

Abstract

High-yield syntheses of the lanthanide dinitrogen complexes [(Cp2tttM)2(μ-1,2-N2)] (1M, M = Gd, Tb, Dy; Cpttt = 1,2,4-C5tBu3H2), in which the [N2]2– ligands solely adopt the rare end-on or 1,2-bridging mode, are reported. The bulk of the tert-butyl substituents and the smaller radii of gadolinium, terbium, and dysprosium preclude formation of the side-on dinitrogen bonding mode on steric grounds. Elongation of the nitrogen-nitrogen bond relative to N2 is observed in 1M, and their Raman spectra show a major absorption consistent with N=N double bonds. Computational analysis of 1Gd identifies that the local symmetry of the metallocene units lifts the degeneracy of two 5dπ orbitals, leading to differing overlap with the π* orbitals of [N2]2–, a consequence of which is that the dinitrogen ligand occupies a singlet ground state. Magnetic measurements reveal antiferromagnetic exchange in 1M and single-molecule magnet (SMM) behavior in 1Dy. Ab initio calculations show that the magnetic easy axis in the ground doublets of 1Tb and 1Dy align with the {M–N=N–M} connectivity, in contrast to the usual scenario in dysprosium metallocene SMMs, where the axis passes through the cyclopentadienyl ligands. The [N2]2– ligands in 1M allow these compounds to be regarded as two-electron reducing agents, serving as synthons for divalent gadolinium, terbium, and dysprosium. Proof of principle for this concept is obtained in the reactions of 1M with 2,2′-bipyridyl (bipy) to give [Cp2M(κ2-bipy)] (2M, M = Gd, Tb, Dy), in which the lanthanide is ligated by a bipy radical anion, with strong metal–ligand direct exchange coupling.
Introduction
Activation of dinitrogen by strongly reducing metal complexes and the subsequent functionalization of N2-derived ligands are reactions of considerable importance, both from a fundamental perspective and because of the insight they provide into a variety of biological and catalytic processes.1,2 Dinitrogen activation by f-elements has been studied for more than a century in the context of ammonia synthesis, with cerium and uranium reportedly being competent catalysts in the Haber-Bosch process.3 More recently, the activation of dinitrogen by well-defined uranium complexes has been demonstrated and, subsequently, used as the basis of non-catalytic routes to ammonia and to other N-functionalized species.4−9 Complexes of rare-earth elements in the divalent oxidation state10−14 and various combinations of trivalent rare earth complexes with alkali metals15−17 also show a propensity to activate dinitrogen via reduction, but subsequent conversion of [N2]2– into hydrazido derivatives has only been accomplished in very few instances.18,19
Much of the fascination with dinitrogen activation by lanthanides stems from the coordination mode of the N2-derived ligand, which can influence the balance of 4f vs 5d orbital usage by the metal. The bridging side-on coordination mode {M2(μ-η2:η2-N2)} is common with rare-earth metals, whereas the end-on variant {M2(μ-1,2-N2)} is uncommon (Scheme 1).1 The difference in coordination mode matters since it impacts the electronic structure of the metal and the dinitrogen ligand, while also influencing the stability of the complex.
Scheme 1. Dinitrogen Coordination Modes toward Rare-Earth Metals, M.

To date, examples of pure end-on dinitrogen-bridged rare-earth complexes are limited to the bulky amido-ligated anions [{(N″)3M}2(μ-1,2-N2)]2– (N″ = N(SiMe3)2, M = Sc, Y, Tb), which form as salts of crown- or cryptand-ligated alkali metal cations.20 The gadolinium and dysprosium analogues form in the solid state as mixtures of two disordered components, defined by end-on and side-on coordination of the [N2]2– ligand.21 The scandium complex was found to eliminate N2 on irradiation with UV light at −78 °C, and the gadolinium and terbium versions were reported to eliminate N2 above −35 °C. The delicate energetic balance between the two coordination modes of [N2]2– was underscored by the neodymium congener, which forms as [{(N″)2Nd}2(μ-1,2-N2)]2– below −90 °C and converts into the side-on version [{(N″)2Nd}2(μ-η2:η2-N2)]2– in the solid-state at higher temperatures.22
The end-on bonding mode of dinitrogen has hitherto not been observed in lanthanide metallocene chemistry, whereas lanthanide metallocenes with side-on bridging dinitrogen are known with [N2]2– and its S = 1/2 radical [N2]3– derivative.23 We were motivated to target end-on dinitrogen-bridged lanthanide metallocenes to gain insight into how {Cp2Ln} fragments influence the electronic structure of the μ-1,2-N2 ligand and phenomena such as magnetic exchange interactions and slow magnetic relaxation. Furthermore, by regarding the [N2]2– ligand as a two-electron reservoir, the dinitrogen complexes themselves might also serve as surrogate divalent reducing agents for lanthanides that do not readily form the oxidation state +2, allowing them to be used in the synthesis of lanthanide complexes of molecular magnets with radical ligands.
Results
Our strategy focused on synthesizing [(Cp2tttM)2(μ-1,2-N2)] (1M, M = Gd, Tb, Dy; Cpttt = 1,2,4-C5tBu3H2) by reduction of [Cp2M(κ2-BH4)] with an excess of potassium graphite under an atmosphere of dinitrogen at room temperature. Combined with the relatively small radii of the lanthanide M3+ cations, the bulky cyclopentadienyl ligand Cpttt was selected assuming that the tert-butyl substituents would hinder close approach of the two metallocene units, discouraging formation of the side-on dinitrogen coordination mode and encouraging formation of the end-on mode. The target compounds 1Gd, 1Tb, and 1Dy were isolated in yields of 87, 83, and 84%, respectively, based on the lanthanide according to Scheme 2.
Scheme 2. Synthesis of 1M (M = Gd, Tb, Dy).

In synthesizing 1M, the initial step probably involves reduction of the trivalent metallocenes [Cp2tttM(κ2-BH4)] to divalent [Cp2M], which then rapidly activates N2 by electron transfer. The formation of divalent lanthanide intermediates is reasonable given the similarities between our reaction conditions and those used to prepare other, related divalent lanthanide metallocenes.14,24,25 Attempts at isolating the putative [Cp2tttM] complexes were made by conducting the KC8 reductions under an atmosphere of argon; however, the dinitrogen complexes 1M still form even under these conditions, implying that the divalent metallocenes are sufficiently reactive to scavenge trace N2 from the high-purity argon.
The molecular structures of 1M were determined by single-crystal X-ray diffraction measurements (Figures 1, S1, Table S1). The three complexes are isostructural, forming as centrosymmetric molecules with two {Cp2tttM} units bridged by an end-on dinitrogen ligand, and with the inversion center coinciding with the midpoint of the N=N bond. The resulting {M–N=N} connectivities are essentially linear at 179.0(3)°, 179.6(3)°, and 179.3(5)° for 1Gd, 1Tb, and 1Dy, respectively. Consistent with the gradual decrease in ionic radius of the M3+ cations, the M–N distances decrease from 2.325(4) Å in 1Gd to 2.296(4) Å in 1Tb and 2.268(7) Å in 1Dy. Concomitantly, the N=N distances increase across the series, being 1.130(8), 1.175(8), and 1.215(13) Å for 1Gd, 1Tb, and 1Dy, respectively. The trend in M–N distances, combined with the apparent flexibility of the N=N bond, produces similar M···M separations of 5.7791(9), 5.7659(9), and 5.7500(9) Å for 1Gd, 1Tb, and 1Dy, respectively. Other pertinent geometric parameters include M–Cpttt distances of 2.4370(16) and 2.4399(17) Å for 1Gd, 2.4146(19) and 2.419(2) Å for 1Tb, and 2.405(3) and 2.406(3) Å for 1Dy, with associated Cpttt–M–Cpttt angles of 144.63(8)°, 143.96(9)°, and 143.86(13)°, respectively.
