

Abstract
The neural circuits governing the induction and progression of neurodegeneration and memory impairment in Alzheimer’s disease (AD) are incompletely understood. The mammillary body (MB), a subcortical node of the medial limbic circuit, is one of the first brain regions to exhibit amyloid deposition in the 5xFAD mouse model of AD. Amyloid burden in the MB correlates with pathological diagnosis of AD in human postmortem brain tissue. Whether and how MB neuronal circuitry contributes to neurodegeneration and memory deficits in AD is unknown. Using 5xFAD mice and postmortem MB samples from individuals with varying degrees of AD pathology, we identified two neuronal cell types in the MB harboring distinct electrophysiological properties and long-range projections: lateral neurons and medial neurons. Lateral MB neurons harbored aberrant hyperactivity and exhibited early neurodegeneration in 5xFAD mice compared to lateral MB neurons in wildtype littermates. Inducing hyperactivity in lateral MB neurons in wildtype mice impaired performance on memory tasks, whereas attenuating aberrant hyperactivity in lateral MB neurons ameliorated memory deficits in 5xFAD mice. Our findings suggest that neurodegeneration may be a result of genetically distinct, projection-specific cellular dysfunction, and that dysregulated lateral MB neurons may be causally linked to memory deficits in AD.
Accessible Summary:
The constellation of circuit dysfunction, pathological insults, and neurodegeneration driving memory deficits in Alzheimer’s disease remains poorly understood. The mammillary body, a subregion of the hypothalamus, exhibits early amyloid deposition in 5xFAD mice, which we hypothesized offered a tractable system to interrogate mechanisms of cell-type specific dysfunction regulating memory loss in Alzheimer’s. Using a combination of single cell RNA-seq of mouse and post-mortem mammillary body, combined with slice electrophysiology and bidirectional circuit manipulation, we identify functional characteristics that confer disproportionate AD related vulnerability to select neurons. These results shed insight on the circuit and molecular logic of neurodegeneration.
One sentence summary:
Lateral mammillary body neurons in mouse brain are disproportionately vulnerable in Alzheimer’s disease and may be involved in memory loss.
INTRODUCTION
Neurodegeneration is a major cause of memory loss and cognitive impairment, but the mechanisms driving susceptibility to cell death in individual neurons of the degenerating brain are incompletely understood. Alzheimer’s disease (AD) is a memory disorder pathologically characterized by extracellular deposits of amyloid plaques and neurofibrillary tangles(1–3), and evidence from human and animal studies indicates that AD impairs circuit connectivity and network function (1, 4–7). Neuronal hyperactivity, including non-convulsive seizure activity in the cortical and hippocampal networks (8–10), is evident in early stages of AD (4, 5, 11) even before amyloid plaque deposition (11, 12). Whereas neuronal hyperactivity and neurodegeneration in the cortical-hippocampal networks are associated with AD, memory systems are distributed throughout the brain (13–16), and contributions to AD dementia from deep brain regions are only vaguely characterized (17). Identifying new brain regions and cell types uniquely vulnerable to hyperexcitability, memory deficits, and neurodegeneration in AD may provide new therapeutic targets for disease-modifying treatments.
We recently examined the sequence of amyloid deposition in 5xFAD mice (18), a mouse model harboring five familial AD mutations that recapitulates AD-related cognitive and cellular dysfunctions (19). We found that the mammillary body (MB) exhibits the highest density of amyloid deposition (18). The MB is a subcortical node of the medial limbic circuit that plays a key role in memory retrieval (20, 21), and lesions in the MB result in anterograde amnesia and memory deficits (22–28). MB volume correlates with episodic memory recall in patients who underwent surgical removal of colloid cysts (25), in which the extent of MB atrophy differs among individuals. The critical role played by the MB in memory recall is recapitulated in lesion studies in monkeys (29–31), rats (32–40) and mice (41–44). Post-mortem brain studies report amyloid deposition and neurofibrillary tangles in the MB of individuals with AD (45–47), and suggest increased MB amyloid is associated with increased likelihood of pathological AD (18). Moreover, MB synaptic alterations are observed before amyloid and neurofibrillary tangle pathology emerge (48), indicating MB synaptic deficits may be an early event leading to disease progression. Reduction of MB volume is also reported in individuals with mild AD (49). Together, these findings suggest a potential role of MB dysfunction in early stages of AD, but whether and how MB circuit dysfunction is related to AD-related cognitive decline is unclear. Here, using single cell transcriptomic profiling of mouse and human MB postmortem tissue, combined with multiple interventions in the 5xFAD mouse model of AD, we provide evidence that a class of MB neurons may be critically involved in AD memory loss.
RESULTS
Single cell transcriptomic profiling of mouse MB brain tissue
To define cell types in the MB of 5xFAD mouse brain, we dissected the hypothalamus, which includes the MB, for single cell RNA-seq (scRNA-seq) (fig. S1A). We pooled hypothalamus tissue from nine female C57BL/6 mice (two months old) into two samples to reduce the variability of the dissected brain regions between samples. We included 7,754 single cells for analysis, which we classified into astrocytes (expressing Slc6a11, Agt, Slc4a4), microglia (Aif1, Csf1r, Cx3cr1), neurons (Snap25, Nrsn2, Syt4), mature oligodendrocytes (Mag, Mal, Mog), oligodendrocyte precursor cells (Pdgfra, Matn4, C1ql1), newly formed oligodendrocytes (Fyn, Gpr17, Bfsp2), tanycytes (Col23a1, Crym, Rax), ependymal cells (Ccdc153, Tmem212, Foxj1), endothelial cells (Cldn5, Itm2a, Ly6c1), and mural cells (Vtn, Higd1b, Ndufa4l2) (fig. S1B, C). All major cell types included cells derived from both samples (fig. S1D). Unsupervised clustering analysis of neurons (n=1,382 cells) revealed twelve clusters (Fig. 1A). We classified neurons into glutamatergic and GABAergic neurons based on the expression of genes necessary for the synthesis and packaging of glutamate and GABA (Fig. 1B) (50). To evaluate which neuronal clusters represented MB neurons, we first determined whether MB neurons are glutamatergic or GABAergic using RNA in situ hybridization of Slc17a6 and Slc32a1. We found expression of Slc17a6 and the absence of Slc32a1 in the mouse MB (Fig. 1C), indicating that the MB is composed of glutamatergic neurons, consistent with previous observations in rats (51, 52). Among the five glutamatergic neuronal clusters, the top marker genes for clusters 4 and 11 were detected in the MB (Fig. 1D), but the other clusters were not (fig. S2A), suggesting that clusters 4 (n=154 cells) and 11 (n=47 cells) represented two separate neuronal cell types in the MB. We found the top marker genes for these two clusters (Rprm and Tac2) labeled neurons in anatomically discrete MB subregions, the medial MB and lateral MB (Fig. 1D). The expression profiles of lateral and medial MB clusters revealed multiple discriminatory marker genes: medial MB marker genes included Rprm, Cck, Cartpt, Lhx1os, and Foxb1, whereas lateral MB marker genes included Tac2, Prlr, Esr1, Nr4a2, and Pdyn (Fig. 1E). To interrogate the functional differences between these two types of neurons, we performed Gene Ontology (GO) enrichment analysis. The top GO terms for medial MB neurons were associated with ion channel activity (fig. S2B), whereas those for lateral MB neurons included structural constituents of ribosomes (fig. S2C). These observations suggest the two types of MB neurons may feature different electrophysiological properties and require specific metabolic demands. To confirm the transcriptional subtype of MB neurons, we micro-dissected the MB and repeated scRNA-seq in wildtype (WT) and 6-month-old 5xFAD mice (fig. S3). We again found consistent evidence for lateral and medial MB neurons in the microdissected MB dataset in both WT and 5xFAD mice (fig. S3). Collectively, these findings indicate that neurons of the MB harbor two distinct neuronal subtypes.
Figure 1. Single cell transcriptomic analysis reveals two anatomically distinct neuronal cell types in the MB.
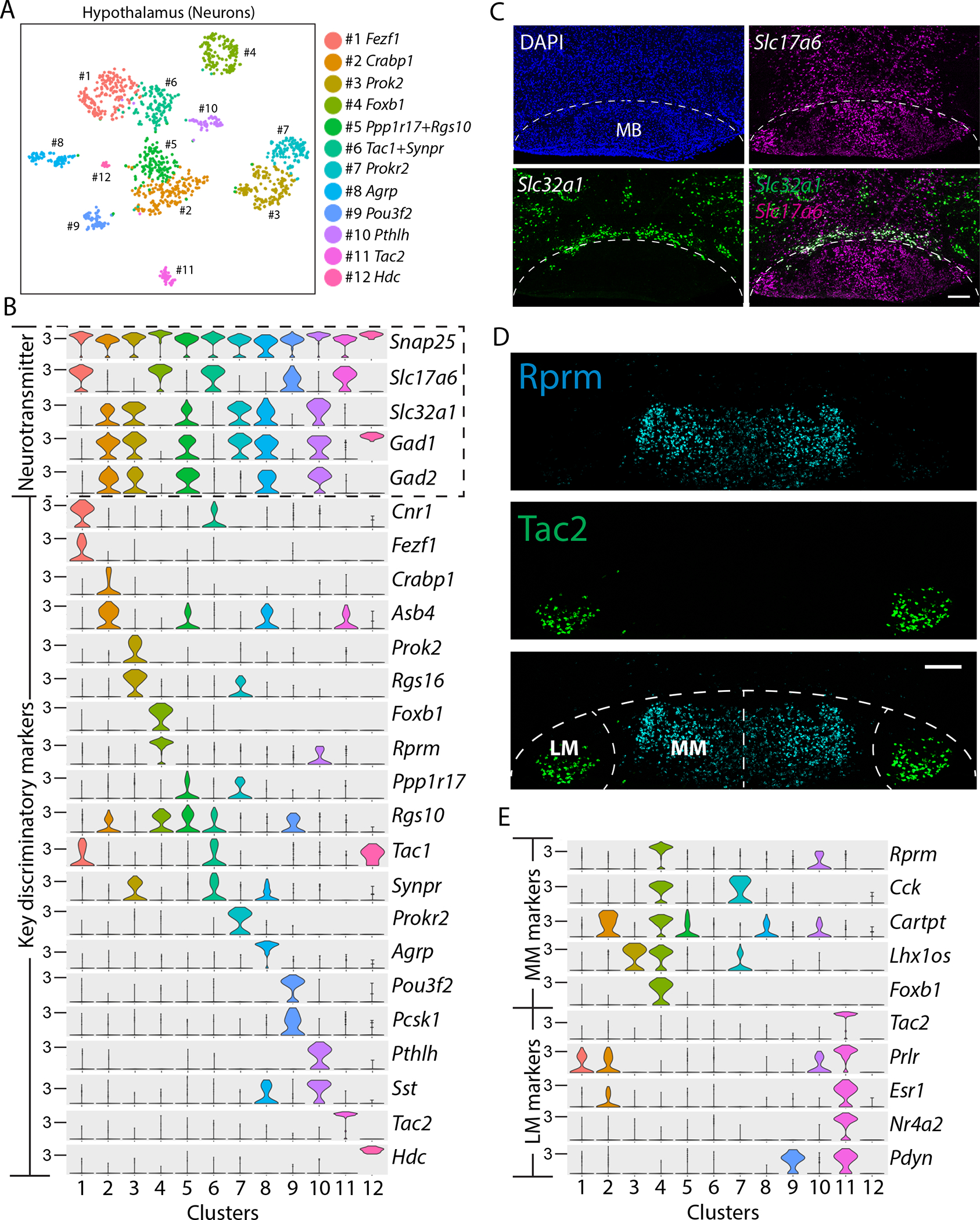
(A) A tSNE plot shows 12 clusters of neuronal subtypes in the hypothalamus of C57BL/6 mice. (B) Violin plots demonstrate the expression of the pan-neuronal gene (Snap25), glutamatergic marker gene (Slc17a6), GABAergic marker genes (Slc32a1, Gad1, and Gad2), and the key discriminatory genes in each neuronal cluster. (C) Confocal images of WT mouse brain show in situ hybridization of Slc17a6 (vesicular glutamate transporter 2; magenta) and Slc32a1 (vesicular GABA transporter; green) in the mammillary body (MB), with DAPI nuclear stain (blue). The dotted line outlines the boundary of the MB. Scale bar: 200μm. (D) Confocal image of in situ hybridization of Tac2 (green) and Rprm (cyan) in the MB of WT mice. The dotted line outlines the boundary of the lateral MB (LM) and medial MB (MM). Scale bar: 200μm. (E) Violin plots show the expression of top marker genes for MM (cluster 4) and LM (cluster 11).
