

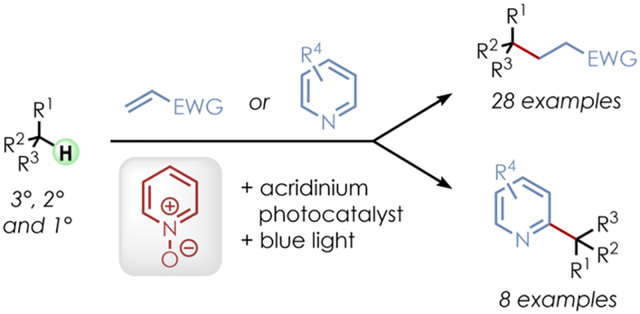
Abstract
The alkylation and heteroarylation of unactivated tertiary, secondary, and primary C(sp3)–H bonds was achieved by employing an acridinium photoredox catalyst along with readily available pyridine Noxides as hydrogen atom transfer (HAT) precursors under visible light. Oxygen-centered radicals, generated by single-electron oxidation of the Noxides, are the proposed key intermediates whose reactivity can be easily modified by structural adjustments. A broad range of aliphatic C–H substrates with electron-donating or -withdrawing groups as well as various olefinic radical acceptors and heteroarenes were well tolerated.
Keywords: photoredox catalysis, acridinium, pyridine N-oxide, C–H functionalization, alkylation, heteroarylation
Graphical Abstract
Visible-light-mediated photoredox catalysis has emerged as a powerful and reliable strategy for the functionalization of aliphatic C–H bonds in the past two decades. Typical approaches employ HAT catalysts that, upon single electron transfer (SET) with an excited photoredox catalyst, generate alkyl radicals by C(sp3)–H abstraction.1 The majority of previous C–H functionalization studies focused on substrates containing C–H bonds alpha to heteroatoms2 or at aldehyde,3 benzylic,4 and allylic5 positions with common bond dissociation energies (BDE) of <95 kcal/mol.6 Although these reactions benefit from high selectivities toward the weakest hydridic bonds, the HAT catalysts employed often lack reactivity for substrates with C–H BDEs ≥ 95 kcal/mol including unactivated tertiary, secondary, and, in particular, primary C–H bonds.7
Aside from halogen-8 and nitrogen-centered radicals,9 oxygen-centered radicals are most widely known to abstract strong unactivated C(sp3)–H bonds because of their high electrophilicity.10 Typical precursors of these reactive species comprise, among others,11 oxo compounds such as aromatic ketones12 and inorganic derivates (e.g., tetrabutyl ammonium decatungstate/TBADT),13 alcohols,14 benzoates,15 and phosphates,16 with the latter ones being employed by our group as well (Figure 1a).17
Figure 1.
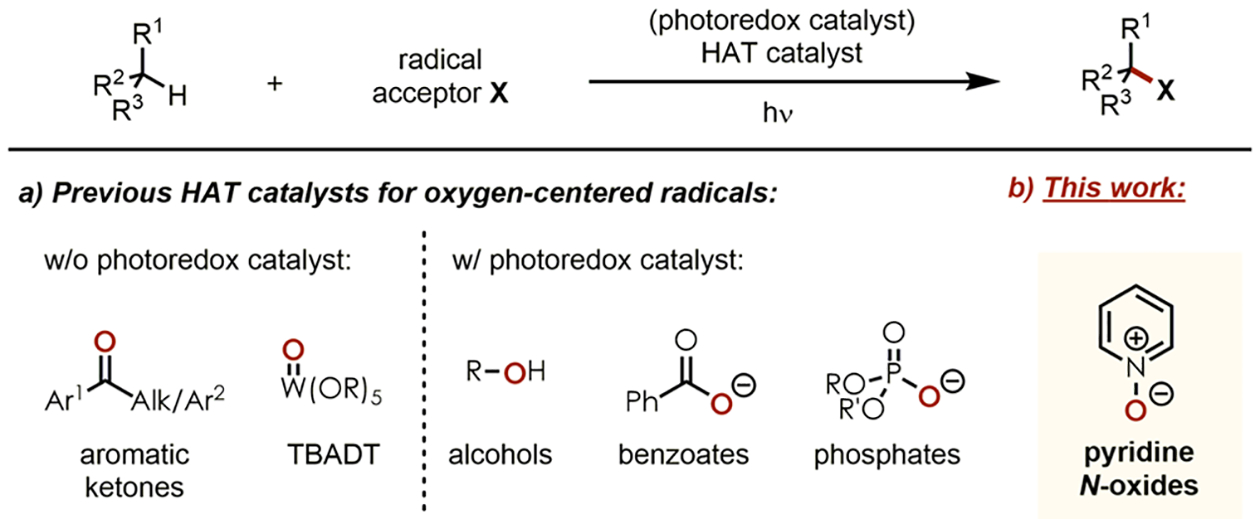
Oxygen-centered radicals in C–H functionalization reactions.
Our continuous interest in developing photoredox catalyzed C–H functionalization reactions that are operationally simple, sustainable, highly efficient and selective, and allow structural fine-tuning of the catalysts led to the discovery of pyridine N-oxides as organic precursors for oxygen-centered radicals that allow HAT processes (Figure 1b).18 Although these compounds had previously been utilized as substrates in a limited number of visible light-mediated transformations,19 their catalytic application under photoredox catalysis remained unprecedented to the best of our knowledge. Wu and coworkers first described the generation of pyridine N-oxy radicals by single electron oxidation with an acridinium photoredox catalyst.19c Based on this report and a study by the Zhang group who demonstrated the oxidation of water with pyridine N-oxides as HAT catalysts using electro-chemitry,20 we hypothesized that they could also abstract H atoms from strong aliphatic C–H bonds using photoredox catalytic conditions. Moreover, pyridine N-oxides are commercially available and inexpensive or can be obtained in one step by simple oxidation of the corresponding pyridines with mCPBA or H2O2,21 resulting in a broad library of readily accessible and fine-tunable organic HAT catalysts.
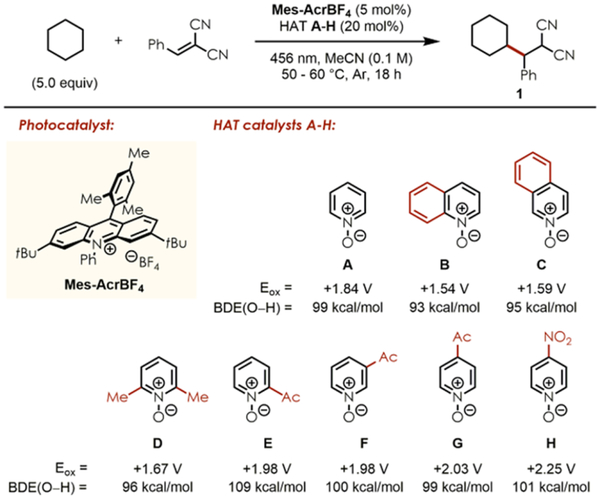
Based on this vision, we found that cyclohexane as C–H substrate reacted with benzylidene malononitrile in the presence of 5 mol % of the photoredox catalyst Mes-AcrBF4 and 20 mol % pyridine N-oxide A under irradiation with blue light (456 nm Kessil), providing product 1 in high NMR yield (Table 1, entry 1). Following this promising result, several other N-oxide derivatives were synthesized or purchased and subjected to the same reaction conditions. In general, electronrich pyridine N-oxides with Eox < +1.8 V showed incomplete conversion of the radical acceptors (entries 2–4), while more electron-deficient ones with Eox > +1.8 V readily provided product 1 in high NMR yields (entries 5–7), except for HAT H which exceeded the redox window of photoexcited Mes-AcrBF4 (E*red = +2.08 V) (entry 8). These results were also supported by DFT calculations22 providing lower BDEs of the O–H bond for the protonated HAT catalysts B-D (93–96 kcal/mol), while A and E-H showed significantly higher BDEs (99–109 kcal/mol) to efficiently abstract an H atom from cyclohexane with BDE(C–H, calc.) = 95 kcal/mol.
