

Abstract

There is an urgent need for improved therapy to better control the ongoing COVID-19 pandemic. The main protease Mpro plays a pivotal role in SARS-CoV-2 replications, thereby representing an attractive target for antiviral development. We seek to identify novel electrophilic warheads for efficient, covalent inhibition of Mpro. By comparing the efficacy of a panel of warheads installed on a common scaffold against Mpro, we discovered that the terminal alkyne could covalently modify Mpro as a latent warhead. Our biochemical and X-ray structural analyses revealed the irreversible formation of the vinyl-sulfide linkage between the alkyne and the catalytic cysteine of Mpro. Clickable probes based on the alkyne inhibitors were developed to measure target engagement, drug residence time, and off-target effects. The best alkyne-containing inhibitors potently inhibited SARS-CoV-2 infection in cell infection models. Our findings highlight great potentials of alkyne as a latent warhead to target cystine proteases in viruses and beyond.
Introduction
The coronavirus disease 2019 (COVID-19), caused by the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has emerged as a global pandemic since its outbreak in December 2019.1,2 As of January 2023, roughly three years after its initial breakout, COVID-19 continues to pose serious threats to human health and public safety, with more than 762-million confirmed cases and 6.8 million deaths worldwide.3 Although vaccination continues to remain the most effective therapeutic strategy to protect people against serious illness or death from COVID-19, the emergence and global spread of highly contagious SARS-CoV-2 variants harboring spike mutations have raised concerns about vaccine effectiveness due to the potential of these variants to escape existing SARS-CoV-2 neutralizing antibodies.4−6 In addition, individuals with immunocompromised conditions often exhibit significantly low seroconversion rates after vaccination, resulting in poor vaccine protection against COVID-19.7,8 The current vaccination approach is also limited in its ability to protect people with historically severe allergic reactions to vaccines by putting this population at an increased risk of life-threatening hypersensitivity adverse events such as anaphylaxis after getting vaccinated, thereby giving rise to hesitation in receiving COVID-19 vaccines and bringing challenges to achieving herd immunity against this infection.9,10 Thus, despite the wide availability of vaccines, there is still an urgent need for the development of other broadly protective interventions to halt the devastation of the evolving pandemic. Among therapeutic interventions, drug discovery efforts in developing specific antiviral agents against SARS-CoV-2 proffer a powerful addition to the host defense mechanisms for combating COVID-19 and eradicating future pandemics.
SARS-CoV-2 is an enveloped positive-sense single-stranded RNA virus of approximately 30 kb in size, belonging to the genus Betacoronavirus of the family Coronaviridae.11,12 Upon host cell entry, the SARS-CoV-2 genomic RNA is translated by the host’s machinery into two large overlapping polyproteins (pp1a and pp1ab), four structural proteins, and other accessory proteins.11,13 One of the key steps in the viral replication cycle involves the proteolytic cleavage of the polyproteins pp1a and pp1ab into 16 highly conserved non-structural proteins (Nsps) for the subsequent formation of the replication–transcription complex.13,14 Together with the papain-like protease, the main protease (Mpro) of SARS-CoV-2, also known as 3-chymotrypsin-like protease (3CLpro) or Nsp5, performs critical proteolytic processing at distinct cleavage sites to yield the 11 mature Nsps required for viral replication.14,15 SARS-CoV-2 Mpro therefore represents one of the most attractive therapeutic targets for the development of antiviral therapy for treating COVID-19.
SARS-CoV-2 Mpro (abbreviated as Mpro in the remainder of the paper) is a cysteine (Cys) protease that comprises three domains that are highly conserved in all known coronaviruses and contains an active site for enzymatic proteolytic function.16,17 Mpro preferentially cleaves substrates with the consensus sequence (P2:Leu/Met/Phe/Val)–P1:Gln↓(P1’:Ser/Ala/Gly) (↓ indicates the cleavage site).18 The absolute requirement of the Gln residue at the P1 position is notably advantageous, as no known human proteases have such unique substrate selectivity, thereby promising high safety profiles for specific antiviral agents against Mpro.19,20 More importantly, the presence of a catalytic Cys145 residue from the catalytic Cys145–His41 dyad in the active site renders Mpro susceptible to targeted covalent inhibition by small-molecule inhibitors featuring reactive electrophilic groups.19,21
Since the onset of the COVID-19 pandemic, a large number of covalent inhibitors containing diverse electrophilic warheads, including α-ketoamides, aldehydes, α,β-unsaturated ketones, hydroxymethylketones, vinyl sulfones, and nitriles, have been reported to covalently inhibit Mpro.19,21−25 Paxlovid (Pfizer), consisting of primarily nirmatrelvir along with ritonavir, has been granted emergency use for the treatment of COVID-19 by the U.S. Food and Drug Administration (FDA) as the first antiviral acting via covalent inhibition of SARS-CoV-2 Mpro.25 Treatment with Paxlovid early in COVID-19 illness has shown an approximately 90% reduction in the risk of progression to severe disease and been associated with a significant decrease in COVID-19-related hospitalizations or deaths.26 Paxlovid owes its potent antiviral activity to the ability of its active ingredient, nirmatrelvir, to reversibly and covalently target the catalytic Cys145 residue via a nitrile warhead.25 Despite the proven effectiveness of nirmatrelvir in disarming SARS-CoV-2, Paxlovid is not recommended for individuals with severe renal or hepatic impairment and contraindicated with CYP3A-dependent medications.27 Furthermore, mutations in Mpro have emerged and conferred resistance against nirmatrelvir in COVID-19 patients.28 These gaps highlight the urgent need to develop novel inhibitors against this viral protease as second-generation anti-COVID therapies.
Electrophilic warheads usually have intrinsic reactivity toward certain nucleophilic groups. For example, the acrylamide that is commonly employed to target the cysteine thiol group can react with thiols in a bi-molecular reaction, which is responsible for nonspecific binding to undesired proteins (off targets), especially abundant cellular proteins.29,30 In contrast, latent warheads do not have intrinsic reactivity but can react with certain functional groups only upon activation at the active site of certain enzymes. Eflornithine, a drug that is used to treat sleeping sickness and excessive hair growth, cannot react with thiols or cysteine on its own.31 However, when bound to ornithine decarboxylase, eflornithine is activated by the catalytic environment to produce a derivative that covalently modifies a cysteine in the enzyme.32 Latent warheads, with their lack of intrinsic chemical reactivity, are often considered advantageous over regular warheads because they tend to be more specific for intended enzyme targets and have much fewer covalent off-targets in cells and organisms.30,33,34
We set out to discover Mpro inhibitors harboring novel electrophilic warheads that may offer new opportunities to treat COVID-19. We began this campaign by installing a panel of cysteine-targeting warheads on a common peptidomimetic scaffold and comparing their inhibition against Mpro. A terminal alkyne, acting as a latent warhead, was found to afford strong inhibition of Mpro in the panel of resulting peptidomimetic derivatives. Installation of the terminal alkyne on more elaborate scaffolds led to the identification of inhibitors with comparable potency of biochemical inhibition against Mpro. The irreversible inhibition of Mpro by these alkyne-containing inhibitors was verified by both biochemical and X-ray structural characterizations and was exploited to generate “clickable” probes for measuring targeting engagement in vitro and in situ. Finally, our alkyne-containing inhibitors exhibited anti-viral activity in cellular models of COVID-19 infection.
Results and Discussion
Design, Synthesis, and Biochemical Characterization of Mpro Inhibitors
Aiming to identify the most attractive warheads in an unbiased manner, we first chose to install a panel of electrophilic warheads on a common dipeptide scaffold. Given that Gln and Leu were found as the highly preferred residues at the P1 and P2 positions, respectively, in peptide substrates of Mpro, this panel of peptidomimetics was designed to consist of three components: a benzyloxycarbonyl (Cbz) cap at the N terminus, a Leu residue, and a Gln analogue linked to a Cys-reactive electrophile at the C terminus. The electrophilic warheads that we selected include α,β-unsaturated esters, vinylsulfones, vinylsulfonamides, terminal alkynes, aldehydes, ketoamides, and nitriles (Figure 1B).21,22,24,25,35,36 Five warheads within the panel were previously employed to covalently target Mpro, whereas vinylsulfonamides and terminal alkynes were not until during the preparation of our manuscript.37
Figure 1.

Clinical-stage inhibitors of Mpro and a panel of initial peptidomimetic inhibitors. (A) Covalent inhibitors of Mpro that are clinically used or underwent clinical evaluation. (B) General design of a panel of peptidomimetic inhibitors featuring various electrophilic warheads, including α,β-unsaturated ester, vinylsulfone, vinylsulfonamide, terminal alkyne, aldehyde, ketoamide, and nitrile.
The general synthesis of these peptidomimetics included the generation of a Weinreb amide from Fmoc-Gln(Trt)-OH, followed by the installation of the second required Leu residue from N-carbobenzyloxy-l-leucine to finally yield a N-protected dipeptide intermediate (Schemes S1–S7). Selective reduction of this Weinreb amide intermediate was then carried out in the presence of lithium aluminum hydride to form an important aldehyde precursor, which was used for synthesizing six compounds (1a–f) within the series (Table 1). Covalent warheads featured in these compounds include α,β-unsaturated ester, vinylsulfone, vinylsulfonamide, terminal alkyne, aldehyde, and ketoamide. The final nitrile-containing compound 1g was synthesized in a similar route but involved key conversion of the primary amide to the target nitrile group.
Table 1. Biochemical IC50 Values (μM) of Covalent Inhibitors against Mpro via a FRET-Based Enzymatic Assaya.
