

Summary
Proteinaceous cysteines function as essential sensors of cellular redox state. Consequently, defining the cysteine redoxome is a key challenge for functional proteomic studies. While proteome-wide inventories of cysteine oxidation state are readily achieved using established, widely adopted proteomic methods such as OxICAT, Biotin Switch, and SP3-Rox, these methods typically assay bulk proteomes and therefore fail to capture protein localization-dependent oxidative modifications. Here we establish the Local Cysteine Capture (Cys-LoC), and Local Cysteine Oxidation (Cys-LOx) methods, which together yield compartment-specific cysteine capture and quantitation of cysteine oxidation state. Benchmarking of the Cys-LoC method across a panel of subcellular compartments revealed more than 3,500 cysteines not previously captured by whole cell proteomic analysis. Application of the Cys-LOx method to LPS-stimulated immortalized murine bone marrow-derived macrophages (iBMDM), revealed previously unidentified, mitochondrially localized cysteine oxidative modifications upon pro-inflammatory activation, including those associated with oxidative mitochondrial metabolism.
Keywords: cysteine, chemoproteomics, proximity labeling, TurboID, cysteine oxidation, LPS, mitochondria, macrophages
Graphical Abstract
eTOC
Yan et al. report the Cys-LoC and Cys-LOx methods, which enable quantitative subcellular cysteine chemoproteomics, including identification of mitochondrial cysteines sensitive to LPS+IFNγ-induced oxidative stress.
Introduction
Distinguished by their sensitivity to oxidative stress, proteinaceous cysteine residues play important roles in physiological and pathophysiological processes1,3,5,7–9. Abnormal levels of reactive oxygen and nitrogen species (ROS and RNS) have been implicated in a number of human diseases, including neurological disorders, cancers, and autoimmune disorders10–13. Cysteine chemoproteomic methods, such as biotin-switch14, OxICAT15, SP3-Rox16, QTRP17 and Oximouse18, enable high throughput quantitation of changes to cysteine oxidation states. Application of these methods have pinpointed cysteines differentially oxidized in association with high levels of ROS and RNS, such as those of TRX19,20, GAPDH21,22 and HBB23. Given the recent advent of cysteine-reactive small molecules as precision therapies for the treatments of cancers and immune disorders24–26, cysteine chemoproteomic methods have also emerged as enabling technology for pinpointing ligandable or potentially ‘druggable’ residues proteome-wide27–36. A central remaining challenge for these studies is the lack of a priori knowledge about the functional impact of covalent modification. Given the functional importance of cysteine oxidative modifications, understanding which cysteines serve as endogenous redox sensors is also of high utility for target prioritization efforts.
Nearly all cysteine redox profiling platforms follow the same general workflow: First, cells are lysed, and the reduced cysteines are capped with a pan-cysteine reactive reagent, such as iodoacetamide alkyne (IAA). After reduction, natively oxidized cysteines are then capped by another cysteine capping reagent, such as isotopically differentiated IAA. Samples are then biotinylated, enriched on avidin resin, subjected to sequence specific proteolysis, and liquid chromatography tandem mass spectrometry analysis (LC-MS/MS). Both absolute and relative changes to cysteine oxidation can be quantified either using precursor ion intensity (MS1-level)27,28,37 or fragment ion intensity (MS2 level)18,38. While such studies provide a global snapshot of cysteine states, they fail to capture subcellular, compartment-specific changes in cysteine oxidation state— and such differences are to be expected given the established spectrum of organelle redox potentials39,40. Notably, by combining OxICAT with biochemical fractionation, recent studies have quantified cysteine oxidation for mitochondrial and endoplasmic reticulum localized proteins4,6. Such studies remain limited by the technical inaccessibility of density gradient centrifugation to many groups, the propensity of cysteines to oxidize during the harsh and prolonged isolation procedure, and incompatibility with membraneless organelles and other compartments for which subcellular fractionation is not feasible.
The emergence of proximity labeling techniques, including APEX41, BioID42, and TurboID43, has enabled high fidelity biotinylation and enrichment of proteins from a range of subcellular compartments, including the cytosol, nucleus, mitochondria, endoplasmic reticulum membrane and endoplasmic reticulum lumen. With the addition of exogenous biotin (or related biotin analogues), biotinylation occurs with spatiotemporal control inside the targeted organelle. Pioneering studies have demonstrated the utility of these proximity based labeling methods in deciphering the protein interactome44,45, the protein composition of membraneless organelles46,47, mapping localization for unannotated proteins48 and interrogation of kinase substrates49. Whether these methods are compatible with capturing the subcellular redoxome remains to be seen.
Across all organelles, the mitochondrial cysteine redoxome is particularly intriguing. In addition to carrying out oxidative phosphorylation (OXPHOS), mitochondria also play key roles in nearly all aspects of cell physiology, including functioning as hubs for biosynthesis, Ca2+ handling, iron homeostasis, and signal transduction50,51. Additionally, mitochondria are also thought to be significant producers of reactive oxygen species (ROS)52, and ROS-sensitive cysteines are known to regulate mitochondrial proteins such as aconitase and respiratory complexes I and III53–55. Mitochondrial ROS is also an emerging hallmark of the innate immune response56,57. For example, in response to lipopolysaccharide (LPS), murine bone marrow-derived macrophages (BMDMs) adopt a pro-inflammatory program which includes profound increases in ROS and RNS levels, due in part to high expression of inducible nitric oxide synthase (iNOS)58. This nitric oxide production is also primarily responsible for the near-total collapse of mitochondrial oxidative phosphorylation in pro-inflammatory macrophages59–61. Chemoproteomic studies have been performed to investigate this pro-inflammatory process62–64. However, the extent to which specific mitochondrial cysteines are oxidized as a result of this mitochondrial reprogramming remains largely unknown.
Here we combine enzymatic (TurboID) proximity-based biotinylation with cysteine redox state analysis to enable in situ subcellular cysteine fractionation and quantitative measures of cysteine oxidation state. We first established the Local Cysteine Capture (Cys-LoC) method, which, when applied to cells expressing TurboID localized to cytosol, endoplasmic reticulum (ER), mitochondria (Mito), golgi, and nucleus, identified >3,500 cysteines not previously captured by whole cell proteomic analysis31. On average, 500 cysteines were captured from each compartment that were not enriched from HEK293T whole cell lysates. Unexpectedly, we observed low subcellular specificity for constructs targeted to a subset of compartments, which we mitigated through simultaneous depletion of endogenous biotin and translation arrest-induced depletion of newly translated TurboID. By combining these two innovations with our SP3 single-pot, solid-phase-enhanced redox sample preparation workflow (SP3-Rox) method16,65, we then established the Local Cysteine Oxidation (Cys-LOx) method. When applied to identify LPS-sensitive cysteines in an immortalized bone marrow derived murine macrophage (iBMDM) cell line, we identified 32 mitochondria-specific cysteines that exhibited cell-state dependent oxidation, including residues in proteins important for bioenergetics, associated with oxidative phosphorylation, and those not captured using bulk SP3-Rox analysis.
Results
Establishing the Local Cysteine Capture (Cys-LoC) method accesses the subcellular cysteineome.
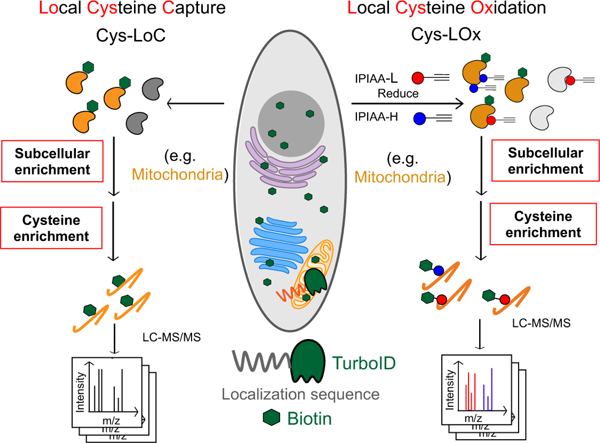
Here we envisioned combining proximity labeling via the ultra-fast biotin ligase TurboID with cysteine chemoproteomics16,27,67,30–36,66 to enable fractionation-free capture of the subcellular cysteinome, for both residue identification and quantification of cysteine oxidation. We were inspired by recent reports of two-step capture for subcellular phosphoproteomics, in which proteins biotinylated by TurboID were first enriched on avidin resin followed by peptide-level capture of phosphopeptides68. As a first step to test the feasibility of an analogous two step enrichment method for cysteine chemoproteomics, we transiently overexpressed a panel of TurboID fusion proteins tagged with localization sequences targeted to cytosol (cyto.), endoplasmic reticulum (ER), golgi, mitochondrial (mito.), and nucleus (nuc.) (Figure S1A)43,69. We then combined expression of these constructs with a customized two-step enrichment strategy, termed Local Cysteine Capture (Cys-LoC) (Figure 1A). In Cys-LoC, TurboID proximal proteins are first biotinylated in situ. Following lysis and cysteine capping with the highly reactive iodoacetamide alkyne (IAA), biotinylated proteins are enriched on streptavidin resin and subjected to sequence specific proteolysis. This digest releases all IAA-tagged peptides derived from the TurboID-modified proteins. Subsequent peptide-level click conjugation to biotin-azide followed by a second enrichment on neutravidin resin affords specific capture of biotinylated cysteine peptides derived from turboID-modified proteins. Demonstrating the utility of the Cys-LoC method, we found that coverage of cysteines substantially increased with two-step biotinylation based Cys-LoC (Figure 1A) compared to one-step TurboID (Figure S1B–C), as detected both in our study in the report by Kisty and coworkers (Figure S1D)2. Furthermore, Cys-LoC showed both high overlap in cysteines identified together with enhanced coverage when compared with subcellular cysteine enrichment with differential centrifugation based organelle fractionation (Figure S1E–F)4,6.
Figure 1. Establishment of Local Cysteine Capture (Cys-LoC) method.
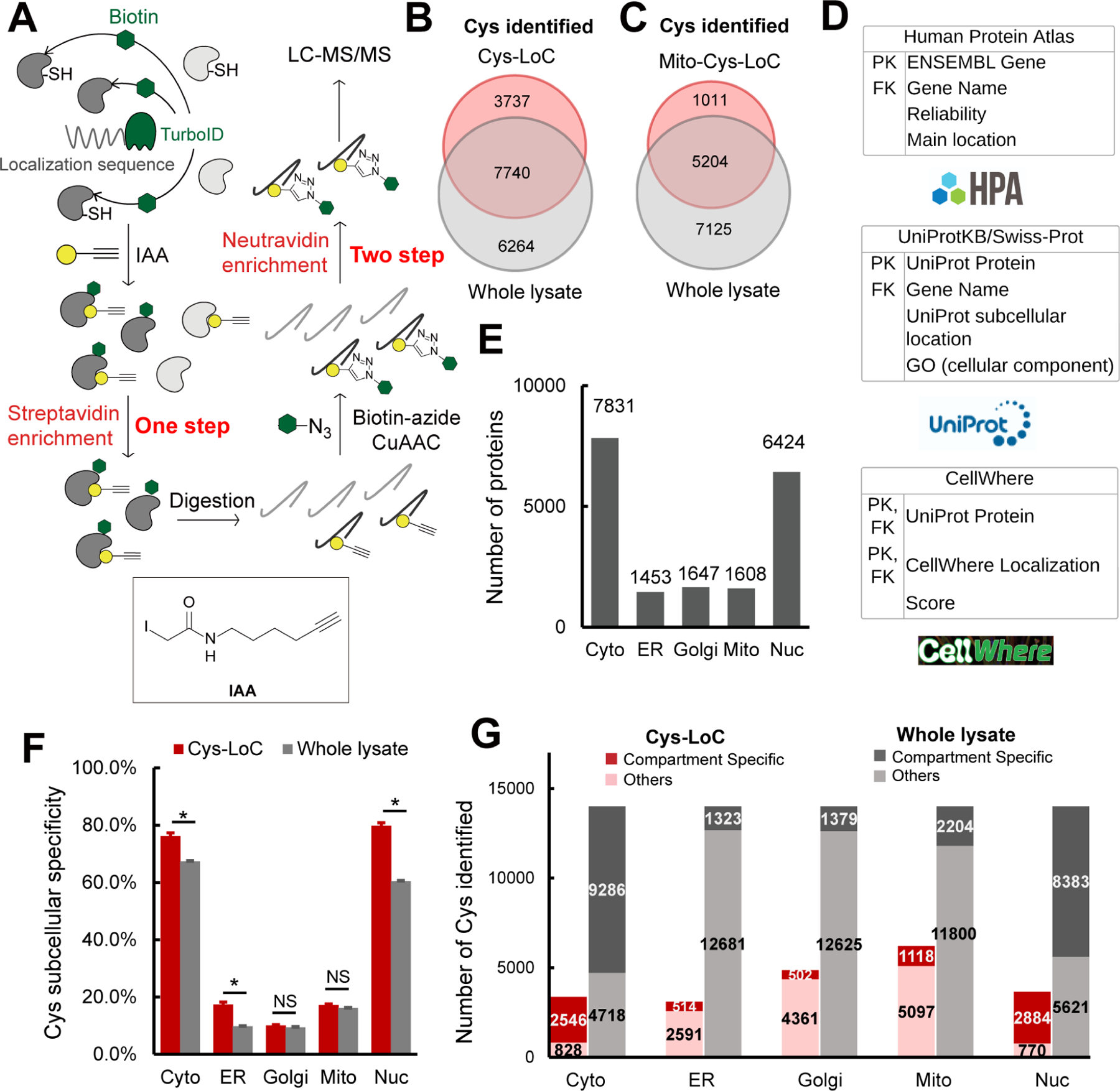
A) Scheme of Cys-LoC Workflow. B) Cysteines identified with Cys-LoC from five compartments aggregated compared to those identified from whole proteome31. C) Cysteines identified with Mito-Cys-LoC compared to those identified from the whole proteome31. D) Scheme of database generation with aggregated protein localization annotations from Human Protein Atlas, UniprotKB and CellWhere. PK and FK represented primary key and foreign key, respectively. E) Number of proteins annotated as localized in cytosol (cyto), endoplasmic reticulum (ER), golgi, mitochondria (mito), and nucleus (nuc). F) Subcellular specificity of cysteines identified with Cys-LoC or from whole lysate31. G) Number of cysteines identified as compartment specific with Cys-LoC or fromwhole lysate31. Statistical significance was calculated with unpaired Student’s t-tests, * p<0.05, NS p>0.05. Experiments were performed in duplicate for B, C, F, G. Data in panel F are represented as mean ± stdev. See also Data S1 and Figure S1.
Implementation of the Cys-LoC method for constructs targeted to all five aforementioned compartments identified in aggregate 11,478 total cysteines, with an average of 3,700 cysteines per construct (Figure 1B). Gratifyingly, more than 450 cysteines were identified from each compartment that had not been previously captured in our previous bulk cysteinome analysis of HEK293T using our SP3-high field asymmetric waveform ion mobility spectrometry (SP3-FAIMS) method31 (Figure S1G–J). Further exemplifying the utility of the Cys-LoC method to capture novel cysteines, for the mitochondrial targeted construct, 1,011 cysteines were identified that were not previously captured by our prior study31 (Figure 1C). When cross referenced with our newly reported CysDB database of 62,888 total identified cysteines, 489 cysteines identified by Cys-LoC had not been previously reported by any of our panel of high coverage cysteine chemoproteomics studies30.
Evaluating the subcellular specificity of the cysteines captured by Cys-LoC
Motivated by the observed expanded portrait of the cysteinome enabled by Cys-LoC, we next asked whether the cysteine-containing proteins captured were representative of the subcellular compartments to which the respective TurboID constructs were targeted. To facilitate the analysis of subcellular proteomes, we generated a comprehensive protein localization database by aggregating protein localization information from the Human Protein Atlas70, UniprotKB71 and CellWhere72 (Data S1). Of the 16,983 proteins with available localization information, 12,835 human proteins were annotated as localized in the cytosol, ER, golgi, mitochondria and nucleus (Figure 1D). 7,831, 1,453, 1,647, 1,608, and 6,424 proteins were annotated as localized in the cytosol, ER, golgi, mitochondria and nucleus, respectively (Figure 1E).
Stratification of our Cys-LoC dataset by TurboID subcellular localization revealed several striking features. For the constructs targeted to the cytosol and nucleus, we observed comparatively high (~80%) localization specificity, calculated as the percentage of cysteines identified with protein localization annotations matching the compartment targeted by the respective TurboID (Figure 1F, red bars). However, when this analysis was extended to the ER, mito and golgi datasets, the specificity dropped dramatically (<20%). While some variability in compartment-specific proximity labeling has been reported previously43, the scale of the difference between compartments was unexpectedly small.
