

Abstract
Mitochondria oxidative phosphorylation is broadly regulated, and mitochondrial Ca2+ is increasingly recognized to impact substrate flux under both physiological and pathological conditions. Under physiologic conditions, mitochondrial Ca2+ enters through the mitochondrial Ca2+ uniporter and boosts ATP production. However, maintaining Ca2+ homeostasis is crucial as too little Ca2+ inhibits adaptation to stress, while Ca2+ overload can trigger cell death. In this review, we discuss new insights obtained over the past several years expanding the relationship between mitochondrial Ca2+ and oxidative phosphorylation, with most data obtained from heart, liver, or skeletal muscle. Two new themes are emerging. First, beyond boosting ATP synthesis, Ca2+ appears to be a critical determinant of fuel substrate choice between glucose and fatty acids. Second, Ca2+ exerts local effects on the electron transport chain indirectly, not via traditional allosteric mechanisms. These depend critically on the transporters involved, such as the uniporter or the Na+-Ca2+ exchanger. Alteration of these new relationships during disease can be either compensatory or harmful and suggest that targeting mitochondrial Ca2+ may be of therapeutic benefit during diseases featuring impairments in oxidative phosphorylation.
1. Introduction
Ca2+ is a ubiquitous divalent ion essential for cellular excitability, mechanical contraction, signaling, and metabolism(1). Studies over several decades have clearly revealed that Ca2+ within the mitochondria is a potent stimulator of NADH production, via its allosteric potentiation of the activity of membrane transporters, tricarboxylic acid (TCA) cycle dehydrogenases, and pyruvate dehydrogenase phosphatase. The net result of such regulation is a two-fold increase in ATP production. These well-established results have been reviewed previously (2–5). The goal of this mini-review is to discuss the most recent discoveries regarding mitochondrial Ca2+ signaling in relation to the electron transport chain (ETC) and oxidative phosphorylation (OXPHOS) in health and disease. These more recent investigations have expanded the mechanism of Ca2+ regulation beyond direct binding and allosteric effects on mitochondrial enzymes, revealing a multitude of indirect mechanisms that alter the stability, function, and fluxes through OXPHOS (Fig. 1). In many cases, these novel forms of regulation depend on local regulation between the Ca2+ transporters and energetic complexes involved, recapitulating the theme that Ca2+ signaling is often a very local phenomenon.
Figure 1 – ETC and Ca2+ signaling overview.
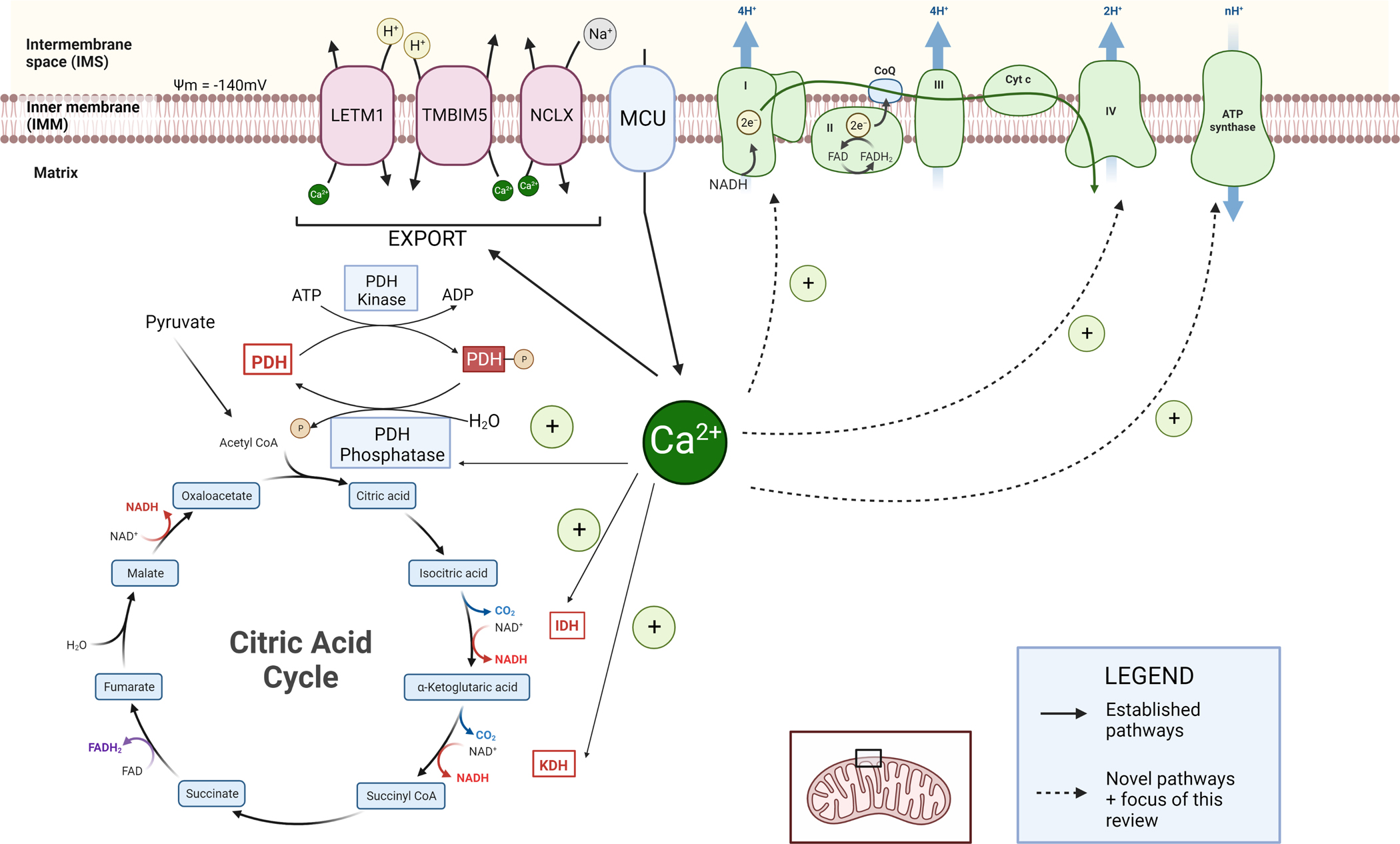
MCU imports Ca2+ which is known to activate the following enzymes: PDH phosphatase, Isocitrate dehydrogenase (IDH), and α-ketoglutarate dehydrogenase (AKGDH). Export mechanisms include LETM1, TMBIM5, and NCLX. Known pathways are shown in solid arrows. Newer mechanisms involve Complex I, CoQ, complex IV, and ATP Synthase, shown in dashed arrows.
2. Ca2+ import into the mitochondria
Ca2+ is normally sequestered in the endoplasmic reticulum (ER) to maintain a low cytoplasmic concentration of ~100 nM (6, 7). In the ER, Ca2+ levels can range from 200 to 500 μM while intra-mitochondrial free Ca2+ levels are typically <100 μM (8, 9). The difference in concentrations within various organelles allow for transient cytoplasmic increases by several orders of magnitude (1–3μM), allowing the cell to utilize Ca2+ as a signaling molecule.
The various Ca2+ transporters involved in mitochondrial Ca2+ uptake have been well defined and reviewed previously(10, 11). Briefly, Ca2+ ions cross the outer mitochondrial membrane (OMM) through voltage-dependent anion channels, then the inner mitochondrial membrane (IMM) primarily through the mitochondrial calcium uniporter (12–15). This Ca2+ channel complex is made up of a pore-forming subunit (MCU), gate-keeping subunits (MICU1–3), an inhibitory subunit (MCUb), and a bridging accessory subunit (EMRE)(15–19). It also associates frequently with other proteins that help regulate Ca2+ influx. To avoid confusion, we refer to the channel complex from here onwards as the uniporter and use subunit abbreviations (e.g. MCU) to refer to specific components of the channel. Through the use of voltage-clamp electrophysiology to isolate mitoplasts (mitochondria devoid of their outer membrane), early studies showed that despite the sea of various cations, the uniporter is highly selective for Ca2+ (20). This is an extreme structural specialization to prevent futile depolarization of the inner mitochondrial membrane potential (ΔΨ) by influx through the uniporter of highly-abundant cytoplasmic ions such as K+, which is ~6 orders of magnitude more abundant than Ca2+ in the cytoplasm. At resting cytosolic Ca2+ levels, MCU activity is kept low by the gate-keeping MICU1–3 subunits which block the pore, while at higher Ca2+ levels, structural rearrangements allow Ca2+ access to the pore and lead to rapid Ca2+ influx into the mitochondrial matrix (16). Other mitochondrial influx pathways have been occasionally reported, but their abundance and relevance to OXPHOS have not been well defined(11).