Figure 1.

Thermal ellipsoid representation (30% probability) of the molecular structure of 1Gd. For clarity, hydrogen atoms are not shown.
Relative to dinitrogen itself, in which the triple bond length is 1.098 Å, considerable lengthening of the nitrogen–nitrogen bond has occurred upon reduction of N2 to give 1M. The extent to which the N2 bond lengthens is, evidently, dependent on the lanthanide, although the supporting Cpttt ligands may also influence this aspect given that the analogous distances in the series [{(N″)2M}2(μ-1,2-N2)]2– are typically longer almost regardless of the metal.20−22 However, it has been pointed out that the nitrogen–nitrogen distance is an imperfect measure of the extent of activation1 and that greater insight can be obtained from vibrational spectroscopy. The Raman spectra of 1M were therefore recorded and found to be very similar (Figure 2), consisting of dominant signals at 1623, 1621, and 1618 cm–1 for 1Gd, 1Tb, and 1Dy, respectively, indicative of N=N double bonds in 1M.26
Figure 2.

Raman spectra of 1Gd, 1Tb, and 1Dy. Spectra are normalized and the baselines are corrected.
Analysis of Bonding in 1Gd
To understand the bonding within the {M2N2} core of 1M, we analyzed 1Gd using density functional theory (DFT) calculations, which were performed on the coordinates obtained from the X-ray structure without optimization, except for the hydrogen atoms. Full computational details are provided in the Supporting Information. The calculations show that the highest occupied molecular orbital (HOMO) corresponds to gadolinium-N2 interactions and is doubly occupied, indicating transfer of two electrons from two putative [Cp2tttGd] complexes to dinitrogen (Figure 3).
Figure 3.

Frontier molecular orbitals in 1Gd.
The π-character of the HOMO arises from overlap between a gadolinium 5d orbital and a π* orbital of the N2 ligand. The lowest unoccupied molecular orbital (LUMO) involves an interaction between the other N2 π* orbital and a gadolinium 5d orbital, but this is weaker due to the relatively poor spatial overlap. An important consequence of this orbital scenario is the HOMO–LUMO gap of 2.03 eV (approximately 196 kJ mol–1), which results in a well-isolated singlet ground state for the bridging [N2]2– ligand. The HOMO–LUMO gap calculated for 1Gd agrees well with the observation of a major absorption in the UV-vis spectrum of 1Gd at λmax = 619 nm (Figure 4), corresponding to the HOMO–LUMO transition.
Figure 4.

UV–vis spectra in toluene for 1Gd (λmax = 619 nm), 1Tb (λmax = 605 nm), and 1Dy (λmax = 562 nm) at −78 °C.
The singlet character of the end-on dinitrogen
ligand in 1Gd is in stark contrast with the
triplet character
of the analogous ligand in [{(N″)2M}2(μ-1,2-N2)]2–, which is a consequence
of the degeneracy of the π* orbitals being preserved.20 In addition, negligible spin density (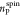 (N) = −0.07) was also calculated
for the dinitrogen ligand in 1Gd along with
a total spin expectation value of ⟨S2⟩ = 56 for the molecule, implying a high-spin configuration
with 14 unpaired electrons. These results further support the singlet
configuration of [N2]2– in 1Gd. The calculations also reveal that the Gd–N
bonds have a 14% weighting of gadolinium, which is composed of 90%
5d character and 9.5% 6p character, with a negligible contribution
from the 4f orbitals (Tables S5 and S6).
Furthermore, the natural electronic configuration of the gadolinium
atoms assigned by the calculations is 4f6.995 5d0.45, which equates to the expected 4f7 5d0 configuration
of Gd3+ and agrees with the formal dianionic charge of
the dinitrogen ligand. The X-band EPR spectrum of polycrystalline 1Gd at 298 K shows extensive fine structure (Figure S5), and a fit of the experimental spectrum
was possible with the inclusion of a small rhombic term with the parameters g = 2.001, D = −0.014 cm–1, and E = −0.0013 cm–1 (E/D = 0.09). The EPR data are consistent
with previously reported Gd3+ complexes of diamagnetic
ligands.27
(N) = −0.07) was also calculated
for the dinitrogen ligand in 1Gd along with
a total spin expectation value of ⟨S2⟩ = 56 for the molecule, implying a high-spin configuration
with 14 unpaired electrons. These results further support the singlet
configuration of [N2]2– in 1Gd. The calculations also reveal that the Gd–N
bonds have a 14% weighting of gadolinium, which is composed of 90%
5d character and 9.5% 6p character, with a negligible contribution
from the 4f orbitals (Tables S5 and S6).
Furthermore, the natural electronic configuration of the gadolinium
atoms assigned by the calculations is 4f6.995 5d0.45, which equates to the expected 4f7 5d0 configuration
of Gd3+ and agrees with the formal dianionic charge of
the dinitrogen ligand. The X-band EPR spectrum of polycrystalline 1Gd at 298 K shows extensive fine structure (Figure S5), and a fit of the experimental spectrum
was possible with the inclusion of a small rhombic term with the parameters g = 2.001, D = −0.014 cm–1, and E = −0.0013 cm–1 (E/D = 0.09). The EPR data are consistent
with previously reported Gd3+ complexes of diamagnetic
ligands.27
Reactivity of Lanthanide Dinitrogen Complexes
The strongly reducing nature of [N2]2– has considerable potential to allow 1M to be developed for bespoke electron-transfer reactivity. For example, adding the two-electron reducing agents 1M to N-heterocyclic ligands with an odd number of binding sites should result in electron transfer and elimination of N2, accompanied by the formation of paramagnetic lanthanide metallocene complexes by heterocyclic radical anions. Achieving reactivity of this nature would provide a new method for the synthesis of molecular magnets such as SMMs, going beyond early studies examining the reactivity of diamagnetic side-on dinitrogen complexes of lanthanum and lutetium.28,29 Indeed, it is noteworthy that bimetallic lanthanidocene complexes of radical N-donor ligands are a well-known type of SMM,30−35 with some examples displaying remarkable magnetic hysteresis properties arising from direct magnetic exchange with the radical ligand.23,36,37 To the best of our knowledge, monometallic lanthanide metallocene SMMs containing radical ligands are unknown. We therefore undertook to synthesize such species by reacting 1M with 2,2′-bipyridyl (bipy), resulting in the formation of [Cp2tttM(κ2-bipy)] (2M, M = Gd, Tb, Dy) according to Scheme 3.
Scheme 3. Synthesis of 2M (M = Gd, Tb, Dy).

Complexes 2M were isolated in excellent yields of 90, 95, and 82% for 2Gd, 2Tb, and 2Dy, respectively. X-ray crystallography confirmed that 2M adopt bent metallocene structures with a κ2-bipy ligand and a mirror plane bisecting the molecules through the metal, oriented perpendicular to the bipy plane (Figures 5, S2 and Tables S2 and S4). The M–N distances of 2.4052(18), 2.384(3), and 2.365(2) Å in 2Gd, 2Tb, and 2Dy, respectively, are much longer than the related distances in 1M to accommodate the larger bipy ligand. In response, the M–Cpttt distances increase by about 0.07 Å relative to 1M to 2.5036(10), 2.4832(14), and 2.4650(11) Å in 2Gd, 2Tb, and 2Dy, respectively, and the associated Cpttt–M–Cpttt angles narrow to 140.17(5)°, 141.04(7)°, and 141.28(5)°.
Figure 5.

Thermal ellipsoid representation (50% probability) of the molecular structure of 2Gd. For clarity, hydrogen atoms are not shown.
