

Abstract
About 5-10% of the human genome remains inaccessible due to the presence of repetitive sequences such as segmental duplications and tandem repeat arrays. We show that existing long read mappers often yield incorrect alignments and variant calls within long, near-identical repeats, as they remain vulnerable to allelic bias. In the presence of a non-reference allele within a repeat, a read sampled from that region could be mapped to an incorrect repeat copy. To address this limitation, we developed a novel long read mapping method Winnowmap2 by using minimal confidently alignable substrings (MCASs). Winnowmap2 computes each read mapping through a collection of confident subalignments. This approach is more tolerant of structural variation and more sensitive to paralog-specific variants (PSVs) within repeats. Our experiments highlight that Winnowmap2 successfully addresses the issue of allelic bias, enabling more accurate downstream variant calls in repetitive sequences.
1. Introduction
Advances in single-molecule sequencing technologies have inspired community efforts to produce high-quality human genome assemblies with accurate resolution of repetitive DNA. The complete, gapless, telomere-to-telomere (T2T) assembly of a human chromosome X is a recent breakthrough that involved assembling a 3.1 Mbp long centromeric satellite DNA array [1]. Similarly, a T2T assembly of human chromosome 8 spanned a 2.1 Mbp long centromere and the 0.6 Mbp long defensin gene cluster for the first time [2]. Such developments are steering genomics into an exciting new era where repeats that were previously thought intractable (e.g., segmental duplications, satellite, and ribosomal DNAs) will no longer remain out of reach. PacBio and Oxford Nanopore (ONT) sequencing, due to their orders of magnitude longer read lengths than Illumina, can easily span many common duplications (e.g., LINEs) in the human genome. However, accurate long read mapping within > 100 kbp-sized repeats remains challenging.
Prior algorithmic developments for long read mapping to a reference sequence have been crucial to resolving many repetitive sequences and complex variants. As such, several specialized methods have been published to improve long-read match seeding and extension [3-14]. The extension stage involves maximizing base-to-base alignment score which rewards matching bases while appropriately penalizing gaps and mismatches. However, these alignment scores do not always favor the correct loci in long near-identical repeats because reads that include non-reference alleles will be penalized, and their true loci may score worse than other copies of the repeat. Occurrence of this allelic bias (a.k.a. reference bias) and its effect on estimates of variation and allele frequencies has been extensively discussed in the literature [15-20]. An analogous problem also occurs during genome assembly validation and polishing when reads are mapped back to a potentially erroneous draft assembly [1,21].
Compared to point mutations or short indels, structural variants (SVs) affect more bases in the genome due to their larger size, and therefore, are bigger contributors to allelic bias. Most existing solutions to address this bias involve modifying the reference sequence, e.g., by adopting a graph-based representation which incorporates known genomic variation [17,22-24]. While this remains a promising and complementary direction, here we seek to address allelic bias by developing a new long read mapping method that is robust to the presence of novel variation. In our proposed method, referred to as Winnowmap2, we introduce the concept of minimal confidently alignable substrings (MCASs), which are minimal-length read substrings that align end-to-end to a reference with mapping quality (confidence) score above a user-specified threshold. Through MCASs, we can identify the correct mapping target of a read by considering the substrings that do not overlap non-reference alleles. In theory, the mapping quality of each substring quantifies the probability that it is correctly placed [25]. This framework draws advantage from paralog-specific variants (PSVs) [26,27] that distinguish near-identical repeat copies from one another. We provide a formal definition of MCAS, an exact dynamic programming algorithm to compute them, as well as fast heuristics to scale this method to large mammalian genomes.
Winnowmap2 was empirically validated using both simulated and real human genome sequencing benchmarks. In both cases, we judge Winnowmap2 along with the currently available long read mappers by the downstream accuracy of SV calls produced by the SV caller Sniffles [7]. The simulation uses SURVIVOR’s SV benchmarking tool [28] which mutates a reference sequence (in our case, the first two completed human chromosomes 8 and X). Winnowmap2 alignments consistently enabled the most accurate SV calls using both ONT and PacBio HiFi data at varying coverage levels, when compared to other commonly used long read mappers (Results). Winnowmap2 showed its biggest gains in highly repetitive human DNA, including those genomic loci that were recently completed for human chromosomes 8 and X but absent from the GRCh38 reference [29]. Consequently, we examined structural variation within these regions for human samples HG004 and HG007, by mapping their respective nanopore reads directly to the T2T human genome assembly [30]. The results of this experiment indicate significant enrichment of SVs across the genomic intervals corresponding to unresolved gaps in GRCh38. This observation corroborates the need for an end-to-end personal human genome sequencing without overlooking complex repeats.
2. Results
An overview of the Winnowmap2 algorithm
If an error-free read is simulated directly from a reference, then its correct mapping to that reference computed using a reasonable pairwise sequence alignment algorithm is naturally guaranteed to have the highest score. However, this guarantee does not hold if the same read is mapped to an alternate reference. Consequently, using a pairwise sequence alignment scoring system to judge the best mapping candidate is sub-optimal, and this is particularly true while mapping reads to highly repetitive sequences. Regardless of the type of scoring function used, e.g., with either a linear or an affine gap penalty, the function would also penalize variant-induced differences between the sequenced individual and the reference sequence. In cases where one of the repeat copies in a reference sequence contains a different allele from the sequenced individual, reads may achieve a better alignment score against an incorrect repeat copy (Figure 1). An ideal scoring system should ignore non-reference bases when computing an optimal alignment, but these are typically unknown a priori.