3. Export of mitochondrial calcium through NCLX, LETM1, TMBIM5, and PTP
To prevent Ca2+ overload and to maintain homeostasis, mitochondria utilize several Ca2+ extrusion strategies. Mitochondrial Ca2+ export is mediated by a Na+-Ca2+ exchanger (NCLX, encoded by the SLC8B1 gene), which couples Ca2+ efflux from the matrix to Na+ influx (21, 22), and Ca2+-H+ exchangers (encoded by LETM1 and TMBIM5, though some controversy exists regarding these) (23–26). NCLX is essential for maintaining mitochondrial homeostasis. Without NCLX, toxic Ca2+ overload leads to organismal lethality (27). Similarly, LETM1 mutations lead to Wolf-Hirschhorn Syndrome, a rare developmental disorder characterized by intellectual disability, seizures, and craniofacial abnormalities (28). Unlike NCLX, the exact mechanism of LETM1 exchange is somewhat controversial. Although studies across multiple labs have clearly established that LETM1 alters Ca2+ efflux (24, 29–31), other research suggests it may be involved in K+-H+ exchange (32). TMBIM5 is a newly discovered potential Ca2+-H+ exchanger, with one possibility being that LETM1 and TMBIM5 are both subunits of a larger Ca2+-H+ exchanger complex.
4. Overview of classical Ca2+ allosteric regulation of the TCA cycle
In classic studies of mitochondrial enzymology, Ca2+ was found to be a key allosteric regulator of multiple mitochondrial enzymes of the TCA cycle, increasing production of NADH and ultimately leading to an approximately two-fold enhancement of ATP synthesis (2, 33–36). Such increases are most evident in cardiac or skeletal muscle during periods of stress or exercise, where increases in cytoplasmic Ca2+ during β-adrenergic stimulation lead to increases in matrix Ca2+. Such a mechanism matches ATP production to energetic demands during stress. In particular, Ca2+ activates several matrix dehydrogenases: isocitrate dehydrogenase (IDH) (37, 38), α-ketoglutarate dehydrogenase (AKGDH) (39, 40), and pyruvate dehydrogenase (PDH) (41). Original descriptions state a K0.5 of IDH at [Ca2+] of 5–50μM (42, 43) while the Km of AKGDH is 0.2μM with ADP or ~2μM with ATP (43). An alternative route of electrons entering the ETC is through the glycerol-3-phosphate shuttle (G3PS). Dihydroxyacetone is converted into glycerol-3-phosphate (G3P) by cystolic glycerol-3-phosphate dehydrogenase (GPD). Mitochondrial GPD2 oxidizes G3P when it diffuses into the intermembrane space. These electrons are transferred to coenzyme Q, providing electrons for OXPHOS. It has been reported that GPD2 is activated by Ca2+-binding (44–46).
Several IMM-embedded metabolite carriers have Ca2+ binding EF-hands facing the intermembrane space, which is similar to the cytoplasm in its Ca2+ levels. Ca2+ binding to these carriers enhances their activity (47–49). Such calcium-binding mitochondrial carriers (CaMC) can be divided into two groups, the aspartate/glutamate exchangers (AGCs, aralar and citrin) or short CaMCs (SCaMC) such as ATP-Mg2+/Pi exchangers. AGCs can be activated by relatively low Ca2+ levels, similar to resting cytosolic levels, whereas SCaMCs can require higher (micromolar) levels of Ca2+ and import ADP into the mitochondria. Deficiency in SCaMC-3/Slc25a23 results in blunted respiration, decreases in ATP, and Ca2+ deregulation(50, 51).
Inside the matrix, the imported aspartate or glutamate can be converted to α-ketoglutarate, the substrate needed for AKGDH, or increase citrate and subsequently isocitrate, used by IDH. Ultimately, enhancing TCA dehydrogenase activity boosts NADH production, feeding the ETC at Complex I and ultimately enhancing ATP production (52). In support of this model, deletion of the Ca2+ uniporter limits the ability of organisms to exercise or increase heart rate during periods of stress.
Although this has been the traditional model, recent analyses across mitochondria, cells, and organs, suggests that allosteric effects of Ca2+ on the TCA cycle may be less prominent than previously thought. In these studies, oxygen consumption, ATP production, or organ function (e.g. muscle contraction) in several cases has been preserved even when components of the uniporter are deleted (53–58). Although Ca2+ is still present in the matrix even after MCU deletion, these data suggest that lack of Ca2+-induced TCA cycle acceleration can perhaps be compensated by other mechanisms. The newer emerging paradigm suggests that though Ca2+ can clearly affect TCA flux and ATP production, its primary effect may occur upstream, in helping determine the efficiency of fuel substrates entering the TCA cycle.
5. Inhibition of the uniporter alters Ca2+ regulation of pyruvate flux, leading to a switch in substrate preference towards fatty acids for OXPHOS.
Recent studies examining cardiac- and skeletal muscle-specific knockouts of MCU, the pore-forming uniporter subunit, reveal that Ca2+ may also be a primary regulator of mitochondrial fuel substrate preference (59–61). This occurs by Ca2+ regulation of the conversion of pyruvate to acetyl-CoA. Pyruvate dehydrogenase (PDH) is inhibited by phosphorylation by PDH kinases, while PDH phosphatases enhance its activity. By enhancing the activity of pyruvate dehydrogenase (PDH) phosphatase, Ca2+ indirectly boosts PDH. Because pyruvate conversion to acetyl CoA is downstream of glycolysis, lowering Ca2+ levels inhibit this pathway. Therefore, in these several models, there is shift of substrate utilization away from glucose towards fatty acids. In particular, after deletion of MCU, investigators found increased rates of fatty acid relative to glucose oxidation, increased PDH phosphorylation, lower levels of malonyl CoA, a key inhibitor of fatty acid oxidation (via CPT1), and activated forms of enzymes involved in β-oxidation, including malonyl CoA decarboxylase and β-hydroxyacyl CoA dehydrogenase. Taken together, these results suggest that, by altering PDH activation, lower levels of Ca2+ produce a profound metabolic rewiring of substrate utilization. Intriguingly, in a separate study, altering pyruvate metabolism also produced a reciprocal change in uniporter activity (62). In several human and mouse cell types, inhibiting glycolysis or mitochondrial pyruvate import led to increased expression of the uniporter gatekeeping subunit MICU1, which apparently inhibited mitochondrial Ca2+ uptake and reduced matrix Ca2+ levels. The authors posited this mechanism may prevent toxic mitochondrial Ca2+ overload during bioenergetic crises caused by nutrient stress.
6. Ca2+ regulation of the ETC and F1-FO-ATP synthase under normal physiology
The data on Ca2+ regulation of flux rates through the ETC and ATP synthase has been somewhat controversial. Multiple early studies on isolated mitochondria or enzyme systems showed Ca2+-dependent boosting of ATP production rates and flux through the ETC (2). However, allosteric binding sites for Ca2+ at physiological concentrations in these systems have been difficult to establish, though there may be Ca2+ induced changes under pathophysiological conditions. Two recent studies of cardiac and skeletal mitochondria using detailed measurements of several intermediate steps in OXPHOS failed to find evidence for Ca2+-induced enhancement of flux through the ETC or ATP synthase (52, 63). In these cases, the Ca2+-induced boost in ATP production was explained by its effects on substrate transport, TCA cycle flux, and changes in mitochondrial membrane potential (ΔΨ), rather than direct effects on the ETC or ATP synthase. In contrast, in another study using skeletal muscle mitochondria, the authors found >2-fold Ca2+-induced increases in conductances through the ETC (distributed across Complexes I, III, and IV) and the ATP synthase (36). Of note, these authors isolated the effects on the ETC by normalizing to NADH production (accounting for effects due to substrate transport or TCA flux); and the effects on the ATP synthase by normalizing to ΔΨ (accounting for effects on the ETC). Explanations for these discrepancies remain open questions, with possibilities including the assays (e.g. respirometry, fluorescent reporters, cytochrome redox absorbance) and the usage of a creatine kinase clamp to mimic physiological constraints, as Ca2+ stimulation of respiration is minimized under the typical assays using saturating levels of ADP (64). Nevertheless, despite the somewhat murky status of direct allosteric regulation of the ETC and ATP synthase by Ca2+, there is a growing body of evidence suggesting mitochondrial Ca2+ is an important indirect regulator of these complexes. We review recent data for these below.
6.1. Complex I
NADH is the substrate utilized by Complex I of the electron transport chain (ETC). Complex I is the first and largest complex of the ETC, consisting of 44-subunit. This L-shaped complex consists of two domains, a transmembrane domain responsible for pumping H+ into the intermembrane space and a matrix arm that protrudes into the matrix space. The matrix arm oxidizes NADH and transfers the electron (e−) down its iron-sulfur (Fe-S) clusters until it reaches coenzyme-Q (CoQ). CoQ shuttles electrons to Complex III (65–67). Current evidence suggests that Ca2+ indirectly stimulates Complex I through increasing the ratio of NADH/NAD+ mostly through the matrix dehydrogenases involved in the TCA.