In contrast to 1Gd, the X-band EPR spectrum of polycrystalline 2Gd at 298 K consists of a single-line absorption at g = 1.98, consistent with the presence of 2,2′-bipy in its S = 1/2 radical anion form (Figure S6). The UV–vis spectra of 2M which are virtually identical, contain more features than the analogous spectra of 1M, including a series of overlapping absorptions in the region 650–1000 nm and higher-energy transitions below approximately 500 nm (Figure 6). A time-dependent DFT (TD-DFT) analysis of the UV–vis spectrum of 2Gd revealed that the group of lower-energy transitions is a combination of ligand-to-metal charge transfer from π* orbitals of the bipy radical anion to gadolinium 5d orbitals and intra-ligand transitions within the π/π* manifold of [bipy]− (Figure S7, Table S7). The higher-energy transitions occur solely within the [bipy]− orbital manifold.
Figure 6.

UV–vis spectra in hexane for 2Gd, 2Tb, and 2Dy complexes at room temperature.
Lanthanide complexes of reduced bipyridyl and related N-heterocyclic ligands are an established class of compounds, yet examples containing metals other than ytterbium or samarium are rare.38,39 The popularity of ytterbium stems from the ready availability of ytterbium(II) metallocenes,40−45 especially [(C5Me5)2Yb], which has been used to reduce N-heterocycles to give complexes such as [(C5Me5)2Yb(κ2-bipy)], which has an unusual electronic ground state with intermediate valence and multiconfigurational character. The rich redox chemistry of bipy-type ligands has also been showcased with trivalent and tetravalent actinide complexes typified by [Cp2tttAn(κ2-bipy)] (trivalent An = U, tetravalent An = Th, U), which were synthesized by in situ reduction of bipy with KC8 in the presence of [Cp2AnCl2].46−48 Actinide metallocenes such as these are notable for their reactivity as one- or two-electron reducing agents in a range of small-molecule activation processes. Similar synthetic routes were also developed for the potassium salt of a radical diazafluorenylidene-substituted phospha-alkene and a boryl-substituted bipy radical, which were used in salt metathesis reactions to give complexes with the radical ligand bound to gadolinium, terbium, and dysprosium.49,50
The availability of 1M as bespoke reducing agents offers numerous synthetic advantages. For example, complexes of 1M allow an extension of lanthanide-based reduction chemistry beyond the classical divalent lanthanides samarium, europium, and ytterbium. In place of large excesses of KC8 as the reductant, the dinitrogen ligand in 1M is a well-defined two-electron source that converts into N2 as a traceless leaving group following reduction, avoiding formation of salt-like bimetallic products. The high-yielding synthesis of 1M is also noteworthy.
Magnetic Properties
The magnetic properties of lanthanide
complexes with end-on bridging dinitrogen ligands have, hitherto,
not been subjected to detailed investigations by DC and AC susceptometry.
To address this gap in understanding, the temperature dependence of
the molar magnetic susceptibility (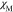 ) in an applied field of 1 kOe and the field
dependence of the magnetization (M) in fields of
0–70 kOe were measured for all compounds. Additionally, the
AC susceptibility and magnetic hysteresis properties of 1Tb, 1Dy, 2Tb, and 2Dy were investigated to check
for SMM behavior.
) in an applied field of 1 kOe and the field
dependence of the magnetization (M) in fields of
0–70 kOe were measured for all compounds. Additionally, the
AC susceptibility and magnetic hysteresis properties of 1Tb, 1Dy, 2Tb, and 2Dy were investigated to check
for SMM behavior.
The χMT value for 1Gd at 300 K is 13.45 cm3 K mol–1, which is somewhat lower than the theoretical value of 15.75 cm3 K mol–1 predicted for two non-interacting Gd3+ ions with g = 2.0 (Figure 7).51 On lowering the temperature to 120 K, χMT decreases gradually before decreasing rapidly to reach 0.83 cm3 K mol–1 at 2 K. The overall temperature-dependence of χMT for 1Gd is consistent with antiferromagnetic exchange via the end-on dinitrogen ligand. The M(H) plot for 1Gd at 2 K shows that the magnetization increases in a near-linear manner up to a value of 6.10 Nβ at the maximum field of 70 kOe attainable with the magnetometer, well below the saturation magnetization value of 14 Nβ expected for two Gd3+ ions (Figure 7). Simultaneous fits of the susceptibility and magnetization data were achieved using the spin Hamiltonian stated in eq 1 as implemented in the PHI software,52 where J is the exchange coupling constant, gGd and SGd denote the g-tensor and total spin associated with the gadolinium centers, respectively, and β is the Bohr magneton. Using g = 1.90, a J-value of −0.81 cm–1 was obtained for 1Gd.
| 1 |
| 2 |
Figure 7.

χMT(T) and M(H) plots for 1Gd, 1Tb, and 1Dy with simulations according to eqs 1 or 2 in the main text.
At 300 K, χMT for 1Tb is 23.13 cm3 K mol–1, which is slightly lower than the value of 23.64 cm3 K mol–1 for two non-interacting Tb3+ ions.51 The gradual decrease in χMT as the temperature decreases toward 100 K is followed by a precipitous drop to 0.52 cm3 K mol–1 at 2 K, most likely due to a combination of antiferromagnetic exchange and depopulation of excited crystal field levels (Figure 7). The M(H) plots in the temperature range 1.9–5.0 K show a gradual increase in magnetization up to 25 kOe before increasing more rapidly toward 8 Nβ, but without reaching saturation at 70 kOe (Figure 7). The data again indicate antiferromagnetic exchange between the Tb3+ centers via the end-on dinitrogen ligand. To simulate the magnetic properties of 1Tb, the SINGLE_ANISO module as implemented in ORCA 5.0.2 was used to compute the g-tensors and crystal field parameters of the low-lying excited states within the 7F6 ground multiplet of Tb3+ (Tables S11–S13).53,54 The axial crystal field parameters Bk0 and Ck (k = 2,4,6) were then included in the spin Hamiltonian in eq 2, leading to good fits of the χMT(T) and M(H) data with J= −0.65 cm–1 (Figure 7).
The real and imaginary components of the AC susceptibility of 1Tb as functions of temperature, i.e., χ′(T) and χ″(T), respectively, were measured in an AC field of 3 Oe and a frequency of 1000 Hz. Slow magnetic relaxation was not observed, and the M(H) magnetic hysteresis measurements produced closed S-shaped traces at 1.9 K (Figures S8 and S9). Since complexes of the non-Kramers ion Tb3+ normally show SMM behavior when occupying a high-symmetry environment or when bound to a radical ligand,36,55 the observations on 1Tb are in line with expectations based on magneto-structural correlations developed for lanthanide metallocene SMMs.56
The DC susceptibility of 1Dy produced a χMT value of 27.27 cm3 K mol–1 at 300 K, slightly lower than the expected value of 28.34 cm3 K mol–1 expected for two non-interacting Dy3+ ions.51 A gradual decrease in the susceptibility on lowering the temperature results in a χMT value of 10.13 cm3 K mol–1 at 2 K (Figure 7), indicating antiferromagnetic exchange and depopulation of excited crystal field levels, as in 1Tb. At 1.9–5.0 K, the magnetization of 1Dy increases rapidly with field up to 10 kOe and then increases more gradually to reach a value of 9.36 Nβ at 70 kOe (Figure 7). A good fit of the susceptibility data was achieved for 1Dy using eq 2 with gDy = 1.33 and the calculated crystal field parameters for Dy3+ (6H15/2 ground state) in Table S11, resulting in an exchange coupling constant of J = – 0.07 cm–1.
In contrast to 1Tb, χ″(ν) for 1Dy in zero DC field above 7 K shows maxima up to 23 K, with the maxima shifting to a higher frequency with increasing temperature (Figures 8 and S10). A Cole–Cole plot of χ″(χ′) was fitted using α-parameters in the range 0.25–0.39, implying a reasonably broad distribution of relaxation times, τ. The plot of ln(τ/s) versus T–1 (Figure S11, Table S9) was fitted with Orbach and Raman terms according to eq 3, where τ0 is the attempt time, Ueff is the effective energy barrier, C is the Raman coefficient, and n is the Raman exponent.
| 3 |
The resulting fit parameters are τ0 = 7.30 × 10–10 s, Ueff = 180(37) cm–1, C = 0.20(5) s–1 K–n, and n = 3.3(3). The observation of SMM behavior in the AC susceptibility is complemented by a narrow opening of the M(H) hysteresis loop at 1.9 K (Figure S12).
