

Abstract
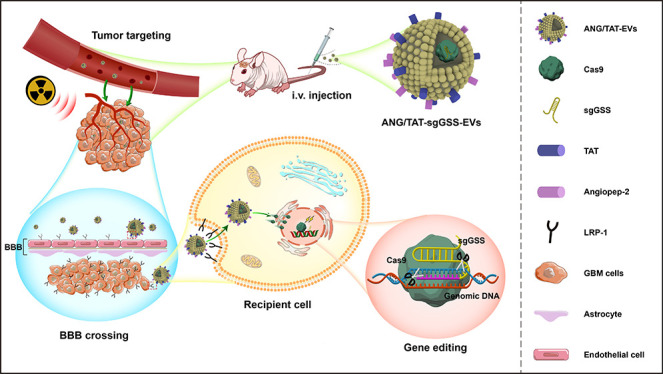
Radiotherapy is a mainstay of glioblastoma (GBM) treatment; however, the development of therapeutic resistance has hampered the efficacy of radiotherapy, suggesting that additional treatment strategies are needed. Here, an in vivo loss-of-function genome-wide CRISPR screen was carried out in orthotopic tumors in mice subjected to radiation treatment to identify synthetic lethal genes associated with radiotherapy. Using functional screening and transcriptome analyses, glutathione synthetase (GSS) was found to be a potential regulator of radioresistance through ferroptosis. High GSS levels were closely related to poor prognosis and relapse in patients with glioma. Mechanistic studies demonstrated that GSS was associated with the suppression of radiotherapy-induced ferroptosis in glioma cells. The depletion of GSS resulted in the disruption of glutathione (GSH) synthesis, thereby causing the inactivation of GPX4 and iron accumulation, thus enhancing the induction of ferroptosis upon radiotherapy treatment. Moreover, to overcome the obstacles to broad therapeutic translation of CRISPR editing, we report a previously unidentified genome editing delivery system, in which Cas9 protein/sgRNA complex was loaded into Angiopep-2 (Ang) and the trans-activator of the transcription (TAT) peptide dual-modified extracellular vesicle (EV), which not only targeted the blood–brain barrier (BBB) and GBM but also permeated the BBB and penetrated the tumor. Our encapsulating EVs showed encouraging signs of GBM tissue targeting, which resulted in high GSS gene editing efficiency in GBM (up to 67.2%) with negligible off-target gene editing. These results demonstrate that a combination of unbiased genetic screens, and CRISPR-Cas9-based gene therapy is feasible for identifying potential synthetic lethal genes and, by extension, therapeutic targets.
Keywords: glioblastoma, GSS, extracellular vesicle, CRISPR-Cas9, radiotherapy
Introduction
Glioblastoma (GBM) is the most common and malignant brain cancer.1 Following advanced standard treatment, which includes maximum surgical resection followed by chemotherapy and radiotherapy, the median survival time of GBM patients is only 12.2 to 18.2 months.2 Although radiotherapy is essential for the treatment of GBM, but its effectiveness is limited, and it endows tumor cells with additional mutations that may contribute to disease progression.3 Genomic investigations of human GBM tumor samples reveals complex genome structure, but it has been challenging to relate these alterations to GBM development and functional cell features.4,5 Identifying potential prospective therapies and providing available techniques that might be combined with radiotherapy need more profound knowledge of the molecular features of growth and therapeutic response throughout this heterogeneous malignancy.
Gene-editing technologies like CRISPR-Cas9, which allow for genome-wide genetic screens, have emerged as a powerful tool for comprehensively characterizing cancer vulnerabilities.6 CRISPR-Cas9 genome-wide screening can be used to identify genes whose deletion increases or decreases the therapeutic effectiveness of anticancer treatments.7 Several large-scale CRISPR loss-of-function experiments incorporating various cancer therapies have been made possible using cell passaging in 2D culture.8,9 By contrast, genome-wide in vivo CRISPR screening using preclinical models that more closely mimic the patient’s tumor microenvironment remains a challenge.
Although CRISPR-Cas9 has been widely used in high-throughput screens, its effectiveness in addressing unmet medical needs has yet to be fully established. Permanent disruption of oncogene by CRISPR-Cas9 gene editing offers the ability to circumvent the repetitive dosage restrictions of conventional cancer therapies, improve treatment effectiveness, and reduce therapeutic costs.10,11 However, an essential missing link in the therapeutic translation of CRISPR editing is the creation of effective and safe delivery systems for cancer. CRISPR/Cas9 therapeutic delivery using viral and nonviral-based delivery vehicles is rapidly expanding.12,13 However, therapeutic delivery of Cas9 protein is currently hampered by the large size of Streptococcus pyogenes, Cas9 exceeding the carrying capacity of viral and nonviral vectors.14 In addition, long exposure time to the Cas9 nuclease and repeated doses may raise the risk for Cas9-related immunogenicity and off-target effects.15,16 Therefore, to reduce this risk, it is necessary to develop a delivery system that may accomplish therapeutically relevant genome editing with reduced doses and a short Cas9 exposure time. Furthermore, there is an intrinsic obstacle of glioma cells to drugs along with the presence of two biological barriers, the blood–brain barrier (BBB) and the blood–brain tumor barrier (BBTB), which significantly hinder the entrance of practically all macromolecules and small-molecule medication into the tumor areas.17,18 As a result, glioma recurrence occurs and the efficacy of existing treatments is diminished. Nanometer-sized particles called extracellular vesicles (EV) are secreted by almost every cell type and play a role in the intercellular transfer of functionally active DNA, RNA, proteins, lipids, and nucleic acids.19 Moreover, EV proteins include CD47, a transmembrane protein linked with integrins that helps shield cells from phagocytosis.20 Several research groups have investigated the feasibility of using EV to transport pharmaceuticals, nucleic acids, and proteins for therapeutic purposes.21,22
In this study, we identified potential candidate genes using an in vivo genetic loss-of-function screening approach based on CRISPR-Cas9 genome editing to identify genes that increase the sensitivity or cause radioresistance in an orthotopic GBM model. Ferroptosis, triggered by the enzyme glutathione synthetase (GSS), has been shown to be a determinant in radiosensitization. Deletion of GSS impairs the synthesis of GSH, which is an important cofactor for GPX4. Impaired GSH synthesis results in the inactivation of GPX4 and iron accumulation, thereby causing lipid peroxidation-mediated ferroptosis. To surmount the existing obstacles, we designed a nonviral CRISPR-Cas9 delivery system that uses Angiopep-2 (Ang) and trans-activator of transcription (TAT) peptides dual-modified functional EVs for encapsulating and protecting the sgRNA and Cas9 protein for targeted and noninvasive gene knockout, thereby overcoming the current challenges of in vivo CRISPR-Cas9 gene editing therapy. By triggering ferroptosis, our innovative CRISPR-Cas9-brain delivery system successfully edited the GSS in vivo and enhanced radiosensitivity. The combination of CRISPR-library screening and CRISPR-Cas9-based gene therapy is a powerful approach to identifying and manipulating therapeutic targets in cancer.