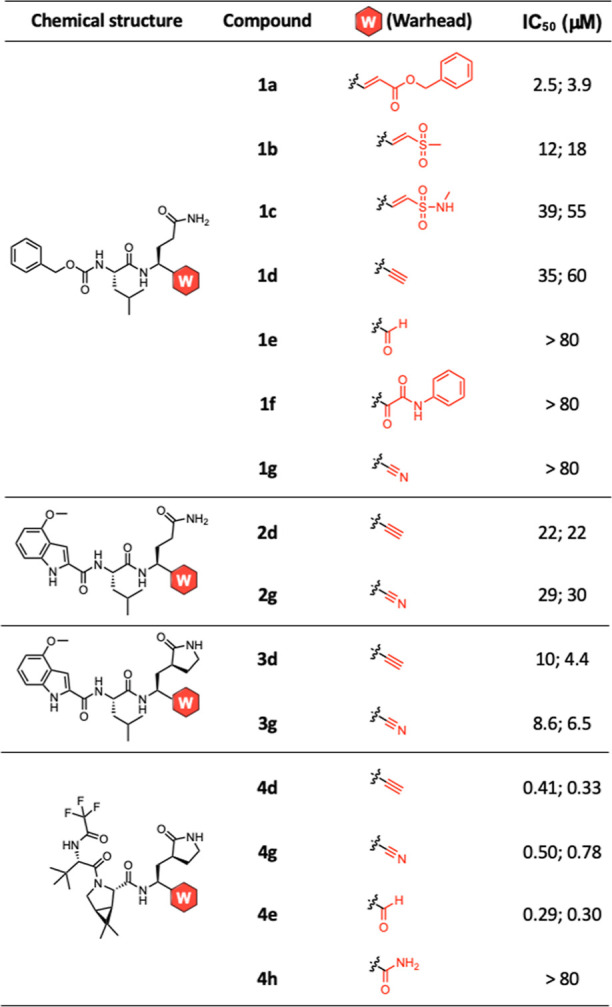
IC50 values were determined in vitro in the presence of the FRET substrate (20 μM) after 15-min treatment of Mpro (0.5 μM) with each compound at various concentrations. Measurements of the inhibitory activities of the compounds were performed in duplicate. IC50 values in duplicates were shown except for those that caused little inhibition at up to 80 μM.
Having synthesized the panel of Cbz-capped peptidomimetics, we screened them for inhibition against Mpro in a fluorescence resonance energy transfer (FRET)-based cleavage assay in vitro.36,38 Mpro was subcloned and expressed in E. coli before affinity purification via a hexa-His tag (Figure S1). Dose response studies revealed the half-maximal inhibitory concentration (IC50) values of compounds 1a–1g. Out of the 7 compounds, 1a, which contains an acrylate warhead, exhibited the strongest inhibitory activity against Mpro with an IC50 of 5.0 μM (average of duplicate, Table 1). 1b and 1c, which contain a warhead of vinyl sulfone and vinyl sulfonamide, have IC50 values of 18 and 47 μM, respectively. It was previously reported that vinyl sulfones reacted with thiols more rapidly than the analogous acrylates due to the stronger electrophilicity of the former group.39,40 However, we found that the acrylate-containing 1a inhibited Mpro more potently than the corresponding vinyl sulfone analogue. This discrepancy may be due to the better fit of the ester, including the benzyl group, than the sulfone group in the S1′ pocket.21 The potency difference between 1b and 1c at inhibiting Mpro is consistent with the thiol reactivity difference between vinyl sulfones and vinyl sulfonamides.
Compounds 1e, 1f, and 1g, which all contain reversible covalent warheads, failed to cause significant inhibition against Mpro up to 80 μM. These results suggest that the efficacy of the reversible warheads requires a scaffold of moderate or high noncovalent binding affinity to Mpro. The Cbz-Leu-Gln scaffold apparently cannot deliver sufficient levels of noncovalent binding to Mpro to manifest the effects of these reversible covalent warheads. Compound 1d, which contains a terminal alkyne, inhibited Mpro with an IC50 of 61 μM. A terminal alkyne has been commonly used as a “clickable” tag because it is largely considered chemically inert toward various compounds in cells.41,42 We chose to include the terminal alkyne in the panel of Cbz-Leu-Gln derivatives because prior studies demonstrated that it served as a latent warhead to covalently target cathepsin K protein and deubiquitinase.33,35,43 In these applications, the terminal alkyne covalently modified the catalytic cysteine of the targeted cysteine protease by forming a vinyl thioether linkage.33,35,43 Unlike nitrile, which acts as a reversible warhead towards cysteine, the thiol–alkyne addition is irreversible, yielding a permanent covalent ligand–protein adduct.44 The advantages of irreversible covalency, the lack of indiscriminate thiol reactivity, and the common applications in targeting diverse cysteine proteases highlight our selection of alkyne as an ideal warhead candidate in the development of covalent inhibitors against Mpro. Attracted by the unique advantages of a latent irreversible warhead, we decided to install terminal alkyne on more sophisticated scaffolds in the hope that more potent inhibition of Mpro could be attained.
We next replaced the Cbz moiety with a 4-methoxy indole cap, which was adopted from a clinical candidate PF-00835231 (Figure 1A), in the Cbz-Leu-Gln derivatives.23 Previous structural analysis revealed extensive van der Waals interactions of the indole group in PF-00835231 with residues in the S3 subsite.23 Both terminal alkyne and its isostere nitrile were installed on the In-Leu-Gln scaffold, yielding 2d and 2g, respectively (Schemes S8 and S9).33 Both 2d and 2g were found to be a few times more potent at inhibiting Mpro than 1d and 1g, confirming that the indole cap afforded improved binding to the protease than Cbz. Importantly, 2d is slightly more potent than 2g, indicating that the terminal alkyne was comparable to or slightly better than the nitrile as the warhead targeting Mpro. The use of a lactam analogue of Gln, first featured in the discovery of rupintrivir (Pfizer),45 has been found to enhance binding to viral proteases compared to the more flexible Gln residue.46,47 We thus substituted the Gln with its lactam analogue (Qla) surrogate in the additional compounds. Installation of alkyne and nitrile on a modified scaffold of In-Leu-Qla led to compounds 3d and 3g (Scheme S10), which showed comparable potency (both ∼7 μM) at inhibiting Mpro. The improved potency of 3d over 2d and over 1d highlights the importance of the scaffold as the foundation of covalent inhibition.
Our further medicinal chemistry efforts involved replacing the nitrile warhead with alkyne in nirmatrelvir, the clinically used Mpro inhibitor that has undergone extensive chemical optimization (Figure 1A).25 In addition to the lactam analogue of Gln, nirmatrelvir also contains a cyclic variant (6,6-di-methyl-3-azabicyclo[3.1.0]hexane) of Leu and a trifluoroacetyl capping group at the N terminus.25 The benefits of this cyclized form of Leu include the removal of a hydrogen bond donor to increase cell permeability and improved fit into Mpro S3 pocket because of preorganization or entropic gain.25 Compound 4d was synthesized following routes that were modified based on the reported synthesis of nirmatrelvir (Scheme S11).48 We also synthesized the nitrile-containing nirmatrelvir and the aldehyde-containing 4e for comparison among the warheads. When tested in Mpro FRET assays, 4d was found to have an IC50 of 0.30 μM, roughly two times lower than that of nirmatrelvir (0.56 μM), which was determined under the same conditions. The potency of 4d is similar to the aldehyde-bearing analogue 4e with an IC50 of 0.30 μM, indicating that the latent alkyne warhead appears to work as well as aldehyde, a warhead with high intrinsic reactivity. To evaluate the non-covalent binding afforded by the nirmatrelvir scaffold, we also synthesized an additional nirmatrelvir analogue that contains a primary amide in place of the warhead. Interestingly, this control compound caused no significant inhibition against Mpro in the FRET assay. These results highlight the effectiveness of the latent alkyne warhead at covalently inhibiting Mpro.
Irreversible Inhibition of Mpro by the Alkyne Inhibitors
Thus, unlike the nitrile warhead in nirmatrelvir, which yields a reversible covalent thioimidate adduct (Figure 2C),25 our alkyne inhibitors are expected to undergo a two-step irreversible binding involving the initial reversible association with Mpro followed by an irreversible modification of the protease at the catalytic Cys145 residue.49 Specifically, upon reversibly binding at the catalytic site that enables precise positioning toward Cys145, the latent alkyne would be no longer bioinert but activated by the catalytic environment toward a hydrothiolation reaction, forming an irreversible vinyl thioether linkage and permanently inactivating Mpro (Figure 2B).
Figure 2.