Figure 8.

Imaginary part of the AC susceptibility as a function of frequency, χ″(ν), in zero DC field for 1Dy at T = 7–23 K (upper) and for 2Dy at T = 1.9–4.0 K (lower).
For 2Gd, χMT at 300 K is 7.48 cm3 K mol–1, close to the expected value of 7.50 cm3 K mol–1 for a Gd3+ ion coupled antiferromagnetically to a bipy radical anion with S = 1/2. The temperature-dependence of χMT decreases steadily down to 75 K before decreasing more rapidly to reach 1.68 cm3 K mol–1 at 2 K (Figure S13). A gradual increase in the magnetization was observed for 2Gd at 2 K up to a field of approximately 30 kOe, before leveling off at higher fields and reaching 5.93 Nβ at 70 kOe but not saturating (Figure S13). A fit of the susceptibility for 2Gd using eq 4, where grad and Srad refer to the g-tensor and total spin of the bipy radical anion, was obtained using g = 2.0 for Gd3+ and the bipy radical anion, yielding an isotropic exchange coupling constant of J= −5.50 cm–1 (Figure S13). This value is comparable in magnitude to those determined for other gadolinium complexes of radical N-heterocyclic ligands.37,49,50
| 4 |
| 5 |
The DC susceptibility of 2Tb is similar to that of 1Tb. From a value of 11.43 cm3 K mol–1 at 300 K, χMT decreases gradually down to 100 K before showing a much sharper decrease at lower temperatures and reaching 1.29 cm3 K mol–1 at 2 K (Figure S14). The room-temperature value of the susceptibility is essentially the same as that of 11.46 cm3 K mol–1 expected for a Tb3+ ion coupled antiferromagnetically to an S = 1/2 organic radical. The coupling is also reflected in the M(H) data at 1.9–5.0 K, with a steady increase in magnetization occurring up to 35 kOe, followed by a sharp upturn at higher fields without approaching saturation at 70 kOe (Figure S14). A reasonable simulation of the χMT(T) data was obtained for 2Tb using the spin Hamiltonian in eq 5 with grad = 2.0, gTb = 1.51, and the crystal field parameters stated in Table S15, which produced a terbium-bipy exchange coupling constant of J = – 4.65 cm–1.
While the χ″(ν) data for 2Tb do not display maxima (Figure S15), openings in the M(H) hysteresis loops were observed up to 6 K with a small remanent magnetization in zero field (Figure S16). Bifurcation in the field-cooled/zero-field-cooled (FC/ZFC) susceptibility also occurred at 5.3 K, broadly consistent with SMM behavior (Figure S17). The absence of maxima in the AC susceptibility likely indicates that fast quantum tunneling of the magnetization (QTM) is the dominant relaxation process in 2Tb in zero field, which is corroborated below with the aid of an ab initio theoretical study.
The DC magnetic susceptibility of 2Dy is consistent with a Dy3+ ion coupled antiferromagnetically to the bipy radical anion, the value of 13.96 cm3 K mol–1 for χMT at 300 K being close to the expected value of 13.80 cm3 K mol–1. Lowering the temperature results in the susceptibility reaching 1.51 cm3 K mol–1 at 2 K (Figure S18). The M(H) data for 2Dy show an increase in the magnetization in low fields, followed by a plateau around 35 kOe, and then a second rapid increase to reach 3.3 Nβ at 70 kOe, but without saturation (Figure S18). Since the magnetization is clearly still increasing at this field value, relatively strong direct antiferromagnetic exchange should be occurring. A fit of the susceptibility of 2Dy using eq 5 with grad = 2.0 and gDy = 1.36 and the crystal field parameters stated in Table S15 gave J = – 3.50 cm–1, which is comparable to the isotropic J-value determined for 2Gd and the J-value for 2Tb. Since J-values for highly anisotropic lanthanide-radical pairs are rarely reported, few systems are available for comparison. However, the couplings in 2Tb and 2Dy are seemingly weaker than those found in the [Bi2]3– radical bridged dimetallic systems [{C5Me5)2M}2(μ-η2:η2-Bi2)]− (M = Tb, Dy),57 most likely because of the relatively diffuse spin density in the bipy radical anion in 2M.
SMM behavior was observed for 2Dy in zero DC field, although this is less pronounced than for 1Dy. Broad maxima in χ″(ν) occur from 1.9 to 4.0 K (Figures 8 and S19), with the relaxation times allowing a fit of the Cole–Cole plot with α= 0.6–0.7, indicating a broad range of relaxation times (Figure S20, Table S10). A fit of the relaxation times was possible using Orbach and Raman terms according to eq 3, which yielded τ0 = 1.40 × 10–8 s, Ueff = 24(5) cm–1, C = 19(4) s–1K–n, and n = 5.2(3). The magnetic hysteresis measurements showed open loops up to 9 K, with two distinct steps around 24 and 59 kOe due to QTM at the magnetic fields corresponding to level crossings (Figure S21). A slight bifurcation of the FC/ZFC data was observed at 7 K (Figure S22).
The energy barrier and hysteresis properties of 1Dy can be interpreted in terms of how the end-on dinitrogen ligand impacts the crystal field at dysprosium relative to the influence of the Cpttt ligands. It is well-known that large energy barriers and open hysteresis loops can occur in dysprosium metallocene SMMs because the bis(cyclopentadienyl) framework provides a dominant axial crystal field.58−63 In SMMs with competing equatorial crystal fields such as [{(η5-Cp*)2Dy}(μ-Fp)]2 (Fp = CpFe(CO)2),56 a large energy barrier of 662 cm–1 and wide butterfly hysteresis loops can be observed, albeit with the latter showing negligible coercivity and remnant magnetization. In contrast, the molecular structure of 1Dy reveals that the Dy–N distances to the equatorial anionic nitrogen donor atoms are shorter by almost 0.14 Å than the Dy–Cp centroid distances, suggesting that the end-on bridging dinitrogen ligand should dominate the crystal field. This structural property of 1Dy also explains why the barrier of 180(37) cm–1 is much lower than those found in purely axial dysprosocenium SMMs, which can exceed 1500 cm–1,58,60,62,64,65 and similar to the barriers of approximately 100–300 cm–1 found in SMMs of the type [{(η5-C5H4Me)2Dy}{μ-ERn}]3 (ERn = MesP(H), MesAs(H), MesSb(H), MesSe, Mes = mesityl).66−68 The same rationale applies to 1Tb, with the added requirement for strict axial symmetry for non-Kramers ions, hence the poorer SMM properties.
To support the empirical observations on 1Tb and 1Dy, multireference ab initio calculations were used to provide deeper insight into their electronic structure. To simplify the calculations, one of the Ln3+ ions in the dinitrogen complexes was replaced by diamagnetic Y3+ and the resulting hypothetical heterobimetallic species subjected to the SINGLE_ANISO routine implemented in ORCA 5.0.2. A complete active space (CAS) of CAS(8,7) was used for 1Tb and CAS(9,7) for 1Dy, with the calculations yielding the g-tensors and crystal field parameters of the low-lying excited states. Full computational details are provided in the Supporting Information (Tables S11–S14).