Results
In vivo CRISPR-Cas9-based screening identifies GSS as a pivotal mediator of radiotherapy resistance
To start deciphering cancer radiosensitizing genes in GBM, we performed a pooled genome-wide CRISPR loss-of-function screen with a lentiviral knockout library. The sgRNA library has 123,411 total sgRNAs, six for each of 19,050 targets and 1,000 negative controls. It was assumed that making GBM cells more vulnerable to radiotherapy (RT)-induced cell death or proliferation suppression was achieved by knocking out a radioresistance driver gene. LN229, a highly tumorigenic GBM cell line, was used in the genome-wide screen. An average multiplicity of infection of 0.3 and a minimum coverage of 500 times per sgRNA were achieved after three separate biological infections of the CRISPR lentiviral library into LN229 cells. Six million puromycin-selected cells were then implanted into immunodeficient nude mice. Animals were randomly allocated to receive either vehicle control or RT for 2 weeks, beginning 1 week after injection (Figure 1A). After 14 days, mice were euthanized, and tumors were harvested 14 days later for high-throughput sequencing of sgRNA libraries (Figure 1B). While the library representation of pretransplanted tumor cells followed a log-normal distribution, the sgRNA representation in post-transplanted cells obtained from tumor masses on RT-treated mice revealed a striking change (Figure 1C). These findings confirm the efficacy of our in vivo whole genome CRISPR/Cas9 screening, indicating that selective stress in vivo leads to functional selection and alters the distribution of sgRNA throughout the tumors. High-scoring resistance genes in these pathways were analyzed by KEGG enrichment, and the findings were ranked according to the number of hits. The studies revealed that genes involved in MAPK signaling, glycine, serine, and threonine metabolism, and ferroptosis were major mediators of radiation resistance in GBM cells (Figure 1D). Of the top strand guides found to be essential for viability were genes that had previously been identified, including ABCC8, HDAC9, SOX8, and EZH223−26 (Figure 1E). Based on the results of our CRISPR/Cas9 knockout library screening, we identified a subset of sgRNAs targeting 342 genes that were substantially downregulated in the RT-treated mice in comparison with the vehicle control, suggesting that these genes might be possible drivers for radioresistance (Figure 1F). In addition to screening a CRISPR/Cas9 knockout library, RNA-seq was used to track the transcriptome changes in LN229 cells while they were subjected to RT for up to 6 weeks. The activation of the DNA damage response system and a decrease in cell death in response to RT therapy are two hallmarks of the effective creation of radioresistant cells (Figures S1–S2). Genes that are differentially expressed under long-term RT from transcriptome analysis and CRISPR/Cas9 knockout screening readouts were then taken at an intersection. On the basis of the intersection analysis, three genes (GSS, RNF8, TGM2) were overlapped and associated with the RT response in GBM (Figure 1G). Subsequently, we found that among the three genes, GSS, the major enzyme of glutathione synthesis, was identified as the gene with the most negative selection under RT conditions in GBM (Figure 1H). Moreover, we also discovered that GSS expression was much higher in the radioresistant PDX model than in the parental mode (Figure 1I). A similar trend was also found in radioresistant LN229 and primary GBM (GBM02) cell lines (Figure 1J). Successfully, we have achieved the establishment of patient-derived GBM organoids (PDOs) to investigate RT responses after treatment. PDOs were sensitive to RT when preincubated with Cas9/sgGSS adenovirus (Figure 1K). To further investigate the effects of GSS on radiosensitivity, we generated stable GSS knockout subclones in LN229 and GBM02 cells and then analyzed their impact on radiosensitivity (Figure S3). It was discovered that GSS depletion enhanced RT-induced cell death synergistically (Figure 1L–M). These findings suggest that GSS may be an essential driver for radioresistance and provide a therapeutic window in which targeting GSS in conjunction with RT may be effective in treating GBM patients.
Figure 1.

In vivo CRISPR-Cas9–based screening identifies GSS as a critical mediator of radiotherapy resistance. (A) Schematics of the experimental design. (B) Tumor volume averaged for groups indicated. n = 5 per group. (C) Cumulative distribution function of library sgRNAs in the three transduced cell replicates. (D) Circos plots displaying CRISPR screen results. Significant screen hits are ranked on the outermost rim (p < 0.05) from most sensitizing to most resistance-associated. (E) Top ten positive hits from the screen according to the robust rank aggregation (RRA) score. (F) Volcano plot displaying the log2 fold change and adjusted P value for all sgRNAs identified in the screen. (G) Venn diagrams show overlapped essential driver genes for radioresistance. (H) Differences in sgRNA counts for GSS, RNF8, and TGM2 between vehicle and RT-treated mice. (I) The expression level of GSS in parental and RT-resistant PDX GBM model. (J) Immunoblots for the sensitive and resistant GBM cell lines, showing expression levels of GSS. (K) The radiosensitization of GSS in PDOs. (L) GSS-KO cells and control sgRNA transfected cells were treated for 72 h with a range of RT doses, after which a CCK-8 assay was used to assess cell viability relative to vehicle-treated cells. (M) Colony formation assays were conducted by plating GSS-KO or control cell lines (500 cells/well) and treating them with RT or vehicle control for 2 weeks (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001; Student’s t test.
GSS expression is correlated with glioma grade and poor prognosis
To seek further insights into the role of GSS in glioma, we first employed Gravendeel, Rembrandt, TCGA, and CGGA databases. The results demonstrated a substantial correlation between GSS expression and tumor grade (Figure 2A). In addition, the GSS expression level was higher in mesenchymal (MES) subtypes than proneural (PN) subtypes, and the MES subtype of glioma often exhibited radioresistance-associated features and had a worse prognosis than those with PN glioma (Figure S4).27 Significantly, in the Gravendeel, TCGA, Rembrandt, and CGGA databases, increased expression of GSS has an important bearing on a worse prognosis (Figure 2B). The protein levels of GSS in glioma patient specimens were assessed to verify the aforementioned findings. Clinical specimens corroborated that GSS was more highly expressed in high-grade glioma than in low-grade glioma (Figure 2C–D). A tissue microarray of 180 glioma tumor samples was stained with immunohistochemistry (IHC) antibodies to determine the level of GSS protein expression in clinical specimens. From the total 180 samples, 93 showed high expression of the GSS protein on tumor cells, whereas the remaining 87 showed low expression. Moreover, GSS levels were elevated in glioma patients who eventually experienced a relapse (Figure 2E). When we divided patients into high and low GSS expression groups (using median expression value as a threshold), the percentage of patients with high GSS expression was considerably lower in the nonrelapsed patient group compared to the relapsed patient group. This information raises the possibility that GSS is linked to relapse in glioma patients. Kaplan–Meier survival analysis demonstrated that overexpression of GSS in both nonrelapsed and relapsed gliomas affected the prognosis of patients (n = 180, P < 0.0001, Figure 2F).
Figure 2.

GSS expression is correlated with glioma grade and poor prognosis. (A) The expression level of GSS was correlated with pathological stages of glioma in Gravendeel, Rembrandt, TCGA, and CGGA databases. (B) Prognostic significance of GSS up-regulation in glioma. (C–D) Immunoblots analysis of GSS expression in different grade glioma tissues. (E) Representative images of IHC staining of GSS in specimens of nonrelapsed and relapsed patients. (F) Kaplan–Meier estimate of survival time for glioma patients with low versus high expression of GSS.
GSS contributes to GBM cell resistance to radiotherapy-induced ferroptosis
As DNA damage and repair are significant effects of radiation therapy, we investigated whether radiosensitization caused by the deletion of GSS involves the response to DNA damage and its subsequent repair. As expected, RT efficiently induced H2AX phosphorylation, a hallmark of DNA damage; nevertheless, GSS deletion did not affect either the baseline level of H2AX phosphorylation or RT-induced H2AX phosphorylation (Figure S5). Base excision repair, nucleotide excision repair, and mismatch repair are DNA repair mechanisms that were not impacted by the absence of GSS (Figure S6). KEGG enrichment analysis was performed on the differentially expressed genes after GSS deletion, and it was found that ferroptosis-related genes were significantly enriched after GSS knockout (Figure 3A–B and Figure S7). GSEA analysis also revealed the enrichment of ferroptosis genomes after GSS knockout (Figure 3C). Similarly, our CRISPR screens have also identified ferroptosis as a regulator of radioresistance (Figure 1D). Recent research has shown that RT induces potent ferroptosis and that this process is a key component of the anticancer effects.28 Indeed, we observed that RT induced lipid peroxidation and ROS generation (Figure S8A). Transmission electron microscopy (TEM) shows that cancer cells treated with RT displayed the morphologic hallmark of ferroptosis—shrinking mitochondria with increased membrane density (Figure S8B). Next, we investigated the presumable role of ferroptosis in the RT induced cell death response. For this purpose, we analyzed the effect of ROS scavenger N-acetyl-l-cysteine (NAC), apoptosis inhibitor ZVAD-fmk, necroptosis inhibitor necrostatin-1, and ferroptosis inhibitor ferrostatin-1 on cell survival in RT-treated LN229 cells. We observed that, in the tested LN229 cells, exposure to RT resulted in impaired clonogenic survival. However, treatment with ferrostatin-1 or NAC partially reversed this impairment. Notably, the restoration of cell survival induced by ferrostatin-1 or NAC was comparable to or even more significant than that induced by Z-VAD-fmk or necrostatin-1 (Figure S9). We also produced LN229 subcloning resistant to ferroptosis through serial exposure to erastin and RSL3. It is noteworthy that RSL3 and erastin-resistant LN229 cells were resistant to radiotherapy (Figure S10). Our data strongly demonstrate that RT induces ferroptosis in GBM cells. Furthermore, our findings indicate that ferroptosis is of momentous significance as a component of the cell death response triggered by RT.
Figure 3.