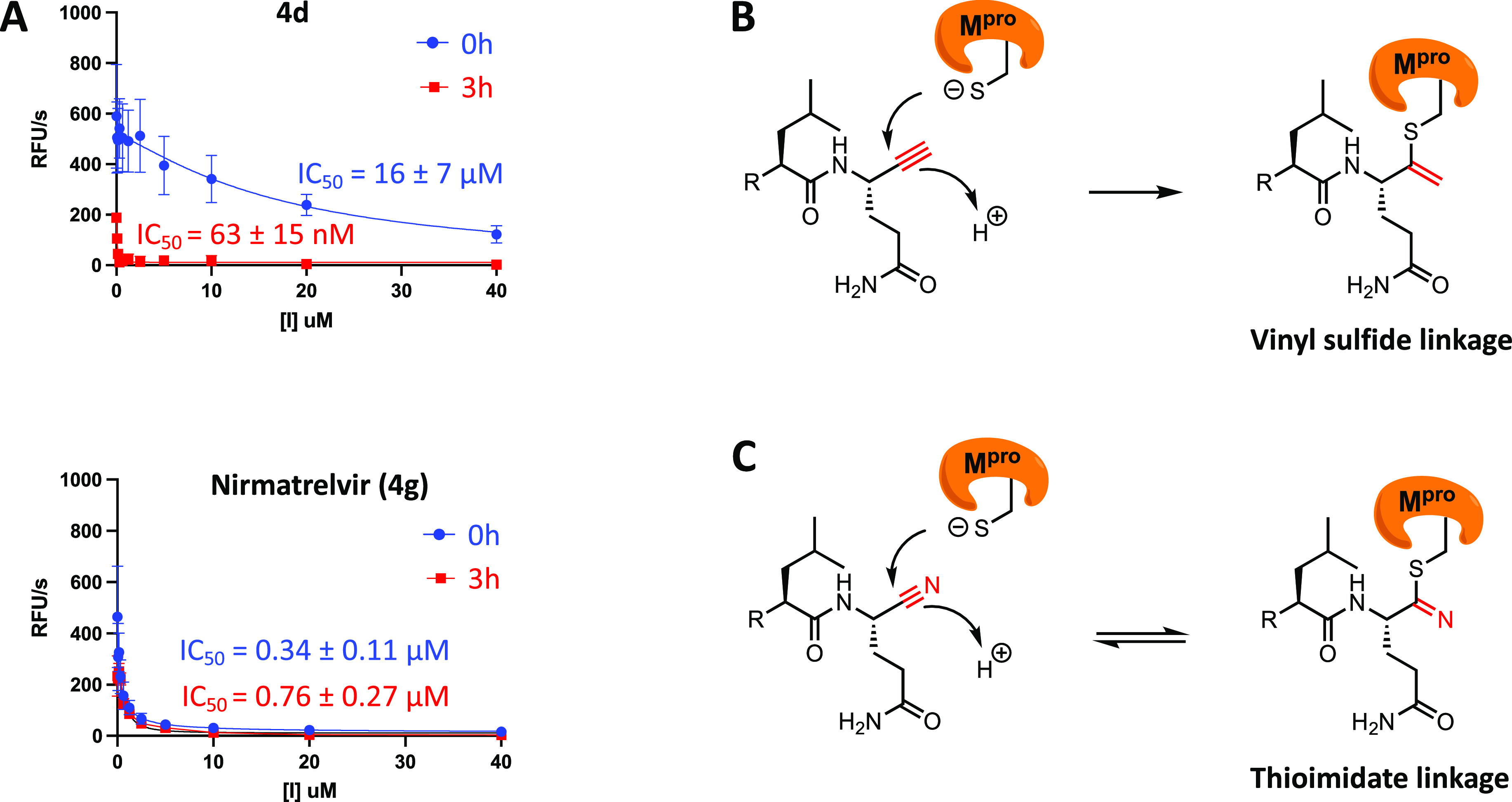
Time-dependent inhibition of Mpro by alkyne 4d and nirmatrelvir (4g). (A) Mpro (0.5 μM) was incubated with increasing concentrations (up to 40 μM) of 4d or nirmatrelvir (4g) in the reaction buffer at 30 °C for 0 or 3 h. The reactions were then initiated by the addition of the FRET substrate (20 μM) followed by continuous measurements of fluorescence for 1 h. IC50 values were determined in duplicates and shown as the mean ± SD. (B) Proposed irreversible thiol–alkyne addition of alkyne-containing inhibitors with Mpro to form a vinyl sulfide linkage. (C) Known reversible reaction of nitrile inhibitors, such as nirmatrelvir, with Mpro to form a thioimidate linkage.
To determine the reversibility of their inhibition, we subjected both 4d and nirmatrelvir (4g) to the same FRET-based cleavage assay and examined their time-dependent inhibition of Mpro (Figure 2A). Dose response inhibition of recombinant Mpro by 4d or nirmatrelvir using the FRET assay was performed either without preincubation or with a 3 h preincubation. The IC50 of nirmatrelvir did not change significantly after the 3 h preincubation, consistent with the reversible inhibition via the nitrile warhead. In contrast, the 3 h preincubation of Mpro with compound 4d lead to a 254-time increase of IC50, supporting the inhibition acting in an irreversible process.50 Furthermore, the dramatic change of IC50 of 4d against Mpro upon alteration of the preincubation time suggests that the rate of its inactivation (kinact) is relatively low, which is consistent with the nature of a latent warhead.
Taking advantage of the irreversibility of alkynes as latent electrophiles,33,43 we went on to identify the amino acid within Mpro that was covalently modified by 4d using liquid chromatography tandem mass spectrometry (LC–MS/MS). Recombinant Mpro was incubated with excess 4d for 4 h, followed by digestion with trypsin and Glu-C prior to LC–MS/MS analysis. LC–MS/MS data revealed that the catalytic Cys (Cys145) was modified by 4d, as predicted (Figure S3). No other residues were found to be modified in the mass spec analysis, highlighting the exquisite selectivity of our alkyne inhibitor for the catalytic Cys residue.
In Vitro and In Situ Labeling of Mpro by a Clickable Analogue of 4d
To further confirm the irreversible binding of 4d to Mpro and allow for measurement of its target engagement, we synthesized Alk-4d, an analogue of 4d, by replacing the tert-butyl group at the P3 position in 4d with a propargyl group (Figure S4A). This replacement was predicted to cause minimal perturbation to 4d′s binding to Mpro since the tert-butyl group was found to protrude toward the solvent in the solved crystal structures of Mpro-nirmatrelvir complexes. Alk-4d was first evaluated for its ability to label recombinant Mpro in an in-gel fluorescence experiment, which involved incubation of the probe with the protein, click conjugation to TAMRA azide, SDS-PAGE resolution, and fluorescence imaging of the gel (Figure S4B). With 1 h of pre-treatment, Alk-4d efficiently labeled Mpro at concentrations above 1 μM, reaching saturation at 3 μM. We next turned to competitive labeling experiments to confirm the target engagement of 4d. Pre-treating recombinant Mpro with 4d dramatically reduced the labeling of Mpro by Alk-4d due to the prior occupancy of Mpro by 4d. Nirmatrelvir also efficiently blocked the labeling of Mpro by Alk-4d in co-treatment experiments.
We net examined the labeling of Mpro by Alk-4d in live cells. By treating HEK293T cells that were transiently transfected with Mpro with Alk-4d, we observed dose-dependent labeling of Mpro by the probe (Figure S4C). The analogous experiment with the C145A mutant of Mpro yielded no labeling, supporting Cys145 being the residue being modified by Alk-4d (Figure S4D). Interestingly, treatment of parental HEK293T cells with Alk-4d with high concentrations of Alk-4d did not label any proteins, as revealed by the in-gel fluorescence experiment, indicating that there were no prominent covalent off-targets of this probe in human cells, including HEK293T and HeLa cells (Figure S5A). To answer the question of whether our alkyne inhibitors might hit host cysteine proteases in view of the precedence of alkyne-containing inhibitors of cathepsin K,33 we tested 4d on three human cysteine proteases—cathepsin B, cathepsin K, and cathepsin L. Despite a 20 min preincubation in the cathepsin enzymatic assays, 4d at up to 40 μM caused little inhibition against the cathepsin proteases (Figure S6). This is consistent with the distinct substrate recognition motif of SARS-CoV-2 Mpro from those of human cysteine proteases.19 Taken together, the proteomic labeling and biochemical data support the high selectivity of our alkyne inhibitors for SARS-CoV-2 Mpro.
While Alk-4d succeeded in labeling Mpro in situ, we noticed that a relatively high concentration (20 or 40 μM) of Alk-4d was often needed to produce consistent labeling. We attribute the poor in situ labeling to the slow kinetics of Mpro modification by Alk-4d, which motivated us to tune up the reactivity of the alkyne group through chemical modifications.
Tuning of Alkyne Reactivity via Substitution with an Electron-Withdrawing Group
The inhibitors and probes that harbor a terminal alkyne were found to have relatively slow kinetics at covalent inactivation of Mpro, as described above. We reasoned that attachment of an electron-withdrawing group (EWD) at the terminal carbon would increase the reactivity of the alkyne toward the catalytic cysteine in Mpro. We chose to install a trifluoromethyl (CF3) group at the alkyne to yield a trifluoromethylated analogue 4i (Figure 3A), inspired by the successful introduction of CF3 on a terminal alkyne to better covalently target deubiquitinases.44
Figure 3.
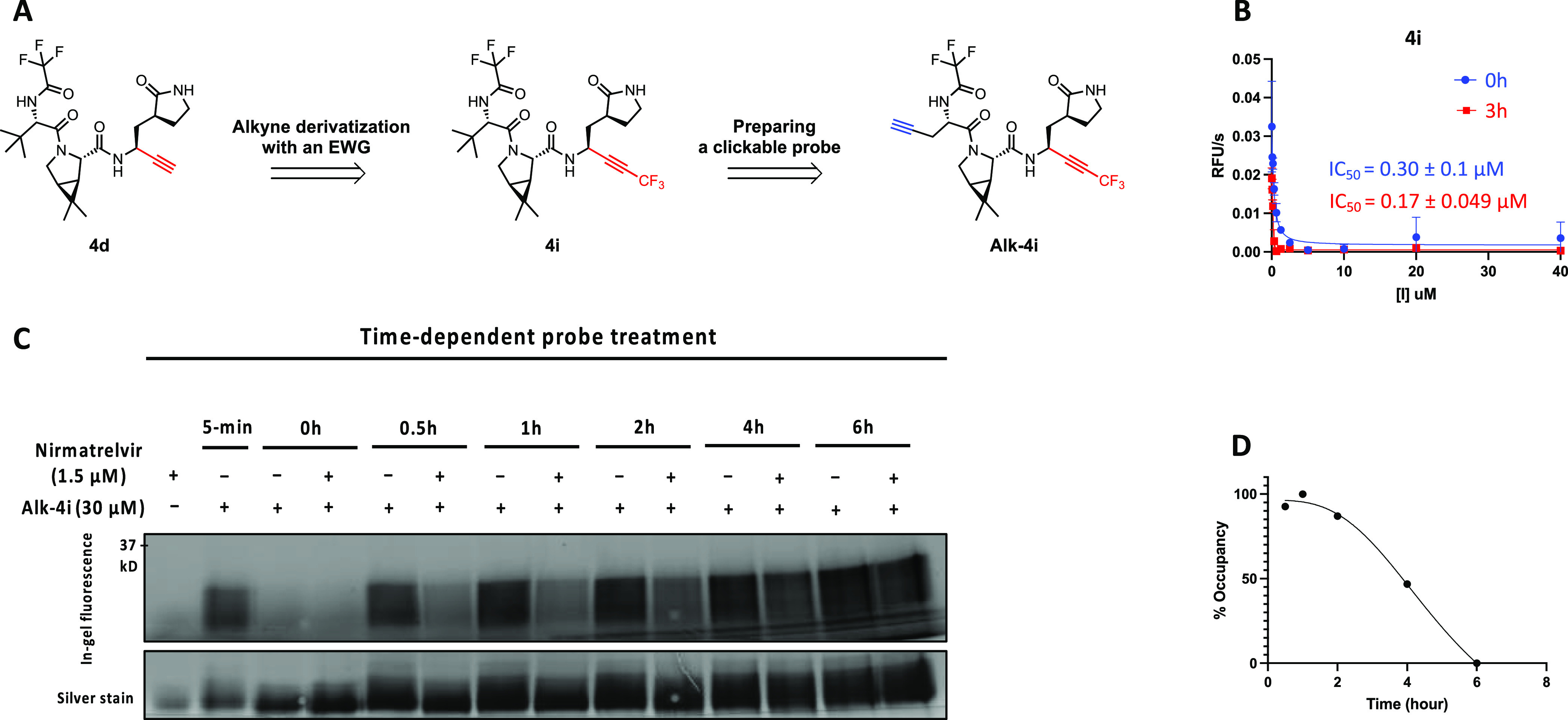
Tuning up alkyne reactivity through a chemical modification. (A) Alkyne substitution with an electron-withdrawing group (EWG), trifluoromethyl (CF3), to tune its warhead reactivity. A clickable probe, Alk-4i, was derived from 4i by replacing the solvent-exposed tert-butyl group with an alkyne reporter tag. (B) 4i inhibited Mpro and dramatically increased the rate of Mpro inactivation in the FRET-based enzymatic assay. (C) Measurements of the residence time of nirmatrelvir (4g) with Mpro by using Alk-4i in a competitive labeling experiment. Treatment of Mpro with a saturating concentration of nirmatrelvir (1.5 μM) for 30 min, followed by treatment with an excessive concentration (30 μM) of Alk-4i, revealed the dissociation kinetics of nirmatrelvir from Mpro. (D) Change of Mpro occupancy by nirmatrelvir over time. The data in (C) were quantified and normalized to generate the temporal curve.