The calculations show that the end-on dinitrogen ligand in 1Tb and 1Dy does indeed dominate the crystal field, with the easy axis of magnetization on each lanthanide center coinciding with the {M–N=N–M} axis (Figure 9). This result is a marked contrast to the commonly observed picture with dysprosium metallocene SMMs, where the easy axis in the ground Kramers doublet (KD) typically passes through both cyclopentadienyl ligands.63,69,70 Constructing a relaxation energy barrier for 1Tb revealed a tunnel splitting in the ground doublet of Δtun = 1.062 cm–1, which reduces the bistability and is consistent with the poor SMM properties of this species. Similar analysis of 1Dy shows that the ground KD is strongly axial, with gx = 0.090, gy = 0.184 and gz = 19.65, with the wavefunction consisting of 97% |MJ| = 15/2 character. In contrast, the first-excited KD at 146 cm–1 has large transverse components with gx = 2.501, gy = 5.153, and gz = 12.90, and is described by a strong admixture of MJ wavefunctions. Since the calculated and experimental energy barriers of 146 and 189(37) cm–1, respectively, are comparable in magnitude, Orbach relaxation in 1Dy should proceed via the first-excited KD.
Figure 9.

Upper: easy axis of magnetization (red lines) in the ground doublets of 1Tb and 1Dy. Middle: relaxation energy barrier for 1Tb. Lower: relaxation energy barrier for 1Dy showing possible transition as red lines, with darker shading indicating a more probable transition.
Discussion
The original rationale for targeting metallocene-capped lanthanide complexes of end-on bridging [N2]2– invoked the idea of steric bulk to preclude formation of the common side-on bonding mode. Since side-on N2-bridged di-lanthanide metallocenes with Ln = Gd, Tb or Dy tend to be capped by relatively small C5Me5 or C5Me4H ligands,23 the bulk of Cpttt is evidently sufficient for the end-on bridging to form exclusively in 1M. The same dinitrogen bonding mode is therefore also likely to be observed in analogous complexes of heavier, smaller lanthanides beyond dysprosium. For the lighter lanthanides with larger radii, it has been shown that the bonding mode of dinitrogen is finely balanced between the two possibilities by steric and other factors, with both end-on and side-on occurring. It is, therefore, of interest to explore the limits and applicability of the chemistry developed for 1M across the full range of the lanthanide series in future work.
The switch from bulky bis(trimethylsilyl)amido as the capping ligand in the dinitrogen-bridged di-lanthanide complexes studied by Evans and co-workers20−22 to Cpttt in 1M has important consequences for the interactions of the lanthanide valence 5d orbitals and [N2]2– and, hence, the spin configuration of the dinitrogen ligand. The local symmetry of the lanthanide sites results in effective overlap of a 5dπ orbital with one of the dinitrogen π* orbitals, and poorer overlap of a second 5dπ orbital with the other dinitrogen π* orbital. The resulting HOMO–LUMO gap produces an energetically isolated singlet configuration for the end-on bridging [N2]2– ligand in 1M, whereas a triplet configuration was proposed for the end-on dinitrogen complexes capped by bis(trimethylsilyl)amido ligands. These contrasting scenarios point toward the possibility of engineering the singlet-triplet energy gap via the capping ligand bound to the lanthanide, which could be used to address magnetic and photophysical properties of lanthanide metallocenes.
The ability to synthesize and isolate 1M in yields reproducibly greater than 80% offers opportunities for new lanthanide-based reduction chemistry without the need to use KC8 in the presence of the target substrate, i.e., 2,2′-bipy in the case of 2M. This is especially valuable for lanthanides that do not readily form a stable +2 oxidation state, such as gadolinium, terbium, and dysprosium. By regarding the [N2]2– ligand as a two-electron reservoir, compounds 1M have potential to act as ‘synthons’ for [Cp2tttM] in bespoke reduction chemistry, potentially across the full 4f series (except promethium). Proof of principle has been achieved with the synthesis of the radical-ligated complexes 2M. Extension of the reactivity to, for example, tri-nucleating N-heterocycles would give access to strongly exchanged-coupled radical-bridged compounds, potentially with SMM properties. Furthermore, noting the recent demonstration of CO homologation by the isolable divalent thulium metallocene [Cp2Tm],24 applications of 1M in small-molecule activation with a broader range of lanthanides can be envisaged. This approach would complement existing methodologies involving stable divalent lanthanide metallocenes and sterically induced reduction.
Conclusions
The targeted synthesis of metallocene-capped lanthanide dinitrogen complexes exclusively with the end-on μ-κ1:κ1-bridging mode has been achieved with 1M. Formation of these compounds is aided by the bulk of the Cpttt ligand substituents in combination with the relatively small radii of gadolinium, terbium, and dysprosium. The high-yielding synthesis and stability at room temperature are noteworthy features of 1M, as is the charge-neutral nature of the complexes, which eliminates considerations relating to alkali metal counter ions and associated chelate ligands. Raman spectroscopy revealed a major stretching vibration for each complex, located at 1623, 1621, and 1618 cm–1 for 1Gd, 1Tb, and 1Dy, respectively, consistent with N=N double bonds in the [N2]2– ligands. Analysis of the bonding in 1Gd shows that the singlet form of the dinitrogen ligand is strongly preferred to the triplet form, which is a consequence of different spatial overlap between gadolinium 5dπ orbitals and the two dinitrogen π* orbitals.
DC magnetic susceptibility and magnetization measurements reveal that [N2]2– mediates antiferromagnetic exchange between the Ln3+ ions, with AC susceptibility and magnetic hysteresis measurements also showing that 1Dy is an SMM. A striking theoretical result from a study of 1Tb and 1Dy is the dominance of the dinitrogen ligand in the lanthanide crystal field, with the easy axis of magnetization being oriented along the {Ln–N=N–Ln} axis as opposed to aligning with the cyclopentadienyl ligands, as is commonly found.
By regarding [N2]2– as a source of two electrons, compounds 1M have been shown to react as synthons for divalent gadolinium, terbium, and dysprosium metallocenes. Proof-of-principle reactions led to the isolation of the radical-ligated complexes [Cp2tttM(κ2-bipy)] (2M, M = Gd, Tb, Dy), which feature appreciable direct exchange coupling between the lanthanide and the bipy radical anion. SMM behavior was also found in 2Dy. The electron-transfer reactivity of 1M introduces possibilities for new directions in lanthanide small-molecule activation, the discovery of molecular magnets, and, potentially, for stoichiometric organic synthesis as alternatives to samarium(II) reductions.
Acknowledgments
The authors thank the EPSRC (grants EP/V003089/1 and EP/V046659/1), the University of Sussex, and the National Natural Science Foundation of China (92261103) for financial support. We also thank the EPSRC UK National Crystallography Service at the University of Southampton for collecting X-ray crystallographic data on 1Gd and Prof. A. B. Dalton (University of Sussex) for providing access to Raman spectroscopy facilities.
Data Availability Statement
Additional research data supporting this publication are available as Supplementary Information at DOI: 10.25377/sussex.23703159.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c07600.