Figure 1:
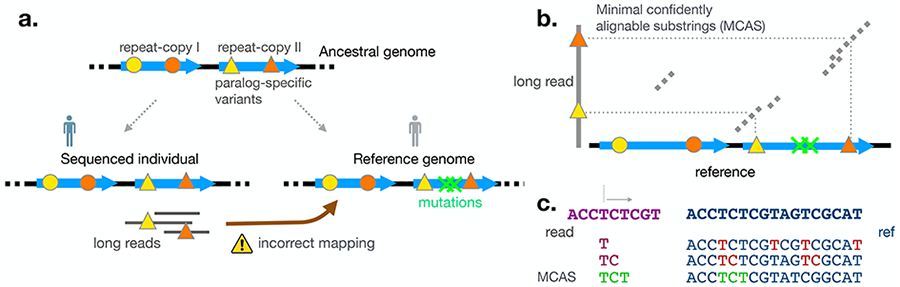
a. Illustration of allelic bias in near-identical genomic repeats. Paralog-specific variants (PSVs), indicated using colored dot and triangle markers, denote variation between two repeat copies in an ancestral human genome . Mutations in the reference sequence are indicated using ‘x’ markers. Long reads can be mapped to an incorrect repeat copy if the best mapping is decided by pairwise sequence alignment score. b. MCAS alignments map to correct loci on the reference. An MCAS is a carefully selected substring of a read. By excluding non-reference alleles, this approach reduces allelic bias. c. A different example is used to illustrate MCAS computation starting from a particular position in a read. To compute MCAS starting from a particular position in a read, we look for the shortest substring that can be uniquely mapped to a reference. Uniqueness of an alignment is determined by using its mapping quality score.
Like most read mappers, Winnowmap2 follows a seed-and-extend workflow. The seeding step reuses Winnowmap’s weighted minimizer sampling [11], which yields an accuracy improvement over the standard minimizer technique [31]. Winnowmap2’s extend stage uses subroutines in minimap2 internally while introducing a novel heuristic to tackle allelic bias. We split the extend stage into two steps. The first step involves identifying minimal confidently alignable substrings (MCASs) from each read to a reference. Informally, an MCAS at position i of a read refers to the minimum length read substring starting at the position i that achieves a sufficiently ‘unique’ end-to-end alignment to a reference locus (see Methods for a formal definition). Here, uniqueness of an alignment is evaluated using a mapping quality (mapQ) score that reflects the score gap between the best and second-best alignment candidates for a substring [25]. Accordingly, we define an MCAS to be valid if its alignment achieves a mapQ score above a user-specified threshold. A read can have as many MCASs as its length. By using MCAS alignments, read bases on either side of a variant can map uniquely to their correct reference loci as they can be scored independently from non-reference bases (Figure 1).
Starting from any position of a read, the minimal length of the MCAS is ensured by iteratively increasing the substring length and checking whether its maximum scoring alignment to a reference satisfies the mapQ cutoff. Suppose a read is sampled from a repetitive region. The frequency of PSVs at its correct mapping loci helps determine the length of an MCAS. The higher the number of PSVs, the smaller the length of the MCAS will be, because its mapQ cutoff will be satisfied at an earlier iteration with fewer aligned bases. Similarly, better raw read accuracy will also lead to shorter MCAS lengths, since more PSVs will be matched by a more accurate sequence. Shorter MCAS lengths help, not only in terms of the runtime with fewer iterations spent, but also in terms of accuracy, as MCASs are less likely to overlap non-reference bases.
Computing all MCAS alignments from a read in an exact manner can be computationally prohibitive (see Methods for complexity analysis). Winnowmap2 is implemented on top of minimap2 codebase, i.e., we rely on banded-alignment and mapQ scoring heuristics from minimap2 to compute each MCAS. For the sake of efficiency, we avoid evaluating MCAS alignments from each consecutive position in a read. Rather, we identify MCAS alignments from a subset of positions that are equally spaced (e.g., 1000 bp apart).
The final step in Winnowmap2 is to consolidate a read’s MCAS alignments into a final alignment output. During the consolidation step, we retrieve all anchors which were part of co-linear anchor chain of an MCAS alignment. Next, we re-execute the co-linear chaining and alignment extension heuristics of minimap2 by using all the anchors to output a final alignment. For various reasons (e.g. sequencing errors, approximation of mapQ computation, and complex sequence variants) some MCASs may be incorrectly mapped. Outlier anchors from false MCAS mappings get filtered out during co-linear chaining assuming a majority of anchors are correct. We will empirically show that the proposed strategy improves mapping accuracy in repetitive DNA while remaining highly scalable.
Evaluation using the β-defensin gene cluster
We visualize the advantage of Winnowmap2 method by using the beta-defensin gene cluster on human chromosome 8 as an example. The 7 Mbp beta-defensin locus (chr8: 6,300,000-13,300,000) of the human genome is known to be a hotspot of copy-number variation [32]. In the sequenced CHM13 human cell line, this locus spans three large (> 500 kbp) segmental duplications [2]. To evaluate long read mapping accuracy at this locus, we simulated ONT reads from chr8 at 40x sequencing coverage by using NanoSim [33] (Methods). In addition, we artificially mutated chr8 by adding a 1 kbp deletion variant at position 12,000,000. This locus was chosen for our illustration as it overlaps with one of the three duplications. If mapped correctly, the 1 kbp simulated deletion in the reference should appear as a 1 kbp-long insertion in the overlapping read alignments.
Figure 2 shows an IGV visualization of primary alignments computed by Winnowmap2 and three other long read mapping tools NGMLR, minimap2 and graphmap. Among the four methods, Winnowmap2 achieved the expected mapping coverage in this region with most read alignments showing the expected insertion call. The other tools mapped fewer reads successfully, resulting in reduced coverage and poor read mapQ scores. When these alignments were used as input to Sniffles, only Winnowmap2 alignments resulted in the true SV call. NGMLR, minimap2 and graphmap rely on pairwise sequence alignment scores across the full length of the read when choosing the best mapping target. Due to the large deletion penalty levied at the mutated (but correct) locus, the majority of reads were incorrectly mapped to the other two duplications. Among the three methods, NGMLR showed the least bias, but most of its correct alignments were associated with poor mapQ scores (< 10). A low mapQ score indicates a marginal alignment score difference between the best and the second-best mapping candidate, and therefore, the read alignment may not be considered by the variant caller. This result illustrates the previously discussed limitation of using pairwise alignment scores to rank candidate alignments in genomic repeats. The use of MCASs in Winnowmap2 enabled correct read placements in this case. A few MCAS alignments computed by Winnowmap2 in this region are visualized as a dot-plot in Supplementary Figure S1. A similar behavior was observed when we simulated an SV within the highly repetitive centromeric satellite DNA array of chromosome 8 (Supplementary Figure S2).