Table 1.
Optimization Reactions with Cyclohexanea
| |||
|---|---|---|---|
| entry | conditions | conversionb [%] | yield 1b [%] |
| 1 | HAT A | >99 | 89 |
| 2 | HAT B | 31 | 18 |
| 3 | HAT C | 12 | 9 |
| 4 | HAT D | 64 | 40 |
| 5 | HAT E | >99 | 95 |
| 6 | HAT F | >99 | 84 |
| 7 | HAT G | >99 | 97 (92)c |
| 8 | HAT H | 21 | 16 |
| 9 | HAT G (10 mol %) | >99 | 91 |
| 10 | HAT G (5 mol %) | >99 | 91 |
| 11 | HAT G, DCMd | >99 | 83 |
| 12 | HAT G, PhCFsd | 66 | 48 |
| 13 | HAT G, no light | 0 | 0 |
| 14 | no HAT | 5 | traces |
| 15 | HAT G, no Mes-AcrBF4 | 0 | 0 |
Scale: 0.100 mmol of benzylidene malononitrile.
Determined by 1H NMR of the crude mixture with HMDSO as internal standard.
Average isolated yield in parentheses (n = 2).
Instead of MeCN as solvent.
4-Acetylpyridine N-oxide G proved to be the most efficient catalyst generating 1 in 92% isolated yield (Table 1, entry 7). The catalytic activity of G was also confirmed by reducing its loading to 10 and 5 mol %, respectively, without loss of reactivity, albeit with slightly lower NMR yields (entries 9–10). Additionally, the choice of solvent is crucial in order to achieve high yields (entries 11–12), and control experiments revealed the necessity of light, the HAT catalyst, and the photocatalyst to observe reactivity (entries 13–15).
Various radical acceptors were investigated next (Chart 1a). Except for 1 and 2, which were synthesized in high yields, less electrophilic radical acceptors such as phenyl vinyl sulfone (product 3) provided poor results under the optimized conditions (Method A). It is noteworthy that previously reported C–H alkylation reactions using HAT and acridinium photoredox catalysts were generally limited to a very narrow radical acceptor scope, with mainly benzylidene malononitrile and other highly electron-deficient olefins being the only acceptors applicable.8d,f,9a After extensive additional optimization (see Table S3 in the Supporting Information (SI)), 50 mol % pyridine N-oxide A in a mixture of MeCN/HFIP (7:3) afforded sulfone 3 with an excellent isolated yield of 85% (91% NMR yield). Although similar results were obtained with only 20 and 10 mol % of A, we settled on 50 mol % due to partial deoxygenation/decomposition of the HAT catalysts during the course of the reactions. This observation including NMR experiments has been discussed in more detail in the SI.
Chart 1.
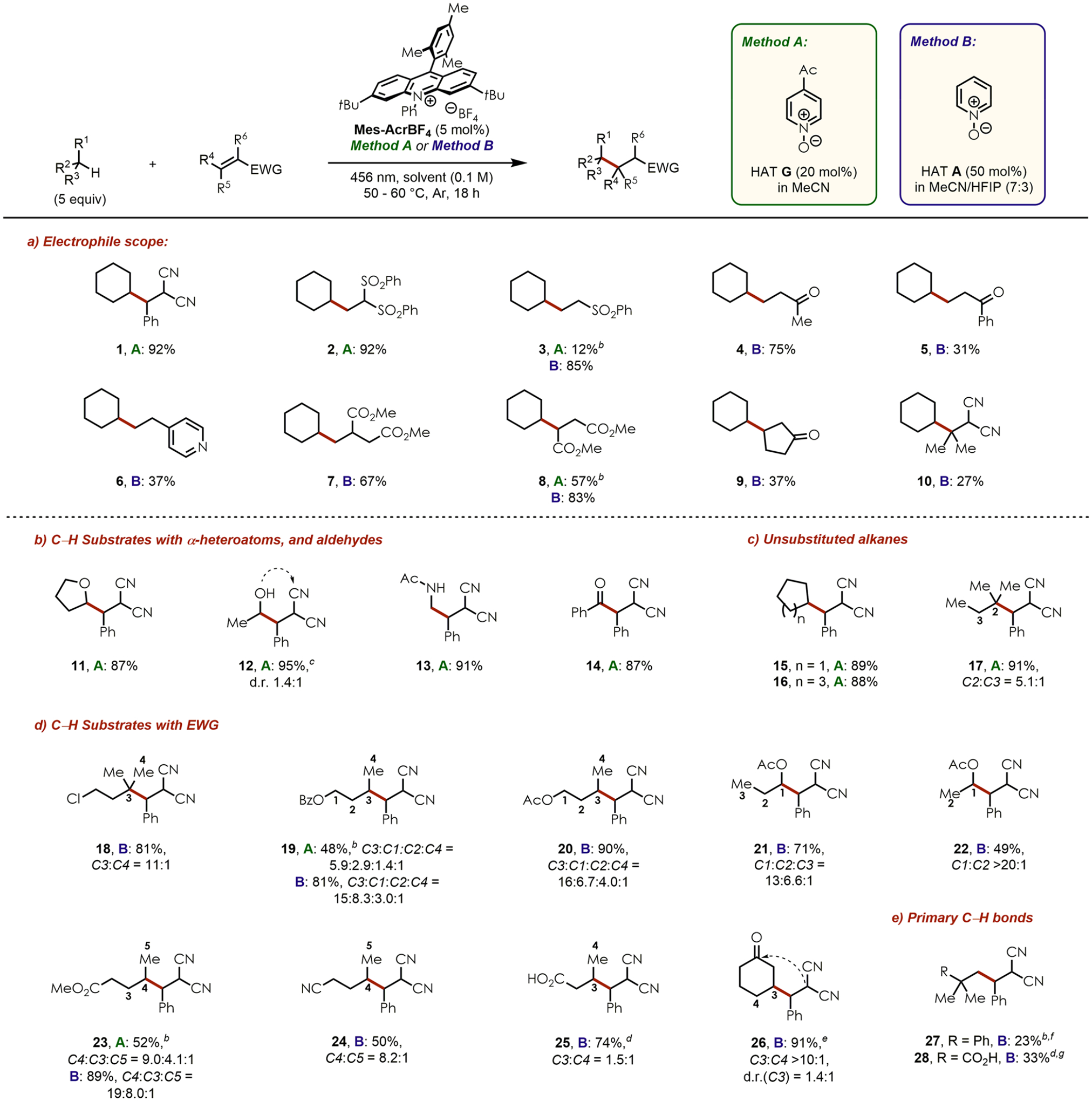
Substrate Scope of the C–H Alkylationa
a Scales: 0.100 or 0.200 mmol of radical acceptor. Average isolated yields are reported (n = 2). Regio- and diastereoselectivities were determined by 1H NMR of the crude mixtures. Only diastereomeric ratios other than d.r. ~1:1 are shown. bYields determined by 1H NMR with HMDSO as internal standard. cWas isolated in its cyclized form 12′ (not shown, indicated by dashed arrow, see SI). dIn CH2Cl2 instead of MeCN/HFIP (7:3), isolated as the methyl ester upon treatment with trimethylsilyldiazomethane (10 equiv). ePartly contains cyclized product 26′ as a single diastereoisomer (not shown, indicated by dashed arrow, see SI). f42 h. g66 h.