Synthesis, spectroscopic characterization, crystallography details, magnetic measurements, computational details (PDF)
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Singh D.; Buratto W. R.; Torres J. F.; Murray L. J. Activation of Dinitrogen by Polynuclear Metal Complexes. Chem. Rev. 2020, 120, 5517–5581. 10.1021/acs.chemrev.0c00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane D. R.; Simonov A. N.; Vu T. M.; Johnston S.; Azofra L. M. Concluding Remarks: Sustainable Nitrogen Activation – Are We There Yet?. Faraday Discuss. 2023, 243, 557–570. 10.1039/D3FD00087G. [DOI] [PubMed] [Google Scholar]
- Haber F. Über Die Synthetische Gewinnung Des Ammoniaks. Angew. Chem. 1914, 27, 473–477. 10.1002/ange.19140276201. [DOI] [Google Scholar]
- Falcone M.; Chatelain L.; Scopelliti R.; Živković I.; Mazzanti M. Nitrogen Reduction and Functionalization by a Multimetallic Uranium Nitride Complex. Nature 2017, 547, 332–335. 10.1038/nature23279. [DOI] [PubMed] [Google Scholar]
- Falcone M.; Barluzzi L.; Andrez J.; Fadaei Tirani F.; Zivkovic I.; Fabrizio A.; Corminboeuf C.; Severin K.; Mazzanti M. The Role of Bridging Ligands in Dinitrogen Reduction and Functionalization by Uranium Multimetallic Complexes. Nat. Chem. 2019, 11, 154–160. 10.1038/s41557-018-0167-8. [DOI] [PubMed] [Google Scholar]
- Jori N.; Barluzzi L.; Douair I.; Maron L.; Fadaei-Tirani F.; Živković I.; Mazzanti M. Stepwise Reduction of Dinitrogen by a Uranium-Potassium Complex Yielding a U(VI)/U(IV) Tetranitride Cluster. J. Am. Chem. Soc. 2021, 143, 11225–11234. 10.1021/jacs.1c05389. [DOI] [PubMed] [Google Scholar]
- Arnold P. L.; Ochiai T.; Lam F. Y. T.; Kelly R. P.; Seymour M. L.; Maron L. Metallacyclic Actinide Catalysts for Dinitrogen Conversion to Ammonia and Secondary Amines. Nat. Chem. 2020, 12, 654–659. 10.1038/s41557-020-0457-9. [DOI] [PubMed] [Google Scholar]
- Wang P.; Douair I.; Zhao Y.; Wang S.; Zhu J.; Maron L.; Zhu C. Facile Dinitrogen and Dioxygen Cleavage by a Uranium(III) Complex: Cooperativity Between the Non-Innocent Ligand and the Uranium Center. Angew. Chem., Int. Ed. 2021, 60, 473–479. 10.1002/anie.202012198. [DOI] [PubMed] [Google Scholar]
- Xin X.; Douair I.; Zhao Y.; Wang S.; Maron L.; Zhu C. Dinitrogen Cleavage and Hydrogenation to Ammonia with a Uranium Complex. Natl. Sci. Rev. 2022, 10, nwac144 10.1093/nsr/nwac144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans W. J.; Ulibarri T. A.; Ziller J. W. Isolation and X-Ray Crystal Structure of the First Dinitrogen Complex of an f-Element Metal, [(C5Me5)2Sm]2N2. J. Am. Chem. Soc. 1988, 110, 6877–6879. 10.1021/ja00228a043. [DOI] [Google Scholar]
- Evans W. J.; Allen N. T.; Ziller J. W. Facile Dinitrogen Reduction via Organometallic Tm(II) Chemistry. J. Am. Chem. Soc. 2001, 123, 7927–7928. 10.1021/ja011282l. [DOI] [PubMed] [Google Scholar]
- Cheng J.; Takats J.; Ferguson M. J.; McDonald R. Heteroleptic Tm(II) Complexes: One More Success for Trofimenko’s Scorpionates. J. Am. Chem. Soc. 2008, 130, 1544–1545. 10.1021/ja0776273. [DOI] [PubMed] [Google Scholar]
- Evans W. J.; Allen N. T.; Ziller J. W. Expanding Divalent Organolanthanide Chemistry: The First Organothulium(II) Complex and the In Situ Organodysprosium(II) Reduction of Dinitrogen. Angew. Chem., Int. Ed. 2002, 41, 359–361. . [DOI] [PubMed] [Google Scholar]
- Jaroschik F.; Momin A.; Nief F.; Le Goff X. F.; Deacon G. B.; Junk P. C. Dinitrogen Reduction and C-H Activation by the Divalent Organoneodymium Complex [(C5H2tBu3)2Nd(μ-I)K([18] Crown-6)]. Angew. Chem., Int. Ed. 2009, 48, 1117–1121. 10.1002/anie.200804934. [DOI] [PubMed] [Google Scholar]
- Evans W. J.; Lee D. S.; Rego D. B.; Perotti J. M.; Kozimor S. A.; Moore E. K.; Ziller J. W. Expanding Dinitrogen Reduction Chemistry to Trivalent Lanthanides via the LnZ3/Alkali Metal Reduction System: Evaluation of the Generality of Forming Ln2(μ-η2:η2-N2) Complexes via LnZ3/K. J. Am. Chem. Soc. 2004, 126, 14574–14582. 10.1021/ja046047s. [DOI] [PubMed] [Google Scholar]
- Evans W. J.; Lee D. S.; Ziller J. W. Reduction of Dinitrogen to Planar Bimetallic M2(μ-η2:η2-N2) Complexes of Y, Ho, Tm, and Lu Using the K/Ln[N(SiMe3)2]3 Reduction System. J. Am. Chem. Soc. 2004, 126, 454–455. 10.1021/ja036923m. [DOI] [PubMed] [Google Scholar]
- Campazzi E.; Solari E.; Floriani C.; Scopelliti R. The Fixation and Reduction of Dinitrogen Using Lanthanides: Praseodymium and Neodymium Meso-Octaethylporphyrinogen–Dinitrogen Complexes. Chem. Commun. 1998, 2603–2604. 10.1039/a807405d. [DOI] [Google Scholar]
- Lv Z. J.; Huang Z.; Zhang W. X.; Xi Z. Scandium-Promoted Direct Conversion of Dinitrogen into Hydrazine Derivatives via N-C Bond Formation. J. Am. Chem. Soc. 2019, 141, 8773–8777. 10.1021/jacs.9b04293. [DOI] [PubMed] [Google Scholar]
- Fang M.; Lee D. S.; Ziller J. W.; Doedens R. J.; Bates J. E.; Furche F.; Evans W. J. Synthesis of the (N2)3– Radical from Y2+ and Its Protonolysis Reactivity to Form (N2H2)2– via the Y[N(SiMe3)2]3/KC8 Reduction System. J. Am. Chem. Soc. 2011, 133, 3784–3787. 10.1021/ja1116827. [DOI] [PubMed] [Google Scholar]
- Woen D. H.; Chen G. P.; Ziller J. W.; Boyle T. J.; Furche F.; Evans W. J. End-On Bridging Dinitrogen Complex of Scandium. J. Am. Chem. Soc. 2017, 139, 14861–14864. 10.1021/jacs.7b08456. [DOI] [PubMed] [Google Scholar]
- Ryan A. J.; Balasubramani S. G.; Ziller J. W.; Furche F.; Evans W. J. Formation of the End-on Bound Lanthanide Dinitrogen Complexes [(R2N)3Ln-N=N-Ln(NR2)3]2– from Divalent [(R2N)3Ln]1– Salts (R = SiMe3). J. Am. Chem. Soc. 2020, 142, 9302–9313. 10.1021/jacs.0c01021. [DOI] [PubMed] [Google Scholar]
- Chung A. B.; Rappoport D.; Ziller J. W.; Cramer R. E.; Furche F.; Evans W. J. Solid-State End-On to Side-On Isomerization of (N=N)2– in {[(R2N)3Nd]2N2}2– (R = SiMe3) Connects In Situ LnIII(NR2)3/K and Isolated [LnII(NR2)3]1– Dinitrogen Reduction. J. Am. Chem. Soc. 2022, 144, 17064–17074. 10.1021/jacs.2c06716. [DOI] [PubMed] [Google Scholar]