To date there is no evidence suggesting Ca2+ binding to Complex I. Instead, our group recently discovered a Complex I-uniporter interaction, through which the uniporter appears to exert a protective effect on Complex I, a phenomenon we termed Complex I Induced Protein Turnover (CLIPT) (Fig. 2A) (68). In normal physiologic conditions, some electrons leak out during transfer from NADH to CoQ at a low rate (~1%), producing potentially toxic reactive oxygen species (ROS) that may locally harm Complex I. We found that the N-terminal domain of MCU interacts with Complex I near the site of electron transport through the Fe-S clusters. Leaking electrons tend to oxidize MCU, leading to its degradation. This constant MCU turnover maintains a basal low rate of uniporter channels, potentially protecting Complex I itself from damage caused by this electron leak. In fact, loss of uniporter components has been associated with a near stoichiometric loss of Complex I in cardiac muscle (59, 69, 70).
FIGURE 2 – New mechanisms involving mitochondrial Ca2+.
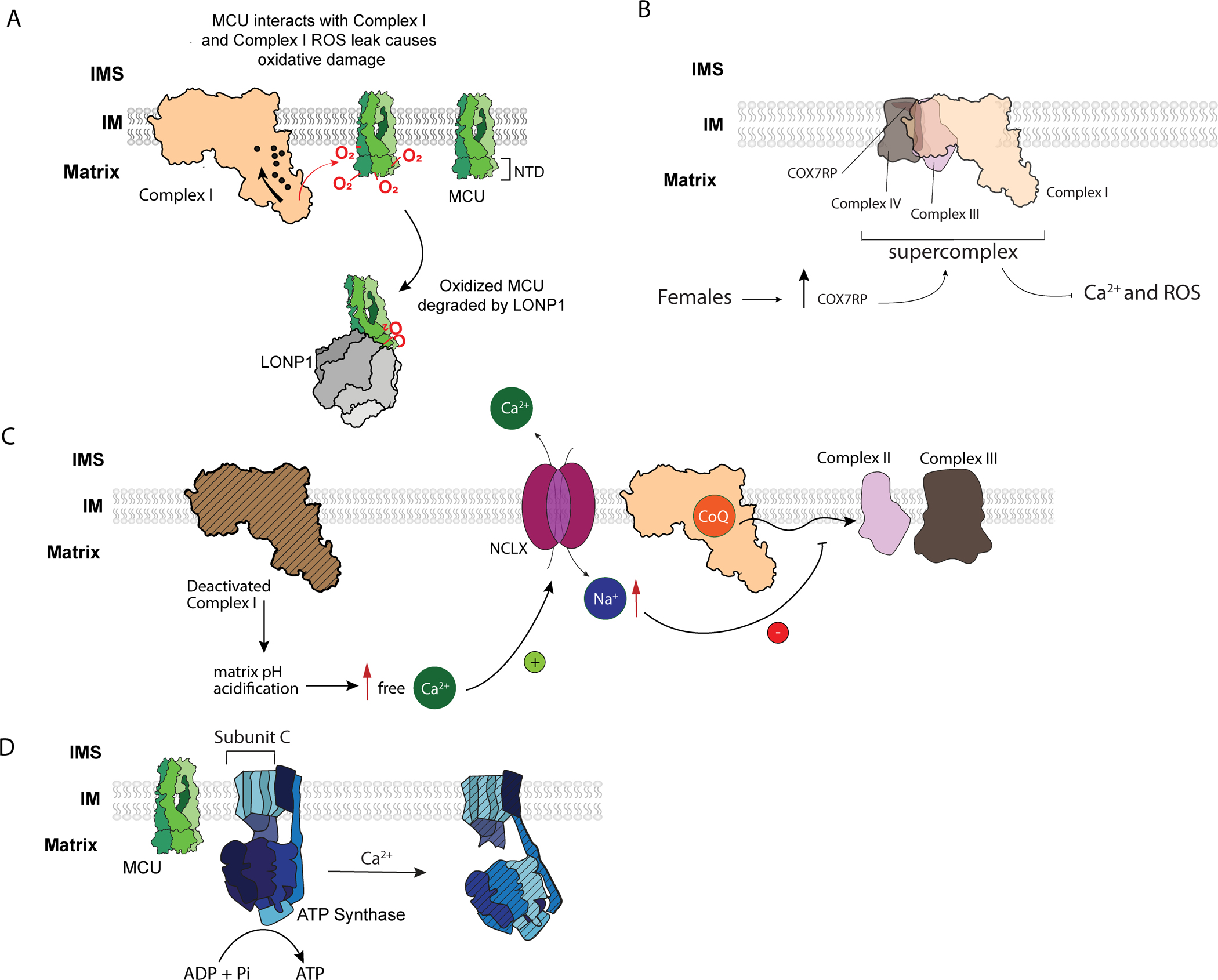
(A) CLIPT – Complex I interacts with MCU and maintains its protein turnover rate. (Adapted from ref. (68))
(B) Complex IV subunit COX7RP is estrogen dependent, sits at the interface between Complex III and IV, and promotes supercomplex assembly and respiratory efficiency. Increasing COX7RP led to reduced mitochondrial Ca2+ and ROS, whereas inhibiting its expression produced the opposite effects (see ref. (86)).
(C) Deactivated complex I promotes matrix acidification which dissolve CaP precipitates. The increased matrix Ca2+ triggers NCLX to exchange Ca2+ for Na+. Increased matrix Na+ decreases IMM fluidity and prevents CoQ diffusion, decreasing oxphos at Complex II + III. (Adapted from ref. (103))
(D) MCU binds to the subunit c of ATP Synthase in Trypanosoma (see ref. (92)). Excess Ca2+ may trigger rearrangement of the F1 component of the ATP synthase to produce channels in the membrane component (see ref. (108)).
6.2. Complex II
Complex II, also known as succinate reductase, is unique in that it is the only one that does not pump protons across the IMM and is directly involved in both the TCA and ETC (71). It contains four protein subunits. It is the second point of electron entry. Complex II couples succinate oxidation to FAD reduction in the TCA cycle, and subsequent FADH2 oxidation to CoQ reduction in the ETC. There is no clear evidence of Ca2+ regulation of Complex II to date.
6.3. Complex III
Complex III, also known as cytochrome c oxidoreductase, is a dimeric structure (72). Electrons from both Complex I and II arrive at Complex III in the form of ubiquinol (CoQH2). At Complex III, a recycling electron loop called the Q-cycle occurs at the two CoQ binding sites. Electrons are ultimately shuttled over to Complex IV by cytochrome c, while 4H+ from 2 CoQH2 are released into the intermembrane space. Although limited older data using Complex III-specific substrates suggested Ca2+ enhancement of flux (73), no specific binding sites or direct allosteric regulation has been confirmed under physiological conditions. Nevertheless, an elegant recent study revealed indirect effects of Ca2+ on Complex III function under pathophysiological conditions (see section 7.2 below).
6.4. Complex IV
The final electron destination is at Complex IV, cytochrome c oxidase, a 13-subunit structure containing 4 redox-active metal centers (74). Two A-type hemes and two copper centers allow electron transfer from cytochrome c to oxygen, the final electron acceptor. Such transfer is coupled to a pumping of 4 H+ to the intermembrane space. Complex IV has a Ca2+ binding site sensitive to Na+ and Ca2+, facing the intermembrane space (75). Although normally bound to Na+ at low cytosolic Ca2+ levels, high Ca2+ ~1μM (previously reported to be 20–30μM (76, 77)) displaces the Na+ and inhibits activity with an IC50 ~0.5μM, though this effect may be evident only during very slow Complex IV activity and may have minimal effect under more rapid physiological rates. This may be an endogenous way to slow down respiration at high Ca2+ levels if substrates are limited (75).
Beyond directly binding Ca2+, Complex IV also may have an interaction with the uniporter. A transmembrane protein of unclear function, MCU regulator 1 (MCUR1), has been identified as both a Complex IV assembly factor as well as a scaffold factor for the uniporter (78–81). Loss of MCUR1 leads to reduced mitochondrial Ca2+ uptake, and in some, though not all studies, to reduced assembly and faster turnover of Complex IV (79). MCUR1 has also been associated with Ca2+-sensing for the permeability transition, and yeast homologs appear to be involved in proline metabolism (79, 82). The mechanism for these various functions remains somewhat mysterious.
6.5. Supercomplex
Complex I, III, and IV are known to exist in a supercomplex form (83, 84). Whether this supercomplex exerts functional effects on energetic flux through the ETC or reflects optimal packing in mitochondrial cristae remains controversial (85). A very recent study explores Ca2+ regulation of supercomplexes in a sex-dependent manner, finding that female rat and healthy human donor ventricular cardiomyocytes displayed lower uniporter, mitochondrial Ca2+, and ROS levels compared to male counterparts though myocardial O2 consumption was unchanged(86). Intriguingly, females appeared to have increased amounts of complexes I, III, and IV assembled into supercomplexes. This depended on the amounts of the estrogen-dependent Complex IV subunit COX7RP, a peptide that sits at the interface between Complex III and IV and promotes supercomplex assembly and respiratory efficiency (87, 88). Increasing COX7RP led to reduced mitochondrial Ca2+ and ROS, whereas inhibiting its expression produced the opposite effects. The combination of increased supercomplex assembly and reduced mitochondrial Ca2+ and ROS was felt to lower the propensity to arrhythmias in pre-menopausal females (Fig 2B).
