

Abstract
Objective
To investigate the relationships between CAG repeat length in the huntingtin gene and cognitive performance in participants above and below the disease threshold for Huntington disease (HD), we performed a cross-sectional analysis of the Enroll-HD database.
Methods
We analyzed data from young, developing adults (≤30 years of age) without a history of depression, apathy, or cognitive deficits. We included participants with and without the gene expansion (CAG ≥36) for HD. All participants had to have a Total Functional Capacity Score of 13, a diagnostic confidence level of zero, and a total motor score of <10 and had to be >28.6 years from their predicted motor onset. We performed regression analyses to investigate the nonlinear relationship between CAG repeat length and various cognitive measures controlling for age, sex, and education level.
Results
There were significant positive relationships between CAG repeat length and the Symbol Digit Modalities, Stroop Color Naming, and Stroop Interference test scores. There were significant negative relationships between CAG repeat length and scores on Parts A and B of the Trails Making Test (p < 0.05), indicating that longer CAG repeat lengths were associated with better performance.
Discussion
An increasing number of CAG repeats in the huntingtin gene below disease threshold and low pathologic CAG ranges were associated with some improvements in cognitive performance. These findings outline the relationship between CAG repeats within the huntingtin gene and cognitive development.
Classification of Evidence
This study provides Class IV evidence that CAG repeat length is positively associated with cognitive function across a spectrum of CAG repeat lengths.
Huntington disease (HD) is a neurodegenerative disorder caused by a pathologic number of CAG repeats (≥40) within the huntingtin gene that leads to a mutated huntingtin protein (HTT).1 The huntingtin gene is highly conserved across species, and there is a direct relationship between the number of CAG repeats within the protein and phylogenetic proximity to humans.2 Numerous genes with triplet repeats are present in the normal, healthy population. In these genes, an abnormal number of triplet repeats often leads to neurodegenerative disorders beyond a specific threshold.3
The wide variation in the number of triplet repeats within specific genes can affect the function of the protein for which they encode4-6 and can lead to broad phenotypic expression above and below the disease threshold. The ability of triplet repeats in specific genes to influence variation in phenotypes has prompted hypotheses that they play a critical role in evolution. Specifically, the huntingtin gene plays a critical role in neurodevelopment and brain maturation.7-11 Therefore, it is possible that the number of repeats within the huntingtin gene may confer neurodevelopmental advantages and disadvantages. Previous studies have examined how the number of repeats within certain genes can affect variation of specific traits in animals,12 but studies in humans are limited.
This study aimed to investigate the relationship between CAG repeat length and cognition in participants from the Enroll-HD database. To evaluate developmental effects of CAG repeat length on cognitive performance, we limited our analysis to subjects far from their predicted motor onset.
Methods
We performed a cross-sectional analysis of the Enroll-HD platform to answer the primary research question of whether CAG repeat length was associated with differences in cognitive measures (Classification of Evidence Level IV). Specifically, “Enroll-HD is a global clinical research platform designed to facilitate research in HD.13 Core datasets are collected annually from all research participants as part of this multi-center longitudinal observational study. Data are monitored for quality and accuracy using a risk-based monitoring approach. All participating sites are required to obtain (and maintain) local ethics committee approvals” (enroll-hd.org, Publication Policy). All participants provide signed informed consent for their data to be included. At the time of this analysis, the fifth periodic dataset was the most up-to-date version of available data. This version of the database includes 21,116 participants from >150 sites around the world; participants may have pre–motor-manifest HD or motor-manifest HD or may be genotype-negative participants, family controls, and healthy controls.