- Demir S.; Gonzalez M. I.; Darago L. E.; Evans W. J.; Long J. R. Giant Coercivity and High Magnetic Blocking Temperatures for N23– Radical-Bridged Dilanthanide Complexes upon Ligand Dissociation. Nat. Commun. 2017, 8, 2144. 10.1038/s41467-017-01553-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simler T.; McCabe K. N.; Maron L.; Nocton G. CO Reductive Oligomerization by a Divalent Thulium Complex and CO2-Induced Functionalization. Chem. Sci. 2022, 13, 7449–7461. 10.1039/D2SC01798A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould C. A.; McClain K. R.; Yu J. M.; Groshens T. J.; Furche F.; Harvey B. G.; Long J. R. Synthesis and Magnetism of Neutral, Linear Metallocene Complexes of Terbium(II) and Dysprosium(II). J. Am. Chem. Soc. 2019, 141, 12967–12973. 10.1021/jacs.9b05816. [DOI] [PubMed] [Google Scholar]
- Fieser M. E.; Woen D. H.; Corbey J. F.; Mueller T. J.; Ziller J. W.; Evans W. J. Raman Spectroscopy of the N-N Bond in Rare Earth Dinitrogen Complexes. Dalton Trans. 2016, 45, 14634–14644. 10.1039/C5DT04547A. [DOI] [PubMed] [Google Scholar]
- Upadhyay A.; Das C.; Shanmugam M.; Langley S. K.; Murray K. S.; Shanmugam M. Electronic and Magnetic Properties of a Gadolinium(III) Schiff Base Complex. Eur. J. Inorg. Chem. 2014, 4320–4325. 10.1002/ejic.201402219. [DOI] [Google Scholar]
- Evans W. J.; Lee D. S.; Ziller J. W.; Kaltsoyannis N. Trivalent [(C5Me5)2(THF)Ln]2(μ-η2:η2-N2) Complexes as Reducing Agents Including the Reductive Homologation of CO to a Ketene Carboxylate, (μ-η4-O2CCCO)2–. J. Am. Chem. Soc. 2006, 128, 14176–14184. 10.1021/ja0640851. [DOI] [PubMed] [Google Scholar]
- Evans W. J.; Lorenz S. E.; Ziller J. W. Investigating Metal Size Effects in the Ln2(μ-η2:η2-N2) Reduction System: Reductive Reactivity with Complexes of the Largest and Smallest Trivalent Lanthanide Ions, La3+ and Lu3+. Inorg. Chem. 2009, 48, 2001–2009. 10.1021/ic801853d. [DOI] [PubMed] [Google Scholar]
- Demir S.; Jeon I. R.; Long J. R.; Harris T. D. Radical Ligand-Containing Single-Molecule Magnets. Coord. Chem. Rev. 2015, 289–290, 149–176. 10.1016/j.ccr.2014.10.012. [DOI] [Google Scholar]
- Guo F.-S.; Layfield R. A. Strong Direct Exchange Coupling and Single-Molecule Magnetism in Indigo-Bridged Lanthanide Dimers. Chem. Commun. 2017, 53, 3130–3133. 10.1039/C7CC01046J. [DOI] [PubMed] [Google Scholar]
- Demir S.; Zadrozny J. M.; Nippe M.; Long J. R. Exchange Coupling and Magnetic Blocking in Bipyrimidyl Radical-Bridged Dilanthanide Complexes. J. Am. Chem. Soc. 2012, 134, 18546–18549. 10.1021/ja308945d. [DOI] [PubMed] [Google Scholar]
- Demir S.; Nippe M.; Gonzalez M. I.; Long J. R. Exchange Coupling and Magnetic Blocking in Dilanthanide Complexes Bridged by the Multi-Electron Redox-Active Ligand 2,3,5,6-Tetra(2-Pyridyl)Pyrazine. Chem. Sci. 2014, 5, 4701–4711. 10.1039/C4SC02154A. [DOI] [Google Scholar]
- Gould C. A.; Darago L. E.; Gonzalez M. I.; Demir S.; Long J. R. A Trinuclear Radical-Bridged Lanthanide Single-Molecule Magnet. Angew. Chem., Int. Ed. 2017, 56, 10103–10107. 10.1002/anie.201612271. [DOI] [PubMed] [Google Scholar]
- Mavragani N.; Errulat D.; Gálico D. A.; Kitos A. A.; Mansikkamäki A.; Murugesu M. Radical-Bridged Ln4 Metallocene Complexes with Strong Magnetic Coupling and a Large Coercive Field. Angew. Chem., Int. Ed. 2021, 60, 24206–24213. 10.1002/anie.202110813. [DOI] [PubMed] [Google Scholar]
- Rinehart J. D.; Fang M.; Evans W. J.; Long J. R. A N23– Radical-Bridged Terbium Complex Exhibiting Magnetic Hysteresis at 14 K. J. Am. Chem. Soc. 2011, 133, 14236–14239. 10.1021/ja206286h. [DOI] [PubMed] [Google Scholar]
- Rinehart J. D.; Fang M.; Evans W. J.; Long J. R. Strong Exchange and Magnetic Blocking in N23– Radical-Bridged Lanthanide Complexes. Nat. Chem. 2011, 3, 538. 10.1038/nchem.1063. [DOI] [PubMed] [Google Scholar]
- Selikhov A. N.; Mahrova T. V.; Cherkasov A. V.; Fukin G. K.; Larionova J.; Long J.; Trifonov A. A. Base-Free Lanthanoidocenes(II) Coordinated by Bulky Pentabenzylcyclopentadienyl Ligands. Organometallics 2015, 34, 1991–1999. 10.1021/acs.organomet.5b00243. [DOI] [Google Scholar]
- Evans W. J.; Drummond D. K. Reductive Coupling of Pyridazine and Benzaldehyde Azine and Reduction of Bipyridine by (C5Me5)2Sm(THF)2. J. Am. Chem. Soc. 1989, 111, 3329–3335. 10.1021/ja00191a034. [DOI] [Google Scholar]
- Booth C. H.; Kazhdan D.; Werkema E. L.; Walter M. D.; Lukens W. W.; Bauer E. D.; Hu Y.-J.; Maron L.; Eisenstein O.; Head-Gordon M.; Andersen R. A. Intermediate-Valence Tautomerism in Decamethylytterbocene Complexes of Methyl-Substituted Bipyridines. J. Am. Chem. Soc. 2010, 132, 17537–17549. 10.1021/ja106902s. [DOI] [PubMed] [Google Scholar]
- Nocton G.; Booth C. H.; Maron L.; Andersen R. A. Influence of the Torsion Angle in 3,3′-Dimethyl-2,2′-Bipyridine on the Intermediate Valence of Yb in (C5Me5)2Yb(3,3′-Me2-Bipy). Organometallics 2013, 32, 5305–5312. 10.1021/om400528d. [DOI] [Google Scholar]
- Walter M. D.; Schultz M.; Andersen R. A. Weak paramagnetism in compounds of the type Cp′2Yb(bipy). New J. Chem. 2006, 30, 238–246. 10.1039/b512865j. [DOI] [Google Scholar]
- Schultz M.; Boncella J. M.; Berg D. J.; Tilley T. D.; Andersen R. A. Coordination of 2,2’-Bipyridyl and 1,10-Phenanthroline to Substituted Ytterbocenes: An Experimental Investigation of Spin Coupling in Lanthanide Complexes. Organometallics 2002, 21, 460–472. 10.1021/om010661k. [DOI] [Google Scholar]
- Walter M. D.; Berg D. J.; Andersen R. A. Coordination Complexes of Decamethylytterbocene with 4,4‘-Disubstituted Bipyridines: An Experimental Study of Spin Coupling in Lanthanide Complexes. Organometallics 2006, 25, 3228–3237. 10.1021/om051051d. [DOI] [Google Scholar]
- Nocton G.; Booth C. H.; Maron L.; Andersen R. A. Thermal Dihydrogen Elimination from Cp*2Yb(4,5-diazafluorene). Organometallics 2013, 32, 1150–1158. 10.1021/om300876b. [DOI] [Google Scholar]
- Wang S.; Wang D.; Li T.; Heng Y.; Hou G.; Zi G.; Walter M. D. Synthesis, Structure, and Reactivity of the Uranium Bipyridyl Complex [{η5-1,2,4-(Me3Si)3C5H2}2U(Bipy)]. Organometallics 2022, 41, 1543–1557. 10.1021/acs.organomet.2c00175. [DOI] [Google Scholar]