Figure 2:
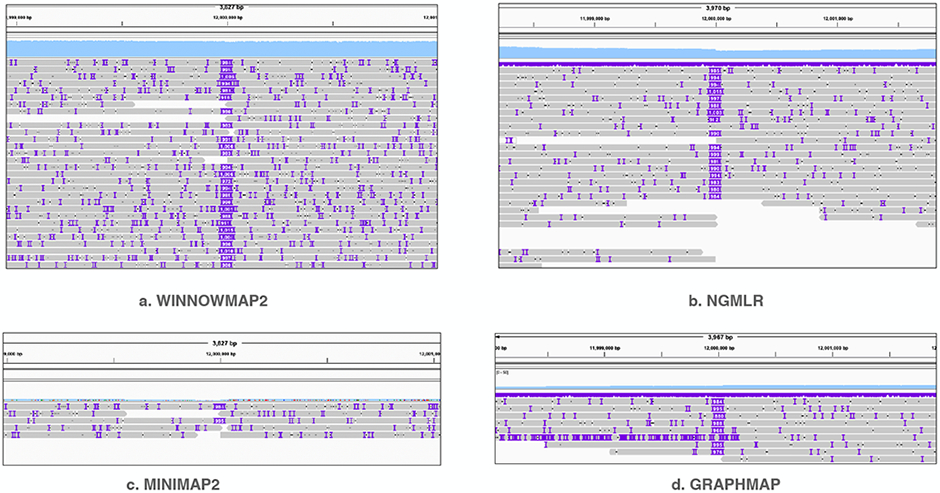
Visualization of alignment pileup near the mutated bases of chromosome 8 by using IGV tool [47]. The sky-blue-colored track on top of each plot shows mapping-coverage using a uniform y-axis scale (0-50). The grey-colored line segments show individual primary read alignments. IGV uses purple markers to indicate presence of indels within read alignments. NGMLR, minimap2, graphmap show reduced coverage due to allelic bias whereas Winnowmap2 shows expected coverage in this region. Consistent large insertions in the middle of each plot are distinctly visible due to simulated SV.
Evaluation using T2T human chromosomes
We simulated long reads, both HiFi (using PBSIM [34]) and ONT (using NanoSim [33]), at coverage levels of 20x and 40x from T2T assemblies of chromosome 8 (146 Mbp) and chromosome X (154 Mbp) respectively (Methods). To evaluate how well Winnowmap2 addressed allelic bias, we also simulated 1100 structural variants, including both indels (1000) and inversions (100) of size ≤ 1 kbp, in each reference chromosome sequence by using the SURVIVOR benchmarking tool [28]. Both the SV simulation and evaluation of variant sets against the ground truth were done using SURVIVOR (Methods).
We evaluated Winnowmap2, Winnowmap, minimap2 and NGMLR in this experiment to check their false-negative and false-positive rates (FNR, FPR), as well as runtime and memory requirements. The long read mappers produced SAM-formatted alignments, which were then fed to Sniffles [7] to compute SVs. A false negative indicates that a true SV is not supported by read alignments whereas a false positive indicates that a false SV is supported. As such, these statistics are good indicators of the correctness of read alignments. We also performed a de novo repeat annotation of each reference sequence (chr8 and chrX) by using Mashmap [35] to identify repetitive sequence intervals of length ≥ 10 kbp and identity ≥ 95% (Supplementary Figure S3). The identified repetitive intervals constitute a notable portion of the two chromosomes; 4.8% in chr8 and 6.9% in chrX. This allowed us to separately evaluate accuracy in near-identical repeats where typical read mappers struggle.
Figures 3a, 3b show the accuracy statistics of the four mapping tools. Winnowmap2 FNR and FPR scores consistently stayed below 3% and 0.3% respectively in this experiment. When compared to the competing methods, Winnowmap2 achieved the best FNR and FPR for both the HiFi and ONT read sets. For instance, using simulated HiFi reads from chromosome 8 (146 Mbp) sampled at 40x coverage, Winnowmap2, minimap2 and NGMLR achieved accuracy scores, i.e., false-negative and false-positive rates (FNR, FPR) of (0.09%, 0.18%), (3.36%, 0.93%) and (3.64%, 2.93%) respectively. Winnowmap2’s improved handling of allelic bias was particularly evident within the repetitive intervals of chromosome 8 (Figures 3c, 3d), achieving (FNR, FPR) scores of (1.89%, 1.89%) in these regions compared to (39.62%, 5.88%) for minimap2 and (56.60%, 36.11%) for NGMLR, respectively. Winnowmap2 succeeds in addressing allelic bias in these regions by preserving good accuracy in complex repeats where the other tools struggle. These gains were made uniformly over all SV types- insertions, deletions and inversions that were simulated (Supplementary Table S1). These complex repetitive intervals also spanned centromeric alpha satellite arrays of both chromosomes 8 and X (Supplementary Figure S3). The Winnowmap2-Sniffles pipeline successfully called all 30 SVs that were simulated by SURVIVOR without any false positives within these regions using both HiFi and ONT read sets.
