

Abstract
Protonation states of ionizable protein residues modulate many essential biological processes. For correct modeling and understanding of these processes, it is crucial to accurately determine their pKa values. Here, we present four tree-based machine learning models for protein pKa prediction. The four models, Random Forest, Extra Trees, eXtreme Gradient Boosting (XGBoost) and Light Gradient Boosting Machine (LightGBM), were trained on three experimental PDB and pKa datasets, two of which included a notable portion of internal residues. We observed similar performance among the four machine learning algorithms. The best model trained on the largest dataset performs 37% better than the widely used empirical pKa prediction tool PROPKA and 15% better than published result from pKa prediction method DelPhiPKa. The overall RMSE for this model is 0.69, with surface and buried RMSE values being 0.56 and 0.78, respectively, considering six residue types (Asp, Glu, His, Lys, Cys and Tyr), and 0.63 when considering Asp, Glu, His and Lys only. We provide pKa predictions for proteins in human proteome from the AlphaFold Protein Structure Database and observed that 1% of Asp/Glu/Lys residues have highly shifted pKa values close to the physiological pH.
Graphical Abstract:
INTRODUCTION
Ionizable residues play crucial roles in protein stability, dynamics, aggregation, binding, and function1,2. Hence, to correctly model proteins, it is essential to correctly determine the protonation states and pKa values of ionizable residues. Many computational methods for prediction of protein pKa values have been introduced3–34. They can be categorized into continuum electrostatic, empirical, molecular dynamics based, hybrid quantum mechanics/molecular mechanics based, and machine learning based. We will discuss them in more detail later in the Introduction. But pKa calculations are often challenging, due to many inherent factors, including the sensitivity of pKa values to local environment, coupling between conformational and protonation changes, and interactions among ionizable groups.
It is particularly challenging to determine the pKa values of ionizable residues found in protein interiors. These residues are sequestered from water, which, in itself alone, favors a pKa shift towards the neutral form. However, internal residues can be exposed to polarity or charges from other ionizable residues. Presence of additional ionizable residues in vicinity of an ionizable group can shift a pKa value towards both the neutral or charged states, depending on the nature of the group and geometric factors. Furthermore, upon change in protonation states of internal ionizable groups, proteins can exhibit conformational changes which are sometimes small and localized, but sometimes large and global. Conformational changes in turn can influence the pKa values, making the determination of pKa values even more challenging. This coupling between protonation/deprotonation and conformational changes is often exploited for function in many biological processes35–40. Engineered internal ionizable residues in variants of Staphylococcal nuclease (SNase) have been studied in the past two decades to catalog both structures and pKa values41–49. For such residues, conformational rearrangements have been shown to be important for their pKa values47,50–55. In addition, these residues were used as targets in the first blind pKa prediction contest, pKa Cooperative, and found challenging for many methodologies2,56.
Various computational methods have been developed for this challenging task of calculating protein pKa values, mainly in two major categories, macroscopic and microscopic. The former ones generally involve less computational cost than the latter ones. Continuum electrostatic (CE) methods, belonging to the class of macroscopic methods, rely on descriptions of electrostatic potentials, which could be calculated through Poisson-Boltzmann equation3,4,15,26,29,57 or generalized Born model30,31. In CE methods, a single value of dielectric constant is usually assumed for describing the dielectric response of a protein. However, a single dielectric constant may not be sufficient, because the dielectric response depends on local polarity and protein flexibility58,59. This is particularly important for internal groups, because the potential structural reorganization could lead to a different local value of the dielectric constant.
The performance of CE methods depends heavily on the choice of the dielectric constant and usually, the value of 4 is invoked for proteins. However, the experimental pKa value of a buried residue L38K in SNase could not be reproduced by CE methods unless the dielectric constant of protein was chosen to be artificially high47. Additionally, for more buried mutant residues of SNase, it was shown that higher than usual dielectric constant values improved the pKa prediction accuracy60. This is consistent with molecular dynamics simulations, which showed that some variants of SNase with buried residues displayed large conformational changes and water penetration could also play a role61–63. Recently a Gaussian-based smooth dielectric function has been introduced into Poisson-Boltzmann equation in some CE methods64,65. This method is an improvement over traditional Poisson-Boltzmann-equation-based methods because it treats proteins as inhomogeneous objects and assigns low values of the dielectric function in highly packed protein interiors and high values in protein cavities and protein surfaces66,67. However, the hyperparameters of the smooth dielectric function, reference dielectric constant for protein and the Gaussian variance, still need to be optimized. CE methods usually utilize only a single protein structure and do not consider conformational changes. A variant of CE, multi-conformation continuum electrostatic (MCCE) method addresses this issue by simultaneously calculating the protonation states and conformation for side chains60,68–71. While MCCE method embraces side chain rotamers, it still cannot deal with the possible structural reorganization of the protein backbone.
Another class of macroscopic methods, empirical methods, are based on energy functions parametrized against protein residues with known experimental pKa values5,6,32–34. Among these empirical methods, PROPKA is the most widely used one5,6, owing to its high speed and good accuracy. However, the residues used for parameterization are mainly near the protein surface and have pKa values close to the model compound values56. Therefore, empirical methods are unlikely to predict large pKa shifts and thus may be only good at residues with small perturbation but not in extreme cases56. However, PROPKA showed the same quality of predictions as the other non-empirical methods in the pKa Cooperative contest where the pKa values of buried engineered residues were predicted2,56.
Microscopic methods model proteins in great details, e.g., by molecular dynamics (MD) simulations with classical force fields7–14,16–20 or hybrid quantum mechanics/molecular mechanics (QM/MM) simulations21–25. QM/MM simulations describe electrostatic energies more accurately, which is important for predicting pKa values. However, they are computationally much more expensive, and thus sampling is a problem. Consequently, classical MD simulations are more widely employed for protein pKa calculations. Free energy perturbation method7,8 calculates free energy difference between two protonation states of a single residue, and the pKa value of this residue is derived from the difference. Constant-pH MD methods9–14,16–20 simulate systems at constant pH and allow changing of protonation states of multiple groups during conformational evolution. Solvents can be modeled implicitly9,16,72–74, explicitly13,17,75–80 or in a hybrid manner19,81. The benefit of MD based methods is that they incorporate conformational changes coupled to protonation states. However, accurate sampling of conformational relaxation in response to changes in protonation state is in some cases challenging, when conformational changes occur on timescale of milliseconds, as is the case for some SNase variants11,12,20,81–91.
In recent years, machine learning (ML) techniques have been applied to many scientific topics, including predictions of pKa values for non-protein molecules92–98 and of protein-ligand binding affinity99–103. These topics are similar to pKa predictions because they both rely on predictions of free energy differences. Cai and coworkers reported a deep learning based pKa predictor, DeepKa, trained on data generated by constant pH simulations27. Reis and coworkers also reported a deep learning based pKa predictor, pKAI, which was trained on pKa values calculated by a continuum electrostatics method104. Another protein pKa prediction paper from Gokcan and Isayev introduced a new empirical scheme based on deep representation learning that was trained on experimental pKa data28. We chose to use the prevalent tree-based ML models in this work, because of their robustness and well-known good performance on various tasks. We noticed that support vector machine and cascade deep forest could perform well on small datasets105,106. However, we chose not to use support vector machine, because it can only perform classification but not regression and thus are not suitable for the task of predicting continuous pKa values. We did not use cascade deep forest because it was not available in the package we used (scikit-learn)107, and it is a relatively lesser-known model than XGBoost and LightGBM. In addition, the method has been mainly used for classification.
Here, we present four tree-based ML models trained with the PKAD database containing experimental pKa values108 and additional pKa data on SNase from Dr. Bertrand Garcia-Moreno’s lab42,43,49. The latter dataset comprises engineered internal ionizable residues with highly shifted pKa values which are usually harder to predict computationally. To assess the performance of our models rigorously, we test our models and report their performances on large test sets, each of which comprises more than 200 residues with diverse residue types, burial extent, and proteins. To gain physical insights from the ML models, we evaluate feature importance and determine the features causing large pKa shifts. We also apply one of our best models to residues in the selectivity filter of a sodium channel exhibiting highly shifted pKa values, and to all ionizable residues in proteins from human proteome category of the AlphaFold Protein Structure Database.
METHODS
Dataset
We used the PKAD database containing the experimental protein pKa measurements108. PKAD includes pKa values for 1350 residues in 157 wildtype proteins and for 232 residues in 45 mutant proteins. Nine of the mutants, however, do not have their own PDB structures reported, and were thus not used as our input. Some entries in PKAD were not included in our datasets, because of one or more of the following reasons: the protein does not have existing PDB structure (as mentioned above); the residue is not captured in the PDB structure, so that structural features cannot be obtained; the pKa value is not a number, but a range, e.g., “<5”; we only consider the ionizable sidechains of Asp, Glu, His, Lys, Cys and Tyr residues (note that, there are no pKa entries for Arg residues in PKAD). Finally, 1189 entries for wildtype proteins and 79 entries for mutant proteins were included in our datasets.
In addition to PKAD, 23 pKa entries for SNase variants (13 for Asp and 10 for Glu) with existing PDB structures were included42,43,49. These residues all have a relative solvent accessible surface area (%SASA) less than 10. Note that, 68 out of a total of 79 entries in the PKAD mutant part are also for SNase variants. Among the 68 entries, 58 of them have a %SASA value less than 50, and 29 of them have a %SASA value less than 10.
We organized three datasets from this data: entries from PKAD wildtype proteins (1189 entries, denoted as “WT” in the following text), entries from PKAD wildtype and mutant proteins (1268 entries, denoted as “WT+MT” in the following text), and all entries (PKAD and additional SNase data, 1291 entries, denoted as “WT+MT+aSN” in the following text).
Three datasets were separately split into training and test sets with a ratio of 80:20 (also 4:1). The random splitting was performed by the train_test_split method of sklearn.model_selection. The distributions of pKa values in each of the three training sets are shown in Figure S1. After splitting, we removed from each of the test sets a few entries which were measured for residues also included in the corresponding training set, or which are duplicates of other entries in the same test set. Specifically, 1 His residue was removed from the test set of “WT” dataset, 1 Glu and 3 His residues were removed from “WT+MT” test set, and 1 Glu, 2 His and 1 Tyr residues were removed from “WT+MT+aSN” test set. The resulted test sets contain 237, 250 and 255 entries, respectively, for “WT”, “WT+MT” and “WT+MT+aSN” datasets. All datasets are attached in Supporting Information and available at https://github.com/adajhu/Protein-pKa-prediction-by-tree-based-machine-learning to facilitate the re-usage by the community.
Features
The target values to predict are the experimental pKa values taken from PKAD or additional SNase data. For each residue, a total of 47 features were generated for trainings, including residue name, B factor, %SASA, numbers of hydrogen bonds, numbers of polar or nonpolar heavy atoms within certain distances of the target residue, and numbers of likely positively or negatively charged or likely neutral ionizable residues within certain distances of the target residue. Definitions of the two types of distances are explained further below. The residue name was encoded by a one-hot encoding scheme. As there are six ionizable amino acid residue types, i.e., Asp, Glu, His, Lys, Cys and Tyr, the residue name was thus represented as a six-dimensional one-hot encoding vector. The B factor of the beta carbon (CB) atom of each target residue was taken from its PDB file. Except for residue name and B factor, the other features were calculated based on the PDB structures.
Before calculating these features, PDB structures were sanitized using PDBFixer109 by adding missing atoms and replacing non-standard residues with their standard equivalents. Also, there are two proteins (PDB ID: 1EH6 and 1NFN), each of which has a missing segment near the target residues. We modeled the missing parts for them by CHARMM-GUI. Based on these sanitized PDB structures, we calculated the following features. We calculated %SASA for each residue by NACCESS110. The %SASA is defined to be the percentage ratio of SASA of that residue in the protein versus SASA in an ALA-X-ALA tripeptide, so in some cases the %SASA values are greater than 100. We counted the numbers of hydrogen bonds involving sidechains and backbones separately, using two methods, PROPKA5 (called “method 1” in Table 1) and the method “baker_hubbard” in Python package MDTraj111 (called “method 2” in Table 1). This provided us with 4 features related to hydrogen bond numbers.
