

Abstract
The tumor suppressor p53 is the most frequently mutated protein in human cancer and tops the list of high-value precision oncology targets. p53 prevents initiation and progression of cancer by inducing cell-cycle arrest and various forms of cell death. Tumors have thus evolved ways to inactivate p53, mainly by TP53 mutations or by hyperactive p53 degradation. This review focuses on two types of p53 targeting compounds, MDM2 antagonists and mutant p53 correctors. MDM2 inhibitors prevent p53 protein degradation, while correctors restore tumor suppressor activity of p53 mutants by enhancing thermodynamic stability. Herein we explore both novel and repurposed p53 targeting compounds, discuss their mode of action, and examine the challenges in advancing them to the clinic.
p53 as a cancer drug target
p53 (encoded by the TP53 gene) is the major tumor suppressor protein in vertebrates and provides a powerful mechanism to protect from cancer initiation and progression [1], Most tumors have developed ways to restrict p53 anticancer activity (Figure 1), making this pathway one of the most attractive pharmaceutical targets for cancer therapy. For many years p53 had been considered undruggable due to lack of obvious drug-binding pockets. Furthermore, therapeutic approaches require restoring or increasing the tumor suppressor function of inactive p53, which remains very challenging. However, during the past few years conventional and computation-driven strategies have identified several small molecules that restore p53 anticancer activity in tumors [2–6]. Some have been or are currently being evaluated in the clinic. The mode of action of these small molecules is diverse and, in some cases, unexpected.
Figure 1. Types and distribution of p53 alterations in cancer.
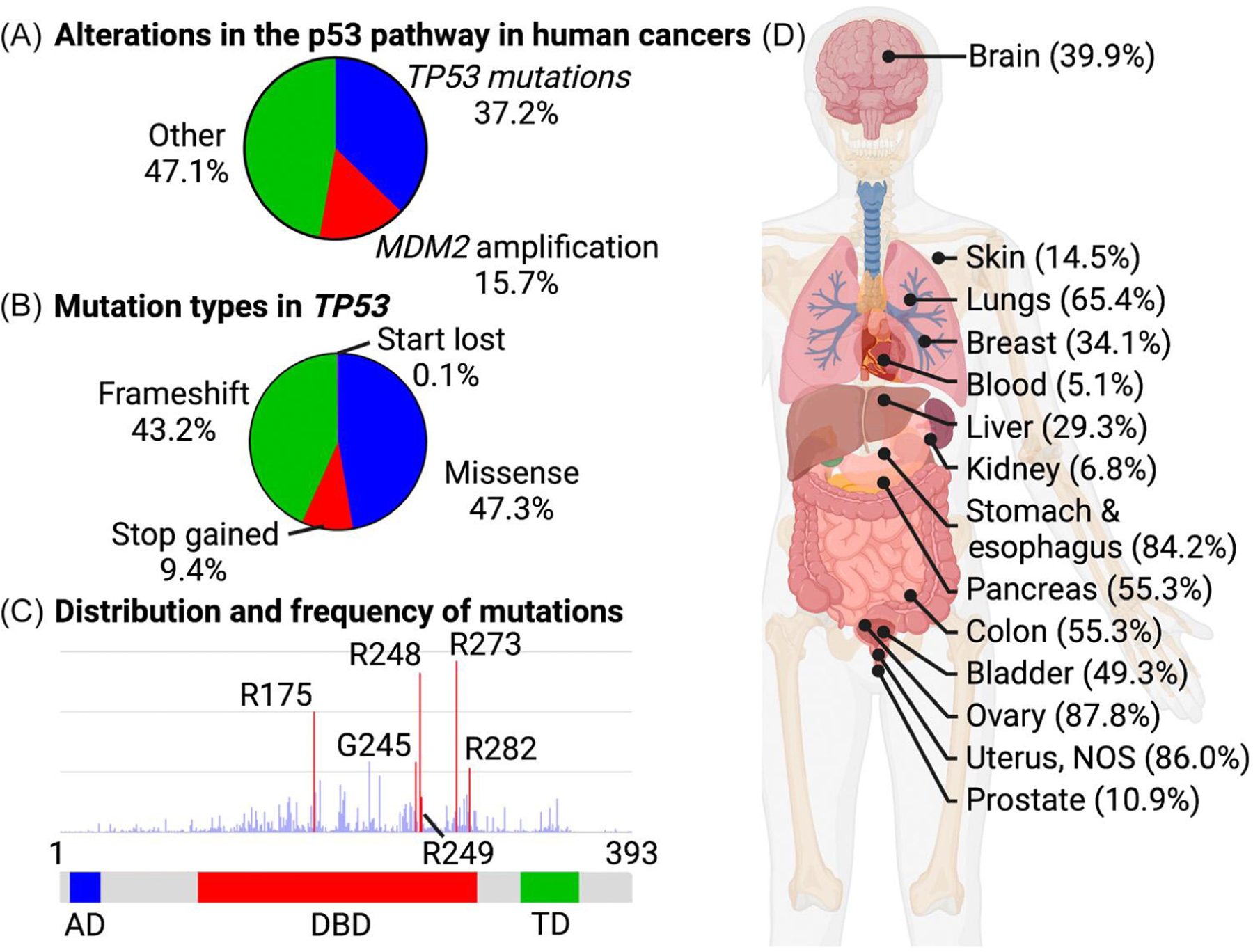
(A) A large fraction of tumors show mutations in p53 or amplification of MDM2, which encodes a ubiquitin ligase that triggers p53 degradation by the proteasome. Other pathways that overcome p53 tumor suppression include degradation of p53 by the high-risk human papilloma protein E6 [93], epigenetic silencing of TP53 [94], and diverse mechanisms that block p53 signaling without directly affecting p53. (B) Type of mutations in p53 that contribute to human cancer. (C) Amino acid position distribution and frequency of p53 missense mutations. (D) Frequency of p53 mutations in various tumor types. Graphs are based on data generated by The Cancer Genome Atlas (TCGA) Research Network: www.cancer.gov/tcga. Abbreviations: AD, activation domain; DBD, DNA-binding domain; TD, tetramerization domain. Created with BioRender.com.
In this review we briefly introduce the p53 pathway and its importance in cancer, describe various strategies for drugging this pathway, and provide a more detalled discussion of compounds that prevent p53 degradation (MDM2 antagonists) and small-molecule reactivators of p53 mutants. We discuss their proposed mode of action and summarize their current clinical status.
p53 function and regulation
p53 is a DNA-binding transcription factor that orchestrates gene expression programs in response to various stress conditions that can compromise genomic integrity and ultimately lead to cancer [1,7], Hence p53 is often referred to as the ‘guardian of the genome’ [8]. While p53 is constitutively expressed in most normal tissues, its protein levels are kept at a very low abundance through constant proteasomal degradation, a pathway initiated by the ubiquitin ligase (see Glossary) MDM2 [9,10]. The remaining p53 activity is further reduced by MDM4/MDMX, an endogenous p53 inhibitor that blocks transcription factor activity by binding to p53. Stress stimuli such as DNA damage, abnormal oncogene activation, and deprivation of oxygen or nutrients block MDM2-mediated p53 degradation and disrupt MDM4/MDMX binding to p53. The now accumulating active p53 induces gene expression programs to halt cell proliferation and allow damage repair, or even cell death if the damage persists or cannot be repaired [11,12].
Cancer cells continuously generate and experience multiple stress stimuli, and p53 activation thus usually triggers their demise. To overcome p53 tumor suppressor function, successful cancer cells find ways to restrict p53 activity by degradation, sequestration, deletion, epigenetic silencing, or – most commonly – mutation [1,7].
Almost 40% of human tumors have p53 mutations with varying frequency across tissues (Figure 1). A smaller proportion of tumors restrict p53 activity by overactive degradation mediated by amplification of MDM2 [13], or in the case of cervical cancer, expression of the viral ubiquitin ligase component E6 that contributes to tumorigenicity of high-risk human papilloma virus [14].
In human cancers, p53 mutations predominantly affect the DNA binding domain and show an interesting pattern [15] (Figure 1 B,C). Not only do missense mutations dominate genetic alterations, but six specific residues (R175, G245, R248, R249, R273, R282) stand out as hotspots. Changes in residues R175, G245, R249, or R282 destabilize p53 folding and are referred to as conformational or structural mutants. Mutation of R248 or R273 leads to DNA contact mutants because these residues directly interact with nucleotides in p53-binding DNA elements [16]. Surprisingly, tumors expressing p53 hotspot mutants are more aggressive than p53NULL tumors, indicating that gain-of-function (GOF) oncomorphic properties are associated with at least some p53 mutants [17–19]. GOF activities vary among different mutants, but have been linked to active stimulation of cell proliferation, increased genomic instability, metastatic potential, metabolic reprogramming, and generation of an immune suppressive microenvironment [20]. These acquired oncomorphic qualities are not well understood, but some mechanistic studies suggest that p53 hotspot mutants gain protein binding to several transcription factors and epigenetic modifiers to stimulate, redirect, or suppress their activities [21]. GOF is an important aspect of cancers driven by mutant p53, and better mechanistic insight is needed.
Drugging the p53 pathway
The large number of TP53 missense mutations in cancers across different tissues presents an attractive target for corrector or reactivation drugs (Figure 1). However, p53 itself has long been considered undruggable, and more indirect approaches that target the p53 pathway have been followed. Probably the most extensively pursued alternative to p53 mutant correctors are MDM2 antagonists. They prevent degradation of p53 by the ubiquitin proteasome pathway [22] (Figure 2A). By contrast, p53 corrector drugs are small molecules that cause enough mutant p53 to gain wild-type (WT) activity to suppress tumor progression (Figure 2B). This is achieved by compounds binding to p53 mutants and structurally stabilizing a WT-like conformation. Note that MDM2 antagonists stabilize protein levels, whereas p53 mutant correctors improve thermodynamic stability. Corrector drug approaches are very challenging for any target. The past few years have, however, seen promising developments for p53 mutant reactivation, with some molecules reaching clinical trial stage (Table 1). A host of additional strategies to drug the p53 pathway have also emerged and have been summarized in a number of excellent reviews [20,23–26]. Approaches include adenovirus as well as RNA/nanoparticle therapeutics to express WT p53 [27–29], induction of mutant p53 degradation to eliminate GOF properties [30], targeting TP53 nonsense mutations with drugs that trigger translational readthrough or block nonsense-mediated mRNA decay [31,32], and stimulating p53 family members p63 and p73 [20]. Furthermore, various p53-based immunotherapies have recently emerged [20], including p53 vaccines [33] and targeted strategies that mark mutant p53 as neoantigens with bispecific antibodies to provoke an immune response [34], Although these diverse approaches are being explored in laboratories around the world, this review will focus on encouraging advancement made with MDM2 antagonists and p53 reactivators.