We next interrogated how the compartment specificity achieved by Cys-LoC compared to datasets generated from unfractionated HEK293T whole cell proteome30,31. We observed modest, yet statistically significant, enrichment for cysteines captured using the Cyto-, Nuc- and ER-Cys-LoC platforms. In contrast, no significant enrichment was observed for the Golgi and Mito-targeted constructs (Figure 1F). Extension of this analysis to consider the total number of cysteines identified revealed a marked decrease in coverage for all constructs assayed via Cys-LoC when compared with whole lysate analysis. For example, cysteine chemoproteomics analysis of whole cell lysate identifies 2,000 mitochondria localized cysteines. The Mito-Cys-LoC platform decreases the number of background cysteines (from 11,800 to 5,097) alongside the number of mitochondria cysteines (from 2,204 to 1,118) (Figure 1G). Nonetheless, 3,737 cysteines were identified by the Cys-LoC platform that had not been identified in our prior SP3-FAIMS analysis of matched HEK293T proteome, including C588 of presequence protease (PITRM1)73, a protease responsible for clearance of accumulated mitochondrial amyloid beta protein, as well as zinc-coordinating cysteine C256 of DnaJ homolog subfamily A member 3 (DNAJ3), a protein regulator of apoptotic signaling in cancer74 (Figure 1B, 1C, S1G–J). These examples highlight the utility of Cys-LoC enabled in situ subcellular fractionation for uncovering novel reactive cysteines.
Investigating established parameters associated with TurboID performance
For most proximity-labeling studies, some background labeling can be accommodated with appropriate controls (e.g. +/− treatment groups). In contrast, implementation of proximity labeling to measure compartment specific changes to the cysteinome, for example for cysteine oxidation, requires comparatively high compartment labeling specificity. This specificity requirement can be rationalized by the following hypothetical cysteine: for a cysteine that is heavily oxidized in the mitochondria but not in the cytoplasm, proximity labeling that captures both subsets of the protein would incorrectly report the average oxidation state across both compartments. With the goal of minimizing non-specific cysteine enrichment, we sought to first pinpoint, and then address, sources of the observed seemingly promiscuous proximity labeling.
To streamline our efforts at method optimization, we established a protein-level proximity labeling workflow (Figure 2A) in which TurboID specificity was assayed by the fraction of total proteins identified in which the localization matched that of the TurboID fusion protein. Using this platform paired with transient overexpression we observed comparable protein localization specificity to that achieved with the Cys-LoC method (Figure 2B and Figure 1E), indicating that the peptide-level enrichment analysis in the Cys-LoC workflow was not a significant contributor to the low specificity.
Figure 2. Pinpointing sources of non-compartment specific TurboID biotinylation.
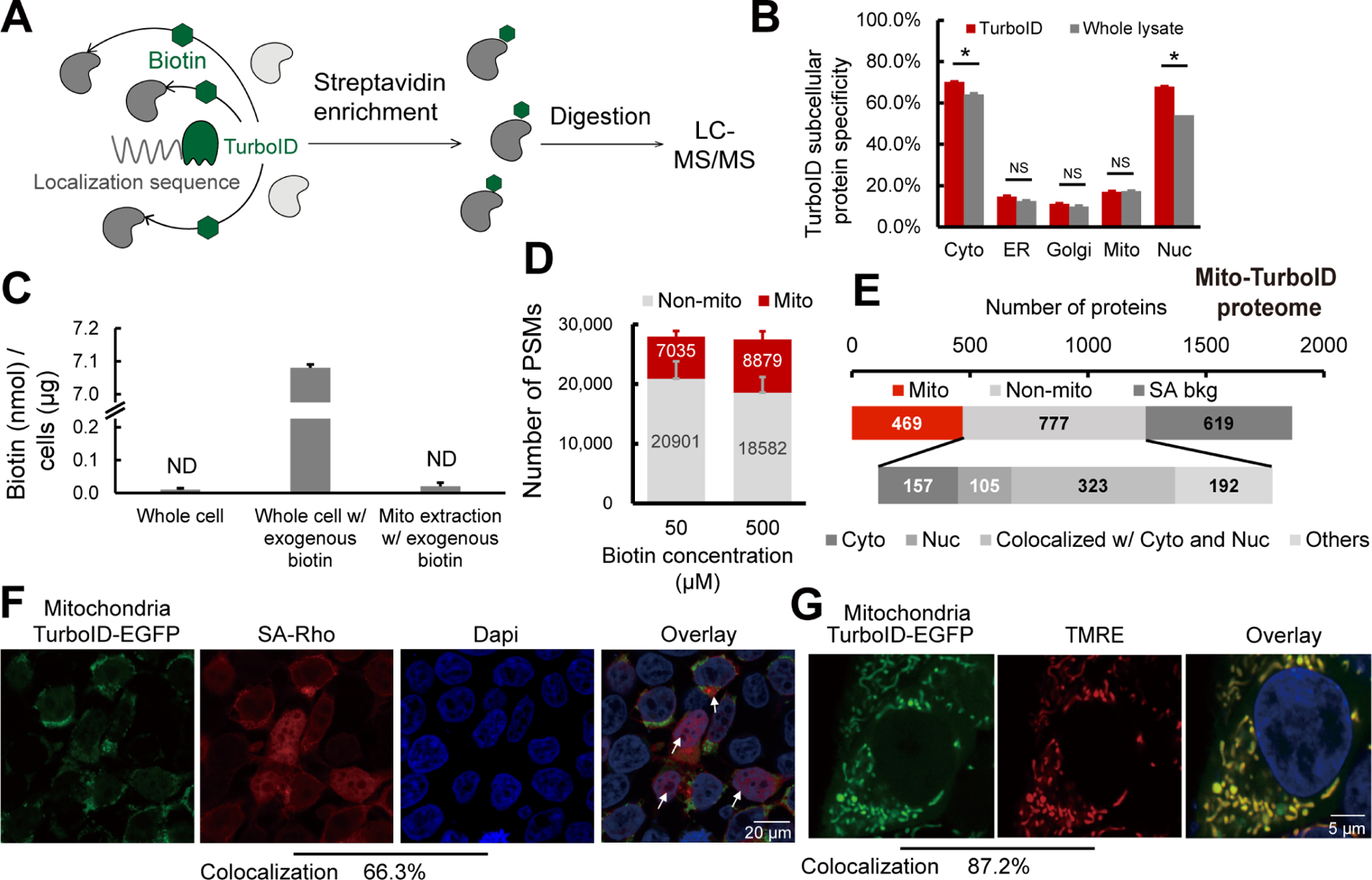
A) Scheme of TurboID based proximity labeling protein enrichment. B) Subcellular specificity of proteins identified with TurboID or with whole lysate. C) Absolute quantification of biotin with or without 500 µM exogenous biotin addition. D) Number of PSMs for proteins identified with mito-TurboID with 50 µM or 500 µM exogenous biotin. E) Distribution of proteins identified with mito-TurboID. SA-bkg: streptavidin background proteome. F) Localization of biotinylation in cells with mito-TurboID indicated by signals of streptavidin-rhodamine (SA-Rho). G) Localization of mito-TurboID overlaid with TMRE. Statistical significance was calculated with unpaired Student’s t-tests, * p<0.05, NS p>0.05. Experiments were performed in duplicates for panels B, C, D, and E. Data in panel B, C, D are represented as mean ± stdev. See also Data S2 and Figure S2.
We then investigated whether our protein localization database might provide insights into the promiscuous proximity labeling. Within our protein localization database, we found that there are more than 6,000 proteins with multi-localization annotations (Figure S2A), with more than 70% of the proteins in each organelle annotated as multi-localized (Figure S2B). Removal of these multi-localized proteins from our datasets did not improve the compartment specificity for cysteines identified by Cys-LoC (Figure S2C–D). As this post-acquisition data filtration did not improve Cys-LoC performance, we turned to our experimental workflow, seeking to increase specificity through methodological optimization.
As one of our primary goals in establishing the Cys-LoC method was to enable streamlined mitochondrial redox proteomics, we opted to use our mitochondrially targeted construct to perform further in-depth analysis of our protocol. Consistent with the previous study75, we found that stable expression of the mito-TurboID compared to transient expression, afforded an increase in mitochondrial protein specificity (22% vs 19%) with only a modest decrease in net proteins identified (571 vs 580, Figure S2E).
Given that previous reports indicated comparatively lower specificity for TurboID-catalyzed labeling of mitochondria43, we hypothesized that low local biotin concentration might contribute to decreased specificity for mitochondrial proteins. While mitochondrial biotin uptake has been suggested to occur through passive diffusion, the prior report of pH-dependent uptake is suggestive of saturable mitochondrial biotin levels76. We find that, after a pulse with 500 μM exogenous biotin, the absolute detectable levels of biotin in the cytoplasm rises rapidly, reaching 7.08 nmol/μg cells. In contrast, biotin remained below the limit of detection in crude mitochondrial extracts (Figure 2C and Figure S2F–G). While TurboID has been shown to proceed efficiently at low biotin concentrations (50 μM), due to its increased affinity for biotin (relative to BioID)43,75, our findings pointed towards the possible requirement for increased biotin concentrations to achieve efficient labeling of mitochondrial proteins. Supporting this premise, comparison of 50 μM and 500 μM biotin revealed increased peptide spectrum matches (PSMs) for mitochondrial proteins with elevated biotin concentrations (Figure 2D).
Given TurboID’s enhanced labeling kinetics relative to BioID43, we investigated labeling time and observed that an improved balance of coverage and specificity could be achieved with 1h labeling time, when compared to 10 min or 3h (Figure S2H). As decreased biotinylation was observed in higher passage (>10) cell lines (Figure S2I), we restricted subsequent analyses to cell lines with <10 passages.
Taken together, implementation of stable expression of the TurboID fusion protein, 1h labeling time and 500 μM biotin increased mito-TurboID specificity to 27% (449/1690).
Promiscuous Mito-TurboID labeling of cytosolic and nuclear proteins
To further understand the factors contributing to our still-marginal compartment specificity, we asked whether insights could be garnered by analyzing the annotated localization of enriched proteins. To eliminate the possibility of non-specific streptavidin binding confounding our analysis, we generated a dataset (Data S2 and methods for details) that defines the HEK293T streptavidin background proteome. Excluding the streptavidin background, we find that >75% (585/777) of the non-mitochondrial proteins enriched by mito-TurboID are annotated as localized in the nucleus or cytosol (Figure 2E). Examples of these non-mitochondrial proteins captured by the mitochondrial-targeted TurboID include histones H2B, H1and H3, ribosomal protein S6 kinase RPS6KA3, eukaryotic translation initiation factors EIF3M, EIF2D, and ELF6. All these proteins are closely related to protein translation, suggestive of proximity labeling by newly translated, yet unlocalized TurboID-fusion proteins. The same trend also extended to the site of labeling analysis (Figure S2J), in which 66% (4,934/7,449) of the TurboID biotinylation sites stemmed from proteins with nuclear localization (Figure S2K–L). This marked labeling of nuclear proteins was also visualized by immunocytochemistry (ICC) in which a substantial accumulation of streptavidin labeling was observed in cellular nuclei (66.3% colocalization with DAPI), indicated by the white arrows (Figure 2F). By comparison, the mitochondrial localization of the Mito-TurboID-EGFP was observed to be high, as indicated by 87.2 % colocalization with the mitochondrially localized dye tetramethylrhodamine ethyl ester (TMRE) (Figure 2G).
Newly translated TurboID is a major cause of promiscuous biotinylation, which can be improved through translation arrest and depletion of endogenous biotin.
Inspired by the modest, yet detectable signal for mito-TurboID detected by ICC outside of the mitochondria (Figure 2F), in addition to the fact that many non-mitochondrial proteins captured by mito-TurboID were closely related to protein translation, we postulated that this trace signal might stem from newly translated protein. To test this hypothesis, we performed subcellular fractionation to quantify the fraction of mito-TurboID localized to mitochondrial versus non-mitochondrial compartments. Roughly 30% of the total mito-TurboID was observed to be localized outside of the mitochondria (Figure 3A). The non-mitochondrial fraction of mito-TurboID protein was nearly completely eliminated in cells subjected to translation arrest (Cycloheximide77, 100 ug/mL, 6h; Figure 3B, 3C), which further implicates newly translated TurboID protein as the likely source of the observed low subcellular labeling specificity. Further supporting this model, we observed a comparatively long (>24h) half-life for the mito-TurboID protein (Figure S3A). Proteomic analysis of CHX-treated cells revealed an increase in the mitochondrial protein specificity to 30% of all proteins enriched (335/1120). Consistent with the CHX treatment primarily impacting newly translated rather than mitochondrial localized proteins, we only observed a modest decrease in total mitochondrial proteins identified (<10%), (Figure 3D). The impact of cycloheximide treatment was also detectable by streptavidin blot, where several bands showed a pronounced decrease is staining upon cycloheximide treatment (Figure S3B, * bands).
Figure 3. Translation arrest improves the subcellular specificity of proteins enriched by TurboID and cysteines captured by Cys-LoC.
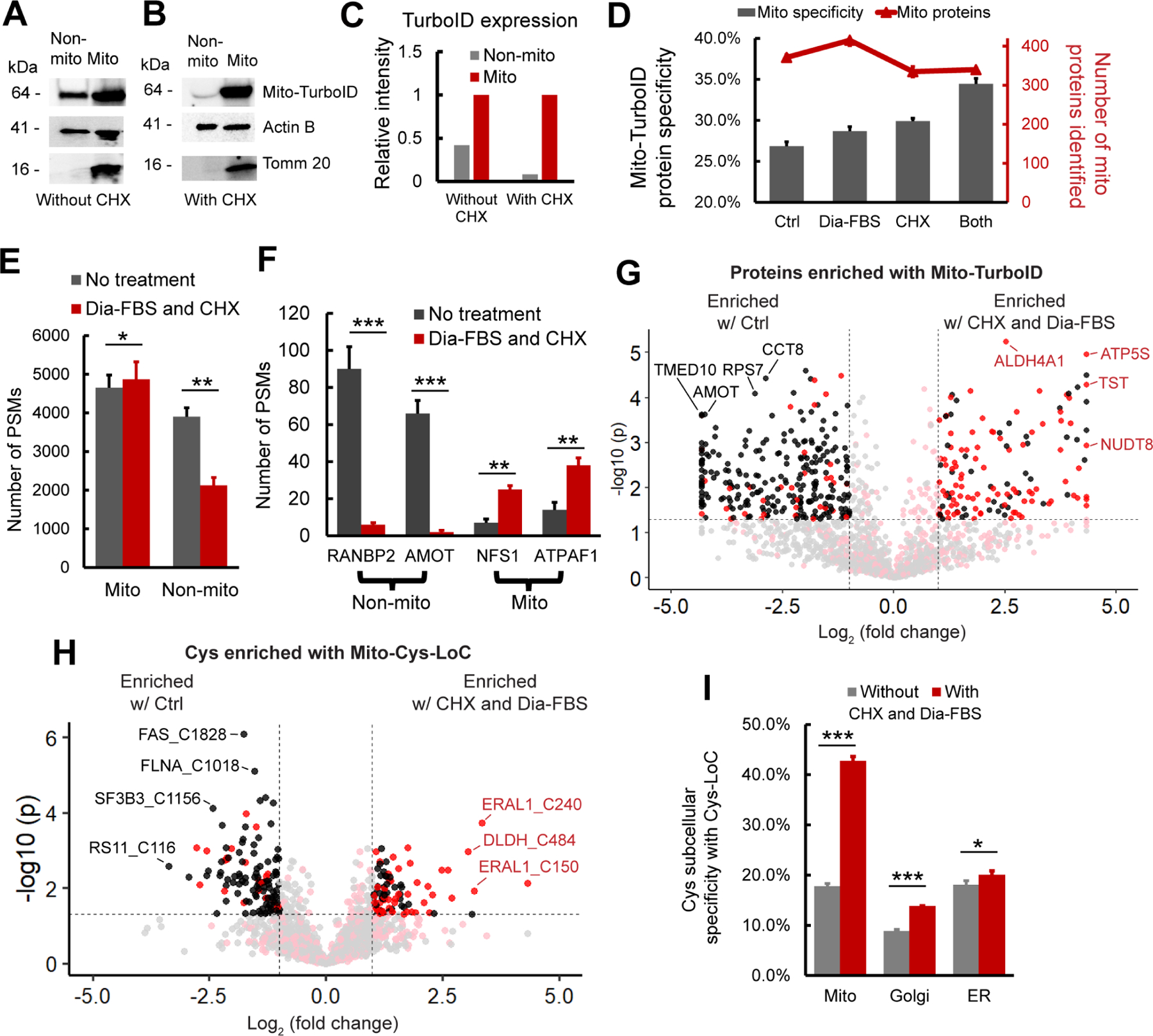
A-C) Abundance of mito-TurboID in mitochondrial vs non-mitochondrial fractions. A, without and B, with CHX treatment as quantified in C. D) Specificity and number of mitochondria localized proteins enriched with mito-TurboID . E) Distribution of PSMs for proteins enriched with mito-TurboID ± dialyzed FBS and CHX. F) PSMs of representative mitochondrial and non-mitochondrial proteins enriched with mito-TurboID ± dialyzed FBS and CHX. G) Intensity differences for proteins enriched with mito-TurboID ± dialyzed FBS and CHX. H) Intensity differences for cysteines enriched with Mito-Cys-LoC. I) Cys-LoC cysteine specificity ± dialyzed FBS and CHX. Statistical significance was calculated with unpaired Student’s t-tests, * p<0.05, ** p<0.01, *** p<0.005. NS p>0.05. Experiments were performed in triplicate for panels D, E, F, G, H, and I. Data in panel D, E, F, and I are represented as mean ± stdev. For G, and H Red: mitochondrial cysteines. Black: non-mitochondrial cysteines. See also Data S3 and Figure S3.