Table 1.
Features used and their descriptions.
| Feature Name | Description of Feature |
|---|---|
| ResName_XXX | “XXX” will be the three-letter code for each residue type, e.g., ASP; this feature contains a value of 0 or 1, meaning “not this type” or “is this type”, respectively |
| B_factor | B factor of CB atom of the target residue |
| %SASA | Percentage ratio of SASA in the protein to that in an ALA-X-ALA tripeptide |
|
#_HB_SC_m1
#_HB_BB_m1 #_HB_SC_m2 #_HB_BB_m2 |
Number of hydrogen bonds (HB) involving sidechains (SC) or backbones (BB), measured by method 1 (m1) or method 2 (m2) |
|
Polar_2 Polar_4 : : Polar_20 |
Number of polar heavy atoms within a radius of the target residue’s CB atom. The radius used is shown after the underscore symbol and ranges from 2 Å to 20 Å with a spacing of 2 Å. |
|
NonPolar_2 NonPolar_4 : : NonPolar_20 |
Number of non-polar heavy atoms within a radius of the target residue’s CB atom. The radius used is shown after the underscore symbol and ranges from 2 Å to 20 Å with a spacing of 2 Å. |
|
Positive_4 Positive_6 : : Positive_12 |
Number of likely positively charged ionizable residues (Arg, His, Lys) within a radius of the target residue, based on distance between centers of charge. The radius used is shown after the underscore symbol and ranges from 4 Å to 12 Å with a spacing of 2 Å. |
|
Negative_4 Negative_6 : : Negative_12 |
Number of likely negatively charged ionizable residues (Asp, Glu) within a radius of the target residue, based on distance between centers of charge. The radius used is shown after the underscore symbol and ranges from 4 Å to 12 Å with a spacing of 2 Å. |
|
Neutral_4 Neutral_6 : : Neutral_12 |
Number of likely neutral ionizable residues (Cys, Tyr) within a radius of the target residue, based on distance between centers of charge. The radius used is shown after the underscore symbol and ranges from 4 Å to 12 Å with a spacing of 2 Å. |
The next 20 features are the numbers of heavy atoms, polar or nonpolar, within a series of distances (2 Å, 4 Å, …, 20 Å) of the CB atom of a target residue, counted by Python scripts utilizing the Biopython package112. The last 15 features are the numbers of ionizable residues (likely positively charged, likely negatively charged or likely neutral), within certain distances (4 Å, 6 Å, …, 12 Å) of the target residue. The distance is defined to be the distance between centers of the charge. We categorized ionizable residues according to their most likely charge states at physiological pH based on their model compound pKa values. The center of charge was defined to be atom CZ for Arg, atom CG for Asp, atom SG for Cys, atom CD for Glu, atom CE1 for His, atom NZ for Lys and atom OH for Tyr. All features are summarized in Table 1.
Model setup
We trained four tree-based regressors: Random Forest (RF), Extra Trees (ExTr), eXtreme Gradient Boosting (XGBoost) and Light Gradient Boosting Machine (LightGBM). We used the RF and ExTr regressors implementations of the scikit-learn library107. We used the LightGBM and XGBoost regressors adopted from the LightGBM113 and XGBoost114 package, respectively. All following calculations/operations were performed with random_state=209 to be reproducible. The hyperparameter tuning was performed using Optuna115 on the training set (80% of the whole dataset). The hyperparameters being tuned and their ranges are listed in Table S1. During the tuning, the average of root mean squared error (RMSE) from 5-fold cross validation was minimized, and the regressors were always instantiated with a random state of 209 to be reproducible. We use the RMSE of test set to assess the quality of models. The workflow of this study is shown in Figure 1.
Figure 1.
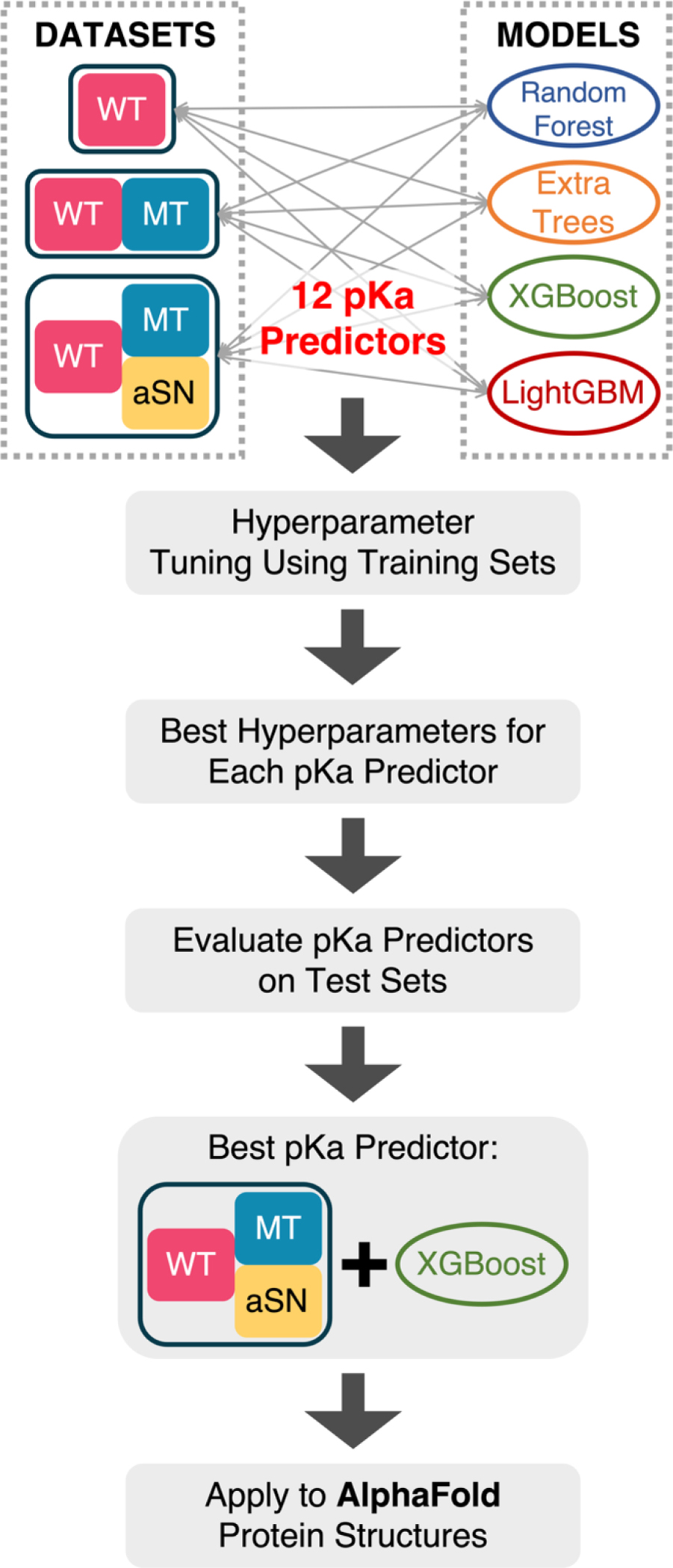
Workflow of this study.
RESULTS
Model performance
All four ML models we trained outperform the null model and PROPKA in terms of predicting pKa values of all, exposed, and buried residues regardless of the dataset used (Figure 2A and Table 2A). The four ML models are showing similar prediction accuracy. Their overall RMSEs’ standard deviation is only 4%, 5% and 3% of the average, respectively with the “WT”, “WT+MT” and “WT+MT+aSN” datasets. Thus, we believe that there is no significant difference in performance between the four ML models for pKa prediction.
Figure 2.
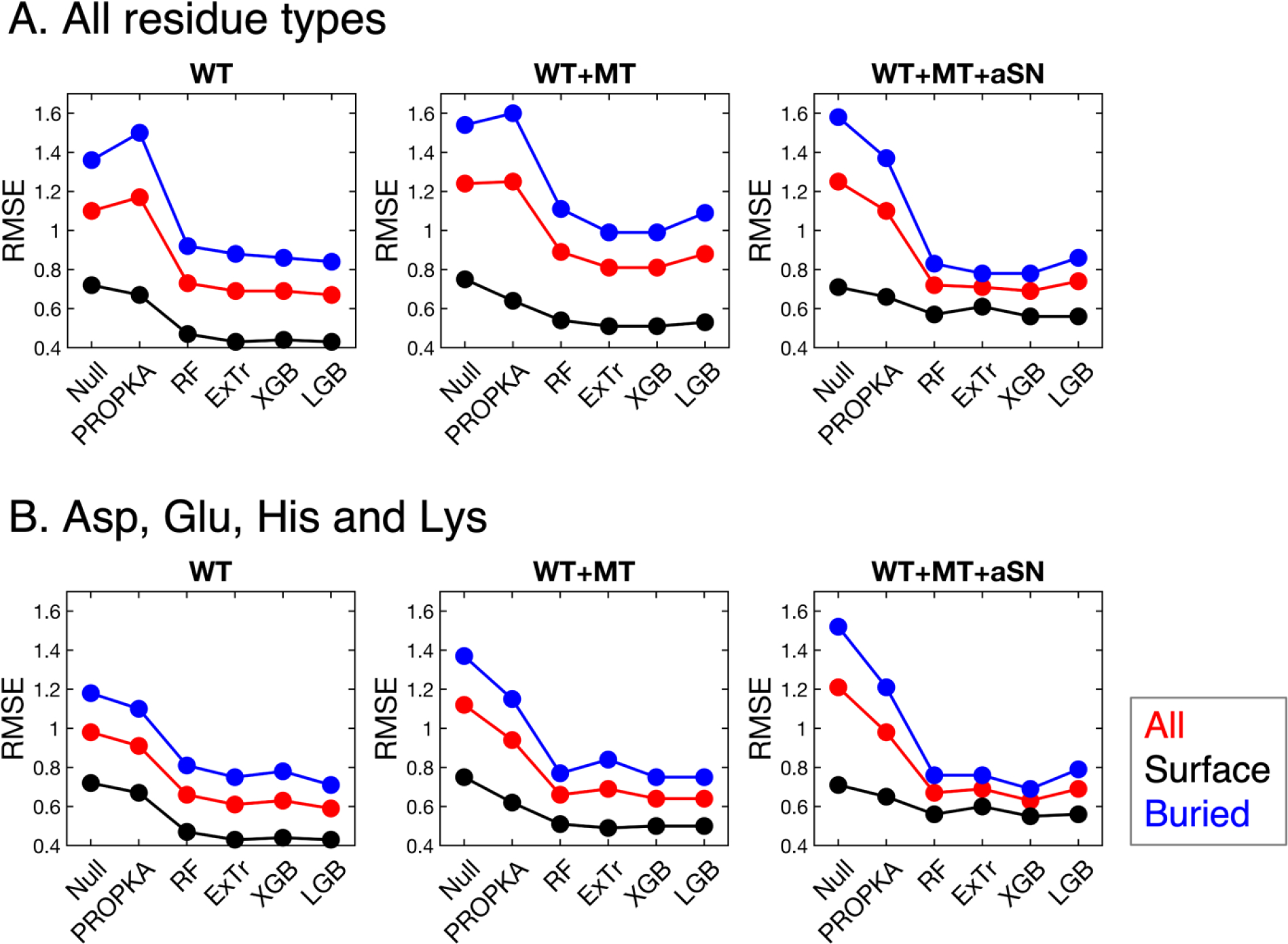
Comparison of different models’ performance which is evaluated on (A) all ionizable residue types (Asp, Glu, His, Lys, Cys and Tyr) or (B) DEHK types (Asp, Glu, His and Lys). Surface groups are those with %SASA greater than 50, and buried groups have %SASA less than 50.