Figure 2. Concepts of wild-type (WT) p53 activation and small-molecule-based mutant p53 reactivation.
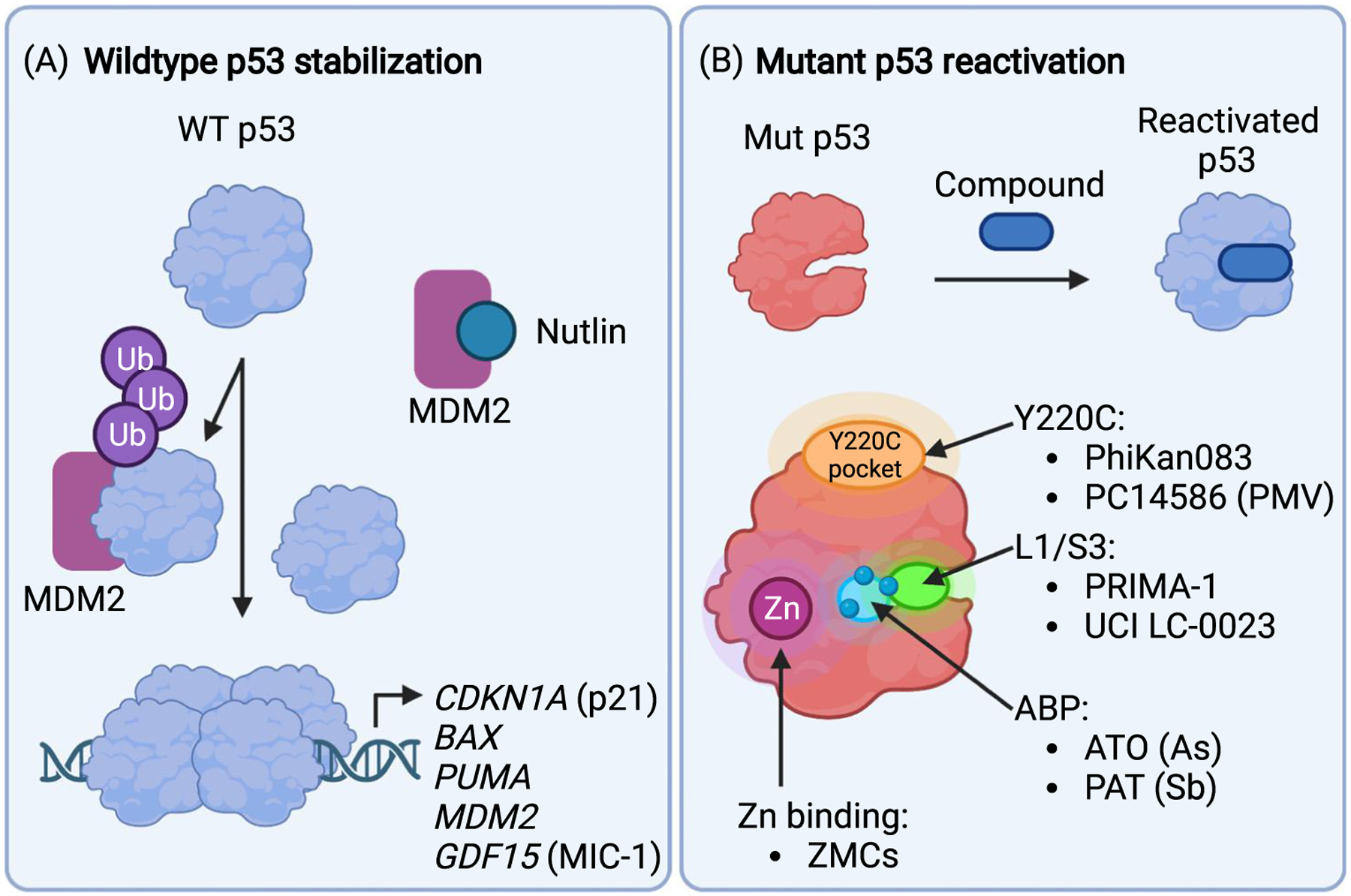
(A) Many tumors with WT p53 overexpress the ubiquitin (Ub) ligase MDM2. This leads to rapid degradation of p53 by the proteasome and inactivation of tumor suppression. Nutlins and other small-molecule MDM2 antagonists block the MDM2/p53 interaction resulting in accumulation of p53, assembly of the active p53 tetramer, and induction of genes that trigger cell-cycle arrest and cell death. (B) Small molecules that bind mutant p53 and stabilize a WT-like conformation are known as corrector or reactivation drugs. Several small molecules with different proposed binding modes and mechanisms have been identified. PhiKan0083 and PC14586 bind to a cryptic crevice created by the Y220C mutation. Zinc metallochaperones (ZMCs) act as zinc ionophores to facilitate zinc binding to p53 mutants with reduced zinc binding capacity. Arsenicals (arsenic trioxide, ATO) and antimonials (potassium antimony tartrate, PAT) release arsenic and antimony, respectively, which are complexed by three specific cysteine residues (three blue circles) that comprise the C124–C135–C141 pocket (light blue region). Arsenic and antimony binding to this pocket (ABP) stabilizes an active conformation of p53 mutants. A degradation product of PRIMA-1 covalently attaches to several cysteines and reactivates p53 mutants, and UCI-LC0023 binds noncovalently to a transiently open pocket (L1/S3 pocket, green area) in the same region to restore activity to p53 mutants. Note that C124 is located in the L1/S3 pocket. Created with BioRender.com.
Table 1.
Clinical trials targeting p53 in cancer (https://clinicaltrials.gov/ct2/home)
| NCT number | Agent | Indication | Phase | Status | Trial title | Mechanism of action |
|---|---|---|---|---|---|---|
| NCT02633059 | Idasanutlin (RG7388, R05503781) | Multiple myeloma | 1/2 | Active, not recruiting | Idasanutlin, ixazomib citrate, and dexamethasone in treating patients with relapsed multiple myeloma | Nutlin MDM2 inhibitor |
| NCT03217266 | AMG 232 (Navtemadlin) | Soft-tissue sarcomas | 1b | Active, not recruiting | Navtemadlin and radiation therapy in treating patients with soft-tissue sarcoma | Piperidinone inhibitor of MDM2-p53 interaction |
| NCT03787602 | AMG 232 (Navtemadlin) | Merkel cell carcinoma | 1b/2 | Recruiting | Navtemadlin (KRT-232) with or without anti-PD-1/anti-PD-L1 for the treatment of patients with Merkel cell carcinoma | Piperidinone inhibitor of MDM2-p53 interaction |
| NCT03611868 | APG-115 (Alrizomadlin) | Melanoma and advanced solid tumors | 1b/2 | Recruiting | A Phase 1b/2 study of APG-115 in combination with pembrolizumab in patients with unresectable or metastatic melanomas or advanced solid tumors | MDM2 inhibitor |
| NCT03781986 | APG-115 (Alrizomadlin) | Salivary gland cancer | 1/2 | Recruiting | APG-115 for the treatment p53 wild-type malignant salivary gland cancer | MDM2 inhibitor |
| NCT05155709 | Siremadlin | Acute myeloid leukemia | 1b/2 | Recruiting | A study of siremadlin in combination with venetoclax plus azacitidine in adult participants with acute myeloid leukemia (AML) who are ineligible for chemotherapy | Imidazopyrro-lidinone scaffold-based inhibitor of p53-MDM2 interaction |
| NCT03975387 | ASTX295 | Advanced solid tumors with wild-type p53 | 1/2 | Recruiting | Study of ASTX295 in patients with solid tumors with wild-type p53 | MDM2 inhibitor |
| NCT03725436 | ALRN-6924 | Advanced solid tumors | 1b | Recruiting | ALRN-6924 and paditaxel in treating patients with advanced, metastatic, or unresectable solid tumors | MDM2/MDMX inhibitor |
| NCT03654716 | ALRN-6924 | Pediatric cancer | 1 | Recruiting | Phase 1 study of the dual MDM2/MDMX Inhibitor ALRN-6924 in pediatric cancer | MDM2/MDMX inhibitor |
| NCT05376800 | Bl 907828 | Glioblastoma | 0/1 a | Recruiting | A study to determine how Bl 907828 is taken up in the tumor and to determine the highest dose of Bl 907828 that could be tolerated in combination with radiation therapy in people with a brain tumor called glioblastoma | MDM2 inhibitor |
| NCT02340117 | SGT-53 | Pancreatic cancer | 2 | Recruiting | Study of combined SGT-53 plus gemcitabine/Nab-paditaxel for metastatic pancreatic cancer | Liposome complex encapsulating normal human wild-type p53 DNA in a plasmid backbone |
| NCT04585750 | PC14586 | Advanced solid tumors with a p53 Y220C mutation | 1/2 | Recruiting | The evaluation of PC14586 in patients with advanced solid tumors harboring a p53 Y220C mutation | Structural corrector of p53 Y220C mutant protein |
| NCT03855371 | Arsenic trioxide (ATO) | AML/MDS | 1 | Recruiting | Combination of decitabine and ATO to treat AML/MDS expressing a classified type of mutant p53 | Structural corrector of conformational p53 mutants |
| NCT04906031 | SSG | AML/MDS | 2 | Recruiting | SS in MDS/AML with one of the 65 defined p53 mutations that can be functionally rescued by SSG | Structural corrector of temperature-sensitive p53 mutants |
Small molecules that block p53 degradation (MDM2 antagonists)
Much of p53 regulation occurs through its MDM2-mediated degradation. MDM2 inhibition was thus considered an attractive therapeutic strategy to stimulate p53 tumor suppressor activity. Primary clinical targets are cancers with MDM2 amplification and WT p53. However, MDM2 is a difficult drug target, because like other E3 ubiquitin ligases it lacks defined enzymatic activities and functions by bridging the interaction between p53 and other parts of the degradation machinery (Figure 2A). Drugging protein-protein interaction surfaces remains challenging, but an attractive hydrophobic pocket involved in p53 binding has been successfully targeted [35]. A series of cis-imidazoline-derived drugs were named nutlins after their discovery location at the Roche Research Center in Nutley, NJ, USA. Many other companies have since developed MDM2 antagonists [36]. Considering that they act as protein interaction disruptors, MDM2 inhibitors perform exceptionally well in experimental models. They dramatically stabilize p53, induce p53-dependent transcription programs that lead to cell cycle arrest and apoptosis, and prevent tumor development in animal models [35]. The first clinical report of the first-generation MDM2 antagonist, RG7112, targeted liposarcoma, which is characterized by MDM2 12q14–15 amplification [37,38]. Day 8 biopsies demonstrated increased concentrations of p53 and p21 (a cell-cycle inhibitor and major p53 target), suggesting that the proposed mode of action could extend to human patients. Significant adverse events prompted the development of next-generation MDM2 inhibitors.