Streptavidin blot analysis additionally indicated the presence of CHX-insensitive bands in the control samples lacking exogenous biotin (Figure S3B). Prior reports have implicated endogenous biotin as a substrate of TurboID and a source of background labeling, and biotin-free dialyzed serum was recently found to decrease this low-level labeling75,78. Consistent with these findings, we observed that proximity labeling using dialyzed fetal bovine serum (Dia-FBS) afforded a modest but significant increase in mitochondrial protein specificity (27% to 29%) together with slight increase in overall mitochondrial proteins identified (415/1448; Figure 3D). More striking, when the dialyzed FBS and CHX treatments were combined nearly 35% of all proteins identified were mitochondrial, and no further decrease in protein coverage (340/987) was observed compared to CHX treatment alone. These findings were further substantiated by streptavidin blot visualization of decreased signal for CHX- and Dia-FBS-sensitive bands both in the presence and absence of exogenous biotin (Figure S3B, * bands).
Stratification of identified PSMs, rather than proteins, by annotated protein localization, more substantially revealed the impact of the combined CHX and Dia-FBS treatment. Excluding PSMs derived from background streptavidin binding, we find that the CHX and Dia-FBS treatment affords a 45% decrease in non-mitochondrial PSMs, from 3898 to 2125 PSMs. In contrast, the mitochondrial PSMs showed a modest increase from 4650 to 4868 with the CHX and Dia-FBS conditions (Figure 3E and S3C). Further exemplifying the impact of the combined translation arrest and biotin treatment, the number of PSMs for some of the most substantially enriched nuclear and cytoplasmic proteins, including angiomotin AMOT, a component of the 40S ribosomal subunit involved in translational repression79 and E3 SUMO-protein ligase RANBP2, which facilitates SUMO1 and SUMO2 conjugation80, decreased by 20-fold, whereas exemplary mitochondrial proteins ATP synthase mitochondrial F1 complex assembly factor 1 ATPAF1, which supports oxidative phosphorylation81 and cysteine desulfurase NFS1, which catalyzes the desulfuration of L-cysteine to L-alanine82, remained unaffected by the treatment (Figure 3F and S3D). Label free quantification (LFQ) comparing relative abundance of proteins captured by TurboID in standard vs CHX-Dia-FBS treated samples revealed preferential enrichment of mitochondrial proteins with CHX-Dia-FBS (90/136 proteins enriched > 2-fold, red dots, Figure 3G) compared with preferential capture of non-mitochondrial proteins under normal treatment conditions (231/265 proteins enriched > 2-fold, black dots, Figure 3G).
Extension of these analyses to Cys-LoC captured cysteine peptides confirmed that the dual CHX/Dia-FBS treatment afforded comparable increased performance to that observed for protein-level analysis. LFQ analysis revealed that 52% (47/90) of cysteines preferentially captured with CHX/Dia-FBS treatment belonged to mitochondrial localized proteins whereas nearly all (97/120) of those preferentially enriched under normal treatment conditions belonged to non-mitochondrial proteins (Figure 3H). Of note, only a handful of mitochondrial cysteines with more pronounced capture under normal conditions, including notably those found in cytochrome c oxidase subunit 4 isoform 1 COX4I1, which drives oxidative phosphorylation83 and receptor of activated protein C kinase 1 RACK1 proteins. These proteins are known to have a comparatively short half-life84. In aggregate, the CHX-Dia-FBS treatment, together with TurboID optimization mentioned beforehand, increased Cys-LoC mitochondrial cysteine specificity from 18% to 43% (Figure 3I).
As our methodological optimization was overwhelmingly focused on improving capture of mitochondrial cysteines, we next asked whether the improvement in specificity afforded by the CHX-Dia-FBS treatment would extend to other low specificity TurboID constructs. Cys-LoC analysis of HEK293T cells stably expressing either a Golgi- or ER-targeted TurboID construct revealed more modest but still significant increases in cysteine localization specificity (Figure 3I). The overlap in cysteines identified comparing the Mito-, Golgi- and ER-TurboID expressing cell lines was comparatively modest, spanning 55%−86% across three replicates analyzed (Figure S3E). Among the cysteines captured, several residues stood out for their lack of detection in prior studies (e.g. CysDB30), including C21 of oxygen-dependent coproporphyrinogen-III oxidase (CPOX), an enzyme that contributes to heme biosynthesis85, and C75 of DNL-type zinc finger protein (DNLZ), which coordinates a zinc in the zinc finger of this mitochondrial chaperone86.
Establishing and applying the Local Cysteine Oxidation (Cys-LOx) method to analyze basal mitochondrial cysteine oxidation
Having achieved a substantial performance increase for Cys-LoC specificity, we returned to our second original objective, quantification of local cysteine oxidation state. To establish the Local Cysteine Oxidation (Cys-LOx) method (Figure 4A), proteins proximal to mitochondria were biotinylated via mito-TurboID. The cells were then lysed and reduced cysteines immediately capped using our custom isotopically labeled isopropyl iodoacetamide alkyne (IPIAA-L) probe16. Subsequently, the samples were subjected to reduction and capping using our isotopically differentiated heavy (IPIAA-H) probe. Supporting that the IPIAA and IAA probes can be used relatively interchangeably, we observed high overlap in cysteines identified with both reagents, which is consistent with our prior study (Figure S4A)16. Fragpipe IonQuant87 was applied to report the ratio of IPIAA-L vs IPIAA-H labeled cysteine peptide precursor ions (Log2(H/L)), from which the cysteine % oxidation state was calculated. As with Cys-LoC, the key innovative step of Cys-LOx is our unprecedented two-step enrichment protocol, which allows for capture of mitochondrial cysteines without conventional biochemical fractionation.
Figure 4. Establishing the Local Cysteine Oxidation (Cys-LOx) method to analyze basal mitochondrial cysteine oxidation states.
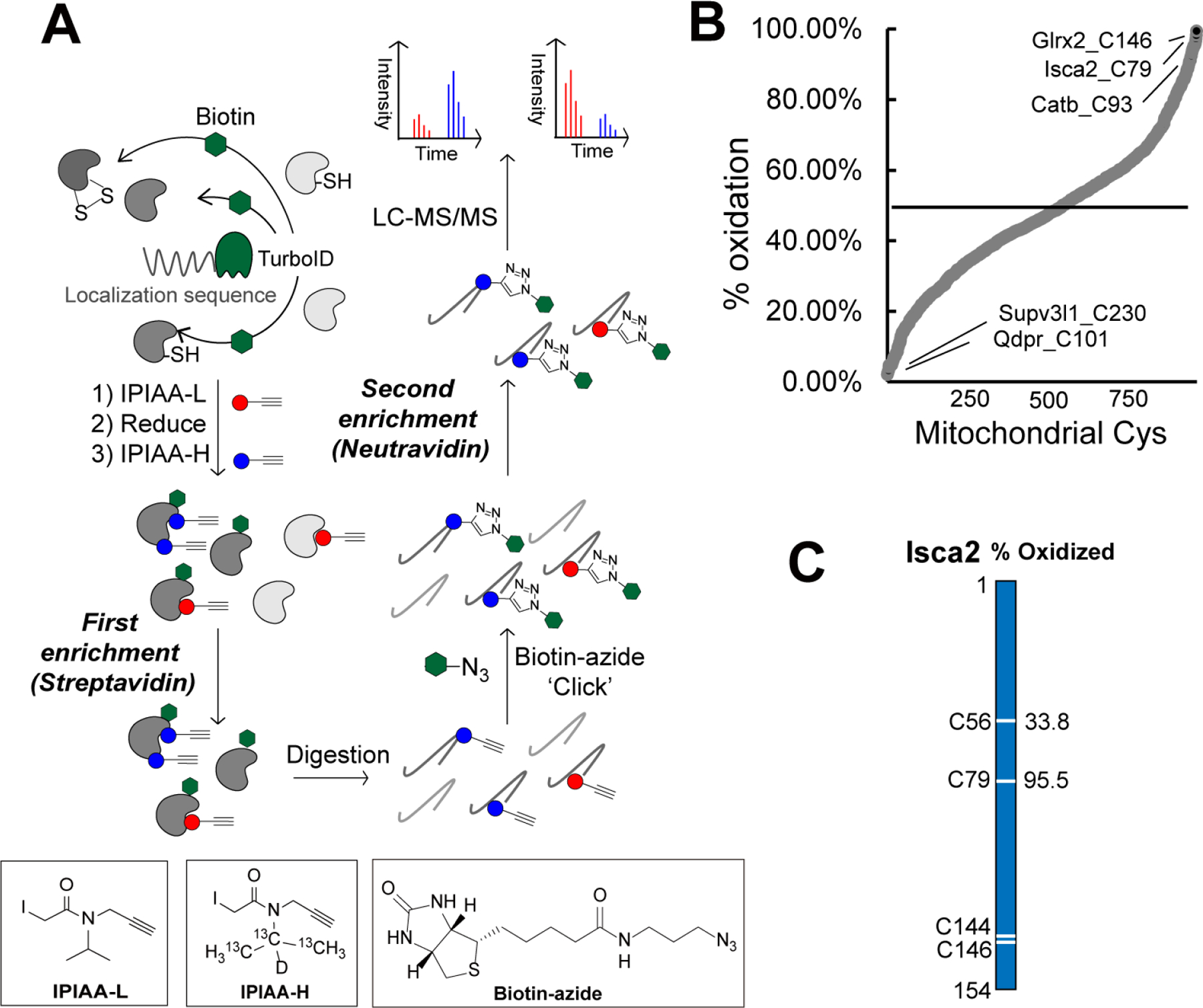
A) Scheme of Cys-LOx method. B) Percent oxidation state of mitochondrial cysteines identified with Mito-Cys-LOx. C) Percent oxidation state of cysteines quantified in an exemplary mitochondrial protein. Experiments were performed in triplicate in iBMDM cells. See also Data S4 and Figure S4.
Cys-LOx analysis of mito-TurboID expressing HEK293T cells quantified 888 total mitochondrial cysteines out of 2739 total cysteines. Of these, 182 had elevated ratios (Log2(H/L) > 1), consistent with % oxidation > 50% (Figure S4B). Exemplary oxidized cysteines included C129 of membrane-associated progesterone receptor component 1 PGRMC1, C47 of peroxiredoxin-6 PRDX6, C229 of peroxiredoxin-3 PRDX3, which have all been reported as either disulfide or redox centers88–90. Reduced cysteines included C46 of Parkinson disease protein 7 PARK7, which was reported as redox sensitive91. For mitochondrial protein aldehyde dehydrogenase 1 family member B1 ALDH1B1, which plays a major role in the detoxification of alcohol-derived acetaldehyde92, the oxidation states of 3 cysteines were quantified (Figure S4C). Both C179 and C386 were found to be > 50% oxidized. By contrast, C169 was calculated to be 9.5% oxidized, indicatively of highly reduced state.
Given our aforementioned demonstration of CHX-enhanced Cys-LoC mitochondrial cysteine specificity, we next incorporated the CHX treatment into the Cys-LOx workflow. Given that CHX treatment can cause cellular stress93,94, we first assessed whether translational arrest would impact cysteine oxidation state (Figure S4D). While we, gratifyingly, observed limited CHX-induced cysteine oxidation, we opted to proceed conservatively with CHX incorporation into Cys-LOx, given the potential for phenotypic changes and cytotoxicity in response to translation arrest. Within the context of mitochondrial physiology, while CHX treatment is not known to inhibit mitochondrial translation95, it does afford decreased gluconeogenesis93, and nearly all mitochondrial proteins are encoded by nuclear genes. To facilitate identification of high confidence mitochondrial oxidation state measurements, we established a “Safe List,” which is comprised of cysteines insensitive to CHX, supporting high confidence mitochondrial-localization not impacted by newly translated mito-TurboID activity. Our Safe List featured 456 total mitochondrial cysteines (Figure 3I, pink dots; Data S4).
Given our interest in mitochondrial cysteines sensitive to LPS-induced inflammation, we next extended the Cys-LOx method to assay basal redox states of cysteines within mitochondria of immortalized bone marrow derived macrophages (iBMDMs)96. We opted to use murine, rather than human BMDMs given the established body of literature demonstrating substantial iNOS induction and ROS production by mouse macrophages relative to human macrophages subjected to the same stimuli97,98. Cys-LOx analysis of iBMDMs quantified 1,156 total mitochondrial cysteines out of 2,998 total cysteines, in the absence of any stimuli. Of these, 182 had elevated ratios (>1) and % oxidation > 50%, indicative of oxidation (Figure 4B). Exemplary oxidized cysteines included C93 of cathepsin B CATB, which has been annotated as a disulfide, C79 of Iron-sulfur cluster assembly 2 Isca2, which is reported as involved in iron-sulfur clusters99 and C146 of Glutaredoxin-2 Glrx2, a glutathione-dependent oxidoreductase that facilitates the maintenance of mitochondrial redox homeostasis100. The most reduced cysteines included C230 of ATP-dependent RNA helicase Supv3l1 and C101 of dihydropteridine reductase Qdpr, with a percentage oxidation as low as 2.2%. Iron-sulfur cluster assembly 2 homolog Isca2 is an exemplary mitochondrial protein with 2 out of 4 cysteines quantified. Isca2 is involved in the iron-sulfur cluster assembly pathway. Notably, C79 in Isca, which is known to be sensitive to oxidative stress and important for reactivating aconitase101, was 95.5% oxidized (Figure 4C).
As with our Cys-LOx analysis of HEK239T cells, integration of CHX into the Cys-LOx analysis of iBMDMs revealed minimal CHX-induced changes to measured oxidation states of mitochondrial cysteines (Figure S4E). As with the HEK293T cysteine redoxome, we also generated a Safe List for the iBMDM mitochondrial cysteine redoxome, which featured 463 total cysteines not impacted by newly translated TurboID (Data S4).
Cys-LOx outperforms SP3-Rox for quantification of LPS-induced changes to cysteine oxidation in immortalized bone marrow derived macrophages (iBMDMs)
Having established the Cys-LOx method, we set out to apply our technology to identify mitochondrial cysteines sensitive to lipopolysaccharide (LPS) and interferon-gamma (IFNγ)-induced macrophage activation. We opted to simultaneously stimulate with LPS+IFNγ given the established synergistic effects both stimuli have on NO production102–104. iBMDMs were treated with either vehicle control or LPS+IFNγ for 24 hours. As expected, LPS+IFNγ treatment ablates mitochondrial respiration (Figure 5A, S5A). Additionally, expression of both inducible nitric oxide synthase (Nos2) and other pro-inflammatory genes increased upon treatment, with a more substantial increase observed for cells treated with both cytokines (Figure 5B, S5B).
Figure 5. Cys-LOx outperforms SP3-Rox for quantification of LPS-induced changes of mitochondrial cysteine oxidation states.