Table 2.
RMSEs evaluated on test set.
| A. All residue types | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Dataset | Residue Type |
Null Model |
PROPKA* | Random Forest |
Extra Trees |
XGBoost | LightGBM | Avg† | 1 - Avg/PROPKA [%] |
| WT | All | 1.10 | 1.17 | 0.73 | 0.69 | 0.69 | 0.67 | 0.70 | 41 |
| Surface | 0.72 | 0.67 | 0.47 | 0.43 | 0.44 | 0.43 | 0.44 | 34 | |
| Buried | 1.36 | 1.50 | 0.92 | 0.88 | 0.86 | 0.84 | 0.88 | 42 | |
|
WT
+MT |
All | 1.24 | 1.25 | 0.89 | 0.81 | 0.81 | 0.88 | 0.85 | 32 |
| Surface | 0.75 | 0.64 | 0.54 | 0.51 | 0.51 | 0.53 | 0.52 | 18 | |
| Buried | 1.54 | 1.60 | 1.11 | 0.99 | 0.99 | 1.09 | 1.05 | 35 | |
|
WT
+MT +aSN |
All | 1.25 | 1.10 | 0.72 | 0.71 | 0.69 | 0.74 | 0.72 | 35 |
| Surface | 0.71 | 0.66 | 0.57 | 0.61 | 0.56 | 0.56 | 0.58 | 13 | |
| Buried | 1.58 | 1.37 | 0.83 | 0.78 | 0.78 | 0.86 | 0.81 | 41 | |
| B. Asp, Glu, His and Lys | |||||||||
| Dataset | Residue Type | Null Model | PROPKA * | Random Forest | Extra Trees | XGBoost | LightGBM | Avg † | 1 - Avg/PROPKA [%] |
| WT | All | 0.98 | 0.91 | 0.66 | 0.61 | 0.63 | 0.59 | 0.62 | 32 |
| Surface | 0.72 | 0.67 | 0.47 | 0.43 | 0.44 | 0.43 | 0.44 | 34 | |
| Buried | 1.18 | 1.10 | 0.81 | 0.75 | 0.78 | 0.71 | 0.76 | 31 | |
|
WT
+MT |
All | 1.12 | 0.94 | 0.66 | 0.69 | 0.64 | 0.64 | 0.66 | 30 |
| Surface | 0.75 | 0.62 | 0.51 | 0.49 | 0.50 | 0.50 | 0.50 | 19 | |
| Buried | 1.37 | 1.15 | 0.77 | 0.84 | 0.75 | 0.75 | 0.78 | 32 | |
|
WT
+MT +aSN |
All | 1.21 | 0.98 | 0.67 | 0.69 | 0.63 | 0.69 | 0.67 | 32 |
| Surface | 0.71 | 0.65 | 0.56 | 0.60 | 0.55 | 0.56 | 0.57 | 13 | |
| Buried | 1.52 | 1.21 | 0.76 | 0.76 | 0.69 | 0.79 | 0.75 | 38 | |
Surface groups are those with %SASA greater than 50, and buried groups have %SASA less than 50.
PROPKA is not capable of processing several residues in test sets: in “WT”, 3 Asp, 1 Glu, 1 Lys and 1 Cys residues, out of a total of 237 residues; in “WT+MT”, one of each of Asp, Glu, Lys and Cys residues, out of a total of 250 residues; in “WT+MT+aSN”, 2 Asp, 1 Glu, 1 Lys and 1 Cys residues, out of a total of 255 residues.
“Avg” means the average over the four ML models.
When the average RMSE values of the four ML models are compared with PROPKA’s RMSEs, the ML models exhibit a large performance boost compared to PROPKA. The overall RMSE decreases by 41%, 32% and 35% respectively for the three datasets. It is remarkable that the ML models have largely enhanced performance for the buried groups (%SASA < 50), while not sacrificing and even improving the performance for the surface groups (%SASA > 50). The RMSE of buried residues decreased by 42%, 35% and 41% respectively for the three datasets than that of PROPKA.
It is also noticeable that, PROPKA failed to improve the null model for the “WT” and “WT+MT” datasets. After a closer examination, we noticed that PROPKA yields highly inaccurate predictions for buried Cys and Tyr residues. Thus, we show the RMSEs only for the Asp, Glu, His and Lys (denoted as DEHK RMSEs in the following text) in Figure 2B and Table 2B. PROPKA shows more accurate predictions than the null model for Asp, Glu, His and Lys. When only these four ionizable residues are considered, the overall RMSEs and the RMSEs for buried residues of our ML models are again both largely decreased by more than or equal to 30% than those of PROPKA. The RMSEs for surface residues are also decreased by 22% on average.
Table 2A is showing that the best ML model among the ones trained on the largest dataset (“WT+MT+aSN”) is XGBoost. We will denote this model as XGB-WMa model in the following text. We compare the RMSEs for individual residue type of this model with those of the null model and PROPKA in Table 3. We will also show the applications of the XGB-WMa model to proteins outside of our datasets in the following sections of Results. Table 3 shows that the XGB-WMa model largely outperforms the null model and PROPKA for Asp, Glu, His, Lys and Cys residues separately. For Tyr residues, the XGB-WMa model performs very similarly to PROPKA, but worse than the null model. Note that, sample sizes of Cys and Tyr residues are too small to draw meaningful conclusions.
Table 3.
RMSE for each residue type evaluated on the test set of the “WT+MT+aSN” dataset.
| Asp | Glu | His | Lys | Cys | Tyr | ||
|---|---|---|---|---|---|---|---|
| Number of residues in training set | 317 | 373 | 185 | 125 | 17 | 15 | |
| Number of residues in test set | 86 | 78 | 51 | 31 | 2 | 7 | |
| RMSE | Null Model | 1.24 | 0.88 | 1.29 | 1.63 | 4.25 | 0.86 |
| PROPKA* | 0.81 | 0.82 | 1.41 | 0.95 | 7.74 | 1.06 | |
| XGB-WMa | 0.61 | 0.5 | 0.74 | 0.76 | 2.79 | 1.07 | |
| Percentage difference between PROPKA and XGB-WMa | −25% | −39% | −48% | −20% | −64% | 1% | |
PROPKA is not capable of processing several residues in this test set: 2 Asp, 1 Glu, 1 Lys and 1 Cys residues, out of a total of 255 residues.
Feature importance ranking
The feature importance is illustrated in Figure 3 and Figure S2. Figure 3 shows that the feature importance rankings of RF, ExTr and XGBoost are more or less similar to each other. In contrast, LightGBM has a very different pattern. We will discuss possible reasons for this in the Discussions section.
Figure 3.

Feature importance ranking excluding the ones representing residue type, averaged over the three datasets. The top 20 most important features for RF, ExTr and XGBoost are shown here, and the rest of features are shown in Figure S2. The features are sorted by the average ranking averaged only over RF, ExTr and XGBoost models. The number in each cell is the ranking, and the darker the cell color is, the more important the feature is.
For RF, ExTr and XGBoost models, “Negative_4” is identified as the most important feature corresponding to the number of negatively charged ionizable residues within 4 Å of the target residue. The “Positive_4”, the number of positively charged ionizable residues within 4 Å of the target residue, is also ranked as the top 4 feature. The high importance of “Negative_4” and “Positive_4” demonstrates that the presence of other ionizable groups nearby have large influence on the pKa values of the target residue. The other three features in the top 5 are “Polar_4”, “NonPolar_4” and “NonPolar_12” corresponding to the number of polar/non-polar/non-polar atoms within 4/4/12 Å of the target residue, respectively.
LightGBM model identified a different set of important features. The top 1 feature for the other three models, “Negative_4”, is only ranked 37 in LightGBM out of 41 non-residue-type features, whereas the most important feature for LightGBM is %SASA. The second most important feature for LightGBM is B factor (shown in Figure S2), which describes the uncertainty of the atomic positions. However, the other three models rank B factor only 31 on average. Interestingly, despite the difference in the ranking of the features, the four models have similar performances.
Application of the XGB-WMa model to an ion channel
The pKa value of Glu residues in the selectivity filter of a bacterial voltage-gated sodium channel, NavMs, has been determined by us through free energy simulations to be 6.4116. The pKa value for a structurally similar sodium channel, NaChBac, was estimated to be 7.6 based on experiments117. This NavMs channel is a tetramer with four repeating chains. Each chain includes a Glu residue sitting in the selectivity filter, as shown in Figure 4. These Glu residues are deeply buried (%SASA = 4.4) and very close to each other, with 7.5 Å between the Cδ atoms of the two Glu residues in adjacent chains. The large extent of burial and the electrostatic interaction between the Glu residues are likely the reasons why their calculated pKa values are much higher than Glu’s normal pKa in water. We note that our previous simulations118 also showed a dependence of pKa values on the number of ions in the selectivity filter, but in equilibrated simulations the number of ions is between 1 and 2. Below we wish to demonstrate the ability of the XGB-WMa model to reproduce the highly shifted pKa value, and potential pitfalls of these sort of predictions.
Figure 4.
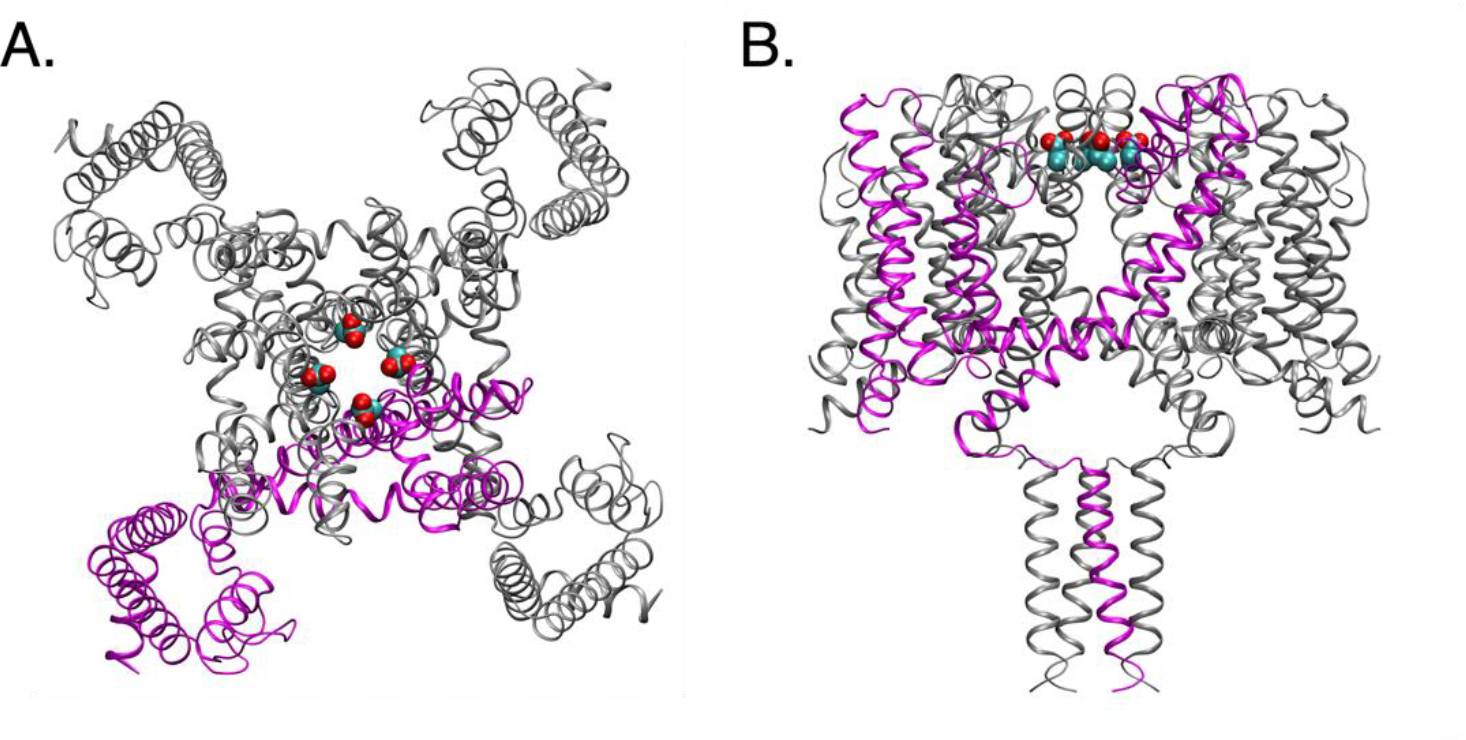
Structure of the sodium channel (PDB: 5HVX), shown in (A) top view and (B) side view. One of the chains is colored magenta, and the other three are colored grey. The sidechain heavy atoms of Glu residues being predicted are shown in spheres.