Figure 3:
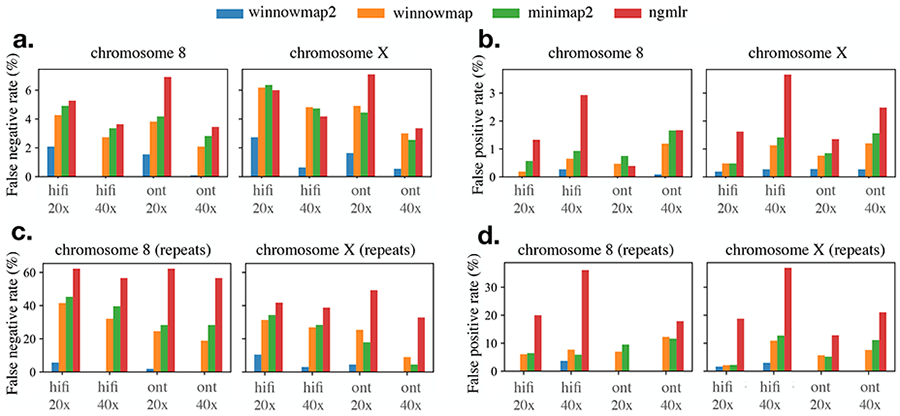
False negative and false positive rates achieved by SV calls of four mapping methods: Winnowmap2, Winnowmap, minimap2 and NGMLR. The top two plots show accuracy statistics over T2T chromosomes 8 and X whereas the bottom two plots show the statistics within only the most repetitive intervals of these chromosomes. Winnowmap2 alignments enabled the most accurate Sniffles SV calls with the least FNR and FPR scores. Note that y-axis scales differ in these plots.
When increasing coverage from 20x to 40x, FNR generally reduces for all methods as better sensitivity is naturally expected with higher sequencing coverage. In a separate benchmark, we also validated Winnowmap2 by measuring fraction of incorrectly mapped reads (Supplementary Table S2). Here we repeated the same benchmark that was used in [11]. Using the complete T2T chromosome X as reference, Winnowmap2 and minimap2 mapped 0.03% and 0.15% reads incorrectly respectively. Using the GRCh38 genome as reference, Winnowmap2 and minimap2 mapped 2.0% and 1.9% reads incorrectly respectively.
Winnowmap2 remains competitive in terms of its runtime and memory usage (Figure 4). As several substring alignments need to be identified from a single read, it requires execution of alignment routines several times rather than just once.
Figure 4:
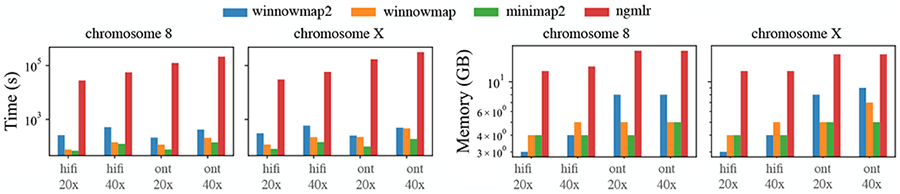
Wall-clock time and memory usage of four mapping methods. Each method was executed using 24 threads on an Intel Xeon processor with 24 physical cores. Y-axis of the above plots is log-scaled.
Evaluation using Genome in a Bottle benchmark
Evaluating mappers on real sequencing data is challenging without a known truth. The Genome in a Bottle (GIAB) Tier1 v0.6 benchmark set provides a high-quality characterization of SVs in the Ashkenazi cell line HG002 relative to the GRCh37 human reference. This call set encompasses 2.51 Gbp of the genome and includes 5262 insertions and 4095 deletions [36]. It excludes SVs overlapping segmental duplications and tandem repeats greater than 10 kbp. Nevertheless, this experiment was useful to validate that Winnowmap2 provides competitive accuracy on real data within the commonly studied regions of the genome. Here we mapped three publicly available HG002 long read sequencing data sets: HiFi (14-15 kbp library, 35x), ONT (Guppy 3.6.0, 35x) and ONT (Guppy 3.6.0, 50x) to GRCh37, and compared results with minimap2. Similar to our simulated benchmark, variants were called using Sniffles. Winnowmap2 achieved slightly better precision and similar recall scores compared to minimap2 (Figure 5), with similar runtime and memory requirements. Using the three data sets, Winnowmap2’s F1-scores were 0.87, 0.92 and 0.91 respectively, and the corresponding numbers using minimap2 were 0.86, 0.91 and 0.90 respectively. We also observed that both Winnowmap2 and minimap2 achieved better SV accuracy using ONT data over HiFi using equal 35x sequencing coverage due to longer ONT read lengths.
Figure 5:
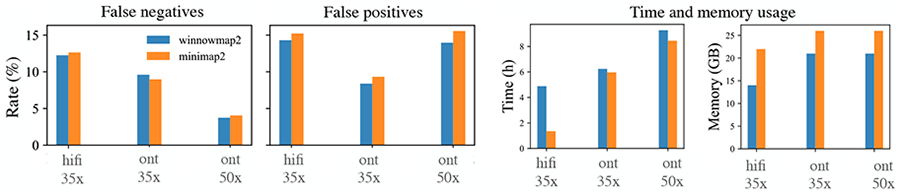
Comparison of Winnowmap2 and minimap2 by using GIAB SV benchmark set defined for HG002 human sample. Current GIAB benchmark set (v0.6) excludes complex repeats of the human genome. Outside the repeats, Winnowmap2 achieves similar FNR scores and slightly better FPR scores compared to minimap2.
Variant discovery in benchmark human genomes
Using Winnowmap2, we examined structural variation within repetitive regions of the human genome by using two publicly available ONT read sets for benchmark human genomes HG004 (coverage: 90x) and HG007 (coverage: 45x). For this experiment, we used both the GRCh38 human genome reference as well as a draft assembly of the human CHM13 genome released by T2T consortium [30]. The CHM13 draft assembly (v1.0) closes hundreds of gaps in GRCh38 that are associated with long segmental duplications and satellite DNAs. This enabled us to assess SVs in these complex repeats. Like CHM13, the HG004 and HG007 samples are both female and do not include chromosome Y. Figure 6 shows the length distribution and count of indel SVs identified by using the Winnowmap2-Sniffles pipeline. Overall, we identified 50,125 and 51,299 SVs in HG004 relative to the GRCh38 reference and CHM13 assemblies, respectively. Similarly, we identified 34,683 and 31,114 SVs in HG007 relative to GRCh38 and CHM13, respectively (Figure 6). 95.0% of bases in the CHM13 assembly (v1.0) were identified to have a one-to-one correspondence with the GRCh38 reference (Methods). This is also expected because 4.9% bases in GRCh38 sequence are annotated as unresolved gaps4. Enrichment of SV calls within these newly resolved regions of the CHM13 assembly was found to be significantly high for both HG004 (22% of total SVs, Figure 6) and HG007 (14% of total SVs) samples. These results suggest the need for a deeper investigation of SVs within these dynamic regions of the genome to understand any functional associations. Winnowmap2 is well-suited for this task due to its superior accuracy when mapping long reads to highly repetitive sequences.