The newly established reaction conditions (Method B) also provided better results for other olefins. Thus, simple vinyl ketones afforded products 4 and 5 in good to moderate yields. Moreover, less reactive 4-vinylpyridine was presumably activated by protonation with HFIP to deliver 6 in 37% yield. Other electron-poor olefins with substituents in the α- or β-position were well tolerated and gave rise to their corresponding products 7–9. However, the additional steric hindrance of two methyl groups at the reactive β-position led to a diminished yield of dinitrile 10.
In order to fully explore the synthetic potential of the oxygen-centered radicals generated from pyridine N-oxides, we subjected several electronically and sterically different C–H substrates to our reaction conditions with benzylidene malononitrile as the radical acceptor. C–H substrates with α-heteroatoms including ethers, alcohols, and amides (11–13), as well as aldehydes (14), unsubstituted cyclic (15–16), and acyclic alkanes (17) readily reacted using Method A in typically excellent yields (Chart 1b,c). As expected, tertiary C–H bonds were favored over secondary ones (17), but only with moderate regioselectivity.
Applying the reaction conditions of Method A to substrates containing short alkyl chains and electron-withdrawing groups such as esters, however, resulted in lower product yields (Chart 1d). Unexpectedly, Method B also provided better results in these cases as shown by comparison of both Methods for benzoate 19 and methyl ester 23.
1-Chloro-3-methylbutane selectively afforded two detectable regioisomers of product 18, with the tertiary position (C3) being favored over the primary ones (C4). In contrast to compound 17, the adjacent secondary position (C2) was not functionalized due to its close proximity to the electron-withdrawing chloride.
Compounds 19 and 20 were synthesized from n-butyl benzoate and n-butyl acetate, respectively, with all four possible regioisomers and with the C3-alkylated product being the major one followed by functionalization at C1. Notably, previous H atom abstraction reactions of O-alkyl esters predominantly occurred at the most remote and least deactivated methylene site, while stronger C–H bonds (e.g., the CH3-group) often remained untouched.23 Thus, our results imply that pyridine N-oxy radicals can be classified as rather strong HAT species. Moreover, a difference in regioselectivity was obtained when ester 19 was synthesized by Method A or B, with the latter one providing higher selectivities toward secondary over primary C–H bonds than under the conditions of Method A. The same trend was also observed for product 23.
Reducing the alkyl chain from n-butyl to n-propyl and ethyl acetate led to slightly lower yields of the corresponding esters 21 and 22 along with a change in regioselectivity favoring the C1–H bonds. Methyl valerate afforded 23 as three regioisomers in excellent overall yield using Method B and with good C4-selectivity. However, valeronitrile, carrying a more electron-withdrawing cyano instead of an ester group, reacted much slower to give rise to 24 in moderate yield. In this case, only two regioisomers (C4+C5) were detected and isolated since no functionalization occurred at the electronically less activated C3-position. Notably, other strong HAT species like TBADT provided a mixture of all three regioisomers (C3, C4 and C5) for valeronitrile, resulting in overall lower site-selectivity than under our conditions.13c
Aliphatic carboxylic acids are typically unsuitable substrates for photoredox catalyzed C–H functionalization reactions with basic HAT catalysts due to their propensity toward decarboxylation upon deprotonation and SET.24 Since pyridine N-oxides are slightly basic (pKa(HAT A in MeCN) = 10),25 mixtures of C–H alkylated and decarboxylated products were generally obtained using both Methods. However, the decarboxylation pathway was completely prevented by switching the solvent to CH2Cl2 for the reaction with n-butanoic acid. Hence, product 25 was isolated in high yield and with a regioselectivity of C3:C4 = 1.5:1. The comparatively higher amount of primary C4–H functionalization in this case implies that the solvent also plays a crucial role and MeCN or MeCN/HFIP (7:3) overall enables better regioselectivities.
Cyclohexanone furnished 26 in excellent yield and with high C3-selectivity.26 Other groups have reported a similar trend regarding the regioselectivity of related transformations with this C–H substrate.9a
The efficiency of pyridine N-oxides as strong HAT catalysts was eventually evaluated by subjecting two substrates with only primary C–H bonds to the reaction conditions of Method B for prolonged reaction times (Chart 1e). Both tert-butyl benzene and pivalic acid provided 27 and 28, respectively, in acceptable yields. The latter one was isolated without any decarboxylated byproduct, again highlighting the high chemoselectivity of this reaction in CH2Cl2.
Despite the major focus of this study toward electron-deficient olefins as radical acceptors, we were able to further extend the substrate scope by employing heteroarenes instead which required the use of a terminal oxidant (Chart 2). Based on reports by Molander and co-workers,27 a quick optimization study proved to be fruitful (see Tables S4 and S5 in the SI). In the presence of K2S2O8 as the terminal oxidant and trifluoroacetic acid (TFA) to activate the heteroarenes by protonation, isoquinoline was alkylated with cyclohexane, toluene, and N-methylacetamide to generate 29–31 in good yields. Benzaldehyde provided alcohol 32 in the absence of K2S2O8 according to a spin-center shift mechanism.28 Additionally, several heteroarenes including quinoline, pyridines, and benzothiazole afforded products 33–36 in acceptable to high yields. Control experiments (see Table S5 in the SI) revealed that sulfate radical anions (SO4•–) generated from K2S2O8 upon homolysis under visible light29 or thermal conditions30 do not act as the HAT species in this approach, at least for cyclohexane as the C–H substrate. However, other possible pathways to generate pyridine N-oxy radicals either by oxidation with SO4•–,31 or by formation of an electron donor–acceptor (EDA) complex between pyridine N-oxide and the protonated heteroarene32 might take place as a background reaction but furnished significantly diminished yields without the photocatalyst under our optimized conditions.
Chart 2.