4i was synthesized following a similar route to that of 4d (Scheme S11). When subjected to the FRET-based cleavage assay (Figure 3B), 4i showed an IC50 of 0.30 μM without pre-incubation with the enzyme. Adding a 3 h preincubation prior to the assay only reduced 4i’s IC50 slightly to 0.17 μM. These results are consistent with the prediction that the introduction of a CF3 group dramatically increased the rate of Mpro inactivation.
We next examined the potency of 4i′s engagement to Mpro in vitro by performing a competitive labeling experiment. A dose response study revealed that 1 μM of 4i was sufficient to abolish the labeling of Mpro by 10 μM of Alk-4d (Figure S7A). Similar dose–response studies were performed for 4d and nirmatrelvir. The data indicate that 4i has comparable potency at engaging Mpro to nirmatrelvir and that 4i is approximately 10-fold more potent at engaging Mpro than 4d in vitro. Kinetic characterizations allowed us to determine the kinact/KI values of 5.3 × 107 (M–1 s–1) for 4d and 4.9 × 108 (M–1 s–1) for 4i, which differ by ∼10 folds. Taken together, these results suggest that the introduction of a CF3 group at alkynes substantially increased the binding affinity of Mpro, apparently through enhancing the rate of enzyme inactivation.
Measurement of Nirmatrelvir’s Mpro Residence Time
We next derivatized the more potent and faster-acting alkyne inhibitor 4i with a propargyl group at P3 to yield a clickable probe Alk-4i (Scheme S13, Figure 3A). In-gel fluorescence analysis of the probe-treated Mpro showed that Alk-4i covalently targeted the main protease in a concentration-dependent manner and exhibited intense labeling at the lowest concentration tested, 300 nM. Moreover, Alk-4i labeled Mpro near saturation after a mere 5 min incubation, demonstrating rapid labeling kinetics as predicted (Figure S7B).
With a more potent and faster-acting clickable probe 4i available, we used it to directly measure the residence time of nirmatrelvir with Mpro. A pulse-chase like labeling experiment was performed (Figure 3C), involving an initial 30 min treatment of recombinant Mpro with nirmatrelvir (1.5 μM) for the drug to occupy the most active sites, followed by adding an excessive concentration (30 μM) of Alk-4i to label sites vacated after nirmatrelvir dissociation at different time points. Without nirmatrelvir pretreatment, the labeling of Mpro by Alk-4i did not change much during 6 h as expected. With nirmatrelvir pretreatment, the labeling of Mpro by Alk-4i increased over 6 h, consistent with the slow dissociation of nirmatrelvir from Mpro. From the curve (Figure 3D), an approximate half-life (t1/2) of 3.5 h was derived, which is the first reported residence time for Mpro-nirmatrelvir complex to our knowledge.
To investigate whether the derivatization of alkynes with an electron-withdrawing trifluoromethyl group would leave Alk-4i with greater non-specific proteomic labeling because of the boosted reactivity of the alkyne group, we turn to in-gel fluorescence experiments. Parental HEK293T cells were treated with either Alk-4d or Alk-4i at various concentrations for 5 h, harvested, and processed for in-gel fluorescence analysis (Figure S5B). While Alk-4d at all tested concentrations caused little proteome labeling, 10 μM of Alk-4i induced significant labeling of the HEK293T proteome. These data suggest that boosting the reactivity of alkynes with a CF3 group resulted in non-specific proteome labeling, which was not significant at low micromolar concentrations.
Structure Basis of Covalent Inhibition of Mpro by Alkyne-Containing Compounds
To elucidate the molecular basis for the covalent inhibition of Mpro by alkyne-containing inhibitors, we solved the X-ray crystal structures of the protease in complex with 3d and 4d. Recombinant Mpro was purified using a RESOURCE Q anion exchange column after affinity purification using the Ni-NTA column. The purified Mpro was then co-crystalized with either 3d or 4d, with the resulting crystals diffracting at synchrotron to resolutions of 1.9 and 2.0 Å, respectively. Molecular replacement using Mpro with nirmatrelvir (PDB: 7RFS) as the search model was then performed to solve the structures.
The complex structures of Mpro-3d and Mpro-4d both confirmed the presence of a covalent bond between the sulfur atom of Cys145 and the internal carbon of alkyne (Figure 4). The indole ring N-terminal capping group within 3d forms extensive contacts with several hydrophobic residues in Mpro, while the leucine residue and the lactam moiety occupy the S2 and S1 pockets of the protease, as predicted. The thiol–alkene adduct resulting from the addition of thiol to alkyne can be clearly visualized based on the electron density. A structural overlay of Mpro in complex with 3d or PF00835231 (PDB: 6XHM), a close analogue containing the same indole-peptidic scaffold, revealed the inhibitors are largely superimposable with the exception of the thiol-modified warheads (thioalkene vs hemithiolketal). The structure of Mpro-4d is very similar to the previously reported Mpro-nirmatrelvir structures. The RMSD between 4d and nirmatrelvir is merely 0.25 Å, consistent with the isosteric nature of the two inhibitors. Despite their different chemical structures, the thiol–alkene, resulting from the reaction of 4d with Mpro, and the imidothiol ester, resulting from the reaction between nirmatrelvir and Mpro, are essentially superimposable in an overlay of our Mpro-4d structure and a Mpro-nirmatrelvir structure (PDB: 7RFS). These observations support the notion that a similar mechanism for thiol addition is shared between a nitrile and an alkyne.
Figure 4.
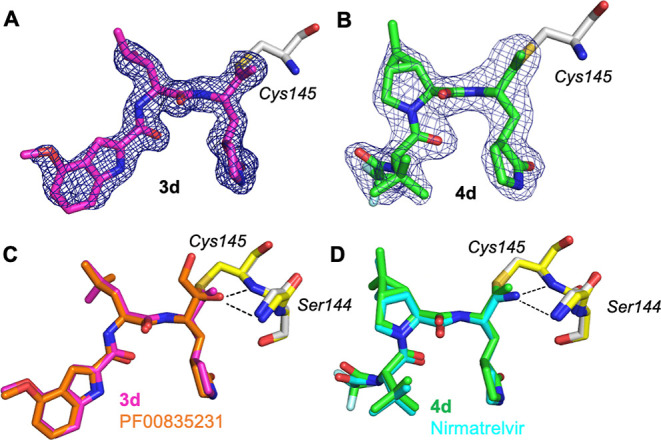
Crystal structures of Mpro in complex with two alkyne-containing inhibitors. (A) Representative OMIT electron density map (mFo – DFc) contoured to 3σ for 3d in complex with Mpro (PDB: 8FY7). (B) Representative OMIT electron density map (mFo – DFc) contoured to 3σ for 4d in complex with Mpro (PDB: 8FY6). (C) Structural overlay of Mpro-3d complex (PDB: 8FY7) with the reported Mpro-PF00835231 complex (orange). (D) Structural overlay of Mpro-4d complex (PDB: 8FY6) with the reported Mpro-nirmatrelvir complex (cyan).
Evaluation of the Anti-COVID Activity of Alkyne-Containing Compounds
We next evaluated the antiviral activity of our most potent Mpro inhibitors in a SARS-CoV-2 in vitro infection model. Specifically, we infected Hela cells constitutively expressing ACE2 (HeLa-ACE2)51 with SARS-CoV-2 at a multiplicity of infection (MOI) of 0.01 for 1 h to allow for cell entry of SARS-CoV-2. The infected cells were then treated with either 4d, 4i, or nirmatrelvir for 24 h, followed by quantification of the viral RNA transcripts in the cell lysates.
Both 4d and 4i showed strong, dose-dependent antiviral activity, as predicted by their potent inhibition against recombinant Mpro in vitro (Figure 5A). At 3 μM, 4d achieved nearly complete inhibition of SARS-CoV-2 infection. 4i at sub-micromolar concentrations afforded significant anti-COVID activity, showing apparent higher potency than 4d under the same conditions. This is consistent with the activating effects on the alkyne group from the trifluoromethyl substituent.
Figure 5.

Alkyne inhibitors exhibit strong anti-COVID activity and minimal cell toxicity. (A) Anti-SARS-CoV-2 efficacy of the compounds in HeLa-ACE2 cells. (B) Cytotoxicity of the compounds in HeLa-ACE2 cells.
Finally, we determined the cytotoxicity of nirmatrelvir, 4d, and 4i by treating non-infected Hela-ACE2 cells with these compounds for 20 h, followed by the collection and analysis of cell viability data (Figure 5B). Similar to nirmatrelvir, both of the alkyne-containing inhibitors, 4d and 4i, showed no notable cytotoxicity up to the highest concentration tested, 10 μM. Taken together, these data suggest that a latent warhead of terminal alkyne has moderate anti-COVID activity and little toxicity at up to 10 μM to cells.