Figure 6:
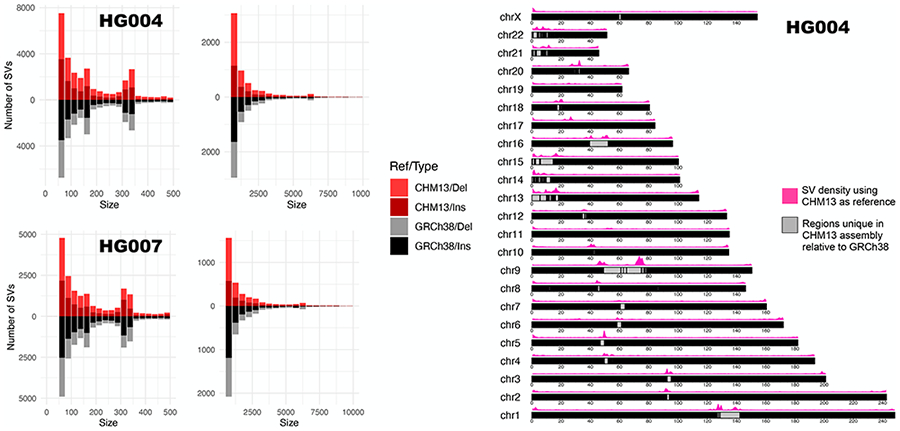
The left plots indicate the size distribution of SVs computed by Winnowmap2-Sniffles pipeline using HG004 and HG007 samples. Here we used both GRCh38 and T2T CHM13 human assembly as reference. The right plot shows the positional density of SVs found in HG004 sample using an ideogram plot [48] of the T2T CHM13 human assembly (v1.0). Significant enrichment of structural variation occurs in unique and newly resolved repetitive portions of the assembly.
3. Discussion and Conclusions
Here we highlighted the advantages of Winnowmap2 by demonstrating its superior downstream variant call accuracy compared to commonly used long read mappers. We demonstrated that Winnowmap2 improves SV calling accuracy within the most repetitive regions of human chromosomes. Prior studies have suggested an enrichment of SVs in near-identical, gene-rich segmental duplications that correspond to unresolved gaps in the human genome reference [37-39]. This underscores the importance of understanding how these regions differ between individuals. In addition to structural variants, it is natural to expect that Winnowmap2’s superior mapping accuracy will also benefit SNP and short indel variant calling. Another application where Winnowmap2 offers an advantage over existing methods is the polishing of draft genome assemblies, where long repeats are a common source of error [40].
Further algorithmic improvements will be needed to improve read alignment accuracy. In particular, it remains challenging to align bases precisely when multiple SVs are clustered in close vicinity. Our simulation made use of SURVIVOR, which simulates SVs at uniformly random positions in a reference sequence and could be an over-simplification of real data. In addition, read mappers and variant callers still remain limited in their ability to handle nested variation and other forms of complex rearrangements [41].
Availability of long-range sequencing technologies makes it feasible to resolve large mega-base sized near-identical duplications in the human genome, a feat that was impossible to achieve using short reads alone [42-46]. These regions include recently diverged segmental duplications, ampliconic gene arrays, rRNA genes, and centromeres, all of which play important functional roles in the genome and all of which go largely unstudied by current variation analyses. As human reference gaps associated with these regions are progressively resolved, this opens up the opportunity to expand the resolution of resequencing approaches. In this work, we highlighted that allelic bias becomes a major challenge for accurately mapping reads to repetitive reference sequences. This challenge affects the accuracy of existing mappers because classic pairwise sequence alignment scoring schemes are not an ideal mechanism to identify the correct mapping target in a repetitive sequence. In Winnowmap2, we have implemented a new idea based on minimal confidently align-able substrings that can be mapped independently of non-reference bases, thus alleviating allelic mapping bias and opening the entire genome to accurate variant calling.
4. Methods
Minimal confidently alignable substring (MCAS)
The use of MCASs distinguishes Winnowmap2 from previous read mapping methods. Prior to defining an MCAS, we formalize when we define an alignment of a substring to be confident. In practice, this confidence is derived using the score difference between the best scoring alignment and other candidate alignments. The mapping quality (mapQ) score was originally defined to address this problem [25], but the existing mathematical definition is restricted to short reads because alignments were assumed to be ungapped. However, the majority of long read sequencing errors are indels and the longer reads are more likely to span structural variants. When allowing for indels, adjacent mapping loci in a reference can no longer be considered independent, as in prior models. Accordingly, we propose the following formulation.
Given a query string and a reference , the top scoring end-to-end (a.k.a. semi-global) alignment candidates of string to can be directly computed in time. From a pairwise alignment of to , we can identify the set of matched base positions between them. For instance, this set would include a tuple if character is matched to character . We say that two alignment candidates do not overlap if and only if their corresponding sets of matched base positions are disjoint. We consider the best-scoring alignment of string to reference to be confident if and only if its second-best non-overlapping alignment candidate has a score , where opt refers to the optimal alignment score and is a user-specified parameter.