First, we used the raw PDB file of 5HVX, including only one chain, and input it into the XGB-WMa model. The pKa value was predicted to be 4.1, far away from the pKa calculated from explicit solvent simulations, and from experiments on similar proteins. This is likely because the Glu will be exposed to water and not surrounded by other ionizable groups (the other three Glu residues) with only one chain (shown as magenta in Figure 4). Because of this incorrect information on the Glu’s local environment, the ML model predicts unshifted pKa value. Later, we predicted the pKa values with the manually assembled functional form of the sodium channel, i.e., a tetramer (shown in Figure 4). The predicted pKa value was then 6.2, showing a large shift from the Glu’s usual pKa value, which is also very close to the previous value (6.4) calculated by free energy methods. This example demonstrates a potential pitfall when applying pKa predictions on many structures. In addition to adding the missing atoms or residues, namely the structures of multimers need to be constructed from the PDBs containing single subunits.
In this model, membrane lipids are not included, which may cause errors of pKa predictions for ionizable groups that are in contact with lipids. However, there are few ionizable groups pointing to lipid tails because of their hydrophobic environment. Regarding ionizable groups close to lipid heads, errors of predicted pKa values will exist. We plan to address this in our future work.
Application of the XGB-WMa model to AlphaFold structures
AlphaFold is a deep learning system which predicts a protein structure based on its amino acid sequence119. It has shown its great accuracy comparable to experiments119. Recently, DeepMind released the AlphaFold-predicted protein structures120. We performed pKa predictions using the XGB-WMa model on all the released structures from the human proteome. These results can be viewed as predictions which could be tested by future experiments. Our model is capable of making predictions for six ionizable residue types, Asp, Glu, His, Lys, Cys and Tyr. However, we cannot predict pKa values for Arg, because there is no experimental pKa data for Arg in the pKa database we used. The results of the pKa predictions are attached in Supporting Information and also available at https://github.com/adajhu/Protein-pKa-prediction-by-tree-based-machine-learning.
Figure 5 is showing the distributions of the predicted pKa values for Asp, Glu, His and Lys. 26% of Asp residues and 39% of Glu residues are predicted to have pKa values higher than their model compound pKa values, 4.0 and 4.3, respectively. For His and Lys residues, 57% and 45% are predicted to have lower pKa values than their model compound pKa values, 6.4 and 10.5, respectively. All four residue types show a notable portion with largely shifted pKa values. Specifically, 1% of Asp and Glu residues have pKa greater than 6, and 8% of His residues have pKa less than 5, and 1% of Lys residues have pKa less than 8.
Figure 5.
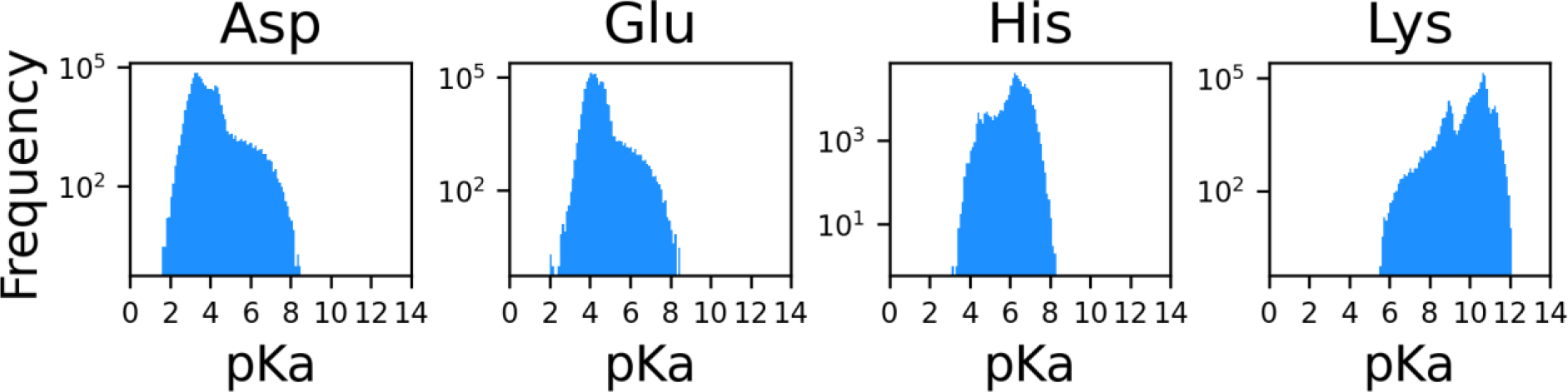
Distributions of pKa values of Asp, Glu, His and Lys residues in proteins in human proteome from the AlphaFold Protein Structure Database. A log scale is used for the y-axes.
To understand which factors are driving the pKa shifts, we plot feature distributions for all residues and the residues with large pKa shifts in Figure S3. We notice that the residues with shifted pKa values are all buried, as evidenced by low %SASA. To demonstrate this clearer, we show the %SASA panels also in Figure 6 in addition to Figure S3. For Asp, Glu and Lys residues, the residues with shifted pKa values also have large B factors and large numbers of polar/non-polar atoms within a greater-than-6 radius of the target residue. Predictions using the LightGBM model exhibit similar distributions of pKa values and features, despite the large difference in feature importance.
Figure 6.
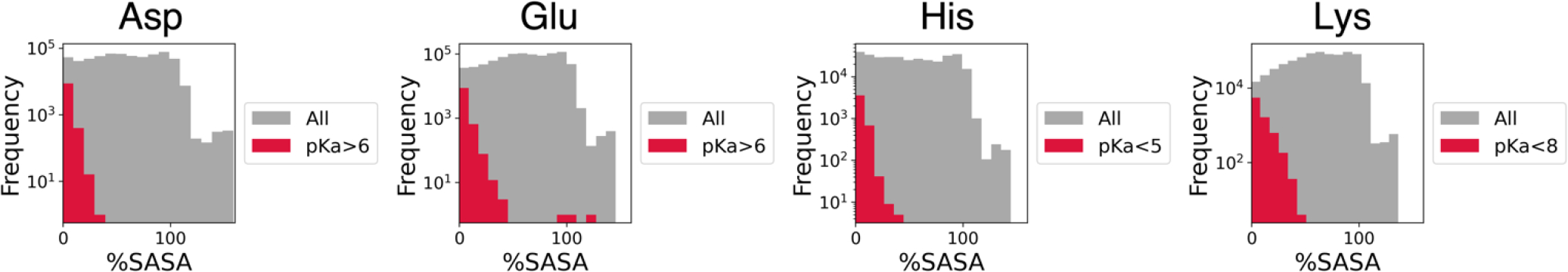
Distributions of %SASA of Asp, Glu, His and Lys residues in proteins in human proteome from the AlphaFold Protein Structure Database. Grey shaded area represents distributions for all residues, and red for residues with large pKa shifts (Asp: pKa > 6, Glu: pKa > 6, His: pKa < 5, Lys: pKa < 8). A log scale is used for the y-axes.
Here, we examine one randomly chosen protein example for each of the shifted Asp, Glu, His and Lys residue types. In all four examples, the target residues are all buried and surrounded by other ionizable residues. Figure 7A shows D445 in chain A from AF-Q96PB7-F1-model_v1.pdb, which is a structure of protein Noelin-3, a neuronal olfactomedin. D445 is highly buried (%SASA = 8.5) and surrounded by two other carboxylic groups, D348 and E396. Its pKa value is shifted to 6.5. Low %SASA and presence of additional carboxylic residues next to a carboxylic residue can shift up the pKa value of a carboxylic group. Figure 7B shows residue E82 in chain A from AF-Q9Y6E0-F1-model_v1.pdb which is a structure of serine/threonine-protein kinase 24. E82 is buried with a %SASA value of 5.5. It is surrounded by two ionizable residues, K65 and D174. Its pKa value was predicted to be 7.1. Figure 7C shows H114 in chain A from AF-Q99675-F1-model_v1.pdb, which is a structure of “cell growth regulator with RING finger domain protein 1”. H114 is also highly buried (%SASA = 0.0), which might be the major reason of having a decreased pKa value of 4.5. In addition, the positively charged R119 is close to it, which could also contribute to the pKa decrease. Figure 7D shows K88 residue in chain A from AF-M5A8F1-F1-model_v1.pdb (a structure of protein suppressyn), which also has a large pKa shift (pKa = 7.4). Like the above residues, K88 is also deeply buried with %SASA being 4.7 and surrounded by other titratable residues. It has two glutamic acids (E46 and E77) and four cysteine residues (C44, C79, C47 and C111) around it. One pair of cysteines is likely involved in a disulfide bond, while that is likely not the case for the other pair. At physiological pH, according to the predicted pKa values of the cysteines, two of the cysteines could protonate/deprotonate, and this could affect the pKa value of K88.
Figure 7.
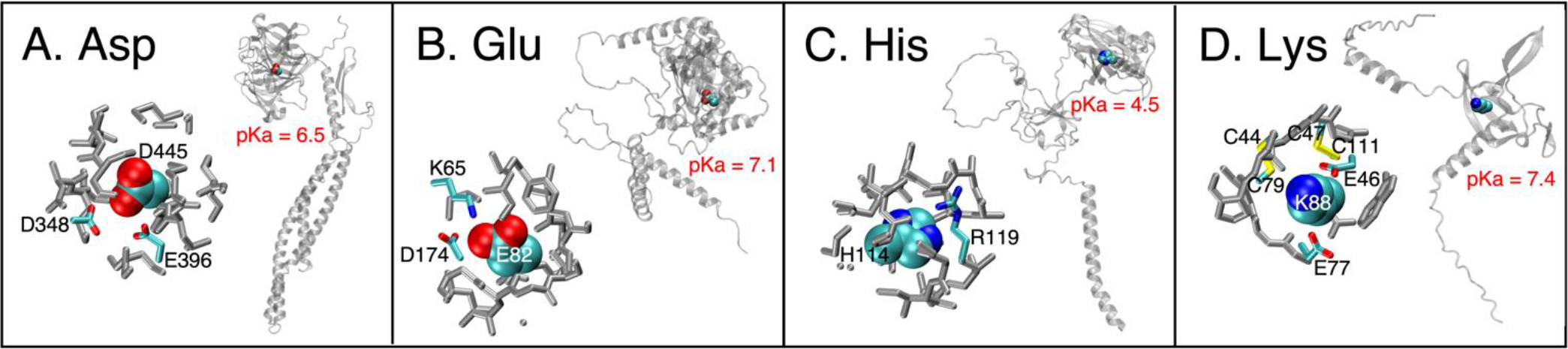
Four example residues with large pKa shifts. In each panel, the right figure shows the residue’s location within the protein, and the left shows its local environment. In left figures of each panel, ionizable residues are colorful while the others are grey; the target residues are shown by VMD’s “VDW” drawing method (atoms as spheres) and the surrounding residues are shown by VMD’s “Licorice” drawing method (atoms as spheres and bonds as cylinders)121.