Two MB neuron subtypes are distinguished by morphology, projection targets, and neuronal activity patterns
Given that the two types of MB neurons were located in anatomically defined subregions, we characterized their morphology, projections, and neuronal activity patterns. To quantify neuronal morphology, we retro-orbitally injected AAV-PHP.eB-CAG-tdTomato (53) into female 2 month-old C57BL/6 mice to sparsely label MB neurons. We found that the soma of lateral MB neurons was significantly (p = 0.0016) larger than that of medial MB neurons (Fig. 2A, B). Given that metabolic activity is associated with cell size (54, 55), this observation was consistent with our finding that lateral MB neurons harbored elevated expression of genes associated with metabolic activity and protein synthesis compared to lateral MB neurons (fig. S2C).
Figure 2. Lateral and medial MB neurons exhibit distinct morphology, electrophysiological properties, and projection targets.
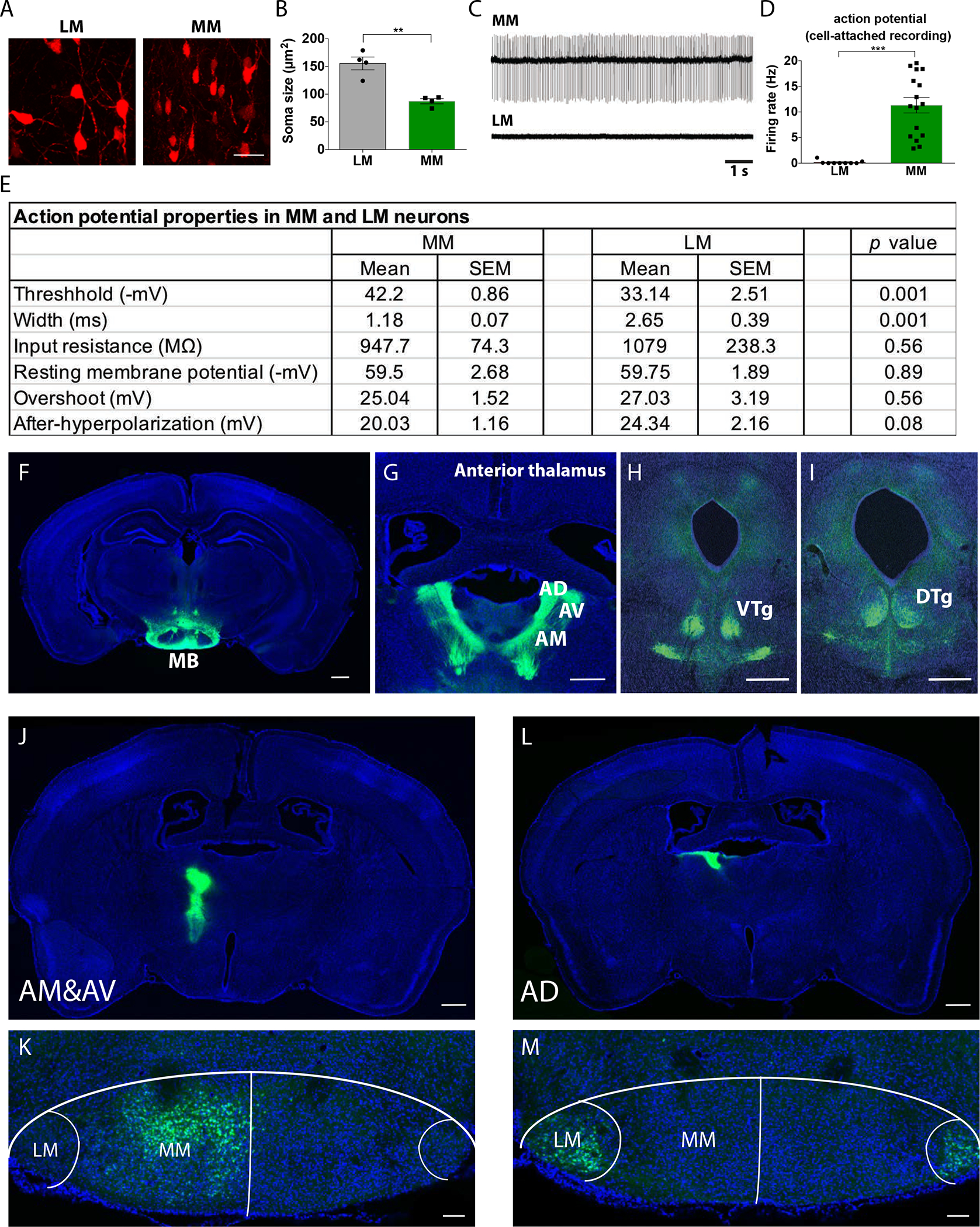
(A) Confocal images of mCherry labelled neurons (red) in the lateral mammillary body (LM) and the medial mammillary body (MM) are shown. Scale bar: 25μm. (B) Cell soma size for neurons in the LM and MM was measured. t(6)=5.506, p=0.0015. **p<0.01 (Unpaired t-test). n=4 mice per group. (C) Representative action potential traces of neurons located in the MM or LM of WT mice. (D) Action potential firing rate for medial MB neurons (MM) and lateral MB neurons (LM) was quantified. t(23)=5.490, p<0.0001, ***p<0.0001 (unpaired t-test). n=9 for LM and n=16 for MM neurons. (E) Action potential properties of MM and LM neurons are shown. n=10–12 cells per group (p values are shown; unpaired t-test). (F-I) Mouse brain images show (F) the mammillary body (MB) projecting to (G) the anterior thalamus, including the anterior dorsal (AD), anterior medial (AM), and anterior ventral (AV) thalamic nuclei, (H) ventral tegmental nuclei (VTg), and (I) dorsal tegmental nuclei (DTg) following AAV8-CAMKII-eGFP injection to MB. DAPI stain (blue), and eGFP (green). Scale bar: 500μm. (J-M) Alexa 488 (green) conjugated cholera toxin B (CTB) was injected into (J) anterior medial (AM) and anterior ventral (AV) thalamic nuclei and (L) anterior dorsal (AD) thalamic nuclei of WT mouse brain. CTB labeled neurons (green) were located in (K) the medial MB (MM) and (M) the lateral MB LM of mouse brain slices. Hoechst stain, blue. For panels J and L, scale bar: 500μm; for panels K and M, scale bar: 100μm.
Given the differences in lateral and medial MB neuronal soma size, we wondered whether these neuronal types harbored different electrophysiological properties. Using cell-attached patch-clamp recordings, we found that medial MB neurons fired more action potentials than did lateral MB neurons (Fig. 2C). The spontaneous firing rate was ~11 Hz for medial MB neurons and ~0.2 Hz for lateral MB neurons (Fig. 2D). The action potential threshold was much lower and the width of action potentials was much shorter in medial MB neurons compared to lateral MB neurons, whereas input resistance, resting membrane potential, and overshoot were not different between the two cell types (Fig. 2E). Action potentials are tightly regulated by sodium and potassium ion channels (56), and many genes encoding these channels, including subunits of sodium channels (Scn1a, Scn1b) and potassium channels (Kcna2), were enriched in medial MB neurons compared to lateral MB neurons (Table S1). Transcripts related to ion channels may therefore be responsible for the increased firing rate of medial compared to lateral MB neurons.
To examine the projection patterns of medial and lateral MB neurons, we began by using anterograde tracing. After injecting AAV8-CAMKII-eGFP into female 2 month-old C57BL/6 mice MB (Fig. 2F), we found abundant axonal eGFP signals in the anterior thalamus (Fig. 2G) and the tegmental nuclei of Gudden (Fig. 2H and I), consistent with previous neural tracing studies in rats and monkeys (51, 52, 57–59). To evaluate whether lateral and medial MB neurons project to distinct brain regions, we performed retrograde tracing using cholera toxin subunit b (CTB), a recombinant protein that binds to ganglioside GM1 and mediates retrograde axonal transport, thereby labeling presynaptic neurons (60). Fluorophore conjugated-CTB injected into the medial and ventral (but not dorsal) regions of the anterior thalamus labeled presynaptic neurons in the medial (but not lateral) mouse MB (Fig. 2J and K). In contrast, CTB injected into the anterior dorsal thalamic nucleus (one hemisphere), labeled presynaptic neurons located in the lateral MB in both hemispheres (Fig. 2L and M), suggesting that lateral MB neurons project both ipsilaterally and contralaterally to the anterior dorsal thalamic nucleus. We also found medial and lateral MB neurons project to distinct nuclei within the tegmental nuclei of Gudden (fig. S4). These observations indicate that medial and lateral MB neurons project to distinct brain regions.
Lateral MB neurons in 5xFAD mice are more vulnerable to neurodegeneration compared to medial MB neurons
Based on our observations that lateral and medial MB neurons are distinct from one another across multiple dimensions, we hypothesized they may be asymmetrically affected by AD. To explore this possibility, we used scRNA-seq from female 6-month-old 5xFAD mice and WT littermates, a time point when these mice suffer memory impairments (19). We pooled posterior hypothalamus from four mice into one sample as one replicate and collected a total of three replicates per genotype (5xFAD and WT). We annotated 16,213 cells into major brain cell types (fig. S4C and D), which were represented across biological replicates (fig. S5E) and genotype (fig. 3C, fig. S6). We confirmed our observation that lateral and medial MB neurons harbor distinct markers (Fig. 3A), including Tac2 as a lateral MB neuronal marker and Rprm as a medial MB neuronal marker (Fig. 3B).
Figure 3. Lateral MB neurons are vulnerable to neurodegeneration in 5xFAD mice.