6.6. ATP Synthase
The F1-FO-ATP Synthase utilizes the ΔΨ produced by the ETC to coupling H+ influx to ATP synthesis from ADP and Pi (89, 90). It is a large (500 kDa) multi-subunit protein mainly divided into the membrane-embedded FO and soluble F1 rotary domains. The two domains are connected by central and side stalks. The catalytic F1 domain protrudes into the matrix and consists of 3 α- and 3 β-subunits and a γδɛ-subunit, with catalytic nucleotide binding sites at the α- and β-subunit interfaces. Within the FO domain, the c-subunit is a highly conserved 75 residue hydrophobic peptide that arranges its 10–12 oligomers into a ring formation, creating the machinery coupling proton movement to rotary movement (91). Although, Ca2+ may bind components of the ATP synthase and alter its behavior during pathophysiological states (see section 7.3 below), direct allosteric regulation has not been shown during normal physiology. Nevertheless, as described above, multiple studies have revealed that Ca2+-induced changes in substrate preference, TCA cycle, and ΔΨ culminate in a two-fold increase in ATP synthesis. Intriguingly, a recent study in trypanosomal parasites showed an interaction between MCU and the membrane-embedded c-subunit of the ATP synthase, while genetic inhibition of trypanosomal MCU reduced ATP production (92). Although an interaction was seen between human MCU and the c-subunit in heterologous systems in that study, confirmation of the interaction in intact mammalian mitochondria or potential functional consequences were not explored. Especially as MCU dimers align in areas of convex membrane curvature (16), contrasted with the convex membrane curvature induced within cristae by ATP synthase dimers, a subpopulation of uniporter channels interacting with the ATP synthase may alter the local environment and modify ATP production.
7. Ca2+ regulation of the ETC and F1-FO-ATP synthase in pathophysiology.
7.1. Complex I impairment triggers compensatory increases in mitochondrial Ca2+ to maintain energetic homeostasis.
Genetically-encoded defects in the OXPHOS machinery belong to a collective group of disorders called mitochondrial diseases. Although rare, mitochondrial disease is the most common in-born error in metabolism occurring in 1 in 5,000 live births, most commonly affecting Complex I of the ETC (93, 94). These diseases can present with a variety of pathologies, often affecting organs with high energetic demand, such as the nervous system, liver, skeletal muscle, and heart. Study of animal and cell models of these diseases have revealed striking new forms of Ca2+ regulation.
During ETC impairment in mitochondrial diseases, our group revealed that increased mitochondrial Ca2+ is a compensatory mechanism needed for survival (95). Previous studies investigating mitochondrial Ca2+ during ETC dysfunction typically found reduced or unchanged Ca2+ uptake and impaired ΔΨ (96–101). Because the level of Ca2+ influx is heavily dependent on ΔΨ, a change to either the ΔΨ or uniporter activity will alter the size of Ca2+ influx and one may mask changes in the other. To overcome confounding results by changes in ΔΨ, pH, or other factors, we used voltage-clamp electrophysiology on mitoplasts (mitochondria devoid of its outer membrane) to fully control ΔΨ, matrix and external solutions. Under these controlled conditions, we found that impairing ETC results in substantial increases in uniporter Ca2+ currents. In fact, closer examination of mitochondria from multiple models of ETC or Complex I impairment reveal an early increase in mitochondrial Ca2+ uptake and uniporter levels, seen by others as well (68, 102). This increase in mitochondrial Ca2+ served to rescue respiration and ATP synthesis to near wild-type levels in animal models of mitochondrial cardiomyopathies (95). The mechanism for this was abrogation of CLIPT, described above in the section on Complex I (section 6.1). Under normal conditions, MCU levels were kept low by transient interactions with Complex I leading to uniporter degradation. However, when Complex I was impaired in these mitochondrial diseases, there was loss of the MCU-Complex I interaction and a striking and sustained increase in uniporter channel levels. Increased channel stability is a post-transcriptional homeostatic mechanism to preserve energetic flux through the remaining ETC. This mechanism was evident across all cell types tested and across species including Drosophila, mice, and humans. In fact, in Drosophila with Complex I deficiency, survival was dependent on the uniporter. Inhibiting uniporter expression caused catastrophic mortality during development, whereas boosting uniporter expression or stability rescued survival to near wild-type levels. This reveals not only the importance of mitochondrial Ca2+ during Complex I dysfunction, but also suggests potential therapeutic targets for these diseases.
7.2. Increases in mitochondrial Ca2+ during hypoxia indirectly limit CoQ diffusion between Complex II and Complex III.
Flux through the Q-cycle of Complex III is thought to depend on the speed of CoQ diffusion from upstream ETC complexes. A recent study elegantly demonstrated how Ca2+ can indirectly regulate CoQ diffusion during pathological states (103). Specifically, during hypoxic injury, Complex I enters a deactivated state which decreases the matrix pH. Within the matrix, Ca2+ is often stored in dense calcium phosphate granules, and matrix acidification is conducive to freeing Ca2+ from these granules. In turn, the increased matrix Ca2+ triggers Ca2+ extrusion through NCLX, which brings more Na+ in. The sudden increase in matrix Na+ during hypoxia was found to decrease the fluidity of the inner mitochondrial membrane, slowing CoQ diffusion from Complex II to Complex III. Ultimately, this causes OXPHOS to slow at this point of the ETC (Fig. 2C). With limited CoQH2 diffusion, the semiquinone already bound within Complex III cannot effectively participate in the Q cycle and excess electrons leak out as potentially toxic superoxide. Thus, unlike the beneficial effects of elevated matrix Ca2+ during Complex I impairment (section 7.1), acute elevations in Ca2+ during hypoxic injury may promote the production of harmful ROS through Complex III.
7.3. Ca2+ binding to the F1 subunit of the ATP synthase may help trigger the permeability transition.
Despite the various physiological roles of Ca2+ listed previously, it has been appreciated for >75 years that excess Ca2+ is deleterious for mitochondrial function (104). Mitochondrial Ca2+ overload opens a mysterious, cyclosporine-sensitive, large-pore channel known as the permeability transition pore (PTP), which leads to catastrophic mitochondrial depolarization, swelling, and structural disruption (105–108). In many cases, this can trigger cell death.
A current leading theory regarding the nature of the PTP is that it occurs via a pathological conformational change in the ATP synthase (109–112). This is based on the finding that a Ca2+-dependent high-conductance channel forms. The data described above suggesting that MCU and the ATP Synthase c-subunit interact in trypanosomes, and perhaps mammals, may further support this theory (92). Mechanistically, the current hypothesis focuses the catalytic site between α and β subunits of the soluble F1 component of the ATP synthase (113). This site typically uses Mg2+ to coordinate ATP or ADP during the synthesis cycle around the central rotor shaft. The current hypothesis posits that at high concentrations, Ca2+ replaces Mg2+ at this nucleotide binding pocket, triggering downstream changes that open a channel within the transmembrane FO component of the ATP synthase (Fig. 2D) (108). In an alternative mechanism, other investigators posited that Ca2+ can trigger the c-subunits to spontaneously fold into a β-sheet conformation that has ion transport activity and resembles amyloidogenic proteins (114). However, because this transition requires extremely high, non-physiological Ca2+ concentrations, (1 mM) its occurrence in intact mitochondria remains questionable.
7.4. Ca2+ regulation of the adenine nucleotide translocator has not been well established.
A problem with the ATP synthase hypothesis for the PTP has been evidence showing the presence of residual PTP-like channels when multiple ATP synthase components have been deleted (115–117). This has led to the resurfacing of an older hypothesis suggesting the existence of a separate class of PTP channels encoded by the adenine nucleotide translocator (ANT) (118), as deletion of multiple ANT isoforms also drastically reduces PTP activity (119) and possible channel-like pores have been seen with crosslinking proteomics and electrophysiology (120, 121). Unfortunately, currently there is little evidence revealing a potential mechanism for Ca2+-induced ANT rearrangement into a PTP-like channel.