DISCUSSION
In this work, we utilize four tree-based ML models trained on three datasets separately to predict pKa values of protein ionizable residues. All four ML models show similar RMSE values and largely outperform the null model and PROPKA with the same test dataset. Specifically, the XGB-WMa model shows the best test set RMSE of 0.69 among models trained on the largest dataset. Evaluated on the same test set, the RMSE values for the null model and PROPKA are 1.25 and 1.10, respectively. Thus, our XGB-WMa model performs 45% and 37% better than these two methods, respectively. In terms of RMSEs for individual residue types, the XGB-WMa model again largely outperforms the null model and PROPKA for Asp, Glu, His and Lys residues, while sample sizes for Cys and Tyr residues are too small to draw any meaningful conclusions.
The RMSE values are summarized in Table 4, which also shows RMSE values for five other pKa predictors: DelPhiPKa, a popular continuum electrostatic pKa prediction method122; PypKa, a python module calculating pKa values by continuum electrostatic method57; DeepKa, a deep learning based pKa predictor trained on pKa values derived from continuous constant pH simulations27; pKAI, a deep learning model trained on pKa values calculated by PypKa104; and a pKa predictor based on deep representation learning and trained on experimental pKa values, which we will refer to as DRL28. Because DelPhiPKa and DeepKa only predict the pKa values of Asp, Glu, His and Lys (DEHK) residues, and PypKa and DRL only predict for Asp, Glu, His, Lys and Tyr (DEHK+Y) residues, we also show DEHK and “DEHK+Y” RMSE values in Table 4. The DEHK RMSE of the XGB-WMa model is 0.63. Considering DEHK RMSEs, similarly to overall RMSEs, our XGB-WMa model performs 48% and 36% better than the null model and PROPKA, respectively. This surpassing is also true in terms of “DEHK+Y” RMSE values. Our XGB-WMa model also performs better than the other five pKa predictors: its DEHK RMSE is lower than DelPhiPKa’s and DeepKa’s by 15% and 40%, respectively; its “DEHK+Y” RMSE is lower than PypKa’s and DRL’s by 21% and 19%, respectively; and its all-residue RMSE is lower than pKAI’s by 30%. The RMSE values for the last five methods were evaluated on their own test sets, which are different from the test set used for XGB-WMa. A comparison of RMSE values evaluated on different test sets may be less significant. We compare our results to DelPhiPKa published results122, obtained for the subsets of our datasets where predictions were available in the reference. Even though DelPhiPKa predictions are overall better than PROPKA, we compare our model with PROPKA throughout this paper, because there was complete availability of PROPKA predictions for our test sets.
Table 4.
Test set RMSE values for several pKa predictors.
| Method | RMSE for all |
RMSE for DEHK+Y residues |
RMSE for DEHK residues |
|---|---|---|---|
| XGB-WMa | 0.69 | 0.65 | 0.63 |
| Null Model † | 1.25 | 1.20 | 1.21 |
| PROPKA † | 1.10 | 0.98 | 0.98 |
| DelPhiPKa * | \ | \ | 0.74 |
| PypKa * | \ | 0.82 | \ |
| DeepKa * | \ | \ | 1.05 |
| pKAI * | 0.98 | \ | \ |
| DRL * | \ | 0.80 | 0.79 |
Evaluated on the test set of “WT+MT+aSN” dataset.
Evaluated on their own test sets containing experimental pKa data, different from the test set used in this work.
We have shown in the Results section that our models have greatly enhanced performance for buried residues while also improving the performance for surface groups. Table 5 is showing DEHK RMSE values separately for surface and buried residues, for our XGB-WMa model, the null model, PROPKA and DelPhiPKa. Our XGB-WMa model outperforms the null model by 23% and 55% in terms of surface and buried RMSEs, respectively. Similarly, the XGB-WMa model outperforms PROPKA by 15% and 43% on surface and buried RMSEs, respectively. While the surface RMSEs are very similar between XGB-WMa and DelPhiPKa, the buried RMSE of XGB-WMa is again much smaller, 26%, than that of DelPhiPKa. Comparisons to these three models clearly demonstrate that the boost in overall performance is mainly coming from the big improvement for buried groups, whose pKa values are often lacking in experimental datasets. This stresses the importance of generating more experimental pKa data on internal residues.
Table 5.
DEHK RMSE values for surface and buried residues.
| All | Surface | Buried | |
|---|---|---|---|
| XGB-WMa | 0.63 | 0.55 | 0.69 |
| Null Model† | 1.21 | 0.71 | 1.52 |
| PROPKA† | 0.98 | 0.65 | 1.21 |
| DelPhiPKa* | 0.74 | 0.53 | 0.93 |
Evaluated on the test set of “WT+MT+aSN” dataset.
Evaluated on its own test set, different from the test set used in this work.
We show that the feature ranking pattern of LightGBM is very different from those of the other three models (Figure 3). RF, ExTr and XGBoost all identified “Negative_4” as the most important feature. However, this feature ranks only 37 out of 41 in LightGBM. In addition, the top two features of LightGBM, “%SASA” and “B_factor”, have average rankings of only 11 and 31, respectively, in the other three models.
We speculate that this big difference of feature ranking may be caused by the different tree growth strategies adopted, which is one of the major differences between LightGBM and the other three models. LightGBM grows trees leaf-wise, while the other three models grow trees level-wise. In level-wise tree growth strategy, every node in the same level gets split into child nodes in the additional lower level, so trees are always symmetric. In contrast, leaf-wise strategy grows asymmetric trees, because it only splits the leaf with maximum loss reduction and does not split the other leaves. A leaf-wise strategy tends to overfit for small datasets, but excel in large datasets. In this work, the datasets are rather small, but there is no significant difference between the performances of the level-wise and leaf-wise models.
The low importance of B-factor is perhaps surprising, and the reasons are not clear. This warrants further work, along with exploring protein flexibility. One possibility is that the effect of B-factor is already accounted in other more important features. Another possibility is that the choice of using the B-factors of CB atoms is suboptimal.
As shown in the pKa prediction results for the ion channel, our XGB-WMa model could give incorrect predictions when the input structure is not a biological assembly and thus does not reflect the real local environment for residues on chain-to-chain interfaces, but XGB-WMa model works better when applied to a functional and correct form of the protein. Thus, users need to input the correct biological assembly into our models to obtain the most accurate pKa predictions. Unfortunately, the current protein structures from AlphaFold are all monomers. Consequently, the predicted pKa values for the residues on chain-to-chain interfaces are likely to be less accurate. DeepMind posted a preprint recently about predicting structures for protein multimers123. In the future, we will re-investigate the pKa predictions when they release the predicted structures for multimers.
We have shown that, in AlphaFold human proteome proteins, 1% of Asp and Glu residues exhibit pKa values greater than 6, 8% of His residues smaller than 5, and 1% of Lys residues smaller than 8. These residues are all buried, with average %SASA being 3, 4, 4 and 6, respectively, for Asp, Glu, His and Lys residues. These residues with shifted pKa values may play important functional roles in proteins. This is particularly true for residues with pKa values close to the physiological pH of 7.4. This implies that such residues can easily change their protonation states at the physiological pH, which may further lead to conformational changes of the protein. Changes in both the protonation state and conformations can be harnessed for function, e.g., during proton transfer, like in cytochrome c oxidase, a Na+/H+ antiporter and a CLC transporter exchanging Cl−/H+ 124–126, or during electron transfer, like in an iron-containing superoxide dismutase127.
To validate our pKa predictions on AlphaFold structures, we compared our predictions with experimental pKa measurements for NUPR1128 which does not have available experimental structures. Our pKa predictions based on the protein’s AlphaFold structure (AF-O60356-F1-model_v1.pdb) exhibit a RMSE of 0.33 with respect to experimental values.
One potential disadvantage of our models is that only a single static structure is considered for pKa prediction. Especially for internal residues, their protonation and deprotonation can be coupled with protein’s conformational changes. Thus, it would be beneficial to incorporate the structural reorganization of protein into the prediction. However, that is far from simple because conformational changes of these residues can in some cases occur on millisecond timescale52, which is still inaccessible to technique such as MD simulations, and particularly constant pH calculations. However, on the other hand, it is possible that potential conformational changes may already be encoded implicitly into the parameters of ML models, especially when properly trained.
Some of SNase variants have shown large pKa value shifts, as well as conformational changes in response to ionization of internal groups, and have already been used as a challenging test set in the blind prediction for pKa Cooperative. To test how our methods performed on predicting just the SNase variants pKa values, we tested how our models trained on “WT” and “WT+MT” training sets performed on a test set containing only the 23 additional entries for SNase variants (“aSN” dataset). Our results show that the models trained on “WT” training set display an average RMSE of 2.75, while the models trained on “WT+MT” training set, which includes some SNase variants pKa entries, show a markedly improved average RMSE of 1.72 (Table S2). In comparison, some methods in the pKa Cooperative contest in 2011 reported the following RMSE values: 4.10 for a method based on Poisson–Boltzmann solver129, 4.3 and 3.14 for two hybrid methods utilizing MCCE130,131. A constant pH method using implicit solvent displayed an average unsigned error of 1.510. Later in 2015, DelPhi reported a RMSE of about 1.6 for the pKa Cooperative dataset, while using a Gaussian variance optimized for this dataset60. However, results of the pKa Cooperative cannot be directly compared to our results, since the dataset used in the pKa Cooperative included only 66% of residues which are greater than 50% buried60, while in our test set (“aSN”) 100% of residues have %SASA less than 10. The pKa values of those highly buried residues are the hardest to predict, thus our results are quite good for such a challenging dataset. The fact that our models trained on “WT+MT” dataset performed much better than those trained on “WT” dataset underscores the need for generating more experimental data on highly buried residues, which can then be used for more accurate pKa predictions.
Though ML models presented here already achieve great performance compared to other pKa predictors, there are still some improvements we can carry out in the future. First, there are only about one thousand experimental pKa values with available protein 3D structures. The small number of data points limits the possibility of employing neural networks, as such techniques could easily overfit. More pKa measurements and/or protein structure determinations would largely accelerate the development of pKa prediction. Second, more features could be added, especially more detailed features, e.g., graph representation describing atomic information of the target residue and its adjacent residues. Also, more ML algorithms could be explored, e.g., graph neural networks132 or 3D point cloud models133, and comparisons to the tree-based models could be made. Lastly, after DeepMind releases the multimer structures in the future, we could apply our models to the multimers, which should lead to more accurate pKa predictions for the residues on interfaces between chains.
SUMMARY
Protonation states of ionizable protein residues modulate many essential biological processes. For correct modeling and understanding of these processes, it is crucial to accurately determine their pKa values. In this work, we report four types of tree-based machine learning pKa predictors trained on experimental pKa values and protein PDB structures. We show that our models outperform all the protein pKa predictors the authors are currently aware of, in terms of both all-residue RMSE and RMSE values of Asp, Glu, His and Lys. Particularly, our best model trained on the largest dataset, XGB-WMa, exhibits an all-residue RMSE 37% smaller than that of PROPKA, while evaluated on an identical test set.
We also show that, the boost in our performance mainly arises from the large enhancement of prediction for buried residues. Application of the XGB-WMa model to residues with highly shifted pKa values in the selectivity filter of a sodium channel shows a great match with our previous pKa calculation by free energy perturbation, but only when the protein is properly modeled as a multimer. We also applied the XGB-WMa model to proteins in the human proteome from the AlphaFold Protein Structure Database and observed 1% of Asp and Glu residues with pKa values greater than 6, and 1% of Lys residues with pKa values less than 8. This suggests that 1% of Asp/Glu/Lys residues may be possibly functionally important, because of potential protonation state changes at physiological pH.