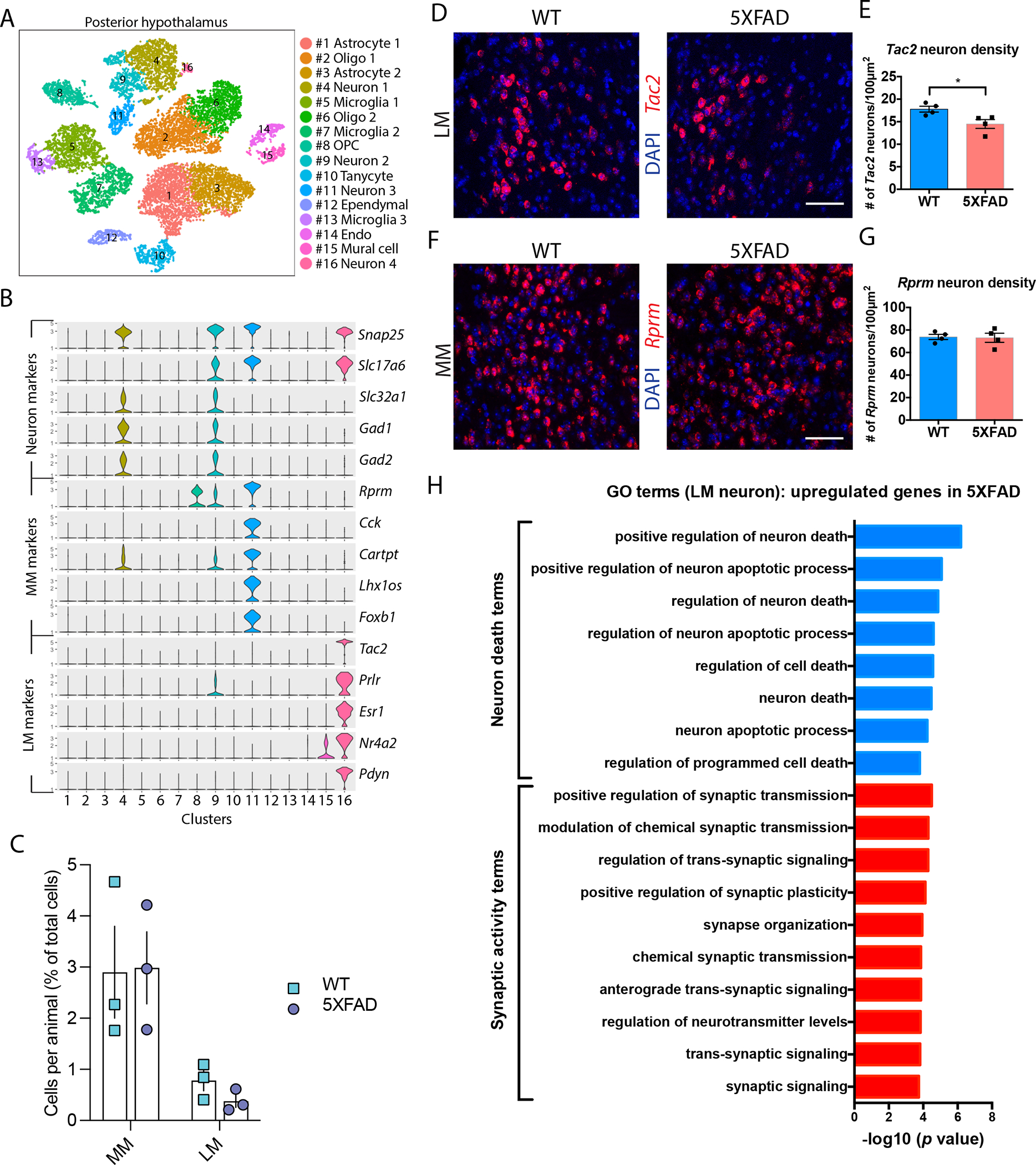
(A) A tSNE plot shows 16 cell clusters composed of cells derived from the posterior hypothalamus of brains from WT and 5xFAD mice. (B) Violin plots show markers for glutamatergic and GABAergic neurons and markers for medial MB neurons (MM) and lateral MB neurons (LM). Cluster 11 and cluster 16 show enrichment for MM and LM markers, respectively. (C) Number of cells per hypothalamus as a percent of total cells recovered (n=3 mice per group; two-way ANOVA). (D) Confocal images show in situ hybridization of Tac2 (red) in the lateral MB of WT and 5xFAD mice at 6 months of age; DAPI stain (blue). Scale bar: 50μm. (E) Quantification of Tac2 positive cells in the lateral MB (LM) of 6-month-old WT and 5xFAD mice. t(6)=2.81, p=0.0308, *p<0.05 (unpaired t test). n=4 mice per genotype. (F) Confocal images show in situ hybridization of Rprm (red) in the medial MB of WT and 5xFAD mice at 6 months of age; DAPI stain (blue). Scale bar: 50μm. (G) Quantification of Rprm positive cells in the medial MB (MM) of 6-month-old WT and 5xFAD mice. n=4 mice per genotype. (H) Gene Ontology (GO) term enrichment for genes upregulated in 5xFAD compared to wildtype mouse brain lateral MB neurons (LM, cluster 16).
To explore if medial and lateral MB neurons are differentially vulnerable to degeneration in 5xFAD mice, we performed in situ hybridization for MB marker genes in 6-month-old 5xFAD mice and WT control littermates. The density of Tac2 neurons, a marker of lateral MB neurons, was reduced in 5xFAD mice (Fig. 3D and E), whereas the density of Rprm neurons, a marker of medial MB neurons, was not altered between genotypes (Fig. 3F and G). We also found reduced expression of the NeuN marker in lateral but not medial MB neurons of 6-month-old 5xFAD mice compared to WT littermates using immunohistochemistry (fig. S7). Furthermore, using stereological techniques and markers for lateral and medial MB neurons (Tac2 and Rprm, respectively), we found reduced lateral MB neuronal density in 6-month-old 5xFAD mice compared to age-matched WT littermates (fig. S8). Collectively, these results suggest lateral MB neurons may be differentially vulnerable to neurodegeneration compared to medial MB neurons in 6-month-old 5xFAD mice.
To gain insights into the potential mechanisms underlying differential degeneration of lateral MB neurons, we compared the transcriptomes of lateral MB neurons between WT and 5xFAD mice. Enriched GO terms for genes upregulated in 5xFAD lateral MB neurons were related to neuronal death (including positive regulation of neuron death, neuron apoptotic process, and regulation of programmed cell death) and synaptic activity (including regulation of synaptic transmission, regulation of neurotransmitter concentrations, and synaptic signaling) (Fig. 3H). We found these terms were not enriched in medial MB neurons (fig. S9). To determine whether lateral MB neuronal dysfunction was unique to MB or shared with other brain regions, we analyzed a previously published snRNA-seq dataset from 7-month old mouse 5xFAD hippocampus (61). We found that genes expressed in the lateral MB neurons were not differentially expressed in the 7-month-old 5xFAD hippocampus (fig. S10), potentially suggesting lateral MB neurodegeneration is associated with a unique transcriptomic state that is not necessarily associated with a brain-wide signature of neurodegeneration associated with the 5xFAD genotype.
Single-nucleus RNA profiling of human postmortem MB samples
To explore whether MB neuron subtypes we identified in mice are conserved in humans, we performed snRNA-seq of postmortem MB samples from 32 individuals in the Religious Order Study or the Rush Memory and Aging Project (ROSMAP), two longitudinal cohort studies of aging and dementia. Information collected in ROSMAP cohorts included clinical data, detailed postmortem pathological evaluations, and multi-omics tissue profiling (62). We selected 13 individuals with AD pathology (8 females and 5 males) (fig. S11A) based on multiple consensus metrics of pathology and 19 individuals as non-AD controls (11 females and 8 males). We did not observe differences in education, age of death, sex, post-mortem interval, fixation interval, and co-morbidities between groups (fig. S11C–H). We confirmed the major differences between the two groups were amyloid burden (fig. S11I) and neurofibrillary tangles (fig. S11J) based on consensus metrics quantifying pathology across multiple brain regions, including hippocampus and multiple cortical regions. Although we cannot rule out the possibility that other pathologies may have influenced our results, these findings suggest that the major differences between the pathologies of our snRNA-seq cohort were due to tau and amyloid. In total, we analyzed 136,636 nuclei in MB samples from 32 individuals, which stratified into major brain cell types, including neurons (expressing SYT1, SNAP25, SYP), astrocytes (GFAP, AQP4, GJA1), microglia (CSF1R, CD74, C3), oligodendrocytes (MOBP, MOG, CNDP1), oligodendrocyte precursor cells (VCAN, MEGF11, PDGFRA), and vascular cells (FLT1, CLDN5, PDGFRB) (fig. S12A).
Within neurons, we identified three major neuronal cell types, of which two expressed glutamatergic markers (SLC17A6) and one expressed GABAergic markers (SLC32A1, GAD1, and GAD2) (Fig. 4A and B). To test whether specific neuronal clusters corresponded to the lateral and medial MB neurons we found in mice, we calculated the enrichment score from the aggregation of the top 15 marker genes identified in the mouse lateral and medial MB neuron clusters (fig. S13A). We found distinct neuronal clusters corresponded to the mouse lateral MB (Fig. 4C, fig. S13C–E) and medial MB (Fig. 4D). RNAscope of the human postmortem MB samples revealed that the mouse lateral MB neuron marker gene KCNC2 was expressed exclusively in lateral MB neurons in human tissue, and the mouse medial MB neuron marker gene FOXB1 was expressed exclusively in medial MB neurons in human tissue, indicating that mouse medial and lateral MB neuronal marker genes are conserved in humans (fig. S14). We also identified a neuronal cell type marked by RELN expression, which we confirmed using in situ hybridization (fig. S12B), that we did not observe in mouse, potentially representing a rare MB neuronal subtype that may be unique to human MB compared to mouse MB.
Figure 4. Single nuclei transcriptomic analysis of human postmortem MB samples reveals lateral and medial MB neurons.