8. Future Directions
First, the current literature provides insights on a range of different tissues such as liver, heart, and skeletal muscle, but there is a clear gap in understanding whether these mechanisms are entirely conserved across organs, or whether other novel tissue-specific mechanisms of Ca2+-dependent OXPHOS regulation exist. Second, although data from tissue- or whole-animal knockout studies has provided substantial insight into Ca2+ regulation of metabolism, the ability to acutely perturb mitochondrial Ca2+ levels to investigate how OXPHOS is affected during physiological activities has been hampered by the lack of highly specific compounds that can be given to intact cells or organisms. Drug screens may hopefully identify novel tools to further investigate the impact of mitochondrial Ca2+ fluxes (122–124). Finally, many studies of metabolism rely on steady-state or endpoint measurements. These assays only partly capture significant changes in metabolic flux through pathways such as metabolite transport, TCA cycle, and the ETC, that are impacted by Ca2+ regulation. The advent of new technologies using isotope-labelled tracers for quantitative flux analysis may offer new ways to address this limitation, allowing more granular assessment of how Ca2+ may alter fluxes and enzymatic activity (125). In addition, as sensors for Ca2+, ATP, NAD+, and other metabolites improve, we will have the opportunity to carry out multi-modal assessments simultaneously, to see how these parameters interact.
9. Conclusion.
Compartmentalizing Ca2+ within the cell is an essential attribute for Ca2+ signaling. It is clear that mitochondrial Ca2+ is necessary for normal function and especially during stress or work while Ca2+ overload can result in disease and cell death. Ca2+ is imported through the uniporter, utilizing the ΔΨ created by the ETC, and exported through NCLX, LETM1, and TMBIM5. In normal physiology, it is clear that mitochondrial Ca2+ (1) augments ATP production by stimulating NADH synthesis through the TCA cycle and (2) shifts preference towards glycolysis by its stimulatory effects on PDH flux. Although Ca2+ has few direct allosteric effects on the ETC and ATP synthase, a newer paradigm is arising suggesting that Ca2+ and its transporters are crucial indirect regulators of OXPHOS by altering the stability, assembly, and conformation of these complexes.
Our understanding of mitochondrial Ca2+ in the context of OXPHOS defects is still expanding. These recent discoveries of new binding partners between ETC complexes and Ca2+ regulators are providing more insight into how cells manage mitochondrial Ca2+, especially during stress. Because mitochondrial damage is a central feature of pathophysiology across many organs and diseases, expanding our understanding of how Ca2+ regulates OXPHOS and may worsen or compensate for ETC dysfunction will have therapeutic implications for the future.
PERSPECTIVES.
Mitochondrial Ca2+ has long been known to be an allosteric activator of metabolite transporters, pyruvate-to-acetyl CoA conversion, and tricarboxylic acid (TCA) cycle dehydrogenases, which ultimately increase ATP synthesis two-fold.
Recent data has shown that mitochondrial Ca2+ is a critical regulator of fuel substrate utilization. Reducing mitochondrial Ca2+ influx produces a profound shift away from glycolysis towards fatty acid oxidation.
Recent investigations have expanded the mechanism of Ca2+ regulation beyond direct allosteric effects to indirect mechanisms that alter the stability and function of OXPHOS complexes, particularly Complex I. These novel forms of regulation may be exploited therapeutically.
ACKNOWLEDGEMENTS
Support is from the National Institutes of Health (HL124070 [D.C.], HL141353 [D.C.], HL165806 [S.L.]) and the Nora Eccles Treadwell Foundation (D.C.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATION
- MCU
Mitochondrial calcium uniporter
- ETC
Electron transport chain
- IMM
Inner mitochondrial membrane
- OXPHOS
Oxidative phosphorylation
- TCA
Tricarboxylic acid cycle
- ROS
Reactive Oxygen Species
- PDH
Pyruvate Dehydrogenase
- IDH
Isocitrate Dehydrogenase
- a-KGH
α-ketoglutarate dehydrogenase
- PTP
Permeability transition pore
- ER
Endoplasmic reticulum
- MCU
Mitochondrial calcium uniporter, pore-forming subunit
- MICU1-3
Mitochondrial uptake 1–3
- MCUb
Mitochondrial calcium uniporter dominant negative subunit beta
- EMRE
Essential component of the mitochondrial calcium uniporter
References
- 1.Clapham DE. Calcium signaling. Cell. 2007;131(6):1047–58. DOI: 10.1016/j.cell.2007.11.028 [DOI] [PubMed] [Google Scholar]
- 2.Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry. 2012;51(14):2959–73. DOI: 10.1021/bi2018909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sterea AM, El Hiani Y. The Role of Mitochondrial Calcium Signaling in the Pathophysiology of Cancer Cells. Adv Exp Med Biol. 2020;1131:747–70. DOI: 10.1007/978-3-030-12457-1_30 [DOI] [PubMed] [Google Scholar]
- 4.Walters GC, Usachev YM. Mitochondrial calcium cycling in neuronal function and neurodegeneration. Front Cell Dev Biol. 2023;11:1094356. DOI: 10.3389/fcell.2023.1094356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams GS, Boyman L, Lederer WJ. Mitochondrial calcium and the regulation of metabolism in the heart. J Mol Cell Cardiol. 2015;78:35–45. DOI: 10.1016/j.yjmcc.2014.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marban E, Rink TJ, Tsien RW, Tsien RY. Free calcium in heart muscle at rest and during contraction measured with Ca2+ -sensitive microelectrodes. Nature. 1980;286(5776):845–50. DOI: 10.1038/286845a0 [DOI] [PubMed] [Google Scholar]
- 7.Tsien RY, Rink TJ. Neutral carrier ion-selective microelectrodes for measurement of intracellular free calcium. Biochim Biophys Acta. 1980;599(2):623–38. DOI: 10.1016/0005-2736(80)90205-9 [DOI] [PubMed] [Google Scholar]
- 8.Fernandez-Sanz C, De la Fuente S, Sheu SS. Mitochondrial Ca(2+) concentrations in live cells: quantification methods and discrepancies. FEBS Lett. 2019;593(13):1528–41. DOI: 10.1002/1873-3468.13427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu X, Ginsburg KS, Kettlewell S, Bossuyt J, Smith GL, Bers DM. Measuring local gradients of intramitochondrial [Ca(2+)] in cardiac myocytes during sarcoplasmic reticulum Ca(2+) release. Circ Res. 2013;112(3):424–31. DOI: 10.1161/CIRCRESAHA.111.300501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garbincius JF, Elrod JW. Mitochondrial calcium exchange in physiology and disease. Physiol Rev. 2022;102(2):893–992. DOI: 10.1152/physrev.00041.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giorgi C, Marchi S, Pinton P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat Rev Mol Cell Biol. 2018;19(11):713–30. DOI: 10.1038/s41580-018-0052-8 [DOI] [PubMed] [Google Scholar]
- 12.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476(7360):341–5. DOI: 10.1038/nature10234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaudhuri D, Sancak Y, Mootha VK, Clapham DE. MCU encodes the pore conducting mitochondrial calcium currents. Elife. 2013;2:e00704. DOI: 10.7554/eLife.00704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476(7360):336–40. DOI: 10.1038/nature10230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamer KJ, Mootha VK. The molecular era of the mitochondrial calcium uniporter. Nat Rev Mol Cell Biol. 2015;16(9):545–53. DOI: 10.1038/nrm4039 [DOI] [PubMed] [Google Scholar]
- 16.Fan M, Zhang J, Tsai CW, Orlando BJ, Rodriguez M, Xu Y, et al. Structure and mechanism of the mitochondrial Ca(2+) uniporter holocomplex. Nature. 2020;582(7810):129–33. DOI: 10.1038/s41586-020-2309-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sancak Y, Markhard AL, Kitami T, Kovacs-Bogdan E, Kamer KJ, Udeshi ND, et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science. 2013;342(6164):1379–82. DOI: 10.1126/science.1242993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Nguyen NX, She J, Zeng W, Yang Y, Bai XC, et al. Structural Mechanism of EMRE-Dependent Gating of the Human Mitochondrial Calcium Uniporter. Cell. 2019;177(5):1252–61.e13. DOI: 10.1016/j.cell.2019.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balderas E, Sommakia S, Eberhardt D, Lee S, Chaudhuri D. The structural era of the mitochondrial calcium uniporter. 2023. In: Calcium Signals [Internet]. IOP Publishing; [12-1--22]. Available from: 10.1088/978-0-7503-2009-2ch12. DOI: 10.1088/978-0-7503-2009-2ch12 [DOI] [Google Scholar]