Participant Selection
For the current study, we aimed to include only young participants who were still developing and who presumably had not begun to undergo significant, measurable neurodegeneration. We included participants who were ≤30 years of age,14 and these participants had to have a total motor score of <10, a total functional score of 13, and a diagnostic confidence level of zero according to the Unified Huntington's Disease Rating Scale.15 Participants were included only if they were >28.6 years from their predicted motor onset according to the Langbehn formula.16 A recent study found that young adults with the gene mutation that causes HD showed subtle signs of neurodegeneration as early as 23.6 years before their predicted motor onset.11 For our analyses, we were interested in evaluating cognition as a function of CAG repeat length independently of neurodegeneration. Therefore, by including participants who were >5 years from the earliest time of the initiation of neurodegeneration, we ensured that our analyses were investigating the effect of CAG repeat on development in participants who were unlikely to have begun the neurodegenerative process. From these criteria, the longest possible CAG repeat length was 43. Last, participants were excluded if they had a history of depression, apathy, or cognitive impairment to ensure that these potential confounders were not significantly affecting our results. We analyzed cross-sectional data from the baseline Enroll visit for each participant. These exclusion criteria are further outlined in figure 1.
Figure 1. Flowchart of Patient Exclusions.

This flowchart shows how many participants were excluded on the basis of the exclusionary criteria outlined in the text. DCL = diagnostic confidence level; TFC = Total Functional Capacity Score; TMS = total motor score; YTO = years to onset.
Cognitive Measures
The cognitive measures used in these analyses were collected by trained clinical personnel. Training includes, but is not limited to, practice videos, test assessments used to certify raters, trainings during investigator meetings, didactic teaching, and online videos. Study personnel undergo periodic recertification (enroll-hd.org). Core assessments are performed during each Enroll-HD visit, and extended assessments are collected to the extent possible on all participants. The core cognitive assessments that are available in the Enroll-HD platform are the Categorical Verbal Fluency Test, Symbol Digit Modalities Test (SDMT), the Stroop Color Naming Test (SCNT), and the Stroop Word Reading Test (SWRT). For the categorical verbal fluency test, performance was measured as the number of correctly generated words within 60 seconds. This test is generally used to investigate semantic memory.17 For the SDMT, the score is the number of numbers that were correctly paired to geometric figures within 90 seconds and is used primarily to assess visual attention and processing speed.18 For the Stroop assessments, the measure of performance is the total number of correctly identified colors or words named by the participant. The SCNT and SWRT are used to assess attention and executive function.18,19 The extended cognitive assessments that are performed as part of the Enroll-HD protocol are the Stroop Interference Test (SIT), Parts A and B of the Trail Making Tests, a Letter Verbal Fluency Test, and the Mini-Mental State Examination (MMSE). We assessed the SIT, Trail Making Tests, and the letter verbal fluency tests, but we did not assess the MMSE. The MMSE is used primarily in older adults to determine whether the examinees have cognitive impairment. Given that we included only young participants who were far from their predicted motor onset of HD and who did not have a history of cognitive impairment, the MMSE was not an appropriate tool to assess this patient population. For the SIT, the measure used was the total number of words accurately named, and this measure assesses response inhibition.18 For the Trail Making Tests, the measure used was the total time required to complete these tasks. The Trail Making Tests measure a number of cognitive functions, most notably speed and fluidity of cognitive abilities, as well as executive function.20 For the letter verbal fluency test, performance was measured as the number of correctly generated words within 60 seconds and assessed primarily semantic memory.17
Enroll-HD assessments are not to last longer than 2.5 hours. This is used to prevent participants from becoming overly fatigued during assessments, which is important in the setting of these analyses. In addition, all participants undergo DNA genotyping at their first Enroll-HD assessment for research purposes to quantify their CAG repeat length. This information is not made available to the participants and is not performed as part of a clinical protocol that includes genetic counseling. Some participants may have already undergone genetic testing at the time of their first Enroll-HD visit, but this information is not made available.21
Statistical Considerations
The Enroll-HD platform includes longitudinal data for participants and is actively enrolling new participants. Consequently, some participants have data available from only 1 visit, while others may have data from multiple time points. To ensure that participants with >1 visit were not falsely inducing significant results, we included only the last known visit for participants who met the inclusions criteria. A cognitive score that was >5 SDs from the group mean was removed to decrease the impact of outliers. We then constructed nonlinear regression models to investigate the relationship between CAG repeat length and cognition. All models included the covariates of age, sex, and education. Specifically, the Enroll-HD study has adopted the International Standard Classification of Education codes to classify educational attainment. The impact of CAG repeat length on cognition was presumed to be nonlinear. Consequently, the independent variable of interest, CAG repeat length, was transformed to a nonlinear variable with the use of natural cubic splines. A separate model was run for all 8 of the cognitive measures investigated. Therefore, all p values were corrected for these multiple comparisons with false discovery rate correction. A false discovery rate–corrected value of p < 0.05 was considered statistically significant for all analyses. Some participants had missing data for specific cognitive measurements. Only participants with the cognitive measure of interest available were included in an individual analysis. RStudio was used to perform all statistical analyses.