Conclusions
We sought novel electrophilic warheads that can be used to covalently target SARS-CoV-2 Mpro, a crucial component of the virus’s replication process. By synthesizing and screening a panel of warheads, we found that the terminal alkyne could serve as a latent warhead to covalently modify Mpro. Biochemical and X-ray structural analyses support the notion that the alkyne forms an irreversible vinyl-sulfide linkage with the catalytic cysteine of Mpro. The best alkyne-containing inhibitors effectively prevented SARS-CoV-2 infection in cell models, indicating the potential of using alkyne as a latent warhead to target cystine proteases in viruses and beyond. The major advantage of latent warheads is that they have little intrinsic chemical reactivity and are only activated for covalent inhibition in the active site of appropriate enzymes.33 A previous study reported that while compounds with a nitrile warhead formed covalent adducts with cysteine, alkyne-bearing counterparts yielded 0 to <1% of adducts after incubation with cysteine.33 As a result, compounds containing latent warheads tend to have fewer covalent off-targets than their counterparts harboring regular warheads. We found that a terminal alkyne was an efficient latent warhead for targeting SARS-CoV-2 Mpro, reflected by the observed potent biochemical inhibition, especially with prolonged incubation. However, the rates of inactivation of Mpro by inhibitors containing a latent alkyne warhead, 4d, are not high, with a preincubation of tens of min being required to attain nanomolar inhibition in vitro. We suspect that the relatively slow inactivation of Mpro disfavors 4d-like compounds in the tight race against SARS-CoV-2 infection and replication, which occurs within minutes, and underlies their moderate potency against SARS-CoV-2 infection in cell culture models. We succeeded in increasing the rate of Mpro inactivation through derivatization of the alkyne warhead with the electron-withdrawing group CF3. Future studies will be required to determine if a terminal or appropriately substituted alkyne can serve as a latent warhead for targeted covalent inhibition in therapeutic development.
Experimental Section
Synthetic Procedures
All of the reagents and solvents were obtained via commercial sources and used without further purification, unless otherwise stated. All anhydrous reactions were carried out under a nitrogen atmosphere. Reactions were monitored by thin-layer chromatography (TLC) on glass TLC plates with silica gel coated with fluorescent indicator F254. UV light and TLC stains, including ninhydrin and 2,4-dinitrophenylhydrazine, were used as visualizing agents. Individual intermediates and final compounds were purified by flash column chromatography using an automated Teledyne CombiFlash system (RF + UV–vis). All 1H and 13C nuclear magnetic resonance (NMR) spectra were obtained on a Varian Mercury 400, 400 MR, or Varian VNMRS-600 spectrometer at ambient temperature. All target compounds were found to be ≥95% pure based on analytical high-performance liquid chromatography analysis. The purities were determined on a Shimadzu LC-20AP system equipped with an SPD-M21A PDA detector set to λ = 254 nm and a Phenomenex C18 (250 × 3.9 mm) column. The analyses were carried out using acetonitrile with 0.1% trifluoroacetic acid (v/v) in the isocratic mode at a flow rate of 1.8 mL/ min for 14 min. The injection peak at ∼2 min was excluded in the quantification of all compounds. The area % of the major peak is ≥95% with respect to the sum of the area % of other detected peaks.
Protease Inhibition Assays
Mpro
The expression and purification of recombinant Mpro were performed as previously described with minor modifications to the protocol.36 Briefly, after expression in E. coli, Mpro was purified using polyHis-Ni affinity chromatography, cleaved with PreScission protease, and then purified by using a ResQ column on an AKTA Pure FPLC system. Purified Mpro was concentrated to about 1 mg/mL and stored in a 20% glycerol solution at −80 °C for later use in biochemical studies.
A fluorescent peptide substrate of the sequence Dabcyl-KTSAVLQSGFRKM-E(Edans)-NH2 and a reaction buffer composed of 20 mM Tris-HCl, 100 mM NaCl, 1 mM EDTA, 1 mM TCEP, pH 7.3 were used in the enzyme assay.36 In the FRET-based cleavage assay, Mpro diluted in the reaction buffer was pre-incubated with compounds at various concentrations at 30 °C for a period (0 min, 15 min, or 3 h) before 20 μM FRET substrate was added to initiate the enzymatic reaction. The fluorescence signal was monitored using a Cytation 5 imaging reader (Thermo Fisher Scientific) or a Spectra MaxR iD5 (Molecular Devices) with excitation at 340 nm and emission at 475 nm at 30 °C every 38 s for 1 h. The linear section of the measurement was used to calculate initial velocity via linear regression in Prism 9.
Cathepsin B, Cathepsin K, and Cathepsin L
Recombinant cathepsin B, cathepsin K, and cathepsin L at 0.5, 0.042, and 0.25 nM, respectively, were pre-incubated with 4d at various concentrations in the appropriate reaction buffers for 20 min. Z-FR-AMC fluorogenic peptide substrate was then added to the pre-treated cathepsin B, cathepsin K, and cathepsin L at 10, 5, and 10 μM, respectively, to initiate the enzymatic reactions. The fluorescence signal was monitored using EnVision with excitation at 355 nm and emission at 460 nm at room temperature every 5 min interval for 2 h. The linear portion of the measurement was used to calculate the initial velocity via Excel. Curve fits were performed using Prism 9. The reaction buffer used in the assay against cathepsin B contained 25 mM MES pH 6, 50 mM NaCl, 0.005% Brij35, 5 mM DTT, and 1% DMSO. The reaction buffer used in the assay against cathepsin K contained 50 mM NaOAc, pH 5.5, 5 mM ethylenediaminetetraacetic acid (EDTA), 0.005% Triton X-100, 5 mM dithiothreitol (DTT), and 1% dimethyl sulfoxide (DMSO). The reaction buffer used in the assay against cathepsin L contained 400 mM NaOAc, pH 5.5, 4 mM EDTA, 8 mM DTT, and 1% DMSO.
LC–MS/MS Analysis
Recombinantly purified Mpro (34 μg, 10 μM) was treated with DMSO or excess 4d (100 μM) for 4 h at 30°C. Chloroform-methanol precipitation was done, followed by resuspension of Mpro in 8 M urea prepared in 50 mM NH4HCO3, pH 7.8. The protein was then reduced with 5 mM DTT at 60 °C for 1 h and alkylated with 15 mM iodoacetamide for 40 min at room temperature in the dark. The reaction was diluted with 50 mM NH4HCO3, pH 7.8, to reduce the urea concentration to 1 M. Mpro was digested by trypsin (V5113, Promega) overnight at 37°C with a trypsin-to-protein ratio of 1:50 (w/w). The digested peptides were desalted using Pierce C18 tips, followed by drying via speedvac. Digestion with Glu-C, Sequencing Grade (Promega, V1651) was carried out in 50 mM sodium phosphate buffer pH 7.8 overnight at 37 °C with a Glu-C to protein ratio of 1:50 (w/w). The same desalting and speedvac steps were done, followed by LC–MS/MS analysis. LC–MS/MS analysis was performed with an EASY-nLC 1200 (Thermo Fisher Scientific, San Jose, CA) coupled to an Orbitrap Eclipse Tribrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA). Peptides were separated on an Aurora UHPLC Column (25 cm × 75 μm, 1.6 μm C18, AUR2-25075C18A, Ion Opticks) with a flow rate of 0.35 μL/min for a total duration of 135 min and ionized at 1.6 kV in the positive ion mode. The gradient was composed of 6% solvent B (7.5 min), 6–25% B (82.5 min), 25–40% B (30 min), and 40–98% B (15 min); solvent A: 0.1% formic acid in water; solvent B: 80% ACN and 0.1% formic acid. MS1 scans were acquired at a resolution of 120,000 from 350 to 2000 m/z, an AGC target of 1e6, and a maximum injection time of 50 ms. MS2 scans were acquired in the ion trap using the fast scan rate on precursors with 2–7 charge states and the quadrupole isolation mode (isolation window: 0.7 m/z) with higher-energy collisional dissociation (HCD, 30%) activation type. Dynamic exclusion was set to 30 s. The temperature of the ion transfer tube was 300 °C and the S-lens RF level was set to 30. MS2 fragmentation spectra were searched with Proteome Discoverer SEQUEST (version 2.5, Thermo Scientific) against the in silico tryptic-digested Uniprot Human herpesvirus 1 (HHV-1) database. The maximum missed cleavages were set to 2. Dynamic modifications were set to oxidation on methionine (M, +15.995 Da), phosphoribosylation (D, E, R and K, +212.009 Da), deamidation (N and Q, +0.984 Da), protein N-terminal acetylation (+42.011 Da), and Met-loss (−131.040 Da). Carbamidomethylation on cysteine residues (C, +57.021 Da) was set as a fixed modification. The maximum parental mass error was set to 10 ppm, and the MS2 mass tolerance was set to 0.6 Da. The false discovery threshold was set strictly to 0.01 using the percolator node validated by q-value. The relative abundance of parental peptides was calculated by integrating the area under the curve of the MS1 peaks using the Minora LFQ node.
Kinetic Measurements of Covalent Inactivation of Mpro
The FRET substrate (25 μM) was first diluted in the reaction buffer containing compounds 4d or 4i at concentrations ranging from 10 nM to 10 μM. Mpro (0.05 μg, 0.015 μM) was then added to initiate the reaction, with the fluorescence changes being continuously monitored for 1 to 3 h. The progress curves were fit to a slow-binding Morrison equation, as described previously.52,53 The kinetic kinact/KI parameter was derived using Prism 9.
Crystallization and Structure Determination
Mpro protein was diluted to ∼7 mg/mL in the same buffer before the inhibitor (3d or 4d) was added to the protein solution at about a 1:5 molar ratio of protein to inhibitor. The protein-inhibitor solution was incubated for 18 h at 4 °C. After incubation, the sample was spun down at 12,000 rpm for 2 min to remove the precipitate. Hanging drop trays were set up in an 18 °C room with 1 μL sample mixed with 1 μL crystallization solution composed of 0.1 M MIB pH 5.5 and 29%(w/v) PEG 1500 (3d). Hanging drop trays were set up in a 4 C room with drops made with 1.5 μL solution and with 1 μL crystallization solution composed of 0.1 MIB pH 6.5, 13% (w/v) PEG 1500, and 10% MPD (4d). Crystals grew overnight and were harvested seven days later using a 20% ethylene glycol cryobuffer.