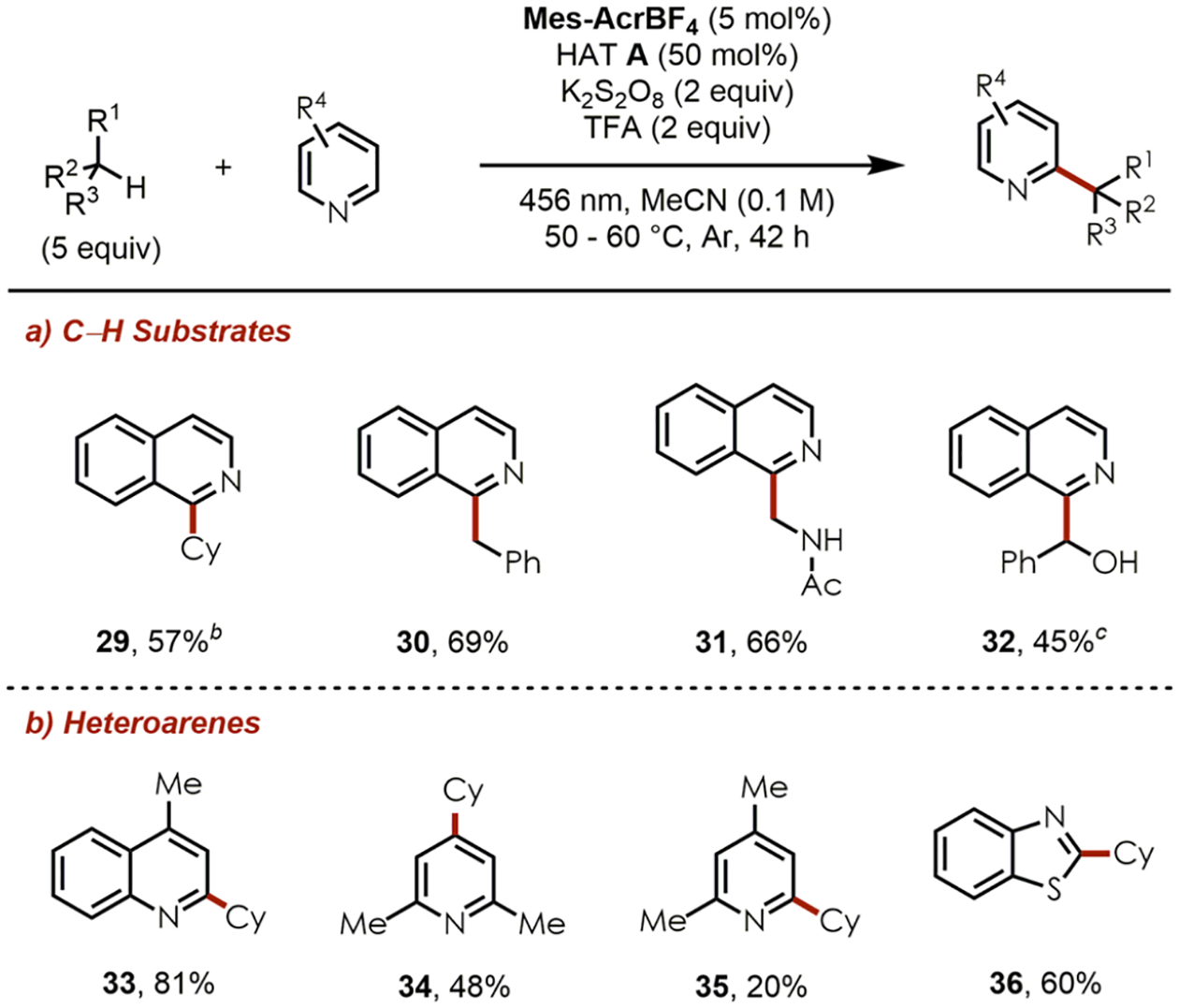
Substrate Scope of the C–H Heteroarylationa
a Scale: 0.200 mmol of heteroarene. Average isolated yields are reported (n = 2). bTFA (1.5 equiv), 18 h. cWithout K2S2O8. Benzaldehyde used as C–H substrate.
In accordance to previous reports8f,17 and kinetic studies (see SI), the proposed mechanism of the C–H alkylation reaction is displayed in Scheme 1.
Scheme 1.
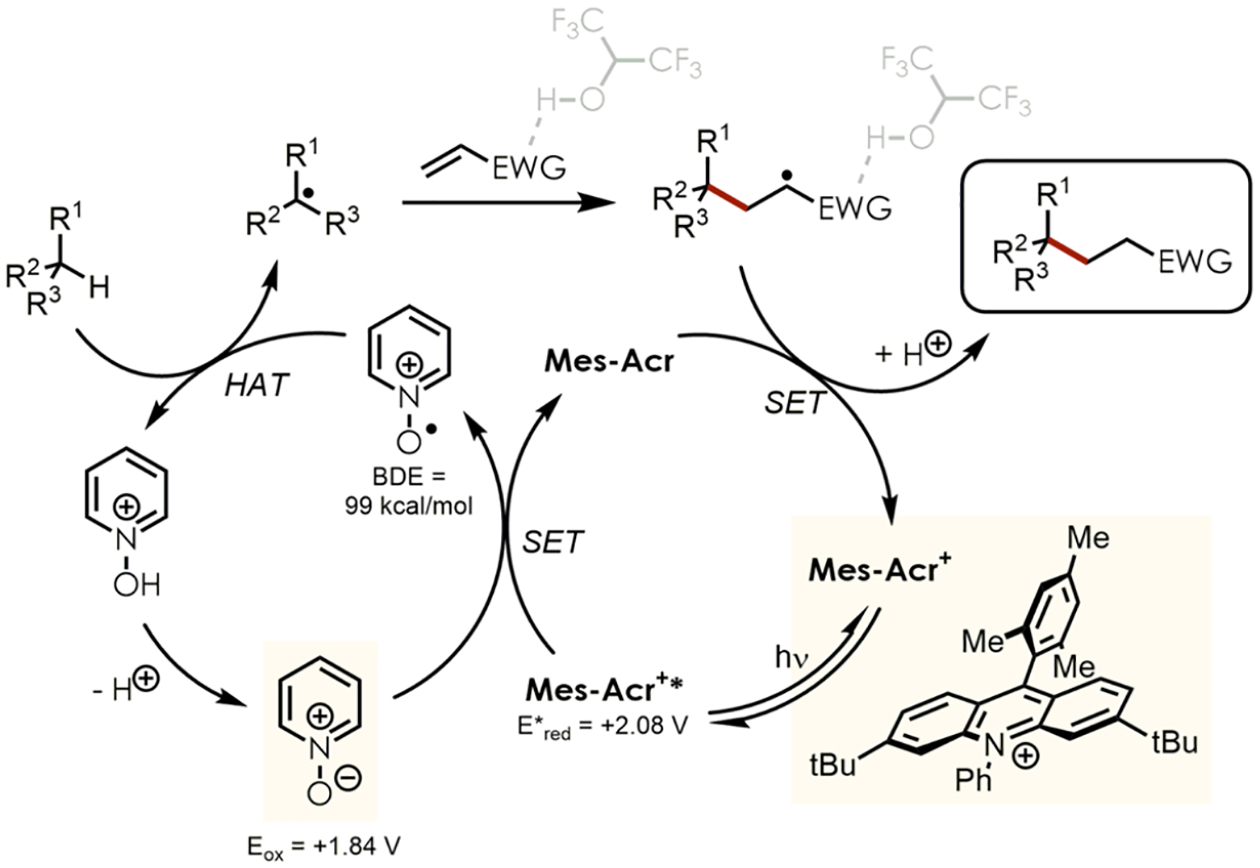
Proposed Mechanism of the C–H Alkylation
After generating highly oxidizing Mes-Acr+* by photoexcitation, pyridine N-oxide undergoes SET to become an N-oxy radical. This electrophilic species (BDE = 99 kcal/mol) can abstract a hydrogen atom from a C–H substrate to afford an alkyl radical, which then reacts with an electron-deficient olefin. In the presence of slightly acidic HFIP, less reactive radical acceptors were preactivated by hydrogen-bonding of the protic cosolvent. The resulting electrophilic radical alpha to the EWG was reduced by the acridine radical Mes-Acr followed by protonation from the N-hydroxy pyridinium to deliver the C–H alkylated product along with the photoredox catalyst Mes-Acr+ and pyridine N-oxide, closing both the photo- and HAT catalytic cycle. These final steps were presumably also enhanced by hydrogen-bonding of HFIP to the EWG which increases the reduction potential of the radical alpha to EWG to oxidize Mes-Acr back to its ground state. A proposed mechanism for the C–H heteroarylation is provided in the SI.