Next-generation nutlin compounds have frequently been evaluated in patients with acute myeloid leukemia (AML) and CD34+ myeloproliferative neoplasms. Orally administered idasanutlin (RG7388) showed a 78% overall response rate in nine evaluated patients (R05503781) [39]. However, adverse effects remained significant and did not improve compared with early-generation nutlins. The pegylated prodrug of idasanutlin (R06839921) was developed to allow for intravenous administration, but no significant improvement in the biological or safety profile was achieved [40].
MDM2 antagonists based on different scaffolds have also been developed. The oral piperidinone-based inhibitor AMG 232 showed improved IC50 in cell proliferation assays (9.1 nM versus RG7112: 580 nM, RG7388: 45 nM), and superior binding affinity to MDM2 (Kd = 45 pM versus RG7112: 2.9 nM, RG7388: 150 pM) [41]. Preclinical studies suggested that MDM2 inhibition synergizes with MEK inhibition against TP53-WT AML cells [42], A Phase 1 study of AMG 232 with or without the oral MEK inhibitor trametinib was therefore performed [43]. Serum MIC-1 and bone-marrow expression of BAX, PUMA, CDKN1A (encoding p21), and MDM2 increased during treatment, demonstrating p53 activation. Of 30 evaluable patients, 20% responded.
Responses appeared significantly different between AMG 232 alone (4/26) and AMG 232 with trametinib (2/10), but numbers are low. Notably, four of 13 TP53-WT patients (31%) and none of three TP53-mutant patients (0%) were responders, suggesting clinical benefit primarily for TP53-WT patients. AMG 232-associated adverse effects remained mainly gastrointestinal but seemed tolerable.
Several MDM2 antagonists (APG-115, siremadlin, milademetan) were also evaluated in the treatment of solid tumors [44–46]. Like outcomes in hematological malignancies, results are encouraging, but so far clinical efficacies did not meet the high expectations. Several companies have since re-evaluated their small-molecule MDM2 antagonist programs in single-agent approaches [47], Alternative approaches to target MDM2 have also been progressing and include stapled peptides that block the p53-MDM2 interaction and proteolysis targeting chimera (PROTAC)-mediated MDM2 depletion [48–51]. Small molecules that bind and activate p53 through a less defined mechanism have also been reported [52].
The general concern remains that MDM2-mediated degradation is a major pathway that represses p53 activity in most tissues. The therapeutic window opened by MDM2 amplification in tumors may not be sufficient to prevent significant on-target adverse effects of MDM2 antagonists. Nevertheless, these classes of compound may be valuable agents in combination therapies, where a lower dose may be effective, as indicated for AMG 232 combined with MEK inhibition [42,43]. MDM2 inhibitors may also prove important in combination with p53 mutant corrector drugs discussed later. Correctors induce a WT-like conformation in p53 mutants, which restores tumor suppressor activities but also triggers MDM2-mediated degradation. MDM2 antagonists may thus potentiate the anticancer effects of p53 corrector drugs.
Small molecules to correct structural defects of p53 mutants
MDM2 antagonists target tumors with MDM2 amplifications and WT p53. However, most tumors with alterations in the p53 pathway already express high levels of p53 but lack tumor suppressor function due to mutations (Figure 1). Efforts to directly target p53 missense mutants have been ongoing for over two decades, only very recently advancing to clinical trials. The slow progress highlights the complexity of developing corrector drugs (also referred to as reactivators) that restore tumor suppressor activity to mutated p53. The first such putative reactivator molecule (CP31398) was identified by Pfizer in 1999 [53]. CP31398 stabilized a WT-like conformation of several p53 mutants in vitro and in vivo, induced p21 expression, and reduced tumor growth in mice. However, the effects on tumor growth were subsequently connected to off-target activities [54], The experience with CP31398 highlights the remaining problem to clearly connect a molecule’s observed effect on mutant p53 with the mode of antitumor action in vivo. Many putative mutant p53 correctors have since been reported (Table 2). Rigorous structural, biophysical, or structure–activity-relationship (SAR) studies are available for only a small number of compounds, and provide a well-defined mode of action for p53Y220C correctors and arsenicals/antimonials [3,4,55]. Most compounds lack this level of understanding regarding their mechanism of action.
Table 2.
Small molecules with proposed mutant p53 corrector activity
| Compounds | Proposed mechanism/comments | Refs |
|---|---|---|
| Antimonials | Deliver antimony, which binds noncovalently to the cysteine triad (C124–C135–C141) to structurally stabilize temperature-sensitive p53 mutants. | [4] |
| APR-246/PRIMA-1 | Prodrug with the active compound being MQ. MQ binds covalently to several cysteine residues in p53 and thermodynamically stabilizes p53 mutants. Off-target thiol reactivity may contribute to anticancer activity. | [70] |
| ATO | Delivers arsenic, which binds covalently to the cysteine triad (C124–C135–C141) to structurally stabilize structural p53 mutants. | [3] |
| Chetomin | Restores activity of p53R175H mutants. Binds Hsp40 and increases Hsp40/p53R175H interaction to restore wild-type-like conformation. | [95] |
| COTI-2 | Thiosemicarbazone that binds p53 in vitro and induces a wild-type-like conformation in p53R175H mutants. | [96] |
| CP-31398 | first identified putative mutant p53 reactivator. Antitumor activity was later attributed to off-target activities. The compound reacts with thiols. | [53,54] |
| KG13 | Covalently binds to C220 in p53Y220C mutants and improves structural stability. Selective for p53Y220C mutants. | [61] |
| MANIO | Activates wild-type and mutant p53. Binds to the p53 core domain and induces thermal stabilization. | [52] |
| MB710, MB725 | Bind to the Y220C-induced surface crevice and structurally stabilize p53Y220C mutants. Selective for p53Y220 mutants. | [59] |
| MIRA-1 | Stabilizes a wild-type like conformation of p53 mutants and induces DNA-binding. It is a maleimide derivative and reacts with thiols and amines. Antiproliferative effect may be due to off-target activity. | [97] |
| P53R3 | Induces DNA binding of p53 mutants in vitro and preferentially inhibits growth of TP53 mutant cell lines. | [98] |
| PC14586 | Clinical compound that binds to the Y220C-induced surface crevice and structurally stabilizes p53Y220C mutants. Selective for p53Y220 mutants. | [6] |
| Phenethyl isothiocyanate | Natural compound that restores wild-type-like conformation to p53R175H. Depletes glutathione and activity is blocked by antioxidants. Mode of action may be indirect. | [99] |
| PhiKan083 (PK083) | The first small molecule targeting the Y220-mutation-induced surface crevice. Structurally stabilizes p53Y220C mutants. Selective for p53Y220 mutants. | [55] |
| PK11007 | One of several sulfonylpyrimidine-derived thiol-reactive agents that lead to thermal stabilization of p53 mutants. Preferentially targets cystine residues in p53. | [100] |
| PK5196 | Binds to the Y220C-induced surface crevice and structurally stabilizes p53Y220C mutants. Selective for p53Y220 mutants. | |
| PK7088 | Binds to the Y220C-induced surface crevice and structurally stabilizes p53Y220C mutants. Selective for p53Y220 mutants. | [58] |
| PK9328 | Improved Y220 pocket binder. Based on PhiKan083. Selective for p53Y220 mutants. | [101] |
| SLMP53-1 | Activates wild-type and mutant p53 by inducing thermal stabilization. | [102] |
| Stictic acid | Natural compound that was selected as a L1/S3 pocket binder. Induces thermal stabilization of pSS02453 and p53R175H mutants in vitro. | [71] |
| STIMA-1 | Enhances DNA binding of mutant p53 and induces p53-dependent gene expression. Compound shows thiol reactivity, and antiproliferative effects may be due to off-target activity. | [103] |
| UCI-LC0019/LC0023 | Binds noncovalently to the L1/S3 pocket. Induces sequence-specific DNA binding of p53R175H mutants in vitro. | [5] |
| ZMC1 (NSC319726) | Thiosemicarbazone that acts as zinc chaperone to structurally stabilize p53 mutants with reduced zinc-binding potential. | [81] |
The past years have seen significant progress in mutant p53 corrector drug research, with several molecules being evaluated in human patients, some of which are discussed in the following sections.
