

Keywords: pancreas, alcohol, autophagy, ATG4B, caspase
Abstract
Excessive alcohol intake is a major risk factor for pancreatitis, sensitizing the exocrine pancreas to stressors by mechanisms that remain obscure. Impaired autophagy drives nonalcoholic pancreatitis, but the effects of ethanol (EtOH) and alcoholic pancreatitis on autophagy are poorly understood. Here, we find that ethanol reduces autophagosome formation in pancreatic acinar cells, both in a mouse model of alcoholic pancreatitis induced by a combination of EtOH diet and cerulein (a CCK ortholog) and in EtOH+CCK-treated acinar cells (ex vivo model). Ethanol treatments decreased pancreatic level of LC3-II, a key mediator of autophagosome formation. This was caused by ethanol-induced upregulation of ATG4B, a cysteine protease that, cell dependently, regulates the balance between cytosolic LC3-I and membrane-bound LC3-II. We show that ATG4B negatively regulates LC3-II in acinar cells subjected to EtOH treatments. Ethanol raised ATG4B level by inhibiting its degradation, enhanced ATG4B enzymatic activity, and strengthened its interaction with LC3-II. We also found an increase in ATG4B and impaired autophagy in a dissimilar, nonsecretagogue model of alcoholic pancreatitis induced by EtOH plus palmitoleic acid. Adenoviral ATG4B overexpression in acinar cells greatly reduced LC3-II and inhibited autophagy. Furthermore, it aggravated trypsinogen activation and necrosis, mimicking key responses of ex vivo alcoholic pancreatitis. Conversely, shRNA Atg4B knockdown enhanced autophagosome formation and alleviated ethanol-induced acinar cell damage. The results reveal a novel mechanism, whereby ethanol inhibits autophagosome formation and thus sensitizes pancreatitis, and a key role of ATG4B in ethanol’s effects on autophagy. Enhancing pancreatic autophagy, particularly by downregulating ATG4B, could be beneficial in mitigating the severity of alcoholic pancreatitis.
NEW & NOTEWORTHY Ethanol sensitizes mice and humans to pancreatitis, but the underlying mechanisms remain obscure. Autophagy is important for maintaining pancreatic acinar cell homeostasis, and its impairment drives pancreatitis. This study reveals a novel mechanism, whereby ethanol inhibits autophagosome formation through upregulating ATG4B, a key cysteine protease. ATG4B upregulation inhibits autophagy in acinar cells and aggravates pathological responses of experimental alcoholic pancreatitis. Enhancing pancreatic autophagy, particularly by down-regulating ATG4B, could be beneficial for treatment of alcoholic pancreatitis.
INTRODUCTION
Pancreatitis is a potentially fatal inflammatory disease of the exocrine pancreas, the pathogenesis of which remains obscure and for which no specific or efficient treatment is available (1, 2). The disease is believed to initiate in injured acinar cells, the main exocrine pancreas cell type. Acute pancreatitis (AP) responses include inappropriate/intrapancreatic trypsinogen activation (its conversion to trypsin), acinar cell vacuolization and death, elevated serum levels of digestive enzymes, and inflammation. Because of limited access to human tissue, the knowledge of the pathogenic mechanism of pancreatitis comes from experimental models, both animal and ex vivo, on isolated acinar cells subjected to pancreatitis stressors (3, 4). Excessive alcohol intake is a common etiology of pancreatitis; however, the risk of developing clinically apparent pancreatic disease is only 10–15% even in heavy drinkers (5, 6). Furthermore, feeding EtOH diet to rodents by itself does not lead to significant pancreas damage (5, 7, 8). Thus, it is believed that ethanol sensitizes exocrine pancreas to the action of other pancreatitis stressors in animal (or ex vivo) models (5, 8). For example, mice or rats treated with supramaximal doses of cholecystokinin-8 (CCK) or its ortholog cerulein (CER) develop pancreatitis—a widely used model of nonalcoholic AP that recapitulates pathological responses of human disease (3). Ethanol feeding sensitizes rodents to the damaging effects of CCK or CER by decreasing the threshold (i.e., CCK/CER dose) for pancreas injury (7, 9). The mechanisms whereby ethanol sensitizes to pancreatitis are poorly understood (5–9).
Macroautophagy/autophagy is the principal catabolic process by which cells eliminate damaged or defective cytoplasmic organelles, long-lived proteins and lipids, and recycle their constituents for energy and biogenesis needs (10–13). Autophagy dysregulation is associated with a variety of diseases (10, 13). Cellular material destined for degradation is sequestered in double-membraned vacuoles called autophagosomes that ultimately fuse with lysosomes, producing single-membraned autolysosomes in which cargo is degraded by lysosomal hydrolases. Autophagosome formation is a multistep process controlled by sequentially recruited complexes of evolutionary conserved ATG (autophagy-related) proteins (11). For example, ATG6/Beclin1 is involved in nucleation/initiation of the autophagosomal membrane, ATG5 and ATG7 mediate its elongation, whereas the ubiquitin-like ATG8 is necessary for its closure. LC3 (microtubule-associated protein-1 light chain 3), the mammalian paralog of ATG8, is present in cytosol as soluble LC3-I protein, which is lipidated (phosphatidylethanolamine conjugated) to become the membrane-bound LC3-II form. This conversion is a critical step in autophagosome closure. LC3 is also involved in autophagy of specific cargo (11, 13), for example, the sequestration of ubiquitinylated protein aggregates (“aggrephagy”) which is mediated through LC3-II interaction with the protein p62/SQSTM1 (sequestosome 1) that serves as an LC3 receptor in this pathway. Because LC3-II localizes almost exclusively to autophagosomal membranes, measurements of LC3-II level (by immunoblot) or LC3-positive vesicular structures (“puncta”; by immunofluorescence) are commonly applied to assess changes in the number of autophagic vacuoles (13–17).
The balance between LC3-I and LC3-II is tightly regulated by the cysteine protease ATG4B which exerts both positive and negative control of LC3-II level (18–21). ATG4B cleaves LC3 precursor to LC3-I, increasing the amount of LC3-I available for conjugation with phosphatidylethanolamine to form LC3-II; on the other hand, ATG4B deconjugates (delipidates) LC3-II, converting it back to LC3-I. The outcome of these opposite effects is cell- and context-dependent (18). Our recent studies, using GFP-LC3 transgenic mice, showed that ATG4B negatively regulates acinar cell LC3-II level and autophagosome formation in nonalcoholic pancreatitis (16, 17).
Autophagy is essential for maintaining pancreatic acinar cell homeostasis, and its dysregulation is a characteristic feature of nonalcoholic pancreatitis (12, 14–17, 22–25). Specifically, in nonalcoholic pancreatitis, autophagosome formation is stimulated but cargo degradation in autolysosomes is inhibited, resulting in reduced autophagic flux and accumulation in acinar cells of abnormally large vacuoles containing poorly degraded cargo (12, 14–16, 22–24). In fact, this phenomenon has been a long-noted, but poorly understood, feature of human disease (14, 24, 26). Genetic alterations that specifically block autophagy in pancreas, such as ablation of ATG5 or ATG7, cause spontaneous pancreatitis in mice, implicating impaired autophagy in disease initiation and development (25, 27–29).
Much less is known about changes in autophagy in models of alcoholic pancreatitis (8, 24, 25). Studies showed pathological alterations in acinar cell lysosomal functions (8, 15, 23, 24, 30, 31); however, the impact of ethanol itself and alcoholic pancreatitis on autophagosome formation in pancreas is poorly understood. The effects of ethanol on acinar cell LC3 and ATG4B, and the role of ATG4B in alcoholic pancreatitis have not been explored.
Here, we find that, in contrast to the effect of nonalcoholic pancreatitis, ethanol inhibits pancreatic autophagosome formation and this inhibition sensitizes mice to acinar cell damage in EtOH+CER/CCK models of alcoholic pancreatitis. The underlying mechanism involves ethanol-induced upregulation of ATG4B, resulting in LC3-II decrease (contrasting ATG4B downregulation and LC3-II increase in nonalcoholic AP models). We also observed ATG4B increase and impaired pancreatic autophagy in a dissimilar, nonsecretagogue model of alcoholic pancreatitis induced by EtOH plus palmitoleic acid. ATG4B knockdown in acinar cells restored autophagosome formation and prevented pathological responses of ex vivo alcoholic pancreatitis, whereas ATG4B overexpression inhibited autophagosome formation and exacerbated acinar cell injury. The results indicate a major role for ATG4B in regulating pancreatic autophagy and pathological responses of alcoholic pancreatitis.