In summary, we have accomplished a highly efficient protocol for C–H alkylation reactions using a synergistic combination of an acridinium photoredox catalyst and readily available pyridine N-oxides as HAT precursors. This purely organic approach allows the abstraction of tertiary, secondary, and even strong primary C–H bonds in the presence of electron-donating and -withdrawing moieties. A broad range of functional groups were tolerated, including aliphatic carboxylic acids that are otherwise prone to readily undergo decarboxylation, and the substrate scope regarding the olefinic radical acceptors was significantly extended compared with previously reported methods. Additionally, the same catalytic system was also applied to Minisci-type reactions demonstrating the high versatility of the established chemistry. The straightforward structural modification of pyridine N-oxides allows the fine-tuning of their electronic and steric properties, including a broad accessible range of BDEs (ca. 90–110 kcal/mol) and, thus, provides the opportunity for highly regioselective transformations which is currently ongoing in our laboratory.
Supplementary Material
ACKNOWLEDGMENTS
This work was generously supported by the German National Academy of Sciences Leopoldina as a postdoctoral fellowship (M.S.) as well as by a National Institutes of Health (NIGMS) Award R35 GM136330 (D.A.N.). We thank Dr. Shubin Liu (UNC) for helpful discussions with the DFT calculations.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.2c02997.
Optimization data, stability tests, CVs, chemical computations, experimental procedures, and characterization data (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acscatal.2c02997
The authors declare no competing financial interest.
Contributor Information
Marcel Schlegel, Department of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3290, United States.
Siran Qian, Department of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3290, United States.
David A Nicewicz, Department of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3290, United States.
REFERENCES
- (1).Reviews:; (a) Holmberg-Douglas N; Nicewicz DA Photoredox-Catalyzed C-H Functionalization Reactions. Chem. Rev 2022, 122, 1925–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cao H; Tang X; Tang H; Yuan Y; Wu J Photoinduced intermolecular hydrogen atom transfer reactions in organic synthesis. Chem. Catalysis 2021, 1, 523–598. [Google Scholar]; (c) Mantry L; Maayuri R; Kumar V; Gandeepan P Photoredox catalysis in nickel-catalyzed C-H functionalization. Beilstein J. Org. Chem 2021, 17, 2209–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Capaldo L; Ravelli D Hydrogen Atom Transfer (HAT): A Versatile Strategy for Substrate Activation in Photocatalyzed Organic Synthesis. Eur. J. Org. Chem 2017, 2017, 2056–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Selected recent examples:; (a) Ryder ASH; Cunningham WB; Ballantyne G; Mules T; Kinsella AG; Turner-Dore J; Alder CM; Edwards LJ; McKay BSJ; Grayson MN; Cresswell AJ Photocatalytic α-Tertiary Amine Synthesis via C-H Alkylation of Unmasked Primary Amines. Angew. Chem., Int. Ed 2020, 59, 14986–14991. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ashley MA; Yamauchi C; Chu JCK; Otsuka S; Yorimitsu H; Rovis T Photoredox-Catalyzed Site-Selective αC(sp3)-H Alkylation of Primary Amine Derivatives. Angew. Chem., Int. Ed 2019, 58, 4002–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang L; Si X; Yang Y; Zimmer M; Witzel S; Sekine K; Rudolph M; Hashmi ASK The Combination of Benzaldehyde and Nickel-Catalyzed Photoredox C(sp3)-H Alkylation/Arylation. Angew. Chem., Int. Ed 2019, 58, 1823–1827. [DOI] [PubMed] [Google Scholar]; (d) Twilton J; Christensen M; DiRocco DA; Ruck RT; Davies IW; MacMillan DWC Selective Hydrogen Atom Abstraction through Induced Bond Polarization: Direct α-Arylation of Alcohols through Photoredox, HAT, and Nickel Catalysis. Angew. Chem., Int. Ed 2018, 57, 5369–5373. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ye J; Kalvet I; Schoenebeck F; Rovis T Direct α-alkylation of primary aliphatic amines enabled by CO2 and electrostatics. Nat. Chem 2018, 10, 1037–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Ide T; Barham JP; Fujita M; Kawato Y; Egami H; Hamashima Y Regio- and chemoselective Csp3-H arylation of benzylamines by single electron transfer/hydrogen atom transfer synergistic catalysis. Chem. Sci 2018, 9, 8453–8460. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Le C; Liang Y; Evans RW; Li X; MacMillan DWC Selective sp3 C-H alkylation via polarity-match-based cross-coupling. Nature 2017, 547, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Shields BJ; Doyle AG Direct C(sp3)-H Cross Coupling Enabled by Catalytic Generation of Chlorine Radicals. J. Am. Chem. Soc 2016, 138, 12719–12722. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Shaw MH; Shurtleff VW; Terrett JA; Cuthbertson JD; MacMillan DWC Native functionality in triple catalytic cross-coupling: sp3 C-H bonds as latent nucleophiles. Science 2016, 352, 1304–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Jin J; MacMillan DWC Direct α-arylation of ethers through the combination of photoredox-mediated C-H functionalization and the Minisci reaction. Angew. Chem., Int. Ed 2015, 54, 1565–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Jeffrey JL; Terrett JA; MacMillan DWC O-H hydrogen bonding promotes H-atom transfer from α C-H bonds for C-alkylation of alcohols. Science 2015, 349, 1532–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Selected recent examples:; (a) Wang L; Wang T; Cheng G-J; Li X; Wei J-J; Guo B; Zheng C; Chen G; Ran C; Zheng C Direct C–H Arylation of Aldehydes by Merging Photocatalyzed Hydrogen Atom Transfer with Palladium Catalysis. ACS Catal 2020, 10, 7543–7551. [Google Scholar]; (b) Jung S; Lee H; Moon Y; Jung H-Y; Hong S Site-Selective C–H Acylation of Pyridinium Derivatives by Photoredox Catalysis. ACS Catal 2019, 9, 9891–9896. [Google Scholar]; (c) Kuang Y; Wang K; Shi X; Huang X; Meggers E; Wu J Asymmetric Synthesis of 1,4-Dicarbonyl