , 14 suggesting that the developed model structure and estimated PK parameters can be used as a starting point for PK modeling of other 2′‐MOE ASOs.
In conclusion, the developed PopPK model was able to simultaneously describe tominersen PKs in plasma and CSF after intrathecal administration, and relevant covariate relationships were identified. The model can be used to predict tominersen concentrations to guide decision making during the clinical development program.
AUTHOR CONTRIBUTIONS
Y.Y., P.S.D., M.B., D.A.N., and H.E.S.B. wrote the manuscript. Y.Y., M.B., and K.S. performed the research. Y.Y. and P.S.D. designed the research. Y.Y., P.S.D., M.B., P.G., S.V., A.P., and B.M. analyzed the data.
FUNDING INFORMATION
This study was funded by F. Hoffmann‐La Roche Ltd. and Ionis Pharmaceuticals Inc.
CONFLICT OF INTEREST STATEMENT
Y.Y., P.S.D., K.S., P.G., S.V., A.P., B.M., and H.E.S.B. are current or former employees of Roche. M.B. is an employee of Pharmetheus AB. D.A.N. is an employee of Ionis Pharmaceuticals Inc.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
The authors thank all the individuals and investigators who participated in the studies. The phase I/IIa study of tominersen was sponsored by Ionis Pharmaceuticals Inc. The open‐label extension of the phase I/IIa study was initially sponsored by Ionis Pharmaceuticals Inc. and transferred to F. Hoffmann‐La Roche Ltd. GENERATION HD1, GEN‐PEAK, and GEN‐EXTEND were sponsored by F. Hoffmann‐La Roche Ltd. The authors thank Chrysalis Medical Communications for providing medical writing support, which was funded by F. Hoffmann‐La Roche Ltd., Basel, Switzerland in accordance with Good Publication Practice (GGP3) guidelines (http://www.ismpp.org/gpp3).
Yamamoto Y, Sanwald Ducray P, Björnsson M, et al. Development of a population pharmacokinetic model to characterize the pharmacokinetics of intrathecally administered tominersen in cerebrospinal fluid and plasma. CPT Pharmacometrics Syst Pharmacol. 2023;12:1213‐1226. doi: 10.1002/psp4.13001
REFERENCES
- 1. Bates GP, Dorsey R, Gusella JF, et al. Huntington disease. Nat Rev Dis Primers. 2015;1:15005. doi: 10.1038/nrdp.2015.5 [DOI] [PubMed] [Google Scholar]
- 2. Roos RA. Huntington's disease: a clinical review. Orphanet J Rare Dis. 2010;5(5):40. doi: 10.1186/1750-1172-5-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ross CA, Aylward EH, Wild EJ, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 2014;10(4):204‐216. doi: 10.1038/nrneurol.2014.24 [DOI] [PubMed] [Google Scholar]
- 4. Keum JW, Shin A, Gillis T, et al. The HTT CAG‐expansion mutation determines age at death but not disease duration in Huntington disease. Am J Hum Genet. 2016;98(2):287‐298. doi: 10.1016/j.ajhg.2015.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Macdonald M. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's disease collaborative research group. Cell. 1993;72(6):971‐983. doi: 10.1016/0092-8674(93)90585-e [DOI] [PubMed] [Google Scholar]
- 6. Novak MJ, Tabrizi SJ. Huntington's disease: clinical presentation and treatment. Int Rev Neurobiol. 2011;98:297‐323. doi: 10.1016/b978-0-12-381328-2.00013-4 [DOI] [PubMed] [Google Scholar]
- 7. Saudou F, Humbert S. The biology of huntingtin. Neuron. 2016;89(5):910‐926. doi: 10.1016/j.neuron.2016.02.003 [DOI] [PubMed] [Google Scholar]
- 8. Tabrizi SJ, Ghosh R, Leavitt BR. Huntingtin lowering strategies for disease modification in Huntington's disease. Neuron. 2019;101(5):801‐819. [DOI] [PubMed] [Google Scholar]
- 9. Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, et al. Targeting huntingtin expression in patients with Huntington's disease. N Engl J Med. 2019;380(24):2307‐2316. doi: 10.1056/NEJMoa1900907 [DOI] [PubMed] [Google Scholar]
- 10. Yu RZ, Grundy JS, Geary RS. Clinical pharmacokinetics of second generation antisense oligonucleotides. Expert Opin Drug Metab Toxicol. 2013;9(2):169‐182. doi: 10.1517/17425255.2013.737320 [DOI] [PubMed] [Google Scholar]
- 11. Miller TM, Pestronk A, David W, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first‐in‐man study. Lancet Neurol. 2013;12(5):435‐442. doi: 10.1016/s1474-4422(13)70061-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chiriboga CA, Swoboda KJ, Darras BT, et al. Results from a Phase 1 study of nusinersen (ISIS‐SMNRx) in children with spinal muscular atrophy. Neurology. 2016;86(10):890‐897. doi: 10.1212/WNL.0000000000002445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Levin AA, Yu RZ, Geary RS. Antisense Drug Technology. CRC Press; 2007. [Google Scholar]
- 14. Geary RS. Antisense oligonucleotide pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol. 2009;5(4):381‐391. doi: 10.1517/17425250902877680 [DOI] [PubMed] [Google Scholar]