EXPERIMENTAL PROCEDURES
Mouse Models of Alcoholic and Nonalcoholic Pancreatitis
In the EtOH+CER model, C57BL/6N (No. 044, Envigo, CA) mice were pair-fed for 6 wk Lieber-DeCarli ethanol-containing or control isocaloric diet, followed by 7 hourly intraperitoneal injections of 5 μg/kg CER or physiological saline; mice were euthanized 1 h after the last injection (9, 15, 16). Thus, animals in the control group received control diet and saline injections. In the second model of alcoholic AP, termed EtOH/POA, mice received 2 hourly intraperitoneal injections of 1.35 g/kg ethanol plus palmitoleic acid (POA; 150 mg/kg) and were euthanized 24 h after the first injection. This combination is administered to sustain a level of the nonoxidative alcohol metabolite, palmitoleic acid ethyl ester, sufficient to cause pancreas toxicity; palmitoleic acid alone does not cause pancreatitis in the dose applied (32, 33). Nonalcoholic CER-AP was induced in male mice by 7 hourly intraperitoneal injections of 50 μg/kg (“high-dose”) CER, and pancreata were harvested 1 h after the last injection (15–17, 22, 34). The development of pancreatitis was confirmed by histopathological changes and increased intrapancreatic trypsin activity.
Isolation of Mouse and Human Pancreatic Acinar Cells
Mouse pancreatic acinar cells were isolated by collagenase digestion as described (9, 15, 22, 34). Human acinar cells were isolated, as described in detail Lugea et al. (35), from deidentified human pancreatic tissues devoid of islets of Langerhans, which were generated as a by-product of isolating islets for clinical transplantation from cadaveric donor pancreata at the Beckman Research Institute of City of Hope (Duarte, CA). Briefly, islet and acinar cells were separated by centrifugation following pancreas digestion; the acinar fractions were washed in Dulbecco’s modified Eagle’s medium (DMEM; No. 30–2002, ATCC) containing 0.1% BSA (No. 10842-662, VWR) and 100 μg/mL soybean trypsin inhibitor (No. LS003571, Worthington, NJ) and further purified by gravity sedimentation through DMEM containing 2% BSA. The resultant acinar cell pellet was resuspended in 199 medium (No. 12340030, Thermo Fisher Scientific) containing 100 μg/mL soybean trypsin inhibitor and used in experiments (35).
Ex Vivo Models of Alcoholic AP
Mouse and human pancreatic acinar cells were incubated for 4 h with 25–100 mM EtOH and treated, where indicated, for the last 30 min of incubation with 100 pM CCK (mouse cells) or 10 µM carbachol (human cells), or vehicle.
Adenoviral Transduction
Freshly isolated mouse acinr cells were transduced with adenoviral vectors (4 × 1010 pfu/mL) bearing recombinant GFP-mATG4B shRNA (Vector Biolabs, shADV-253222) or “scrambled” (control) GFP-shRNA (Vector Biolabs, 1122) followed by 38–40h incubation at 37°C; or with recombinant adenovirus overexpressing mouse ATG4B. To produce thelatter, we used the plasmid encoding mouse ATG4B-mCherry (Ex-Mm21901-M55, GeneCopoeia, Rockville, MD). The gene was subcloned into the pShuttle-CMV vector and transferred into the pAdEasy adenoviral vector, followed by vector purification and concentration using AdEasy virus purification kit (all from Agilent Technologies). The ATG4B-mCherry or control (mock) mCherry adenoviruses were added at 2.5–5 × 1011 pfu/mL titer to the culture medium followed by 16-h incubation with acinar cells at 37°C. In these prolonged-culture experiments (14), acinar cells were cultured on collagen IV in DMEM medium containing 10% FBS (No. 30–2020, ATCC, VA), 5 ng/mL EGF (No. NEX160, Perkin Elmer, Boston, MA), 200 μg/mL soybean trypsin inhibitor, 100 U/mL penicillin, and 100 µg/mL streptomycin (No. AA-40, Omega Scientific). The efficiency of ATG4B protein knockdown or overexpression was assessed by IB analysis with anti-ATG4B antibody.
Immunoblot Analysis
Frozen tissue or cells were homogenized on ice in radioimmunoprecipitation assay buffer supplemented with 1 mM phenylmethylsulfonyl fluoride and protease inhibitors cocktail (No. 11836153001, Roche). Supernatants were collected and stored at −80°C. Protein concentration in the supernatants was determined by Bradford assay (No. 5000006, Bio-Rad, CA). Proteins in pancreatic tissue and cell lysates were separated by SDS-PAGE and subjected to immunoblot (IB) analysis as described (9, 14, 15, 22, 34, 35). Blots were developed for visualization using enhanced chemiluminescence detection kit (No. 32106, Thermo Fisher Scientific); band intensities were quantified by densitometry using the FluorChem HD2 imaging system (Alpha Innotech/ProteinSimple).
Immunoprecipitation
LC3 was immunoprecipitated from pancreatic tissue lysate using same total protein input (500 μg). The immunoprecipitates were collected using the Pierce Immunoprecipitation kit (No. 26149, Thermo Fisher Scientific) according to the manufacturer’s protocol. LC3 and ATG4B levels in the immunoprecipitates were measured by IB.
Fluorescence and Light Microscopy
Formalin-fixed paraffin-embedded pancreatic tissue sections were analyzed by immunofluorescence (IF) or immunohistochemistry (IHC). IF images were acquired with a Zeiss LSM710 confocal microscope using ×63 objective. Nuclei were counterstained with DAPI. Differential interference contrast (DIC) microscopy was used to display zymogen granules area in acinar cells (15, 22). The signal was detected using secondary antibodies conjugated with Alexa Fluor 488 (green; A-21206, Thermo Fisher Scientific) or Alexa Fluor 633 (red; A-21070, Thermo Fisher Scientific). To measure proteins’ co-localization, IF images were analyzed with ImageJ or Volocity (Perkin-Elmer) software. Light microscopy images were acquired with a Nikon Eclipse TE2000-S microscope equipped with a CCD camera, using the SPOT imaging software. Quantification of IHC images was performed as described (15, 22).
qPCR
Total RNA was extracted from mouse pancreatic acinar cells using RNeasy Plus (No. 74134, Qiagen, CA) and converted to cDNA using iScript Reverse Transcription Supermix kit (No. 1708840, Bio-Rad, CA). Quantitative PCR was done using SYBR Green in a 7500 Fast Real-Time PCR system (Applied Biosystems). qPCR data were normalized to those for the reference Rplp0/36B4 gene encoding mouse acidic ribosomal protein P0 (ARP) or 18S ribosomal RNA (18S). The primers (5′ to 3′) used were as follows: Atg4B-F (forward), TATGATACTCTCCGGTTTGCTGA; Atg4B-R (reverse), GTTCCCCCAATAGCTGGAAAG; ARP-F, CGTCCTGGCATTGTCTGTGG; ARP-R, CATCTGATTCCTCCGACTCTTCC; 18S-F, CGCCGCTAGAGGTGAAATTCT; 18S-R, CATTCTTGGCAAATGCTTTCG.
ATG4B Activity
ATG4B activity was measured in pancreatic tissue lysates by using an active site-directed probe composed of recombinant LC3 protein conjugated to vinyl sulfone (LC3-VS; No. UL-451, Boston Biochem, MA). The probe interacts with a cysteine residue in the catalytic center of ATG4B active form, resulting in the formation of a stable covalent complex (36). The reaction was initiated by the addition of 1 µM LC3-VS to tissue lysate and stopped after 1-h incubation at 37°C by the addition of 4× SDS sample buffer (No. 12340030, Thermo Fisher Scientific). Complex formation between LC3-VS and endogenous ATG4B was manifest by the appearance of ∼65-kDa protein adduct, detected by immunoblot with anti-ATG4B antibody. In control experiments, complex formation between recombinant human ATG4B (No. E-400, Boston Biochem, MA) and LC3-VS was measured in the same way. Formation of the 65-kDa adduct was completely abolished by 30 min preincubation on ice of the reaction mixture with N-ethylmaleimide (NEM; 10 mM), which prevents S-S bonding and thus abolishes ATG4B interaction with LC3-VS (see Fig. 4).
Protease Activities
Trypsin and caspase-3-like (DEVDase) activities were measured in tissue or cell lysates as described (14, 15, 22, 34, 37), by fluorogenic assays using specific substrates Boc-Gln-Ala-Arg-AMC (No. 4017019, Bachem, CA) and Ac-DEVD-AMC (No. 14986, Cayman Chemical, MI), respectively.
Acinar Cell Necrosis
Acinar cell necrosis was determined by the release of the cytosolic enzyme glucose-6-phosphate dehydrogenase (G6PD) into the incubation medium using Vybrant Cytotoxicity Assay kit (No. V-23111, Invitrogen, CA). Cell necrosis was quantified as a ratio of G6PD released into the incubation medium to the total amount of G6PD.
Antibodies and Other Reagents
Antibodies against LC3 (No. 2775), p62/SQSTM1(No. 5114), ATG4B (No. 13507), ATG5) (No. 260), ATG7 (No. 8558), Beclin1 (No. 3459), and p44/42 MAP kinase (ERK1/2) (No. 9102) were from Cell Signaling Technology, MA; against LDH (No. sc-33781), from Santa Cruz Biotechnology, CA. Cholecystokinin-8 (CCK; No. 471-47) was from Echelon Bioscience, UT; cerulein (CER; No. 4030451) from Bachem, CA; other reagents were from Sigma-Aldrich.