Y220C correctors
Amino acid changes of tyrosine at position 220 (mostly to cysteine, Y220C) are not considered classical hotspot mutations (Figure 1C) but are the most frequent p53 cancer mutations outside the DNA-binding surface [15]; p53Y220 mutations affect about 1% of solid tumors and present a valuable target for precision oncology. Y220 is part of the hydrophobic core of the central β-sheet in p53. Mutation to cysteine and other residues (H, N, S, D, or F) thermodynamically destabilizes the p53 core domain and thereby indirectly prevents DNA binding [56,57], Furthermore, mutation of Y220 creates a solvent-accessible cleft that represents the first druggable pocket identified in p53 [56] (Figure 2B). Subsequently, pioneering work led to the development of the methylmethanamine-derived PhiKan083. This compound is anchored to the Y220C-induced surface crevice by several hydrophobic and hydrogen-bond interactions that result in thermal stabilization of p53Y220C mutants [55]. Several additional molecules were subsequently developed that bind to this Y220-mutation-induced cleft and compensate for the mutation’s conformational destabilization in vitro and in vivo [55,58–60]. Molecules restored p53-dependent transcription in cell lines carrying the TP53-Y220C allele, albeit high micromolar concentrations were required for pyrazole-based PhiKan7088 and its iodophenol derivative PhiKan5196 [58,60]. Further development resulted in the aminobenzothiazole derivative MB725 with a much improved dissociation constant and cell-based activity [59].
PMV Pharma adopted a similar strategy to target the mutation-induced crevice in p53Y220C leading to the development of the orally available compound PC14586 [6]. Progress was recently reported for a first-in-human Phase 1 dose-escalation study in patients with advanced or metastatic TP53-Y220C mutant solid tumors [6]. The most common adverse effects were nausea, vomiting, fatigue, and increased aspartate aminotransferase. Dosing reached 1500 mg twice daily without dose-limiting toxicities, and enrollment onto the trial is ongoing. Notably, five partial responses (21 patients evaluated) were observed in small-cell lung, breast, colorectal, and prostate cancers. Pharmacodynamic studies demonstrated decreasing TP53-Y220C circulating tumor DNA, consistent with the known mechanism of action. Recently, covalent compounds that directly target the unique cysteine of p53Y220C have been developed, which may indicate future strategies to develop more potent molecules [61].
The importance of work focused on p53Y220C correctors cannot be overestimated, despite the comparably small representation among TP53 missense mutations. Compounds targeting the Y220C-induced surface crevice act through a well-defined mode of action that is supported by rigorous structural biology, and allow detalled evaluation of mechanism and clinical potential of mutant p53 corrector drugs. Other compounds (discussed later) target a broader panel of TP53 missense mutations, but apart from arsenicals and antimonials, their modes of action are less defined. Compounds targeting p53Y220C also show promise in combination with cancer immunotherapy in animal models [62], and may suggest a general stimulatory role of p53 mutant correctors on tumor immunogenicity.
Arsenicals and antimonials
Compounds containing arsenic (arsenicals) or antimony (antimonials) have very recently been identified as p53 mutant correctors [3,4], Cysteine reactivity of the most widely studied p53 mutant reactivator PRIMA-1 (discussed later) inspired a computational approach that led to the identification of arsenicals [3]. Compounds predicted to bind at least two cysteines were mined from a set of small molecules that showed selective growth inhibition of cancer cells expressing conformational p53 mutants [3]. The FDA approved drug arsenic trioxide (ATO) was among several identified compounds containing thiol-reactive metals. A biochemical approach led to identification of the antiparasitic potassium antimony tartrate (PAT) as a mutant p53 reactivator [4], PAT was selected from a differential scanning fluorometry (DSF) screen of clinical and preclinical compounds by its virtue of increasing thermal stability of the temperature-sensitive p53V272M mutant. Cocrystal structures demonstrated that a single arsenic or antimony atom is coordinated to C124, C135, and C141 in the DNA-binding domain of p53 (Figure 3). These additional contacts stabilize an active conformation of p53 mutants. Importantly, mutation of any of these arsenic/antimony-coordinating cysteines to alanine blocks reactivation of p53 mutants by ATO or PAT, providing strong support for the proposed mode of action [3,4]. Amino-acid interactions in the C124–C135–C141 pocket have previously been described as critical components of p53 structural stability [63]. This region’s amino acid sequences exhibit variability through evolution, and this variability was suggested to coordinate thermodynamic stability of p53 with organismal body temperatures. This ensures that p53 maintains a half-life for spontaneous unfolding in vivo of 10–20 min across different homeotherm organisms [64]. Arsenic and antimony atoms delivered by ATO and PAT, respectively, act as intramolecular glues that strengthen the C124–C135–C141 pocket. This ATO/PAT-induced increased structural stability compensates for the destabilizing effects of p53 cancer mutations, leading to reactivation of structural p53 cancer mutants [3,4].
Figure 3. Cysteine residues in the p53 DNA-binding domain involved in mutant p53 reactivation.
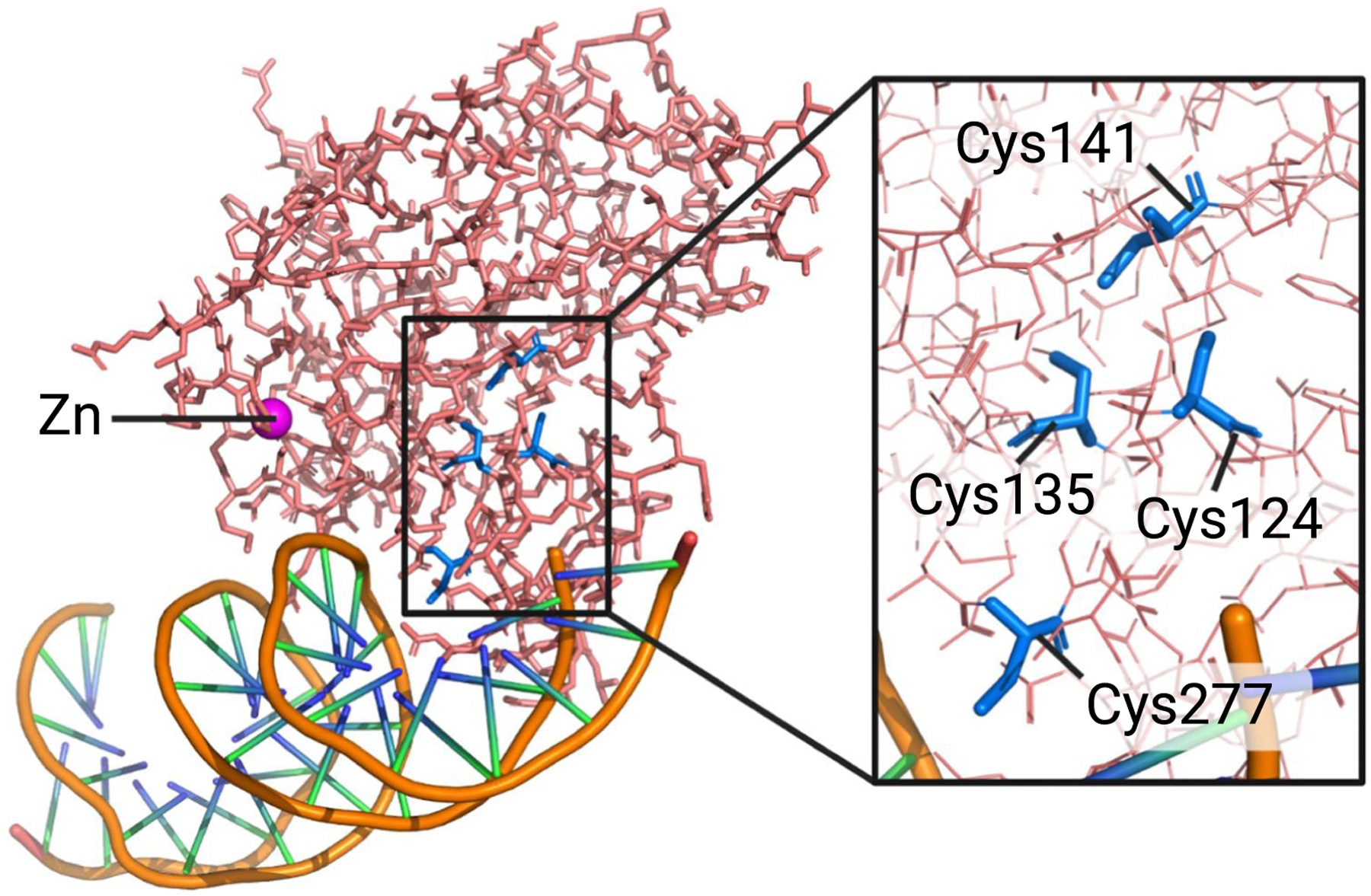
The p53 DNA binding domain bound to DNA (PDB ID: 1TSR) with four cysteine residues involved in p53 reactivation. Cys124 and Cys277 have been linked to PRIMA-1/APR-246-mediated reactivation of p53 mutants [71,72]. Cys124, Cys135, and Cys141 coordinate antimony and arsenic as part of the corrector drug mode of action for antimonials and arsenic trioxide (ATO), respectively [3,4]. Note that arsenic covalently binds to these cysteines, whereas antimony interacts noncovalently. Figure was created in PyMOL.
In contrast to the covalent binding of arsenic to p53, antimony binds in a noncovalent fashion. The difference in binding modes is manifested in the varying degrees of structural stabilization conferred by arsenic and antimony. The weaker interactions formed by antimony with the C124–C135–C141 triad can only effectively reactivate moderately structurally destabilized mutants [4]. PAT is therefore selective for correction of temperature-sensitive p53 mutants, which are inactive at 37°C, but gain activity at lower temperature [16,65,66]. Temperature-sensitive p53 variants comprise up to 10% of all p53 mutants and represent a significant precision oncology target. The covalent binding mode of arsenic makes ATO a strong stabilizing agent that can also restore activity of thermodynamically severely destabilized p53 mutants, as exemplified by p53R175H [3]. Both ATO and PAT show efficacy in preclinical animal models. ATO significantly delayed tumor growth in xenografts expressing TP53-R175H or TP53-R282W, in a p53-dependent manner [3], and PAT treatment improved the survival of an AML TP53-V272M mouse model from 17 to 24 days [4].