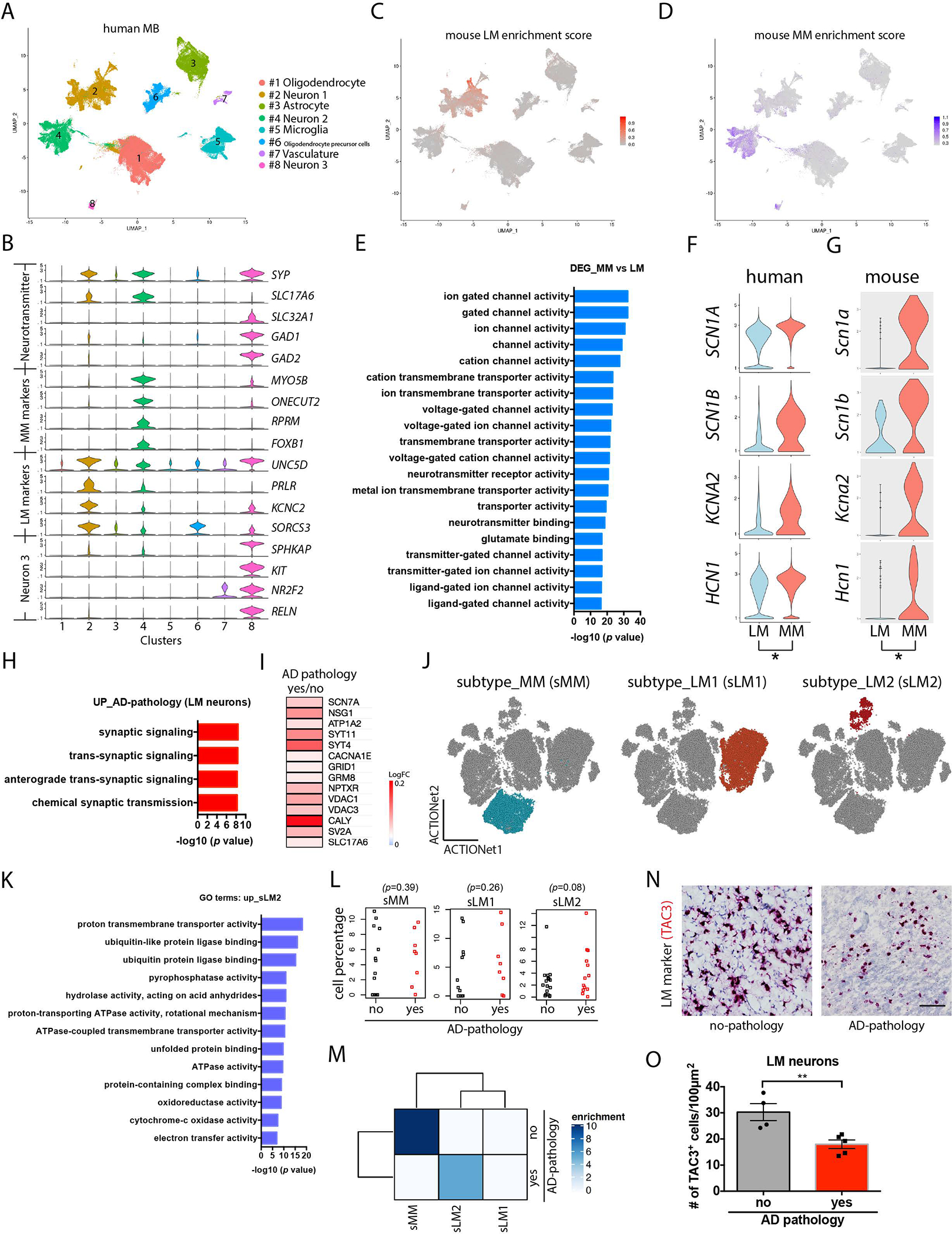
(A) Shown is a Uniform Manifold Approximation and Projection (UMAP) of single nuclei transcriptomes in postmortem MB samples from individuals with and without AD pathology. (B) Violin plots of markers for glutamatergic and GABAergic neurons and markers for the 3 neuronal cell types. (C). Enrichment score analysis of human MB with mouse lateral MB neurons. (LM) (D) Enrichment score analysis of human MB with mouse lateral MB neuron markers . (E) Enriched GO terms for differentially expressed genes (DEG) between human medial and lateral MB neurons. (F, G) Violin plots showing medial and lateral MB neuron expression of subunits of sodium and potassium ion channels and hyperpolarization-activated cyclic nucleotide-gated channels in MB samples from human (F) and mouse (G). (H) Selection of GO terms for genes upregulated in human LM neurons in postmortem MB samples from individuals with AD pathology. (I) A heatmap showing a selection of upregulated synaptic genes in human lateral MB neurons in postmortem MB samples from individuals with AD pathology. (J) Shown are dimension plots indicating subtypes of human medial and lateral MB neurons that have the most transcriptional similarity to mouse medial and lateral MB neurons. (K) Enriched GO terms are presented for genes upregulated in a lateral MB neuron sub cluster (sLM2, stressed lateral MB cluster 2). (L) Shown is the percentage of cells in human medial MB (sMM, stressed medial MB) and in two lateral MB clusters (sLM1, stressed lateral MB cluster 1, and sLM2, stressed lateral MB neuron cluster 2) for postmortem MB samples from individuals with or without AD pathology. Wilcoxon tests were used to compare cell percentage between AD pathology and no AD pathology. (M) Overrepresentation analysis for subtypes of human medial and lateral MB neurons (sMM, sLM1, sLM2). Color scale indicates the significance of overrepresentation. (N) Example images of immunohistochemical staining of human postmortem MB sections from individuals with and without AD pathology showing RNA in situ hybridization of TAC3 (red), a lateral MB neuron marker gene. Scale bar: 50μm. (O) Quantification of TAC3+ lateral MB neurons (LM) in postmortem MB samples from individuals with and without AD pathology. t(7)=3.614. p=0.0086. **p<0.01 by unpaired t-test.
Mouse lateral and medial MB neurons generated distinct firing patterns in our slice electrophysiology experiments, which was reflected by their differential expression of ion channel transcripts. Enriched GO terms for differentially expressed genes (DEGs) between human lateral and medial MB neurons were also associated with ion channel activity (Fig. 4E). As expected, compared to human lateral MB neurons, human medial MB neurons harbored higher expression of the genes encoding the subunits of ion channels, including SCN1A, SCN1B, KCNA2, and HCN1 (Fig. 4F and G), potentially suggesting that distinct spiking properties of medial and lateral MB neurons might be conserved across mice and humans.
To determine whether human lateral MB neurons also upregulate genes associated with synaptic activity in individuals with AD as we observed in 5xFAD mice, we performed DEG analysis between MB samples from individuals with AD pathology and non-AD controls. Individuals with AD pathology showed upregulation of genes related to synaptic activity in human lateral MB neurons (Fig. 4H), such as SCN7A (voltage-gated sodium channel), NSG1 (neuronal vesicle trafficking protein), ATP1A2 (a ATPase Na+/K+ transporter subunit), SYT4 and SYT11 (synaptotagmins), CACNA1E (voltage-gated calcium channel), GRID1 (glutamate ionotropic receptor), and GRM8 (glutamate metabotropic receptor), NPTXR (neuronal pentraxin receptor), VDAC1 and VDAC3 (voltage dependent anion channels), CALY (calcyon neuron-specific vesicular protein), SV2A (a synaptic vesicle glycoprotein), SLC17A6 (vesicular glutamate transporter) (Fig. 4I). This observation suggests lateral MB neurons may become more active in individuals with AD compared to non-AD controls.
To further resolve transcriptomic responses to AD in human MB samples, we identified sub-clusters within medial and lateral MB neurons (Fig. 4J). Lateral MB neurons clustered into two subpopulations (Fig. 4J), one of which showed enrichment for GO terms associated with cellular stress responses (including unfolded protein binding, ubiquitin protein ligase binding, and electron transfer activity) (Fig. 4K–L). Overrepresentation analysis revealed that individuals with AD pathology were overrepresented in this cluster (Fig. 4M). Together, these findings suggest that human lateral MB neurons mount a unique stress-associated transcriptional response to AD compared to medial MB neurons. In situ hybridization of human MB samples further confirmed that individuals with AD pathology harbored increased expression of CALY (Calcyon Neuron Specific Vesicular Protein) (fig. S15) and SYT11 (Synaptotagmin-11) (fig. S16) in the lateral MB compared to individuals with no AD pathology. These results suggest the lateral MB neurons in AD individuals and 6-month-old 5xFAD mice harbored similar gene expression signatures related to neuronal hyperexcitability and cellular stress (64–66). To directly test whether lateral MB neurons degenerate in individuals with AD, we performed RNA in situ hybridization of the lateral MB marker gene TAC3, the human homologue of the rodent Tac2, and quantified TAC3 cells in the postmortem MB samples of individuals with AD pathology. We found that the number of TAC3 cells in the MB was reduced in individuals with AD pathology compared to no pathology (Fig. 4 N and O). These findings suggest lateral MB neurons exhibit degeneration in individuals with AD, similar to our observations in the 5xFAD mice.
LM neurons of 5xFAD mice become hyperactive in an age-dependent manner
Our transcriptional findings in both mice and humans suggested that lateral MB neurons increase neuronal firing in AD compared to medial MB neurons. To directly test this hypothesis, we used electrophysiology and recorded lateral and medial MB neurons in brain slices from female 2-month-old 5xFAD mice and WT control littermates, an age we chose because neuronal hyperactivity may occur in the early stages of the disease (12, 66). Using cell-attached patch-clamp recordings, we found the frequency of spontaneous action potentials was increased in mouse lateral MB neurons (Fig. 5A and B), but not in medial MB neurons (Fig. 5C and D), indicating that lateral MB neurons exhibited increased firing in the MB of 5xFAD mice compared to WT littermates. We also recorded spontaneous action potentials of lateral MB neurons in brain slices from the tau P301S mouse model of AD (67), which recapitulates AD tau pathology. Similar to our findings in 5xFAD mice, lateral MB neurons also showed increased spiking activity in tau P301S mouse brains compared to mouse brains from WT control littermates (fig. S17). To gain insights into the synaptic mechanisms underlying increased firing of lateral MB neurons, we performed whole-cell patch-clamp recording. By measuring the spontaneous postsynaptic excitatory currents (sEPSC) in medial and lateral MB neurons, we found the frequency but not the amplitude of sEPSC was increased in lateral MB neurons of 5xFAD mice (Fig. 5 E–G), whereas neither the frequency nor the amplitude of sEPSC were altered between genotypes in medial MB neurons (Fig. 5H–J). These observations suggest lateral MB neurons in the 5xFAD mouse model may increase firing activity in part because they receive more excitatory synaptic inputs than lateral MB neurons of WT littermates.
Figure 5. Lateral MB neurons of 5xFAD mice are hyperactive.