Statistical Analysis
Statistical analysis of the results was performed with Prism 9 (GraphPad software) using a two-tailed Student’s t test for comparison between two groups and one-way ANOVA with Tukey’s post hoc test for comparison between multiple groups. Values are expressed as means ± SE; a P value of less than 0.05 was considered significant.
Study Approval
All experimental protocols were approved by the animal research committee of Veterans Affairs Greater Los Angeles Healthcare System following the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Experimental protocols using human acinar cells (detailed in Ref. 35) were approved by the institutional review boards of the Veterans Affairs Greater Los Angeles Healthcare System and the Beckman Research Institute of City of Hope.
RESULTS
Ethanol Treatments Inhibit Autophagosome Formation in Exocrine Pancreas
To examine ethanol’s effects on autophagy, we primarily used the EtOH+CER mouse model of experimental alcoholic AP induced by a combination of ethanol-containing Lieber-DeCarli diet and intraperitoneal injections of a “low-dose” (3–5 µg/kg) CER (9, 14, 16). A hallmark of secretagogue-induced digestive enzyme secretion from pancreatic acinar cells is its biphasic dose dependence curve: secretion is stimulated by CCK or CER until it reaches a maximal level (at 10–30 pM CCK in the mouse); secretion then diminishes with a further increase in CCK/CER concentration (38). Administration of supramaximal (i.e., above maximally stimulating) doses of CCK/CER to rodents causes AP; in particular, pancreatitis induced in mice or rats by intraperitoneal injections of “high-dose” (50–100 µg/kg) CER is the most widely used and best-studied model of nonalcoholic AP (3, 14, 15, 22, 24, 34). Ethanol diet sensitizes mice or rats to CER-induced pancreas damage so that low doses of CER that do not cause pancreas damage in animals fed control diet elicit AP responses in animals consuming alcohol (7, 9). Figure 1, A and B shows that the combination of EtOH diet and 5 µg/kg CER caused characteristic histopathological changes in the pancreas and an approximately fivefold increase in intrapancreatic trypsin activity, a signature response of AP. Neither treatment alone caused a significant increase in intrapancreatic trypsin activity (Fig. 1A).
Figure 1.

LC3-II level and the number of autophagic vacuoles in pancreas are reduced in mouse models of alcoholic pancreatitis, compared with no-alcohol conditions. A–E, G–K: in EtOH+CER model of alcoholic pancreatitis, mice were fed Lieber-DeCarli ethanol-containing or isocaloric control diet for 6 wk followed by 7 hourly intraperitoneal injections of 5 µg/kg (low-dose) cerulein (CER) or saline. In the EtOH/POA model (F), mice received 2 hourly intraperitoneal injections of 1.35 g/kg ethanol plus palmitoleic acid (POA; 150 mg/kg). The nonalcoholic CER-AP (K) was induced by 7 hourly intraperitoneal injections of 50 µg/kg (high-dose) CER in mice on standard chow. Details on the animal models are given in experimental procedures. A: trypsin activity in pancreas was measured with a fluorogenic enzymatic assay. B: histopathological changes in pancreas were assessed by H&E staining; scale bars, 10 µm. C and F: IB analysis of pancreatic LC3 and p62/SQSTM1 levels. In this and other figures, ERK1/2 is the loading control; each lane represents an individual animal; a narrow white space indicates that the lanes are on the same gel but not contiguous. D and E: densitometric IB band intensities for LC3-II and p62 in the EtOH+CER model were normalized to that of ERK in the same sample, and the mean ratios further normalized to that in control (i.e., no EtOH, no CER) group. G–J: IF analysis of LC3 (green) and p62 (red) in pancreas. Nuclei were stained with DAPI (blue); differential interference contrast (DIC) microscopy was used to display zymogen granules area in acinar cells. Scale bars, 10 µm. The numbers of LC3- and p62-positive puncta were expressed per number of nuclei (DAPI) in the field and further normalized to those in control group. Colocalization of p62 with LC3 was analyzed using Manders-Costes coefficient (Volocity image analysis software). At least 700 acinar cells were analyzed for each animal. K: electron micrographs of pancreas of mice from indicated AP models. Scale bars, 2 µm. Values are means ± SE. In A, D, and E, each symbol represents an individual mouse (n = 3–7 mice/condition). In H–J, each symbol corresponds to 20–30 cells in a different field (n = 4–9 fields from 3 mice/condition). **P < 0.01; ***P < 0.001. Significance was determined by one-way ANOVA followed by Tukey’s multiple comparison test. DAPI, 4′,6-diamidino-2-phenylindole; ERK, extracellular signal-regulated kinase; EtOH, ethanol; IB, immunoblot; LC3, microtubule-associated protein-1 light chain 3.
We found that EtOH diet and the low-dose CER alone had opposite effects on autophagic vacuole formation (Fig. 1, C–E): ethanol reduced, whereas 5 µg/kg CER increased the LC3-II protein level in pancreas. Furthermore, the low-dose CER caused a much lesser increase in LC3-II in ethanol-fed mice than in mice fed control diet (Fig. 1, C and D). Similarly, the number of LC3-positive puncta in pancreas, measured with IF, was approximately two times less in mice with EtOH+CER pancreatitis than in CER-treated mice on control diet (Fig. 1, G and H).
Autophagic efficiency can be assessed by measuring the level of p62, a protein that is required for aggrephagy and, on the other hand, is specifically degraded through autophagy (11, 13). Stimulation of autophagic activity enhances p62 degradation, resulting in its decreased level, whereas impaired/inefficient autophagy manifests itself by p62 accumulation. Both IB (Fig. 1, C and E) and IF (Fig. 1, G and I) analyses showed that the combination of EtOH and CER increased the pancreatic p62 level five- to sevenfold—much greater than with the low-dose CER alone (i.e., in mice on control diet); further indicating that ethanol diet “sensitized” pancreas to CER-induced autophagy impairment.
Of note, we observed similar effects of alcoholic pancreatitis on LC3 and p62 in a different, nonsecretagogue model induced by EtOH plus palmitoleic acid (Fig. 1F). The concomitant increase in both proteins indicates impairment of pancreatic autophagy in this model as well. Interestingly, EtOH/POA pancreatitis predominantly increased the cytosolic LC3-I level in the pancreas (Fig. 1F).
Although the number of p62-positive puncta increased several-fold in pancreas of mice with EtOH+CER pancreatitis (Fig. 1I), p62 colocalization with LC3-II decreased almost to baseline, contrasting the ∼2.5-fold increase with CER alone (Fig. 1J). The data suggest accumulation of p62-decorated protein aggregates, which are not sequestered by autophagosomes and thus are not degraded through autophagy. One reason for this could be decreased formation of autophagosomes with ethanol treatments. Decreased autophagosome formation could also explain the paucity of large autophagic vacuoles on electron micrographs from pancreas of EtOH+CER-treated mice, which are so prominent in the nonalcoholic CER-AP (14–16) (Fig. 1K). As stated in the INTRODUCTION, accumulation of such vacuoles in acinar cells is a hallmark of both human disease and experimental models of nonalcoholic AP (14–16, 22–24, 26).
To gain further insight into the mechanisms whereby ethanol affects autophagy in pancreas, we analyzed the effect of ethanol alone and ex vivo models of alcoholic pancreatitis on LC3-II levels in mouse and human acinar cells (Fig. 2). Like animal models, the ex vivo models are based on ethanol’s ability to sensitize acinar cells to the damaging effects of stressors (4, 5, 9). In the first model (detailed in experimental procedures), mouse acinar cells were treated with the combination of EtOH and low-dose (100 pM) CCK (Fig. 2, A–D); in the second (Fig. 2, E and F), human acinar cells were treated with the combination of EtOH and another secretagogue, carbachol (CCh; an agonist of acetylcholine receptor). We used CCh as pancreatitis stressor because the expression of CCK-A receptor in human acinar cells is very low and variable (38). Ethanol (25–100 mM) dose-dependently reduced LC3-II levels in mouse acinar cells (Fig. 2, A and B), as well as in human cells (Fig. 2, C and D). In contrast, the secretagogues CCK or CCh increased LC3-II levels in both mouse and human acinar cells (Fig. 2, C–F). Furthermore, cells subjected to combined EtOH+CCK or EtOH+CCh treatments had significantly less LC3-II than with secretagogue-only treatments (Fig. 2, C–F). Thus, effects of ethanol in ex vivo models are similar to those we found in vivo (Fig. 1).
Figure 2.