ATO is an FDA-approved drug used for the treatment of acute promyelocytic leukemia through induction of apoptosis and differentiation of leukemic promyelocytes [67], and the approved antimonial sodium stibogluconate (SSG) has been used extensively for the treatment of the parasitic disease leishmaniasis, with limited side effects [68]. The exciting potential of repurposing ATO and SSG for the targeted treatment of cancers with different p53 mutants is currently being evaluated in several clinical trials. Results are anxiously awaited. ATO is being tested in Phase 1 clinical trials for refractory cancer patients with TP53 mutations (NCT04695223 and CT04869475), and patients with metastatic ovarian and endometrial cancers (NCT04489706). A Phase 2 clinical trial was initiated to evaluate the safety and effectiveness of SSG in the treatment of myelodysplastic syndrome (MDS)/AML with any of the 65 SSG-treatable TP53 mutations (NCT04906031). General toxicity of ATO may limit efficacy in solid tumors, but SSG is generally well tolerated and may not face such dose limitations.
The discovery of FDA-approved compounds with activity as p53 mutant correctors and their repurposing represents a particularly exciting development. These findings have dramatically accelerated the pathway for p53-targeting drugs to the bedside and encourage other studies that may discover additional clinical-grade compounds as mutant p53 reactivators.
PRIMA-1 and eprenetapopt (APR-246)
PRIMA-1 was identified in 2002 in a cell-based screen selecting for compounds with differential effects on SAOS-2 osteosarcoma cells (p53NULL) and engineered SAOS-2 cells expressing p53R273H [69]. Subsequent work showed that the compound is active towards a broad spectrum of TP53 missense mutations and blocks tumor progression in 53R273H-expressing xenograft models [69,70]. PRIMA-1 and the methylated derivative APR-246, developed by APREA Therapeutics for clinical use, are the most widely studied p53 mutant corrector compounds today. It is thanks to this pioneering work that we appreciate the complexity of mutant p53 reactivation. PRIMA-1 and APR-246 are prodrugs, which under physiological conditions are spontaneously converted to methylene quinuclidinone (MQ) [70]. Owing to its Michael acceptor activity, MQ covalently attaches to cysteines in proteins. 0124 as well as 0277 in p53 (Figure 3) were not only shown to be targeted by MQ, but also to be necessary for PRIMA-1-mediated mutant p53 reactivation in cells [71–73]. Several crystal structures of the p53 core domain bound to MQ have been reported recently [73]. p53 mutant and WT cocrystals with MQ were obtained by both cocrystallization and crystal soaking with MQ. Depending on the approach, various accessible cysteine residues were found to be targeted by MQ in vitro, with 0124, 0229, and 0277 being the most consistent MQ acceptors. The cocrystal structures imply that MQ conjugation to the these three cysteine residues may contribute in different ways to stabilization of the p53 structure [73]. Direct assessment of MQ-induced thermal stability changes in vitro identified conjugation to 0277 as having the main stabilizing effect, although 0229 was not analyzed in these experiments [72], However, in vivo studies demonstrated that both 0124 and 0277 are crucial for PRIMA-1/APR-246/MQ-mediated reactivation of p53 mutants. Substituting either of these cysteine residues with alanine is sufficient to block reactivation of p53R175H transcriptional activity [71,72].
An elegant reactivation experiment further supported a direct mode of action for PRIMA-1. Recombinant p53R175H or p53G248Q was treated in vitro with PRIMA-1 and transfected into SAOS-2 (p53NULL) cells. p53 mutants were as effective in blocking cell proliferation and inducing p53-dependent target genes as WT p53, but only after in vitro incubation with PRIMA-1 [70]. PRIMA-1/APR-246 show many other hallmarks of mutant p53 reactivation, such as stabilization of a WT-like conformation in p53 mutants, stimulation of p53MUT association with cognate promoters, and expression of p53-dependent genes [69].
Despite the compelling evidence, alternative mechanisms for the primary mode of action, especially in vivo, were proposed recently [73–76]. These activities of PRIMA-1/APR-246 are related to their thiol reactivity. Depletion of glutathione emerged as a main contributor to the anticancer effect of these compounds [74]. The differential sensitivity of p53 missense-mutant-expressing cells and TP53-NULL cells to PRIMA-1/APR-246 was rationalized by a GOF activity associated with mutant p53. According to this model, mutant p53 binds to and inhibits the antioxidant transcription factor NRF2, leading to repression of SLC7A11, a component of the cystine/glutamate antiporter (Figure 4). Consequently, cysteine import is reduced, limiting glutathione production and causing hypersensitivity to reactive oxygen species specifically in TP53 missense mutants [74]. MQ reacts with the central cysteine in glutathione to form a thioether, and further reduces functional glutathione to a level that compromises cancer cell survival (Figure 4). Consistent with this mode of action for PRIMA-1/APR-246, decreasing SLC7A11 expression in TP53-NULL cells sensitizes them to APR-246, and expressing SLC7A11 in TP53 mutant ceils renders them more resistant [74], Furthermore, SLC7A11 levels are a superior predictor to TP53 mutation status for response to APR-246 [77]. Nevertheless, as discussed earlier, there is compelling evidence that PRIMA-1/APR-246 can function as mutant p53 reactivators. In particular, this evidence includes diminished p53R175H reactivation when the MQ acceptor sites C124 or C277 are mutated [71,72], Efficient p53 mutant reactivation may require simultaneous modification of multiple cysteines to achieve sufficient thermodynamic stabilization. A ‘two-hit’ model would likely require very high concentrations of PRIMA-1/APR-246 given the relatively inefficient alkylation yields when p53 is treated with APR-246 in vitro [72]. The high abundance in vivo of thiols such as glutathione would compete for modification by MQ and further reduce p53-MQ complex formation efficiency [74], Furthermore, the thermodynamic stabilization mechanism does not explain reactivation of the DNA contact mutant p53R273H [69]. It is thus currently unclear to what extent APR-246-mediated reactivation of p53 mutants contributes to the therapeutic efficacy. These findings highlight the importance of considering contributions from off-target activities such as induction of redox imbalance (Figure 4) to p53 corrector drug efficacy, and the intricacies to unambiguously define a direct mode of action for corrector drugs in vivo.
Figure 4. Proposed off-target mechanism for thiol-reactive p53 correctors.
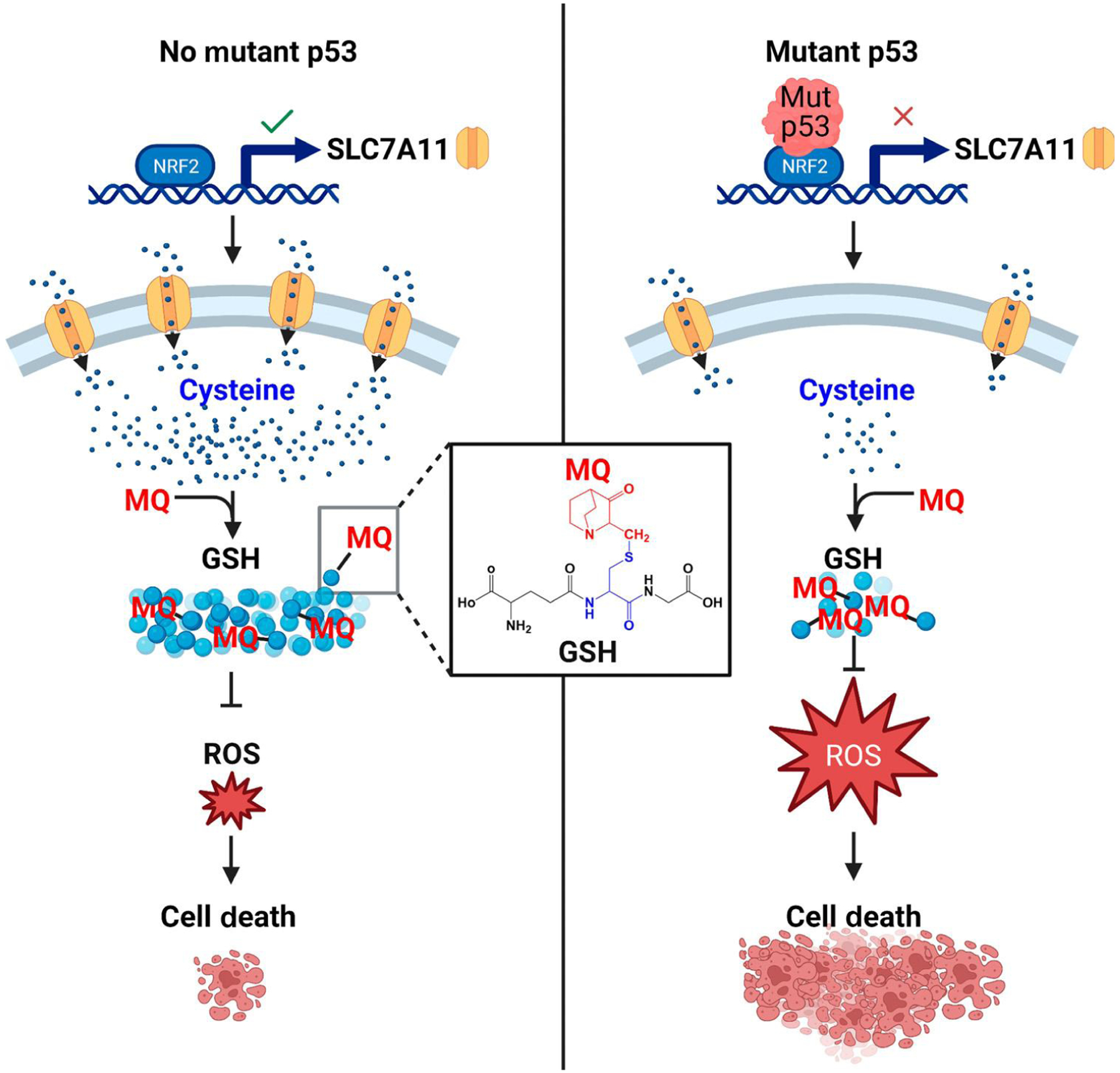
Glutathione (GSH) depletion by thiol-reactive compounds such as the PRIMA-1/APR-246 degradation product methylene quinudidinone (MQ) has been suggested as an important off-target activity that contributes to the In vivo mode of action of these compounds. Some p53 mutants gain the ability to bind and inhibit the major antioxidant transcription factor NRF2. This gain-of-function activity reduces expression of the NRF2 target SLC7A11 (a cysteine/glutamate antiporter), resulting in reduced GSH levels specifically in p53 mutants. MQ covalently reacts with the thiol in GSH and further reduces the pool of active antioxidants. Cells with p53 mutants that bind to NRF2 are already sensitized to oxidative stress and cannot tolerate the additional PRIMA-1/APR-246-mediated GSH depletion. TP53 missense mutant cell lines are thus hypersensitive to these compounds. Abbreviation: ROS, reactive oxygen species. Created with BioRender.com.