Supplementary Material
ACKNOWLEDGEMENTS
This work was performed on the National Institutes of Health high-performance computing cluster Biowulf (http://hpc.nih.gov). A.D. acknowledges National Heart, Lung, and Blood Institute (NHLBI) grants: 75N92019P00048 and 75N92020P00042. This research was supported by the Intramural Research Program of the National Institutes of Health, NHLBI (ZIA HL001051). This work was also supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) (No. 2019M3E5D4066897, 2019M3E5D4066898) and the National Research Foundation funded by the Korean government (MSIT) (No. 2018R1C1B600543513, No. 2020R1F1A1075998).
Footnotes
ASSOCIATED CONTENT
Supporting Information.
Hyperparameters being tuned and their ranges; and distribution of pKa values in training sets; and complete feature importance ranking; and distribution of features for proteins in the human proteome from the AlphaFold Protein Structure Database (PDF)
Training set of “WT” dataset (txt)
Test set of “WT” dataset (txt)
Training set of “WT+MT” dataset (txt)
Test set of “WT+MT” dataset (txt)
Training set of “WT+MT+aSN” dataset (txt)
Test set of “WT+MT+aSN” dataset (txt)
Predicted pKa values for proteins in the human proteome from the AlphaFold Protein Structure Database (txt)
This information is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Warshel A; Sharma PK; Kato M; Parson WW Modeling Electrostatic Effects in Proteins. Biochim. Biophys. Acta - Proteins Proteomics 2006, 1764 (11), 1647–1676. [DOI] [PubMed] [Google Scholar]
- (2).Nielsen JE; Gunner MR; García-Moreno E, The PKa Cooperative B: A Collaborative Effort to Advance Structure-Based Calculations of PKa Values and Electrostatic Effects in Proteins. Proteins Struct. Funct. Bioinforma. 2011, 79 (12), 3249–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Rocchia W; Alexov E; Honig B Extending the Applicability of the Nonlinear Poisson−Boltzmann Equation: Multiple Dielectric Constants and Multivalent Ions. J. Phys. Chem. B 2001, 105 (28), 6507–6514. [Google Scholar]
- (4).Holst M; Baker N; Wang F Adaptive Multilevel Finite Element Solution of the Poisson–Boltzmann Equation I. Algorithms and Examples. J. Comput. Chem. 2000, 21 (15), 1319–1342. [Google Scholar]
- (5).Olsson MHM; Søndergaard CR; Rostkowski M; Jensen JH PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical PKa Predictions. J. Chem. Theory Comput. 2011, 7 (2), 525–537. [DOI] [PubMed] [Google Scholar]
- (6).Søndergaard CR; Olsson MHM; Rostkowski M; Jensen JH Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of PKa Values. J. Chem. Theory Comput. 2011, 7 (7), 2284–2295. [DOI] [PubMed] [Google Scholar]
- (7).Sakipov SN; Flores-Canales JC; Kurnikova MG A Hierarchical Approach to Predict Conformation-Dependent Histidine Protonation States in Stable and Flexible Proteins. J. Phys. Chem. B 2019, 123 (24), 5024–5034. [DOI] [PubMed] [Google Scholar]
- (8).Yu H; Ratheal IM; Artigas P; Roux B Protonation of Key Acidic Residues Is Critical for the K+-Selectivity of the Na/K Pump. Nat. Struct. Mol. Biol. 2011, 18 (10), 1159–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Mongan J; Case DA; McCammon JA Constant PH Molecular Dynamics in Generalized Born Implicit Solvent. J. Comput. Chem. 2004, 25 (16), 2038–2048. [DOI] [PubMed] [Google Scholar]
- (10).Arthur EJ; Yesselman JD; Brooks III CL Predicting Extreme PKa Shifts in Staphylococcal Nuclease Mutants with Constant PH Molecular Dynamics. Proteins Struct. Funct. Bioinforma. 2011, 79 (12), 3276–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Meng Y; Roitberg AE Constant PH Replica Exchange Molecular Dynamics in Biomolecules Using a Discrete Protonation Model. J. Chem. Theory Comput. 2010, 6 (4), 1401–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Swails JM; Roitberg AE Enhancing Conformation and Protonation State Sampling of Hen Egg White Lysozyme Using PH Replica Exchange Molecular Dynamics. J. Chem. Theory Comput. 2012, 8 (11), 4393–4404. [DOI] [PubMed] [Google Scholar]
- (13).Goh GB; Hulbert BS; Zhou H; Brooks CL Constant PH Molecular Dynamics of Proteins in Explicit Solvent with Proton Tautomerism. Proteins Struct. Funct. Bioinforma. 2014, 82 (7), 1319–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Khandogin J; Brooks CL Constant PH Molecular Dynamics with Proton Tautomerism. Biophys. J. 2005, 89 (1), 141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jo S; Vargyas M; Vasko-Szedlar J; Roux B; Im W PBEQ-Solver for Online Visualization of Electrostatic Potential of Biomolecules. Nucleic Acids Res. 2008, 36 (suppl_2), W270–W275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Baptista AM; Teixeira VH; Soares CM Constant-PH Molecular Dynamics Using Stochastic Titration. J. Chem. Phys. 2002, 117 (9), 4184–4200. [Google Scholar]
- (17).Bürgi R; Kollman PA; van Gunsteren WF Simulating Proteins at Constant PH: An Approach Combining Molecular Dynamics and Monte Carlo Simulation. Proteins Struct. Funct. Bioinforma. 2002, 47 (4), 469–480. [DOI] [PubMed] [Google Scholar]
- (18).Lee MS; Salsbury FR; Brooks CL Constant-PH Molecular Dynamics Using Continuous Titration Coordinates. Proteins Struct. Funct. Bioinforma. 2004, 56 (4), 738–752. [DOI] [PubMed] [Google Scholar]
- (19).Wallace JA; Shen JK Continuous Constant PH Molecular Dynamics in Explicit Solvent with PH-Based Replica Exchange. J. Chem. Theory Comput. 2011, 7 (8), 2617–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Khandogin J; Brooks CL Toward the Accurate First-Principles Prediction of Ionization Equilibria in Proteins. Biochemistry 2006, 45 (31), 9363–9373. [DOI] [PubMed] [Google Scholar]
- (21).Riccardi D; Schaefer P; Cui Q PKa Calculations in Solution and Proteins with QM/MM Free Energy Perturbation Simulations: A Quantitative Test of QM/MM Protocols. J. Phys. Chem. B 2005, 109 (37), 17715–17733. [DOI] [PubMed] [Google Scholar]
- (22).Li H; Hains AW; Everts JE; Robertson AD; Jensen JH The Prediction of Protein PKa’s Using QM/MM: The PKa of Lysine 55 in Turkey Ovomucoid Third Domain. J. Phys. Chem. B 2002, 106 (13), 3486–3494. [Google Scholar]
- (23).Jensen JH; Li H; Robertson AD; Molina PA Prediction and Rationalization of Protein PKa Values Using QM and QM/MM Methods. J. Phys. Chem. A 2005, 109 (30), 6634–6643. [DOI] [PubMed] [Google Scholar]
- (24).Kamerlin SCL; Haranczyk M; Warshel A Progress in Ab Initio QM/MM Free-Energy Simulations of Electrostatic Energies in Proteins: Accelerated QM/MM Studies of PKa, Redox Reactions and Solvation Free Energies. J. Phys. Chem. B 2009, 113 (5), 1253–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ghosh N; Cui Q PKa of Residue 66 in Staphylococal Nuclease. I. Insights from QM/MM Simulations with Conventional Sampling. J. Phys. Chem. B 2008, 112 (28), 8387–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Lu B; Cheng X; Huang J; McCammon JA An Adaptive Fast Multipole Boundary Element Method for Poisson−Boltzmann Electrostatics. J. Chem. Theory Comput. 2009, 5 (6), 1692–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cai Z; Luo F; Wang Y; Li E; Huang Y Protein PKa Prediction with Machine Learning. 2021. ChemRxiv. 10.26434/chemrxiv-2021-7gk5l-v2 (accessed December 4, 2021). [DOI] [Google Scholar]
- (28).Gokcan H; Isayev O Prediction of Protein PKa with Representation Learning. 2021. ChemRxiv. 10.26434/chemrxiv-2021-tcn0f (accessed November 11, 2021). [DOI] [Google Scholar]
- (29).Li C; Jia Z; Chakravorty A; Pahari S; Peng Y; Basu S; Koirala M; Panday SK; Petukh M; Li L; Alexov E DelPhi Suite: New Developments and Review of Functionalities. J. Comput. Chem. 2019, 40 (28), 2502–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Feig M; Brooks CL Recent Advances in the Development and Application of Implicit Solvent Models in Biomolecule Simulations. Curr. Opin. Struct. Biol. 2004, 14 (2), 217–224. [DOI] [PubMed] [Google Scholar]
- (31).Feig M; Onufriev A; Lee MS; Im W; Case DA; Brooks III CL Performance Comparison of Generalized Born and Poisson Methods in the Calculation of Electrostatic Solvation Energies for Protein Structures. J. Comput. Chem. 2004, 25 (2), 265–284. [DOI] [PubMed] [Google Scholar]
- (32).Cvitkovic JP; Pauplis CD; Kaminski GA PKA17—A Coarse-Grain Grid-Based Methodology and Web-Based Software for Predicting Protein PK a Shifts. J. Comput. Chem. 2019, 40 (18), 1718–1726. [DOI] [PubMed] [Google Scholar]
- (33).Milletti F; Storchi L; Cruciani G Predicting Protein PKa by Environment Similarity. Proteins Struct. Funct. Bioinforma. 2009, 76 (2), 484–495. [DOI] [PubMed] [Google Scholar]
- (34).Tan KP; Nguyen TB; Patel S; Varadarajan R; Madhusudhan MS Depth: A Web Server to Compute Depth, Cavity Sizes, Detect Potential Small-Molecule Ligand-Binding Cavities and Predict the PKa of Ionizable Residues in Proteins. Nucleic Acids Res. 2013, 41 (W1), W314–W321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Rastogi VK; Girvin ME Structural Changes Linked to Proton Translocation by Subunit c of the ATP Synthase. Nature 1999, 402 (6759), 263–268. [DOI] [PubMed] [Google Scholar]
- (36).Nakano T; Ikegami T; Suzuki T; Yoshida M; Akutsu H A New Solution Structure of ATP Synthase Subunit c from Thermophilic Bacillus PS3, Suggesting a Local Conformational Change for H+-Translocation. J. Mol. Biol. 2006, 358 (1), 132–144. [DOI] [PubMed] [Google Scholar]
- (37).Brown LS; Kamikubo H; Zimányi L; Kataoka M; Tokunaga F; Verdegem P; Lugtenburg J; Lanyi JK A Local Electrostatic Change Is the Cause of the Large-Scale Protein Conformation Shift in Bacteriorhodopsin. Proc. Natl. Acad. Sci. 1997, 94 (10), 5040–5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hartmut L; Brigitte S; Hans-Thomas R; Jean-Philippe C; K. LJ Structural Changes in Bacteriorhodopsin During Ion Transport at 2 Angstrom Resolution. Science (80-. ). 1999, 286 (5438), 255–260. [DOI] [PubMed] [Google Scholar]
- (39).Lanyi JK Bacteriorhodopsin. Annu. Rev. Physiol. 2004, 66 (1), 665–688. [DOI] [PubMed] [Google Scholar]
- (40).Shinya Y; Kyoko S-I; Ryosuke N; Rieko Y; Eiki Y; Noriko I; Min Y; Jie FM; Peters LC; Tsunehiro M; Hiroshi Y; Takashi T; Tomitake T Redox-Coupled Crystal Structural Changes in Bovine Heart Cytochrome c Oxidase. Science (80-. ). 1998, 280 (5370), 1723–1729. [DOI] [PubMed] [Google Scholar]
- (41).Stites WE; Gittis AG; Lattman EE; Shortle D In a Staphylococcal Nuclease Mutant the Side-Chain of a Lysine Replacing Valine 66 Is Fully Buried in the Hydrophobic Core. J. Mol. Biol. 1991, 221 (1), 7–14. [DOI] [PubMed] [Google Scholar]