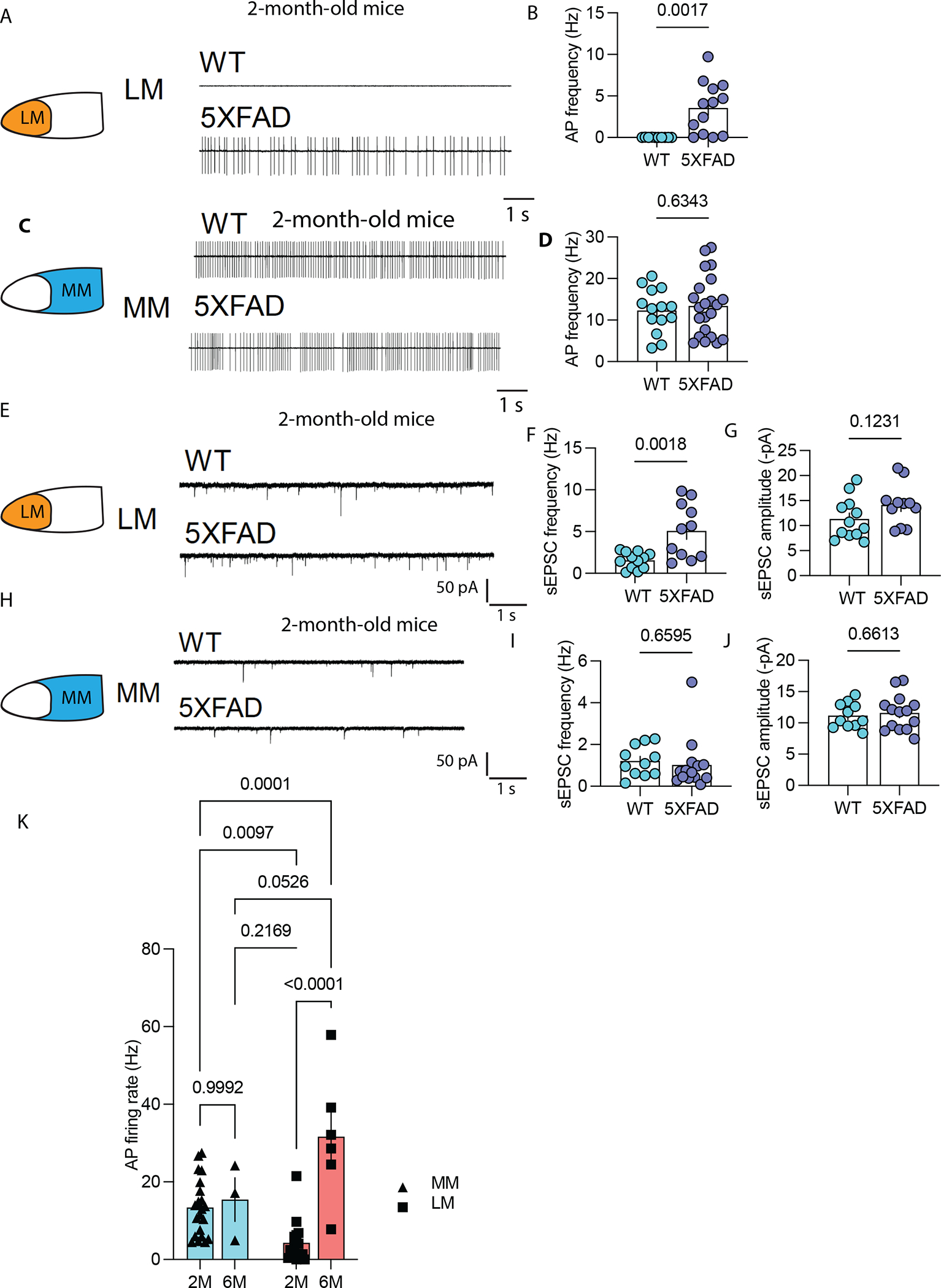
(A) Shown are representative action potential (AP) traces of the lateral MB neurons (LM) of a 2-month-old 5xFAD mouse and a WT littermate control using slice electrophysiology. (B) Shown is the summary of the firing frequency of LM neurons in a 2-month-old 5xFAD mouse and a WT littermate control using slice electrophysiology. t(20)=3.403, p=0.0017. **p<0.01 (unpaired t test). n=9 in WT group and n=13 in 5xFAD mouse group. (C) Shown are representative action potential traces from the medial MB neurons (MM) of a 2-month 5xFAD and a WT littermate control. (D) Shown is the summary of the firing frequency of medial MB neurons in a 2-month 5xFAD mouse and a WT littermate control. n=14 in the WT group and n=22 in 5xFAD group. (E) Shown are representative traces of spontaneous excitatory postsynaptic currents (sEPSC) in the lateral MB neurons of a 2-month 5xFAD mouse and a WT littermate control. (F, G) Quantification of sEPSC frequency (F) and amplitude (G) in the lateral MB neurons of a 2-month-old 5xFAD mouse and a WT littermate control. t(21)=3.57, p=0.0018 (F) (unpaired t-test). n=12 in WT group and n=11 in 5xFAD group. (H) Shown are representative traces of sEPSCs of medial MB neurons (MM) from a two-month-old 5xFAD mouse and a WT littermate control. (I, J) Quantification of sEPSC frequency (I) and amplitude (J) in the medial MB neurons of a 2-month-old 5xFAD mouse and a WT littermate control. n=11 in WT group and n=13 in 5xFAD group. (K) Quantification of action potential frequency in the lateral and medial MB neurons of 2-month-old and 6-month-old 5xFAD mice is shown. Two-way ANOVA with Sidak’s post hoc test F(1,14)=6.64, p=0.022 (interaction).
To determine if the hyperactivity of LM neurons observed in 2-month-old 5xFAD mice persists in later stages of AD, we recorded neuronal spiking in 6-month-old 5xFAD mice. We found that the frequency of spontaneous action potentials was further increased in lateral MB neurons of 6-month-old mice compared to that of lateral MB neurons in 2-month-old 5xFAD mice (Fig. 5K), whereas the firing frequency of medial MB neurons was not altered between 2 months and 6 months of age (Fig. 5K). The baseline firing frequency of lateral MB neurons in 5xFAD mice increased from ~4 Hz in 2-month-old mice to ~32 Hz in 6-month-old mice (Fig. 5K). This suggests that mouse lateral MB neurons show increased neuronal hyperactivity in an age-dependent manner.
LM neuronal hyperactivity drives memory deficits in mice
To determine if lateral MB neuron hyperactivity and memory deficits in AD are causally linked, we used a designer receptor exclusively activated by designer drug (DREADD) to increase activity in lateral MB neurons of female C57BL/6 mice. We injected AAV8-CaMKIIα-hM3Dq into the MB of 2-month-old C57BL/6 female mice and implanted a bilateral cannula above the lateral MB neurons, enabling the delivery of clozapine-N-oxide (CNO) to activate Gq, into the lateral MB neurons (Fig. 6A, B). We confirmed that Gq-expressing lateral MB neurons increased firing activity in response to bath application of CNO (Fig. 6C). AAV was injected 3 weeks before the implantation of a bilateral cannula, allowing the expression of Gq in the mouse MB. We started the behavioral tests 3 days after cannula implantation. We delivered CNO through the cannula to activate lateral MB neurons 1 hour prior to the behavioral tests. CNO was delivered to both control and Gq-expressing mice to control for CNO-dependent behavioral effects. To assess spatial working memory, we tested spontaneous alternation in the Y maze task (68). Control (AAV8-CamKIIα-mCherry only) mice alternated arms during ~75% of trails, whereas the Gq-expressing mice alternated in ~57% of trials (Fig. 6D). Additionally, we performed trace fear conditioning. Training comprised five tone-interval-shock sequences (15s tone + 30s interval + 1s shock) and fear memory recall was tested 24 hours later. Gq-expressing mice showed reduced freezing compared to control mice during the recall tests (Fig. 6E). The effect of chemogenetic inhibition on memory performance was not confounded by changes in anxiety or motor activity measures because we found no difference in freezing behavior in the pre-tone habituation period (fig. S18). Although we cannot rule out the possibility that non-lateral MB neurons may have been affected by our viral strategy and contributed to memory deficits, these observations suggest that lateral MB neuronal hyperactivity may be sufficient to induce memory deficits.
Figure 6. Lateral MB neuronal hyperactivity contributes memory deficits in mice.
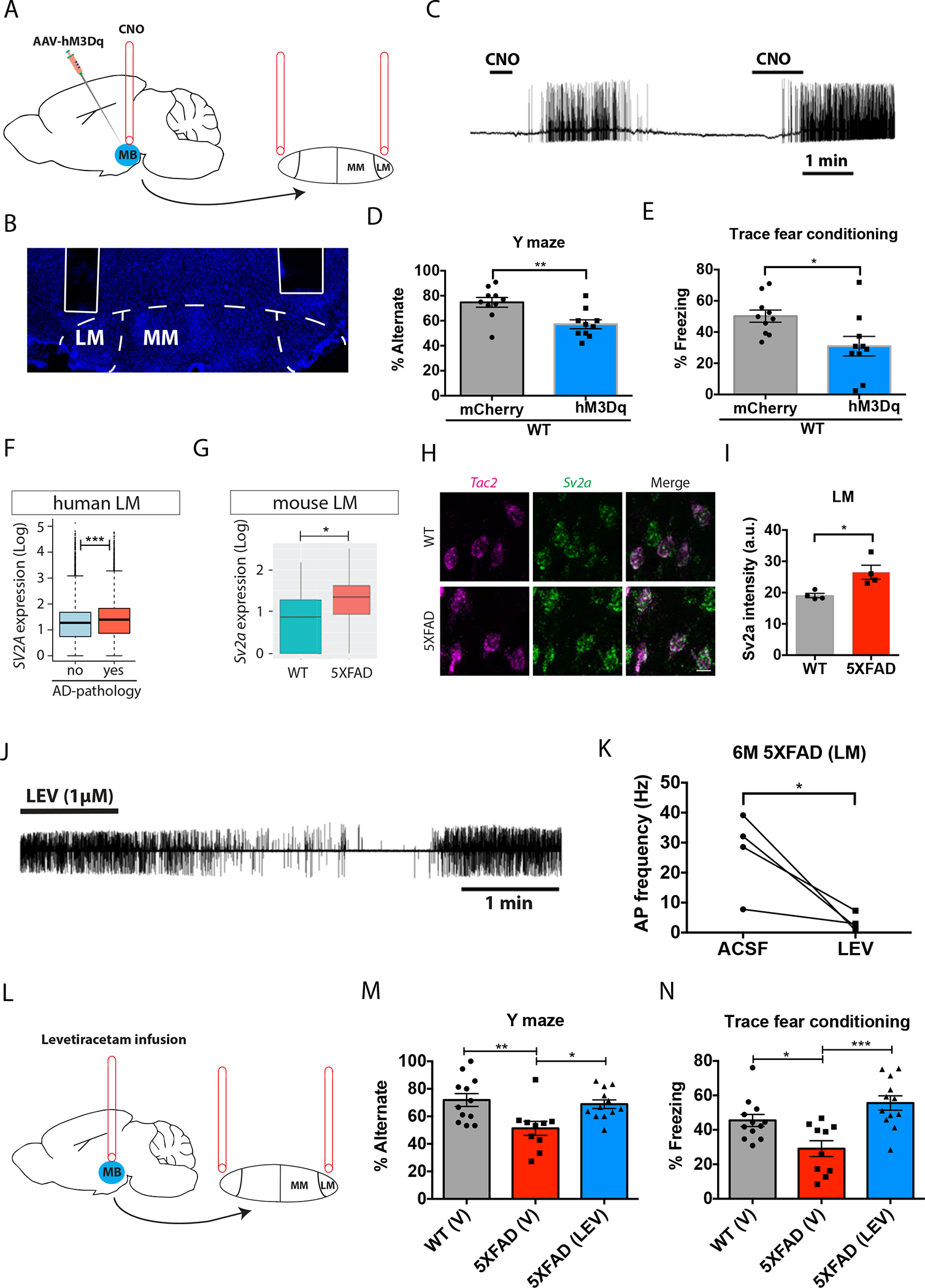
(A) Experimental design for increasing neuronal activity specifically in neurons located in the lateral mammillary body (LM) of mice. An AAV vector carrying the DREADD hM3Dq (Gq) was injected into the mammillary body (MB) and a cannula was implanted above the LM, allowing the delivery of clozapine-N-oxide (CNO) into the mouse LM. (B) A confocal image shows the location of the cannula just above the LM in mouse brain. (C) Shown is an action potential trace induced by mouse LM neurons expressing the hM3Dq DREADD in response to 10 μM CNO. (D) WT mice expressing hM3Dq showed reduced performance in the Y maze task compared to control mice (expressing mCherry). t(18)=3.319, p=0.0038 (unpaired t-test). n=10 mice per group. (E) WT mice expressing hM3Dq showed reduced freezing time in the trace fear conditioning test compared to control mice (expressing mCherry). t(18)=2.612, p=0.0176 (unpaired t-test). n=10 mice per group. (F, G) SV2A expression in (F) human lateral MB neurons (LM) and (G) mouse LM neurons. (H) In situ hybridization of Tac2 (magenta) and Sv2a (green) in brain sections from the lateral MB of a 6-month-old 5xFAD mouse and a WT littermate control. Scale bar: 10μm. (I) Quantification of Sv2a intensity in Tac2+ LM neurons in a 6-month-old 5xFAD mouse and a WT littermate control. t(6)=3.161, p=0.0195. *p<0.05 (unpaired t-test). n=4 mice per group. (J) A representative trace of spontaneous action potentials acquired using cell-attached patch-clamp recording in LM neurons of 5xFAD mice at 6 months of age before, during, and after bath application of levetiracetam (1μM). (K) Quantification of action potential (AP) frequency in LM neurons from 6 month-old 5xFAD mice before and after bath application of levetiracetam (LEV) or artificial cerebrospinal fluid (ACSF) as control. t(3)=3.286, p=0.0462 (paired t-test). n=4 neurons from 3 mice. (L) Experimental design for levetiracetam infusion specifically into the lateral MB of 6 month-old 5xFAD mice. (M) Percentage of spontaneous alternation in the Y maze task in WT mice treated with vehicle (V), and 6-month old 5xFAD mice treated with vehicle or levetiracetam (LEV). F(2, 31)= 6.238, p=0.0053 (One-way ANOVA with Tukey’s post hoc test). n=12 mice in WT (V) group, 10 mice in 5xFAD (V) group, and 12 mice in 5xFAD (LEV) group. (N) Percentage of freezing responses during the cued trace fear conditioning test in WT mice treated with vehicle (V), and 6-month old 5xFAD mice treated with vehicle or levetiracetam (LEV). F(2, 31)=10.35, p=0.0004 (One-way ANOVA with Tukey’s post hoc test; N=12 mice in WT (V) group, 10 mice in 5xFAD (V) group, and 12 mice in 5xFAD (LEV) group. *p<0.05, **p<0.01, ***p<0.001).