Clinical studies with APR-246 are conducted mainly as combination therapies. APR-246 has synergistic cytotoxicity when combined with the DNA hypomethylating agent azacitidine in TP53-mutant MDS and AML cancer models [78]. A Phase 1 b/2 trial evaluating combined treatment with APR-246 and azacitidine was conducted in patients with TP53-mutant MDS and AML [79], Azacitidine alone resulted in an overall response rate of 31% (complete response 24%). The combination of APR-246 and azacitidine improved the response rate to 71% (44% complete response). Notably, responding patients had significant reductions in TP53 variant allele frequency and p53 expression, with 38% achieving complete molecular remission. The treatment was well tolerated, the most frequent adverse effects being febrile neutropenia, leukopenia, and neutropenia. These promising results led to a Phase 3 trial of APR-246 plus azacitidine for the frontline treatment of patients with TP53 mutant MDS (NCT03745716).
Thiosemicarbazones (TSCs)
TSCs are metal chelators that have been explored as antivirals, antimicrobials, and cancer therapeutics [80]. They show a broad spectrum of activities due to their metal-chelating properties and generation of reactive oxygen species [80]. Two classes of TSCs – zinc metallochaperones (ZMCs) and COTI-2 – have been linked to mutant p53 reactivation. ZMC1 was identified in an elegant data-mining approach that correlated compound activity with mutant p53 status [2], ZMC1 and related compounds show selectivity for a class of conformational p53 mutants that are deficient in zinc binding, most notably the hotspot mutant p53R175H [81]. A single zinc ion is coordinated by C176, H179, C238, and C242 in the p53 DNA-binding domain and is critical to stabilize the active conformation (Figure 3). ZMC1 has a remarkably low nanomolar IC50 in cell proliferation assays and shows many hallmarks of a mutant p53 corrector. In cells treated with ZMCs, p53R175H adopts a more WT-like conformation, binds to target promotors in living cells and cell lysates, and induces p53-dependent gene expression programs [2,81,82], ZMC1 is very effective in preventing tumor progression in xenograft models, with selectivity for p53R175H tumors [2].
Thiosemicarbazones are frequently identified in screening campaigns because their metal-chelating activity is thought to inhibit a spectrum of metal-dependent biological functions. A very different mechanism is proposed for ZMCs in p53 reactivation. Here, ZMCs act as zinc ionophores to increase intracellular zinc concentrations and facilitate zinc binding to p53 mutants. ZMCs thereby compensate for zinc-binding deficiencies of some p53 mutants [81]. p53-independent mechanisms are likely contributing to antitumor activities of ZMCs. For example, a panel of patient-derived glioblastoma cell lines was growth-inhibited with picomolar IC50 independently of the TP53 status [83]. This effect was dependent on ZMC1 or other TSCs compiexing with the redox-active metal copper and generation of reactive oxygen species. No clinical trials have been reported for ZMCs, but preclinical studies demonstrated synergistic effects of ZMC1 with MDM2 and B-cell lymphoma 2 (BCL2) antagonists in ovarian cancer xenograft models [84].
COTI-2 is another TSC with anticancer activity proposed to reactivate p53 mutants. However, its corrector drug function is less well characterized than that of ZMCs [85,86]. Further studies are necessary to link COTI-2 to mutant p53 reactivation and exclude pleiotropic effects on redox balance and metal chelation as the primary mode of action. COTI-2 is currently being evaluated for ovarian cancer and head-and-neck squamous-cell cancers in Phase 2 studies (NCT02433626).
Compounds targeting the L1/S3 pocket
Reactivation of mutant p53 has been extensively investigated using intragenic compensatory mutation to study feasibility and mechanisms of the reactivation process [87–89]. A number of these single-residue reactivation mutations cluster at the L1/S3 region of p53, marking it as a potential reactivation site [71]. Molecular dynamic simulation revealed a transiently open, relatively shallow pocket in this region. This pocket was targeted in a computational docking approach using a dynamic structural model [5,71], Biochemical and cell-based assays subsequently selected the benzimidazole-derived compound series UCI-LC0023 as putative p53 mutant correctors [5]. The molecules stabilize a WT-like conformation of p53R175H and bind noncovalently to the L1/S3 pocket. Notably, this pocket expands around C124, a residue involved in APR-246 and arsenic/antimony-mediated p53 mutant reactivation [3–5] (Figures 2B and 3). UCI-LC0023 was subsequently shown to restore sequence-specific DNA binding of p53R175H in a recombinant reconstituted in vitro system, indicating a direct mode of action. Mouse models using isogenic ovarian cancer xenografts demonstrated TP53-R175H -dependent inhibition of tumor progression [5]. The UCI-LC0023 series of compounds contains a putative Michael acceptor, but its reactivity is uncertain because no covalent attachment to p53 was observed, and glutathione levels were not reduced in tumors after compound treatment [5]. These results exclude indirect effects on redox balance as proposed for the mode of action of PRIMA-1/APR-246 (Figure 4). However, other off-target effects may contribute to the observed anticancer activity of these compounds.
Concluding remarks and future perspectives
The p53 pathway is among the most attractive precision oncology targets. More than two decades have seen great efforts to develop small-molecule drugs to stabilize WT p53 in MDM2-amplified cancers and to reactivate p53 missense mutants with corrector compounds. MDM2 antagonists have been extensively evaluated in humans, but despite their potent anticancer activities in preclinical models, efficacy at tolerable dose ranges in humans was underwhelming in monotherapies. There will, however, be value in MDM2 antagonists in combination therapies with current cancer therapeutics as well as p53 mutant correctors. The latter will benefit because reactivated mutated p53 induces MDM2 expression and often also restores MDM2 binding that triggers degradation of reactivated p53 [2–5,69]. A combination therapy with MDM2 antagonists may thus significantly enhance p53 mutant correctors. A similar dual drug strategy that employs correctors and potentiators in the treatment of cystic fibrosis led to wonderful clinical outcomes [90] and may guide the way for the treatment of cancers with TP53 mutations.
Mutated p53 has long been considered undruggable, but the past few years have seen dramatic progress in the development of p53 mutant corrector compounds. Compounds targeting the cryptic crevice in p53Y220C mutants have spearheaded clinical translation and have already delivered promising results [6]. The discovery of ATO and the antimonial SSG as p53 mutant correctors led to several clinical trials that repurpose these compounds for the treatment of human cancers with TP53 missense mutations [3,4], Exciting times are ahead for p53 mutant correctors, but several outstanding questions need to be addressed (see Outstanding questions).
Outstanding questions.
What is the target specificity of mutant p53 corrector compounds?
Can DNA contact hotspot mutants be pharmacologically reactivated in the near future?
Do p53 correctors repress p53 mutant gain-of-function activities?
How quickly will resistance mechanisms for p53 mutant correctors develop?
Despite the recent progress in understanding mechanisms of mutant p53 reactivation, target specificity of p53 mutant correctors has received limited attention. This is particularly surprising because many of the compounds that have advanced to clinical trials are prone to off-target activities because they target thiols [3,4,70] or chelate metals [86]. Therefore, it will be important to evaluate their target specificity to determine the contribution of off-target activities to therapeutic efficacy.
There is strong experimental support for compound-mediated structural stabilization as a mechanism that leads to reactivation of conformational and temperature-sensitive p53 mutants [3,4,58,81]. Several compounds have also been suggested to reactivate p53 DNA contact mutants (residues R248 and R273), albeit direct biochemical evidence is missing and mechanisms are not apparent [5,69]. It is thus possible, if not likely, that indirect mechanisms contribute to the observed ‘reactivation’ phenotypes of p53 DNA contact mutants. Intragenic compensatory mutations have been described that overcome defects of DNA contact mutants [89,91,92], Some do so by introducing alternative contact sites such as S240R, but other reactivating mutations act through mechanisms that are little understood [92], Better understanding of the concepts by which DNA contact mutants can be reactivated will be important to target this class of p53 hotspot mutants.
GOF activities associated with p53 missense mutations play an active role in cancer development, and it will be interesting to test whether corrector drugs reverse GOF properties [21]. Some evidence has been observed for the L1/S3 pocket-targeting UCI-LC0023 compound, which suppressed expression of genes suggested to be associated with GOF activities [5].
Last, as with all cancer therapeutics, emergence of resistance will likely become a problem in the clinic. Dependence of tumors on the GOF activities of p53 mutants may, however, reduce the spectrum of resistance mechanisms.
The coming years will bring answers to these outstanding questions and present a particularly exciting period for p53 corrector drugs. We anxiously await the results from several clinical trials and expect further mechanistic insights into strategies to reactivate mutated proteins.
Highlights.
The tumor suppressor p53 is among the most attractive precision oncology targets. The two main small-molecule-based approaches for targeting p53 are blocking degradation of wild-type p53 and reactivating mutant p53 with corrector drugs.
MDM2 antagonists efficiently increase p53 protein levels and activate tumor suppression in preclinical settings, but clinical use may need combination therapy.