Compounds from Aldehydes by Hydrogen Atom Transfer Photocatalysis and Chiral Lewis Acid Catalysis. Angew. Chem., Int. Ed 2019, 58, 16859–16863. [DOI] [PubMed] [Google Scholar]; (d) Meanwell M; Lehmann J; Eichenberger M; Martin RE; Britton R Synthesis of acyl fluorides via photocatalytic fluorination of aldehydic C-H bonds. Chem. Commun 2018, 54, 9985–9988. [DOI] [PubMed] [Google Scholar]; (e) Mukherjee S; Patra T; Glorius F Cooperative Catalysis: A Strategy To Synthesize Trifluoromethyl-thioesters from Aldehydes. ACS Catal 2018, 8, 5842–5846. [Google Scholar]; (f) Kawaai K; Yamaguchi T; Yamaguchi E; Endo S; Tada N; Ikari A; Itoh A Photoinduced Generation of Acyl Radicals from Simple Aldehydes, Access to 3-Acyl-4-arylcoumarin Derivatives, and Evaluation of Their Antiandrogenic Activities. J. Org. Chem 2018, 83, 1988–1996. [DOI] [PubMed] [Google Scholar]; (g) Zhang X; MacMillan DWC Direct Aldehyde C-H Arylation and Alkylation via the Combination of Nickel, Hydrogen Atom Transfer, and Photoredox Catalysis. J. Am. Chem. Soc 2017, 139, 11353–11356. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Vu MD; Das M; Liu X-W Direct Aldehyde Csp2–H Functionalization through Visible-LightMediated Photoredox Catalysis. Chem-Eur. J 2017, 23, 15899–15902. [DOI] [PubMed] [Google Scholar]
- (4).Selected recent examples:; (a) Kawasaki T; Ishida N; Murakami M Dehydrogenative Coupling of Benzylic and Aldehydic C-H Bonds. J. Am. Chem. Soc 2020, 142, 3366–3370. [DOI] [PubMed] [Google Scholar]; (b) Meng Q-Y; Schirmer TE; Berger AL; Donabauer K; König B Photocarboxylation of Benzylic C-H Bonds. J. Am. Chem. Soc 2019, 141, 11393–11397. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dewanji A; Krach PE; Rueping M The Dual Role of Benzophenone in Visible-Light/Nickel Photoredox-Catalyzed C-H Arylations: Hydrogen-Atom Transfer and Energy Transfer. Angew. Chem., Int. Ed 2019, 58, 3566–3570. [DOI] [PubMed] [Google Scholar]; (d) Pandey G; Laha R; Singh D Benzylic C(sp(3))-H Functionalization for C-N and C-O Bond Formation via Visible Light Photoredox Catalysis. J. Org. Chem 2016, 81, 7161–7171. [DOI] [PubMed] [Google Scholar]; (e) Nodwell MB; Bagai A; Halperin SD; Martin RE; Knust H; Britton R Direct photocatalytic fluorination of benzylic C-H bonds with N-fluorobenzenesulfonimide. Chem. Commun 2015, 51, 11783–11786. [DOI] [PubMed] [Google Scholar]; (f) Pandey G; Laha R Visible-Light-Catalyzed Direct Benzylic C(sp(3))-H Amination Reaction by Cross-Dehydrogenative Coupling. Angew. Chem., Int. Ed 2015, 54, 14875–14879. [DOI] [PubMed] [Google Scholar]
- (5).Selected recent examples:; (a) Kim J; Kang B; Hong SH Direct Allylic C(sp 3)–H Thiolation with Disulfides via Visible Light Photoredox Catalysis. ACS Catal 2020, 10, 6013–6022. [Google Scholar]; (b) Vu MD; Das M; Guo A; Ang Z-E; Đokić M; Soo HS; Liu X-W Visible-Light Photoredox Enables Ketone Carbonyl Alkylation for Easy Access to Tertiary Alcohols. ACS Catal 2019, 9, 9009–9014. [Google Scholar]; (c) Huang L; Rueping M Direct Cross-Coupling of Allylic C(sp3)-H Bonds with Aryl- and Vinylbromides by Combined Nickel and Visible-Light Catalysis. Angew. Chem., Int. Ed 2018, 57, 10333–10337. [DOI] [PubMed] [Google Scholar]; (d) Cuthbertson JD; MacMillan DWC The direct arylation of allylic sp(3) C-H bonds via organic and photoredox catalysis. Nature 2015, 519, 74–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Luo Y-R Handbook of Bond Dissociation Energies in Organic Compounds; CRC Press, 2002; pp 11–95. DOI: 10.1201/9781420039863. [DOI] [Google Scholar]
- (7).(a) Yu J; Zhao C; Zhou R; Gao W; Wang S; Liu K; Chen S; Hu K; Mei L; Yuan L; Chai Z; Hu H; Shi W Visible-Light-Enabled C-H Functionalization by a Direct Hydrogen Atom Transfer Uranyl Photocatalyst. Chem.-Eur. J 2020, 26, 16521–16529. [DOI] [PubMed] [Google Scholar]; (b) Fan X-Z; Rong J-W; Wu H-L; Zhou Q; Deng H-P; Tan JD; Xue C-W; Wu L-Z; Tao H-R; Wu J Eosin Y as a Direct Hydrogen-Atom Transfer Photocatalyst for the Functionalization of C-H Bonds. Angew. Chem., Int. Ed 2018, 57, 8514–8518. [DOI] [PubMed] [Google Scholar]; (c) Heitz DR; Tellis JC; Molander GA Photochemical Nickel-Catalyzed C-H Arylation: Synthetic Scope and Mechanistic Investigations. J. Am. Chem. Soc 2016, 138, 12715–12718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Dai Z-Y; Zhang S-Q; Hong X; Wang P-S; Gong L-Z A practical FeCl3/HCl photocatalyst for versatile aliphatic C–H functionalization. Chem. Catalysis 2022, 2 (5), 1211. [Google Scholar]; (b) Huang CY; Li J; Li C-J A cross-dehydrogenative C(sp3)-H heteroarylation via photo-induced catalytic chlorine radical generation. Nat. Commun 2021, 12, 4010. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Treacy SM; Rovis T Copper Catalyzed C(sp3)-H Bond Alkylation via Photoinduced Ligand-to-Metal Charge Transfer. J. Am. Chem. Soc 2021, 143, 2729–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jia P; Li Q; Poh WC; Jiang H; Liu H; Deng H; Wu J Light-Promoted Bromine-Radical-Mediated Selective Alkylation and Amination of Unactivated C(sp3)–H Bonds. Chem 2020, 6, 1766–1776. [Google Scholar]; (e) Rohe S; Morris AO; McCallum T; Barriault L Hydrogen Atom Transfer Reactions via Photoredox Catalyzed Chlorine Atom Generation. Angew. Chem., Int. Ed 2018, 57, 15664–15669. [DOI] [PubMed] [Google Scholar]; (f) Deng H-P; Zhou Q; Wu J Microtubing-Reactor-Assisted Aliphatic C-H Functionalization with HCl as a Hydrogen-AtomTransfer Catalyst Precursor in Conjunction with an Organic Photoredox Catalyst. Angew. Chem., Int. Ed 2018, 57, 12661–12665. [DOI] [PubMed] [Google Scholar]