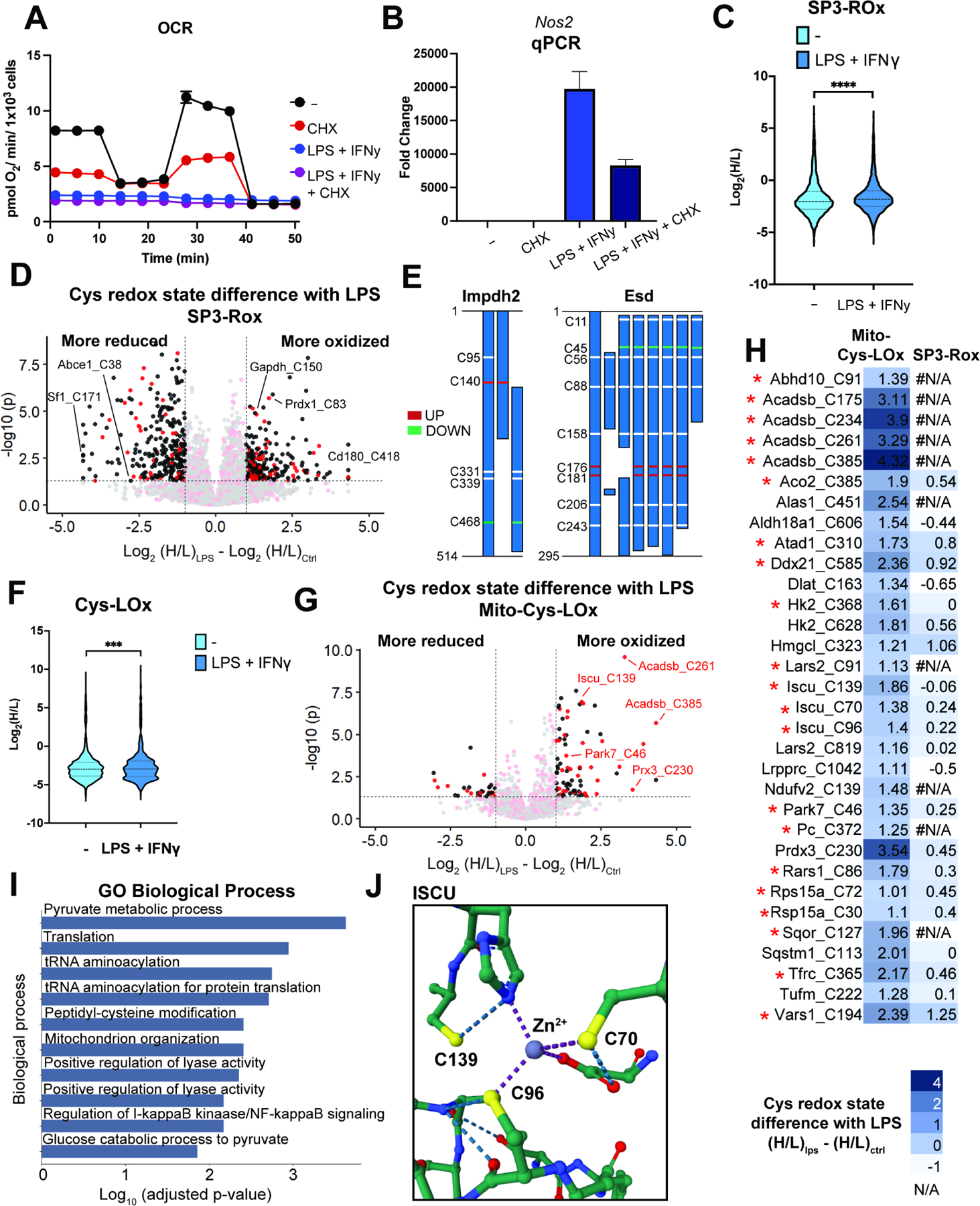
A) Oxygen consumption rate (OCR) of mitochondria for control, LPS+IFNγ, CHX or both. Representative trace of one biological replicate with 5 technical replicates. For some timepoints, symbols obscure error bars. B) qCPR analysis of expression of Nos2 with control, LPS+IFNγ, CHX or both. C) SP3-Rox data ± LPS+IFNγ treatment. D) Difference of redox states for cysteines quantified with SP3-Rox ± LPS+IFNγ treatment. E) SP3-Rox data ± LPS+IFNγ treatment in exemplary proteins with splice isoforms. F) Mito-Cys-Lox ± LPS+IFNγ treatment. G) Difference of redox states for cysteines quantified with Mito-Cys-Lox ± LPS+IFNγ treatment. H) Comparison of Mito-Cys-LOx and SP3-Rox with or without LPS+IFNγ treatment. * indicates Safe List cysteines. I) GO biological process analysis of mitochondrial cysteines quantified with Mito-Cys-LOx that showed more oxidized redox states upon LPS+IFNγ treatment. J) Crystal structure of Iscu with cysteines more oxidized with LPS+IFNγ treatment (PDB ID: 1WFZ). Statistical significance was calculated with unpaired Student’s t-tests, *** p<0.005, **** p<0.0001. Experiments all performed in biological triplicate with additional acquisition of technical replicates. Data in panel B are represented as mean ± stdev. For D, and G, Red: mitochondrial cysteines. Black: non-mitochondrial cysteines. See also Data S5 and Figure S5.
Having previously established the SP3-Rox method16, which reports proteome-wide cell-state- dependent changes to cysteine oxidation, we next subjected the iBMDMs to SP3-Rox analysis. Our goals were to establish a baseline for LPS-induced whole-cell cysteine oxidation from which we could compare the Cys-LOx method, and to assess whether the Cys-LOx method could capture mitochondrial cysteines not readily quantified by established, bulk proteomic methods. Additionally, we hypothesized that Cys-LOx analysis would reveal mitochondrial specific redox changes that get masked by the bulk SP3-Rox analysis. In aggregate, SP3-Rox analysis captured 7,523 total cysteines and identified 290 cysteines that showed increased oxidation upon LPS+IFNγ treatment (Figure 5D). Quantified cysteine ratios for the bulk cellular proteome analyzed by SP3-Rox were significantly increased after LPS+IFNγ treatment, indicative of substantial cell-wide oxidation (Figure 5C). This finding is to be expected given the marked increase in iNOS expression and resulting widespread cysteine nitrosylation. Overall, 1742 total mitochondrial cysteines were identified and 70 were found to exhibit increased oxidation in response to LPS+IFNγ (Figure 5D). Of the LPS+IFNγ oxidation-sensitive cysteines identified in the bulk proteome Sp3-Rox analysis, several have been previously identified as redox active, including C150 of glyceraldehyde-3-phosphate dehydrogenase Gapdh, C147 of superoxide dismutase [Cu-Zn] Sod1, C54 of peroxiredoxin-4 Prdx4, C83 of peroxiredoxin-1 Prdx1, and C1557 of fatty acid synthase Fasn16,30.
Curiously, our bulk redox analysis also revealed a marked population of 293 total cysteines with significantly reduced oxidation state upon LPS+IFNγ treatment, as analyzed by SP3-Rox of bulk proteomes. Given the large burst of ROS and RNS produced upon macrophage stimulation, this cysteine population was unexpectedly large. Therefore, we opted to further investigate the extent to which Consistent with prior reports105,106, we see a decrease in total glutathione concentration upon activation with LPS + IFNγ, excluding changes in glutathionine from rationalizing the reduced subset (Figure S5C). Pathway analysis for the reduced population revealed a marked enrichment for genes involved in RNA and protein biogenesis, including splicing, NFkB gene targets, and translation in both Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis (Figure S5D–E). We postulated that post-stimulation, translation of LPS+IFNγ responsive genes was likely responsible for the observed population of highly reduced cysteines. Supporting this finding, we observed a comparatively short (2h) half-life protein ABCE1 (Figure S5F) that contained more reduced cysteines C38 and C65, with oxidation states reduced from 80.7% to 39.5% and 87.8% to 58.7%, respectively upon LPS+IFNγ treatment. Further implicating altered gene expression, we observed 14 proteins which harbored cysteines with both increased and decreased oxidation states, including Inosine-5’-monophosphate dehydrogenase 2 (Impdh2 C140, C468), S-formylglutathione hydrolase (Esd C176, C181, C45), and Valine–tRNA ligase (Vars C194, C1051) (Figure 5E, S5G), all of which encode multiple splice forms, suggestive that LPS+IFNγ-dependent alternative splicing may play a role in production of this population of proteins (Data S5). Gratifyingly, only 49 mitochondrial cysteines were found to be more reduced, indicating the mitochondrial proteome is relatively insensitive to the observed global reducing response to LPS+IFNγ.
Application of Mito-Cys-LOx to our LPS+IFNγ iBMDM system quantified, in aggregate, 1,451 total and 559 mitochondrial cysteines. Similar to SP3-Rox, a marked increase in the ratios of identified cysteines was observed after LPS+IFNγ treatment (Figure 5F). 32 mitochondrial cysteines exhibited significant increase in the H:L ratio upon LPS treatment (Log2(H/L)LPS — Log2(H/L)Ctrl > 1), indicating increased oxidation (Figure 5G, 5H). 23 of the 32 cysteines that showed increased oxidation state in the Cys-LOx analysis were identified in the SP3-Rox analysis. Of these, only 2 residues (Hmgcl_C323 and Vars1_C194) also showed increased oxidation upon LPS+IFNγ activation in the SP3-Rox analysis, while the oxidation state of the other 21 was not significantly changed (Figure 5H). This result highlights the value of subcellular redox state analysis in capturing compartment-specific changes in cysteine oxidation, as a global analysis masks the redox changes that are specific to a single compartment.
We observed a pronounced decrease in mitochondrial respiration following 6h CHX treatment, substantiating our prior concerns for potential CHX-dependent alterations in mitochondrial cysteine redox states. (Figure 5A, S5A). Additionally, qPCR analysis of genes associated with response to LPS+IFNγ revealed both comparatively CHX-insensitive (Irg1 and Il1b) and highly CHX-sensitive (Tfna and Il6) changes to gene expression (Figure S5B). We referenced our previously generated iBMDM cysteine safe list (Data S4) to delineate high confidence mitochondrial cysteines that exhibit increased oxidation in response to LPS+IFNγ. Within the 32 mitochondrial cysteines exhibiting more elevated H:L ratios, indicating increased oxidation upon LPS treatment, we identified 21 of them to be insensitive to translational arrest, which are indicated by the red asterisks (Figure 5H).
Pathway analysis of the oxidized subset revealed peptidyl cysteine modification (GO: 0018198) as a major enriched Gene Ontology biological process (Figure 5I), with glycolysis (while a cytosolic process, this enrichment stems from oxidation of cysteines in hexokinase, which is tethered to the outer mitochondrial membrane107) and HIF-1 signaling as significantly enriched KEGG pathways (Figure S5H). Supporting the robustness of our LPS+IFNγ dataset, many of the cysteines identified by Cys-LOx are residues previously characterized as sensitive to oxidative modification. Examples include Iron-sulfur cluster assembly enzyme Iscu C139 and C70 (cysteine persulfide)108, Parkinson disease protein 7 homolog Park7 C46 (cysteine palmitoyl)91, Hydroxymethylglutaryl-CoA lyase Hmgcl C323 (disulfide)109, Thioredoxin-dependent peroxide reductase Prdx3 C230 (disulfide)110, and Aconitate hydratase Aco2 C38554. All three cysteines we identified as sensitive to LPS+IFNγ in Iscu are proximal to or serve as ligands for the coordinated Zn2+ ion (Figure 5J), suggesting that these residues may play a role in sensing oxidative stress. Intriguingly, C385 of Aco2 binds the active site Fe-S cluster that is essential for catalytic activity, and oxidation of this cysteine has been suggested to render the protein inactive54,111. The result suggested that modification of aconitase – in addition to those of isocitrate dehydrogenase and succinate dehydrogenase112 – may result in cellular respiration defects caused by pro-inflammatory activation. We additionally identified a number of LPS+IFNγ sensitive cysteines that have not been previously annotated as sites of oxidative modification (e.g. C96 in Iron-sulfur cluster assembly enzyme Iscu and C91 and C819 in Isoleucine—tRNA ligase Iars2), which likely represent novel sites of redox regulation. Taken together these residues, highlights the general utility of Cys-LOx for studying the subcellular cysteinome, including in the context of mitochondrial sensitivity to cellular pro-inflammatory processes.
Discussion
Here we establish two novel cysteine chemoproteomic platforms, Local Cysteine Capture (Cys-LoC) and Local Cysteine Oxidation (Cys-LOx), which enable compartment-specific capture of cysteines and quantification of changes to local cysteine oxidation state, respectively. Both Cys-LoC and Cys-LOx implement a customized two-step biotinylation workflow that features sequential enrichment of subcellular-localized proteins (e.g. cytosol, ER, golgi, mitochondria, and nucleus, as labeled by targeted TurboID constructs) followed by peptide-level click-conjugation biotinylation and a subsequent second round of enrichment to capture cysteine-containing peptides derived from the TurboID-labeled proteins. While our methods are in many ways conceptually similar to TurboID-based subcellular phosphoproteomics68, to our knowledge, such sequential rounds of biotin-avidin capture, first at the protein level and then at the peptide level, have not been reported previously. Application of Cys-LOx to iBMDMs quantified 559 total mitochondrial cysteines and 32 sensitive to LPS+IFNγ treatment. Notably, a number of these residues are found in proteins involved in iron sulfur cluster biogenesis and respiration, including aconitase and iron-sulfur cluster assembly enzyme Iscu. Given the importance of these proteins to mitochondrial function, we expect that oxidative modifications to some of these cysteines may, in part, explain the marked respiration defects caused by LPS+IFNγ (Figure 5A).
Showcasing the utility of Cys-LOx, we found that bulk SP3-Rox proteome cysteine redox analysis of LPS+IFNγ stimulated iBMDMs was complicated by the appearance of a substantial fraction of cysteines that showed decreased ratios, indicative of decreased oxidation, after treatment. We expect that this increased pool of newly reduced cysteines stems from the increase in cytoplasmic reduced nicotinamide adenine dinucleotide cofactors, including NADH and NADPH, the latter of which is particularly noteworthy due to its essentiality for NADPH oxidase activity that generating antibacterial ROS. LPS+IFNγ-induced production of new proteinacious cysteines, derived from protein synthesis and alternative splicing is also expected to also contribute to the curious appearance of more reduced residues after oxidative assault. As the SP3-Rox method relies on a ratio difference calculation, it is relatively insensitive to protein abundance changes. However, in this case, we expect that turnover of NO-damaged protein together with production of newly synthesized protein could in part rationalize the ratios observed. This model is consistent with prior studies that reported substantial remodeling of the macrophage proteome in response to LPS+IFNγ113. Alongside altered protein expression, an increased pool of reduced nicotinamide-based cofactors may also contribute to the LPS+IFNγ-induced decreased cysteine oxidation. Taken together, the observed differences in the bulk and Cys-LOx analyzed proteomes highlight the value in subcellular stratification of the cysteine redoxome, particularly given the compartmentalization of cellular metabolite pools.
As mitochondrial isolation is feasible using established biochemical fractionation methods, we expect that some of our findings could be corroborated by fractionation-based OxICAT platforms4,6. Given that mitochondrial isolation, particularly when performed on cultured cells, can be complicated by organelle damage accrued during isolation and by contamination of isolates with non-mitochondrial proteins114, we expect that Cys-LOx may prove particularly useful for reducing these potential experimental confounders. Looking beyond organelles that can be separated by density gradient centrifugation methods, we expect that the Cys-LoC and Cys-LOx methods will also prove highly useful for chemoproteomic analysis of subcellular compartments, protein complexes, and cell types that are not readily amenable to biochemical fractionation. Examples of such applications include membraneless organelles, such as stress granules and P-bodies46,47,115, and proteins localized to neuronal axons116. Fully realizing the utility of Cys-LoC and Cys-LOx for membraneless organelles will require implementation using TurboID fusion proteins that are known to undergo phase separation, comparable to the previously reported BioID46 and APEX247 constructs. To ensure cysteine specificity, analysis of additional control datasets is likely warranted, including, for example in samples prepared without stimulation for membraneless organelles that only assemble in response to a stimulant, like SGs, and cytoplasm or nucleus-localized TurboID controls, for membraneless organelles that exist in the cytoplasm (e.g stress granules and P-bodies) or the nucleus (nucleoli, Cajal bodies, paraspeckles).
One challenge that we encountered while establishing the Cys-LoC and Cys-LOx methods was the seemingly low compartment specificity of proteins and cysteines captured by TurboID proximity labeling. Through our in-depth analysis of sources of aberrant TurboID performance, we first pinpointed endogenous biotin derived from serum as a source of non-compartment specific labeling, consistent with prior studies75,78. While depletion of endogenous biotin using dialyzed serum afforded a modest increase in subcellular specificity, a more pronounced effect was observed after treatment with cycloheximide. These findings are consistent with newly translated TurboID as a major source of the observed seemingly non-specific biotinylation. CHX treatment proved particularly useful in our study for establishing Safe Lists of cysteines not impacted by this unwanted TurboID activity. As shown by the toxicity of CHX to mitochondrial respiration, both measured in our study and reported previously93, CHX likely will prove suboptimal as a universal solution to the TurboID localization conundrum. Further exemplifying the need for additional methods, we also found that the CHX treatment afforded the greatest improvement in localization specificity for the mito-TurboID construct, with more modest enhancement observed for other compartments analyzed. While a clear rationale for these construct-dependent differences remains to be fully explored, we expect that the previously reported CHX-dependent impact on subcellular RNA localization, as assayed by APEX-Seq117, may implicate a confluence of RNA- and protein-localization factors. Differences in biotin uptake across organelles and across cell types is another area that warrants further investigation, which we expect to be particularly relevant for studies aiming to achieve optimal compartment-specific proximity-labeling.