- Yang P.; Zhou E.; Fang B.; Hou G.; Zi G.; Walter M. D. Preparation of (η5-C5Me5)2Th(Bipy) and Its Reactivity toward Small Molecules. Organometallics 2016, 35, 2129–2139. 10.1021/acs.organomet.6b00357. [DOI] [Google Scholar]
- Wang D.; Ding W.; Hou G.; Zi G.; Walter M. D. Experimental and Computational Studies on a Base-Free Terminal Uranium Phosphinidene Metallocene. Chem. – Eur. J. 2020, 26, 16888–16899. 10.1002/chem.202003465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.; Hu Z.; Li J.; Ruan H.; Zhao Y.; Tan G.; Song Y.; Wang X. Isolable Lanthanide Metal Complexes of a Phosphorus-Centered Radical. Inorg. Chem. 2020, 59, 2111–2115. 10.1021/acs.inorgchem.9b01950. [DOI] [PubMed] [Google Scholar]
- Chen C.; Hu Z.-B.; Ruan H.; Zhao Y.; Zhang Y.-Q.; Tan G.; Song Y.; Wang X. Tuning the Single-Molecule Magnetism of Dysprosium Complexes by a Redox-Noninnocent Diborane Ligand. Organometallics 2020, 39, 4143–4148. 10.1021/acs.organomet.9b00819. [DOI] [Google Scholar]
- Benelli C.; Gatteschi D.. Introduction to Molecular Magnetism. In Introduction to Molecular Magnetism; John Wiley & Sons, Ltd., 2015; pp 1–23. [Google Scholar]
- Chilton N. F.; Anderson R. P.; Turner L. D.; Soncini A.; Murray K. S. PHI: A Powerful New Program for the Analysis of Anisotropic Monomeric and Exchange-Coupled Polynuclear d- and f-Block Complexes. J. Comput. Chem. 2013, 34, 1164–1175. 10.1002/jcc.23234. [DOI] [PubMed] [Google Scholar]
- Neese F.; Wennmohs F.; Becker U.; Riplinger C. The ORCA Quantum Chemistry Program Package. J. Chem. Phys. 2020, 152, 224108. 10.1063/5.0004608. [DOI] [PubMed] [Google Scholar]
- Chibotaru L. F.; Ungur L. Ab Initio Calculation of Anisotropic Magnetic Properties of Complexes. I. Unique Definition of Pseudospin Hamiltonians and Their Derivation. J. Chem. Phys. 2012, 137, 064112 10.1063/1.4739763. [DOI] [PubMed] [Google Scholar]
- Rinehart J. D.; Long J. R. Exploiting Single-Ion Anisotropy in the Design of f-Element Single-Molecule Magnets. Chem. Sci. 2011, 2, 2078–2085. 10.1039/c1sc00513h. [DOI] [Google Scholar]
- Pugh T.; Chilton N. F.; Layfield R. A. A Low-Symmetry Dysprosium Metallocene Single-Molecule Magnet with a High Anisotropy Barrier. Angew. Chem., Int. Ed. 2016, 55, 11082–11085. 10.1002/anie.201604346. [DOI] [PubMed] [Google Scholar]
- Zhang P.; Nabi R.; Staab J. K.; Chilton N. F.; Demir S. Taming Super-Reduced Bi23– Radicals with Rare Earth Cations. J. Am. Chem. Soc. 2023, 145, 9152–9163. 10.1021/jacs.3c01058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F.-S.; Day B. M.; Chen Y.-C.; Tong M.-L.; Mansikkamäki A.; Layfield R. A. Magnetic Hysteresis up to 80 Kelvin in a Dysprosium Metallocene Single-Molecule Magnet. Science 2018, 362, 1400–1403. 10.1126/science.aav0652. [DOI] [PubMed] [Google Scholar]
- Guo F.-S.; Day B. M.; Chen Y.-C.; Tong M.-L.; Mansikkamäki A.; Layfield R. A. A Dysprosium Metallocene Single-Molecule Magnet Functioning at the Axial Limit. Angew. Chem., Int. Ed. 2017, 56, 11445–11449. 10.1002/anie.201705426. [DOI] [PubMed] [Google Scholar]
- Randall McClain K.; Gould C. A.; Chakarawet K.; Teat S. J.; Groshens T. J.; Long J. R.; Harvey B. G. High-Temperature Magnetic Blocking and Magneto-Structural Correlations in a Series of Dysprosium(III) Metallocenium Single-Molecule Magnets. Chem. Sci. 2018, 9, 8492–8503. 10.1039/C8SC03907K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F.-S.; He M.; Huang G.-Z.; Giblin S.; Billington D.; Heinemann F.; Tong M.-L.; Mansikkamäki A.; Layfield R. Discovery of a Dysprosium Metallocene Single-Molecule Magnet with Two High-Temperature Orbach Processes. Inorg. Chem. 2022, 61, 6017–6025. 10.1021/acs.inorgchem.1c03980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin C. A. P.; Ortu F.; Reta D.; Chilton N. F.; Mills D. P. Molecular Magnetic Hysteresis at 60 Kelvin in Dysprosocenium. Nature 2017, 548, 439. 10.1038/nature23447. [DOI] [PubMed] [Google Scholar]
- Day B.; Guo F.-S.; Layfield R. Cyclopentadienyl Ligands in Lanthanide Single-Molecule Magnets: One Ring to Rule Them All?. Acc. Chem. Res. 2018, 51, 1880–1889. 10.1021/acs.accounts.8b00270. [DOI] [PubMed] [Google Scholar]
- Vincent A. H.; Whyatt Y. L.; Chilton N. F.; Long J. R. Strong Axiality in a Dysprosium(III) Bis(borolide) Complex Leads to Magnetic Blocking at 65 K. J. Am. Chem. Soc. 2023, 145, 1572–1579. 10.1021/jacs.2c08568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanjak J. C.; Wilkins B. O.; Vieru V.; Bhuvanesh N. S.; Reibenspies J. H.; Martin C. D.; Chibotaru L. F.; Nippe M. A High-Performance Single-Molecule Magnet Utilizing Dianionic Aminoborolide Ligands. J. Am. Chem. Soc. 2022, 144, 17743–17747. 10.1021/jacs.2c06698. [DOI] [PubMed] [Google Scholar]
- Pugh T.; Tuna F.; Ungur L.; Collison D.; McInnes E. J. L.; Chibotaru L. F.; Layfield R. A. Influencing the Properties of Dysprosium Single-Molecule Magnets with Phosphorus Donor Ligands. Nat. Commun. 2015, 6, 7492. 10.1038/ncomms8492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh T.; Vieru V.; Chibotaru L. F.; Layfield R. A. Magneto-Structural Correlations in Arsenic- and Selenium-Ligated Dysprosium Single-Molecule Magnets. Chem. Sci. 2016, 7, 2128–2137. 10.1039/C5SC03755G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh T.; Chilton N. F.; Layfield R. A. Antimony-Ligated Dysprosium Single-Molecule Magnets as Catalysts for Stibine Dehydrocoupling. Chem. Sci. 2017, 8, 2073–2080. 10.1039/C6SC04465D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harriman K. L. M.; Murugesu M. An Organolanthanide Building Block Approach to Single-Molecule Magnets. Acc. Chem. Res. 2016, 49, 1158–1167. 10.1021/acs.accounts.6b00100. [DOI] [PubMed] [Google Scholar]
- Errulat D.; Gabidullin B.; Mansikkamäki A.; Murugesu M. Two Heads Are Better than One: Improving Magnetic Relaxation in the Dysprosium Metallocene upon Dimerization by Use of an Exceptionally Weakly-Coordinating Anion. Chem. Commun. 2020, 56, 5937–5940. 10.1039/D0CC01980A. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Additional research data supporting this publication are available as Supplementary Information at DOI: 10.25377/sussex.23703159.