- (9).Selected examples:; (a) Matsumoto A; Yamamoto M; Maruoka K Cationic DABCO-Based Catalyst for Site-Selective C–H Alkylation via Photoinduced Hydrogen-Atom Transfer. ACS Catal 2022, 12, 2045–2051. [Google Scholar]; (b) Ohmatsu K; Suzuki R; Furukawa Y; Sato M; Ooi T Zwitterionic 1,2,3-Triazolium Amidate as a Catalyst for Photoinduced Hydrogen-Atom Transfer Radical Alkylation. ACS Catal 2020, 10, 2627–2632. [Google Scholar]; (c) Yang H-B; Feceu A; Martin DBC Catalyst-Controlled C–H Functionalization of Adamantanes Using Selective H-Atom Transfer. ACS Catal 2019, 9, 5708–5715. [Google Scholar]; (d) Choi GJ; Zhu Q; Miller DC; Gu CJ; Knowles RR Catalytic alkylation of remote C-H bonds enabled by proton-coupled electron transfer. Nature 2016, 539, 268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Schmidt VA; Quinn RK; Brusoe AT; Alexanian EJ Site-selective aliphatic CH bromination using N-bromoamides and visible light. J. Am. Chem. Soc 2014, 136, 14389–14392. [DOI] [PubMed] [Google Scholar]; (f) Kee CW; Chin KF; Wong MW; Tan C-H Selective fluorination of alkyl C-H bonds via photocatalysis. Chem. Commun 2014, 50, 8211–8214. [DOI] [PubMed] [Google Scholar]
- (10).(a) Zhang J; Liu D; Chen Y 1.9 Oxygen-Centered Radicals. In Free Radicals: Fundamentals and Applications in Organic Synthesis 1; Fensterbank L, Ollivier C, Eds.; Georg Thieme Verlag KG, 2021; p 1469. DOI: 10.1055/sos-SD-234-00177;. [DOI] [Google Scholar]; (b) Hartung J; Gottwald T; Špehar K Selectivity in the Chemistry of Oxygen-Centered Radicals - The Formation of Carbon-Oxygen Bonds. Synthesis 2002, 1469–1498. [Google Scholar]
- (11).(a) Ueda M; Kamikawa K; Fukuyama T; Wang Y-T; Wu Y-K; Ryu I Site-Selective Alkenylation of Unactivated C(sp3)-H Bonds Mediated by Compact Sulfate Radical. Angew. Chem., Int. Ed 2021, 60, 3545–3550. [DOI] [PubMed] [Google Scholar]; (b) Vasilopoulos A; Krska SW; Stahl SS C(sp3)-H methylation enabled by peroxide photosensitization and Ni-mediated radical coupling. Science 2021, 372, 398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tian H; Yang H; Tian C; An G; Li G Cross-Dehydrogenative Coupling of Strong C(sp3)-H with N-Heteroarenes through Visible-Light-Induced Energy Transfer. Org. Lett 2020, 22, 7709–7715. [DOI] [PubMed] [Google Scholar]; (d) Ouyang X-H; Li Y; Song R-J; Hu M; Luo S; Li J-H Intermolecular dialkylation of alkenes with two distinct C(sp3)–H bonds enabled by synergistic photoredox catalysis and iron catalysis. Sci. Adv 2019, 5, eaav9839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Selected examples:; (a) Lee W; Jung S; Kim M; Hong S Site-Selective Direct C-H Pyridylation of Unactivated Alkanes by Triplet Excited Anthraquinone. J. Am. Chem. Soc 2021, 143, 3003–3012. [DOI] [PubMed] [Google Scholar]; (b) Shen Y; Gu Y; Martin R sp3 C-H Arylation and Alkylation Enabled by the Synergy of Triplet Excited Ketones and Nickel Catalysts. J. Am. Chem. Soc 2018, 140, 12200–12209. [DOI] [PubMed] [Google Scholar]; (c) Kamijo S; Takao G; Kamijo K; Hirota M; Tao K;Murafuji T Photo-induced Substitutive Introduction of the Aldoxime Functional Group to Carbon Chains: A Formal Formylation of Non-Acidic C(sp(3))-H Bonds. Angew. Chem., Int. Ed 2016, 55, 9695–9699. [DOI] [PubMed] [Google Scholar]; (d) Xia J-B; Zhu C; Chen C Visible light-promoted metalfree sp(3)-C-H fluorination. Chem. Commun 2014, 50, 11701–11704. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Hoshikawa T; Inoue M Photoinduced direct 4pyridination of C(sp3)–H Bonds. Chem. Sci 2013, 4, 3118. [Google Scholar]; (f) Doohan RA; Hannan JJ; Geraghty NWA The photomediated reaction of alkynes with cycloalkanes. Org. Biomol. Chem 2006, 4, 942–952. [DOI] [PubMed] [Google Scholar]
- (13).Selected recent examples:; (a) Xu S; Chen H; Zhou Z; Kong W Three-Component Alkene Difunctionalization by Direct and Selective Activation of Aliphatic C-H Bonds. Angew. Chem., Int. Ed 2021, 60, 7405–7411. [DOI] [PubMed] [Google Scholar]; (b) Laudadio G; Deng Y; van der Wal K; Ravelli D; Nuño M; Fagnoni M; Guthrie D; Sun Y; Noël T C(sp3)-H functionalizations of light hydrocarbons using decatungstate photocatalysis in flow. Science 2020, 369, 92–96. [DOI] [PubMed] [Google Scholar]; (c) Ravelli D; Fagnoni M; Fukuyama T; Nishikawa T; Ryu I Site-Selective C–H Functionalization by Decatungstate Anion Photocatalysis: Synergistic Control by Polar and Steric Effects Expands the Reaction Scope. ACS Catal 2018, 8, 701–713. [Google Scholar]; (d) Laudadio G; Govaerts S; Wang Y; Ravelli D; Koolman HF; Fagnoni M; Djuric SW; Noël T Selective C(sp3)-H Aerobic Oxidation Enabled by Decatungstate Photocatalysis in Flow. Angew. Chem., Int. Ed 2018, 57, 4078–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Schultz DM; Lévesque F; DiRocco DA; Reibarkh M; Ji Y; Joyce LA; Dropinski JF; Sheng H; Sherry BD; Davies IW Oxyfunctionalization of the Remote C-H Bonds of Aliphatic Amines by Decatungstate Photocatalysis. Angew. Chem., Int. Ed 2017, 56, 15274–15278. [DOI] [PubMed] [Google Scholar]; (f) West JG; Bedell TA; Sorensen EJ The Uranyl Cation as a Visible-Light Photocatalyst for C(sp(3))-H Fluorination. Angew. Chem., Int. Ed 2016, 55, 8923–8927. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Yamada K; Okada M; Fukuyama T; Ravelli D; Fagnoni M; Ryu I Photocatalyzed site-selective C-H to C-C conversion of aliphatic nitriles. Org. Lett 2015, 17, 1292–1295. [DOI] [PubMed] [Google Scholar]
- (14).(a) An Q; Wang Z; Chen Y; Wang X; Zhang K; Pan H; Liu W; Zuo Z Cerium-Catalyzed C-H Functionalizations of Alkanes Utilizing Alcohols as Hydrogen Atom Transfer Agents. J. Am. Chem. Soc 2020, 142, 6216–6226. [DOI] [PubMed] [Google Scholar]; (b) Li G-X; Hu X; He G; Chen G Photoredox-mediated remote C(sp3)-H heteroarylation of free alcohols. Chem. Sci 2019, 10, 688–693. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hu A; Guo J-J; Pan H; Zuo Z Selective functionalization of methane, ethane, and higher alkanes by cerium photocatalysis. Science 2018, 361, 668–672. [DOI] [PubMed] [Google Scholar]; (d) Hu A; Guo J-J; Pan H; Tang H; Gao Z; Zuo Z δ-Selective Functionalization of Alkanols Enabled by Visible-Light-Induced Ligand-to-Metal Charge Transfer. J. Am. Chem. Soc 2018, 140, 1612–1616. [DOI] [PubMed] [Google Scholar]; (e) Wu X; Wang M; Huan L; Wang D; Wang J; Zhu C Tertiary-Alcohol-Directed Functionalization of Remote C(sp3)-H Bonds by Sequential Hydrogen Atom and Heteroaryl Migrations. Angew. Chem., Int. Ed 2018, 57, 1640–1644. [DOI] [PubMed] [Google Scholar]