Let be a long read sequence. A minimal confidently alignable substring of read refers to the shortest substring starting at position that has a confident end-to-end alignment to reference . For a given read , we seek , and their corresponding alignments. MCASs can have variable lengths and can overlap one another. Note that the existence of depends on whether it is possible to satisfy the confidence criteria. In the worst case, where two repetitive regions lack any PSVs (i.e., 100% identical duplicates), a read sampled from either repeat copy will not contain an MCAS. The rationale for introducing the MCAS idea is to address allelic bias; whereas a non-reference SV allele will cause mis-alignment in the traditional approach, the MCASs are treated independently and those neighboring the SV will remain unaffected (Supplementary Figure S4).
Considering the issue of allelic bias, it is also desirable to enforce a maximum length parameter for valid MCASs because long MCASs again become vulnerable to allelic bias. By default, we set the maximum length parameter to 8 kbp for HiFi reads and 16 kbp for ONT reads based on our experimental observations. As such, the maximum length of a valid MCAS is a constant. We prove that the asymptotic runtime and space complexity for computing MCASs is and respectively (Supplementary Note S1). An time complexity resembles the complexity of DP-based alignment algorithms. As such, the exact algorithm does not offer desired scalability. In Winnowmap2, we make use of fast heuristics and make careful accuracy-performance trade-offs to address this. First, we perform the MCAS computation from a subset of equally spaced starting positions, i.e., every 1000th base for HiFi and every 2000th base for ONT sequences. Next, while computing an MCAS, we reduce the alignment search space by making use of known minimizer seeding and clustering ideas [9, 11]. Starting from a small substring length, our iterative method exponentially grows the substring (rather than growing linearly). In each iteration, we check its mapping to reference . This is done until the substring either satisfies the alignment confidence criteria or cannot be extended further. While computing each mapping, we rely on efficiently engineered anchor chaining, banded-alignment, and mapQ computation code from minimap2. In a way, the mapQ scoring heuristic in minimap2 approximates our definition for confidence assessment.
Heuristic to compute mapping quality
In Winnowmap2 implementation, we use the same heuristic as minimap2 to compute the mapping quality score of a read alignment. For completeness, we also mention it here. Once the anchors between a read and a reference are identified, minimap2 runs a co-linear chaining algorithm to locate alignment candidates. The chaining procedure ensures that alignment candidates use a disjoint set of anchors to prevent overlaps. To compute mapQ, minimap2 compares the anchor chaining score of the best-scoring chain relative to the second-best. Suppose their scores are denoted as and respectively. Also, let be the count of anchors chained along the best alignment. Minimap2 uses the following empirical formula to calculate mapQ score of the best alignment candidate:
The above score is readjusted by minimap2 to fall within the range of 0 to 60. By default, we use mapQ cutoff of 5 in Winnowmap2 to mark an MCAS alignment as confident. This cutoff can be modified by users. In practice, a higher cutoff typically leads to longer MCASs, as expected. A lower cutoff increases the probability of an incorrect alignment to be considered as the best.
Consolidating MCASs into a single alignment output
Once we compute MCAS alignments from a read, these need to be aggregated into a single alignment output. At this step, we extract anchors that were co-linearly chained in each MCAS alignment. Subsequently, the union of all anchor sets is passed as input to chaining and alignment routines to output the final set of best-scoring alignments. Typically, there are only a few anchors to process at this step, which does not require significant time. In a few cases, presence of frequent sequencing errors or lack of PSVs within repeats may prevent Winnowmap2 from identifying any MCAS. If this happens, Winnowmap2 falls back to the default Minimap2 algorithm to recompute anchors instead of leaving a read unmapped.
Simulation and evaluation of structural variant calls
In our simulation benchmark, we made use of T2T chromosome assemblies for chromosome 8 (v9) and chromosome X (v0.7) that are available from https://github.com/nanopore-wgs-consortium/CHM13. SURVIVOR (v1.0.6) was used to simulate 1,100 SVs of length ranging from 50 bp to 1000 bp in each chromosome sequence. We also simulated PacBio HiFi reads as well as ONT reads using PBSIM (commit:e014b1) and NanoSim (v2.6.0) respectively. Command line parameters provided to these tools are listed in Supplementary Table S3. NanoSim requires real data for training its error model. Training was executed using a publicly available R10.3 Guppy 3.4.5 ONT sequencing data of the Escherichia coli K12 genome (ENA:PRJEB36648). PBSIM command line parameters were adjusted to achieve PacBio HiFi data characteristics with an indel error rate of about 1%. Supplementary Table S4 specifies the read length statistics. Long read mappers were tested using two sequencing coverage levels, 20x and 40x. In our mapping evaluation, we compared Winnowmap2 (v2.03), Winnowmap (v1.01), minimap2 (v2.18), ngmlr (v0.2.7) and graphmap (v0.5.2). Each mapper was executed using their recommended parameters and 24 CPU threads (Supplementary Table S3). SV calling from BAM alignment file outputs was done using Sniffles (v1.0.11). The SV call sets were evaluated using SURVIVOR against its own simulated ground truth. We also evaluated SV calling accuracy within repetitive reference intervals. For this, de novo repeat annotation of reference sequences was computed by using Mashmap (commit:ffeef4) to approximately identify all duplications of ≥10 kbp length and ≥ 95% identity. SV evaluation within the repeats was done by intersecting variant coordinates and repeat intervals using bedtools (v2.29.2) [49].
Evaluation using GIAB SV calls
We evaluated Winnowmap2 and minimap2 using the GIAB Tier1 (v0.6) SV call set [36] available for the HG002 human sample relative to the GRCh37 human genome reference. In this experiment, we utilized HG002 ONT and PacBio HiFi sequencing data [50, 51] made available through the precision FDA site https://precision.fda.gov/challenges/10/. Sniffles SV call sets were evaluated using SVanalyzer (v0.36).