Standard Protocol Approvals, Registrations, and Patient Consents
All sites were required to obtain and maintain local Ethics Committee approvals. Participants must have signed informed consent forms for their data to be included in the datasets.21
Data Availability
These results were generated with the Enroll-HD database (enroll-hd.org), which is funded by CHDI, Inc. This dataset is made widely available to any interested researcher working at a recognized research institution through a straightforward approval process. Researchers requesting this database will be asked to sign the respective agreements governing access and use of these resources. Data not provided in the article because of space limitations may be shared at the request of any qualified investigator (defined as an investigator with granted access to the Enroll-HD database) for purposes of replicating procedures and results.
Results
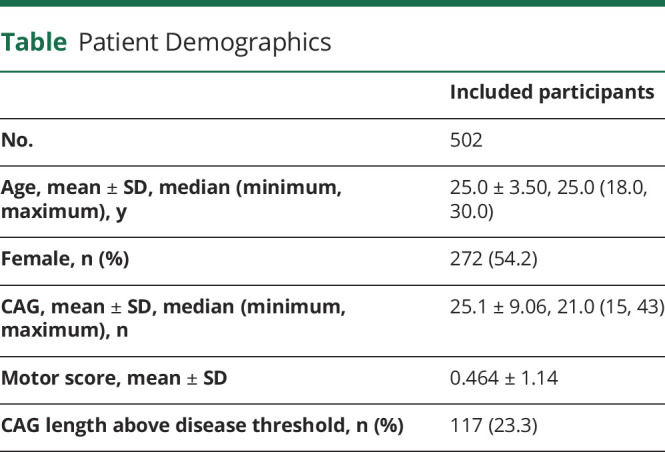
There were 502 participants included in these analyses, and mean baseline demographics are outlined in the table. The majority of participants (n = 385) had a CAG repeat length below the HD disease threshold of ≥36. The most common CAG repeat length among the entire group was 17 (n = 84). The most common CAG repeat length among the participants with a CAG repeat above the disease threshold was 41 (n = 39), followed by 40 (n = 28).
Table.
Patient Demographics
There was a significant, nonlinear relationship between CAG repeat length and SDMT score (F2,359.5 = 3.62, pfdr = 0.044; figure 2A). Specifically, there was a subtle, linear increase in SDMT scores across the spectrum of CAG repeat lengths. Next, we found that among 431 participants (68 missing observations and 3 outlier values), there was a significant relationship between CAG repeat length and Part A of the Trails Making Test (F2,295.9 = 5.54, pfdr = 0.0085; figure 2B). For this task, the time to complete the task seemed to decrease from 15 to ≈30 CAG repeats. Above a CAG repeat length of 30, the total time to complete the task was fairly unchanged (figure 2C). There were data available for Part B of the Trails Making Test for 431 participants (69 missing observations and 2 outlier values). Again, the relationship between CAG repeat length and time to complete this test was statistically significant (F2, 2,547.0 = 6.74, pfdr = 0.0053; figure 2C); however, the trajectory of change across the spectrum of CAG repeats appeared to be relatively linear.