Looking beyond CHX, we can envision that two-step cysteine capture methods with enhanced performance could be achieved using several relevant alternative approaches, including, for example, the previously reported Split TurboID45, TurboID caged using unnatural amino acid (UAA) incorporation68 and potentially alternative genetic approaches that afford tighter control over protein expression (e.g. Flp-In™ T-Rex™ system; Invitrogen Life Technologies) that would reduce the fraction of newly synthesized protein. As BioID is less active43, we expect that newly translated BioID fusion proteins should less substantially impact proximity labeling performance. However, this low catalytic activity may prove insufficient to achieve high coverage two-step cysteine capture. Furthermore, as shown by our absolute quantitation of mitochondrial and whole cell biotin concentrations achieved after exogenous biotin addition, limited biotin entry into organelles may continue to confound such improved platforms. Alternative proximity labeling methods that utilize reagents with enhanced membrane solubility may be required.
Limitations of this study
Comparatively modest proteomic coverage and subcellular specificity are observed limitations of both the Cys-LoC and Cys-LOx methods. Our proteomic coverage was limited to ~1,000–2,000 unique cysteines identified per sample, which is almost undoubtably due to the two rounds of enrichment. We expect that further methodological optimization will enhance cysteine coverage, which will be of particular value for redox analyses that require high cysteine coverage to calculate difference values between treated and control groups. While our proof-of-concept studies using CHX demonstrated enhanced compartment specificity, we expect that CHX’s toxicity will preclude widespread use. Therefore, approaches that further improve specificity will decrease the likelihood of spurious identification of non-compartment specific cysteines. Lastly, as our study has relied on stable overexpression in immortalized cell lines, our methodology is currently incompatible with primary cells and in vivo studies. We do expect that, once in vivo proximity labeling systems emerge that demonstrate highly efficient protein labeling, our methodology should translate into these physiologically meaningful systems.
Star methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Keriann M. Backus (kbackus@mednet.ucla.com).
Materials availability
Plasmids generated in this study have been deposited to Addgene, deposit # 82589.
All unique reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
All MS data was deposited to the ProteomeXchange Consortium via the PRIDE118,119 partner repository and are publicly available as of the date of publication. Dataset identifiers are listed in the key resources table.
All code used in this paper is reported in the previous study30.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell lines
HEK 293T (CRL-3216, Homo sapiens, female, embryonic kidney) cells were cultured in DMEM supplemented with 10% Fetal Bovine Serum (FBS) and 1% antibiotics (Penn/Strep, 100 U/mL). Immortalized bone marrow derived macrophages (iBMDMs) were generated by immortalizing murine BMDMs via overexpression of V-fraf and V-myc with J2 virus to generate immortalized BMDB (iBMDM)96. iBMDMs were cultured in DMEM supplemented with 10% FBS, 1% antibiotics (Penn/Strep, 100 U/mL) and 5% (v/v) conditioned media containing macrophage colony stimulating factor (M-CSF)120 to induce differentiation to BMDMs. Media was filtered (0.22 μm) prior to use. Cells were maintained in a humidified incubator at 37 °C with 5% CO2.
METHOD DETAILS
Cloning of different TurboID constructs.
List of plasmids with detailed information used in this study can be found in the key resources table. PCR fragments of TurboID with different localization sequences were amplified using Phusion polymerase (Berkeley). The destination vectors and PCR products were both digested using standard enzymatic restriction and cleaned up, followed by T4 DNA ligation. Ligated products were transformed into TOP10 competent cells and sent for sequence to confirm the cloning.
Transient transfection of TurboID constructs.
Cells were transfected at 70–80% confluency. For a 10-cm plate, plasmid (5 ug), serum-free DMEM (350 uL) and PEI (25 uL of 1 mg/mL) were mixed and incubated for 15 min at room temperature (RT), followed by adding dropwise to the cells.
Generation of cell lines stably expressing TurboId constructs.
For preparation of lentiviruses, HEK 293FT cells in 10 cm plates were transfected at ~80–90% confluency with lentiviral vector FUGW containing the gene of interest with the lentiviral packaging plasmids pVSVG (4 ug; Addgene #8454) and Δ8.9 (8 ug; Addgene #8455)121 and 66 uL of Turbo DNAfectin3000 (Lamda Biotech Inc.) in antibiotic-free media for 6 h. The DNAfectin-containing media was replaced with fresh antibiotic-free media and the cells were left to incubate for 48 hours for lentiviral generation. The media was collected and cells were allowed to incubate for another 24 hours in fresh media. After 24 h, the lentivirus-containing media was collected, and added to the previously harvested media. All collected lentivirus-containing media was stored at 4 °C. 1/3 volume of Lenti-X concentrator (Takara Bio) was added to the total harvested media and incubated 16 h at 4 C. The lentivirus was pelleted at 1500 g for 45 mins at 4 °C and resuspended in 500 uL plain DMEM and stored in 100 uL aliquots at −80 °C.
To generate the stable cell lines, cells were infected at 75 % confluency, passaged 5 times, and selected via flow cytometry for positive EGFP signal.
Database Construction.
Subcellular location annotations from CellWhere Atlas (accessed 2208), Human Protein Atlas (HPA) version 21.1 and UniProtKB/Swiss-Prot (2208_release) were aggregated. Unique proteins were established using UniProt protein identifiers. CellWhere localization, HPA main location and UniProt subcellular location columns were mined for specific location keywords (ex. ‘golgi’). Proteins containing these keywords are reported in Data S1.
Biotinylation with TurboID.
For transiently expressed TurboID, biotin labeling was initiated 24 h after transfection. 100 mM biotin stock was made in dimethyl sulfoxide (DMSO). Biotin was directly added to cells at a final concentration of 500 μM and incubated for 1 h at 37 °C. After washing with cold DPBS for 3 times, cells were harvested by centrifugation (4,500 g, 5 min, 4 °C), washed twice with cold DPBS, lysed in RIPA buffer (Fisher, Cat# AAJ62885AE), and clarified by centrifuging (21,000 g, 10 min, 4 °C). Protein concentrations were determined using a BCA protein assay kit (Thermo Fisher, Cat# 23227) and the lysate diluted to the working concentrations indicated below.
SDS-PAGE gels and western blots.
Lysate was normalized to 2 mg/mL and separated on a 4–20% SDS-PAGE gel. Gels were transferred to nitrocellulose membrane (Bio-Rad) and blocked in 5% (w/v) milk in TBS-T (Tris-buffered saline, 0.1% Tween 20) for 1 h at RT. Membranes were incubated with primary antibodies overnight (14–16 h) at 4 °C then washed 3 times with TBS for 5 mins. Membranes were then incubated with secondary antibodies for 1 h at RT and washed 3 times with TBS. For blots assessing biotin signal, membranes were incubated with a streptavidin-fluorophore conjugate overnight at 4 °C. Membrane was imaged on Bio-Rad ChemiDoc. Antibodies used were listed in the key resources table. ImageJ was used to normalize and quantify the band intensity122.
Proteomic sample preparation for Cys-LoC and Cys-LOx, protein level enrichment.
Biotinylated lysates (500 μL of 1 mg/mL, prepared as described above) were labeled with either 2 mM IAA for Cys-LoC or 2 mM IPIAA-L for Cys-LOx for 1h at RT. 50 uL Pierce streptavidin agarose beads were washed with RIPA and incubated with lysates for 2 h at RT. The proteins bound to beads were washed once with 1 mL 2M urea in RIPA, twice with 1 mL RIPA, and 3 times with 1 mL PBS. The beads were resuspended in 200 μL 6 M urea, reduced with 1 mM DTT for 15 min at 65 °C, and labeled with 2 mM IAA for Cys-LoC or 2 mM IPIAA-H for Cys-LOx for 1h at RT. Then, beads were washed with PBS and resuspended in 200 μL 2 M urea. 3 μL of 1 mg/mL trypsin solution (Wathington) was added. Proteins were digested off the bead overnight at 37 ºC with shaking.
Proteomic sample preparation for Cys-LoC and Cys-LOx, peptide level enrichment.
After digestion, CuAAC was performed with biotin-azide (4 μL of 200 mM stock in DMSO, final concentration = 4 mM), TCEP (4 μL of fresh 50 mM stock in water, final concentration = 1 mM), TBTA (12 μL of 1.7 mM stock in DMSO/t-butanol 1:4, final concentration = 100 μM), and CuSO4 (4 μL of 50 mM stock in water, final concentration = 1 mM) for 1h at RT. 20 μL Sera-Mag SpeedBeads Carboxyl Magnetic Beads, hydrophobic (GE Healthcare, 65152105050250, 50 μg/μL, total 1 mg) and 20 μL Sera-Mag SpeedBeads Carboxyl Magnetic Beads, hydrophilic (GE Healthcare, 45152105050250, 50 μg/μL, total 1 mg) were mixed and washed with water three times. The bead slurries were then transferred to the CuAAC samples, incubated for 5 min at RT with shaking (1000 rpm). Approximately 4 mL acetonitrile (> 95% of the final volume) was added to each sample and the mixtures were incubated for 10 min at RT with shaking (1000 rpm). The beads were then washed (3 × 1 mL acetonitrile) with a magnetic rack. Peptides were eluted from SP3 beads with 100 μL of 2% DMSO in MB water for 30 min at 37 °C with shaking (1000 rpm). The elution was repeated with 100 μL of 2% DMSO in MB water. For each sample, 50 μL of NeutrAvidin Agarose resin slurry (Thermo Fisher, 29200) was washed three times in 10 mL IAP buffer (50 mM MOPS pH 7.2, 10 mM sodium phosphate, and 50 mM NaCl buffer) and then resuspended in 800 μL IAP buffer. Peptide solutions eluted from SP3 beads were then transferred to the NeutrAvidin Agarose resin suspension, and the samples were rotated for 2 h at RT. After incubation, the beads were pelleted by centrifugation (21,000 g, 1 min) and washed (3 × 1 mL PBS, 6 × 1 mL water). Bound peptides were eluted twice with 60 μL of 80% acetonitrile in MB water containing 0.1% FA. The first 10 min incubation at RT and the second one at 72 °C. The combined eluants were dried (SpeedVac), then reconstituted with 5% acetonitrile and 1% FA in MB water and analyzed by LC-MS/MS.
Proteomic sample preparation for TurboID-labeled samples.
With Biotinylated lysates (500 μL of 1 mg/mL, prepared as described above, 50 uL Pierce streptavidin agarose beads were washed with RIPA and incubated with lysates for 2 h at RT. The proteins bound to beads were washed once with 1 mL 2M urea in RIPA, twice with 1 mL RIPA, and 3 times with 1 mL PBS. The beads were resuspended in 200 μL 6 M urea, reduced with 10 mM DTT for 15 min at 65 °C, and labeled with 20 mM IA for 30 min at 37 °C. Then, beads were washed with PBS and resuspended in 200 μL 2 M urea. 3 μL of 1 mg/mL trypsin solution (Washington) was added. Proteins were digested off the bead overnight at 37 ºC with shaking. After digestion, peptides were desalted with C18 column (Pierce, 87784) and analyzed by LC-MS/MS.
Crude mitochondria fractionation.
HEK293T cells stably expressing mito-TurboID were plated in four 15-cm dishes. At 90% confluency, cells were harvested by centrifugation (4,500 g, 5 min, 4°C), washed twice with cold DPBS. The pellets were suspended in the MSHE buffer (70 mM sucrose, 210 mM mannitol, 5.0 mM HEPES, 1.0 mM EGTA) and homogenized in a glass grinder by 25 up-and-down passes of the pestle. The homogenate was then pelleted (1,100 g, 10 min, 4°C). The supernatant was transferred to a new tube and ultracentrifuged. (14,000 g, 10 min, 4°C). The supernatant was then saved as non-mito portion and the pellet was saved as mito portion.
GC-MS biotin quantification.
HEK293T cells were plated in 15-cm dishes. At 90% confluency, 500 μM Biotin was added and incubated for 1 h at 37°C. Intact HEK293T cells or mitochondria isolated from HEK293T cells as described above were extracted and analyzed using GC/MS to quantify biotin levels. For extraction, 15 mL tubes containing 293T cells or isolated mitochondria were placed on ice. To remove residual biotin - containing culture medium, tissue culture cell pellets were first quickly washed with ice-cold 0.9% (w/v) NaCl. The cells or mitochondria pellets were immediately treated with 500 μL of ice-cold MeOH and 200 μL water containing 1 μg of the internal standard norvaline. Next, 500 μL of chloroform was added, after which samples were vortexed for 1 min and then spun at 10,000 g for 5 min at 4°C. The aqueous layer was transferred to a GC-MS sample vial and dried overnight using a refrigerated CentriVap. Once dry, samples were resuspended in 20 μL of 2% (w/v) methoxyamine in pyridine and incubated at 37°C for 45 min. This was followed by addition of 20 μL of MTBSTFA + 1% TBDMSCl (Ntert-Butyldimethylsilyl-N-methyltrifluoroacetamide with 1% tertButyldimethylchlorosilane), mixing, and incubation for an additional 45 min at 37°C. Samples were run as previously described 123, and analyzed using Agilent MassHunter software.
Immunocytochemistry.
Mito-TurboID-EGFP stably expressed HEK293T cells were grown on glass coverslips in a 24-well plate. At 75% confluency, 500 μM Biotin was added and incubated for 1 h at 37°C. Cells were washed with PBS 3 times. Cells were fixed in 5% formalin in PBS for 15 min at RT and permeabilized by 0.1% Triton X-100 in PBS for 6 min at RT. Cells were blocked with 1% BSA for 1 hr at RT. For biotinylation detection, cells were treated with Streptavidin Alexa Fluor™ 594 (1:500, Thermo Fisher, S11227) for 1 hr at RT. For mitochondrial localization, cells were treated with TMRE (1:1000, Invitrogen, T669) for 10 min at 37°C. Nucleus was detected with Dapi dye. Coverslips were mounted. Confocal images were obtained using Zeiss LSM 800 Confocal Laser Scanning Microscope. For localization analysis, the Coloc2 module in ImageJ was used with default settings122.
Generating streptavidin background dataset.
To generate the streptavidin background dataset, 2 individual negative control experiments for the TurboID workflow were prepared as described in Proteomic sample preparation for TurboID. In the negative control experiments, no TurboID fusion protein was expressed, and no exogenous biotin was added. Proteins were identified as “streptavidin background” if they were present in the streptavidin background dataset. The dataset contained a total of 966 proteins in aggregate.
Respirometry.
Rates of oxygen consumption and extracellular acidification were measured using an Agilent Seahorse XFe96 Analyzer. Briefly, iBMDMs were plated at 7.5×103 per well in a 96-well Seahorse XF cell plate. After a 48-hour incubation, the cells were treated with 100 ng/mL lipopolysaccharide (LPS) and 20 ng/mL interferon gamma (IFNγ) for 24 hours. To mimic cysteine profiling experiments, 100 µg/mL cycloheximide was added to the cells for 6 hours prior to conducting respirometry experiments. At the time of experiment, iBMDM growth media was replaced with respirometry assay medium which consisted of unbuffered DMEM (Sigma #5030) supplemented with 2 mM pyruvate, 10 mM glucose, 2 mM glutamine, and 5 mM HEPES. Respiration was measured at baseline and in response to acute treatment with 2 µM oligomycin, FCCP (two sequential pulses of 0.75 µM), and 0.2 µM rotenone with 1 µM antimycin A. All respiratory parameters were calculated as previously described 124.
RNA Isolation and qPCR analysis of iBMDMs.
Immortalized iBMDMs were plated at 1×105 per well in 12-well plates. After a 48-hour incubation, cells were treated with 100 ng/mL LPS and 20 ng/mL IFNγ for 24 hours. To maintain consistency with cysteine profiling experiments, 100 µg/mL cycloheximide was added to the cells for 6 hours prior to cell lysis. Following cell lysis, RNA was isolated using the RNeasy Mini kit (Qiagen). cDNA was synthesized with the high-capacity cDNA reverse transcription kit (Applied Biosystems). RT-qPCR assay was performed using PowerUp SYBR Green qPCR Master Mix kit (Applied Biosystems) on a QuantStudio 5 RT-PCR (Applied Biosystems). Relative gene expression values were calculated using the delta-delta Ct methods and Rplp0 was used as the control gene.
Proteomic sample preparation for SP3-Rox of iBMDMs.
iBMDM cells were treated either with or without 100 ng/mL LPS and 20 ng/mL IFNγ for 24 h at 37 °C. Cells were then harvested and SP3-Rox procedure was carried out as reported16.
Liquid-chromatography tandem mass-spectrometry (LC-MS/MS) analysis.