Next, we tested whether reducing activity in lateral MB neurons of 5xFAD mice could ameliorate memory deficits. We used the anti-epileptic drug levetiracetam to attenuate the increased activity of lateral MB neurons in female 6-month-old 5xFAD mice. Levetiracetam is known to treat seizures by reducing neuronal activity in part by binding to SV2A, a synaptic vesicle glycoprotein (69–72). We found that the expression of SV2A was elevated in individuals with AD pathology as well as in 5xFAD mice in the corresponding human and mouse lateral MB neuronal clusters (Fig. 6F, G). In situ hybridization of Sv2a and Tac2 further revealed that the intensity of Sv2a puncta in Tac2 neurons in the lateral MB was increased in 6-month-old 5xFAD mice (Fig. 6H, I). Bath application of 1μM levetiracetam to 5xFAD mouse brain slices reduced lateral MB neuronal spiking activity 5xFAD (Fig. 6J), as the frequency of spontaneous action potentials was decreased after the application of levetiracetam (Fig. 6K). To test if reducing lateral MB neuronal activity rescued memory deficits in female 6-month-old 5xFAD mice, we delivered levetiracetam specifically into the lateral MB via a bilateral cannula (Fig. 6L). We infused levetiracetam daily for 7 days before behavioral testing, and on the day of behavioral testing, levetiracetam was infused 1 hour before the tests. We again used spontaneous alternation in the Y maze task and trace fear conditioning tests in which 5xFAD mice show deficits by 6 months of age (19, 73). In the Y maze task, we found that while 5xFAD mice receiving vehicle alternated in ~51% of the trials, less than WT control littermates receiving vehicle (~72%), 5xFAD mice receiving levetiracetam alternated in ~69% of the trials (Fig. 6M). In the trace fear conditioning tests, the freezing percentage of 5xFAD mice receiving vehicle was reduced compared to that of WT control littermates receiving vehicle, and levetiracetam rescued this deficit (Fig. 6N). Additionally, we found levetiracetam attenuated the intensity of Sv2a puncta in Tac2 neurons in the lateral MB neurons to WT levels, suggesting the transcriptional signature relating to neuronal hyperactivity was also alleviated by levetiracetam (fig. S19). Although we cannot rule out effects of levetiracetam on other cell types, we found levetiracetam had no effect on the firing activity of medial MB neurons (fig. S20), suggesting the effect of levetiracetam on memory within the MB was due to changes in lateral MB neurons. Together, these observations provide evidence that hyperactivity of lateral MB neurons contributes to memory deficits, and that manipulating lateral MB neuronal activity may represent a therapeutic strategy to improve cognition in AD.
DISCUSSION
The mechanisms predisposing individual neuronal circuits to degeneration in the AD brain are poorly understood. One possibility is that aging neurons die in a stochastic, erratic manner. A competing possibility—one with critical therapeutic implications—is that specific neurons become vulnerable to degeneration in a predictable manner based on a convergence of genetic identity, anatomic connectivity, and cellular milieu. Here, we found that the MB contains two neuronal cell types with distinct molecular markers, neuronal morphology, long-range projection targets, and electrophysiological properties. The transcriptional profiles of the two types of MB neurons were conserved across mice and humans. Lateral MB neurons upregulated genes associated with neuronal activity in both an AD mouse model and postmortem human brain tissue with AD pathology compared to non-AD controls. Lateral MB neurons—but not medial MB neurons—exhibited age-dependent progression of neuronal hyperactivity in 5xFAD mice. Increasing spiking activity in lateral MB neurons of WT mice impaired performance in two tests of memory, and selectively reducing lateral MB neuronal hyperactivity in 5xFAD mice rescued memory deficits, suggesting that lateral MB neuronal hyperactivity is both necessary and sufficient to drive memory deficits.
Previous studies suggest that neurodegeneration in the cerebral cortex and subiculum is not observed before 9 months of age in 5xFAD mice (19, 74). Our findings suggest lateral MB neurons degenerate as early as 6 months of age in 5xFAD mice, potentially one of the earliest brain regions to degenerate in this AD mouse model5xFAD. These findings more broadly suggest some neuronal types may harbor distinct cellular properties, including circuit connectivity and cell-autonomous properties, that increase their risk for dysfunction and degeneration. The asymmetric reduction of neurons in the MB may induce desynchronized circuit assembly and brain-wide network dysfunction. Loss of lateral MB neurons may also redirect MB presynaptic inputs to the remaining lateral MB neurons, leading to excessive synaptic connections and neuronal hyperactivity. Indeed, our data indicate that lateral MB neurons increased spontaneous firing frequency in 5xFAD mice, suggesting lateral MB neurons might receive more excitatory presynaptic inputs in 5xFAD mice compared to control littermates. Given that behavioral deficits in 5xFAD mice do not emerge until ~6 months of age (19), lateral MB neuronal hyperactivity may critically induce memory deficits. Reducing lateral MB neuronal hyperexcitability in 6-month-old 5xFAD mice using levetiracetam rescued memory deficits. Of note, levetiracetam reduced neuronal hyperactivity in lateral MB neurons, but did not affect the firing of medial MB neurons, suggesting that levetiracetam may correct pathological hyperactivity without affecting physiological neuronal firing, making it a potential approach to treat AD associated with neuronal hyperactivity (75–77).
Our study has several limitations. We acknowledge that some of our behavioral data had broad overlap between groups and therefore the subtle changes observed should be interpreted with caution. However, transcriptomic overlap between mouse and human MB neuronal populations suggest increased neuronal activity in lateral MB neurons may represent an inflection point in the progressive loss of memory in AD. We found upregulation of neuronal activity in 5xFAD LM neurons at 2 months and 6 months of age compared to age-matched wildtype littermates, and we also found upregulation of LM neuronal activity in Tau P301S mice at 9 months of age compared to age-matched wildtype littermates, and additional models may be helpful to further test the effect of age, genotype, and AD pathology on neuronal activity in MB neurons. Additionally, while the primary goal of our study was to explore contributions of MB neuronal heterogeneity to AD pathology and memory dysfunction, we did not elucidate the cellular mechanisms linking neuronal hyperactivity to cell death. Hyperactivity may be associated with DNA damage and a cellular state leading to neuronal senescence (78). Furthermore, pinpointing how subtypes of MB neurons are coordinated within broader medial limbic memory circuits might contribute to additional insight into memory impairments in AD. Further defining hypothalamic neurons (79–83) might help to unravel the complex physiology of hypothalamic contributions to memory deficits in AD, particularly given that lesion studies suggest the MB governs spatial working memory, navigational memory, and episodic memory (35). Future studies using transgenic mouse lines to specifically label distinct MB neuronal subtypes will enable the exploration of functional contributions of each subtype to particular aspects of learning, memory, and cognition. For example, previous studies have found head-direction and angular-velocity cells in the LM (84, 85), and our study provides a strategy to test their functional contributions to specific memory processes. Collectively, our data suggest that subcortical contributions to memory may play important roles in AD and neurodegeneration.
MATERIALS AND METHODS
Study design
The goal of this study was to define the cellular heterogeneity of the mammillary body (MB) in mouse brain and its possible roles in regulating memory in AD. We used scRNA-seq to define the cell types of the MB in WT mice. 5XFAD (Tg 6799) breeding pairs were acquired from the Mutant Mouse Resource and Research Center (MMRRC) (Stock No. 034848-JAX). 5XFAD mice were crossed with WT mice to generate offspring for this study. All animal experiments were conducted in accordance with NIH guidelines and were approved by The Committee for Animal Care of the Division of Comparative Medicine at the Massachusetts Institute of Technology. Female mice were used in the study, unless otherwise specified. Two-month-old WT (C57BL/6: JAX Stock No. 000664) mice were used for initial single cell RNA sequencing (scRNAseq). Six-month-old 5XFAD and littermate control mice were further used for scRNAseq and the subsequent validation experiments. Mice for slice electrophysiology contains both 2-month-old and 6-month-old 5XFAD and littermate control. To compare differences in MB neurons in AD, we performed scRNAseq of 6-month-old 5xFAD mice versus WT littermates and snRNA-seq in postmortem MB samples from individuals with varying stages of AD pathology. For human sequencing experiments, postmortem samples were processed in batches that included both AD and non AD pathology in case of batch effects. Our human mammillary body single nuclei RNA sequencing dataset contained a total of 32 individuals from the Religious Order Study (ROS) or the Rush Memory and Aging Project (MAP), two longitudinal cohort studies of aging and dementia. The studies include clinical data collected annually, detailed postmortem pathological evaluation, and extensive genetic, epigenetic, transcriptomic, proteomic, and metabolomic bulk-tissue profiling (62). ROSMAP data and biospecimens can be requested at www.radc.rush.edu). Details of the clinical and pathological data collection methods has been previously reported (87–91). Informed consent was obtained from each subject, and the ROSMAP were approved by an Institutional Review Board (IRB) of Rush University Medical Center. Further, all participants signed a repository consent to allow their data and biospecimens to be repurposed. NIA-Regan pathologic diagnosis of intermediate- or high-likelihood was used to dichotomize individuals pathologic AD or not pathologic AD. This study contains 13 individuals with AD-pathology (8 females and 5 males) and 19 individuals with no-pathology (11 females and 8 males). AD-pathology and no-pathology groups were balanced for years of education and age of death. Inclusion criteria for AD diagnosis included multiple metrics of AD pathology.