Ethanol inhibits autophagosome formation in mouse and human pancreatic acinar cells, both in basal conditions and ex vivo alcoholic pancreatitis. A–F: acinar cells isolated from mouse or human pancreas (see experimental procedures) were preincubated for 30 min with and without bafilomycin A1 (BafA1; 20 µM) and then treated with (A and B) indicated concentrations or (C–F) 100 mM EtOH for 4 h. Where indicated, 100 pM CCK or 10 µM carbachol (CCh) was added to the cells for the last 30 min of incubation. LC3 level was measured by IB. B, D, and F: densitometric quantification of LC3-II band was performed as described in Fig. 1. Values are means ± SE. Each symbol represents individual acinar cell preparation (n = 3–7 cell preparations/condition). *P < 0.05, **P < 0.01, ***P < 0.001; one-way ANOVA with Tukey’s multiple comparison test. CCK, cholecystokinin; EtOH, ethanol; IB, immunoblot; LC3, microtubule-associated protein-1 light chain 3.
Ethanol-induced decrease in LC3-II could be caused by inhibition of autophagosome formation or by an increase in autophagic degradation. To discriminate between these two possibilities, we treated cells with the vATPase inhibitor bafilomycin A1 (BafA1), which blocks lysosomal degradation. The magnitude of LC3-II increase elicited by BafA1 in a given condition (e.g., in cells without ethanol) is a measure of the efficiency of autophagic degradation (flux) in this condition, the blockade of which by BafA1 causes LC3-II increase (13). BafA1 elevated LC3-II levels in control/untreated mouse and, importantly, human acinar cells by ∼3.8- and ∼3.4-fold, respectively (Fig. 2, C–F), indicating efficient basal autophagic flux the blockade of which causes accumulation of LC3-positive autophagic vacuoles (13, 16). On the other hand, in the presence of BafA1, the changes in LC3-II level between two conditions (e.g., with and without ethanol) are due solely to the changes in autophagosome formation, as autophagic flux is blocked (13, 16). In the presence of BafA1, ethanol decreased LC3-II in both mouse (Fig. 2, C and D) and human (Fig. 2, E and F) cells, providing direct evidence for reduced autophagosome formation. The effect was especially pronounced in ex vivo pancreatitis conditions (EtOH+CCK vs. CCK alone in mouse cells or EtOH+CCh vs. CCh alone in human cells; all in the presence of BafA1) in which ethanol decreased LC3-II level almost to the baseline (∼4 times) in both models.
Ethanol Increases ATG4B Protein Level and Activity in Pancreas
To elucidate the mechanisms mediating the observed inhibition of autophagosome formation by ethanol, we measured the effects of ethanol treatments on pancreatic levels of key ATG proteins (Fig. 3). Ethanol itself and EtOH+CER pancreatitis caused no significant changes in Beclin1, ATG7, ATG5 (measured as the ATG5-ATG12 conjugate); but both treatments markedly increased the level of ATG4B in pancreas. Of note, EtOH+CER pancreatitis increased pancreatic ATG4B to the same level as ethanol diet itself, whereas the low-dose CER had no significant effect (Fig. 3, A and B). Pancreatic ATG4B level was also increased in the EtOH/POA model of alcoholic pancreatitis (Fig. 3, B and D). In accord with the IB results, IF showed the appearance of ATG4B-positive puncta colocalized with LC3 in the pancreas of mice fed ethanol diet, whereas in control pancreas, ATG4B immunostaining had mostly a diffuse cytosolic pattern (Fig. 3C). Similarly, incubation of mouse acinar cells with ethanol did not affect Beclin1, ATG7 or ATG5-12, but it dose-dependently increased ATG4B (Fig. 3, E–G). The combined EtOH+CCK treatment (ex vivo model of alcoholic AP) increased ATG4B in acinar cells to about the same level as EtOH alone (Fig. 3, F and G).
Figure 3.

Ethanol upregulates pancreatic ATG4B level. A–D: mice were fed EtOH-containing or control diet followed by intraperitoneal injections of 5 µg/kg CER (or saline); or received intraperitoneal injections of EtOH plus POA. The animal models were performed as in Fig. 1. E–G: mouse pancreatic acinar cells were incubated for 4 h with and without 100 mM (or indicated concentrations) EtOH; 100 pM CCK was added, where shown, to the cells for the last 30 min of incubation. A, B, D–G: IB analysis of autophagy-mediating proteins in pancreatic tissue or cells. C: IF analysis of LC3 (green) and ATG4B (red) in pancreas of mice fed control or EtOH-containing diet. Nuclei were stained with DAPI (blue). Scale bars, 2 µm. Values are means ± SE; each symbol represents an individual mouse or acinar cell preparation (n = 4–7/condition). *P < 0.05, **P < 0.01, ***P < 0.001; one-way ANOVA with Tukey’s multiple comparison test. CCK, cholecystokinin; CER, cerulein; DAPI, 4′,6-diamidino-2-phenylindole; EtOH, ethanol; IB, immunoblot; LC3, microtubule-associated protein-1 light chain 3; POA, palmitoleic acid.
Of note, ATG4B increases by ethanol alone and in experimental alcoholic pancreatitis contrast the decrease in pancreatic ATG4B that we observed in nonalcoholic AP induced with high-dose CER/CCK (16, 17).
We next measured the effect of ethanol on ATG4B activity (Fig. 4) by applying a probe directed toward ATG4B active site (36), namely, the recombinant LC3 protein modified with vinyl sulfone, a COOH-terminal electrophilic trap (LC3-VS). Cysteine residue in the catalytic center of the active form of ATG4B covalently binds to the LC3-VS active site-directed probe, resulting in the formation of ∼65-kDa protein adduct ATG4B:LC3-VS (36). We validated that the protein adduct formation in acinar cells was abolished by N-ethylmaleimide, an irreversible inhibitor of cysteine peptidases (Fig. 4A), indicating the complete dependence of adduct formation on the active site cysteine residue of pancreatic ATG4B. Thus, the amount of ATG4B:LC3-VS adduct directly correlates with enzymatic activity of ATG4B (36). Ethanol diet itself and EtOH+CER pancreatitis both markedly stimulated the adduct formation, indicating an increase in ATG4B enzymatic activity, whereas the low-dose CER alone had no effect (Fig. 4, B and C). Similar to their effect on ATG4B level (Fig. 3, A and B), ethanol alone and the combined EtOH+CER treatment increased ATG4B activity to about the same level (Fig. 4C).
Figure 4.

Ethanol enhances pancreatic ATG4B activity and complex formation with LC3. To assess ATG4B enzymatic activity, cytosolic fractions obtained from pancreata of untreated mice (A) and mice subjected to indicated treatments were preincubated (or not) with cysteine peptidase inhibitor NEM (10 mM) followed by incubation with LC3 vinyl sulfone (LC3-VS) active site-directed probe (B and C). The levels of ATG4B protein (∼45 kDa; numbers on the right) and its covalent adduct with LC3-VS probe (∼65 kDa) were measured by IB using anti-ATG4B antibody. Lactate dehydrogenase (LDH) served as loading control. C: ATG4B activity was assessed by densitometry of the ATG4B:LC3-VS adduct IB band. D: LC3 was immunoprecipitated from pancreas of mice subjected to indicated treatments; LC3 and ATG4B levels in the immunoprecipitates (IP) were analyzed by IB and quantified by densitometry. Numbers on the bottom show densitometric band intensities normalized to those for control (i.e., no EtOH, no CER) mice. In C, values are means ± SE from three mice per condition. ***P < 0.001; one-way ANOVA with Tukey’s multiple comparison test. CER, cerulein; EtOH, ethanol; IB, immunoblot; LC3, microtubule-associated protein-1 light chain 3; NEM, N-ethylmaleimide.
Finally, we applied coimmunoprecipitation assay, using anti-LC3 antibody, to examine whether ethanol-induced increase in ATG4B protein boosted ATG4B interaction with LC3. In all conditions, ATG4B was present in the LC3 immunoprecipitate, indicating complex formation between these proteins (Fig. 4D). With equal input (500 µg total protein), there was less LC3 in pancreas of mice fed ethanol diet (see Fig. 1, C and D) and, correspondingly, less immunoprecipitated LC3 in these samples (Fig. 4D). Similar to the effect observed in vivo (Fig. 1) and in isolated acinar cells (Fig. 2), ethanol decreased the LC3-II amount in the immunoprecipitate, compared with corresponding no-alcohol conditions: by ∼40% with EtOH diet itself and by ∼76% with EtOH+CER pancreatitis (Fig. 4D). Importantly, the amount of ATG4B coimmunoprecipitated with LC3 significantly increased with both ethanol treatments (Fig. 4D), indicating that ethanol facilitated complex formation between ATG4B and LC3-II in pancreas. There was no such increase with low-dose CER alone.
Collectively, the results in Figs. 3 and 4 indicate that ethanol treatments increased pancreatic ATG4B protein level, enzymatic activity, and complex formation with LC3-II.