Diffraction data for Mpro in complex with 3d and 4d were collected at beamlines 23ID-D and 23ID-B, respectively, at the National Institute of General Medical Sciences and the National Cancer Institute Structural Biology Facility at the Advanced Photon Source. A complete dataset was collected for each structure and processed using the DIALS data processing pipeline on the APS server, including indexing, integration, and scaling. Molecular replacement was performed using the Phaser-MR program from the PHENIX package, while model rebuilding and refinement were carried out using COOT and Phenix for simulated annealing and refinement, respectively. Ligands were generated using the eLBOW program in Phenix based on their SMILES code. The search model for molecular replacement was Mpro with nirmatrelvir (PDB: 7RFS) with the ligand removed. The initial phases of the MR protein model were improved by cyclic model building and refinement until a good model for each of the complexes was achieved. The final models for Mpro in complex with 3d and 4d were solved at a resolution range of 1.94 and 2.00 Å, respectively.
In Vitro Labeling of Recombinant Mpro by Clickable Probes
Recombinant Mpro was incubated with probes Alk-4d or Alk-4i at indicated concentrations at 30 °C for 1 h or with the incubation time indicated for the time-course experiment. Next, click chemistry was performed at a final concentration of 25 μM TAMRA-azide, 1 mM tris(2-carboxyethyl)phosphine (TCEP, Thermo Scientific), 100 μM Tris-hydroxypropyltriazolylmethylamine (THPTA, Sigma-Aldrich), and 1 mM CuSO4 (Sigma-Aldrich) in a total volume of 21 μL. The reactions were carried out at room temperature for 1 h in the dark. Click reactions were terminated by the addition of 7 μL of 4× Laemmli sample loading buffer (Bio-Rad), boiled for 7 min, and resolved onto 4–20% sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel (Bio-Rad). Fluorescence was visualized at 532 nm for excitation and at 600 nm for emission on a Typhoon 9400 Variable Mode Imager (GE Healthcare), and images were displayed as grayscale. After fluorescence scanning, protein loadings were visualized by silver staining using the Thermo Scientific Pierce Silver Stain Kit. Competition labeling involved a cotreatment of recombinant Mpro (1 μg) with a competitor (nirmatrelvir, 4d, or 4i) and a clickable probe (Alk-4d or Alk-4i) at indicated concentrations at 30 °C for 1 h. All subsequent steps on click reaction and gel imaging were the same as those above.
Pulse-Chase Style Competition Labeling for Measuring Nirmatrelvir Residence Time
Recombinant Mpro was first incubated with 1.5 μΜ of nirmatrelvir at 30 °C for 1 h, followed by treatment with the Alk-4i probe at 30 μM for different periods of time. All subsequent steps on click reaction and gel imaging were the same as those above.
Measurement of Mpro Engagement Using Clickable Probes in Cells
HEK293T or HeLa cells were grown in 6-well plates and transfected with an appropriate plasmid harboring Mpro. At 24 h post-transfection, the growth media [Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS)] was aspirated off, and the cells were treated with fresh media containing various concentrations of probe (1000× stock solution in DMSO) or vehicle control for the indicated time. For competitive labeling experiments, cells were first incubated with the inhibitor at various concentrations for 1 h, washed with fresh, warm medium three times, and then treated with the probe at the appropriate concentration for another hour. After probe treatment, the medium was aspirated off, and the cells were washed twice with ice-cold Dulbecco’s phosphate-buffered saline. The cells were harvested, and the pellet was resuspended in 100 μL of NP40 lysis buffer (50 mM HEPES, pH 7.4, 1% NP-40, 150 mM NaCl) with a protease inhibitor cocktail (Roche). The lysate was incubated on ice for 20 min and fractionated by centrifugation at 18,000g for 10 min. The protein concentration was measured from each of the supernatant samples by a BCA assay (Pierce) and normalized to 1 mg/mL. All subsequent steps on click reaction and gel imaging were the same as those above.
Cell Viability and Cell-Based Antiviral Assays
SARS-CoV-2 infection was performed at the UCLA BSL3 facility. The recombinant SARS-CoV-2 (icSARS-CoV-2-mNG) expressing mNeonGreen54 was a kind gift from the World Reference Center for Emerging Viruses and Arboviruses (WRCEVA) at the University of Texas Medical Branch. Hela-ACE2 cells were grown in standard DMEM with 10% FBS and 1% penicillin/streptomycin (GIBCO) at 37 °C in a 5% CO2 humidified atmosphere. SARS-CoV-2 infection was performed, as described previously.51 Briefly, Hela-ACE2 cells were infected with SARS-CoV-2 (MOI: 0.01) and incubated at 37 °C. The viral inoculum was removed after 1 h and replaced with fresh, complete DMEM media, followed by treating cells with the serially diluted compounds. At 24 h post-infection, the media was collected for evaluating the viral particles released to the media, and cell lysate was evaluated for quantification of the viral RNA transcripts by using RT-qPCR. Cells were collected in TRIzol, and RNA was isolated by standard isopropanol precipitation. 1 μg of RNA was reverse transcribed using iScript (BioRad) according to the manufacturer’s protocols by using random hexamers as primers. RT-qPCR analysis was done using the iCycler thermocycler (Bio-Rad). RT-qPCR was conducted in a final volume of 20 μL. Amplification conditions were 95 °C (3 min), 40 cycles of 95 °C (20 s), 55 °C (30 s), 72 °C (20 s). The expression values from untreated control cells were used to obtain the relative fold change, which was normalized to the ribosomal RNA L32 values. For detection of SARS-CoV-2 genomic RNA, the below primers targeting SARS-CoV-2 nucleocapsid protein (NP) were used: NP-fwd, 5′-TAATCAGACAAGGAACTGATTA-3′; NP rev, 5′-CGAAGGTGTGACTTCCATG-3′ RT-qPCR cycling conditions were 95 °C for 30 s and 40 cycles of 95 °C for 5 s, followed by 55 °C for 30 s.
Evaluating the 50% cytotoxic concentration (CC50) by treating cells with serially diluted compounds. Twenty hours prior to the cytotoxicity assay, 2 × 104 Hela-ACE-2 cells were seeded in 96 Well white/clear bottom plate, TC Surface (Thermo Fisher). Cells were treated with serially diluted compounds. The cell viability was determined by using the Cell Titer-Glo Luminescent Cell Viability Assay (Promega). IC50 and CC50 values were calculated by non-linear regression analysis using GraphPad 5, where applicable.
Acknowledgments
This study was supported by the Zumberge Epidemic and Virus Related Research Awards at the University of Southern California (C.Z., X.J.C., and P.F.) and the US National Institute of Health (AI150524 to X.J.C., AI158154 to G.C., and AG070904 to P.F.). C.N. was supported by the Dornsife Graduate School Fellowship at USC.
Glossary
Abbreviation
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- COVID-19
coronavirus disease 2019
- Mpro
severe acute respiratory syndrome coronavirus 2 main protease
- RNA
ribonucleic acid
- kb
kilobase
- CYP3A
cytochrome P450, family 3, subfamily A
- FRET
fluorescence resonance energy transfer
- EWD
electron withdrawing group
- Ni-NTA
nickle-nitrilotriacetic acid
- PDB
protein data bank
- RMSD
root mean square deviation
- HEK293T
human embryonic kidney 293 cells containing SV40 large T antigen
- HeLa
A cancer cell line from Henrietta Lacks, a cervical cancer patient
- HeLa-ACE2
Hela cells constitutively expressing ACE2
- MOI
multiplicity of infection
- NMR
nuclear magnetic resonance
- HPLC
high-performance liquid chromatography
- LC–MS/MS
liquid chromatography tandem mass spectrometry
- DTT
dithiothreitol
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c00810.
Representative gel image of fractions after the purification of Mpro with RESOURCE Q column, dose responses of the compounds’ inhibition of Mpro in FRET-based cleavage assays, LC–MS/MS analysis and identification of site-specific modification of Mpro using 4d, measurements of target engagement of Mpro in vitro and in situ using clickable probes, chemoproteomic labeling of 4d in mammalian cell lines, measurement of drug-target residence time using clickable probe of 4d, synthetic methods including detailed procedures and synthetic schemes, and 1H NMR characterization of all compounds prepared for this study, purity analyses of representative compounds via HPLC detailing HPLC chromatograms with ≥95% purity (PDF)
Molecular formula strings of compounds 1a-g, 2d, 2g, 3d, 3g, 4d-i, Alk-4d, and Alk-4i(CSV)
Protein Data Bank (PDB) IDs for the crystal structures of Mpro in complex with 3d and 4d are 8FY7 and 8FY6, respectively. Authors will release the atomic coordinates upon article publication
The authors declare the following competing financial interest(s): C.Z. is a stockholder and consultant to BridGene Biosciences and Myelogene.
Supplementary Material
References
- Wu Y.-C.; Chen C.-S.; Chan Y.-J. The Outbreak of COVID-19: An Overview. J. Chin. Med. Assoc. 2020, 83, 217–220. 10.1097/jcma.0000000000000270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B.; Guo H.; Zhou P.; Shi Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. 10.1038/s41579-020-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO . WHO Coronavirus (COVID-19) Dashboard. https://covid19.who.int (accessed April 6, 2023).