We characterized distinct neuronal populations of the MB in mouse and human brain using histology. Based on our observation that lateral MB neurons are associated with hyperactivity in AD using slice electrophysiology and single cell transcriptomic profiling, we manipulated lateral MB neurons in WT and 5xFAD mice. For all studies, mice were controlled by age and sex across groups. For sequencing, immunohistochemistry, and RNAscope, all groups were processed, stained, and imaged at the same time. Imaging quantification was carried out in a blinded fashion by a researcher who did not perform the imaging, and no mice were excluded from analysis.
Statistical analysis
Prior studies using similar experiments or power analysis were used to determine sample size. Student t-tests (two-sided) were used for comparison between two groups (e.g., lateral versus medial MB; WT versus 5xFAD). One-way analysis of variance (ANOVA) was used in experiments containing more than two groups. Tukey’s post hoc tests were used where appropriate. Two-way analyses of variance (ANOVAs) were used in experiments containing two variables (genotypes and ages). All statistics, except single cell RNA sequencing analysis, which was performed using R, were performed using GraphPad Prism, with significance set at p<0.05. Throughout the paper, values represent means; error bars indicate SEMs; N refers to biological replicates. Only positive statistics were reported. All measurements and testing were performed blind to the genotypes or experimental manipulations.
Supplementary Material
Acknowledgements:
We thank the study participants and staff of the Rush Alzheimer’s Disease Center. We thank all members of the Tsai laboratory for technical assistance and helpful comments.
Funding:
This research was supported in part by the JBP foundation, Ludwig family foundation, and National Institute of Health (NIH) grants RF1AG054321, RF1AG062377, RF1AG054012, U01NS110453, R01AG062335, and R01AG058002 (to L-HT); P30AG10161, R01AG15819, and R01AG1717 (DAB); RF1AG054012, U01NS110453, R01AG062335, and R01AG058002 (to MK).
Footnotes
Competing interests: All authors declare that they have no competing interests.
Data and materials availability: All data are present in the main paper or supplementary materials. Mouse MB sequencing data can be found in GEO with accession code: GSE224647. Human MB sequencing data can be found in Synapse at https://www.synapse.org/#!Synapse:syn50670858 using the SynID syn50670858.
References and Notes
- 1.Canter RG, Penney J, Tsai L-H, The road to restoring neural circuits for the treatment of Alzheimer’s disease. Nature 539, 187–196 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Graham WV, Bonito-Oliva A, Sakmar TP, Update on Alzheimer’s Disease Therapy and Prevention Strategies. Annu. Rev. Med. 68, 413–430 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Long JM, Holtzman DM, Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 179, 312–339 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Busche MA, Konnerth A, Neuronal hyperactivity--A key defect in Alzheimer’s disease? BioEssays News Rev. Mol. Cell. Dev. Biol. 37, 624–632 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Palop JJ, Mucke L, Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 17, 777–792 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palop JJ, Chin J, Mucke L, A network dysfunction perspective on neurodegenerative diseases. Nature 443, 768–773 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Zott B, Busche MA, Sperling RA, Konnerth A, What Happens with the Circuit in Alzheimer’s Disease in Mice and Humans? Annu. Rev. Neurosci. 41, 277–297 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinez-Losa M, Tracy TE, Ma K, Verret L, Clemente-Perez A, Khan AS, Cobos I, Ho K, Gan L, Mucke L, Alvarez-Dolado M, Palop JJ, Nav1.1-Overexpressing Interneuron Transplants Restore Brain Rhythms and Cognition in a Mouse Model of Alzheimer’s Disease. Neuron 98, 75–89.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu G-Q, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L, Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55, 697–711 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verret L, Mann EO, Hang GB, Barth AMI, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, Mucke L, Palop JJ, Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149, 708–721 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zott B, Simon MM, Hong W, Unger F, Chen-Engerer H-J, Frosch MP, Sakmann B, Walsh DM, Konnerth A, A vicious cycle of β amyloid–dependent neuronal hyperactivation. Science 365, 559–565 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, Konnerth A, Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A. 109, 8740–8745 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vann SD, Re-evaluating the role of the mammillary bodies in memory. Neuropsychologia 48, 2316–2327 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Vann SD, Aggleton JP, The mammillary bodies: two memory systems in one? Nat. Rev. Neurosci. 5, 35–44 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Vetere G, Kenney JW, Tran LM, Xia F, Steadman PE, Parkinson J, Josselyn SA, Frankland PW, Chemogenetic Interrogation of a Brain-wide Fear Memory Network in Mice. Neuron 94, 363–374.e4 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Wheeler AL, Teixeira CM, Wang AH, Xiong X, Kovacevic N, Lerch JP, McIntosh AR, Parkinson J, Frankland PW, Identification of a functional connectome for long-term fear memory in mice. PLoS Comput. Biol 9, e1002853 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aggleton JP, Pralus A, Nelson AJD, Hornberger M, Thalamic pathology and memory loss in early Alzheimer’s disease: moving the focus from the medial temporal lobe to Papez circuit. Brain J. Neurol. 139, 1877–1890 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gail Canter R, Huang W-C, Choi H, Wang J, Ashley Watson L, Yao CG, Abdurrob F, Bousleiman SM, Young JZ, Bennett DA, Delalle I, Chung K, Tsai L-H, 3D mapping reveals network-specific amyloid progression and subcortical susceptibility in mice. Commun. Biol. 2, 1–12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R, Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. Off. J. Soc. Neurosci. 26, 10129–10140 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhattacharyya KB, James Wenceslaus Papez, His Circuit, and Emotion. Ann. Indian Acad. Neurol. 20, 207–210 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bubb EJ, Kinnavane L, Aggleton JP, Hippocampal - diencephalic - cingulate networks for memory and emotion: An anatomical guide. Brain Neurosci. Adv. 1, 2398212817723443 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teuber H-L, Milner B, Vaughan HG, Persistent anterograde amnesia after stab wound of the basal brain. Neuropsychologia 6, 267–282 (1968). [Google Scholar]
- 23.Dusoir H, Kapur N, Byrnes DP, McKinstry S, Hoare RD, The role of diencephalic pathology in human memory disorder. Evidence from a penetrating paranasal brain injury. Brain J. Neurol. 113 (Pt 6), 1695–1706 (1990). [DOI] [PubMed] [Google Scholar]
- 24.Carlesimo GA, Serra L, Fadda L, Cherubini A, Bozzali M, Caltagirone C, Bilateral damage to the mammillo-thalamic tract impairs recollection but not familiarity in the recognition process: a single case investigation. Neuropsychologia 45, 2467–2479 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Tsivilis D, Vann SD, Denby C, Roberts N, Mayes AR, Montaldi D, Aggleton JP, A disproportionate role for the fornix and mammillary bodies in recall versus recognition memory. Nat. Neurosci. 11, 834–842 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Vann SD, Tsivilis D, Denby CE, Quamme JR, Yonelinas AP, Aggleton JP, Montaldi D, Mayes AR, Impaired recollection but spared familiarity in patients with extended hippocampal system damage revealed by 3 convergent methods. Proc. Natl. Acad. Sci. U. S. A. 106, 5442–5447 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenbaum RS, Gao F, Honjo K, Raybaud C, Olsen RK, Palombo DJ, Levine B, Black SE, Congenital absence of the mammillary bodies: a novel finding in a well-studied case of developmental amnesia. Neuropsychologia 65, 82–87 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Tedder J, Miller L, Tu S, Hornberger M, Lah S, Into the future with little past: exploring mental time travel in a patient with damage to the mammillary bodies/fornix. Clin. Neuropsychol 30, 351–366 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Aggleton JP, Mishkin M, Mamillary-body lesions and visual recognition in monkeys. Exp. Brain Res. 58, 190–197 (1985). [DOI] [PubMed] [Google Scholar]
- 30.Holmes EJ, Jacobson S, Stein BM, Butters N, Ablations of the mammillary nuclei in monkeys: effects on postoperative memory. Exp. Neurol. 81, 97–113 (1983). [DOI] [PubMed] [Google Scholar]
- 31.Parker A, Gaffan D, Mamillary Body Lesions in Monkeys Impair Object-in-Place Memory: Functional Unity of the Fornix-Mamillary System. J. Cogn. Neurosci. 9, 512–521 (1997). [DOI] [PubMed] [Google Scholar]
- 32.Aggleton JP, Neave N, Nagle S, Hunt PR, A comparison of the effects of anterior thalamic, mamillary body and fornix lesions on reinforced spatial alternation. Behav. Brain Res. 68, 91–101 (1995). [DOI] [PubMed] [Google Scholar]
- 33.Field TD, Rosenstock J, King EC, Greene E, Behavioral role of the mammillary efferent system. Brain Res. Bull. 3, 451–456 (1978). [DOI] [PubMed] [Google Scholar]
- 34.Gaffan EA, Bannerman DM, Warburton EC, Aggleton JP, Rats’ processing of visual scenes: effects of lesions to fornix, anterior thalamus, mamillary nuclei or the retrohippocampal region. Behav. Brain Res. 121, 103–117 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Neave N, Nagle S, Aggleton JP, Evidence for the involvement of the mammillary bodies and cingulum bundle in allocentric spatial processing by rats. Eur. J. Neurosci. 9, 941–955 (1997). [DOI] [PubMed] [Google Scholar]
- 36.Nelson AJD, Vann SD, Mammilliothalamic Tract Lesions Disrupt Tests of Visuo-Spatial Memory. Behav. Neurosci. 128, 494–503 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosenstock J, Field TD, Greene E, The role of mammillary bodies in spatial memory. Exp. Neurol. 55, 340–352 (1977). [DOI] [PubMed] [Google Scholar]
- 38.Sziklas V, Petrides M, Memory impairments following lesions to the mammillary region of the rat. Eur. J. Neurosci. 5, 525–540 (1993). [DOI] [PubMed] [Google Scholar]
- 39.Vann SD, Dismantling the Papez circuit for memory in rats. eLife 2, e00736 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vann SD, Aggleton JP, Evidence of a spatial encoding deficit in rats with lesions of the mammillary bodies or mammillothalamic tract. J. Neurosci. Off. J. Soc. Neurosci. 23, 3506–3514 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beracochea DJ, Jaffard R, Effects of ibotenic lesions of mammillary bodies on spontaneous and rewarded spatial alternation in mice. J. Cogn. Neurosci. 2, 133–140 (1990). [DOI] [PubMed] [Google Scholar]
- 42.Béracochéa DJ, Jaffard R, Impairment of spontaneous alternation behavior in sequential test procedures following mammillary body lesions in mice: evidence for time-dependent interference-related memory deficits. Behav. Neurosci 101, 187–197 (1987). [DOI] [PubMed] [Google Scholar]