Corrector molecules act through diverse mechanisms to stabilize a wild-type-like conformation in p53 mutants. Several cysteine residues in p53 play a crucial role in the mode of action for a number of compounds.
Molecules binding to the same binding site can reactivate a different spectrum of p53 mutants.
Early clinical reports using mutant p53 corrector drugs are promising, and the recent discovery of mutant p53 reactivator activities in FDA-approved drugs has accelerated clinical translation.
Acknowledgments
We would like to thank Marcus Seldin for help with bioinformatics analyses and Karin Hick for suggestions on the manuscript. Work in the Kaiser laboratory is supported by National Institutes of Health (NIH) (R35GM148350 and R21CA267495-A1).
Glossary
- Correctors and potentiators in the treatment of cystic fibrosis
mutations in the cystic fibrosis transmembrane conductance regulator (CTFR) cause the disease cystic fibrosis. Correctors that support the active structure of mutated CTFR proteins and potentiator drugs that further stimulate activity of corrected CTFR provide an effective therapy for cystic fibrosis patients.
- Differential scanning fluorometry (DSF)
a method for measuring protein unfolding as a function of temperature.
- Gain-of-function (GOF)
refers to an activity or function that is not detected in the WT protein but is gained by the mutated protein. Note that GOF is distinct from dominant negative activities.
- Intragenic compensatory mutation
an additional amino add change in the same protein that can overcome defects caused by the primary amino add change. For example, the p53 hotspot mutant p53G245S is inactive, but the compensatory mutation N239Y that changes residue 239 from asparagine to tyrosine creates the reactivated p53G245S,N23gy double mutant.
- Neoantigen
a new protein that forms on cancer cells when certain mutations that occur in tumor DNA or splice variants are expressed. Neoantigens may play an important role in helping the body mount an immune response against cancer cells.
- Proteolysis targeting chimera (PROTAC)
a small molecule that hijacks the cellular protein degradation system to induce proteasomal degradation of a target protein. PROTACs are bifunctional molecules that bind the target protein and a ubiquitin ligase – usually von Hippd-Lindau (vHL) and cereblon (CRBN) proteins – and thereby induce ubiquitylation and degradation of the target protein by generating proximity.
- Stapled peptides
peptide-based drugs that are covalently linked by one or more hydrocarbon chains to stabilize the helical fold and increase biological stability.
- Ubiquitin ligase (E3 ubiquitin ligase)
a protein or protein complex that binds a ubiquitylation substrate and a ubiquitin-conjugating enzyme (E2) to allow covalent modification of the substrate protein with ubiquitin.
- Variant allele frequency
the percentage of sequence reads observed matching a specific DNA variant divided by the overall coverage at that locus.
Footnotes
Declaration of interests
P.K. is listed as inventor on the following patents, which describe molecules involved in p53 reactivation. Amaro, R.E. et al.: Small molecules to enhance p53 activity. US20160193214 A1. Luecke, H. et al. Small molecules for restoring function to p53 cancer mutants, US20150307519 A1. Kaiser, P. et al. Methods and compositions for treating cancer using small molecules that reactivate p53, provisional patent, UC Case 2020-611 (pending).
References
- 1.Levine AJ (2020) p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 20, 471–480 [DOI] [PubMed] [Google Scholar]
- 2.Yu X et al. (2012) Allele-specific p53 mutant reactivation. Cancer Cell 21, 614–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen S et al. (2021) Arsenic trioxide rescues structural p53 mutations through a cryptic alosteric site. Cancer Cell 39, 225–239.e8 [DOI] [PubMed] [Google Scholar]
- 4.Tang Y et al. (2022) Repurposing antiparasitb antimonials to noncovalently rescue temperature-sensitive p53 mutations. Cell Rep. 39, 110622. [DOI] [PubMed] [Google Scholar]
- 5.Durairaj G et al. (2022) Discovery of compounds that reactivate p53 mutants in vivo and in vitro. Cell Chem. Biol 29, 1381–1395.e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dumbrava EE et al. (2022) First-in-human study of PC14586, a small molecule structural corrector of Y220C mutant p53, in patients with advanced solid tumors harboring a TP53 Y220C mutation. J. Clin. Oncol 40, 300335594490 [Google Scholar]
- 7.Vogelstein B et al. (2000) Surfing the p53 network. Nature 408, 307–310 [DOI] [PubMed] [Google Scholar]
- 8.Kastenhiber ER and Lowe SW (2017) Putting p53 in context. Cell 170, 1062–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haupt Y et al. (1997) Mdm2 promotes the rapid degradation of p53. Nature 387, 296–299 [DOI] [PubMed] [Google Scholar]
- 10.Kubbutat MH et al. (1997) Regulation of p53 stability by Mdm2. Nature 387,299–303 [DOI] [PubMed] [Google Scholar]
- 11.Klein AM et al. (2021) The roles and regulation of MDM2 and MDMX: it is not just about p53. Genes Dev. 35, 575–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wade M et al. (2013) MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 13, 83–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oliner JD et al. (2016) The role of MDM2 amplification and overexpression in tumorigenesis. Cold Spring Harb. Perspect. Med 6, a026336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scheffner M et al. (1990) The E6 oncoproter encoded by human papillomavirus types 16 and 18 promotes the dagradatbn of p53. Caff 63,1129–1136 [DOI] [PubMed] [Google Scholar]
- 15.Olivier M et al. (2002) The IARC TP53 database: new online mutation analysis and recommendations to users. Hum. Mutat 19, 607–614 [DOI] [PubMed] [Google Scholar]
- 16.Joerger AC and Fersht AR (2007) Structure–function–rescue: the diverse nature of common p53 cancer mutants. Oncogene 26, 2226–2242 [DOI] [PubMed] [Google Scholar]
- 17.Aubrey BJ et al. (2018) Mutant TRP53 exerts a target gene-selective dominant-negative effect to drive tumor development. Genes Dev. 32, 1420–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu J et al. (2015) Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 525, 206–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stein Y et al. (2020) Mutant p53 oncogenicity: dominant-negative or gain-of-functbn? Carcinogenesis 41,1635–1647 [DOI] [PubMed] [Google Scholar]
- 20.Hassin O and Oren M (2022) Drugging p53 in cancer: one protein, many targets. Nat. Rev. Drug Discov 22,127–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bargonetti J and Prives C (2019) Gain-of-function mutant p53: history and speculation. J. Mol. Cell Biol 11, 605–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu H et al. (2022) Targeting p53-MDM2 interaction by small-molecule inhibitors: learning from MDM2 inhibitors in clinical trials. J. Hematol. Oncol 15,91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoe KK et al. (2014) Drugging the p53 pathway: understanding the route to clinical efficacy. Nat. Rev. Drug Discov 13, 217–236 [DOI] [PubMed] [Google Scholar]
- 24.Sabapathy K and Lane DP (2018) Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol 15,13–30 [DOI] [PubMed] [Google Scholar]
- 25.Bykov VJN et al. (2018) Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 18, 89–102 [DOI] [PubMed] [Google Scholar]
- 26.Zhang S et al. (2022) Advanced strategies for therapeutic targeting of wild-type and mutant p53 in cancer. Biomolecules 12, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gabrilovich D.l. (2006) INGN 201 (Advexin): adenoviral p53 gene therapy for cancer. Expert. Opin. Biol. Ther 6, 823–832 [DOI] [PubMed] [Google Scholar]
- 28.Atencio IA et al. (2006) Bblogical activities of a recombinant adenovirus p53 (SCH 58500) administered by hepatic arterial infusion in a Phase 1 colorectal cancer trial. Cancer Gene Ther. 13,169–181 [DOI] [PubMed] [Google Scholar]
- 29.Kong N et al. (2019) Synthetic mRNA nanoparticle-mediated restoration of p53 tumor suppressor sensitizes p53-deficient cancers to mTOR inhibition. Sci. Transl. Med 11, eaaw1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alvarado-Ortiz E et al. (2020) Mutant p53 gain-of-function: role in cancer development, progression, and therapeutic approaches. Front. Cell Dev. Biol 8,607670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Floquet C et al. (2011) Rescue of non-sense mutated p53 tumor suppressor gene by aminoglycosides. Nucleic Acids Res. 39, 3350–3362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin L et al. (2014) Identification and characterization of small molecules that inhibit nonsense-mediated RNA decay and suppress nonsense p53 mutations. Cancer Res. 74, 3104–3113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou S et al. (2021) Clinical and immunological effects of p53-targeting vaccines. Front. Cell Dev. Bbl 9, 762796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsiue EH-C et al. (2021) Targeting a neoantigen derived from a common TP53 mutation. Science 371, eabc8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vassilev LT et al. (2004) in vivo actK/ation of the p53 patlway by small-moteaie antagonists of MDM2. Science 303, 844–848 [DOI] [PubMed] [Google Scholar]
- 36.Wang S and Chen F-E (2022) Small-molecule MDM2 inhibitors in clinical trials for cancer therapy. Eur. J. Med. Chem 236, 114334. [DOI] [PubMed] [Google Scholar]
- 37.Ley TJ et al. (2013) Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engi. J. Med 368, 2059–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ray-Coquard l. et al. (2012) Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. 13, 1133–1140 [DOI] [PubMed] [Google Scholar]
- 39.Mascarenhas J et al. (2019) Oral idasanutlin in patients with polycythemia vera. Blood 134, 525–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uy GL et al. (2020) Phase 1 study of the MDM2 antagonist R06839921 in patients with acute myeloid leukemia. Investig. New Drugs 38, 1430–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun D et al. (2014) Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J. Med. Chem 57, 1454–1472 [DOI] [PubMed] [Google Scholar]
- 42.Zhang W et al. (2010) Blockade of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase and murine double minute synergistically induces apoptosis in acute myeloid leukemia via BH3-only proteins Puma and Bim. Cancer Res. 70, 2424–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Erba HP et al. (2019) Phase 1 b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv. 3,1939–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stein EM et al. (2022) Results from a first-in-human Phase I study of siremadlin (HDM201) in patients with advanced wild-type TP53 solid tumors and acute leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 28, 870–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dumbrava EE et al. (2022) A Phase 2 study of the MDM2 inhibitor milademetan in patients with TP53-wild type and MDM2-amplified advanced or metastatic solid tumors (MANTRA-2). J. Clin. Oncol 40, 7PS3165–TPS3165 [Google Scholar]