The samples were analyzed by liquid chromatography tandem mass spectrometry using a Thermo Scientific™ Orbitrap Eclipse™ Tribrid™ mass spectrometer. Peptides were fractionated online using a 18 cm long, 100 μM inner diameter (ID) fused silica capillary packed in-house with bulk C18 reversed phase resin (particle size, 1.9 μm; pore size, 100 Å; Dr. Maisch GmbH). The 70-minute water-acetonitrile gradient was delivered using a Thermo Scientific™ EASY-nLC™ 1200 system at different flow rates (Buffer A: water with 3% DMSO and 0.1% formic acid and Buffer B: 80% acetonitrile with 3% DMSO and 0.1% formic acid). The detailed gradient includes 0 – 5 min from 3 % to 10 % at 300 nL/min, 5 – 64 min from 10 % to 50 % at 220 nL/min, and 64 – 70 min from 50 % to 95 % at 250 nL/min buffer B in buffer A (Table S1). Data was collected with charge exclusion (1, 8,>8). Data was acquired using a Data-Dependent Acquisition (DDA) method consisting of a full MS1 scan (Resolution = 120,000) followed by sequential MS2 scans (Resolution = 15,000) to utilize the remainder of the 1 second cycle time. Precursor isolation window was set as 1.6 and normalized collision energy were set as 30%. Details of MS data can be found in Table S2. All MS data was deposited to the ProteomeXchange Consortium via the PRIDE118,119 partner repository with the dataset identifier PXD039626.
Protein, peptide, and cysteine identification.
Raw data collected by LC-MS/MS were searched with MSFragger (v3.3) and FragPipe (v19.0)125,126. The proteomic workflow and its collection of tools was set as default. Precursor and fragment mass tolerance was set as 20 ppm. Missed cleavages were allowed up to 1. Peptide length was set 7 – 50 and peptide mass range was set 500 – 5000. For Cys-LoC, Cysteine residues were searched with differential modification C+463.2366. For Cys-LOx and SP3-Rox, MS1 labeling quant was enabled with Light set as C+463.2366 and Heavy set as C+467.2529. MS1 intensity ratio of heavy and light labeled cysteine peptides were reported with Ionquant (v1.8.9)87. Calibrated and deisotoped spectrum files produced by FragPipe were retained and reused for this analysis. Custom python scripts were implemented to compile labeled peptide datasets. Unique proteins, unique cysteines, and unique peptides were quantified for each dataset. Unique proteins were established based on UniProt protein IDs. Unique peptides were found based on sequences containing a modified cysteine residue. Unique cysteines were classified by an identifier consisting of a UniProt protein ID and the amino acid number of the modified cysteine (ProteinID_C#); residue numbers were found by aligning the peptide sequence to the corresponding UniProt protein sequence. When there are multiple cysteines in one peptide, all the modified cysteine residue numbers will be reported as ProteinID_C#_C#.
MS Data analysis.
For the subcellular annotation, our customized localization database was used to cross referenced with the proteins or cysteines identified. Proteins were counted as localized in the compartment that the TurboID fusion protein targeted even if they contained multiple localization annotations. For Cys-LOx, the medium of heavy to light ratios for the same cysteine residue from cysteine peptides of different charges and miss cleavages in the same sample was calculated. Means of reported logged ratio values for each condition (+/− LPS+IFNγ or +/− CHX-Dia-FBS) were calculated for all replicates per condition. % oxidation for a cysteine was calculation based on heavy to light ratio via the following formula: (R/(1+R))*100, using unlogged ratios. When calculating oxidation difference, relative oxidation changes between two cellular conditions were reported by calculating the change of heavy to light ratios between treated and untreated samples.
Spliceform analysis.
Candidate proteins for spliceform analysis were obtained by filtering the SP3-Rox dataset for proteins containing at both lower and higher ratio cysteines, indicative of decreased and increased oxidation states, in response the LPS+IFNγ treatment. Splice variants were identified by querying gene names on the Ensembl genome browser. Ensembl translation protein sequences for all variants were aligned using ensemble’ multiple alignment tool, Clustal Omega. Alignments were exported and used to map Sp3-Rox-identified cysteines and their redox states.
QUANTIFICATION AND STATISTICAL ANALYSIS
For bar plots and violin plots in Figure 1F, 2B, 2D, 3D, 3E, 3F, 3I, 5B, 5C, 5F, S2C–E, S2K–L, and S5A–C, average of replicates was reported as indicated. Error bars were calculated using standard deviation (stdev). Statistical significance was calculated with paired Student’s t-tests using R stats (v 3.6.2) if applicable. * p<0.05, ** p<0.01, *** p<0.005, **** p<0.0001, NS p>0.05. For volcano plots in Figure 3G, 3H, 5C, 5F and S4D–E, variances were calculated for each sample-condition pairing and a corresponding two-sample t-test was performed using R stats (v 3.6.2) to generate p-values. p-values were adjusted for multiple comparisons using Benjamini-Hochberg procedure.
Supplementary Material
Data S1. MS data related to Figure 1.
Data S2. MS data related to Figure 2.
Data S3. MS data related to Figure 3.
Data S4. MS data related to Figure 4.
Data S5. MS data related to Figure 5.
Document S1. Figures S1–S5 and Table S1-S2
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| V5 Mouse mAb (1:3000) | Invitrogen | Cat# R960-25, RRID: AB_2556564 |
| β-Actin Rabbit mAb (1:10000) | Abclonal | Cat# AC038, RRID: AB_2863784 |
| Tomm 20 Rabbit mAb (1:2000) | Invitrogen | Cat# MA5-32148, RRID: AB_2809438 |
| ABCE1 Rabbit mAb (1:1000) | Abclonal | Cat# A9135, RRID: AB_2863666 |
| IRDye® 800CW Goat anti-Mouse IgG (1:5000) | LI-COR | Cat# 92632210, RRID: AB_621842 |
| IRDye® 800CW Goat anti-Rabbit IgG (1:5000) | LI-COR | Cat# 92632211, RRID: AB_621843 |
| IRDye® 800CW Streptavidin (1:5000) | LI-COR | Cat# 92632230 |
| Streptavidin Alexa Fluor™ 594 | Thermo Fisher | Cat# S11227 |
| Tetramethylrhodamine, ethyl ester (TMRE) | Invitrogen | Cat# T669 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| DMEM | Fisher Scientific | Cat# 11-995-073 |
| Fetal Bovine Serum | Avantor Seradigm | Cat# 1500-500 |
| Fetal Bovine Serum, Dialyzed | Thermo Fisher | Cat# 26400044 |
| Penicillin-Streptomycin (10,000 U/mL) | Thermo Fisher | Cat# 15140122 |
| Recombinant M-CSF generated from CMG14-12 culture supernatant | Takeshita et al.120 | N/A |
| Phusion polymerase | QB3 MacroLab | N/A |
| T4 ligase | Fisher Scientific | Cat# 50-811-605 |
| PEI MAX | Polysciences | 24765-1 |
| Turbo DNAfectin3000 | Fisher Scientific | Cat# 50-204-5796 |
| Lenti-X concentrator | Takara Bio | Cat# 631231 |
| D-Biotin | Combi-Blocks | Cat# SS-7910 |
| DMSO Cell culture grade | Fisher Scientific | Cat# MT25950CQC |
| DPBS, no Calcium, no Magnesium | Fisher Scientific | Cat# 14190250 |
| RIPA buffer | Fisher Scientific | Cat# AAJ62885AE |
| 10x TBS | Bioland | Cat# TBS03-03 |
| Tween™ 20 | Fisher Scientific | Cat# BP337-500 |
| Iodoacetamide alkyne (IAA) | Yan et al.28 | N/A |
| IPIAA-L | Desai et al.13 | N/A |
| IPIAA-H | Desai et al.13 | N/A |
| Pierce streptavidin agarose beads | Fisher Scientific | Cat# PI20353 |
| Urea | Fisher Scientific | Cat# U15-3 |
| 10X PBS | Bioland | Cat# PBS01-02 |
| DL-Dithiothreitol | Fisher Scientific | Cat# D107125G |
| Trypsin, TPCK Treated | Worthington-biochemical. | Cat# LS003740 |
| Biotin-azide | Yan et al.28 | N/A |
| Tris (2-carboxyethyl) phosphine (TCEP) | Fisher Scientific | Cat# 50-121-6649 |
| Tris((1-benzyl-4-triazolyl)methyl)amine (TBTA) | Click Chemistry Tools | Cat# 1061-1G |
| CuSO4 | Fisher Scientific | Cat# BP346-500 |
| Sera-Mag SpeedBeads Carboxyl Magnetic Beads, hydrophobic | GE Healthcare | Cat# 65152105050250 |
| Sera-Mag SpeedBeads Carboxyl Magnetic Beads, hydrophilic | GE Healthcare | Cat# 45152105050250 |
| Acetonitrile | Fisher Scientific | Cat# A18-20 |
| Water, Molecular Biology Grade | Fisher Scientific | Cat# BP28191 |
| Pierce™ NeutrAvidin™ Agarose | Thermo Fisher | Cat# 29200 |
| MOPS | Fisher Scientific | Cat# AAA1291422 |
| Sodium Phosphate | Fisher Scientific | Cat# S397-500 |
| NaCl | Fisher Scientific | Cat# S271-500 |
| Formic Acid | Fisher Scientific | Cat# A117-50 |
| Iodoacetamide | Fisher Scientific | Cat# AAA1471506 |
| Sucrose | Sigma-Aldrich | Cat# 84097 |
| Mannitol | Sigma-Aldrich | Cat# M9546 |
| HEPES solution | Sigma-Aldrich | Cat# H3537-500ML |
| Egtazic acid (EGTA) | Sigma-Aldrich | Cat# E3889 |
| Methanol | Sigma-Aldrich | Cat# 34860-4L-R |
| Norvaline | Sigma-Aldrich | Cat# N7502-25G |
| Chloroform | Sigma-Aldrich | Cat# 366927-4L |
| Methoxyamine hydrochloride | Fisher Scientific | Cat# ICN15540 |
| Pyridine | Sigma-Aldrich | Cat# 270407-1L |
| Ntert-Butyldimethylsilyl-N-methyltrifluoroacetamide with 1% tert-Butyldimethyl-chlorosilane | Sigma-Aldrich | Cat# 00942-10X1ML |
| Formaldehyde | Fisher Scientific | Cat# F79-1 |
| Triton X-100 | Sigma-Aldrich | Cat# 93443 |
| DAPI | Invitrogen | Cat# D1306 |
| Lipopolysaccharide (LPS) | InvivoGen | Cat# tlrl-smlps |
| Recombinant Murine IFN-γ | Peprotech | Cat# 315-05 |
| Cycloheximide | Fisher Scientific | Cat# AC357420010 |
| Unbuffered DMEM | Sigma-Aldrich | Cat# 5030 |
| Pyruvic acid | Sigma-Aldrich | Cat# 107360 |
| D-(+)-Glucose solution | Sigma-Aldrich | Cat# G8769-100ML |
| Glutamic acid | Sigma-Aldrich | Cat# G1251 |
| Oligomycin | Sigma-Aldrich | Cat# 75351 |
| Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) | Sigma-Aldrich | Cat# C290 |
| Rotenone | Sigma-Aldrich | Cat# R8875 |
| Antimycin A | Sigma-Aldrich | Cat# A8674 |
| Critical commercial assays | ||
| BCA protein assay kit | Thermo Fisher | Cat# 23227 |
| Trans-Blot Turbo Transfer System RTA Transfer Kit | BioRad | Cat# 1704272 |
| Pierce™ C18 Spin Tips & Columns | Thermo Fisher | Cat# 89873 |
| RNeasy Mini Kit | Qiagen | Cat# 74104 |
| High-capacity cDNA reverse transcription kit | Applied Biosystems | Cat# 4368814 |
| PowerUp SYBR Green qPCR Master Mix kit | Applied Biosystems | Cat# A25741 |
| Deposited data | ||
| PRIDE | This paper | PRIDE identifier PXD039626 |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | Cat# CRL-3216 RRID:CVCL_0045 |
| Cre-J2 viral supernatant Immortalized bone marrow-derived macrophages | Laboratory of Stephen Smale | N/A |
| FUGW-Cytosol-TurboID HEK293T | This paper | N/A |
| FUGW-ER-TurboID HEK293T | This paper | N/A |
| FUGW-Golgi-TurboID HEK293T | This paper | N/A |
| FUGW-Mito-TurboID HEK293T | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| Cytosol-TurboID | Branon et al.42 | Addgene Cat# 107169 |
| ER-TurboID | Branon et al.42 | Addgene Cat# 107173 |
| Golgi-TurboID | This paper | Addgene Deposit# 82589 |
| Mito-TurboID | This paper | Addgene Deposit# 82589 |
| Nucleus-TurboID | Branon et al.42 | Addgene Cat# 107171 |
| FUGW-Cytosol-TurboID | This paper | Deposit# 82589 |
| FUGW-ER-TurboID | This paper | Deposit# 82589 |
| FUGW-Golgi-TurboID | This paper | Deposit# 82589 |
| FUGW-Mito-TurboID | This paper | Deposit# 82589 |
| FUGW-Nucleus-TurboID | This paper | Deposit# 82589 |
| VSV-G | Stewart et al.121 | Addgene Cat# 8454 |
| pCMV-dR8.2 dvpr | Stewart et al.121 | Addgene Cat# 8455 |
| Software and algorithms | ||
| Image J | Schneider et al.122 | https://imagej.net/ij/index.html |
| Fragpipe | Kong et al.125, da Veiga Leprevost et al.126, Yu et al.87 |
https://fragpipe.nesvilab.org/ |
| R-4.2.3 | The Comprehensive R Archive Network (CRAN) | https://cran.rstudio.com/ |
| Thermo XCalibur | Thermo Fisher | RRID:SCR_014593 |
| ChemDraw Professional 18.0 | PerkinElmer | https://www.perkinelmer.com/category/chemdraw |
| Other | ||
| Orbitrap Eclipse™ Tribrid™ mass spectrometer | Thermo Fisher | Cat# FSN04-10000 |
| EASY-nLC™ 1200 System | Thermo Fisher | Cat# LC140 |
| Seahorse XFe96 Analyzer | Agilent | Cat# 740879 |
Significance.
Mass spectrometry-based cysteine chemoproteomics has emerged as a useful strategy to generate proteome-wide inventories of functional, ligandable, and redox sensitive cysteine residues. A central remaining challenge for these studies is the lack of stratification of cysteine residues by corresponding protein subcellular localization. Toward this end, we describe herein two novel cysteine chemoproteomics methods, Cys-LoC and Cys-LOx, that harness TurboID-catalyzed proximity labeling to enable subcellular cysteine identification and quantification of local oxidation state. Application of the Cys-LoC method, which enables straightforward subcellular fractionation, allowed for annotation of the cysteine proteomes of five subcellular compartments. Cys-LOx analysis of immortalized murine bone marrow-derived macrophages (BMDMs) uncovered mitochondrial cysteines sensitive to LPS+IFNγ stimulation. Further showcasing the value of our methods, we identify critical differences in cysteine oxidation states generated from bulk proteomes and Cys-LOx fractionated proteomes.
Highlights.
TurboID combined with cysteine chemoproteomics to capture the subcellular cysteinome
Enhance proximity labeling method to improve compartment specificity
Local Cysteine Capture (Cys-LoC) method captures the subcellular cysteineome
Oxidation (Cys-LOx) method captures the subcellular redoxome.