- 43.Béracochéa DJ, Jaffard R, The effects of mammillary body lesions on delayed matching and delayed non-matching to place tasks in the mice. Behav. Brain Res. 68, 45–52 (1995). [DOI] [PubMed] [Google Scholar]
- 44.Célérier A, Pierard C, Beracochea D, Effects of ibotenic acid lesions of the dorsal hippocampus on contextual fear conditioning in mice: comparison with mammillary body lesions. Behav. Brain Res. 151, 65–72 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Grossi D, Lopez OL, Martinez AJ, Mamillary bodies in Alzheimer’s disease. Acta Neurol. Scand. 80, 41–45 (1989). [DOI] [PubMed] [Google Scholar]
- 46.McDuff T, Sumi SM, Subcortical degeneration in Alzheimer’s disease. Neurology 35, 123–126 (1985). [DOI] [PubMed] [Google Scholar]
- 47.Stief A, über die anatomischen Grundlagen der vegetativen Störungen bei Geisteskrankheiten. Dtsch. Z. Für Nervenheilkd. 97, 112–132 (1927). [Google Scholar]
- 48.Baloyannis SJ, Mavroudis I, Baloyannis IS, Costa VG, Mammillary Bodies in Alzheimer’s Disease: A Golgi and Electron Microscope Study. Am. J. Alzheimers Dis. Dementias® 31, 247–256 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Copenhaver BR, Rabin LA, Saykin AJ, Roth RM, Wishart HA, Flashman LA, Santulli RB, McHugh TL, Mamourian AC, The fornix and mammillary bodies in older adults with Alzheimer’s disease, mild cognitive impairment, and cognitive complaints: a volumetric MRI study. Psychiatry Res. 147, 93–103 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Blandina P, Munari L, Provensi G, Passani MB, Histamine neurons in the tuberomamillary nucleus: a whole center or distinct subpopulations? Front. Syst. Neurosci. 6, 33 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gv A, Da H, Topography and synaptology of mamillary body projections to the mesencephalon and pons in the rat. J. Comp. Neurol. 301 (1990), doi: 10.1002/cne.903010206. [DOI] [PubMed] [Google Scholar]
- 52.Gonzalo-Ruiz A, Morte L, Sanz JM, Glutamate/aspartate and leu-enkephalin immunoreactivity in mammillothalamic projection neurons of the rat. Brain Res. Bull. 47, 565–574 (1998). [DOI] [PubMed] [Google Scholar]
- 53.Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu W-L, Sánchez-Guardado L, Lois C, Mazmanian SK, Deverman BE, Gradinaru V, Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 20, 1172–1179 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miettinen TP, Björklund M, Cellular Allometry of Mitochondrial Functionality Establishes the Optimal Cell Size. Dev. Cell 39, 370–382 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pende M, Kozma SC, Jaquet M, Oorschot V, Burcelin R, Le Marchand-Brustel Y, Klumperman J, Thorens B, Thomas G, Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature 408, 994–997 (2000). [DOI] [PubMed] [Google Scholar]
- 56.Kress GJ, Mennerick S, Action potential initiation and propagation: upstream influences on neurotransmission. Neuroscience 158, 211–222 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cruce JA, An autoradiographic study of the descending connections of the mammillary nuclei of the rat. J. Comp. Neurol. 176, 631–644 (1977). [DOI] [PubMed] [Google Scholar]
- 58.Seki M, Zyo K, Anterior thalamic afferents from the mamillary body and the limbic cortex in the rat. J. Comp. Neurol. 229, 242–256 (1984). [DOI] [PubMed] [Google Scholar]
- 59.Veazey RB, Amaral DG, Cowan WM, The morphology and connections of the posterior hypothalamus in the cynomolgus monkey (Macaca fascicularis). II. Efferent connections. J. Comp. Neurol. 207, 135–156 (1982). [DOI] [PubMed] [Google Scholar]
- 60.Stoeckel K, Schwab M, Thoenen H, Role of gangliosides in the uptake and retrograde axonal transport of cholera and tetanus toxin as compared to nerve growth factor and wheat germ agglutinin. Brain Res. 132, 273–285 (1977). [DOI] [PubMed] [Google Scholar]
- 61.Habib N, McCabe C, Medina S, Varshavsky M, Kitsberg D, Dvir-Szternfeld R, Green G, Dionne D, Nguyen L, Marshall JL, Chen F, Zhang F, Kaplan T, Regev A, Schwartz M, Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 23, 701–706 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA, Religious Orders Study and Rush Memory and Aging Project. J. Alzheimers Dis. JAD 64, S161–S189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lezi E, Swerdlow RH, Mitochondria in neurodegeneration. Adv. Exp. Med. Biol. 942, 269–286 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leak RK, Heat shock proteins in neurodegenerative disorders and aging. J. Cell Commun. Signal. 8, 293–310 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fulda S, Gorman AM, Hori O, Samali A, Cellular stress responses: cell survival and cell death. Int. J. Cell Biol. 2010, 214074 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G, Hegde M, Cornes SB, Henry ML, Nelson AB, Seeley WW, Geschwind MD, Gorno-Tempini ML, Shih T, Kirsch HE, Garcia PA, Miller BL, Mucke L, Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 70, 1158–1166 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yoshiyama Y, Higuchi M, Zhang B, Huang S-M, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM-Y, Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Kraeuter A-K, Guest PC, Sarnyai Z, The Y-Maze for Assessment of Spatial Working and Reference Memory in Mice. Methods Mol. Biol. Clifton NJ 1916, 105–111 (2019). [DOI] [PubMed] [Google Scholar]
- 69.Abou-Khalil B, Levetiracetam in the treatment of epilepsy. Neuropsychiatr. Dis. Treat 4, 507–523 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Deshpande LS, Delorenzo RJ, Mechanisms of levetiracetam in the control of status epilepticus and epilepsy. Front. Neurol 5, 11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lynch BA, Lambeng N, Nocka K, Kensel-Hammes P, Bajjalieh SM, Matagne A, Fuks B, The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc. Natl. Acad. Sci. U. S. A. 101, 9861–9866 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vogl C, Mochida S, Wolff C, Whalley BJ, Stephens GJ, The synaptic vesicle glycoprotein 2A ligand levetiracetam inhibits presynaptic Ca2+ channels through an intracellular pathway. Mol. Pharmacol. 82, 199–208 (2012). [DOI] [PubMed] [Google Scholar]
- 73.Ohno M, Chang L, Tseng W, Oakley H, Citron M, Klein WL, Vassar R, Disterhoft JF, Temporal memory deficits in Alzheimer’s mouse models: rescue by genetic deletion of BACE1. Eur. J. Neurosci. 23, 251–260 (2006). [DOI] [PubMed] [Google Scholar]
- 74.Eimer WA, Vassar R, Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Aβ42 accumulation and Caspase-3 activation. Mol. Neurodegener 8, 2 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bakker A, Albert MS, Krauss G, Speck CL, Gallagher M, Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. NeuroImage Clin 7, 688–698 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, Yassa MA, Bassett SS, Shelton AL, Gallagher M, Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron 74, 467–474 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, Devidze N, Ho K, Yu G-Q, Palop JJ, Mucke L, Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. U. S. A. 109, E2895–2903 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Welch GM, Boix CA, Schmauch E, Davila-Velderrain J, Victor MB, Dileep V, Bozzelli PL, Su Q, Cheng JD, Lee A, Leary NS, Pfenning AR, Kellis M, Tsai L-H, Neurons burdened by DNA double-strand breaks incite microglia activation through antiviral-like signaling in neurodegeneration. Sci. Adv. 8, eabo4662 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Campbell JN, Macosko EZ, Fenselau H, Pers TH, Lyubetskaya A, Tenen D, Goldman M, Verstegen AMJ, Resch JM, McCarroll SA, Rosen ED, Lowell BB, Tsai LT, A molecular census of arcuate hypothalamus and median eminence cell types. Nat. Neurosci. 20, 484–496 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen R, Wu X, Jiang L, Zhang Y, Single-Cell RNA-Seq Reveals Hypothalamic Cell Diversity. Cell Rep. 18, 3227–3241 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim D-W, Yao Z, Graybuck LT, Kim TK, Nguyen TN, Smith KA, Fong O, Yi L, Koulena N, Pierson N, Shah S, Lo L, Pool A-H, Oka Y, Pachter L, Cai L, Tasic B, Zeng H, Anderson DJ, Multimodal Analysis of Cell Types in a Hypothalamic Node Controlling Social Behavior. Cell 179, 713–728.e17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mickelsen LE, Bolisetty M, Chimileski BR, Fujita A, Beltrami EJ, Costanzo JT, Naparstek JR, Robson P, Jackson AC, Single-cell transcriptomic analysis of the lateral hypothalamic area reveals molecularly distinct populations of inhibitory and excitatory neurons. Nat. Neurosci. 22, 642–656 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Romanov RA, Zeisel A, Bakker J, Girach F, Hellysaz A, Tomer R, Alpár A, Mulder J, Clotman F, Keimpema E, Hsueh B, Crow AK, Martens H, Schwindling C, Calvigioni D, Bains JS, Máté Z, Szabó G, Yanagawa Y, Zhang M-D, Rendeiro A, Farlik M, Uhlén M, Wulff P, Bock C, Broberger C, Deisseroth K, Hökfelt T, Linnarsson S, Horvath TL, Harkany T, Molecular interrogation of hypothalamic organization reveals distinct dopamine neuronal subtypes. Nat. Neurosci. 20, 176–188 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stackman RW, Taube JS, Firing properties of rat lateral mammillary single units: head direction, head pitch, and angular head velocity. J. Neurosci. Off. J. Soc. Neurosci. 18, 9020–9037 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Blair HT, Cho J, Sharp PE, Role of the lateral mammillary nucleus in the rat head direction circuit: a combined single unit recording and lesion study. Neuron 21, 1387–1397 (1998). [DOI] [PubMed] [Google Scholar]
- 86.Bennett DA, Wilson RS, Schneider JA, Evans DA, Beckett LA, Aggarwal NT, Barnes LL, Fox JH, Bach J, Natural history of mild cognitive impairment in older persons. Neurology 59, 198–205 (2002). [DOI] [PubMed] [Google Scholar]
- 87.Bennett DA, Wilson RS, Schneider JA, Evans DA, Aggarwal NT, Arnold SE, Cochran EJ, Berry-Kravis E, Bienias JL, Apolipoprotein E epsilon4 allele, AD pathology, and the clinical expression of Alzheimer’s disease. Neurology 60, 246–252 (2003). [DOI] [PubMed] [Google Scholar]
- 88.Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE, Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch. Neurol. 61, 378–384 (2004). [DOI] [PubMed] [Google Scholar]
- 89.Bennett DA, Schneider JA, Aggarwal NT, Arvanitakis Z, Shah RC, Kelly JF, Fox JH, Cochran EJ, Arends D, Treinkman AD, Wilson RS, Decision rules guiding the clinical diagnosis of Alzheimer’s disease in two community-based cohort studies compared to standard practice in a clinic-based cohort study. Neuroepidemiology 27, 169–176 (2006). [DOI] [PubMed] [Google Scholar]
- 90.Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, Wilson RS, Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 66, 1837–1844 (2006). [DOI] [PubMed] [Google Scholar]
- 91.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, Martorell AJ, Ransohoff RM, Hafler BP, Bennett DA, Kellis M, Tsai L-H, Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R, Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mohammadi S, Davila-Velderrain J, Kellis M, A multiresolution framework to characterize single-cell state landscapes. Nat. Commun. 11, 5399 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.