Figure 2. CAG Relationship With Cognitive Tests.

Thick black regression lines represent the nonlinear relationship across all CAG repeat lengths and the cognitive tasks. This line was produced from the individual models described. Dashed lines represent the 95% confidence interval of those models. For panels A–H, the y-axis represents the total correct of the listed cognitive tasks. Category VF = Category Verbal Fluency Task; Letter VF = Letter Verbal Fluency Task; SCNT = Stroop Color Naming Test; SDMT = Symbol Digit Modalities Test; SIT = Stroop Interference Test; SWRT = Stroop Word Reading Test; Trails A = Part A of the Trail Making Test; Trails B = Part B of the Trail Making Test.
There were 501 available observations for the SCNT (1 missing observation), and the relationship with CAG repeat length was also significant (F2, 1,296.3 = 7.89, pfdr = 0.0034; figure 2D). Again, the SCNT scores improved in a fairly linear fashion across all CAG repeat lengths (figure 2D).
Data for the SIT were available for 484 participants (18 missing observations). The pattern of change for this task was similar to that of the SCNT and also was statistically significant (F2,581.6 = 5.75, pfdr = 0.0085; figure 2E). There was a somewhat positively linear pattern of change among the 502 observations of SWRT, but these results also did not reach statistical significance (F2, 265.0 = 1.15, pfdr = 0.36; figure 2F). There were no distinguishable patterns between CAG repeat length and the letter fluency task (F2,50.1 = 0.41, pfdr = 0.67; figure 2G) or the categorical fluency task (F2,50.7 = 2.04, pfdr = 0.18; figure 2H).
Discussion
These results expand on previous reports that before the onset of neurodegeneration, increasing CAG repeats may be associated with improved cognitive performance.22-25 Here, we have expanded on these reports by identifying specific cognitive domains that seem to be associated with CAG repeat length and identifying other domains that do not seem to be influenced by CAG repeat length. Specifically, there were significant associations between CAG repeat length and SDMT, Trail Making Tests, SCNT, and SIT scores. These tasks all assess executive-attentional functions to some extent. This is important because these functions are known to be mediated by frontostriatal circuits,26 and HTT has been linked to developmental changes in the striatum.9 Conversely, verbal fluency tasks are often used to assess memory and verbal functioning.17 In our present study, there did not seem to be any association between CAG repeat length and performance on the verbal fluency tasks or SWRT. These functions may be more closely related to medial temporal regions of the brain,27 but widespread, predictable degeneration in these areas of the brain is not common in HD.
The HTT is known to affect neurodevelopment below the HD disease threshold.7-11 Furthermore, mutant HTT has been associated with neurodevelopmental changes that were associated with CAG repeat length.9 Previously, we reported the effects of CAG repeat in children across the entire spectrum of repeats from 15 through 59 in a unique study of children at risk for HD. We showed that higher CAG repeat lengths were associated with higher IQ scores with a peak around at ≈40 to 41 repeats.22 However, at CAG repeat lengths >41, the detrimental effects of the mutant HTT led to lower IQ measures.28 The study of children at risk for HD is unique in being able to evaluate children far before onset and therefore being able to minimize any effect of the disease process itself on IQ, underscoring that the findings support the notion that HTT throughout the spectrum is affecting brain development and function. Our results seem to indicate similar findings that a higher number of CAG repeats within the huntingtin gene (independently of disease threshold) is associated with improvements in cognition. More important, the cognitive skills that seem to be affected by the number of CAG repeats are cognitive skills that may be associated with regions of the brain such as the striatum that are known to be developmentally influenced by CAG repeat length.9
It is important to interpret these results in the correct context. Specifically, the participants who were evaluated in this study were decades from their predicted motor onset and were completely asymptomatic according to the available information. Consequently, these results demonstrate how CAG repeat length may affect neurodevelopment and cognition before neurodegeneration. There is no doubt that the participants with a CAG repeat length above the disease threshold (CAG ≥36) will eventually experience decline in these same cognitive scores as a direct result of later neurodegeneration caused by HD.29,30 However, early in life, before the degenerative portion of the disease, a CAG repeat length that is longer, even past the disease threshold, seems to be associated with improved attentional-executive function.