Ethanol Upregulates Acinar Cell ATG4B by Inhibiting Its Proteolytic Degradation
Ethanol did not affect pancreatic ATG4B mRNA level (Fig. 5A), implying that the observed effects are not due to changes in ATG4B synthesis. Instead, we found that ethanol increased ATG4B protein level by inhibiting its proteolytic degradation. The pan-caspase inhibitor Z-VAD-fmk (zVAD) increased ATG4B level in control/untreated acinar cells and abrogated the increases in ATG4B caused by EtOH or EtOH+CCK; that is, in the presence of zVAD, there was no additional increase in ATG4B with ethanol treatments (Fig. 5, B and C). zVAD did not change ATG4B level when added into the cell lysate, indicating no direct effect on ATG4B. Using the caspase-3 substrate DEVD, we measured that CCK increased DEVDase activity, whereas ethanol treatments inhibited it (Fig. 5D). zVAD canceled the CCK-induced increase, but it did not cause an additional decrease in caspase activity in cells treated with EtOH or EtOH+CCK (Fig. 5D). The data in Fig. 5, B–D show a correlation between reduced caspase activity and increased ATG4B protein level in acinar cells. These results are supported by the data in other cell types that ATG4B is proteolytically degraded by caspases (39–41). Also, the results are in accord with our previous report indicating that ethanol reduces caspase activity in rat pancreas (37).
Figure 5.

Ethanol upregulates ATG4B through inhibiting its degradation by caspases. Mouse pancreatic acinar cells were incubated with and without 100 mM EtOH for 4 h in the presence or absence of the pan-caspase inhibitor zVAD-fmk (50 µM), proteasomal inhibitor MG-132 (20 µM), apoptosis activator Ridaifen B (RidB; 5 µM), or calpain inhibitor ALLN (50 µM). CCK (100 pM) was added to the cells for the last 30 min of incubation. A–C, E, and F: ATG4B mRNA and protein levels were analyzed by qPCR and IB. Densitometric quantification of ATG4B band (C) was done as described in Fig. 1. D and G: Caspase-3-like (DEVDase) activity was measured with a fluorogenic assay. Values are means ± SE; each symbol represents an individual acinar cell preparation (n = 3–9/condition). *P < 0.05, **P < 0.01, ***P < 0.001; one-way ANOVA with Tukey’s multiple comparison test. CCK, cholecystokinin; EtOH, ethanol; IB, immunoblot.
In contrast, calpain inhibitor ALLN did not increase ATG4B level in control/untreated acinar cells and did not prevent CCK-induced decrease in ATG4B (Fig. 5E), which would be expected if calpains mediated ATG4B degradation. Proteasomal inhibitor MG-132 also failed to increase ATG4B level (Fig. 5F), suggesting that proteasomal degradation does not mediate ATG4B decrease. In fact, both ALLN and MG-132 decreased ATG4B protein in acinar cells (Fig. 5, E and F), which can be explained by their stimulatory effect on apoptosis, as shown in other cells (42, 43). Indeed, MG-132 and ALLN increased DEVDase activity in acinar cells (Fig. 5G), indicating that both inhibitors caused ATG4B degradation (Fig. 5, E and F) through stimulating caspase activity. In support of this conclusion, the small-molecule apoptosis activator Ridaifen B (44, 45) reduced ATG4B level and stimulated DEVDase activity; both effects were abrogated by zVAD (Fig. 5, F and G).
The results in Fig. 5 indicate that acinar cell ATG4B level is negatively regulated by zVAD-sensitive caspases and that ethanol-induced increase in ATG4B is through caspase inhibition.
ATG4B shRNA Knockdown Enhances Autophagy and Ameliorates Ex Vivo EtOH+CCK Pancreatitis, Whereas ATG4B Overexpression Reduces Autophagosome Formation and Worsens Acinar Cell Damage
To further assess the role of ATG4B in regulation of pancreatic autophagy by ethanol, we measured the effects of modulating ATG4B level on LC3-II and p62 in mouse acinar cells (Fig. 6). Transduction of acinar cells with adenovirus bearing shRNA against ATG4B reduced the basal ATG4B level by ∼70% and increased LC3-II ∼3.0-fold (Fig. 6, A and B). Importantly, ATG4B knockdown abrogated the effects of EtOH, CCK, or their combination on LC3-II. That is, acinar cells transduced with ATG4B shRNA had the same elevated (by ∼3.0-fold) LC3-II level in all experimental conditions, without any effect of the treatments on LC3-II (Fig. 6 A and B). In accord with the IB data, ATG4B knockdown markedly increased the number of LC3 puncta, evident in both control and EtOH-treated cells (Fig. 6C). shRNA ATG4B knockdown had little effect on p62 in control and CCK-treated cells, but it abrogated the increases in p62 caused by EtOH or EtOH+CCK (Fig. 6A). Interestingly, the effects of EtOH itself on p62 were different between pancreatic tissue and cells: mice fed ethanol and control diet had the same level of pancreatic p62 (Fig. 1D), whereas ex vivo EtOH increased p62 in acinar cells (Fig. 6A). A putative explanation for the latter effect is that acinar cells are stressed during isolation from the pancreas (46) and therefore are more sensitive to the damaging effect of ethanol.
Figure 6.

ATG4B overexpression reduces LC3-II in acinar cells, whereas ATG4B knockdown restores autophagosome formation in EtOH+CCK ex vivo pancreatitis. Mouse pancreatic acinar cells were transduced with adenoviral vectors expressing shRNA against ATG4B (ATG4B-shRNA) or control (“scrambled”) shRNA (Ctrl-shRNA); or vectors bearing mCherry-ATG4B or mCherry alone (with no transgene). Cells were incubated for 4 h with and without 100 mM EtOH; 100 pM CCK was added to the cells, as indicated, for the last 30 min of incubation. A, B, and D: IB analysis of indicated proteins. Densitometric quantification of LC3-II band (B) was done as described in Fig. 1; values were normalized to control (i.e., untreated cells transduced with control shRNA). C, E, and F: immunostaining for LC3 and p62; nuclei stained with DAPI. Scale bars, 10 µm. p62 co-localization with LC3 (F) was quantified using Manders-Costes coefficient (Volocity software). Values are means ± SE from five to nine acinar cell preparations; each symbol represents an individual cell preparation (B) or at least 150 cells assessed for p62 colocalization with LC3 (F). *P < 0.05, ***P < 0.001; one-way ANOVA with Tukey’s multiple comparison test (B) or two-tailed Student’s t test (F). CCK, cholecystokinin; DAPI, 4′,6-diamidino-2-phenylindole; EtOH, ethanol; IB, immunoblot; LC3, microtubule-associated protein-1 light chain 3.
In the opposite direction, ATG4B overexpression with adenovirus bearing ATG4B-mCherry dramatically decreased LC3-II and increased p62 compared with control cells (Fig. 6D). In line with the IB results, cells overexpressing ATG4B showed marked decrease in LC3-positive vacuoles, increased number of p62 puncta, and approximately four times less p62 colocalization with LC3 (Fig. 6, E and F). Thus, ATG4B overexpression mimics the effects of alcoholic pancreatitis on autophagy, namely the decreased LC3-II levels, accumulation of p62, and decreased amount of p62 sequestered by autophagic vacuoles (i.e., colocalized with LC3-II).
Taken together, the results provide further evidence that ethanol-induced increase in ATG4B reduces LC3-II and thus acts to suppress autophagosome formation in acinar cells.
We next examined the effects of modulating ATG4B level on key parameters of acinar cell damage caused by EtOH, CCK, or their combination (Fig. 7). shRNA ATG4B knockdown prevented trypsinogen activation in acinar cells treated with EtOH, CCK, or EtOH+CCK (i.e., ex vivo alcoholic pancreatitis) (Fig. 7A). Similarly, the ATG4B knockdown abrogated necrosis caused by EtOH and EtOH+CCK treatments (Fig. 7B). Conversely, ATG4B overexpression potentiated acinar cell damage induced by EtOH, CCK, or their combination (Fig. 7, C and D); moreover, ATG4B overexpression itself caused some necrosis in control cells (Fig. 7D). Thus, the decrease in autophagosome formation caused by ATG4B overexpression promotes acinar cell damage.
Figure 7.

ATG4B promotes pathological responses of ex vivo alcoholic pancreatitis. Same as in Fig. 6, mouse pancreatic acinar cells were transduced with adenoviral vectors expressing shRNA against ATG4B (shATG4B) or control/“scrambled” shRNA (shCtrl) (A and B); or vectors bearing mCherry-ATG4B or mCherry alone (with no transgene) (C and D). Cells were incubated for 4 h with and without 100 mM EtOH; 100 pM CCK was added to the cells, as indicated, for the last 30 min of incubation. A and C: trypsin activity was measured in cell homogenates with a fluorogenic assay. B and D: necrosis was quantified as glucose-6-phosphate dehydrogenase (G6PD) releases into the extracellular medium. Values are means ± SE from three individual cell preparations, normalized to control (i.e., untreated cells transduced with control shRNA or “empty” mCherry adenoviruses). *P < 0.05, **P < 0.01, ***P < 0.001; one-way ANOVA with Tukey’s multiple comparison test. CCK, cholecystokinin; EtOH, ethanol.