- Garcia-Beltran W. F.; Lam E. C.; St. Denis K.; Nitido A. D.; Garcia Z. H.; Hauser B. M.; Feldman J.; Pavlovic M. N.; Gregory D. J.; Poznansky M. C.; Sigal A.; Schmidt A. G.; Iafrate A. J.; Naranbhai V.; Balazs A. B. Multiple SARS-CoV-2 Variants Escape Neutralization by Vaccine-Induced Humoral Immunity. Cell 2021, 184, 2372–2383.e9. 10.1016/j.cell.2021.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey W. T.; Carabelli A. M.; Jackson B.; Gupta R. K.; Thomson E. C.; Harrison E. M.; Ludden C.; Reeve R.; Rambaut A.; Peacock S. J.; Robertson D. L.; SARS-CoV-2 Variants, Spike Mutations and Immune Escape. Nat. Rev. Microbiol. 2021, 19, 409–424. 10.1038/s41579-021-00573-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y.; Wang J.; Jian F.; Xiao T.; Song W.; Yisimayi A.; Huang W.; Li Q.; Wang P.; An R.; Wang J.; Wang Y.; Niu X.; Yang S.; Liang H.; Sun H.; Li T.; Yu Y.; Cui Q.; Liu S.; Yang X.; Du S.; Zhang Z.; Hao X.; Shao F.; Jin R.; Wang X.; Xiao J.; Wang Y.; Xie X. S. Omicron Escapes the Majority of Existing SARS-CoV-2 Neutralizing Antibodies. Nature 2022, 602, 657–663. 10.1038/s41586-021-04385-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton A.; Embi P. J.; Levy M. E.; Gaglani M.; DeSilva M. B.; Dixon B. E.; Dascomb K.; Patel P.; Schrader K. E.; Klein N. P.; Ong T. C.; Natarajan K.; Hartmann E.; Kharbanda A. B.; Irving S. A.; Dickerson M.; Dunne M. M.; Raiyani C.; Grannis S. J.; Stenehjem E.; Zerbo O.; Rao S.; Han J.; Sloan-Aagard C.; Griggs E. P.; Weber Z. A.; Murthy K.; Fadel W. F.; Grisel N.; McEvoy C.; Lewis N.; Barron M. A.; Nanez J.; Reese S. E.; Mamawala M.; Valvi N. R.; Arndorfer J.; Goddard K.; Yang D.-H.; Fireman B.; Ball S. W.; Link-Gelles R.; Naleway A. L.; Tenforde M. W. Effectiveness of COVID-19 MRNA Vaccines Against COVID-19–Associated Hospitalizations Among Immunocompromised Adults During SARS-CoV-2 Omicron Predominance — VISION Network, 10 States, December 2021—August 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 1335–1342. 10.15585/mmwr.mm7142a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A. R. Y. B.; Wong S. Y.; Chai L. Y. A.; Lee S. C.; Lee M. X.; Muthiah M. D.; Tay S. H.; Teo C. B.; Tan B. K. J.; Chan Y. H.; Sundar R.; Soon Y. Y. Efficacy of Covid-19 Vaccines in Immunocompromised Patients: Systematic Review and Meta-Analysis. Br. Med. J. 2022, 376, e068632 10.1136/bmj-2021-068632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castells M. C.; Phillips E. J. Maintaining Safety with SARS-CoV-2 Vaccines. N. Engl. J. Med. 2021, 384, 643–649. 10.1056/nejmra2035343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdiabadi S.; Rezaei N. Anaphylaxis and Allergic Reactions to COVID-19 Vaccines: A Narrative Review of Characteristics and Potential Obstacles on Achieving Herd Immunity. Health Sci. Rep. 2022, 5, e787 10.1002/hsr2.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.; Lee J.-Y.; Yang J.-S.; Kim J. W.; Kim V. N.; Chang H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. 10.1016/j.cell.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal M.; Berhanu G.; Desalegn C.; Kandi V. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2): An Update. Cureus 2020, 12, e7423 10.7759/cureus.7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naqvi A. A. T.; Fatima K.; Mohammad T.; Fatima U.; Singh I. K.; Singh A.; Atif S. M.; Hariprasad G.; Hasan G. M.; Hassan M. I. Insights into SARS-CoV-2 Genome, Structure, Evolution, Pathogenesis and Therapies: Structural Genomics Approach. Biochim. Biophys. Acta, Mol. Basis Dis. 2020, 1866, 165878. 10.1016/j.bbadis.2020.165878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohaim M. A.; El Naggar R. F.; Clayton E.; Munir M. Structural and Functional Insights into Non-Structural Proteins of Coronaviruses. Microb. Pathog. 2021, 150, 104641. 10.1016/j.micpath.2020.104641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto F. K. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS CoV-2 or n-COV19), the Cause of COVID-19. Protein J. 2020, 39, 198–216. 10.1007/s10930-020-09901-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J.; Gorbalenya A. E.; Snijder E. J. Virus-Encoded Proteinases and Proteolytic Processing in the Nidovirales. J. Gen. Virol. 2000, 81, 853–879. 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- Roe M. K.; Junod N. A.; Young A. R.; Beachboard D. C.; Stobart C. C. Targeting Novel Structural and Functional Features of Coronavirus Protease Nsp5 (3CLpro, Mpro) in the Age of COVID-19. J. Gen. Virol. 2021, 102, 001558. 10.1099/jgv.0.001558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Worrall L. J.; Vuckovic M.; Rosell F. I.; Gentile F.; Ton A.-T.; Caveney N. A.; Ban F.; Cherkasov A.; Paetzel M.; Strynadka N. C. J. Crystallographic Structure of Wild-Type SARS-CoV-2 Main Protease Acyl-Enzyme Intermediate with Physiological C-Terminal Autoprocessing Site. Nat. Commun. 2020, 11, 5877. 10.1038/s41467-020-19662-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Lin D.; Sun X.; Curth U.; Drosten C.; Sauerhering L.; Becker S.; Rox K.; Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S.; Chen Y.; Lockbaum G. J.; Sen S.; Chaudhuri S.; Reyes A. C.; Lee J. M.; Kaur A. N.; Sultana N.; Cameron M. D.; Shaffer S. A.; Schiffer C. A.; Fitzgerald K. A.; Thompson P. R. Dual Inhibitors of Main Protease (M Pro ) and Cathepsin L as Potent Antivirals against SARS-CoV2. J. Am. Chem. Soc. 2022, 144, 21035–21045. 10.1021/jacs.2c04626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z.; Du X.; Xu Y.; Deng Y.; Liu M.; Zhao Y.; Zhang B.; Li X.; Zhang L.; Peng C.; Duan Y.; Yu J.; Wang L.; Yang K.; Liu F.; Jiang R.; Yang X.; You T.; Liu X.; Yang X.; Bai F.; Liu H.; Liu X.; Guddat L. W.; Xu W.; Xiao G.; Qin C.; Shi Z.; Jiang H.; Rao Z.; Yang H. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582, 289–293. 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Vuong W.; Khan M. B.; Fischer C.; Arutyunova E.; Lamer T.; Shields J.; Saffran H. A.; McKay R. T.; van Belkum M. J.; Joyce M. A.; Young H. S.; Tyrrell D. L.; Vederas J. C.; Lemieux M. J. Feline Coronavirus Drug Inhibits the Main Protease of SARS-CoV-2 and Blocks Virus Replication. Nat. Commun. 2020, 11, 4282. 10.1038/s41467-020-18096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman R. L.; Kania R. S.; Brothers M. A.; Davies J. F.; Ferre R. A.; Gajiwala K. S.; He M.; Hogan R. J.; Kozminski K.; Li L. Y.; Lockner J. W.; Lou J.; Marra M. T.; Mitchell L. J.; Murray B. W.; Nieman J. A.; Noell S.; Planken S. P.; Rowe T.; Ryan K.; Smith G. J.; Solowiej J. E.; Steppan C. M.; Taggart B. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rut W.; Groborz K.; Zhang L.; Sun X.; Zmudzinski M.; Pawlik B.; Wang X.; Jochmans D.; Neyts J.; Młynarski W.; Hilgenfeld R.; Drag M. SARS-CoV-2 Mpro Inhibitors and Activity-Based Probes for Patient-Sample Imaging. Nat. Chem. Biol. 2021, 17, 222–228. 10.1038/s41589-020-00689-z. [DOI] [PubMed] [Google Scholar]
- Owen D. R.; Allerton C. M. N.; Anderson A. S.; Aschenbrenner L.; Avery M.; Berritt S.; Boras B.; Cardin R. D.; Carlo A.; Coffman K. J.; Dantonio A.; Di L.; Eng H.; Ferre R.; Gajiwala K. S.; Gibson S. A.; Greasley S. E.; Hurst B. L.; Kadar E. P.; Kalgutkar A. S.; Lee J. C.; Lee J.; Liu W.; Mason S. W.; Noell S.; Novak J. J.; Obach R. S.; Ogilvie K.; Patel N. C.; Pettersson M.; Rai D. K.; Reese M. R.; Sammons M. F.; Sathish J. G.; Singh R. S. P.; Steppan C. M.; Stewart A. E.; Tuttle J. B.; Updyke L.; Verhoest P. R.; Wei L.; Yang Q.; Zhu Y. An Oral SARS-CoV-2 M pro Inhibitor Clinical Candidate for the Treatment of COVID-19. Science 2021, 374, 1586–1593. 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- Hammond J.; Leister-Tebbe H.; Gardner A.; Abreu P.; Bao W.; Wisemandle W.; Baniecki M.; Hendrick V. M.; Damle B.; Simón-Campos A.; Pypstra R.; Rusnak J. M. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with Covid-19. N. Engl. J. Med. 2022, 386, 1397–1408. 10.1056/nejmoa2118542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S.; Tignanelli C. J.; Hoertel N.; Boulware D. R.; Usher M. G. Prevalence of Medical Contraindications to Nirmatrelvir/Ritonavir in a Cohort of Hospitalized and Nonhospitalized Patients With COVID-19. Open Forum Infect. Dis. 2022, 9, ofac389. 10.1093/ofid/ofac389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padhi A. K.; Tripathi T. Hotspot Residues and Resistance Mutations in the Nirmatrelvir-Binding Site of SARS-CoV-2 Main Protease: Design, Identification, and Correlation with Globally Circulating Viral Genomes. Biochem. Biophys. Res. Commun. 2022, 629, 54–60. 10.1016/j.bbrc.2022.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Péczka N.; Orgován Z.; Ábrányi-Balogh P.; Keserű G. M. Electrophilic Warheads in Covalent Drug Discovery: An Overview. Expert Opin. Drug Discovery 2022, 17, 413–422. 10.1080/17460441.2022.2034783. [DOI] [PubMed] [Google Scholar]