The first phase 3 study investigating the effectiveness of an antisense oligonucleotide as a disease-modifying therapy for patients with HD is underway (NCT03761849). Additional studies are also underway or in planning stages.31,32 These studies are primarily recruiting patients with early-manifest HD. If successful at modifying the disease course of HD, however, there will be an inevitable flood of participants requesting to be treated with these medications. One primary question that still remains is how early in the disease course of HD can treatment be safely initiated. The brain continues to develop until ≈30 years of age.14 Therefore, if HTT is critically involved in neurodevelopment, the current results may provide preliminary evidence that practitioners should consider the long-term neurodevelopmental implications of treating, for example, a 19-year-old with a CAG repeat of 40. The scenarios outlined above are hypothetical, and further research into the efficacy of HTT-lowering strategies is warranted, as well as further research regarding how HTT affects neurodevelopment. However, the results presented provide a basis for considering the implications of these treatments. Previous studies have outlined the necessity for providers to consider the potential tradeoffs associated with using disease-modifying therapies in HD,33 and these results may add to those future considerations. In addition, HTT has been implicated in various functions such as DNA repair. Therefore, it is important to consider the possibility that HTT is affecting cognition through a nonneurodevelopmental pathway.
There are several limitations to our work. First, we aimed to select participants who were not yet experiencing neurodegeneration; however, neuroimaging studies are not conducted as part of the Enroll-HD protocol. Therefore, there is no way to determine with certainty that the included participants were not undergoing neurodegenerative changes at the time of assessment. It would be possible to use stricter restrictions on the sample based on the estimated years until motor onset; however, this would reduce statistical power. Furthermore, neuroanatomic changes seem to occur as early as 6 years of age in participants carrying the gene mutation that causes HD.9 This analysis aimed to analyze the relationship between CAG repeat length and cognition independently of neurodegeneration in those participants with a CAG repeat length of ≥36. Consequently, our strict inclusion criteria for this analysis may have introduced selection bias. It is also important to note that the observed changes in cognitive scores are quite subtle. For example, the mean difference in SDMT score between a participant with a CAG of 15 and a participant with a CAG of 40 is ≈5 to 10 points. While this difference is statistically significant, it is difficult to determine whether that difference is clinically significant. The ability to determine clinical significance depends on the conduct of a full neuropsychological assessment that includes a patient interview in the context of how cognition affects functionality in patients. The reported results were collected in the setting of a research study, not for clinical purposes. Consequently, these results are not meant to be interpreted clinically. Rather, the data are presented to demonstrate a relationship between CAG repeat length and cognition, which may provide important information on the biology that may underpin how the HTT affects neurodevelopment. Most important, this study describes associations between CAG repeat length and cognitive scores, and the results should not be interpreted as outlining causation between these factors. Despite these limitations, a major strength of this study is the large sample size. The Enroll-HD platform is the most robust collection of observational data of patients with HD in the world. In addition, we used strict inclusion and exclusion criteria to eliminate the presence of significant confounders. Still, it is nearly impossible to account for all confounders when using observational data.
This study demonstrates that higher CAG repeats are associated with better cognitive outcomes during the neurodevelopmental phase of HD and before neurodegeneration. Participants carrying an expanded CAG repeat will certainly undergo neurodegeneration in the future, but our study provides further evidence that HTT plays a specific role in neurodevelopment, conferring possible advantages and disadvantages. Therefore, further research is warranted to better understand the role of HTT in neurodevelopment. This may prove to be extremely valuable as more huntingtin-lowering treatment strategies are tested for disease modification of HD.