- 20.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427(6972):360–4. DOI: 10.1038/nature02246 [DOI] [PubMed] [Google Scholar]
- 21.Carafoli E, Tiozzo R, Lugli G, Crovetti F, Kratzing C. The release of calcium from heart mitochondria by sodium. J Mol Cell Cardiol. 1974;6(4):361–71. DOI: 10.1016/0022-2828(74)90077-7 [DOI] [PubMed] [Google Scholar]
- 22.Crompton M, Capano M, Carafoli E. The Sodium-Induced Efflux of Calcium from Heart Mitochondria. European Journal Biochemistry. 1976;69:453–62. DOI: [Google Scholar]
- 23.Austin S, Mekis R, Mohammed SEM, Scalise M, Wang WA, Galluccio M, et al. TMBIM5 is the Ca(2+) /H(+) antiporter of mammalian mitochondria. EMBO Rep. 2022;23(12):e54978. DOI: 10.15252/embr.202254978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang D, Zhao L, Clapham DE. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science. 2009;326(5949):144–7. DOI: 10.1126/science.1175145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patron M, Tarasenko D, Nolte H, Kroczek L, Ghosh M, Ohba Y, et al. Regulation of mitochondrial proteostasis by the proton gradient. Embo j. 2022;41(16):e110476. DOI: 10.15252/embj.2021110476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L, Dietsche F, Seitaj B, Rojas-Charry L, Latchman N, Tomar D, et al. TMBIM5 loss of function alters mitochondrial matrix ion homeostasis and causes a skeletal myopathy. Life Sci Alliance. 2022;5(10). DOI: 10.26508/lsa.202201478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, et al. The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability. Nature. 2017;545(7652):93–7. DOI: 10.1038/nature22082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang D, Zhao L, Clish CB, Clapham DE. Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf-Hirschhorn syndrome. Proc Natl Acad Sci U S A. 2013;110(24):E2249–54. DOI: 10.1073/pnas.1308558110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doonan PJ, Chandramoorthy HC, Hoffman NE, Zhang X, Cardenas C, Shanmughapriya S, et al. LETM1-dependent mitochondrial Ca2+ flux modulates cellular bioenergetics and proliferation. FASEB J. 2014;28(11):4936–49. DOI: 10.1096/fj.14-256453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang E, Qu D, Huang T, Rizzi N, Boonying W, Krolak D, et al. PINK1-mediated phosphorylation of LETM1 regulates mitochondrial calcium transport and protects neurons against mitochondrial stress. Nat Commun. 2017;8(1):1399. DOI: 10.1038/s41467-017-01435-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsai MF, Jiang D, Zhao L, Clapham D, Miller C. Functional reconstitution of the mitochondrial Ca2+/H+ antiporter Letm1. J Gen Physiol. 2014;143(1):67–73. DOI: 10.1085/jgp.201311096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Froschauer E, Nowikovsky K, Schweyen RJ. Electroneutral K+/H+ exchange in mitochondrial membrane vesicles involves Yol027/Letm1 proteins. Biochim Biophys Acta. 2005;1711(1):41–8. DOI: 10.1016/j.bbamem.2005.02.018 [DOI] [PubMed] [Google Scholar]
- 33.McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70(2):391–425. DOI: 10.1152/physrev.1990.70.2.391 [DOI] [PubMed] [Google Scholar]
- 34.Territo PR, French SA, Dunleavy MC, Evans FJ, Balaban RS. Calcium activation of heart mitochondrial oxidative phosphorylation: rapid kinetics of mVO2, NADH, AND light scattering. J Biol Chem. 2001;276(4):2586–99. DOI: 10.1074/jbc.M002923200 [DOI] [PubMed] [Google Scholar]
- 35.Territo PR, Mootha VK, French SA, Balaban RS. Ca(2+) activation of heart mitochondrial oxidative phosphorylation: role of the F(0)/F(1)-ATPase. Am J Physiol Cell Physiol. 2000;278(2):C423–35. DOI: 10.1152/ajpcell.2000.278.2.C423 [DOI] [PubMed] [Google Scholar]
- 36.Glancy B, Willis WT, Chess DJ, Balaban RS. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry. 2013;52(16):2793–809. DOI: 10.1021/bi3015983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Denton RM, Richards DA, Chin JG. Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem J. 1978;176(3):899–906. DOI: 10.1042/bj1760899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCormack JG, Denton RM. Role of calcium ions in the regulation of intramitochondrial metabolism. Properties of the Ca2+-sensitive dehydrogenases within intact uncoupled mitochondria from the white and brown adipose tissue of the rat. Biochem J. 1980;190(1):95–105. DOI: 10.1042/bj1900095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Denton RM, McCormack JG, Edgell NJ. Role of calcium ions in the regulation of intramitochondrial metabolism. Effects of Na+, Mg2+ and ruthenium red on the Ca2+-stimulated oxidation of oxoglutarate and on pyruvate dehydrogenase activity in intact rat heart mitochondria. Biochem J. 1980;190(1):107–17. DOI: 10.1042/bj1900107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Panov AV, Scaduto RC Jr. Substrate specific effects of calcium on metabolism of rat heart mitochondria. Am J Physiol. 1996;270(4 Pt 2):H1398–406. DOI: 10.1152/ajpheart.1996.270.4.H1398 [DOI] [PubMed] [Google Scholar]
- 41.Denton RM, Randle PJ, Martin BR. Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase. Biochem J. 1972;128(1):161–3. DOI: 10.1042/bj1280161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rutter GA, Denton RM. The binding of Ca2+ ions to pig heart NAD+-isocitrate dehydrogenase and the 2-oxoglutarate dehydrogenase complex. Biochem J. 1989;263(2):453–62. DOI: 10.1042/bj2630453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rutter GA, Denton RM. Regulation of NAD+-linked isocitrate dehydrogenase and 2-oxoglutarate dehydrogenase by Ca2+ ions within toluene-permeabilized rat heart mitochondria. Interactions with regulation by adenine nucleotides and NADH/NAD+ ratios. Biochem J. 1988;252(1):181–9. DOI: 10.1042/bj2520181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beleznai Z, Szalay L, Jancsik V. Ca2+ and Mg2+ as modulators of mitochondrial L-glycerol-3-phosphate dehydrogenase. Eur J Biochem. 1988;170(3):631–6. DOI: 10.1111/j.1432-1033.1988.tb13744.x [DOI] [PubMed] [Google Scholar]
- 45.Wohlrab H The divalent cation requirement of the mitochondrial glycerol-3-phosphate dehydrogenase. Biochim Biophys Acta. 1977;462(1):102–12. DOI: 10.1016/0005-2728(77)90192-x [DOI] [PubMed] [Google Scholar]
- 46.MacDonald MJ, Brown LJ. Calcium activation of mitochondrial glycerol phosphate dehydrogenase restudied. Arch Biochem Biophys. 1996;326(1):79–84. DOI: 10.1006/abbi.1996.0049 [DOI] [PubMed] [Google Scholar]
- 47.Contreras L, Gomez-Puertas P, Iijima M, Kobayashi K, Saheki T, Satrustegui J. Ca2+ Activation kinetics of the two aspartate-glutamate mitochondrial carriers, aralar and citrin: role in the heart malate-aspartate NADH shuttle. J Biol Chem. 2007;282(10):7098–106. DOI: 10.1074/jbc.M610491200 [DOI] [PubMed] [Google Scholar]
- 48.Del Arco A, Contreras L, Pardo B, Satrustegui J. Calcium regulation of mitochondrial carriers. Biochim Biophys Acta. 2016;1863(10):2413–21. DOI: 10.1016/j.bbamcr.2016.03.024 [DOI] [PubMed] [Google Scholar]
- 49.Hajnóczky G, Booth D, Csordás G, Debattisti V, Golenár T, Naghdi S, et al. Reliance of ER-mitochondrial calcium signaling on mitochondrial EF-hand Ca2+ binding proteins: Miros, MICUs, LETM1 and solute carriers. Curr Opin Cell Biol. 2014;29:133–41. DOI: 10.1016/j.ceb.2014.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Amigo I, Traba J, Gonzalez-Barroso MM, Rueda CB, Fernandez M, Rial E, et al. Glucagon regulation of oxidative phosphorylation requires an increase in matrix adenine nucleotide content through Ca2+ activation of the mitochondrial ATP-Mg/Pi carrier SCaMC-3. J Biol Chem. 2013;288(11):7791–802. DOI: 10.1074/jbc.M112.409144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rueda CB, Traba J, Amigo I, Llorente-Folch I, Gonzalez-Sanchez P, Pardo B, et al. Mitochondrial ATP-Mg/Pi carrier SCaMC-3/Slc25a23 counteracts PARP-1-dependent fall in mitochondrial ATP caused by excitotoxic insults in neurons. J Neurosci. 2015;35(8):3566–81. DOI: 10.1523/JNEUROSCI.2702-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wescott AP, Kao JPY, Lederer WJ, Boyman L. Voltage-energized Calcium-sensitive ATP Production by Mitochondria. Nat Metab. 2019;1(10):975–84. DOI: 10.1038/s42255-019-0126-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, et al. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep. 2015;12(1):15–22. DOI: 10.1016/j.celrep.2015.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15(12):1464–72. DOI: 10.1038/ncb2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015;12(1):23–34. DOI: 10.1016/j.celrep.2015.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kosmach A, Roman B, Sun J, Femnou A, Zhang F, Liu C, et al. Monitoring mitochondrial calcium and metabolism in the beating MCU-KO heart. Cell Rep. 2021;37(3):109846. DOI: 10.1016/j.celrep.2021.109846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu JC, Syder NC, Ghorashi NS, Willingham TB, Parks RJ, Sun J, et al. EMRE is essential for mitochondrial calcium uniporter activity in a mouse model. JCI Insight. 2020;5(4). DOI: 10.1172/jci.insight.134063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Holmström KM, Pan X, Liu JC, Menazza S, Liu J, Nguyen TT, et al. Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J Mol Cell Cardiol. 2015;85:178–82. DOI: 10.1016/j.yjmcc.2015.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Altamimi TR, Karwi QG, Uddin GM, Fukushima A, Kwong JQ, Molkentin JD, et al. Cardiac-specific deficiency of the mitochondrial calcium uniporter augments fatty acid oxidation and functional reserve. J Mol Cell Cardiol. 2019;127:223–31. DOI: 10.1016/j.yjmcc.2018.12.019 [DOI] [PubMed] [Google Scholar]