- 46.Zhang X et al. (2019) A Phase I study of a novel MDM2-P53 antagonist APG-115 in Chinese patients with advanced soft tissue sarcomas. J. Clin. Oncol 37,312431449470 [Google Scholar]
- 47.Mullard A (2020) p53 programmes plough on. Nat. Rev. Drug Discov 19, 497–500 [DOI] [PubMed] [Google Scholar]
- 48.Li Y et al. (2019) Discovery of MD-224 as a first-in-class, highly potent, and efficacious proteolysis targeting chimera murine double minute 2 degrader capable of achieving complete and durable tumor regression. J. Med. Chem 62, 448–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bernal F et al. (2010) A stapled p53 helix overcomes HDMX-mediated suppression of p53. Cancer Cell 18, 411–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang B et al. (2019) Development of selective small molecule MDM2 degraders based on nutlin. Eur. J. Med. Orem 176, 476–491 [DOI] [PubMed] [Google Scholar]
- 51.Chen S et al. (2022) Design of stapled peptide-based PROTACs for MDM2/MDMX atypical degradation and tumor suppression. Theranostics 12, 6665–6681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramos H et al. (2021) A selective p53 actK/ator and anticancer agent to improve colorectal cancer therapy. Cell Rep. 35,108982. [DOI] [PubMed] [Google Scholar]
- 53.Foster BA et al. (1999) Pharmacological rescue of mutant p53 conformation and function. Science 286, 2507–2510 [DOI] [PubMed] [Google Scholar]
- 54.Rippin TM et al. (2002) Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene 21, 2119–2129 [DOI] [PubMed] [Google Scholar]
- 55.Boeckler FM et al. (2008) Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. U. S. A 105,10360–10365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Joerger AC et al. (2006) Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl. Acad. Sci. U. S. A 103,15056–15061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bauer MR et al. (2020) Targeting cavity-creating p53 cancer mutations with small-molecule stabilizers: the Y220X paradigm. ACS Chem. Bbl 15, 657–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu X et al. (2013) Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 41, 6034–6044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baud MGJ et al. (2018) Aminobenzothiazole derivatives stabilize the thermolabile p53 cancer mutant Y220C and show anticancer activity in p53-Y220C cell lines. Eur. J. Med. Chem 152,101–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wilcken R et al. (2012) Halogen-enriched fragment libraries as leads for drug rescue of mutant p53. J. Am. Chem. Soc 134, 6810–6818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guiley KZ and Shokat KM (2023) A small molecule reacts with the p53 somatic mutant Y220C to rescue wild-type thermal stability. Cancer Discov. 13, 56–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Puzio-Kuter AM et al. (2022) Small molecule reactivators of Y220C mutant p53 modulate tumor infiltrating leukocytes and synergize with immune checkpoint inhibitors. Cancer Res. 82, 1295 [Google Scholar]
- 63.Cho Y et al. (1994) Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science 265, 346–355 [DOI] [PubMed] [Google Scholar]
- 64.Khoo KH et al. (2009) Adaptive evolution of p53 thermodynamic stability. J. Mol. Bbl 393,161–175 [DOI] [PubMed] [Google Scholar]
- 65.Aiken CT et al. (2011) Oxidative stress-mediated regulation of proteasome complexes. Mol. Cell. Proteomics 10, R110 006924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shiraishi K et al. (2004) Isolation of temperature-sensitive p53 mutations from a comprehensive missense mutation library. J. Bbl. Chem 279, 348–355 [DOI] [PubMed] [Google Scholar]
- 67.de Thé H et al. (2017) Acute promyelocytic leukemia: a paradigm for oncoprotein-targeted cure. Cancer Cell 32, 552–560 [DOI] [PubMed] [Google Scholar]
- 68.Berman JD (1988) Chemotherapy for leishmaniasis: biochemical mechanisms, clinical efficacy, and future strategies. Rev. Infect. Dis 10, 560–586 [DOI] [PubMed] [Google Scholar]
- 69.Bykov VJ et al. (2002) Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 8, 282–288 [DOI] [PubMed] [Google Scholar]
- 70.Lambert JMR et al. (2009) PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 15, 376–388 [DOI] [PubMed] [Google Scholar]
- 71.Wassman CD et al. (2013) Computational identificatton of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun 4,1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang Q et al. (2018) APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 9,439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Degtjarik O et al. (2021) Structural basis of reactivation of oncogenic p53 mutants by a small molecule: methylene quinuclidinone (MQ). Nat. Commun 12, 7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu DS et al. (2017) Inhibiting the system Xc/glutathbne axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun 8,14844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Peng X et al. (2013) APR-246/PRIMA-1 MET inhibits thiore-doxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 4, e881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ceder S et al. (2021) A thiol-bound drug reservoir enhances APR-246-induced mutant p53 tumor cell death. EMBO Mol. Med 13, e10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fujihara KM et al. (2021) SLC7A11 is a superior determinant of APR-246 (Eprenetapopt) response than TP53 mutation status. Mol. Cancer Ther 20,1858–1867 [DOI] [PubMed] [Google Scholar]
- 78.Maslah N et al. (2020) Synergistic effects of PRIMA-1 (Met) (APR-246) and 5-azacitidine in TP53-mutated myebdysplastic syndromes and acute myeloid leukemia. Haematobgca 105, 1539–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sallman DA et al. (2021) Eprenetapopt (APR-246) and azacitidine in TP53-mutant myebdysplastic syndromes. J. Clin. Oncol 39, 1584–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shakya B and Yadav PN (2020) Thiosemicarbazones as potent anticancer agents and their modes of actbn. Mini Rev. Med. Chem 20, 638–661 [DOI] [PubMed] [Google Scholar]
- 81.Blanden AR et al. (2020) Zinc shapes the folding landscape of p53 and establishes a pathway for reactivating structurally diverse cancer mutants. Bite 9, e61487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yu X et al. (2014) Small molecule restoration of widtype structure and function of mutant p53 using a novel zinc-met allochaperone based mechanism. Oncotarget 5, 8879–8892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shimada K et al. (2018) Copper-binding small molecule induces oxidative stress and cell-cycle arrest in glioblastoma-patient-derived cells. Cell Chem. Biol 25, 585–594.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zaman S et al. (2019) Combinatorial therapy of zinc metallochaperones with mutant p53 reactivation and diminished copper binding. Mol. Cancer Ther 18,1355–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lindemann A et al. (2019) COTI-2, a novel thiosemicarbazone derivative, exhibits antitumor activity in HNSCC through p53-dependent and -independent mechanisms. Clin. Cancer Res 25, clincanres.0096.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Salim KY et al. (2016) COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget 7, 41363–41379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nikolova PV et al. (2000) Mechanism of rescue of common p53 cancer mutations by second-site suppressor mutations. EMBO J. 19, 370–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brachmann RK et al. (1998) Genetb selection of intragenic suppressor mutations that reverse the effect of common p53 cancer mutations. EMBO J. 17,1847–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Baronio R et al. (2010) All-codon scanning identifies p53 cancer rescue mutations. Nucleic Acids Res. 38, 7079–7088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lopes-Pacheco M et al. (2021) Discovery of CFTR modulators for the treatment of cystic fibrosis. Expert Opin. Drug Discov 16,897–913 [DOI] [PubMed] [Google Scholar]
- 91.Baroni TE et al. (2004) A global suppressor motif for p53 cancer mutants. Proc. Natl. Acad. Sci. U. S. A 101, 4930–4935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Eldar A et al. (2013) Structural studies of p53 inactivation by DNA-contact mutations and its rescue by suppressor mutations via alternative protein-DNA interactions. Nucleic Acids Res. 41,8748–8759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Howley PM (2006) Warts, cancer and ubiquitylation: lessons from the papillomaviruses. Trans. Am. Clin. Climatol. Assoc 117,113–117 [PMC free article] [PubMed] [Google Scholar]
- 94.Sun X et al. (2022) BRD8 maintains glioblastoma by epigenetic reprogramming of the p53 network. Nature 613,195–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hiraki M et al. (2015) Small-molecule reactivation of mutant p53 to wild-type-like p53 through the p53-Hsp40 regulatory axis. Chem. Biol 22, 1206–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Synnott NC et al. (2020) COTI-2 reactivates mutant p53 and inhibits growth of triple-negative breast cancer cells. Breast Cancer Res. Treat 179, 47–56 [DOI] [PubMed] [Google Scholar]
- 97.Bykov VJ et al. (2005) Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J. Biol. Chem 280, 30384–30391 [DOI] [PubMed] [Google Scholar]
- 98.Weinmann L et al. (2008) A novel p53 rescue compound induces p53-dependent growth arrest and sensitises glioma cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ. 15, 718–729 [DOI] [PubMed] [Google Scholar]
- 99.Aggarwal M et al. (2016) Reactivation of mutant p53 by a dietary-related compound phenethyl isothiocyanate inhibits tumor growth. Cell Death Differ. 23,1615–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bauer MR et al. (2016) 2-Sulfonylpyrimidines: mild alkylating agents with anticancer activity toward p53-compromised cells. Proc. Natl. Acad. Sci. U. S. A 113, E5271–E5280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bauer MR et al. (2019) A structure-guided molecular chaperone approach for restoring the transcriptional activity of the p53 cancer mutant Y220C. Future Med. Orem 11, 2491–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gomes AS et al. (2020) SLMP53–1 interacts with wild-type and mutant p53 DNA-binding domain and reactivates multiple hotspot mutations. Biochim. Biophys. Acta Gen. Subj 1864, 129440. [DOI] [PubMed] [Google Scholar]
- 103.Zache N et al. (2008) Mutant p53 targeting by the low molecular weight compound STIMA-1. Mol. Oncol 2, 70–80 [DOI] [PMC free article] [PubMed] [Google Scholar]