Structural variant calling in HG004 and HG007 human samples
HG004 ONT sequencing data was accessed from precision FDA site https://precision.fda.gov/challenges/10/, whereas HG007 ONT sequencing data was accessed from a prior study [50]. Here we made use of both GRCh38 and T2T CHM13 assembly (v1.0). The CHM13 assembly was accessed from https://github.com/nanopore-wgs-consortium/CHM13. Long read mapping and SV calling was done using Winnowmap2 and Sniffles respectively. Approximately 5% of bases in this assembly correspond to unresolved gaps in GRCh38 human genome reference. A one-to-one homology map between CHM13 assembly and GRCh38 reference was computed using Mashmap. This map was used to quantify SV enrichment within newly resolved regions of CHM13 assembly.
Supplementary Material
Acknowledgements
We would like to thank Kishwar Shafin, Alla Mikheenko, Melanie Kirsche and Sergey Nurk for providing useful feedback regarding Winnowmap2. We also acknowledge Heng Li for responding to our queries regarding minimap2 code. Winnowmap2 and Winnowmap were developed on top of minimap2 code. This research was supported in part by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health and funding from the Indian Institute of Science.
Footnotes
Competing interests
The authors declare no competing interests.
Data availability
This study used publicly available data for evaluation. Complete gapless genome assembly of CHM13 human cell line (v1.0), chromosome 8 (v9) and chromosome X (v0.7) can be accessed from https://github.com/marbl/CHM13#downloads. ONT and PacBio HiFi sequencing data for HG002, HG003 and HG004 samples are available at https://precision.fda.gov/challenges/10. Escherichia coli K-12 nanopore sequencing data used for training NanoSim simulator is available in European Nucleotide Archive (PRJEB36648). Genome in a Bottle (GIAB) SV call set (v0.6) for HG002 human sample is available at https://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/release/AshkenazimTrio.
Code availability
Winnowmap2 code is available at https://github.com/marbl/Winnowmap. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
References
- 1.Miga KH, Koren S, Rhie A, Vollger MR, Gershman A, Bzikadze A, et al. : Telomere-to-telomere assembly of a complete human X chromosome. Nature (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Logsdon GA, Vollger MR, Hsieh P, Mao Y, Liskovykh MA, Koren S, Nurk S, Mercuri L, Dishuck PC, Rhie A, et al. : The structure, function and evolution of a complete human chromosome 8. Nature pp. 1–7 (2021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chaisson MJ, Tesler G: Mapping single molecule sequencing reads using basic local alignment with successive refinement (blasr): application and theory. BMC bioinformatics 13(1), 238 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sović I, Šikić M, Wilm A, Fenlon SN, Chen S, Nagarajan N: Fast and sensitive mapping of nanopore sequencing reads with graphmap. Nature communications 7(1), 1–11 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin HN, Hsu WL: Kart: a divide-and-conquer algorithm for NGS read alignment. Bioinformatics 33(15), 2281–2287 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suzuki H, Kasahara M: Introducing difference recurrence relations for faster semi-global alignment of long sequences. BMC bioinformatics 19(1), 33–47 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, Von Haeseler A, Schatz MC: Accurate detection of complex structural variations using single-molecule sequencing. Nature methods 15(6), 461–468 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jain C, Dilthey A, Koren S, Aluru S, Phillippy A: A fast approximate algorithm for mapping long reads to large reference databases. Journal of Computational Biology 25(7), 766 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H.: Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34(18), 3094–3100 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haghshenas E, Sahinalp SC, Hach F: lordfast: sensitive and fast alignment search tool for long noisy read sequencing data. Bioinformatics 35(1), 20–27 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jain C, Rhie A, Zhang H, Chu C, Walenz BP, Koren S, Phillippy AM: Weighted minimizer sampling improves long read mapping. Bioinformatics 36(Supplement_1), i111–i118 (07 2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeni A, Guidi G, Ellis M, Ding N, Santambrogio MD, Hofmeyr S, Buluç A, Oliker L, Yelick K: Logan: High-performance gpu-based x-drop long-read alignment. In: 2020 IEEE International Parallel and Distributed Processing Symposium (IPDPS). pp. 462–471. IEEE; (2020) [Google Scholar]
- 13.Prodanov T, Bansal V: Sensitive alignment using paralogous sequence variants improves long-read mapping and variant calling in segmental duplications. Nucleic acids research 48(19), e114–e114 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marco-Sola S, Moure JC, Moreto M, Espinosa A: Fast gap-affine pairwise alignment using the wavefront algorithm. Bioinformatics 37(4), 456–463 (2021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Degner JF, Marioni JC, Pai AA, Pickrell JK, Nkadori E, Gilad Y, Pritchard JK: Effect of readmapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics 25(24), 3207–3212 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwartz S, Oren R, Ast G: Detection and removal of biases in the analysis of next-generation sequencing reads. PloS one 6(1), e16685 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vijaya Satya R, Zavaljevski N, Reifman J: A new strategy to reduce allelic bias in RNA-seq readmapping. Nucleic acids research 40(16), e127–e127 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stevenson KR, Coolon JD, Wittkopp PJ: Sources of bias in measures of allele-specific expression derived from RNA-seq data aligned to a single reference genome. BMC genomics 14(1), 536 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brandt DY, Aguiar VR, Bitarello BD, Nunes K, Goudet J, Meyer D: Mapping bias overestimates reference allele frequencies at the HLA genes in the 1000 genomes project phase i data. G3: Genes, Genomes, Genetics 5(5), 931–941 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Günther T, Nettelblad C: The presence and impact of reference bias on population genomic studies of prehistoric human populations. PLoS genetics 15(7), e1008302 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mikheenko A, Bzikadze AV, Gurevich A, Miga KH, Pevzner PA: Tandemtools: mapping long reads and assessing/improving assembly quality in extra-long tandem repeats. Bioinformatics 36(Supplement_1), i75–i83 (07 2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dilthey A, Cox C, Iqbal Z, Nelson MR, McVean G: Improved genome inference in the MHC using a population reference graph. Nature genetics 47(6), 682–688 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paten