- (15).(a) Qiu Y; Hartwig JF Mechanism of Ni-Catalyzed Oxidations of Unactivated C(sp3)-H Bonds. J. Am. Chem. Soc 2020, 142, 19239–19248. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mukherjee S; Maji B; Tlahuext-Aca A; Glorius F Visible-Light-Promoted Activation of Unactivated C(sp3)-H Bonds and Their Selective Trifluoromethylthiolation. J. Am. Chem. Soc 2016, 138, 16200–16203. [DOI] [PubMed] [Google Scholar]
- (16).(a) Li D-S; Liu T; Hong Y; Cao C-L; Wu J; Deng H-P Stop-Flow Microtubing Reactor-Assisted Visible Light-Induced Hydrogen-Evolution Cross Coupling of Heteroarenes with C(sp 3)–H Bonds. ACS Catal 2022, 12, 4473–4480. [Google Scholar]; (b) Wakaki T; Sakai K; Enomoto T; Kondo M; Masaoka S; Oisaki K; Kanai M C(sp3)-H Cyanation Promoted by Visible-Light Photoredox/Phosphate Hybrid Catalysis. Chem.Eur. J 2018, 24, 8051–8055. [DOI] [PubMed] [Google Scholar]
- (17).Margrey KA; Czaplyski WL; Nicewicz DA; Alexanian EJ A General Strategy for Aliphatic C-H Functionalization Enabled by Organic Photoredox Catalysis. J. Am. Chem. Soc 2018, 140, 4213–4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).The Deng group cosubmitted their approach along with us, see:; Deng Y; Wang B; Ascenzi Pettenuzzo C; Singh J; Mccabe G; Clark L; Young R; Pu J Photoinduced Site-Selective C-H Functionalization by Pyridine N-oxide Based HAT Catalysts. ChemRxiv, June 22, 2022. DOI: 10.26434/chemrxiv-2022-0rb4h. [DOI] [Google Scholar]
- (19).(a) Deng Y; Zhang J; Bankhead B; Markham JP; Zeller M Photoinduced oxidative cyclopropanation of ene-ynamides: synthesis of 3-azan.1.0bicycles via vinyl radicals. Chem. Commun 2021, 57, 5254–5257. [DOI] [PubMed] [Google Scholar]; (b) Markham JP; Wang B; Stevens ED; Burris SC; Deng Y ortho-Alkylation of Pyridine N-Oxides with Alkynes by Photocatalysis: Pyridine N-Oxide as a Redox Auxiliary. Chem.Eur. J 2019, 25, 6638–6644. [DOI] [PubMed] [Google Scholar]; (c) Xu J-H; Wu W-B; Wu J Photoinduced Divergent Alkylation/Acylation of Pyridine N-Oxides with Alkynes under Anaerobic and Aerobic Conditions. Org. Lett 2019, 21, 5321–5325. [DOI] [PubMed] [Google Scholar]; (d) Zhou W; Miura T; Murakami M Photocatalyzed ortho-Alkylation of Pyridine N-Oxides through Alkene Cleavage. Angew. Chem., Int. Ed 2018, 57, 5139–5142. [DOI] [PubMed] [Google Scholar]; (e) Zhang W-M; Dai J-J; Xu J; Xu H-J Visible-Light-Induced C2 Alkylation of Pyridine N-Oxides. J. Org. Chem 2017, 82, 2059–2066. [DOI] [PubMed] [Google Scholar]
- (20).Li H; Xie F; Zhang M-T Metal-Free Electrocatalyst for Water Oxidation Initiated by Hydrogen Atom Transfer. ACS Catal 2021, 11, 68–73. [Google Scholar]
- (21).Youssif S Recent trends in the chemistry of pyridine N-oxide. Arkivoc 2001, 2001, 242–268. [Google Scholar]
- (22). See the Supporting Information for more details about the DFT calculations.
- (23).(a) Saito M; Kawamata Y; Meanwell M; Navratil R; Chiodi D; Carlson E; Hu P; Chen L; Udyavara S; Kingston C; Tanwar M; Tyagi S; McKillican BP; Gichinga MG; Schmidt MA; Eastgate MD; Lamberto M; He C; Tang T; Malapit CA; Sigman MS; Minteer SD; Neurock M; Baran PS N-Ammonium Ylide Mediators for Electrochemical C-H Oxidation. J. Am. Chem. Soc 2021, 143, 7859–7867. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kawamata Y; Yan M; Liu Z; Bao D-H; Chen J; Starr JT; Baran PS Scalable, Electrochemical Oxidation of Unactivated C-H Bonds. J. Am. Chem. Soc 2017, 139, 7448–7451. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Canta M; Font D; Gómez L; Ribas X; Costas M The Iron(II) Complex [Fe(CF 3 SO 3) 2 (mcp)] as a Convenient, Readily Available Catalyst for the Selective Oxidation of Methylenic Sites in Alkanes. Adv. Synth. Catal 2014, 356, 818–830. [Google Scholar]; (d) Asensio G; Castellano G; Mello R; González Núñez ME Oxyfunctionalization of Aliphatic Esters by Methyl-(trifluoromethyl)dioxirane. J. Org. Chem 1996, 61, 5564–5566. [Google Scholar]
- (24).(a) Ramirez NP; Gonzalez-Gomez JC Decarboxylative Giese-Type Reaction of Carboxylic Acids Promoted by Visible Light: A Sustainable and Photoredox-Neutral Protocol. Eur. J. Org. Chem 2017, 2017, 2154–2163. [Google Scholar]; (b) Griffin JD; Zeller MA; Nicewicz DA Hydrodecarboxylation of Carboxylic and Malonic Acid Derivatives via Organic Photoredox Catalysis: Substrate Scope and Mechanistic Insight. J. Am. Chem. Soc 2015, 137, 11340–11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Mech P; Bogunia M; Nowacki A; Makowski M Calculations of pKa Values of Selected Pyridinium and Its N-Oxide Ions in Water and Acetonitrile. J. Phys. Chem. A 2020, 124, 538–551. [DOI] [PubMed] [Google Scholar]
- (26). Additional information about the determined regioselectivity of 26 and the identification of the cyclized product 26′ are summarized in the SI.
- (27).Matsui JK; Primer DN; Molander GA Metal-free C-H alkylation of heteroarenes with alkyltrifluoroborates: a general protocol for 1°, 2° and 3° alkylation. Chem. Sci 2017, 8, 3512–3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Fuse H; Nakao H; Saga Y; Fukatsu A; Kondo M; Masaoka S; Mitsunuma H; Kanai M Photocatalytic redox-neutral hydroxyalkylation of N-heteroaromatics with aldehydes. Chem. Sci 2020, 11, 12206–12211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Review:; Saha S; Bagdi AK Visible light-promoted photocatalyst-free activation of persulfates: a promising strategy for C-H functionalization reactions. Org. Biomol. Chem 2022, 20, 3249–3262. [DOI] [PubMed] [Google Scholar]
- (30).McCallum T; Jouanno L-A; Cannillo A; Barriault L Persulfate-Enabled Direct C–H Alkylation of Heteroarenes with Unactivated Ethers. Synlett 2016, 27, 1282–1286. [Google Scholar]
- (31).Pitre SP; Muuronen M; Fishman DA; Overman LE Tertiary Alcohols as Radical Precursors for the Introduction of Tertiary Substituents into Heteroarenes. ACS Catal 2019, 9, 3413–3418. [Google Scholar]
- (32).During the preparation of this manuscript, related Minisci-type reactions, that require different conditions without photocatalyst or terminal oxidants, have been published on a preprint-server:; Ciszewski Ł; Gryko D Pyridine N-oxides as HAT reagents for photochemical C-H functionalization of electron-deficient heteroarenes. ChemRxiv, June 13, 2022, version 1. DOI: 10.26434/chemrxiv-2022-j348w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.