GSS contributes to GBM cell resistance to radiotherapy-induced ferroptosis. (A) Heatmap indicating the top 10 upregulated and downregulated genes between sgNC and sgGSS LN229 cells. (B) KEGG enrichment analysis of genes differentially expressed after GSS deletion. (C) GSEA demonstrating that ferroptosis-related genes were significantly enriched following GSS deletion. (D) Representative images of 4-HNE, PTGS2, GPX4, and GSS IHC staining of matched PDX GBM samples before and after radiotherapy. (E) Immunoblots analysis of ACSL4, SLC7A11, GPX4 and GSS expression in LN229 and GBM02 cell lines at 24, 48, and 72 h after exposure to 12 Gy of RT. (F) The effect of GSS deletion on RSL3/Erastin-resistant LN229 cells. (G) Detection of living and dead cells. (H) TEM images of LN229 cells (GSS deletion) with or without RT. Nu, nucleus; red arrows, mitochondria; yellow arrows, autophagosomes; black arrows, necrosis-related vacuoles. (I) The ratio of oxidized to nonoxidized lipids. (J) Confocal microscopy visualized the alterations in lipid peroxidation in LN229 cells after C11-BODIPY probe staining. Scale bar = 10 μm. (K) Liperfluo staining visualized lipid ROS in cells after treatment. (L) GSEA analysis of differentially abundant metabolites identified in GSS deletion LN229 cells. (M) The glutathione (GSH)-to-oxidized glutathione (GSSG) ratio was detected by flow cytometry. (N) The expression level of lipid peroxidation products (MDA). (O) The ratio of oxidized to nonoxidized lipids was assessed by flow cytometry. (P) Detection the activity of GPX4. (Q) The level of intracellular ferrous ions (Fe2+) was measured by FerroOrange probes. Scale bar = 10 μm. (R) Mechanism Diagram of GSS.
Then, we made further efforts to dig into whether GSS modulates radiosensitivity in GBM cells by regulating ferroptosis. In GBM PDX models, RT induced ferroptosis as evidenced by elevated expression of PTGS2 and 4-HNE (Figure 3D). Intriguingly, we demonstrated that RT also significantly suppressed the production of GPX4, which reduced glutathione to convert lipid hydroperoxides to lipid alcohols, lowering the lipid peroxidation rate and preventing ferroptosis (Figure 3D). Then, 12 Gy of RT was applied to the GBM cells, the threshold at which all wild-type cells die. We found that RT-treated GBM cells exhibited a similar tendency with the PDX model in several critical components involved in ferroptosis (Figure 3E). Of note, GSS was significantly decreased in ferroptosis cells caused by RT in vitro and in vivo. Subsequently, we confirmed that GSS deletion elevated the radiotherapy efficacy in erastin and RSL3-resistant LN229 cells (Figure 3F). We showed that GSS deletion significantly increased cell death induced by RT; interestingly, deletion of GSS had no additional effect on cell death in cells treated with ferrostatin-1, indicating that GSS increases radiosensitivity primarily through modulation of ferroptosis (Figure 3G). TEM revealed a distinctive morphological characteristic of ferroptosis in cells subjected to GSS deletion (Figure 3H). The percentage of oxidized lipids relative to total lipids was significantly higher after GSS deletion, as shown by C11-BODIPY probe staining (Figure 3I). The alterations in lipid peroxidation in LN229 cells were observed by confocal microscopy (Figure 3J). Flow cytometry also showed that after GSS deletion lipid ROS gradually accumulated on the cytomembrane (Figure 3K).
In order to better understand how GSS regulates ferroptosis vulnerability, we conducted a metabolomics analysis to examine the effect of GSS deletion on the levels of metabolites in GBM cells. GSEA analysis of identified variable-abundance metabolites showed that they were associated with amino acid metabolism, Glycine, serine, and threonine metabolism (Figure 3L). Reduced GSH is produced when λ-glutamyl cysteine reacts with glycine, catalyzed by GSS. GSH, an important antioxidant, is a cofactor for the enzyme selenium GPX4, a GSH-dependent lipid hydroperoxidase that is critical for the elimination of lipid ROS.29 It is perfectly understandable that the GSH deficiency can lead to ferroptosis. Notably, knockout GSS led to a significant drop in GSH and increased oxidized glutathione levels (GSSG) (Figure 3M). Additionally, GSS-deficient cells had a significantly increased level of the lipid peroxidation product (MDA) (Figure 3N). The addition of exogenous GSH mitigated lipid peroxidation caused by RT. The ratio of GSH to GSSG was suppressed using 2-AAPA, a dithiocarbamate GR inhibitor. As expected, 2-AAPA induces the accumulation of GSSG, which elevates lipid peroxidation once RT is administered (Figure 3O). In addition, as GSH is a cofactor for GPX4, impaired GSH synthesis caused by GSS depletion results in reduced GPX4 activity, thereby causing the accumulation of lipid peroxidation products (Figure 3P). Consistently, the expression of GPX4 was strongly linked with the GSS level in the Gravendeel, Rembrandt, TCGA, and CGGA databases (Figure S11). GSH also has a role in detecting and regulating iron levels, iron trafficking, and the production of iron cofactors, making it an essential molecule in iron metabolism.30 Then, we used FerroOrange probes to determine the concentration of ferrous ions (Fe2+) within the cells. The labile iron pool increased after GSS deletion, which authenticated the function of GSS in iron homeostasis (Figure 3Q). Accumulation of the labile iron pool can lead to the Fenton reaction, by which excessive hydroxyl radicals, a kind of ROS, can be produced. Our findings showed that GSS deletion drastically reduces GSH synthesis and Fe2+ efflux efficiency, increasing lipid peroxidation and accumulating labile iron pools (Figure 3R).
Development and Characterization of EVs Encapsulating Cas9 Protein and sgRNA
Although CRISPR-Cas9 marks a significant step forward in gene editing technology, there are several obstacles to overcome before it can be safely delivered to the brain using existing delivery techniques. Therefore, a crucial gap in the therapeutic translation of CRISPR editing is the development of an effective and safe delivery system for glioma. To overcome the limitation of delivery of the Cas9/sgRNA complex, we developed a method where Cas9/sgRNA can be loaded into extracellular vesicles to treat different brain disorders. Cas9 protein, rather than plasmid DNA, was chosen because it limits the nuclease’s exposure time, reducing the likelihood of immunogenicity and off-target effects. Chemical modification of sgRNA with 5-methoxyuridine improved the RNA stability and reduced immunogenicity. Peptides like Angiopep-2(Ang) have a high affinity for LRP1 and can easily cross the blood–brain barrier.31 Nanocapsules modified with Ang peptides may have improved ability to penetrate the BBB and deliver genetic drugs to the brain. The TAT peptide is a potent cell-penetrating peptide that can enter the nucleus of most live cells through the plasma membrane.32 TAT can stimulate internalization through an unsaturated and receptor/transporter-independent mechanism, allowing it to cross BBB biofilm barriers and access dense tumor tissues readily. The dual peptide-modified EVs not only benefit from the effective cell membrane penetration mediated by TAT and the targeting capacity to glioma mediated by Ang but also circumvent Ang receptor saturation due to the synergistic effects of Ang and TAT. In order to gain stable expressions of Ang-Lamp2b-FLAG and TAT-Lamp2b-HA fusion proteins, the HEK293T cells were infected with the lentivirus particles (Figure 4A). Ang-Lamp2b-FLAG and TAT-Lamp2b-HA were significantly expressed in transfected HEK293T, as shown by Immunoblot (Figure 4B). Nanoparticle tracking analysis (NTA) showed that the morphology and size distribution of the EVs were unaffected by the peptide changes (Figure 4C). TEM revealed that all EVs took the shape of spherical vesicles, each of which had a lipid bilayer and a size distribution of 125 ± 40 nm (Figure 4D).
Figure 4.

Development and characterization of EVs encapsulating Cas9 protein and sgRNA. (A) Ang and TAT peptide are modified to EVs membrane surface to obtain engineered EVs with glioma targeting and tumor-penetrating functions. (B) Biomarkers of EVs detected by Immunoblot. (C–D) DLS and TEM the image of purified EVs. (E) Loading capabilities of various exogenous approaches. (F) Immunoblot analysis to determine the loading efficiency of Cas9 protein in EVs. (G) Frequency of GSS indel mutation detected by T7EI assay. (H) DNA sequencing results of GSS gene editing in LN229 cells treated with Ang/TAT-sgGSS-EVs.
In the preliminary experiment, we attempted to load the Cas9 protein/sgRNA complex into purified EVs by electroporation, freeze–thawing, and sonication. Immunoblot showed that Cas9 protein was effectively electroporated into the isolated EVs. However, the other two approaches showed ineffective loading of Cas9 protein into the EVs, as shown by the low-intensity band of Cas9 (Figure 4E). When it comes to loading large molecules like nucleic acids and protein drugs, electroporation has the potential to outperform other approaches while remaining relatively inexpensive. After electroporation-assisted Cas9/sgRNA complex loading, no apparent EV size or morphology alterations were seen (Figure S12A). Following this, the Ang/TAT-EVs were analyzed for their stability using DLS. Extended incubation in either serum-free or serum-containing solution did not lead to a growth in particle size, as illustrated in Figure S12B. Immunoblot study revealed that the Cas9 protein loading efficiency of Ang/TAT-EVs was almost 36.99% (Figure 4F).