DISCUSSION
The pathogenic mechanism of alcoholic pancreatitis, a common and sometimes fatal disease, remains obscure. Here, we examined the effects of ethanol alone and alcoholic pancreatitis on pancreatic autophagy using primarily the well-established mouse EtOH+CER and cellular/ex vivo models; the latter being induced by the combination of EtOH+CCK in mouse and EtOH+CCh in human acinar cells (5, 9, 30, 47, 48). These models are based on the finding that ethanol sensitizes exocrine pancreas to pathological effects of stressors (5, 7); the EtOH+CER model, in particular, utilizes a combination of ethanol diet and low-dose CER which does not cause pancreas damage in animals fed control diet. We also used a dissimilar, nonsecretagogue model of alcoholic AP induced by EtOH plus palmitoleic acid. This combined treatment is applied to maintain an injurious level of the nonoxidative alcohol metabolite, palmitoleic acid ethyl ester (32, 33). EtOH concentrations used in cellular models are within the range of blood alcohol levels (BAL) occurring in rodent models of ethanol-mediated pancreatitis (7, 49). They also are within the range of BALs reported in drinkers cited for driving under the influence (50), the mean and the highest BAL levels being 0.17 and 0.52%, respectively (equivalent to 40 and 112 mM ethanol).
We find that ethanol alone and alcoholic pancreatitis suppress autophagosome formation in the pancreas, and that the underlying mechanism involves upregulation of ATG4B resulting in LC3-II decrease (Fig. 8). The most direct evidence for this action of ethanol is that EtOH alone and the EtOH+CCK and EtOH+CCh treatments all markedly decreased acinar cell level of LC3-II in the presence of BafA1, which blocks autophagy downstream of autophagosome formation. Of note, ATG4B can regulate LC3-II level both positively, by cleaving LC3 pro-protein to create a pool of LC3-I available for lipidation to become LC3-II; and negatively, by deconjugating membrane-associated LC3-II back to the cytosolic LC3-I (18–20). The outcome of this dual action varies in different cells and conditions (52, 53). Our recent studies using nonalcoholic AP models in GFP-LC3 transgenic mice (16) found that ATG4B negatively regulates acinar cell LC3-II level and autophagosome formation. The present study shows similar negative regulation of pancreatic LC3-II by ATG4B in mouse and ex vivo models of alcoholic pancreatitis. This effect was particularly pronounced in the EtOH/POA model where ATG4B increase was associated with marked accumulation of the cytosolic LC3-I form.
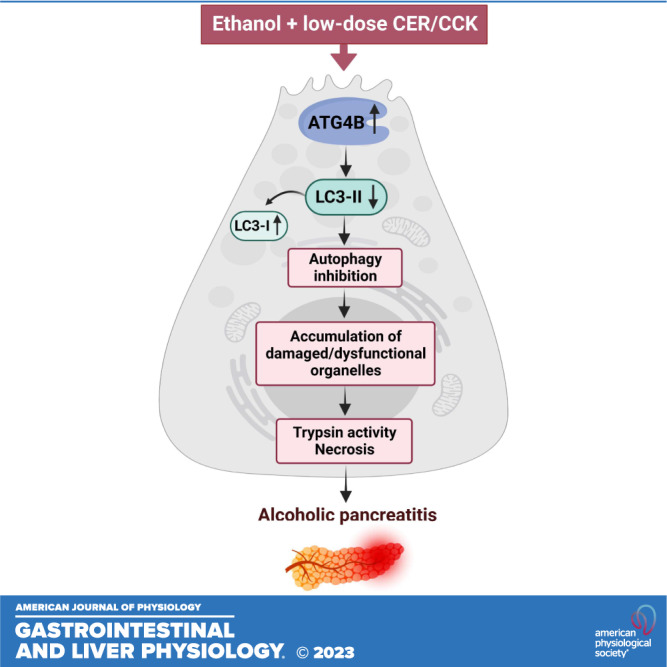
Figure 8.

Schematic illustrating the mechanisms whereby ethanol inhibits autophagy and thus sensitizes pancreas to pancreatitis induced by stressors such as supramaximal CER/CCK. Dashed line indicates ethanol’s effects not explored in this study but characterized in detail by us and other groups (5, 8, 9, 12, 14,15, 23, 33, 37, 51). CCK, cholecystokinin-8; CER, cerulein.
Of note, opposite to the effect of ethanol treatments, pancreatic ATG4B level decreases in nonalcoholic AP models and that of LC3-II increases (16, 17). Thus, there is an interesting dichotomy between the effects of nonalcoholic versus alcoholic pancreatitis on ATG4B and LC3-II levels in pancreas; the mechanisms underlying these opposing effects remain to be elucidated.
Ethanol augmented ATG4B protein level in pancreas, stimulated enzymatic activity, and facilitated its complex formation with LC3 resulting in LC3-II decrease. The mediatory role of ATG4B in ethanol-induced LC3-II decrease is further supported by the findings that ATG4B overexpression reduced LC3-II level, whereas ATG4B knockdown with shRNA elevated LC3-II and thus stimulated autophagosome formation in ethanol-treated cells. In fact, ATG4B knockdown caused concomitant increase in LC3-II and decrease in p62, indicating enhanced autophagic activity. Several lines of evidence demonstrate that ethanol upregulates pancreatic ATG4B by inhibiting its proteolytic degradation by caspases. Ethanol treatments did not change ATG4B mRNA expression in pancreas; the pan-caspase inhibitor zVAD markedly increased ATG4B protein level; proteasomal inhibitor MG-132 and calpain inhibitor ALLN did not prevent but instead caused ATG4B decrease (which is likely due to the fact that both stimulated acinar cell caspase activity). In other cells, ATG4B level is regulated via cleavage by caspases-1 and -3 (54); and the data in this and our previous study (37) indicate that ethanol inhibits caspase activity in acinar cells. The results suggest that caspases regulate pancreatic autophagy by modulating ATG4B level: caspase activation stimulates autophagosome formation (through ATG4B degradation and resultant increase in LC3-II), whereas caspase inhibition downregulates autophagy as well as apoptosis (34).
Impaired autophagy has emerged as a key pathogenic mechanism of both nonalcoholic and alcoholic experimental pancreatitis (25). In particular, genetic mouse models targeting different steps in autophagic pathway in the pancreas revealed that autophagy is critical for maintaining acinar cell homeostasis and its blockade or impairment initiates and drives pancreatitis (12, 15, 25, 27–29, 31, 51, 55). The secretory function of acinar cells relies on coordinated actions of organelles including mitochondria, endoplasmic reticulum, and the endolysosomal system (25). Pancreatitis stressors cause mitochondrial depolarization and loss of ATP, endoplasmic reticulum stress, and endolysosomal dysfunction; in turn, organellar dysfunction mediates pancreatitis responses such as intra-acinar trypsin activity, parenchymal necrosis, and inflammation (8, 9, 14–17, 22–25, 33, 51, 56). An essential role of autophagy is the removal of damaged cellular organelles (10–12); impaired autophagy causes accumulation in acinar cells of damaged/dysfunctional organelles, thus predisposing to pancreatitis (Fig. 8). In particular, failure to remove organelles in the lysosomal/autophagic pathway that contain active trypsin results in increased intra-acinar trypsin activity in models of pancreatitis (14, 24, 51).
Notably, our results reveal different mechanisms of autophagy impairment in alcoholic versus nonalcoholic pancreatitis. The main defect in nonalcoholic AP is impaired lysosomal degradation, resulting in accumulation of abnormally large autolysosomes with poorly degraded cargo, whereas autophagosome formation is enhanced (e.g., in CER-AP), likely as a compensatory response (14–16, 24, 25). Differently, the present study shows that ethanol and alcoholic pancreatitis increase ATG4B level in acinar cells, resulting in the suppression of autophagosome formation and reduced cell capacity for cargo delivery to autolysosomes. The effects of ethanol were mimicked by ATG4B overexpression, which decreased LC3-II and worsened acinar cell necrosis and trypsinogen activation. Conversely, stimulation of autophagosome formation with ATG4B shRNA alleviated acinar cell damage caused by ethanol treatments. The detailed mechanisms mediating the effects of ATG4B on pancreatitis responses, as well as organellar damage, require further studies.
In summary, the results reveal a novel mechanism, whereby ethanol inhibits autophagy and thus sensitizes to pancreatitis (Fig. 8). ATG4B upregulation and the resultant LC3-II decrease and inhibition of autophagosome formation play a key role in ethanol’s effects on pancreatic autophagy. The findings further underscore the importance of efficient autophagy in maintaining homeostasis of pancreatic acinar cells. Enhancing pancreatic autophagy, in particular by down-regulating ATG4B, could be beneficial in mitigating the severity of alcoholic pancreatitis.