- Kim H.; Hwang Y. S.; Kim M.; Park S. B. Recent Advances in the Development of Covalent Inhibitors. RSC Med. Chem. 2021, 12, 1037–1045. 10.1039/d1md00068c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milord F.; Pépin J.; Ethier L.; Milord F.; Loko L.; Ethier L.; Mpia B. Efficacy and Toxicity of Eflornithine for Treatment of Trypanosoma Brucei Gambiense Sleeping Sickness. Lancet 1992, 340, 652–655. 10.1016/0140-6736(92)92180-n. [DOI] [PubMed] [Google Scholar]
- Poulin R.; Lu L.; Ackermann B.; Bey P.; Pegg A. E. Mechanism of the Irreversible Inactivation of Mouse Ornithine Decarboxylase by Alpha-Difluoromethylornithine. Characterization of Sequences at the Inhibitor and Coenzyme Binding Sites. J. Biol. Chem. 1992, 267, 150–158. 10.1016/s0021-9258(18)48472-4. [DOI] [PubMed] [Google Scholar]
- Mons E.; Jansen I. D. C.; Loboda J.; van Doodewaerd B. R.; Hermans J.; Verdoes M.; van Boeckel C. A. A.; van Veelen P. A.; Turk B.; Turk D.; Ovaa H. The Alkyne Moiety as a Latent Electrophile in Irreversible Covalent Small Molecule Inhibitors of Cathepsin K. J. Am. Chem. Soc. 2019, 141, 3507–3514. 10.1021/jacs.8b11027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortenson D. E.; Brighty G. J.; Plate L.; Bare G.; Chen W.; Li S.; Wang H.; Cravatt B. F.; Forli S.; Powers E. T.; Sharpless K. B.; Wilson I. A.; Kelly J. W. “Inverse Drug Discovery” Strategy To Identify Proteins That Are Targeted by Latent Electrophiles As Exemplified by Aryl Fluorosulfates. J. Am. Chem. Soc. 2018, 140, 200–210. 10.1021/jacs.7b08366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekkebus R.; van Kasteren S. I.; Kulathu Y.; Scholten A.; Berlin I.; Geurink P. P.; de Jong A.; Goerdayal S.; Neefjes J.; Heck A. J. R.; Komander D.; Ovaa H. On Terminal Alkynes That Can React with Active-Site Cysteine Nucleophiles in Proteases. J. Am. Chem. Soc. 2013, 135, 2867–2870. 10.1021/ja309802n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Lin D.; Sun X.; Curth U.; Drosten C.; Sauerhering L.; Becker S.; Rox K.; Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewitz L.; Dumjahn L.; Zhao Y.; Owen C. D.; Laidlaw S. M.; Malla T. R.; Nguyen D.; Lukacik P.; Salah E.; Crawshaw A. D.; Warren A. J.; Trincao J.; Strain-Damerell C.; Carroll M. W.; Walsh M. A.; Schofield C. J. Alkyne Derivatives of SARS-CoV-2 Main Protease Inhibitors Including Nirmatrelvir Inhibit by Reacting Covalently with the Nucleophilic Cysteine. J. Med. Chem. 2023, 66, 2663–2680. 10.1021/acs.jmedchem.2c01627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Sacco M. D.; Hurst B.; Townsend J. A.; Hu Y.; Szeto T.; Zhang X.; Tarbet B.; Marty M. T.; Chen Y.; Wang J. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddick J. J.; Cheng J.; Roush W. R. Relative Rates of Michael Reactions of 2′-(Phenethyl)Thiol with Vinyl Sulfones, Vinyl Sulfonate Esters, and Vinyl Sulfonamides Relevant to Vinyl Sulfonyl Cysteine Protease Inhibitors. Org. Lett. 2003, 5, 1967–1970. 10.1021/ol034555l. [DOI] [PubMed] [Google Scholar]
- Chatani S.; Nair D. P.; Bowman C. N. Relative Reactivity and Selectivity of Vinyl Sulfones and Acrylates towards the Thiol–Michael Addition Reaction and Polymerization. Polym. Chem. 2013, 4, 1048–1055. 10.1039/c2py20826a. [DOI] [Google Scholar]
- Devaraj N. K. The Future of Bioorthogonal Chemistry. ACS Cent. Sci. 2018, 4, 952–959. 10.1021/acscentsci.8b00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright M. H.; Sieber S. A. Chemical Proteomics Approaches for Identifying the Cellular Targets of Natural Products. Nat. Prod. Rep. 2016, 33, 681–708. 10.1039/c6np00001k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer S.; Weikart N. D.; Linne U.; Mootz H. D. Covalent Inhibition of SUMO and Ubiquitin-Specific Cysteine Proteases by an in Situ Thiol–Alkyne Addition. Bioorg. Med. Chem. 2013, 21, 2511–2517. 10.1016/j.bmc.2013.02.039. [DOI] [PubMed] [Google Scholar]
- Mons E.; Kim R. Q.; van Doodewaerd B. R.; van Veelen P. A.; Mulder M. P. C.; Ovaa H. Exploring the Versatility of the Covalent Thiol–Alkyne Reaction with Substituted Propargyl Warheads: A Deciding Role for the Cysteine Protease. J. Am. Chem. Soc. 2021, 143, 6423–6433. 10.1021/jacs.0c10513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragovich P. S.; Prins T. J.; Zhou R.; Webber S. E.; Marakovits J. T.; Fuhrman S. A.; Patick A. K.; Matthews D. A.; Lee C. A.; Ford C. E.; Burke B. J.; Rejto P. A.; Hendrickson T. F.; Tuntland T.; Brown E. L.; Meador J. W.; Ferre R. A.; Harr J. E. V.; Kosa M. B.; Worland S. T. Structure-Based Design, Synthesis, and Biological Evaluation of Irreversible Human Rhinovirus 3C Protease Inhibitors. 4. Incorporation of P 1 Lactam Moieties as l -Glutamine Replacements. J. Med. Chem. 1999, 42, 1213–1224. 10.1021/jm9805384. [DOI] [PubMed] [Google Scholar]
- Matthews D. A.; Dragovich P. S.; Webber S. E.; Fuhrman S. A.; Patick A. K.; Zalman L. S.; Hendrickson T. F.; Love R. A.; Prins T. J.; Marakovits J. T.; Zhou R.; Tikhe J.; Ford C. E.; Meador J. W.; Ferre R. A.; Brown E. L.; Binford S. L.; Brothers M. A.; DeLisle D. M.; Worland S. T. Structure-Assisted Design of Mechanism-Based Irreversible Inhibitors of Human Rhinovirus 3C Protease with Potent Antiviral Activity against Multiple Rhinovirus Serotypes. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 11000–11007. 10.1073/pnas.96.20.11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J.; George S.; Kusov Y.; Perbandt M.; Anemüller S.; Mesters J. R.; Norder H.; Coutard B.; Lacroix C.; Leyssen P.; Neyts J.; Hilgenfeld R. 3C Protease of Enterovirus 68: Structure-Based Design of Michael Acceptor Inhibitors and Their Broad-Spectrum Antiviral Effects against Picornaviruses. J. Virol. 2013, 87, 4339–4351. 10.1128/jvi.01123-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D. R.; Allerton C. M. N.; Anderson A. S.; Aschenbrenner L.; Avery M.; Berritt S.; Boras B.; Cardin R. D.; Carlo A.; Coffman K. J.; Dantonio A.; Di L.; Eng H.; Ferre R.; Gajiwala K. S.; Gibson S. A.; Greasley S. E.; Hurst B. L.; Kadar E. P.; Kalgutkar A. S.; Lee J. C.; Lee J.; Liu W.; Mason S. W.; Noell S.; Novak J. J.; Obach R. S.; Ogilvie K.; Patel N. C.; Pettersson M.; Rai D. K.; Reese M. R.; Sammons M. F.; Sathish J. G.; Singh R. S. P.; Steppan C. M.; Stewart A. E.; Tuttle J. B.; Updyke L.; Verhoest P. R.; Wei L.; Yang Q.; Zhu Y. An Oral SARS-CoV-2 M pro Inhibitor Clinical Candidate for the Treatment of COVID-19. Science 2021, 374, 1586–1593. 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- Baillie T. A. Targeted Covalent Inhibitors for Drug Design. Angew. Chem., Int. Ed. 2016, 55, 13408–13421. 10.1002/anie.201601091. [DOI] [PubMed] [Google Scholar]
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. The Resurgence of Covalent Drugs. Nat. Rev. Drug Discovery 2011, 10, 307–317. 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Aliyari S. R.; Ghaffari A. A.; Pernet O.; Parvatiyar K.; Wang Y.; Gerami H.; Tong A.-J.; Vergnes L.; Takallou A.; Zhang A.; Wei X.; Chilin L. D.; Wu Y.; Semenkovich C. F.; Reue K.; Smale S. T.; Lee B.; Cheng G. Suppressing Fatty Acid Synthase by Type I Interferon and Chemical Inhibitors as a Broad Spectrum Anti-Viral Strategy against SARS-CoV-2. Acta Pharm. Sin. B 2022, 12, 1624–1635. 10.1016/j.apsb.2022.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musharrafieh R.; Ma C.; Zhang J.; Hu Y.; Diesing J. M.; Marty M. T.; Wang J. Validating Enterovirus D68-2A pro as an Antiviral Drug Target and the Discovery of Telaprevir as a Potent D68-2A pro Inhibitor. J. Virol. 2019, 93, e022211-18 10.1128/jvi.02221-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison J. F.; Walsh C. T.. The Behavior and Significance of Slow-Binding Enzyme Inhibitors. In Advances in Enzymology and Related Areas of Molecular Biology; Meister A., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; pp 201–301. [DOI] [PubMed] [Google Scholar]
- Xie X.; Muruato A.; Lokugamage K. G.; Narayanan K.; Zhang X.; Zou J.; Liu J.; Schindewolf C.; Bopp N. E.; Aguilar P. V.; Plante K. S.; Weaver S. C.; Makino S.; LeDuc J. W.; Menachery V. D.; Shi P.-Y. An Infectious CDNA Clone of SARS-CoV-2. Cell Host Microbe 2020, 27, 841–848.e3. 10.1016/j.chom.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.