The assays of T7 endonuclease I (T7EI) cleavage were used for quantifying the GSS gene editing efficiency and the capacity of our CRISPR-Cas9 EVs to shield sgRNA from RNase degradation. The capacity of free Cas9/sgRNA to cleave target DNA was inhibited, however, by the presence of RNase. Ang/TAT-sgGSS-EVs had superior gene editing efficiency, regardless of RNase presence (Figure 4G–H). This finding demonstrated the ability of Ang/TAT-sgGSS-EVs to protect the Cas9/sgRNA complex from enzymatic hydrolysis, indicating that these EVs might be an appropriate protective delivery method for performing gene in vivo gene editing. Functionally, Ang/TAT-sgGSS-EVs mediated gene editing could decrease the GSH level and lead to GPX4 inactivation in vitro (Figure S12C–D).
In vitro Assessments of Targeting Efficiency, Cellular Uptake, and Genome-Editing Efficiency
To evaluate the BBB transportation ability of the peptide-modified EVs, an in vitro BBB model was constructed by culturing a monolayer of bEnd.3 cells. The confocal microscopy imaging demonstrated that Ang/TAT-EVs possess the highest targeting efficiency compared to that of single peptide-modified EVs and unmod-EVs (Figure 5A–B). Moreover, subcellular analyses revealed colocalization of eSpCas9-GFP and sgGSS-cy3 in LN229 cells (Figure 5B). Cellular internalization of the EVs described above was detected by semiquantitative flow cytometry. The findings demonstrated that Ang/TAT modification enhanced intracellular GFP fluorescence compared to that of monopeptide modification groups (Figure 5C). Interestingly, DID-labeled EVs were significantly accumulated in the interior area of PDOs receiving Ang/TAT-EVs treatment, supporting Ang/TAT-EVs effectively penetrated into GBM tissues. In contrast, PDOs receiving the treatment of unmod-EVs, TAT-EVs, and Ang-EVs displayed DiD fluorescence only in the superficial area (Figure 5D). Using the T7EI assay, we found that mutations in the target GSS gene occurred with a frequency of 58.6% for Ang/TAT-sgGSS-EVs, 14.7% for TAT-EVs, 10.8% for Ang-EVs, and 2.5% for unmod-EVs (Figure 5E). The level of gene editing achieved using Ang/TAT-sgGSS-EVs was much more significant than EVs edited with a single peptide. NGS sequencing was used to analyze the frequency of insertions, deletions, and substitutions at GSS target sites in LN229 cells treated with Ang/TAT-sgGSS-EVs. This analysis revealed a mutation frequency of 67.2%, consisting of single base substitutions and large deletions (Figure 5F).
Figure 5.

In vitro assessments of targeting efficiency, cellular uptake, and genome-editing efficiency. (A) Schematic image of the BBB model in vitro. (B) Immunofluorescence images detected unmod-EVs, Ang-EVs, TAT-EVs and Ang/TAT-EVs uptake into LN229 cells after passing through a bEnd.3 monolayer. Scale bar, 10 μm. (C) The cellular internalization of the EVs were detected by flow cytometry. (D) The penetrating efficacy of Ang/TAT-sgGSS-EVs was evaluated in PDOs. (E) Frequency of GSS indel mutation. (F) Sequencing results of GSS gene editing in PDOs. (G) Detection of living and dead cells in PDOs. (H) The ratio of oxidized to nonoxidized lipids was assessed by flow cytometry. (I) The expression level of lipid peroxidation products (MDA). (J) Liperfluo staining visualized lipid ROS in cells after treatment. (K) The mitochondrial membrane potential MMP (Δψm) after treatment.
To test the possibility that Ang/TAT-sgGSS-EVs regulate the susceptibility of GBM cells to radiotherapy, calcein AM/propidium iodide (PI) was used to detect live and dead cells in PDOs. The PI-positive (PI+) population increased following Ang-sgGSS-EVs and RT treatment in the PDOs (Figure 5G). Further, Ang/TAT-sgGSS-EVs counterparts with RT induced massive lipid peroxidation (Figure 5H). In addition, Ang/TAT-sgGSS-EVs induced a dramatic increase in ROS accumulation and MDA levels induced by RT (Figure 5I–J). JC-1 staining detects mitochondrial dysfunction. The results showed that after treatment with Ang/TAT-sgGSS-EVs, the mitochondrial membrane potential MMP (Δψm) decreased significantly, which was manifested by an increase in the ratio of green-red fluorescence (Figure 5K). Thus, Ang/TAT-sgGSS-EVs efficiently disrupted the targeted gene and aggravated ferroptosis caused by RT.
Safety Evaluation of CRISPR-Cas9 EVs in Vivo
The three major concerns about CRISPR-Cas9 therapeutic—off-target effects, potential toxicity, and immunogenicity—needed to be addressed before we could evaluate the therapeutic potential of Ang/TAT-sgGSS-EVs for glioma. As a result, we pinpointed the tumor areas most likely to contain off-target GSS sequences (Table S2). After administration of Ang/TAT-sgGSS-EVs to LN229-bearing mice, genetic disruption at these putative loci within the tumor tissue was not apparent on NGS examination. Across all five putative target sites in these models, the mutation frequency was below 0.5%. Off-target effects in normal brain tissue must also be analyzed to guarantee the security of EVs. According to data derived from NGS, mutation frequencies were lower than 0.5%, indicating negligible effects on normal brain tissues. Since EVs tend to accumulate in the heart, liver, and kidney, we investigated possible off-target effects on these organs. Reassuringly, we discovered that the mutation frequency in the LN229-bearing mice’s heart, liver, and kidneys were below 0.5% in five most likely off-target sites (Figure S13). The low off-targets of Ang/TAT-sgGSS-EVs may be due to decreased expression of LRP-1 receptor in 18 normal systemic organs as detected by human multiple organ normal tissue chip (Figure S14A). In addition, we also analyzed the expression of LRP-1 in a public database of The Human Protein Atlas. The results demonstrated that LRP-1 is rarely expressed in kidney, colon, testis, lymph, liver, and brain (Figure S14B). Subsequently, the healthy BALB/c mice were intravenously given Ang/TAT-sgGSS-EVs every other day three times to evaluate the immune response and toxicity induced by Ang/TAT-sgGSS-EVs. Throughout treatment, mice given Ang/TAT-sgGSS-EVs exhibited blood parameters and biochemical profiles identical to those of mice given saline (Figure S15A–B). Mice also kept their weight, suggesting that Ang/TAT-sgGSS-EVs had little to no effect on hematological parameters or kidney and liver function (Figure S15C). Comparing plasma levels of a panel of cytokines—including IL-10, IL-1β, TNF-α, IFN-γ—revealed no discernible variations (Figure S15D). These results suggest that Ang/TAT-sgGSS-EVs are neither toxic nor immunogenic when administered systemically at therapeutically relevant doses. However, further evaluation of the potential toxicity is required for preclinical development.
Targeted delivery of the Ang/TAT-sgGSS-EVs sensitizes GBM to radiotherapy
Orthotopic LN229-bearing mice were developed to study the biodistribution of Ang/TAT-EVs and their potential to traverse the BBB and BBTB in vivo (Figure 6A). Fluorescence images were captured at different time points using an IVIS Spectrum system after intravenous administration of saline or EVs (TAT-EVs, Ang-EVs, Ang/TAT-EVs, and Unmod-EVs = 1 mg/kg for each mouse). The Ang/TAT-EVs group had stronger brain fluorescence signals than the TAT-EVs, Ang-EVs and Unmod-EVs groups (Figure 6B). After in vivo imaging, the organs and brain of each group of mice are taken out for ex imaging. Luminescence channels indicate the region where the tumor is located, AF647 channels reveal the distribution of EVs (labeled with DID) in different organs, and results showed that AF647 fluorescent signal was the brightest in the Ang/TAT-sgGSS-EVs group compared with other groups. Then, the brains of each group were made into frozen sections (10um) and observed under a confocal microscope. Cas9 is labeled with GFP, sgGSS is labeled with Cy3, and EV is labeled with DID. There are more GFP, Cy3, and DID fluorescence in tumor tissue in the Ang/TAT-sgGSS-EVs treatment group, suggesting that Ang/TAT-sgGSS-EVs exhibited superior penetration of the BBB and accumulation within brain tumor tissue compared to the other three groups. More importantly, Ang/TAT-sgGSS-EVs were distributed mainly inside the tumor boundary, suggesting their superior tumor penetration ability (Figure 6C). Compared with Unmod-EVs, TAT-EVs are much more effective in penetrating the glioma area. It is highly probable that the transmembrane peptide TAT enhances EVs’ high BBB permeability and tumor penetration.
Figure 6.