DATA AVAILABILITY
Data will be made available upon reasonable request.
GRANTS
This work was supported by National Institutes of Health Grants R01AA19730 (to I.G. and O.A.M.), P01DK098108 (to A.S.G.), and P50AA11999 (a project in the Southern California Research Center for ALPD and Cirrhosis; to A.S.G., I.G., and O.A.M.); and by the Department of Veterans Affairs Merit award I01BX004306 and BLR&D Research Career Scientist Award IK6BX005793 (both, to A.S.G.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
O.A.M., A.S.G., and I.G. conceived and designed research; O.A.M., S.R.G., G.E.L., M.P., Y.Q., J.M.E., J.N., and Z.R. performed experiments; O.A.M., A.S.G., and I.G. analyzed data; O.A.M., A.S.G., and I.G. interpreted results of experiments; O.A.M., A.S.G., and I.G. prepared figures; O.A.M., A.S.G., and I.G. drafted manuscript; O.A.M., A.S.G., and I.G. edited and revised manuscript; O.A.M., A.S.G., and I.G. approved final version of manuscript.
ACKNOWLEDGMENTS
Ethanol feeding studies were performed by the Southern California Research Center Animal Core. The authors thank Drs Janet Treger and Emmanuelle Faure-Kumar for generating the adenoviral vectors used in the study (UCLA Integrated Molecular Technologies/Vector Core supported by National Institutes of Health Grant P30DK041301 to CURE: Digestive Diseases Research Center).
REFERENCES
- 1. Pandol SJ, Saluja AK, Imrie CW, Banks PA. Acute pancreatitis: bench to the bedside. Gastroenterology 132: 1127–1151, 2007. [Erratum in Gastroenterology ibid, 133: 1056, 2007]. doi: 10.1053/j.gastro.2007.01.055. [DOI] [PubMed] [Google Scholar]
- 2. Peery AF, Crockett SD, Murphy CC, Jensen ET, Kim HP, Egberg MD, Lund JL, Moon AM, Pate V, Barnes EL, Schlusser CL, Baron TH, Shaheen NJ, Sandler RS. Burden and cost of gastrointestinal, liver, and pancreatic diseases in the United States: update 2021. Gastroenterology 162: 621–644, 2022. doi: 10.1053/j.gastro.2021.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lerch MM, Gorelick FS. Models of acute and chronic pancreatitis. Gastroenterology 144: 1180–1193, 2013. doi: 10.1053/j.gastro.2012.12.043. [DOI] [PubMed] [Google Scholar]
- 4. Mareninova O, Orabi AI, Husain S, Sohail Z. Experimental acute pancreatitis: in vitro models. In: Williams JA, ed. Pancreatitis. Mountain View, CA: Michigan Publishing, 2016, p. 3–14. [Google Scholar]
- 5. Pandol SJ, Lugea A, Mareninova OA, Smoot D, Gorelick FS, Gukovskaya AS, Gukovsky I. Investigating the pathobiology of alcoholic pancreatitis. Alcohol Clin Exp Res 35: 830–837, 2011. doi: 10.1111/j.1530-0277.2010.01408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Apte MV, Wilson JS. Alcohol-induced pancreatic injury. Best Pract Res Clin Gastroenterol 17: 593–612, 2003. doi: 10.1016/s1521-6918(03)00050-7. [DOI] [PubMed] [Google Scholar]
- 7. Pandol SJ, Periskic S, Gukovsky I, Zaninovic V, Jung Y, Zong Y, Solomon TE, Gukovskaya AS, Tsukamoto H. Ethanol diet increases the sensitivity of rats to pancreatitis induced by cholecystokinin octapeptide. Gastroenterology 117: 706–716, 1999. doi: 10.1016/s0016-5085(99)70465-8. [DOI] [PubMed] [Google Scholar]
- 8. Clemens DL, Schneider KJ, Arkfeld CK, Grode JR, Wells MA, Singh S. Alcoholic pancreatitis: new insights into the pathogenesis and treatment. World J Gastrointest Pathophysiol 7: 48–58, 2016. doi: 10.4291/wjgp.v7.i1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shalbueva N, Mareninova OA, Gerloff A, Yuan J, Waldron RT, Pandol SJ, Gukovskaya AS. Effects of oxidative alcohol metabolism on the mitochondrial permeability transition pore and necrosis in a mouse model of alcoholic pancreatitis. Gastroenterology 144: 437–446 e436, 2013. doi: 10.1053/j.gastro.2012.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 368: 651–662, 2013. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 11. Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 20: 460–473, 2014. doi: 10.1089/ars.2013.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gukovskaya AS, Gukovsky I, Algul H, Habtezion A. Autophagy, inflammation, and immune dysfunction in the pathogenesis of pancreatitis. Gastroenterology 153: 1212–1226, 2017. doi: 10.1053/j.gastro.2017.08.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S, Abeliovich H, Abildgaard MH, Abudu YP, Acevedo-Arozena A, Adamopoulos IE, Adeli K, Adolph TE, Adornetto A, Aflaki E, Agam G, Agarwal A, Aggarwal BB, Agnello M, Agostinis P, Agrewala JN, Agrotis A, Aguilar PV, Ahmad ST, Ahmed ZM, Ahumada-Castro U, Aits S, Aizawa S, Akkoc Y, Akoumianaki T. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 17: 1–382, 2021. doi: 10.1080/15548627.2020.1797280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mareninova OA, Hermann K, French SW, O'Konski MS, Pandol SJ, Webster P, Erickson AH, Katunuma N, Gorelick FS, Gukovsky I, Gukovskaya AS. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J Clin Invest 119: 3340–3355, 2009. [Erratum in J Clin Invest 123: 1844, 2013]. doi: 10.1172/JCI38674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mareninova OA, Sendler M, Malla SR, Yakubov I, French SW, Tokhtaeva E, Vagin O, Oorschot V, Lullmann-Rauch R, Blanz J, Dawson D, Klumperman J, Lerch MM, Mayerle J, Gukovsky I, Gukovskaya AS. Lysosome associated membrane proteins maintain pancreatic acinar cell homeostasis: LAMP-2 deficient mice develop pancreatitis. Cell Mol Gastroenterol Hepatol 1: 678–694, 2015. doi: 10.1016/j.jcmgh.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mareninova OA, Jia W, Gretler SR, Holthaus CL, Thomas DDH, Pimienta M, Dillon DL, Gukovskaya AS, Gukovsky I, Groblewski GE. Transgenic expression of GFP-LC3 perturbs autophagy in exocrine pancreas and acute pancreatitis responses in mice. Autophagy 16: 2084–2097, 2020. doi: 10.1080/15548627.2020.1715047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mareninova OA, Dillon DL, Wightman CJM, Yakubov I, Takahashi T, Gaisano HY, Munson K, Ohmuraya M, Dawson D, Gukovsky I, Gukovskaya AS. Rab9 mediates pancreatic autophagy switch from canonical to noncanonical, aggravating experimental pancreatitis. Cell Mol Gastroenterol Hepatol 13: 599–622, 2022. doi: 10.1016/j.jcmgh.2021.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yu ZQ, Ni T, Hong B, Wang HY, Jiang FJ, Zou S, Chen Y, Zheng XL, Klionsky DJ, Liang Y, Xie Z. Dual roles of Atg8-PE deconjugation by Atg4 in autophagy. Autophagy 8: 883–892, 2012. doi: 10.4161/auto.19652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fernández ÁF, López-Otín C. The functional and pathologic relevance of autophagy proteases. J Clin Invest 125: 33–41, 2015. doi: 10.1172/JCI73940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kauffman KJ, Yu S, Jin J, Mugo B, Nguyen N, O'Brien A, Nag S, Lystad AH, Melia TJ. Delipidation of mammalian Atg8-family proteins by each of the four ATG4 proteases. Autophagy 14: 992–1010, 2018. doi: 10.1080/15548627.2018.1437341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhou Y, Wang Z, Huang Y, Bai C, Zhang X, Fang M, Ju Z, Liu B. Membrane dynamics of ATG4B and LC3 in autophagosome formation. J Mol Cell Biol 13: 853–863, 2022. doi: 10.1093/jmcb/mjab059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Biczo G, Vegh ET, Shalbueva N, Mareninova OA, Elperin J, Lotshaw E, Gretler S, Lugea A, Malla SR, Dawson D, Ruchala P, Whitelegge J, French SW, Wen L, Husain SZ, Gorelick FS, Hegyi P, Rakonczay Z Jr, Gukovsky I, Gukovskaya AS. Mitochondrial dysfunction, through impaired autophagy, leads to endoplasmic reticulum stress, deregulated lipid metabolism, and pancreatitis in animal models. Gastroenterology 154: 689–703, 2018. doi: 10.1053/j.gastro.2017.