Acknowledgements
This study was supported by a Packard Fellowship (K.M.B.), National Institutes of Health DP2 OD030950–01 (K.M.B.), National Institutes of Health R35GM138003 and P30DK063491 (A.S.D.), W.M. Keck Foundation Award 995337 (A.S.D.), TRDRP T31DT1800 (T.Y.), NIGMS System and Integrative Biology 5T32GM008185‐33 (L.M.B.), NIGMS UCLA Chemistry Biology Interface T32GM136614 (A.R.J, M.V., and A.B.B). iBMDM cells were graciously provided by the laboratory of Dr. Stephen T. Smale. We thank all members of the Divakaruni and Backus labs for helpful suggestions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
K.M.B. is a member of the advisory board at Oncovalent Therapeutics and Matchpoint Therapeutics.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
References
- 1.Fra A, Yoboue ED, and Sitia R (2017). Cysteines as Redox Molecular Switches and Targets of Disease. Front. Mol . Neurosci 10. 10.3389/fnmol.2017.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kisty EA, Falco JA, and Weerapana E (2023). Redox proteomics combined with proximity labeling enables monitoring of localized cysteine oxidation in cells. Cell Chem. Biol 30, 321–336.e6. 10.1016/j.chembiol.2023.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang T, Gaffrey MJ, Li X, and Qian W-J (2021). Characterization of cellular oxidative stress response by stoichiometric redox proteomics. Am. J. Physiol. Physiol 320, C182–C194. 10.1152/ajpcell.00040.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bak DW, Pizzagalli MD, and Weerapana E (2017). Identifying Functional Cysteine Residues in the Mitochondria. ACS Chem. Biol 12, 947–957. 10.1021/acschembio.6b01074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abo M, and Weerapana E (2019). Chemical Probes for Redox Signaling and Oxidative Stress. Antioxid. Redox Signal 30, 1369–1386. 10.1089/ars.2017.7408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bechtel TJ, Li C, Kisty EA, Maurais AJ, and Weerapana E (2020). Profiling Cysteine Reactivity and Oxidation in the Endoplasmic Reticulum. ACS Chem. Biol 15, 543–553. 10.1021/acschembio.9b01014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paulsen CE, and Carroll KS (2013). Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem. Rev 113, 4633–4679. 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang J (2022). Oxidize me to the space. Nat. Chem. Biol 18, 680–681. 10.1038/s41589-022-00986-9. [DOI] [PubMed] [Google Scholar]
- 9.Kovalyova Y, Bak DW, Gordon EM, Fung C, Shuman JHB, Cover TL, Amieva MR, Weerapana E, and Hatzios SK (2022). An infection-induced oxidation site regulates legumain processing and tumor growth. Nat. Chem. Biol 18, 698–705. 10.1038/s41589-022-00992-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun L, Wang X, Saredy J, Yuan Z, Yang X, and Wang H (2020). Innate-adaptive immunity interplay and redox regulation in immune response. Redox Biol 37, 101759. 10.1016/j.redox.2020.101759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D’Autréaux B, and Toledano MB (2007). ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol 8, 813–824. 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 12.Bae B-I, Hara MR, Cascio MB, Wellington CL, Hayden MR, Ross CA, Ha HC, Li X-J, Snyder SH, and Sawa A (2006). Mutant Huntingtin: Nuclear translocation and cytotoxicity mediated by GAPDH. Proc. Natl. Acad. Sci 103, 3405–3409. 10.1073/pnas.0511316103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrante RJ, Shinobu LA, Schulz JB, Matthews RT, Thomas CE, Kowall NW, Gurney ME, and Beal MF (1997). Increased 3-nitrotyrosine and oxidative damage in mice with a human copper/zinc superoxide dismutase mutation. Ann. Neurol 42, 326–334. 10.1002/ana.410420309. [DOI] [PubMed] [Google Scholar]
- 14.Jaffrey SR, and Snyder SH (2001). The Biotin Switch Method for the Detection of S -Nitrosylated Proteins. Sci. STKE 2001 10.1126/stke.2001.86.pl1. [DOI] [PubMed]
- 15.Leichert LI, Gehrke F, Gudiseva HV, Blackwell T, Ilbert M, Walker AK, Strahler JR, Andrews PC, and Jakob U (2008). Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc. Natl. Acad. Sci 105, 8197–8202. 10.1073/pnas.0707723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desai HS, Yan T, Yu F, Sun AW, Villanueva M, Nesvizhskii AI, and Backus KM (2022). SP3-Enabled Rapid and High Coverage Chemoproteomic Identification of Cell-State–Dependent Redox-Sensitive Cysteines. Mol. Cell. Proteomics 21, 100218. 10.1016/j.mcpro.2022.100218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu L, Li Z, Liu K, Tian C, He J, He J, He F, Xu P, and Yang J (2020). A quantitative thiol reactivity profiling platform to analyze redox and electrophile reactive cysteine proteomes. Nat. Protoc 15, 2891–2919. 10.1038/s41596-020-0352-2. [DOI] [PubMed] [Google Scholar]
- 18.Xiao H, Jedrychowski MP, Schweppe DK, Huttlin EL, Yu Q, Heppner DE, Li J, Long J, Mills EL, Szpyt J, et al. (2020). A Quantitative Tissue-Specific Landscape of Protein Redox Regulation during Aging. Cell 180, 968–983.e24. 10.1016/j.cell.2020.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Go Y-M, Roede JR, Walker DI, Duong DM, Seyfried NT, Orr M, Liang Y, Pennell KD, and Jones DP (2013). Selective Targeting of the Cysteine Proteome by Thioredoxin and Glutathione Redox Systems. Mol. Cell. Proteomics 12, 3285–3296. 10.1074/mcp.M113.030437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arts IS, Vertommen D, Baldin F, Laloux G, and Collet J-F (2016). Comprehensively Characterizing the Thioredoxin Interactome In Vivo Highlights the Central Role Played by This Ubiquitous Oxidoreductase in Redox Control. Mol. Cell. Proteomics 15, 2125–2140. 10.1074/mcp.M115.056440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peralta D, Bronowska AK, Morgan B, Dóka É, Van Laer K, Nagy P, Gräter F, and Dick TP (2015). A proton relay enhances H2O2 sensitivity of GAPDH to facilitate metabolic adaptation. Nat. Chem. Biol 11, 156–163. 10.1038/nchembio.1720. [DOI] [PubMed] [Google Scholar]
- 22.van der Reest J, Lilla S, Zheng L, Zanivan S, and Gottlieb E (2018). Proteome-wide analysis of cysteine oxidation reveals metabolic sensitivity to redox stress. Nat. Commun 9, 1581. 10.1038/s41467-018-04003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kettisen K, Strader MB, Wood F, Alayash AI, and Bülow L (2018). Site-directed mutagenesis of cysteine residues alters oxidative stability of fetal hemoglobin. Redox Biol 19, 218–225. 10.1016/j.redox.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Backus KM, Cao J, and Maddox SM (2019). Opportunities and challenges for the development of covalent chemical immunomodulators. Bioorg. Med. Chem 27, 3421–3439. 10.1016/j.bmc.2019.05.050. [DOI] [PubMed] [Google Scholar]
- 25.Maurais AJ, and Weerapana E (2019). Reactive-cysteine profiling for drug discovery. Curr. Opin. Chem. Biol 10.1016/j.cbpa.2019.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Visscher M, Arkin MR, and Dansen TB (2016). Covalent targeting of acquired cysteines in cancer. Curr. Opin. Chem. Biol 30, 61–67. 10.1016/j.cbpa.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Backus KM, Correia BE, Lum KM, Forli S, Horning BD, González-Páez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, et al. (2016). Proteome-wide covalent ligand discovery in native biological systems. Nature 534, 570–574. 10.1038/nature18002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MBD, Bachovchin DA, Mowen K, Baker D, and Cravatt BF (2010). Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795. 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vinogradova EV, Zhang X, Remillard D, Lazar DC, Suciu RM, Wang Y, Bianco G, Yamashita Y, Crowley VM, Schafroth MA, et al. (2020). An Activity-Guided Map of Electrophile-Cysteine Interactions in Primary Human T Cells. Cell 182, 1009–1026.e29. 10.1016/j.cell.2020.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boatner LM, Palafox MF, Schweppe DK, and Backus KM (2022). CysDB: A Human Cysteine Database based on Experimental Quantitative Chemoproteomics. ChemRxiv 10.26434/chemrxiv-2022-w4h3. [DOI] [PMC free article] [PubMed]
- 31.Yan T, Desai HS, Boatner LM, Yen SL, Cao J, Palafox MF, Jami‐Alahmadi Y, and Backus KM (2021). SP3‐FAIMS Chemoproteomics for High‐Coverage Profiling of the Human Cysteinome**. ChemBioChem 22, 1841–1851. 10.1002/cbic.202000870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuljanin M, Mitchell DC, Schweppe DK, Gikandi AS, Nusinow DP, Bulloch NJ, Vinogradova EV, Wilson DL, Kool ET, Mancias JD, et al. (2021). Reimagining high-throughput profiling of reactive cysteines for cell-based screening of large electrophile libraries. Nat. Biotechnol 39, 630–641. 10.1038/s41587-020-00778-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H, Remsberg JR, Won SJ, Zhao KT, Huang TP, Lu B, Simon GM, Liu DR, and Cravatt BF (2022). Assigning functionality to cysteines by base editing of cancer dependency genes. BioRxiv 10.1101/2022.11.17.516964. [DOI] [PMC free article] [PubMed]
- 34.Bar-Peled L, Kemper EK, Suciu RM, Vinogradova EV, Backus KM, Horning BD, Paul TA, Ichu T-A, Svensson RU, Olucha J, et al. (2017). Chemical Proteomics Identifies Druggable Vulnerabilities in a Genetically Defined Cancer. Cell 171, 696–709.e23. 10.1016/j.cell.2017.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lazear MR, Remsberg JR, Jaeger MG, Rothamel K, Demeester KE, Njomen E, Hogg SJ, Rahman J, Landon R, Won SJ, et al. (2022). Proteomic discovery of chemical probes that perturb protein complexes in human cells. BioRxiv 10.1101/2022.12.12.520090. [DOI] [PMC free article] [PubMed]
- 36.He J-X, Fei Z-C, Fu L, Tian C-P, He F-C, Chi H, and Yang J (2022). A modification-centric assessment tool for the performance of chemoproteomic probes. Nat. Chem. Biol 18, 904–912. 10.1038/s41589-022-01074-8. [DOI] [PubMed] [Google Scholar]
- 37.Yang F, Gao J, Che J, Jia G, and Wang C (2018). A Dimethyl-Labeling-Based Strategy for Site-Specifically Quantitative Chemical Proteomics. Anal. Chem 90, 9576–9582. 10.1021/acs.analchem.8b02426. [DOI] [PubMed] [Google Scholar]
- 38.Zhang T, Zhu M, Zhu N, Strul JM, Dufresne CP, Schneider JD, Harmon AC, and Chen S (2016). Identification of thioredoxin targets in guard cell enriched epidermal peels using cysTMT proteomics. J. Proteomics 133, 48–53. 10.1016/j.jprot.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 39.Go Y-M, and Jones DP (2008). Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta - Gen. Subj 1780, 1273–1290. 10.1016/j.bbagen.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones DP, and Go Y-M (2010). Redox compartmentalization and cellular stress. Diabetes, Obes. Metab 12, 116–125. 10.1111/j.1463-1326.2010.01266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lam SS, Martell JD, Kamer KJ, Deerinck TJ, Ellisman MH, Mootha VK, and Ting AY (2015). Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 12, 51–54. 10.1038/nmeth.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roux KJ, Kim DI, Raida M, and Burke B (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol 196, 801–810. 10.1083/jcb.201112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Branon TC, Bosch JA, Sanchez AD, Udeshi ND, Svinkina T, Carr SA, Feldman JL, Perrimon N, and Ting AY (2018). Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol 36, 880–887. 10.1038/nbt.4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vilen Z, Joeh E, Critcher M, Parker CG, and Huang ML (2021). Proximity Tagging Identifies the Glycan-Mediated Glycoprotein Interactors of Galectin-1 in Muscle Stem Cells. ACS Chem. Biol 16, 1994–2003. 10.1021/acschembio.1c00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cho KF, Branon TC, Rajeev S, Svinkina T, Udeshi ND, Thoudam T, Kwak C, Rhee H-W, Lee I-K, Carr SA, et al. (2020). Split-TurboID enables contact-dependent proximity labeling in cells. Proc. Natl. Acad. Sci 117, 12143–12154. 10.1073/pnas.1919528117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Youn J-Y, Dunham WH, Hong SJ, Knight JDR, Bashkurov M, Chen GI, Bagci H, Rathod B, MacLeod G, Eng SWM, et al. (2018). High-Density Proximity Mapping Reveals the Subcellular Organization of mRNA-Associated Granules and Bodies. Mol. Cell 69, 517–532.e11. 10.1016/j.molcel.2017.12.020. [DOI] [PubMed] [Google Scholar]
- 47.Markmiller S, Soltanieh S, Server KL, Mak R, Jin W, Fang MY, Luo E-C, Krach F, Yang D, Sen A, et al. (2018). Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 172, 590–604.e13. 10.1016/j.cell.2017.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Na Z, Dai X, Zheng S-J, Bryant CJ, Loh KH, Su H, Luo Y, Buhagiar AF, Cao X, Baserga SJ, et al. (2022). Mapping subcellular localizations of unannotated microproteins and alternative proteins with MicroID. Mol. Cell 82, 2900–2911.e7. 10.1016/j.molcel.2022.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang T, Fassl A, Vaites LP, Fu S, Sicinski P, Paulo JA, and Gygi SP (2022). Interrogating Kinase–Substrate Relationships with Proximity Labeling and Phosphorylation Enrichment. J. Proteome Res 21, 494–506. 10.1021/acs.jproteome.1c00865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murphy MP, and Hartley RC (2018). Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov 17, 865–886. 10.1038/nrd.2018.174. [DOI] [PubMed] [Google Scholar]
- 51.Chandel NS (2015). Evolution of Mitochondria as Signaling Organelles. Cell Metab 22, 204–206. 10.1016/j.cmet.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 52.Murphy MP (2009). How mitochondria produce reactive oxygen species. Biochem. J 417, 1–13. 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Riemer J, Schwarzländer M, Conrad M, and Herrmann JM (2015). Thiol switches in mitochondria: operation and physiological relevance. Biol. Chem 396, 465–482. 10.1515/hsz-2014-0293. [DOI] [PubMed] [Google Scholar]
- 54.Han D, Canali R, Garcia J, Aguilera R, Gallaher TK, and Cadenas E (2005). Sites and Mechanisms of Aconitase Inactivation by Peroxynitrite: Modulation by Citrate and Glutathione. Biochemistry 44, 11986–11996. 10.1021/bi0509393. [DOI] [PubMed] [Google Scholar]
- 55.Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S, James AM, Cochemé HM, Reinhold J, Lilley KS, et al. (2013). Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med 19, 753–759. 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Däbritz JHM, Gottlieb E, Latorre I, et al. (2016). Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 167, 457–470.e13. 10.1016/j.cell.2016.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, and Ghosh S (2011). TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472, 476–480. 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bailey JD, Diotallevi M, Nicol T, McNeill E, Shaw A, Chuaiphichai S, Hale A, Starr A, Nandi M, Stylianou E, et al. (2019). Nitric Oxide Modulates Metabolic Remodeling in Inflammatory Macrophages through TCA Cycle Regulation and Itaconate Accumulation. Cell Rep 28, 218–230.e7. 10.1016/j.celrep.2019.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al. (2013). Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242. 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang SC-C, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY, O’Neill CM, et al. (2014). Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol 15, 846–855. 10.1038/ni.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, Luque-Martin R, Chen H-J, Boshuizen MCS, Ahmed M, et al. (2016). Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep 17, 684–696. 10.1016/j.celrep.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 62.Qin W, Zhang Y, Tang H, Liu D, Chen Y, Liu Y, and Wang C (2020). Chemoproteomic Profiling of Itaconation by Bioorthogonal Probes in Inflammatory Macrophages. J. Am. Chem. Soc 10.1021/jacs.9b11962. [DOI] [PubMed]
- 63.Qin W, Qin K, Zhang Y, Jia W, Chen Y, Cheng B, Peng L, Chen N, Liu Y, Zhou W, et al. (2019). S-glycosylation-based cysteine profiling reveals regulation of glycolysis by itaconate. Nat. Chem. Biol 10.1038/s41589-019-0323-5. [DOI] [PubMed]
- 64.Gordon EM, and Hatzios SK (2020). Chemical tools for decoding redox signaling at the host–microbe interface. PLoS Pathog 10.1371/journal.ppat.1009070. [DOI] [PMC free article] [PubMed]
- 65.Hughes CS, Moggridge S, Müller T, Sorensen PH, Morin GB, and Krijgsveld J (2019). Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc 14, 68–85. 10.1038/s41596-018-0082-x. [DOI] [PubMed] [Google Scholar]
- 66.Yan T, Palmer AB, Geiszler DJ, Polasky DA, Boatner LM, Burton NR, Armenta E, Nesvizhskii AI, and Backus KM (2022). Enhancing Cysteine Chemoproteomic Coverage through Systematic Assessment of Click Chemistry Product Fragmentation. Anal. Chem 94, 3800–3810. 10.1021/acs.analchem.1c04402. [DOI] [PubMed] [Google Scholar]