- 60.Kwong JQ, Huo J, Bround MJ, Boyer JG, Schwanekamp JA, Ghazal N, et al. The mitochondrial calcium uniporter underlies metabolic fuel preference in skeletal muscle. JCI Insight. 2018;3(22). DOI: 10.1172/jci.insight.121689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gherardi G, Nogara L, Ciciliot S, Fadini GP, Blaauw B, Braghetta P, et al. Loss of mitochondrial calcium uniporter rewires skeletal muscle metabolism and substrate preference. Cell Death Differ. 2019;26(2):362–81. DOI: 10.1038/s41418-018-0191-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nemani N, Dong Z, Daw CC, Madaris TR, Ramachandran K, Enslow BT, et al. Mitochondrial pyruvate and fatty acid flux modulate MICU1-dependent control of MCU activity. Sci Signal. 2020;13(628). DOI: 10.1126/scisignal.aaz6206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fisher-Wellman KH, Davidson MT, Narowski TM, Lin CT, Koves TR, Muoio DM. Mitochondrial Diagnostics: A Multiplexed Assay Platform for Comprehensive Assessment of Mitochondrial Energy Fluxes. Cell Rep. 2018;24(13):3593–606 e10. DOI: 10.1016/j.celrep.2018.08.091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rutter GA, McCormack JG, Halestrap AP, Denton RM. The roles of cytosolic and intramitochondrial Ca(2+) and the mitochondrial Ca(2+)-uniporter (MCU) in the stimulation of mammalian oxidative phosphorylation. J Biol Chem. 2020;295(30):10506. DOI: 10.1074/jbc.L120.013975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hunte C, Zickermann V, Brandt U. Functional modules and structural basis of conformational coupling in mitochondrial complex I. Science. 2010;329(5990):448–51. DOI: 10.1126/science.1191046 [DOI] [PubMed] [Google Scholar]
- 66.Baradaran R, Berrisford JM, Minhas GS, Sazanov LA. Crystal structure of the entire respiratory complex I. Nature. 2013;494(7438):443–8. DOI: 10.1038/nature11871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhu J, Vinothkumar KR, Hirst J. Structure of mammalian respiratory complex I. Nature. 2016;536(7616):354–8. DOI: 10.1038/nature19095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Balderas E, Eberhardt DR, Lee S, Pleinis JM, Sommakia S, Balynas AM, et al. Mitochondrial calcium uniporter stabilization preserves energetic homeostasis during Complex I impairment. Nat Commun. 2022;13(1):2769. DOI: 10.1038/s41467-022-30236-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chapoy Villanueva H, Sung JH, Stevens JA, Zhang MJ, Nelson PM, Denduluri LS, et al. Distinct effects of cardiac mitochondrial calcium uniporter inactivation via EMRE deletion in the short and long term. J Mol Cell Cardiol. 2023;181:33–45. DOI: 10.1016/j.yjmcc.2023.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Balderas E, Chaudhuri D. Absence (of the uniporter) makes the heart grow fonder: The cardiac response to injury adapts after prolonged EMRE inhibition. J Mol Cell Cardiol. 2023;181:31–2. DOI: 10.1016/j.yjmcc.2023.05.006 [DOI] [PubMed] [Google Scholar]
- 71.Sun F, Huo X, Zhai Y, Wang A, Xu J, Su D, et al. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell. 2005;121(7):1043–57. DOI: 10.1016/j.cell.2005.05.025 [DOI] [PubMed] [Google Scholar]
- 72.Zhang Z, Huang L, Shulmeister VM, Chi YI, Kim KK, Hung LW, et al. Electron transfer by domain movement in cytochrome bc1. Nature. 1998;392(6677):677–84. DOI: 10.1038/33612 [DOI] [PubMed] [Google Scholar]
- 73.Murphy AN, Kelleher JK, Fiskum G. Submicromolar Ca2+ regulates phosphorylating respiration by normal rat liver and AS-30D hepatoma mitochondria by different mechanisms. J Biol Chem. 1990;265(18):10527–34. DOI: [PubMed] [Google Scholar]
- 74.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, et al. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A. Science. 1996;272(5265):1136–44. DOI: 10.1126/science.272.5265.1136 [DOI] [PubMed] [Google Scholar]
- 75.Vygodina T, Kirichenko A, Konstantinov AA. Direct regulation of cytochrome c oxidase by calcium ions. PLoS One. 2013;8(9):e74436. DOI: 10.1371/journal.pone.0074436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wikstrom M, Saari H. A spectral shift in cytochrome a induced by calcium ions. Biochim Biophys Acta. 1975;408(2):170–9. DOI: 10.1016/0005-2728(75)90009-2 [DOI] [PubMed] [Google Scholar]
- 77.Saari H, Penttila T, Wikstrom M. Interactions of Ca2+ and H+ with heme A in cytochrome oxidase. J Bioenerg Biomembr. 1980;12(3–4):325–38. DOI: 10.1007/BF00744692 [DOI] [PubMed] [Google Scholar]
- 78.Vais H, Tanis JE, Müller M, Payne R, Mallilankaraman K, Foskett JK. MCUR1, CCDC90A, Is a Regulator of the Mitochondrial Calcium Uniporter. Cell Metab. 2015;22(4):533–5. DOI: 10.1016/j.cmet.2015.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, Hoffman NE, et al. MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell Rep. 2016;15(8):1673–85. DOI: 10.1016/j.celrep.2016.04.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Paupe V, Prudent J, Dassa EP, Rendon OZ, Shoubridge EA. CCDC90A (MCUR1) is a cytochrome c oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metab. 2015;21(1):109–16. DOI: 10.1016/j.cmet.2014.12.004 [DOI] [PubMed] [Google Scholar]
- 81.Mallilankaraman K, Cárdenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenár T, et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol. 2012;14(12):1336–43. DOI: 10.1038/ncb2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chaudhuri D, Artiga DJ, Abiria SA, Clapham DE. Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc Natl Acad Sci U S A. 2016;113(13):E1872–80. DOI: 10.1073/pnas.1602264113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Letts JA, Fiedorczuk K, Sazanov LA. The architecture of respiratory supercomplexes. Nature. 2016;537(7622):644–8. DOI: 10.1038/nature19774 [DOI] [PubMed] [Google Scholar]
- 84.Gu J, Wu M, Guo R, Yan K, Lei J, Gao N, et al. The architecture of the mammalian respirasome. Nature. 2016;537(7622):639–43. DOI: 10.1038/nature19359 [DOI] [PubMed] [Google Scholar]
- 85.Milenkovic D, Blaza JN, Larsson NG, Hirst J. The Enigma of the Respiratory Chain Supercomplex. Cell Metab. 2017;25(4):765–76. DOI: 10.1016/j.cmet.2017.03.009 [DOI] [PubMed] [Google Scholar]
- 86.Clements RT, Terentyeva R, Hamilton S, Janssen PML, Roder K, Martin BY, et al. Sexual dimorphism in bidirectional SR-mitochondria crosstalk in ventricular cardiomyocytes. Basic Res Cardiol. 2023;118(1):15. DOI: 10.1007/s00395-023-00988-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ikeda K, Shiba S, Horie-Inoue K, Shimokata K, Inoue S. A stabilizing factor for mitochondrial respiratory supercomplex assembly regulates energy metabolism in muscle. Nat Commun. 2013;4:2147. DOI: 10.1038/ncomms3147 [DOI] [PubMed] [Google Scholar]
- 88.Cogliati S, Calvo E, Loureiro M, Guaras AM, Nieto-Arellano R, Garcia-Poyatos C, et al. Mechanism of super-assembly of respiratory complexes III and IV. Nature. 2016;539(7630):579–82. DOI: 10.1038/nature20157 [DOI] [PubMed] [Google Scholar]
- 89.Abrahams JP, Leslie AG, Lutter R, Walker JE. Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria. Nature. 1994;370(6491):621–8. DOI: 10.1038/370621a0 [DOI] [PubMed] [Google Scholar]