B, Novak AM, Eizenga JM, Garrison E: Genome graphs and the evolution of genome inference. Genome research 27(5), 665–676 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Feng X, Chu C: The design and construction of reference pangenome graphs with minigraph. Genome biology 21(1), 1–19 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H, Ruan J, Durbin R: Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome research 18(11), 1851–1858 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, Tsalenko A, Sampas N, Bruhn L, Shendure J, Eichler EE, et al. : Diversity of human copy number variation and multicopy genes. Science 330(6004), 641–646 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vollger MR, Dishuck PC, Sorensen M, Welch AE, Dang V, Dougherty ML, Graves-Lindsay TA, Wilson RK, Chaisson MJ, Eichler EE: Long-read sequence and assembly of segmental duplications. Nature methods 16(1), 88–94 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeffares DC, Jolly C, Hoti M, Speed D, Shaw L, Rallis C, Balloux F, Dessimoz C, Bähler J, Sedlazeck FJ: Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nature communications 8(1), 1–11 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen HC, Kitts PA, Murphy TD, Pruitt KD, Thibaud-Nissen F, Albracht D, et al. : Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome research 27(5), 849–864 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, Vollger MR, Altemose N, Uralsky L, Gershman A, et al. : The complete sequence of a human genome. bioRxiv (2021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roberts M, Hayes W, Hunt BR, Mount SM, Yorke JA: Reducing storage requirements for biological sequence comparison. Bioinformatics 20(18), 3363–3369 (2004) [DOI] [PubMed] [Google Scholar]
- 32.Hollox EJ, Armour JA, Barber JC: Extensive normal copy number variation of a β-defensin antimicrobial-gene cluster. The American Journal of Human Genetics 73(3), 591–600 (2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang C, Chu J, Warren RL, Birol I: Nanosim: nanopore sequence read simulator based on statistical characterization. GigaScience 6(4), gix010 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ono Y, Asai K, Hamada M: Pbsim: Pacbio reads simulator—toward accurate genome assembly. Bioinformatics 29(1), 119–121 (2013) [DOI] [PubMed] [Google Scholar]
- 35.Jain C, Koren S, Dilthey A, Phillippy AM, Aluru S: A fast adaptive algorithm for computing whole-genome homology maps. Bioinformatics 34(17), i748–i756 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zook JM, Hansen NF, Olson ND, Chapman L, Mullikin JC, Xiao C, Sherry S, Koren S, Phillippy AM, Boutros PC, et al. : A robust benchmark for detection of germline large deletions and insertions. Nature biotechnology pp. 1–9 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, Pertz LM, Clark RA, Schwartz S, Segraves R, et al. : Segmental duplications and copy-number variation in the human genome. The American Journal of Human Genetics 77(1), 78–88 (2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chaisson MJ, Sanders AD, Zhao X, Malhotra A, Porubsky D, Rausch T, Gardner EJ, Rodriguez OL, Guo L, Collins RL, et al. : Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nature communications 10(1), 1–16 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Audano PA, Sulovari A, Graves-Lindsay TA, Cantsilieris S, Sorensen M, Welch AE, Dougherty ML, Nelson BJ, Shah A, Dutcher SK, et al. : Characterizing the major structural variant alleles of the human genome. Cell 176(3), 663–675 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mc Cartney AM, Shafin K, Alonge M, Bzikadze AV, Formenti G, Fungtammasan A, Howe K, Jain C, Koren S, Logsdon GA, et al. : Chasing perfection: validation and polishing strategies for telomere-to-telomere genome assemblies. biorxiv (2021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ho SS, Urban AE, Mills RE: Structural variation in the sequencing era. Nature Reviews Genetics pp. 1–19 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kamath GM, Shomorony I, Xia F, Courtade TA, David NT: Hinge: long-read assembly achieves optimal repeat resolution. Genome research 27(5), 747–756 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bzikadze AV, Pevzner PA: Automated assembly of centromeres from ultra-long error-prone reads. Nature Biotechnology 38(11), 1309–1316 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nurk S, Walenz BP, Rhie A, Vollger MR, Logsdon GA, Grothe R, Miga KH, Eichler EE, Phillippy AM, Koren S: Hicanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Research 30(9), 1291–1305 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bankevich A, Bzikadze AV, Kolmogorov M, Antipov D, Pevzner PA: Multiplex de bruijn graphs enable genome assembly from long, high-fidelity reads. Nature Biotechnology pp. 1–7 (2022) [DOI] [PubMed] [Google Scholar]
- 46.Cheng H, Concepcion GT, Feng X, Zhang H, Li H: Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature methods 18(2), 170–175 (2021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP: Integrative genomics viewer. Nature biotechnology 29(1), 24–26 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gel B, Serra E: karyoploter: an r/bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics 33(19), 3088–3090 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quinlan AR, Hall IM: Bedtools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26(6), 841–842 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shafin K, Pesout T, Lorig-Roach R, Haukness M, Olsen HE, Bosworth C, Armstrong J, Tigyi K, Maurer N, Koren S, et al. : Nanopore sequencing and the shasta toolkit enable efficient de novo assembly of eleven human genomes. Nature Biotechnology pp. 1–10 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wenger AM, Peluso P, Rowell WJ, Chang PC, Hall RJ, Concepcion GT, Ebler J, Fungtammasan A, Kolesnikov A, Olson ND, et al. : Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nature biotechnology 37(10), 1155–1162 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study used publicly available data for evaluation. Complete gapless genome assembly of CHM13 human cell line (v1.0), chromosome 8 (v9) and chromosome X (v0.7) can be accessed from https://github.com/marbl/CHM13#downloads. ONT and PacBio HiFi sequencing data for HG002, HG003 and HG004 samples are available at https://precision.fda.gov/challenges/10. Escherichia coli K-12 nanopore sequencing data used for training NanoSim simulator is available in European Nucleotide Archive (PRJEB36648). Genome in a Bottle (GIAB) SV call set (v0.6) for HG002 human sample is available at https://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/release/AshkenazimTrio.
Winnowmap2 code is available at https://github.com/marbl/Winnowmap. Further information on research design is available in the Nature Research Reporting Summary linked to this article.