- 15. Smith RA, Miller TM, Yamanaka K, et al. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest. 2006;116(8):2290‐2296. doi: 10.1172/jci25424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mazur C, Powers B, Zasadny K, et al. Brain pharmacology of intrathecal antisense oligonucleotides revealed through multimodal imaging. JCI Insight. 2019;4(20):e129240. doi: 10.1172/jci.insight.129240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Monine M, Norris D, Wang Y, Nestorov I. A physiologically‐based pharmacokinetic model to describe antisense oligonucleotide distribution after intrathecal administration. J Pharmacokinet Pharmacodyn. 2021;48(5):639‐654. doi: 10.1007/s10928-021-09761-0 [DOI] [PubMed] [Google Scholar]
- 18. Wurster CD, Winter B, Wollinsky K, et al. Intrathecal administration of nusinersen in adolescent and adult SMA type 2 and 3 patients. J Neurol. 2019;266(1):183‐194. doi: 10.1007/s00415-018-9124-0 [DOI] [PubMed] [Google Scholar]
- 19. Finkel RS, Chiriboga CA, Vajsar J, et al. Treatment of infantile‐onset spinal muscular atrophy with nusinersen: a phase 2, open‐label, dose‐escalation study. Lancet. 2016;388(10063):3017‐3026. [DOI] [PubMed] [Google Scholar]
- 20. Biliouris K, Gaitonde P, Yin W, et al. A semi‐mechanistic population pharmacokinetic model of Nusinersen: an antisense oligonucleotide for the treatment of spinal muscular atrophy. CPT Pharmacometrics Syst Pharmacol. 2018;7(9):581‐592. doi: 10.1002/psp4.12323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luu KT, Norris DA, Gunawan R, Henry S, Geary R, Wang Y. Population pharmacokinetics of nusinersen in the cerebral spinal fluid and plasma of pediatric patients with spinal muscular atrophy following intrathecal administrations. J Clin Pharmacol. 2017;57(8):1031‐1041. doi: 10.1002/jcph.884 [DOI] [PubMed] [Google Scholar]
- 22. Sanwald Ducray P, Frances N, Smart K, et al. Translational pharmacokinetic/pharmacodynamic (PK/PD) modeling strategy to support RG6042 dose selection in Huntington's disease (HD). Neurology. 2019;92(15 Suppl):S16.005. [Google Scholar]
- 23. Ducray PS, Wild EJ, Portron A, et al. Design of an open‐label, adaptive multiple‐dose study to investigate the PK/ PD of RG6042 in CSF and plasma following intrathecal administration in patients with HD. Eur J Neurol. 2019;26(Suppl. 1):16‐111. [Google Scholar]
- 24. Schobel S, Palermo G, Trundell D, et al. A global development program testing RG6042, an anti‐sense oligonucleotide, for the treatment of early manifest Huntington's disease (HD). J Neurol Neurosurg Psychiatry. 2018;89:A98. [Google Scholar]
- 25. Clinicaltrials.gov . NCT03842969. Accessed May, 2019. https://clinicaltrials.gov/ct2/show/NCT03842969
- 26. Jonsson EN, Karlsson MO. Automated covariate model building within NONMEM. Pharm Res. 1998;15(9):1463‐1468. doi: 10.1023/a:1011970125687 [DOI] [PubMed] [Google Scholar]
- 27. Lindbom L, Ribbing J, Jonsson EN. Perl‐speaks‐NONMEM (PsN)‐a Perl module for NONMEM related programming. Comput Methods Programs Biomed. 2004;75(2):85‐94. doi: 10.1016/j.cmpb.2003.11.003 [DOI] [PubMed] [Google Scholar]
- 28. Lindbom L, Pihlgren P, Jonsson EN. PsN‐toolkit‐a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79(3):241‐257. doi: 10.1016/j.cmpb.2005.04.005 [DOI] [PubMed] [Google Scholar]
- 29. Jonsson E, Harling K. Increasing the Efficiency of the Covariate Search Algorithm in the SCM. Population Approach Group Europe (PAGE) meeting in 2018. 2018. Accessed August, 2022. https://www.page‐meeting.org/. [Google Scholar]
- 30. Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 2011;13(2):143‐151. doi: 10.1208/s12248-011-9255-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Beal S, Boeckmann A, Sheiner L. NONMEM User's Guides (1989–2014). Icon Development Solutions; 2018. [Google Scholar]
- 32. R Development Core Team . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2007. [Google Scholar]
- 33. Irby DJ, Ibrahim ME, Dauki AM, et al. Approaches to handling missing or “problematic” pharmacology data: pharmacokinetics. CPT Pharmacometrics Syst Pharmacol. 2021;10(4):291‐308. doi: 10.1002/psp4.12611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28(5):481‐504. doi: 10.1023/a:1012299115260 [DOI] [PubMed] [Google Scholar]
- 35. Barkhof F, Kouwenhoven M, Scheltens P, Sprenger M, Algra P, Valk J. Phase‐contrast cine MR imaging of normal aqueductal CSF flow. Effect of aging and relation to CSF void on modulus MR. Acta Radiol. 1994;35(2):123‐130. [PubMed] [Google Scholar]
- 36. Geary RS, Norris D, Yu R, Bennett CF. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev. 2015;87:46‐51. doi: 10.1016/j.addr.2015.01.008 [DOI] [PubMed] [Google Scholar]
- 37. Yu RZ, Collins JW, Hall S, et al. Population pharmacokinetic‐Pharmacodynamic modeling of Inotersen, an antisense oligonucleotide for treatment of patients with hereditary transthyretin amyloidosis. Nucleic Acid Ther. 2020;30(3):153‐163. doi: 10.1089/nat.2019.0822 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1