- (42).Isom DG; Cannon BR; Castañeda CA; Robinson A; García-Moreno E, High B Tolerance for Ionizable Residues in the Hydrophobic Interior of Proteins. Proc. Natl. Acad. Sci. 2008, 105 (46), 17784–17788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Isom DG; Castañeda CA; Cannon BR; Velu PD; García-Moreno E, Charges B in the Hydrophobic Interior of Proteins. Proc. Natl. Acad. Sci. 2010, 107 (37), 16096–16100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Isom DG; Castañeda CA; Cannon BR; García-Moreno E, Large B Shifts in PKa Values of Lysine Residues Buried inside a Protein. Proc. Natl. Acad. Sci. 2011, 108 (13), 5260–5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Fitch CA; Karp DA; Lee KK; Stites WE; Lattman EE; García-Moreno EB Experimental PKa Values of Buried Residues: Analysis with Continuum Methods and Role of Water Penetration. Biophys. J. 2002, 82 (6), 3289–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Karp DA; Gittis AG; Stahley MR; Fitch CA; Stites WE; García-Moreno E, High B Apparent Dielectric Constant Inside a Protein Reflects Structural Reorganization Coupled to the Ionization of an Internal Asp. Biophys. J. 2007, 92 (6), 2041–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Harms MJ; Castañeda CA; Schlessman JL; Sue GR; Isom DG; Cannon BR; García-Moreno E, The B PKa Values of Acidic and Basic Residues Buried at the Same Internal Location in a Protein Are Governed by Different Factors. J. Mol. Biol. 2009, 389 (1), 34–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Harms MJ; Schlessman JL; Sue GR; García-Moreno E, Arginine B Residues at Internal Positions in a Protein Are Always Charged. Proc. Natl. Acad. Sci. 2011, 108 (47), 18954–18959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Cannon BR Thermodynamic Consequences of Substitutions of Internal Positions in Proteins with Polar and Ionizable Residues, Johns Hopkins University, 2008. [Google Scholar]
- (50).Chimenti MS; Khangulov VS; Robinson AC; Heroux A; Majumdar A; Schlessman JL; García-Moreno B Structural Reorganization Triggered by Charging of Lys Residues in the Hydrophobic Interior of a Protein. Structure 2012, 20 (6), 1071–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Damjanović A; Brooks BR; García-Moreno E, Conformational Relaxation B and Water Penetration Coupled to Ionization of Internal Groups in Proteins. J. Phys. Chem. A 2011, 115 (16), 4042–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Richman DE; Majumdar A; García-Moreno E, Conformational B Reorganization Coupled to the Ionization of Internal Lys Residues in Proteins. Biochemistry 2015, 54 (38), 5888–5897. [DOI] [PubMed] [Google Scholar]
- (53).Sarkar A; Roitberg AE PH-Dependent Conformational Changes Lead to a Highly Shifted PKa for a Buried Glutamic Acid Mutant of SNase. J. Phys. Chem. B 2020, 124 (49), 11072–11080. [DOI] [PubMed] [Google Scholar]
- (54).Sarkar A; Gupta PL; Roitberg AE PH-Dependent Conformational Changes Due to Ionizable Residues in a Hydrophobic Protein Interior: The Study of L25K and L125K Variants of SNase. J. Phys. Chem. B 2019, 123 (27), 5742–5754. [DOI] [PubMed] [Google Scholar]
- (55).Zheng Y; Cui Q Microscopic Mechanisms That Govern the Titration Response and PKa Values of Buried Residues in Staphylococcal Nuclease Mutants. Proteins Struct. Funct. Bioinforma. 2017, 85 (2), 268–281. [DOI] [PubMed] [Google Scholar]
- (56).Alexov E; Mehler EL; Baker N; Baptista, A. M; Huang Y; Milletti F; Erik Nielsen J; Farrell D; Carstensen T; Olsson MHM; Shen JK; Warwicker J; Williams S; Word JM Progress in the Prediction of PKa Values in Proteins. Proteins Struct. Funct. Bioinforma. 2011, 79 (12), 3260–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Reis PBPS; Vila-Viçosa D; Rocchia W; PypKa Machuqueiro, M.: A Flexible Python Module for Poisson–Boltzmann-Based PKa Calculations. J. Chem. Inf. Model. 2020, 60 (10), 4442–4448. [DOI] [PubMed] [Google Scholar]
- (58).Muegge I; Qi PX; Wand AJ; Chu ZT; Warshel A The Reorganization Energy of Cytochrome c Revisited. J. Phys. Chem. B 1997, 101 (5), 825–836. [Google Scholar]
- (59).Simonson T; Carlsson J; Case DA Proton Binding to Proteins: PKa Calculations with Explicit and Implicit Solvent Models. J. Am. Chem. Soc. 2004, 126 (13), 4167–4180. [DOI] [PubMed] [Google Scholar]
- (60).Wang L; Li L; Alexov E PKa Predictions for Proteins, RNAs, and DNAs with the Gaussian Dielectric Function Using DelPhi PKa. Proteins Struct. Funct. Bioinforma. 2015, 83 (12), 2186–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Damjanović A; García-Moreno B; Lattman EE; García AE Molecular Dynamics Study of Water Penetration in Staphylococcal Nuclease. Proteins Struct. Funct. Bioinforma. 2005, 60 (3), 433–449. [DOI] [PubMed] [Google Scholar]
- (62).Damjanović A; Wu X; García-Moreno E,B; Brooks BR Backbone Relaxation Coupled to the Ionization of Internal Groups in Proteins: A Self-Guided Langevin Dynamics Study. Biophys. J. 2008, 95 (9), 4091–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Wu X; Lee J; Brooks BR Origin of PKa Shifts of Internal Lysine Residues in SNase Studied Via Equal-Molar VMMS Simulations in Explicit Water. J. Phys. Chem. B 2017, 121 (15), 3318–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Jia Z; Li L; Chakravorty A; Alexov E Treating Ion Distribution with Gaussian-Based Smooth Dielectric Function in DelPhi. J. Comput. Chem. 2017, 38 (22), 1974–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Chakravorty A; Jia Z; Peng Y; Tajielyato N; Wang L; Alexov E Gaussian-Based Smooth Dielectric Function: A Surface-Free Approach for Modeling Macromolecular Binding in Solvents. Front. Mol. Biosci. 2018, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Li L; Li C; Zhang Z; Alexov E On the Dielectric “Constant” of Proteins: Smooth Dielectric Function for Macromolecular Modeling and Its Implementation in DelPhi. J. Chem. Theory Comput. 2013, 9 (4), 2126–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Chakravorty A; Panday S; Pahari S; Zhao S; Alexov E Capturing the Effects of Explicit Waters in Implicit Electrostatics Modeling: Qualitative Justification of Gaussian-Based Dielectric Models in DelPhi. J. Chem. Inf. Model. 2020, 60 (4), 2229–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Georgescu RE; Alexov EG; Gunner MR Combining Conformational Flexibility and Continuum Electrostatics for Calculating PKas in Proteins. Biophys. J. 2002, 83 (4), 1731–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Antosiewicz J; McCammon JA; Gilson MK Prediction of Ph-Dependent Properties of Proteins. J. Mol. Biol. 1994, 238 (3), 415–436. [DOI] [PubMed] [Google Scholar]
- (70).Alexov EG; Gunner MR Incorporating Protein Conformational Flexibility into the Calculation of PH-Dependent Protein Properties. Biophys. J. 1997, 72 (5), 2075–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Song Y; Mao J; Gunner MR MCCE2: Improving Protein PKa Calculations with Extensive Side Chain Rotamer Sampling. J. Comput. Chem. 2009, 30 (14), 2231–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Dlugosz M; Antosiewicz JM; Robertson AD Constant-PH Molecular Dynamics Study of Protonation-Structure Relationship in a Heptapeptide Derived from Ovomucoid Third Domain. Phys. Rev. E 2004, 69 (2), 21915. [DOI] [PubMed] [Google Scholar]
- (73).Dlugosz M; Antosiewicz JM Constant-PH Molecular Dynamics Simulations: A Test Case of Succinic Acid. Chem. Phys. 2004, 302 (1), 161–170. [Google Scholar]
- (74).Schaefer M; Karplus M A Comprehensive Analytical Treatment of Continuum Electrostatics. J. Phys. Chem. 1996, 100 (5), 1578–1599. [Google Scholar]
- (75).Donnini S; Tegeler F; Groenhof G; Grubmüller H Constant PH Molecular Dynamics in Explicit Solvent with λ-Dynamics. J. Chem. Theory Comput. 2011, 7 (6), 1962–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Chen Y; Roux B Constant-PH Hybrid Nonequilibrium Molecular Dynamics–Monte Carlo Simulation Method. J. Chem. Theory Comput. 2015, 11 (8), 3919–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Radak BK; Chipot C; Suh D; Jo S; Jiang W; Phillips JC; Schulten K; Roux B Constant-PH Molecular Dynamics Simulations for Large Biomolecular Systems. J. Chem. Theory Comput. 2017, 13 (12), 5933–5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Lee J; Miller BT; Damjanović A; Brooks BR Constant PH Molecular Dynamics in Explicit Solvent with Enveloping Distribution Sampling and Hamiltonian Exchange. J. Chem. Theory Comput. 2014, 10 (7), 2738–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Wu X; Brooks BR A Virtual Mixture Approach to the Study of Multistate Equilibrium: Application to Constant PH Simulation in Explicit Water. PLOS Comput. Biol. 2015, 11 (10), 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Stern HA Molecular Simulation with Variable Protonation States at Constant PH. J. Chem. Phys. 2007, 126 (16), 164112. [DOI] [PubMed] [Google Scholar]
- (81).Swails JM; York DM; Roitberg AE Constant PH Replica Exchange Molecular Dynamics in Explicit Solvent Using Discrete Protonation States: Implementation, Testing, and Validation. J. Chem. Theory Comput. 2014, 10 (3), 1341–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).May ER; Arora K; Brooks CL PH-Induced Stability Switching of the Bacteriophage HK97 Maturation Pathway. J. Am. Chem. Soc. 2014, 136 (8), 3097–3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Laricheva EN; Goh GB; Dickson A; Brooks CL PH-Dependent Transient Conformational States Control Optical Properties in Cyan Fluorescent Protein. J. Am. Chem. Soc. 2015, 137 (8), 2892–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Ahlstrom LS; Law SM; Dickson A; Brooks CL Multiscale Modeling of a Conditionally Disordered PH-Sensing Chaperone. J. Mol. Biol. 2015, 427 (8), 1670–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Itoh SG; Damjanović A; Brooks BR PH Replica-Exchange Method Based on Discrete Protonation States. Proteins Struct. Funct. Bioinforma. 2011, 79 (12), 3420–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Williams SL; de Oliveira CAF; McCammon JA Coupling Constant PH Molecular Dynamics with Accelerated Molecular Dynamics. J. Chem. Theory Comput. 2010, 6 (2), 560–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Meng Y; Sabri Dashti D; Roitberg AE Computing Alchemical Free Energy Differences with Hamiltonian Replica Exchange Molecular Dynamics (H-REMD) Simulations. J. Chem. Theory Comput. 2011, 7 (9), 2721–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Lee J; Miller BT; Damjanović A; Brooks BR Enhancing Constant-PH Simulation in Explicit Solvent with a Two-Dimensional Replica Exchange Method. J. Chem. Theory Comput. 2015, 11 (6), 2560–2574. [DOI] [PubMed] [Google Scholar]