Acknowledgment
Enroll-HD is a longitudinal observational study for families with HD intended to accelerate progress toward therapeutics; it is sponsored by CHDI Foundation, a nonprofit biomedical research organization dedicated exclusively to developing therapeutics for HD. Enroll-HD would not be possible without the vital contribution of the research participants and their families.
Glossary
- HD
Huntington disease
- MMSE
Mini-Mental State Examination
- SCNT
Stroop Color Naming Test
- SDMT
Symbol Digit Modalities Test
- SIT
Stroop Interference Test
- SWRT
Stroop Word Reading Test
Appendix. Authors

Footnotes
Class of Evidence: NPub.org/coe
Podcast: NPub.org/lw0vx9
Study Funding
No targeted funding reported.
Disclosure
Dr. Schultz, Dr. Saft, and Dr. Nopoulos report no disclosures. Go to Neurology.org/N for full disclosures.
References
- 1.A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes: the Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–983. [DOI] [PubMed] [Google Scholar]
- 2.Tartari M, Gissi C, Lo Sardo V, et al. . Phylogenetic comparison of huntingtin homologues reveals the appearance of a primitive polyQ in sea urchin. Mol Biol Evol. 2008;25:330–338. [DOI] [PubMed] [Google Scholar]
- 3.Budworth H, McMurray CT. A brief history of triplet repeat diseases. Methods Mol Biol. 2013;1010:3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hannan AJ. Tandem repeat polymorphisms: modulators of disease susceptibility and candidates for “missing heritability”. Trends Genet. 2010;26:59–65. [DOI] [PubMed] [Google Scholar]
- 5.Nithianantharajah J, Hannan AJ. Dynamic mutations as digital genetic modulators of brain development, function and dysfunction. Bioessays. 2007;29:525–535. [DOI] [PubMed] [Google Scholar]
- 6.La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. [DOI] [PubMed] [Google Scholar]
- 7.Nopoulos PC, Aylward EH, Ross CA, et al. Smaller intracranial volume in prodromal Huntington's disease: evidence for abnormal neurodevelopment. Brain. 2011;134:137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tereshchenko AV, Schultz JL, Bruss JE, Magnotta VA, Epping EA, Nopoulos PC. Abnormal development of cerebellar-striatal circuitry in Huntington disease. Neurology. 2020;94:e1908–e1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van der Plas E, Langbehn DR, Conrad AL, et al. Abnormal brain development in child and adolescent carriers of mutant huntingtin. Neurology. 2019;93:e1021–e1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barnat M, Capizzi M, Aparicio E, et al. Huntington's disease alters human neurodevelopment. Science. 2020;369:787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scahill RI, Zeun P, Osborne-Crowley K, et al. Biological and clinical characteristics of gene carriers far from predicted onset in the Huntington's Disease Young Adult Study (HD-YAS): a cross-sectional analysis. Lancet Neurol. 2020;19:502–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fondon JW III, Hammock EA, Hannan AJ, King DG. Simple sequence repeats: genetic modulators of brain function and behavior. Trends Neurosci. 2008;31:328–334. [DOI] [PubMed] [Google Scholar]
- 13.Landwehrmeyer GB, Fitzer-Attas CJ, Giuliano JD, et al. Data analytics from Enroll-HD, a global clinical research platform for Huntington's disease. Mov Disord Clin Pract. 2017;4:212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lebel C, Beaulieu C. Longitudinal development of human brain wiring continues from childhood into adulthood. J Neurosci. 2011;31:10937–10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Unified Huntington's Disease Rating Scale: reliability and consistency: Huntington Study Group. Mov Disord. 1996;11:136–142. [DOI] [PubMed] [Google Scholar]
- 16.Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR; International Huntington's Disease Collaborative Group. A new model for prediction of the age of onset and penetrance for Huntington's disease based on CAG length. Clin Genet. 2004;65:267–277. [DOI] [PubMed] [Google Scholar]
- 17.Quaranta D, Piccininni C, Caprara A, Malandrino A, Gainotti G, Marra C. Semantic relations in a categorical verbal fluency test: an exploratory investigation in mild cognitive impairment. Front Psychol. 2019;10:2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mills JA, Long JD, Mohan A, Ware JJ, Sampaio C. Cognitive and motor norms for Huntington's disease. Arch Clin Neuropsychol. 2020;35:671–682. [DOI] [PubMed] [Google Scholar]
- 19.Homack S, Riccio CA. A meta-analysis of the sensitivity and specificity of the Stroop Color and Word Test with children. Arch Clin Neuropsychol. 2004;19:725–743. [DOI] [PubMed] [Google Scholar]
- 20.Salthouse TA. What cognitive abilities are involved in Trail-Making performance? Intelligence. 2011;39:222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Enroll-HD Investigators. Enroll-HD clinical protocol [online]. Available at: enroll-hd.org. Accessed November 9, 2020.