Targeted delivery of the Ang/TAT-sgGSS-EVs sensitizes GBM to radiotherapy. (A) Schematic illustration of EVs for in vivo delivery of Cas9 protein and sgRNA for the treatment of brain tumor. (B) Fluorescence images of orthotopic LN229-bearing nude mice following injection of different types of EVs. (C) Luciferase luminescence, sgGSS-cy3, Cas9-GFP and DID-labeled EVs fluorescence from major organs in orthotopic mice model after intravenous injection of different types of EVs. (D) Schematic illustration of the timeline of the LN229 orthotopic tumor model study. (E) Quantified luminescence levels of mice following the indicated treatments. (F) Tumor growth and volume quantification. (G) Survival curves of LN229-bearing mice following the indicated treatments. n = 8 animals per treatment group. (H) Indel frequency of GSS gene in tumor tissues excised from mice on day 28. (I) Sequencing results of GSS gene editing in LN229-bearing mice treated with Ang/TAT-sgGSS-EVs. (J) IHC analysis of GSS and 4-HNE expression in tumor tissues.
Next, we evaluated whether Ang/TAT-sgGSS-EVs can promote radiosensitivity in an orthotopic LN229 model. Tumor signal was detected by bioluminescence imaging, and mice with comparable tumor sizes were randomly split into four groups. Bioluminescence images were captured every 3 days to track the development of the tumor (Figure 6D). We found that systemic administration of Ang/TAT-sgGSS-EVs (Once a week through the tail vein) improved the therapeutic benefit of radiotherapy, and the effectiveness of the combination regimen of RT plus Ang/TAT-sgGSS-EVs was found to be more potent than that of RT and Ang/TAT-sgGSS-EVs alone (Figure 6E). Importantly, our study revealed a statistically significant decrease in tumor volume when subjected to the combination treatment (Figure 6F). Notably, Ang/TAT-sgGSS-EVs combined with RT resulted in a longer survival than other groups (Figure 6G). Tumor tissues were collected from mice with different treatment, and evaluated using T7EI assay and NGS to demonstrate that the tumor growth suppression was caused by GSS gene disruption. Treatment with Ang/TAT-sgGSS-EVs resulted in a 61.8% indel frequency, but treatment with RT or saline resulted in no detectable cleavage of GSS (Figure 6H). NGS data also verified GSS gene disruption; the overall mutation frequency (including single base substitutions, large deletions, and base deletions) was 65.8%, consistent with the results of T7EI (Figure 6I). Thus, loss of GSS function results in a dramatic decrease in GSH levels accompanied by an increase in lipid peroxidation products (Figure S16). Consistent with the observed suppression of tumor development, IHC staining revealed that the combination RT and Ang/TAT-sgGSS-EVs resulted in a supraadditive decrease in GSS protein level. Furthermore, 4-HNE staining intensity was dramatically improved by the combination therapy (Figure 6J). Collectively, our findings suggest that radiotherapy could benefit from the induction of ferroptosis by Ang/TAT-sgGSS-EVs.
Assessment of the Effect of Ang/TAT-sgGSS-EVs on Patient-Derived GSC Xenografts
Before developing a patient-derived xenograft mouse model, the gene and protein expression of GSS was first confirmed in patient-derived glioma stem cell-05 (GSC05). Patient-derived GSCs had the GSS target sequence, as determined by DNA sequencing, and GSS protein expression was found to be on par with that of the LN229 cells, validating the suitability of the GSC05 model (Figure 7A–B and Figure S17). In addition, the LRP-1 protein, which is required for enhanced recognition by targeted EVs, was overexpressed in GSC05 cells (Figure 7B). To investigate the therapeutic potential of our CRISPR/Cas9 EVs, we first developed stable luciferase-expressing GSC05 cells (GSC05-Luc) to establish a facile bioluminescence-based orthotopic GSC mouse model. Treatment with Ang/TAT-sgGSS-EVs plus RT significantly suppressed tumor development, as evidenced by a reduced luminescence intensity (Figure 7C). The powerful anticancer effect of Ang/TAT-sgGSS-EVs was further validated by H&E staining of removed brain tissue (Figure 7C–D). The median survival time of mice who received combined therapy was 46 days, substantially greater than that of mice given monotherapy (Figure 7E). Immunohistochemical analysis revealed lower GSS expression but higher 4-HNE expression in tumor tissue slices excised from mice receiving combined treatment compared to other groups (Figure 7F). Significantly decreased GSH levels and increased lipid peroxidation products were found in the combined treatment group (Figure 7G). As measured by the T7EI assay, GSS gene disruption was triggered by Ang/TAT-sgGSS-EVs, and lower GSS protein expression in GSC tumor tissue corroborated this finding (Figure 7H). Consistent with the T7EI assay, GSS gene disruption was confirmed by NGS, revealing a mutation rate of 57.2% (Figure 7I). These findings were similar to those from the LN229 xenograft study. Collectively, the above data show that Ang/TAT-sgGSS-EVs can effectively cross the BBB, increase tumor accumulation and retention, and activate Cas9/sgRNA uptake and intracellular release in tumor cells, leading to excellent gene editing efficiency and radiosensitization efficacy in vivo.
Figure 7.

Therapeutic effect of Ang/TAT-sgGSS-EVs on patient-derived GSC xenografts. (A) Schematic of patient-derived xenograft (PDX)–derived GSC orthotopic model establishment. (B) GSS and LRP-1 protein expression in LN229, GSC05 and NHA, respectively. (C) Luminescence images of orthotopic GSC05-Luc GSC tumor–bearing mice following treatment. (D) Tumor growth and volume quantification. Survival curves of LN229-bearing mice. n = 8 animals per treatment group. (E) IHC analysis of GSS and 4-HNE expression in tumor tissues. (F) Relative level of MDA and GSH. (H) Indel frequency of GSS gene in tumor tissues excised from mice on day 28. (I) Sequencing results of GSS gene editing in GSC05-bearing mice treated with Ang/TAT-sgGSS-EVs.
Discussion
Since the limited BBB permeability to current chemo- and immunotherapies, the clinical course of GBM has not changed for over a decade.33 As radiotherapy has been shown to be an effective treatment for GBM, our primary goal in this research was to uncover potential pharmacological targets that might synergistically boost the therapeutic effectiveness of current radiotherapy in this disease.34 To achieve this, a library of sgRNAs was used for in vivo CRISPR loss-of-function screening to identify radiosensitizing genes whose depletion might lead to synthetic lethality with radiotherapy. GSS was found to be a promising therapeutic target for treating GBM by combining findings from genome-wide CRISPR screening with assessments of clinical cohorts. It was also indicated that GSS expression is significantly elevated in HGG glioma patients compared to LGG glioma patients in the TCGA, CGGA, Rembrandt, and Gravendeel databases; more critically, higher GSS expression is related to considerably lower survival in patients with glioma. Since HGG gliomas tend to have greater levels of GSS expression, this suggests that GSS has a potentially more potent selective effect on glioma cells when combined with radiotherapy.
Mechanistically, GSS was found to be closely related to the regulation of ferroptosis, and the knockout of GSS resulted in the up-regulation of ferroptosis-related genes in glioma cells. Indeed, our research and that of others have revealed that RT can effectively induce lipid peroxidation and ferroptosis in cell lines, xenograft tumors, and glioma patients, indicating that ferroptosis represents an important part of RT-mediated anticancer effects. We further investigated whether GSS regulates the radiosensitivity of glioma cells by regulating ferroptosis. Our results showed that GSS deletion significantly increased RT treatment-induced lipid peroxidation and ROS production; importantly, GSS depletion did not further promote cell death in ferrostatin-1-treated cells, suggesting that GSS mainly promotes ferroptosis to increase radiosensitivity. GPX4 is a major detoxification protein involved in ferroptosis and functions as a lipid peroxide scavenger.35 When GPX4 is inactivated via either direct or indirect targeting, this can result in the induction of ferroptotic cancer cell death.36 GSH acts as a cofactor that allows GPX4 to scavenge intracellular lipid peroxides. Our comprehensive RNA-seq and metabolomics analysis show that GSS was able to sustain GSH synthesis, thereby resulting in inactivation of GPX4. Meanwhile, GSH has a crucial role in sensing and regulation iron levels, iron trafficking, and biosynthesis of iron cofactors.37 As we have shown, GSS plays an extremely important role in maintaining the cellular iron balance in ferroptosis and its deletion adds to the pool of free intracellular iron. Excess iron in cells triggers the Fenton reaction, which produces large amounts of hydroxyl radicals, which are a type of reactive oxygen species (ROS). These findings suggested that GSS had a causal effect on the cancer cell susceptibility to ferroptosis. Our findings point to GSS as a major cause and biomarker of radiotherapy-induced ferroptosis.