- (89).Barroso daSilva FL; Dias LG Development of Constant-PH Simulation Methods in Implicit Solvent and Applications in Biomolecular Systems. Biophys. Rev. 2017, 9 (5), 699–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Liu J; Swails J; Zhang JZH; He X; Roitberg AE A Coupled Ionization-Conformational Equilibrium Is Required To Understand the Properties of Ionizable Residues in the Hydrophobic Interior of Staphylococcal Nuclease. J. Am. Chem. Soc. 2018, 140 (5), 1639–1648. [DOI] [PubMed] [Google Scholar]
- (91).Damjanovic A; Miller BT; Okur A; Brooks BR Reservoir PH Replica Exchange. J. Chem. Phys. 2018, 149 (7), 72321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Li M; Zhang H; Chen B; Wu Y; Guan L Prediction of PKa Values for Neutral and Basic Drugs Based on Hybrid Artificial Intelligence Methods. Sci. Rep. 2018, 8 (1), 3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Chen B; Zhang H; Li M Prediction of PK(a) Values of Neutral and Alkaline Drugs with Particle Swarm Optimization Algorithm and Artificial Neural Network. Neural Comput. Appl. 2019, 31 (12), 8297–8304. [Google Scholar]
- (94).Lu Y; Anand S; Shirley W; Gedeck P; Kelley BP; Skolnik S; Rodde S; Nguyen M; Lindvall M; Jia W Prediction of PKa Using Machine Learning Methods with Rooted Topological Torsion Fingerprints: Application to Aliphatic Amines. J. Chem. Inf. Model. 2019, 59 (11), 4706–4719. [DOI] [PubMed] [Google Scholar]
- (95).Mansouri K; Cariello NF; Korotcov A; Tkachenko V; Grulke CM; Sprankle CS; Allen D; Casey WM; Kleinstreuer NC; Williams AJ Open-Source QSAR Models for PKa Prediction Using Multiple Machine Learning Approaches. J. Cheminform. 2019, 11 (1), 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Baltruschat M; Czodrowski P Machine Learning Meets PKa [Version 2; Peer Review: 2 Approved]. F1000Research 2020, 9 (113). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Yang Q; Li Y; Yang J-D; Liu Y; Zhang L; Luo S; Cheng J-P Holistic Prediction of the PKa in Diverse Solvents Based on a Machine-Learning Approach. Angew. Chemie Int. Ed. 2020, 59 (43), 19282–19291. [DOI] [PubMed] [Google Scholar]
- (98).Pan X; Wang H; Li C; Zhang JZH; Ji C MolGpka: A Web Server for Small Molecule PKa Prediction Using a Graph-Convolutional Neural Network. J. Chem. Inf. Model. 2021, 61 (7), 3159–3165. [DOI] [PubMed] [Google Scholar]
- (99).Kwon Y; Shin W-H; Ko J; Lee J AK-Score: Accurate Protein-Ligand Binding Affinity Prediction Using an Ensemble of 3D-Convolutional Neural Networks. Int. J. Mol. Sci. 2020, 21 (22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Wee J; Xia K Ollivier Persistent Ricci Curvature-Based Machine Learning for the Protein–Ligand Binding Affinity Prediction. J. Chem. Inf. Model. 2021, 61 (4), 1617–1626. [DOI] [PubMed] [Google Scholar]
- (101).Wang DD; Zhu M; Yan H Computationally Predicting Binding Affinity in Protein–Ligand Complexes: Free Energy-Based Simulations and Machine Learning-Based Scoring Functions. Brief. Bioinform. 2021, 22 (3), bbaa107. [DOI] [PubMed] [Google Scholar]
- (102).Liu X; Feng H; Wu J; Xia K Persistent Spectral Hypergraph Based Machine Learning (PSH-ML) for Protein-Ligand Binding Affinity Prediction. Brief. Bioinform. 2021, 22 (5), bbab127. [DOI] [PubMed] [Google Scholar]
- (103).Son J; Kim D Development of a Graph Convolutional Neural Network Model for Efficient Prediction of Protein-Ligand Binding Affinities. PLoS One 2021, 16 (4), e0249404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Reis P; Bertolini M; Montanari F; Rocchia W; Machuqueiro M; Clevert D-A PKAI: A Fast and Interpretable Deep Learning Approach for Accurate Electrostatics-Driven PKa Predictions. 2021. Research Square. 10.21203/rs.3.rs-949180/v1 (accessed December 8, 2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Xiong Y; Liu J; Wei D-Q An Accurate Feature-Based Method for Identifying DNA-Binding Residues on Protein Surfaces. Proteins Struct. Funct. Bioinforma. 2011, 79 (2), 509–517. [DOI] [PubMed] [Google Scholar]
- (106).Chu Y; Kaushik AC; Wang X; Wang W; Zhang Y; Shan X; Salahub DR; Xiong Y; Wei D-Q DTI-CDF: A Cascade Deep Forest Model towards the Prediction of Drug-Target Interactions Based on Hybrid Features. Brief. Bioinform. 2021, 22 (1), 451–462. [DOI] [PubMed] [Google Scholar]
- (107).Pedregosa F; Varoquaux G; Gramfort A; Michel V; Thirion B; Grisel O; Blondel M; Prettenhofer P; Weiss R; Dubourg V; Vanderplas J; Passos A; Cournapeau D; Brucher M; Perrot M; Duchesnay E Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- (108).Pahari S; Sun L; Alexov E PKAD: A Database of Experimentally Measured PKa Values of Ionizable Groups in Proteins. Database 2019, 2019, baz024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Eastman P PDBFixer Documentation. https://htmlpreview.github.io/?https://github.com/openmm/pdbfixer/blob/master/Manual.html (accessed December 10, 2021). [Google Scholar]
- (110).Hubbard SJ; Thornton JM NACCESS, version 2.1.1; 1996. [Google Scholar]
- (111).McGibbon RT; Beauchamp KA; Harrigan MP; Klein C; Swails JM; Hernández CX; Schwantes CR; Wang L-P; Lane TJ; Pande VS MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109 (8), 1528–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Cock PJA; Antao T; Chang JT; Chapman BA; Cox CJ; Dalke A; Friedberg I; Hamelryck T; Kauff F; Wilczynski B; de Hoon MJL Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics 2009, 25 (11), 1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Ke G; Meng Q; Finley T; Wang T; Chen W; Ma W; Ye Q; Liu T-Y LightGBM: A Highly Efficient Gradient Boosting Decision Tree. In Proceedings of the 31st International Conference on Neural Information Processing Systems; NIPS’17; Curran Associates Inc.: Red Hook, NY, USA, 2017; pp 3149–3157. [Google Scholar]
- (114).Chen T; Guestrin C XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining; KDD ‘16; ACM: New York, NY, USA, 2016; pp 785–794. [Google Scholar]
- (115).Akiba T; Sano S; Yanase T; Ohta T; Koyama M Optuna: A Next-Generation Hyperparameter Optimization Framework. In Proceedings of the 25rd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining; 2019. [Google Scholar]
- (116).Chen AY; Brooks BR; Damjanovic A Determinants of Conductance of a Bacterial Voltage-Gated Sodium Channel. Biophys. J. 2021, 120 (15), 3050–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).DeCaen PG; Takahashi Y; Krulwich TA; Ito M; Clapham DE Ionic Selectivity and Thermal Adaptations within the Voltage-Gated Sodium Channel Family of Alkaliphilic Bacillus. Elife 2014, 3, e04387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Damjanovic A; Chen AY; Rosenberg RL; Roe DR; Wu X; Brooks BR Protonation State of the Selectivity Filter of Bacterial Voltage-Gated Sodium Channels Is Modulated by Ions. Proteins Struct. Funct. Bioinforma. 2020, 88 (3), 527–539. [DOI] [PubMed] [Google Scholar]
- (119).Jumper J; Evans R; Pritzel A; Green T; Figurnov M; Ronneberger O; Tunyasuvunakool K; Bates R; Žídek A; Potapenko A; Bridgland A; Meyer C; Kohl SAA; Ballard AJ; Cowie A; Romera-Paredes B; Nikolov S; Jain R; Adler J; Back T; Petersen S; Reiman D; Clancy E; Zielinski M; Steinegger M; Pacholska M; Berghammer T; Bodenstein S; Silver D; Vinyals O; Senior AW; Kavukcuoglu K; Kohli P; Hassabis D Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596 (7873), 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).AlphaFold Protein Structure Database. https://alphafold.ebi.ac.uk (accessed December 10, 2021). [Google Scholar]
- (121).Humphrey W; Dalke A; Schulten K VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14 (1), 33–38. [DOI] [PubMed] [Google Scholar]
- (122).Pahari S; Sun L; Basu S; Alexov E DelPhiPKa: Including Salt in the Calculations and Enabling Polar Residues to Titrate. Proteins Struct. Funct. Bioinforma. 2018, 86 (12), 1277–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Evans R; O’Neill M; Pritzel A; Antropova N; Senior A; Green T; Zidek A; Bates R; Blackwell S; Yim J; Ronneberger O; Bodenstein S; Zielinski M; Bridgland A; Potapenko A; Cowie A; Tunyasuvunakool K; Jain R; Clancy E; Kohli P; Jumper J; Hassabis D Protein Complex Prediction with AlphaFold-Multimer. 2021. bioRxiv. 10.1101/2021.10.04.463034 (accessed December 10, 2021). [DOI] [Google Scholar]
- (124).Iwata S; Ostermeier C; Ludwig B; Michel H Structure at 2.8 Å Resolution of Cytochrome c Oxidase from Paracoccus Denitrificans. Nature 1995, 376 (6542), 660–669. [DOI] [PubMed] [Google Scholar]
- (125).Hunte C; Screpanti E; Venturi M; Rimon A; Padan E; Michel H Structure of a Na+/H+ Antiporter and Insights into Mechanism of Action and Regulation by PH. Nature 2005, 435 (7046), 1197–1202. [DOI] [PubMed] [Google Scholar]
- (126).Liang F; B. CE; Yichun H; Roderick M Structure of a Eukaryotic CLC Transporter Defines an Intermediate State in the Transport Cycle. Science (80-. ). 2010, 330 (6004), 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Yikilmaz E; Rodgers DW; Miller A-F The Crucial Importance of Chemistry in the Structure−Function Link: Manipulating Hydrogen Bonding in Iron-Containing Superoxide Dismutase,. Biochemistry 2006, 45 (4), 1151–1161. [DOI] [PubMed] [Google Scholar]
- (128).Neira JL; Rizzuti B; Iovanna JL Determinants of the PKa Values of Ionizable Residues in an Intrinsically Disordered Protein. Arch. Biochem. Biophys. 2016, 598, 18–27. [DOI] [PubMed] [Google Scholar]
- (129).Word JM; Nicholls A Application of the Gaussian Dielectric Boundary in Zap to the Prediction of Protein PKa Values. Proteins Struct. Funct. Bioinforma. 2011, 79 (12), 3400–3409. [DOI] [PubMed] [Google Scholar]
- (130).Song Y Exploring Conformational Changes Coupled to Ionization States Using a Hybrid Rosetta-MCCE Protocol. Proteins Struct. Funct. Bioinforma. 2011, 79 (12), 3356–3363. [DOI] [PubMed] [Google Scholar]
- (131).Witham S; Talley K; Wang L; Zhang Z; Sarkar S; Gao D; Yang W; Alexov E Developing Hybrid Approaches to Predict PKa Values of Ionizable Groups. Proteins Struct. Funct. Bioinforma. 2011, 79 (12), 3389–3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Wu Z; Pan S; Chen F; Long G; Zhang C; Yu PS A Comprehensive Survey on Graph Neural Networks. IEEE Trans. Neural Networks Learn. Syst. 2021, 32 (1), 4–24. [DOI] [PubMed] [Google Scholar]
- (133).Bello SA; Yu S; Wang C; Adam JM; Li J Review: Deep Learning on 3D Point Clouds. Remote Sens. 2020, 12 (11). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.