- 67.Cao J, Boatner LM, Desai HS, Burton NR, Armenta E, Chan NJ, Castellón JO, and Backus KM (2021). Multiplexed CuAAC Suzuki–Miyaura Labeling for Tandem Activity-Based Chemoproteomic Profiling. Anal. Chem 93, 2610–2618. 10.1021/acs.analchem.0c04726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu Y, Zeng R, Wang R, Weng Y, Wang R, Zou P, and Chen PR (2021). Spatiotemporally resolved subcellular phosphoproteomics. Proc. Natl. Acad. Sci 118. 10.1073/pnas.2025299118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Navarro AP, and Cheeseman IM (2022). Identification of a Golgi-localized peptide reveals a minimal Golgi-targeting motif. Mol. Biol. Cell 33. 10.1091/mbc.E22-03-0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thul PJ, Åkesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Björk L, Breckels LM, et al. (2017). A subcellular map of the human proteome. Science (80-. ) 356. 10.1126/science.aal3321. [DOI] [PubMed] [Google Scholar]
- 71.Bateman A, Martin M-J, Orchard S, Magrane M, Ahmad S, Alpi E, Bowler-Barnett EH, Britto R, Bye-A-Jee H, Cukura A, et al. (2023). UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res 51, D523–D531. 10.1093/nar/gkac1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhu L, Malatras A, Thorley M, Aghoghogbe I, Mer A, Duguez S, Butler-Browne G, Voit T, and Duddy W (2015). CellWhere: graphical display of interaction networks organized on subcellular localizations. Nucleic Acids Res 43, W571–W575. 10.1093/nar/gkv354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Falkevall A, Alikhani N, Bhushan S, Pavlov PF, Busch K, Johnson KA, Eneqvist T, Tjernberg L, Ankarcrona M, and Glaser E (2006). Degradation of the Amyloid β-Protein by the Novel Mitochondrial Peptidasome, PreP. J. Biol. Chem 281, 29096–29104. 10.1074/jbc.M602532200. [DOI] [PubMed] [Google Scholar]
- 74.Ahn BY, Trinh DLN, Zajchowski LD, Lee B, Elwi AN, and Kim SW (2010). Tid1 is a new regulator of p53 mitochondrial translocation and apoptosis in cancer. Oncogene 29, 1155–1166. 10.1038/onc.2009.413. [DOI] [PubMed] [Google Scholar]
- 75.May DG, Scott KL, Campos AR, and Roux KJ (2020). Comparative Application of BioID and TurboID for Protein-Proximity Biotinylation. Cells 9, 1070. 10.3390/cells9051070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Said HM, McAlister-Henn L, Mohammadkhani R, and Horne DW (1992). Uptake of biotin by isolated rat liver mitochondria. Am. J. Physiol. Liver Physiol 263, G81–G86. 10.1152/ajpgi.1992.263.1.G81. [DOI] [PubMed] [Google Scholar]
- 77.Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, Green R, Shen B, and Liu JO (2010). Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat. Chem. Biol 6, 209–217. 10.1038/nchembio.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chua XY, Aballo T, Elnemer W, Tran M, and Salomon A (2021). Quantitative Interactomics of Lck-TurboID in Living Human T Cells Unveils T Cell Receptor Stimulation-Induced Proximal Lck Interactors. J. Proteome Res 20, 715–726. 10.1021/acs.jproteome.0c00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Anger AM, Armache J-P, Berninghausen O, Habeck M, Subklewe M, Wilson DN, and Beckmann R (2013). Structures of the human and Drosophila 80S ribosome. Nature 497, 80–85. 10.1038/nature12104. [DOI] [PubMed] [Google Scholar]
- 80.Pichler A, Gast A, Seeler JS, Dejean A, and Melchior F (2002). The Nucleoporin RanBP2 Has SUMO1 E3 Ligase Activity. Cell 108, 109–120. 10.1016/S0092-8674(01)00633-X. [DOI] [PubMed] [Google Scholar]
- 81.Zhou Z, Zhang K, Liu Z, Gao X, Huang K, Chen C, Wang D, Yang Q, and Long Q (2021). ATPAF1 deficiency impairs ATP synthase assembly and mitochondrial respiration. Mitochondrion 60, 129–141. 10.1016/j.mito.2021.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Boniecki MT, Freibert SA, Mühlenhoff U, Lill R, and Cygler M (2017). Structure and functional dynamics of the mitochondrial Fe/S cluster synthesis complex. Nat. Commun 8, 1287. 10.1038/s41467-017-01497-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Čunátová K, Reguera DP, Vrbacký M, Fernández-Vizarra E, Ding S, Fearnley IM, Zeviani M, Houštěk J, Mráček T, and Pecina P (2021). Loss of COX4I1 Leads to Combined Respiratory Chain Deficiency and Impaired Mitochondrial Protein Synthesis. Cells 10, 369. 10.3390/cells10020369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen W, Smeekens JM, and Wu R (2016). Systematic study of the dynamics and half-lives of newly synthesized proteins in human cells. Chem. Sci 7, 1393–1400. 10.1039/C5SC03826J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Breckau D, Mahlitz E, Sauerwald A, Layer G, and Jahn D (2003). Oxygen-dependent Coproporphyrinogen III Oxidase (HemF) from Escherichia coli Is Stimulated by Manganese. J. Biol. Chem 278, 46625–46631. 10.1074/jbc.M308553200. [DOI] [PubMed] [Google Scholar]
- 86.Vu MT, Zhai P, Lee J, Guerra C, Liu S, Gustin MC, and Silberg JJ (2012). The DNLZ/HEP zinc-binding subdomain is critical for regulation of the mitochondrial chaperone HSPA9. Protein Sci 21, 258–267. 10.1002/pro.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yu F, Haynes SE, and Nesvizhskii AI (2021). IonQuant Enables Accurate and Sensitive Label-Free Quantification With FDR-Controlled Match-Between-Runs. Mol. Cell. Proteomics 20, 100077. 10.1016/j.mcpro.2021.100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Neubauer H, Clare SE, Wozny W, Schwall GP, Poznanović S, Stegmann W, Vogel U, Sotlar K, Wallwiener D, Kurek R, et al. (2008). Breast cancer proteomics reveals correlation between estrogen receptor status and differential phosphorylation of PGRMC1. Breast Cancer Res 10, R85. 10.1186/bcr2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fisher AB (2011). Peroxiredoxin 6: A Bifunctional Enzyme with Glutathione Peroxidase and Phospholipase A 2 Activities. Antioxid. Redox Signal 15, 831–844. 10.1089/ars.2010.3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cunniff B, Newick K, Nelson KJ, Wozniak AN, Beuschel S, Leavitt B, Bhave A, Butnor K, Koenig A, Chouchani ET, et al. (2015). Disabling Mitochondrial Peroxide Metabolism via Combinatorial Targeting of Peroxiredoxin 3 as an Effective Therapeutic Approach for Malignant Mesothelioma. PLoS One 10, e0127310. 10.1371/journal.pone.0127310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kim KS, Kim JS, Park J-Y, Suh YH, Jou I, Joe E-H, and Park SM (2013). DJ-1 Associates with lipid rafts by palmitoylation and regulates lipid rafts-dependent endocytosis in astrocytes. Hum. Mol. Genet 22, 4805–4817. 10.1093/hmg/ddt332. [DOI] [PubMed] [Google Scholar]
- 92.Jackson BC, Reigan P, Miller B, Thompson DC, and Vasiliou V (2015). Human ALDH1B1 Polymorphisms may Affect the Metabolism of Acetaldehyde and All-trans retinaldehyde—In Vitro Studies and Computational Modeling. Pharm. Res 32, 1648–1662. 10.1007/s11095-014-1564-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jomain-Baum M, Garber AJ, Farber E, and Hanson RW (1973). The Effect of Cycloheximide on the Interaction between Mitochondrial Respiration and Gluconeogenesis in Guinea Pig and Rat Liver. J. Biol. Chem 248, 1536–1543. 10.1016/S0021-9258(19)44223-3. [DOI] [PubMed] [Google Scholar]
- 94.. Babu D, Soenen, S. J, Raemdonck K, Leclercq G De Backer O, Motterlini R, and Lefebvre AR (2012). TNF-α/Cycloheximide-Induced Oxidative Stress and Apoptosis in Murine Intestinal Epithelial MODE-K Cells. Curr. Pharm. Des 18, 4414–4425. 10.2174/138161212802481291. [DOI] [PubMed] [Google Scholar]
- 95.Chakrabarti S, Dube DK, and Roy SC (1972). Effects of emetine and cycloheximide on mitochondrial protein synthesis in different systems. Biochem. J 128, 461–462. 10.1042/bj1280461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.De Nardo D, Kalvakolanu DV, and Latz E (2018). Immortalization of Murine Bone Marrow-Derived Macrophages. In Methods in Molecular Biology, pp. 35–49. 10.1007/978-1-4939-7837-3_4. [DOI] [PubMed]
- 97.Vijayan V, Pradhan P, Braud L, Fuchs HR, Gueler F, Motterlini R, Foresti R, and Immenschuh S (2019). Human and murine macrophages exhibit differential metabolic responses to lipopolysaccharide - A divergent role for glycolysis. Redox Biol 22, 101147. 10.1016/j.redox.2019.101147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dorresteijn MJ, Paine A, Zilian E, Fenten MGE, Frenzel E, Janciauskiene S, Figueiredo C, Eiz-Vesper B, Blasczyk R, Dekker D, et al. (2015). Cell-type-specific downregulation of heme oxygenase-1 by lipopolysaccharide via Bach1 in primary human mononuclear cells. Free Radic. Biol. Med 78, 224–232. 10.1016/j.freeradbiomed.2014.10.579. [DOI] [PubMed] [Google Scholar]
- 99.Nasta V, Da Vela S, Gourdoupis S, Ciofi-Baffoni S, Svergun DI, and Banci L (2019). Structural properties of [2Fe-2S] ISCA2-IBA57: a complex of the mitochondrial iron-sulfur cluster assembly machinery. Sci. Rep 9, 18986. 10.1038/s41598-019-55313-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Johansson C, Kavanagh KL, Gileadi O, and Oppermann U (2007). Reversible Sequestration of Active Site Cysteines in a 2Fe-2S-bridged Dimer Provides a Mechanism for Glutaredoxin 2 Regulation in Human Mitochondria. J. Biol. Chem 282, 3077–3082. 10.1074/jbc.M608179200. [DOI] [PubMed] [Google Scholar]
- 101.Gourdoupis S, Nasta V, Calderone V, Ciofi-Baffoni S, and Banci L (2018). IBA57 Recruits ISCA2 to Form a [2Fe-2S] Cluster-Mediated Complex. J. Am. Chem. Soc 140, 14401–14412. 10.1021/jacs.8b09061. [DOI] [PubMed] [Google Scholar]
- 102.Lorsbach RB, Murphy WJ, Lowenstein CJ, Snyder SH, and Russell SW (1993). Expression of the nitric oxide synthase gene in mouse macrophages activated for tumor cell killing. Molecular basis for the synergy between interferon-gamma and lipopolysaccharide. J. Biol. Chem 268, 1908–1913. [PubMed] [Google Scholar]
- 103.Lowenstein CJ, Alley EW, Raval P, Snowman AM, Snyder SH, Russell SW, and Murphy WJ (1993). Macrophage nitric oxide synthase gene: two upstream regions mediate induction by interferon gamma and lipopolysaccharide. Proc. Natl. Acad. Sci 90, 9730–9734. 10.1073/pnas.90.20.9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Held TK, Weihua X, Yuan L, Kalvakolanu DV, and Cross AS (1999). Gamma Interferon Augments Macrophage Activation by Lipopolysaccharide by Two Distinct Mechanisms, at the Signal Transduction Level and via an Autocrine Mechanism Involving Tumor Necrosis Factor Alpha and Interleukin-1. Infect. Immun 67, 206–212. 10.1128/IAI.67.1.206-212.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Al-Amin MM, Choudhury MFR, Chowdhury AS, Chowdhury TR, Jain P, Kazi M, Alkholief M, Alshehri SM, and Reza HM (2018). Pretreatment With Risperidone Ameliorates Systemic LPS-Induced Oxidative Stress in the Cortex and Hippocampus. Front. Neurosci 12. 10.3389/fnins.2018.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yamada H, Arai T, Endo N, Yamashita K, Fukuda K, Sasada M, and Uchiyama T (2006). LPS-induced ROS generation and changes in glutathione level and their relation to the maturation of human monocyte-derived dendritic cells. Life Sci 78, 926–933. 10.1016/j.lfs.2005.05.106. [DOI] [PubMed] [Google Scholar]
- 107.Ehsani-Zonouz A, Golestani A, and Nemat-Gorgani M (2001). Interaction of hexokinase with the outer mitochondrial membrane and a hydrophobic matrix. Mol. Cell. Biochem 223, 81–87. 10.1023/a:1017952827675. [DOI] [PubMed] [Google Scholar]
- 108.Parent A, Elduque X, Cornu D, Belot L, Le Caer J-P, Grandas A, Toledano MB, and D’Autréaux B (2015). Mammalian frataxin directly enhances sulfur transfer of NFS1 persulfide to both ISCU and free thiols. Nat. Commun 6, 5686. 10.1038/ncomms6686. [DOI] [PubMed] [Google Scholar]
- 109.Tuinstra RL, Burgner II JW, and Miziorko HM (2002). Investigation of the oligomeric status of the peroxisomal isoform of human 3-hydroxy-3-methylglutaryl-CoA lyase. Arch. Biochem. Biophys 408, 286–294. 10.1016/S0003-9861(02)00584-2. [DOI] [PubMed] [Google Scholar]
- 110.Cao Z, Roszak AW, Gourlay LJ, Lindsay JG, and Isaacs NW (2005). Bovine Mitochondrial Peroxiredoxin III Forms a Two-Ring Catenane. Structure 13, 1661–1664. 10.1016/j.str.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 111.Lushchak OV, Piroddi M, Galli F, and Lushchak VI (2014). Aconitase post-translational modification as a key in linkage between Krebs cycle, iron homeostasis, redox signaling, and metabolism of reactive oxygen species. Redox Rep 19, 8–15. 10.1179/1351000213Y.0000000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Van den Bossche J, O’Neill LA, and Menon D (2017). Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol 38, 395–406. 10.1016/j.it.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 113.Shalek AK, Satija R, Shuga J, Trombetta JJ, Gennert D, Lu D, Chen P, Gertner RS, Gaublomme JT, Yosef N, et al. (2014). Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 510, 363–369. 10.1038/nature13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Divakaruni AS, and Jastroch M (2022). A practical guide for the analysis, standardization and interpretation of oxygen consumption measurements. Nat. Metab 4, 978–994. 10.1038/s42255-022-00619-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Marmor-Kollet H, Siany A, Kedersha N, Knafo N, Rivkin N, Danino YM, Moens TG, Olender T, Sheban D, Cohen N, et al. (2020). Spatiotemporal Proteomic Analysis of Stress Granule Disassembly Using APEX Reveals Regulation by SUMOylation and Links to ALS Pathogenesis. Mol. Cell 80, 876–891.e6. 10.1016/j.molcel.2020.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mathew B, Bathla S, Williams KR, and Nairn AC (2022). Deciphering Spatial Protein–Protein Interactions in Brain Using Proximity Labeling. Mol. Cell. Proteomics 21, 100422. 10.1016/j.mcpro.2022.100422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fazal FM, Han S, Parker KR, Kaewsapsak P, Xu J, Boettiger AN, Chang HY, and Ting AY (2019). Atlas of Subcellular RNA Localization Revealed by APEX-Seq. Cell 178, 473–490.e26. 10.1016/j.cell.2019.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Deutsch EW, Csordas A, Sun Z, Jarnuczak A, Perez-Riverol Y, Ternent T, Campbell DS, Bernal-Llinares M, Okuda S, Kawano S, et al. (2017). The ProteomeXchange consortium in 2017: Supporting the cultural change in proteomics public data deposition. Nucleic Acids Res 45, D1100–D1106. 10.1093/nar/gkw936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Perez-Riverol Y, Bai J, Bandla C, García-Seisdedos D, Hewapathirana S, Kamatchinathan S, Kundu DJ, Prakash A, Frericks-Zipper A, Eisenacher M, et al. (2022). The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res 50, D543–D552. 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, and Teitelbaum SL (2000). TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Invest 106, 1481–1488. 10.1172/JCI11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.STEWART SA, DYKXHOORN DM, PALLISER D, MIZUNO H, YU EY, AN DS, SABATINI DM, CHEN ISY, HAHN WC, SHARP PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cordes T, and Metallo CM (2019). Quantifying Intermediary Metabolism and Lipogenesis in Cultured Mammalian Cells Using Stable Isotope Tracing and Mass Spectrometry. In Methods in Molecular Biology, pp. 219–241. 10.1007/978-1-4939-9236-2_14. [DOI] [PubMed]
- 124.Divakaruni AS, Paradyse A, Ferrick DA, Murphy AN, and Jastroch M (2014). Analysis and Interpretation of Microplate-Based Oxygen Consumption and pH Data. In Methods in Enzymology, pp. 309–354. 10.1016/B978-0-12-801415-8.00016-3. [DOI] [PubMed]
- 125.Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, and Nesvizhskii AI (2017). MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry–based proteomics. Nat. Methods 14, 513–520. 10.1038/nmeth.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.da Veiga Leprevost F, Haynes SE, Avtonomov DM, Chang H-Y, Shanmugam AK, Mellacheruvu D, Kong AT, and Nesvizhskii AI (2020). Philosopher: a versatile toolkit for shotgun proteomics data analysis. Nat. Methods 17, 869–870. 10.1038/s41592-020-0912-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. MS data related to Figure 1.
Data S2. MS data related to Figure 2.
Data S3. MS data related to Figure 3.
Data S4. MS data related to Figure 4.
Data S5. MS data related to Figure 5.
Document S1. Figures S1–S5 and Table S1-S2
Data Availability Statement
All MS data was deposited to the ProteomeXchange Consortium via the PRIDE118,119 partner repository and are publicly available as of the date of publication. Dataset identifiers are listed in the key resources table.
All code used in this paper is reported in the previous study30.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.