Although significant progress has been made in improving the efficiency and safety of CRISPR-Cas9 gene editing, safe delivery methods that can edit effectively in vivo would help broad clinical translation.38 Three approaches are available for the delivery cargos, namely, viral vector encoding Cas9 and sgRNA, Cas9 mRNA plus sgRNA, and Cas9 protein complexed with sgRNA.39 Among them, Cas9 protein delivery avoids many of the pitfalls encountered by DNA and mRNA during transcription and translation, allowing for rapid initiation of editing with minimal immune response and limited off-target activity. However, the large size of the Cas9 protein (160kd) exceeds the loading capacity of nonviral and viral vectors, which is presently a bottleneck in the therapeutic delivery of Cas9 protein. Furthermore, degradation or denaturation of Cas9 protein-based genome editing systems during formulation and circulation further restricts their therapeutic applications.40 Although some nonviral vectors can prevent Cas9/sgRNA protein degradation in blood, tissue-specific genome editing after systemic administration remains a challenge. To address these issues, we engineered and tested a nonviral extracellular vesicle (EV) system for CRISPR-Cas9 gene editing called Ang/TAT-EVs (Cas9/sgRNA), which effectively can protect and encapsulate sgRNA and Cas9 protein for targeted and noninvasive gene knockdown, achieving gene editing efficiencies of 67.2% in vivo. Functional EVs modified with Ang peptides can efficiently cross the BBB and target glioma. In the meantime, TAT peptide-modified EVs can traverse the cell membrane and overcome Ang receptor saturation on the cell membrane, which boosts the BBB permeability and promotes tumor penetration. We show that our design system (up to 67% gene editing efficiency) significantly improved over previous nanoparticle-based drug delivery systems, which only manage an editing efficiency of 30–40% in target organs.41 These encouraging results probably account for the aggravated ferroptosis and increased median survival seen in patient-derived GSC xenografts and the LN229-orthotopic model after treatment with Ang/TAT-sgGSS-EVs plus RT.
Given the additional safety risk of off-target gene editing of bystander cells in converting CRISPR technologies to clinical applications, it has been crucial to thoroughly investigate the possible off-target impact of Ang/TAT-sgGSS-EVs in normal tissues as well as glioma. The encapsulation of Cas9 protein rather than plasmid or mRNA and particular targeting modification accounted for the low rate of off-target side effects (<0.5%) seen after systemic delivery of Ang/TAT-sgGSS-EVs. Another element acting to decrease off-target effects is that expression of GSS and LRP-1 is much lower in normal brain tissue compared to glioma. Immunogenicity represents another bottleneck restriction for bringing CRISPR technology to the clinic; our results suggest that Ang/TAT-sgGSS-EVs are not immunogenic when supplied systemically at therapeutically relevant dosages.
Conclusion
In conclusion, our research employed in vivo unbiased whole genome CRISPR-library screening to comprehensively identify GSS and ferroptosis as the most important gene and pathways associated with radioresistance. By validating and characterizing GSS, we showed that perturbation of this gene in glioma cells could obtain significant antitumor activity combined with radiotherapy. Low delivery efficiency and lack of tissue selectivity are significant barriers to using CRISPR-Cas9-based genome editing. We developed an Ang/TAT dual decorated EVs as a noninvasive brain delivery system. We systemically showed its superior properties, such as improved CRISPR-Cas9 loading efficiency, excellent glioma targeting and penetration potentials, negligible off-target side effects, and outstanding gene editing efficiency in preclinical animal models, thus efficiently addressing the bottlenecks (weak tumor tissue targeting, low BBB/BBTB penetration, unwanted off-target effects, and low in vivo gene editing efficiency) in CRISPR-Cas9 brain delivery toward a safe and effective method for glioma gene therapy. Our research demonstrates a strategy for integrating CRISPR library screening with CRISPR-Cas9 genome editing therapy; this system is a comprehensive and effective platform for the treatment of GBM and all other types of cancer.
Methods
Cell Culture
Human GBM cell line LN229 was obtained from ATCC and cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin (NCM, Suzhou, China). Patient-derived GBM cell lines (GBM02) and glioma stem cells (GSC05) were isolated from GBM surgical specimens and cultured in DMEM/F12 medium supplemented with 10% FBS and 1% penicillin/streptomycin.
Genome-wide CRISPR Screen
The LN229 cells were transduced with the GeCKO v2A library, which consists of 123,411 sgRNA sequences targeting 19,050 human genes and 1864 miRNAs. The library contains 6 sgRNAs per gene, 4 sgRNAs per miRNA, and 1000 nontargeting controls. Transduction was performed at a low MOI (approximately 0.3) to ensure the efficient barcoding of single cells. The workflow for this targeted genetic screening is shown in Figure 1A.
Antibodies and Drugs
Anti-GSS (sc-166882, RRID: AB_10611504) antibody was obtained from Santa Cruz, anti-GSS (15712-1-AP, RRID: AB_2878171); anti-GAPDH (10494-1-AP, RRID: AB_2263076), anti-PTGS2 (66351-1-Ig, RRID: AB_2881731) antibody were obtained from Proteintech; and anti-GPX4 (ab125066, RRID: AB_10973901), anti-4-HNE (ab48506, RRID: AB_867452) were obtained from Abcam. Erastin (T1765, 571203-78-6), RSL3 (T3646, 1219810-16-8) were obtained from TagerMol.
Transfection and Infection
Cas9/sgRNAs and plasmids were designed and synthesized by Tsingke Biotechnology Co., Ltd. (Beijing, China), and sequences of Cas9/sgRNAs are shown in the Supporting Information. The Lipofectamine 3000 (Invitrogen, USA) was used for transfection of plasmids or Cas9/sgRNA.
Establishment of PDOs
PDOs were established and cultivated as described previously.42
ROS and Lipid Peroxidation Assay
Cells were seeded in 6-well plates or a 20 mm Glass Bottom Cell Culture Dish (NEST, China) overnight and then treated with irradiation or drugs. The cells were incubated for 48 h, 72 h, or other time, and the C11 BODIPY 581/591 assay kit (GLPBIO, USA, GC40165) was used following the manufacturer’s manual. The ROS Assay Kit (GOYOO, China, GY044) was used following the manufacturer’s manual. Lipid peroxidation levels and ROS levels were detected by flow cytometry through a BD Accuri C6 Plus (BD, USA).
Measurement of Mitochondrial Membrane Potential
The cells were treated with irradiation or drugs and then incubated for 48 h, 72 h, or other time, and the JC-1 assay kit (MCE, USA, HY15534) was used to detect mitochondrial membrane potential.
Measurement of Malondialdehyde (MDA)
Cells were inoculated in cell culture flasks overnight and then treated by irradiation. The cells were incubated for 72 h, and the MDA Assay Kit (Dojindo, Japan, M496) was used following the manufacturer’s instructions.
Measurement of Reduced Glutathione/Oxidized Glutathione Activity
Cells were inoculated in 75 cm2 cell culture flasks overnight and then treated by irradiation. The cells were incubated for 72 h, and the GSSG/GSH Quantification Kit II (G263, Dojindo) was used following the manufacturer’s manual. The ratio of GSH/GSSG was measured by luminescence using a microplate reader (Biotek, USA).
Cell Viability
Cells were inoculated in a 20 mm confocal dish (NEST, China) overnight and then treated with radiation or drugs. The Live & Dead Assay Kit (Proteintech, China, PF00007) consisting of Calcein AM/PI was used following the manufacturer’s manual. On the other hand, 5000 cells per well were inoculated in 96-well plates and incubated overnight and then treated with irradiation or drugs. The cells were incubated for 48 h, 72 h or other time, and Cell Counting Kit-8 (CCK8) reagent (Dojindo, Japan) was used following the manufacturer’s manual. Each assay was independently repeated three times.
Transmission Electron Microscopy
Cells were seeded in 75 cm2 cell culture flasks overnight and then treated by radiation. The cells were incubated for 72 h and harvested with a cell scraper. Next, 2.5% glutaraldehyde was used to fix at 4 °C for 2 h and 0.1 M PBS (pH 7.4) to wash 3 times. Then 1% osmium acid was used to treat for 2 h and 0.1 M PBS to wash 3 times. The dehydrated cells were then embedded in resin after undergoing an alcohol gradient. Samples were incubated in a 60 °C for 48 h after dehydration and embedding. The samples were detected using transmission electron microscopy (HITACHI-HT7700, Japan) after double staining with lead citrate and uranyl acetate.
Bioinformatics Analyses of Public Glioma Database
Analysis of gene expression and patient survival information in human glioma was performed using gene profiling data from TCGA, Gravendeel, Rembrandt, and CGGA databases.
Orthotopic Mouse Xenografts
GBM cells expressing luciferase were implanted intracranially using a stereotactic frame into nude mice. In brief, a burr hole was created with a dental drill measuring 0.7 mm in diameter, 2.5 mm to the left of the sagittal suture, and 0.5 mm anterior to the bregma. The injection was made at a depth of 2.5 mm. To analyze tumor growth, d-Luciferin potassium salt (Abcam, USA) was administered intraperitoneally to animals followed by anesthesia with isoflurane for imaging analysis.