- 90.Stock D, Leslie AG, Walker JE. Molecular architecture of the rotary motor in ATP synthase. Science. 1999;286(5445):1700–5. DOI: 10.1126/science.286.5445.1700 [DOI] [PubMed] [Google Scholar]
- 91.Yoshida M, Muneyuki E, Hisabori T. ATP synthase--a marvellous rotary engine of the cell. Nat Rev Mol Cell Biol. 2001;2(9):669–77. DOI: 10.1038/35089509 [DOI] [PubMed] [Google Scholar]
- 92.Huang G, Docampo R. The Mitochondrial Calcium Uniporter Interacts with Subunit c of the ATP Synthase of Trypanosomes and Humans. mBio. 2020;11(2). DOI: 10.1128/mBio.00268-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126(Pt 8):1905–12. DOI: 10.1093/brain/awg170 [DOI] [PubMed] [Google Scholar]
- 94.Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, et al. Mitochondrial diseases. Nat Rev Dis Primers. 2016;2:16080. DOI: 10.1038/nrdp.2016.80 [DOI] [PubMed] [Google Scholar]
- 95.Sommakia S, Houlihan PR, Deane SS, Simcox JA, Torres NS, Jeong MY, et al. Mitochondrial cardiomyopathies feature increased uptake and diminished efflux of mitochondrial calcium. J Mol Cell Cardiol. 2017;113:22–32. DOI: 10.1016/j.yjmcc.2017.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Karamanlidis G, Lee CF, Garcia-Menendez L, Kolwicz SC Jr., Suthammarak W, Gong G, et al. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 2013;18(2):239–50. DOI: 10.1016/j.cmet.2013.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Granatiero V, Giorgio V, Cali T, Patron M, Brini M, Bernardi P, et al. Reduced mitochondrial Ca(2+) transients stimulate autophagy in human fibroblasts carrying the 13514A>G mutation of the ND5 subunit of NADH dehydrogenase. Cell Death Differ. 2016;23(2):231–41. DOI: 10.1038/cdd.2015.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Aydin J, Andersson DC, Hanninen SL, Wredenberg A, Tavi P, Park CB, et al. Increased mitochondrial Ca2+ and decreased sarcoplasmic reticulum Ca2+ in mitochondrial myopathy. Hum Mol Genet. 2009;18(2):278–88. DOI: 10.1093/hmg/ddn355 [DOI] [PubMed] [Google Scholar]
- 99.Brini M, Pinton P, King MP, Davidson M, Schon EA, Rizzuto R. A calcium signaling defect in the pathogenesis of a mitochondrial DNA inherited oxidative phosphorylation deficiency. Nat Med. 1999;5(8):951–4. DOI: 10.1038/11396 [DOI] [PubMed] [Google Scholar]
- 100.Willems PH, Valsecchi F, Distelmaier F, Verkaart S, Visch HJ, Smeitink JA, et al. Mitochondrial Ca2+ homeostasis in human NADH:ubiquinone oxidoreductase deficiency. Cell Calcium. 2008;44(1):123–33. DOI: 10.1016/j.ceca.2008.01.002 [DOI] [PubMed] [Google Scholar]
- 101.McKenzie M, Duchen MR. Impaired Cellular Bioenergetics Causes Mitochondrial Calcium Handling Defects in MT-ND5 Mutant Cybrids. PLoS One. 2016;11(4):e0154371. DOI: 10.1371/journal.pone.0154371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kuhl I, Miranda M, Atanassov I, Kuznetsova I, Hinze Y, Mourier A, et al. Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. Elife. 2017;6. DOI: 10.7554/eLife.30952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hernansanz-Agustin P, Choya-Foces C, Carregal-Romero S, Ramos E, Oliva T, Villa-Pina T, et al. Na(+) controls hypoxic signalling by the mitochondrial respiratory chain. Nature. 2020;586(7828):287–91. DOI: 10.1038/s41586-020-2551-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Potter VR. The assay of animal tissues for respiratory enzymes; further studies on oxidative phosphorylation. J Biol Chem. 1947;169(1):17–37. DOI: [PubMed] [Google Scholar]
- 105.Hunter DR, Haworth RA, Southard JH. Relationship between configuration, function, and permeability in calcium-treated mitochondria. J Biol Chem. 1976;251(16):5069–77. DOI: [PubMed] [Google Scholar]
- 106.Strzelecki T, Kumar S, Khauli R, Menon M. Impairment by cyclosporine of membrane-mediated functions in kidney mitochondria. Kidney Int. 1988;34(2):234–40. DOI: 10.1038/ki.1988.170 [DOI] [PubMed] [Google Scholar]
- 107.Fournier N, Ducet G, Crevat A. Action of cyclosporine on mitochondrial calcium fluxes. J Bioenerg Biomembr. 1987;19(3):297–303. DOI: 10.1007/bf00762419 [DOI] [PubMed] [Google Scholar]
- 108.Bernardi P, Carraro M, Lippe G. The mitochondrial permeability transition: Recent progress and open questions. Febs j. 2022;289(22):7051–74. DOI: 10.1111/febs.16254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Urbani A, Giorgio V, Carrer A, Franchin C, Arrigoni G, Jiko C, et al. Purified F-ATP synthase forms a Ca(2+)-dependent high-conductance channel matching the mitochondrial permeability transition pore. Nat Commun. 2019;10(1):4341. DOI: 10.1038/s41467-019-12331-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Neginskaya MA, Solesio ME, Berezhnaya EV, Amodeo GF, Mnatsakanyan N, Jonas EA, et al. ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore. Cell Rep. 2019;26(1):11–7 e2. DOI: 10.1016/j.celrep.2018.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci U S A. 2014;111(29):10580–5. DOI: 10.1073/pnas.1401591111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A. 2013;110(15):5887–92. DOI: 10.1073/pnas.1217823110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Giorgio V, Burchell V, Schiavone M, Bassot C, Minervini G, Petronilli V, et al. Ca(2+) binding to F-ATP synthase β subunit triggers the mitochondrial permeability transition. EMBO Rep. 2017;18(7):1065–76. DOI: 10.15252/embr.201643354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Amodeo GF, Lee BY, Krilyuk N, Filice CT, Valyuk D, Otzen DE, et al. C subunit of the ATP synthase is an amyloidogenic calcium dependent channel-forming peptide with possible implications in mitochondrial permeability transition. Sci Rep. 2021;11(1):8744. DOI: 10.1038/s41598-021-88157-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Carroll J, He J, Ding S, Fearnley IM, Walker JE. Persistence of the permeability transition pore in human mitochondria devoid of an assembled ATP synthase. Proc Natl Acad Sci U S A. 2019;116(26):12816–21. DOI: 10.1073/pnas.1904005116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.He J, Carroll J, Ding S, Fearnley IM, Walker JE. Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase. Proc Natl Acad Sci U S A. 2017;114(34):9086–91. DOI: 10.1073/pnas.1711201114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.He J, Ford HC, Carroll J, Ding S, Fearnley IM, Walker JE. Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc Natl Acad Sci U S A. 2017;114(13):3409–14. DOI: 10.1073/pnas.1702357114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Brustovetsky N The Role of Adenine Nucleotide Translocase in the Mitochondrial Permeability Transition. Cells. 2020;9(12). DOI: 10.3390/cells9122686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Karch J, Bround MJ, Khalil H, Sargent MA, Latchman N, Terada N, et al. Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci Adv. 2019;5(8):eaaw4597. DOI: 10.1126/sciadv.aaw4597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Caudal A, Tang X, Chavez JD, Keller A, Mohr JP, Bakhtina AA, et al. Mitochondrial interactome quantitation reveals structural changes in metabolic machinery in the failing murine heart. Nat Cardiovasc Res. 2022;1(9):855–66. DOI: 10.1038/s44161-022-00127-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Neginskaya MA, Solesio ME, Berezhnaya EV, Amodeo GF, Mnatsakanyan N, Jonas EA, et al. ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore. Cell Rep. 2019;26(1):11–7.e2. DOI: 10.1016/j.celrep.2018.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Márta K, Hasan P, Rodríguez-Prados M, Paillard M, Hajnóczky G. Pharmacological inhibition of the mitochondrial Ca(2+) uniporter: Relevance for pathophysiology and human therapy. J Mol Cell Cardiol. 2021;151:135–44. DOI: 10.1016/j.yjmcc.2020.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Arduino DM, Wettmarshausen J, Vais H, Navas-Navarro P, Cheng Y, Leimpek A, et al. Systematic Identification of MCU Modulators by Orthogonal Interspecies Chemical Screening. Mol Cell. 2017;67(4):711–23.e7. DOI: 10.1016/j.molcel.2017.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Di Marco G, Vallese F, Jourde B, Bergsdorf C, Sturlese M, De Mario A, et al. A High-Throughput Screening Identifies MICU1 Targeting Compounds. Cell Rep. 2020;30(7):2321–31.e6. DOI: 10.1016/j.celrep.2020.01.081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bartman CR, TeSlaa T, Rabinowitz JD. Quantitative flux analysis in mammals. Nat Metab. 2021;3(7):896–908. DOI: 10.1038/s42255-021-00419-2 [DOI] [PMC free article] [PubMed] [Google Scholar]