- 22.Lee JK, Ding Y, Conrad AL, et al. Sex-specific effects of the Huntington gene on normal neurodevelopment. J Neurosci Res. 2017;95:398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skodda S, Gronheit W, Lukas C, et al. Two different phenomena in basic motor speech performance in premanifest Huntington disease. Neurology. 2016;86:1329–1335. [DOI] [PubMed] [Google Scholar]
- 24.Saft C. What is the course of Huntington's disease? Lancet Neurol. 2014;13:1165–1166. [DOI] [PubMed] [Google Scholar]
- 25.Lo Sardo V, Zuccato C, Gaudenzi G, et al. An evolutionary recent neuroepithelial cell adhesion function of huntingtin implicates ADAM10-Ncadherin. Nat Neurosci. 2012;15:713–721. [DOI] [PubMed] [Google Scholar]
- 26.Leh SE, Petrides M, Strafella AP. The neural circuitry of executive functions in healthy subjects and Parkinson's disease. Neuropsychopharmacology. 2010;35:70–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pihlajamaki M, Tanila H, Hanninen T, et al. Verbal fluency activates the left medial temporal lobe: a functional magnetic resonance imaging study. Ann Neurol. 2000;47:470–476. [PubMed] [Google Scholar]
- 28.Lee JK, Conrad A, Epping E, et al. Effect of trinucleotide repeats in the Huntington's gene on intelligence. EBioMedicine. 2018;31:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paulsen JS, Long JD, Ross CA, et al. Prediction of manifest Huntington's disease with clinical and imaging measures: a prospective observational study. Lancet Neurol. 2014;13:1193–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tabrizi SJ, Scahill RI, Owen G, et al. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington's disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol. 2013;12:637–649. [DOI] [PubMed] [Google Scholar]
- 31.Paulsen JS, Coffey CS. Antisense oligonucleotides might change the therapeutic landscape for Huntington's disease. Lancet Neurol. 2019;18:911–912. [DOI] [PubMed] [Google Scholar]
- 32.Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, et al. Targeting huntingtin expression in patients with Huntington's disease. N Engl J Med. 2019;380:2307–2316. [DOI] [PubMed] [Google Scholar]
- 33.Albin RL, Burke JF. Potential trade-offs in treatment of premanifest Huntington's disease. Mov Disord. 2015;30:1319–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
These results were generated with the Enroll-HD database (enroll-hd.org), which is funded by CHDI, Inc. This dataset is made widely available to any interested researcher working at a recognized research institution through a straightforward approval process. Researchers requesting this database will be asked to sign the respective agreements governing access and use of these resources. Data not provided in the article because of space limitations may be shared at the request of any qualified investigator (defined as an investigator with granted access to the Enroll-HD database) for purposes of replicating procedures and results.