In Vitro BBB Model
An in vitro BBB model was established using LN229 and bEnd.3 cells as described previously.40 Seed LN229 cells were placed in the lower transwell chamber. Unmod-EVs, TAT-EVs, Ang-EVs, and Ang/TAT-EVs were added to the upper chamber to assess their efficiency of penetration into the blood–brain region.
Statistical Analysis
All quantification results are presented as mean ± SD (n = 3, unless further indicated). Statistical analyses were performed with a two-tailed unpaired Student’s t test or one- or two-way ANOVA as indicated. Blinding during analysis was implemented in all in vivo experiments for the animal survival studies.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (82003220, 81803053, and 81872049). This study was approved by the FMMU Ethics Committee (Xian, China). All data generated or analyzed during the course of this research will be included in the published article, or upon reasonable request from the relevant authors.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.2c12857.
Supplementary experimental methods, sgRNA and off-target sequences and supplementary figures. (PDF)
Author Contributions
∥ X.L., Z.C., and W.W. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Singh M. M.; Johnson B.; Venkatarayan A.; Flores E. R.; Zhang J.; Su X.; Barton M.; Lang F.; Chandra J. Preclinical activity of combined HDAC and KDM1A inhibition in glioblastoma. Neuro Oncol 2015, 17 (11), 1463–73. 10.1093/neuonc/nov041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karachi A.; Dastmalchi F.; Mitchell D. A.; Rahman M. Temozolomide for immunomodulation in the treatment of glioblastoma. Neuro Oncol 2018, 20 (12), 1566–1572. 10.1093/neuonc/noy072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janjua T. I.; Rewatkar P.; Ahmed-Cox A.; Saeed I.; Mansfeld F. M.; Kulshreshtha R.; Kumeria T.; Ziegler D. S.; Kavallaris M.; Mazzieri R.; Popat A. Frontiers in the treatment of glioblastoma: Past, present and emerging. Adv. Drug Deliv Rev. 2021, 171, 108–138. 10.1016/j.addr.2021.01.012. [DOI] [PubMed] [Google Scholar]
- Shergalis A.; Bankhead A. 3rd; Luesakul U.; Muangsin N.; Neamati N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol Rev. 2018, 70 (3), 412–445. 10.1124/pr.117.014944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran S.; Hu Y.; Canella A.; Peterson C.; Gross A.; Cam M.; Nazzaro M.; Haffey A.; Serin-Harmanci A.; Distefano R.; Nigita G.; Wang W.; Kreatsoulas D.; Li Z.; Sepeda J. A.; Sas A.; Hester M. E.; Miller K. E.; Elemento O.; Roberts R. D.; Holland E. C.; Rao G.; Mardis E. R.; Rajappa P. Single-cell RNA sequencing reveals immunosuppressive myeloid cell diversity during malignant progression in a murine model of glioma. Cell Rep 2023, 42 (3), 112197. 10.1016/j.celrep.2023.112197. [DOI] [PubMed] [Google Scholar]
- Zeng X.; Zhao F.; Cui G.; Zhang Y.; Deshpande R. A.; Chen Y.; Deng M.; Kloeber J. A.; Shi Y.; Zhou Q.; Zhang C.; Hou J.; Kim W.; Tu X.; Yan Y.; Xu Z.; Chen L.; Gao H.; Guo G.; Liu J.; Zhu Q.; Cao Y.; Huang J.; Wu Z.; Zhu S.; Yin P.; Luo K.; Mer G.; Paull T. T.; Yuan J.; Tao K.; Lou Z. METTL16 antagonizes MRE11-mediated DNA end resection and confers synthetic lethality to PARP inhibition in pancreatic ductal adenocarcinoma. Nat. Cancer 2022, 3 (9), 1088–1104. 10.1038/s43018-022-00429-3. [DOI] [PubMed] [Google Scholar]
- Wei X.; Yang J.; Adair S. J.; Ozturk H.; Kuscu C.; Lee K. Y.; Kane W. J.; O’Hara P. E.; Liu D.; Demirlenk Y. M.; Habieb A. H.; Yilmaz E.; Dutta A.; Bauer T. W.; Adli M. Targeted CRISPR screening identifies PRMT5 as synthetic lethality combinatorial target with gemcitabine in pancreatic cancer cells. Proc. Natl. Acad. Sci. U. S. A. 2020, 117 (45), 28068–28079. 10.1073/pnas.2009899117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S.; Zhan M.; Jiang C.; He M.; Yang L.; Shen H.; Huang S.; Huang X.; Lin R.; Shi Y.; Liu Q.; Chen W.; Mohan M.; Wang J. Genome-wide CRISPR screen identifies ELP5 as a determinant of gemcitabine sensitivity in gallbladder cancer. Nat. Commun. 2019, 10 (1), 5492. 10.1038/s41467-019-13420-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; W J.; Chen H.; Yang Y.; Xiao C.; Yi X.; Shi C.; Zhong K.; He H.; Li Y.; Wu Z.; Zhou G.; Rao Q.; Wang X.; Zhou X.; Lomberk G.; Liu B.; Zhao J.; Ge J.; Zhou W.; Chu X.; Chen C.; Zhou X.; Wang L.; Guan K.; Qu L. Genome-wide CRISPR-Cas9 screen identified KLF11 as a druggable suppressor for sarcoma cancer stem cells. Sci. Adv. 2021, 7 (5), 3445. 10.1126/sciadv.abe3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiyaboonchai A.; Vonada A.; Posey J.; Pelz C.; Wakefield L.; Grompe M. Self-cleaving guide RNAs enable pharmacological selection of precise gene editing events in vivo. Nat. Commun. 2022, 13 (1), 7391. 10.1038/s41467-022-35097-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Qian X.; Wang B.; Xia Y.; Zheng Y.; Du L.; Xu D.; Xing D.; DePinho R. A.; Lu Z. Programmable base editing of mutated TERT promoter inhibits brain tumour growth. Nat. Cell Biol. 2020, 22 (3), 282–288. 10.1038/s41556-020-0471-6. [DOI] [PubMed] [Google Scholar]
- Ren X. H.; He X. Y.; Xu C.; Han D.; Cheng S. X. Functional Tumor Targeting Nano-Systems for Reprogramming Circulating Tumor Cells with In Situ Evaluation on Therapeutic Efficiency at the Single-Cell Level. Adv. Sci. (Weinh) 2022, 9 (21), 2105806 10.1002/advs.202105806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J.; Chen Y.; Zhang L.; Jin Q.; Wang L.; Xu S.; Chen K.; Li L.; Zeng T.; Fan X.; Liu T.; Li J.; Wang J.; Han C.; Gao F.; Yang Y.; Wang Y. i-CRISPR: a personalized cancer therapy strategy through cutting cancer-specific mutations. Mol. Cancer 2022, 21 (1), 164. 10.1186/s12943-022-01612-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S. A.; Senger K.; Madireddi S.; Akhmetzyanova I.; Ishizuka I. E.; Tarighat S.; Lo J. H.; Shaw D.; Haley B.; Rutz S.. High-efficiency nonviral CRISPR/Cas9-mediated gene editing of human T cells using plasmid donor DNA. J. Exp Med. 2022, 219 ( (5), ). 10.1084/jem.20211530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley J. A.; Kim S.; Lou L.; Ye C.; Alsaidan O. A.; Sulejmani E.; Cai J.; Desrochers E. G.; Beharry Z.; Rickman C. B.; Klingeborn M.; Liu Y.; Xie Z. R.; Cai H. Encapsulating Cas9 into extracellular vesicles by protein myristoylation. J. Extracell Vesicles 2022, 11 (4), e12196 10.1002/jev2.12196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deligne C.; Hachani J.; Duban-Deweer S.; Meignan S.; Leblond P.; Carcaboso A. M.; Sano Y.; Shimizu F.; Kanda T.; Gosselet F.; Dehouck M. P.; Mysiorek C. Development of a human in vitro blood-brain tumor barrier model of diffuse intrinsic pontine glioma to better understand the chemoresistance. Fluids Barriers CNS 2020, 17 (1), 37. 10.1186/s12987-020-00198-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Cao Z.; Liu N.; Gao G.; Du M.; Wang Y.; Cheng B.; Zhu M.; Jia B.; Pan L.; Zhang W.; Jiang Y.; He W.; Xu L.; Zhang W.; An Q.; Guo Q.; Gu J. Kill two birds with one stone: Engineered exosome-mediated delivery of cholesterol modified YY1-siRNA enhances chemoradiotherapy sensitivity of glioblastoma. Front Pharmacol 2022, 13, 975291. 10.3389/fphar.2022.975291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J.; Mu N.; Jia B.; Guo Q.; Pan L.; Zhu M.; Zhang W.; Zhang K.; Li W.; Li M.; Wei L.; Xue X.; Zhang Y.; Zhang W. Targeting radiation-tolerant persister cells as a strategy for inhibiting radioresistance and recurrence in glioblastoma. Neuro Oncol 2022, 24 (7), 1056–1070. 10.1093/neuonc/noab288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzas E. I. The roles of extracellular vesicles in the immune system. Nat. Rev. Immunol 2023, 23, 236. 10.1038/s41577-022-00763-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Wu Y.; Federzoni E. A.; Wang X.; Dharmawan A.; Hu X.; Wang H.; Hawley R. J.; Stevens S.; Sykes M.; Yang Y. G. CD47 cross-dressing by extracellular vesicles expressing CD47 inhibits phagocytosis without transmitting cell death signals. Elife 2022, 11, e73677 10.7554/eLife.73677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Huang J.; Xia J.; Liang Y.; Li G. Tracking tools of extracellular vesicles for biomedical research. Front Bioeng Biotechnol 2022, 10, 943712. 10.3389/fbioe.2022.943712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horbay R.; Hamraghani A.; Ermini L.; Holcik S.; Beug S. T.; Yeganeh B. Role of Ceramides and Lysosomes in Extracellular Vesicle Biogenesis, Cargo Sorting and Release. Int. J. Mol. Sci. 2022, 23 (23), 15317. 10.3390/ijms232315317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou K.; Liu Y.; Zhao Z.; Wang Y.; Huang L.; Chai R.; Li G.; Jiang T. ABCC8 mRNA expression is an independent prognostic factor for glioma and can predict chemosensitivity. Sci. Rep 2020, 10 (1), 12682. 10.1038/s41598-020-69676-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milde T.; Oehme I.; Korshunov A.; Kopp-Schneider A.; Remke M.; Northcott P.; Deubzer H. E.; Lodrini M.; Taylor M. D.; von Deimling A.; Pfister S.; Witt O. HDAC5 and HDAC9 in medulloblastoma: novel markers for risk stratification and role in tumor cell growth. Clin. Cancer Res. 2010, 16 (12), 3240–52. 10.1158/1078-0432.CCR-10-0395. [DOI] [PubMed] [Google Scholar]
- Han B.; Meng X.; Wu P.; Li Z.; Li S.; Zhang Y.; Zha C.; Ye Q.; Jiang C.; Cai J.; Jiang T. ATRX/EZH2 complex epigenetically regulates FADD/PARP1 axis, contributing to TMZ resistance in glioma. Theranostics 2020, 10 (7), 3351–3365. 10.7150/thno.41219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z.; Chen X.; Zhu P.; Sun W.; Lv Q.; Cheng S.; Yang X.; Yu X. SOX8 Knockdown Overcomes Enzalutamide Resistance in Castration-Resistant Prostate Cancer by Inhibiting the Notch Signaling Pathway. Biomed Res. Int. 2022, 2022, 9235837. 10.1155/2022/9235837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat K. P. L.; Balasubramaniyan V.; Vaillant B.; Ezhilarasan R.; Hummelink K.; Hollingsworth F.; Wani K.; Heathcock L.; James J. D.; Goodman L. D.; Conroy S.; Long L.; Lelic N.; Wang S.; Gumin J.; Raj D.; Kodama Y.; Raghunathan A.; Olar A.; Joshi K.; Pelloski C. E.; Heimberger A.; Kim S. H.; Cahill D. P.; Rao G.; Den Dunnen W. F. A.; Boddeke H.; Phillips H. S.; Nakano I.; Lang F. F.; Colman H.; Sulman E. P.; Aldape K. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24 (3), 331–46. 10.1016/j.ccr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei G.; Zhang Y.; Koppula P.; Liu X.; Zhang J.; Lin S. H.; Ajani J. A.; Xiao Q.; Liao Z.; Wang H.; Gan B. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020, 30 (2), 146–162. 10.1038/s41422-019-0263-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt C.; Lillig C. H. Glutathione, Glutaredoxins, and Iron. Antioxid Redox Signal 2017, 27 (15), 1235–1251. 10.1089/ars.2017.7132. [DOI] [PubMed] [Google Scholar]
- Hider R. C.; Kong X. L. Glutathione: a key component of the cytoplasmic labile iron pool. Biometals 2011, 24 (6), 1179–87. 10.1007/s10534-011-9476-8. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Li D.; Zhao J.; Song J.; Zhao Y. The role of the low-density lipoprotein receptor-related protein 1 (LRP-1) in regulating blood-brain barrier integrity. Rev. Neurosci 2016, 27 (6), 623–34. 10.1515/revneuro-2015-0069. [DOI] [PubMed] [Google Scholar]
- Parrasia S.; Szabo I.; Zoratti M.; Biasutto L. Peptides as Pharmacological Carriers to the Brain: Promises, Shortcomings and Challenges. Mol. Pharmaceutics 2022, 19 (11), 3700–3729. 10.1021/acs.molpharmaceut.2c00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delello Di Filippo L.; Hofstatter Azambuja J.; Paes Dutra J. A.; Tavares Luiz M.; Lobato Duarte J.; Nicoleti L. R.; Olalla Saad S. T.; Chorilli M. Improving Temozolomide biopharmaceutical properties in glioblastoma multiforme (GBM) treatment using GBM-targeting nanocarriers. Eur. J. Pharm. Biopharm 2021, 168, 76–89. 10.1016/j.ejpb.2021.08.011. [DOI] [PubMed] [Google Scholar]
- Minniti G.; Niyazi M.; Alongi F.; Navarria P.; Belka C. Current status and recent advances in reirradiation of glioblastoma. Radiat Oncol 2021, 16 (1), 36. 10.1186/s13014-021-01767-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei G.; Mao C.; Yan Y.; Zhuang L.; Gan B. Ferroptosis, radiotherapy, and combination therapeutic strategies. Protein Cell 2021, 12 (11), 836–857. 10.1007/s13238-021-00841-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Zheng L.; Shang W.; Yang Z.; Li T.; Liu F.; Shao W.; Lv L.; Chai L.; Qu L.; Xu Q.; Du J.; Liang X.; Zeng J.; Jia J. Wnt/beta-catenin signaling confers ferroptosis resistance by targeting GPX4 in gastric cancer. Cell Death Differ. 2022, 29 (11), 2190–2202. 10.1038/s41418-022-01008-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floros K. V.; Cai J.; Jacob S.; Kurupi R.; Fairchild C. K.; Shende M.; Coon C. M.; Powell K. M.; Belvin B. R.; Hu B.; Puchalapalli M.; Ramamoorthy S.; Swift K.; Lewis J. P.; Dozmorov M. G.; Glod J.; Koblinski J. E.; Boikos S. A.; Faber A. C. MYCN-Amplified Neuroblastoma Is Addicted to Iron and Vulnerable to Inhibition of the System Xc-/Glutathione Axis. Cancer Res. 2021, 81 (7), 1896–1908. 10.1158/0008-5472.CAN-20-1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J.; Ran D.; Xie C.; Shen Q.; Wang S.; Lu W. EGFR/EGFRvIII Dual-Targeting Peptide-Mediated Drug Delivery for Enhanced Glioma Therapy. ACS Appl. Mater. Interfaces 2017, 9 (29), 24462–24475. 10.1021/acsami.7b05617. [DOI] [PubMed] [Google Scholar]
- Doench J. G.; Fusi N.; Sullender M.; Hegde M.; Vaimberg E. W.; Donovan K. F.; Smith I.; Tothova Z.; Wilen C.; Orchard R.; Virgin H. W.; Listgarten J.; Root D. E. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34 (2), 184–191. 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. B.; Zhang S.; Kos P.; Xiong H.; Zhou K.; Perelman S. S.; Zhu H.; Siegwart D. J. Non-Viral CRISPR/Cas Gene Editing In Vitro and In Vivo Enabled by Synthetic Nanoparticle Co-Delivery of Cas9 mRNA and sgRNA. Angew. Chem., Int. Ed. Engl. 2017, 56 (4), 1059–1063. 10.1002/anie.201610209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y.; Sun X.; Yang Q.; Zheng M.; Shimoni O.; Ruan W.; Wang Y.; Zhang D.; Yin J.; Huang X.; Tao W.; Park J. B.; Liang X. J.; Leong K. W.; Shi B. Blood-brain barrier-penetrating single CRISPR-Cas9 nanocapsules for effective and safe glioblastoma gene therapy. Sci. Adv. 2022, 8 (16), eabm8011 10.1126/sciadv.abm8011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamboa C. M.; Jara K.; Pamarthy S.; Liu L.; Aiken R.; Xiong Z.; Danish S.; Sabaawy H. E. Generation of glioblastoma patient-derived organoids and mouse brain orthotopic xenografts for drug screening. STAR Protoc 2021, 2 (1), 100345. 10.1016/j.xpro.2021.100345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.