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fortunato F, Bürgers H, Bergmann F, Rieger P, Büchler MW, Kroemer G, Werner J. Impaired autolysosome formation correlates with Lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology 137: 350–360, 2009. 360 e351-355 doi: 10.1053/j.gastro.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 24. Gukovskaya AS, Gukovsky I. Autophagy and pancreatitis. Am J Physiol Gastrointest Liver Physiol 303: G993–G1003, 2012. doi: 10.1152/ajpgi.00122.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gukovskaya AS, Gorelick F, Groblewski GE, Mareninova O, Lugea A, Antonucci L, Waldron RT, Habtezion A, Karin M, Pandol S, Gukovsky I. Recent insights into the pathogenic mechanism of pancreatitis: role of acinar cell organelle disorders. Pancreas 48: 459–470, 2019. doi: 10.1097/MPA.0000000000001298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klöppel G, Dreyer T, Willemer S, Kern HF, Adler G. Human acute pancreatitis: its pathogenesis in the light of immunocytochemical and ultrastructural findings in acinar cells. Virchows Arch A Pathol Anat Histopathol 409: 791–803, 1986. doi: 10.1007/BF00710764. [DOI] [PubMed] [Google Scholar]
- 27. Antonucci L, Fagman JB, Kim JY, Todoric J, Gukovsky I, Mackey M, Ellisman MH, Karin M. Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc Natl Acad Sci USA 112: E6166–E6174, 2015. doi: 10.1073/pnas.1519384112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Diakopoulos KN, Lesina M, Wörmann S, Song L, Aichler M, Schild L, Artati A, Römisch-Margl W, Wartmann T, Fischer R, Kabiri Y, Zischka H, Halangk W, Demir IE, Pilsak C, Walch A, Mantzoros CS, Steiner JM, Erkan M, Schmid RM, Witt H, Adamski J, Algül H. Impaired autophagy induces chronic atrophic pancreatitis in mice via sex- and nutrition-dependent processes. Gastroenterology 148: 626–638.e17, 2015. [Erratum in Gastroenterology ibid, 160: 2230, 2021]. doi: 10.1053/j.gastro.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 29. Gukovsky I, Gukovskaya AS. Impaired autophagy triggers chronic pancreatitis: lessons from pancreas-specific atg5 knockout mice. Gastroenterology 148: 501–505, 2015. doi: 10.1053/j.gastro.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dolai S, Liang T, Orabi AI, Holmyard D, Xie L, Greitzer-Antes D, Kang Y, Xie H, Javed TA, Lam PP, Rubin DC, Thorn P, Gaisano HY. Pancreatitis-induced depletion of syntaxin 2 promotes autophagy and increases basolateral exocytosis. Gastroenterology 154: 1805–1821.e5, 2018. doi: 10.1053/j.gastro.2018.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang S, Ni HM, Chao X, Ma X, Kolodecik T, De Lisle R, Ballabio A, Pacher P, Ding WX. Critical role of TFEB-mediated lysosomal biogenesis in alcohol-induced pancreatitis in mice and humans. Cell Mol Gastroenterol Hepatol 10: 59–81, 2020. doi: 10.1016/j.jcmgh.2020.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang W, Booth DM, Cane MC, Chvanov M, Javed MA, Elliott VL, Armstrong JA, Dingsdale H, Cash N, Li Y, Greenhalf W, Mukherjee R, Kaphalia BS, Jaffar M, Petersen OH, Tepikin AV, Sutton R, Criddle DN. Fatty acid ethyl ester synthase inhibition ameliorates ethanol-induced Ca2+-dependent mitochondrial dysfunction and acute pancreatitis. Gut 63: 1313–1324, 2014. 2014. doi: 10.1136/gutjnl-2012-304058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mukherjee R, Mareninova OA, Odinokova IV, Huang W, Murphy J, Chvanov M, Javed MA, Wen L, Booth DM, Cane MC, Awais M, Gavillet B, Pruss RM, Schaller S, Molkentin JD, Tepikin AV, Petersen OH, Pandol SJ, Gukovsky I, Criddle DN, Gukovskaya AS, Sutton R; NIHR Pancreas Biomedical Research Unit. Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: inhibition prevents acute pancreatitis by protecting production of ATP. Gut 65: 1333–1346, 2016. doi: 10.1136/gutjnl-2014-308553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mareninova OA, Sung KF, Hong P, Lugea A, Pandol SJ, Gukovsky I, Gukovskaya AS. Cell death in pancreatitis: caspases protect from necrotizing pancreatitis. J Biol Chem 281: 3370–3381, 2006. doi: 10.1074/jbc.M511276200. [DOI] [PubMed] [Google Scholar]
- 35. Lugea A, Waldron RT, Mareninova OA, Shalbueva N, Deng N, Su HY, Thomas DD, Jones EK, Messenger SW, Yang J, Hu C, Gukovsky I, Liu Z, Groblewski GE, Gukovskaya AS, Gorelick FS, Pandol SJ. Human pancreatic acinar cells: proteomic characterization, physiologic responses, and organellar disorders in ex vivo pancreatitis. Am J Pathol 187: 2726–2743, 2017. doi: 10.1016/j.ajpath.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hemelaar J, Lelyveld VS, Kessler BM, Ploegh HL. A single protease, Apg4B, is specific for the autophagy-related ubiquitin-like proteins GATE-16, MAP1-LC3, GABARAP, and Apg8L. J Biol Chem 278: 51841–51850, 2003. doi: 10.1074/jbc.M308762200. [DOI] [PubMed] [Google Scholar]
- 37. Wang YL, Hu R, Lugea A, Gukovsky I, Smoot D, Gukovskaya AS, Pandol SJ. Ethanol feeding alters death signaling in the pancreas. Pancreas 32: 351–359, 2006. doi: 10.1097/01.mpa.0000220859.93496.e1. [DOI] [PubMed] [Google Scholar]
- 38. Williams JA. Regulation of acinar cell function in the pancreas. Curr Opin Gastroenterol 26: 478–483, 2010. doi: 10.1097/MOG.0b013e32833d11c6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Norman JM, Cohen GM, Bampton ET. The in vitro cleavage of the hAtg proteins by cell death proteases. Autophagy 6: 1042–1056, 2010. doi: 10.4161/auto.6.8.13337. [DOI] [PubMed] [Google Scholar]
- 40. Li M, Tan J, Miao Y, Lei P, Zhang Q. The dual role of autophagy under hypoxia-involvement of interaction between autophagy and apoptosis. Apoptosis 20: 769–777, 2015. doi: 10.1007/s10495-015-1110-8. [DOI] [PubMed] [Google Scholar]
- 41. Tsapras P, Nezis IP. Caspase involvement in autophagy. Cell death Differ 24: 1369–1379, 2017. doi: 10.1038/cdd.2017.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guo N, Peng Z. MG132, a proteasome inhibitor, induces apoptosis in tumor cells. Asia Pac J Clin Oncol 9: 6–11, 2013. doi: 10.1111/j.1743-7563.2012.01535.x. [DOI] [PubMed] [Google Scholar]
- 43. Hamidi R, Ataei F, Hosseinkhani S. Inhibition of noncaspase proteases, calpain and proteasome, via ALLN and Bortezomib contributes to cell death through low degradation of pro-/anti-apoptotic proteins and apoptosis induction. Med Oncol 39: 125, 2022. doi: 10.1007/s12032-022-01716-w. [DOI] [PubMed] [Google Scholar]
- 44. Nagahara Y, Shiina I, Nakata K, Sasaki A, Miyamoto T, Ikekita M. Induction of mitochondria-involved apoptosis in estrogen receptor-negative cells by a novel tamoxifen derivative, ridaifen-B. Cancer Sci 99: 608–614, 2008. doi: 10.1111/j.1349-7006.2007.00709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Louhimo J, Steer ML, Perides G. Necroptosis is an important severity determinant and potential therapeutic target in experimental severe pancreatitis. Cell Mol Gastroenterol Hepatol 2: 519–535, 2016. doi: 10.1016/j.jcmgh.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Blinman TA, Gukovsky I, Mouria M, Zaninovic V, Livingston E, Pandol SJ, Gukovskaya AS. Activation of pancreatic acinar cells on isolation from tissue: cytokine upregulation via p38 MAP kinase. Am J Physiol Cell Physiol 279: C1993–C2003, 2000. doi: 10.1152/ajpcell.2000.279.6.C1993. [DOI] [PubMed] [Google Scholar]
- 47. Gukovsky I, Reyes CN, Vaquero EC, Gukovskaya AS, Pandol SJ. Curcumin ameliorates ethanol and nonethanol experimental pancreatitis. Am J Physiol Gastrointest Liver Physiol 284: G85–G95, 2003. doi: 10.1152/ajpgi.00138.2002. [DOI] [PubMed] [Google Scholar]
- 48. Satoh A, Gukovskaya AS, Reeve JR, Shimosegawa T, Pandol SJ. Ethanol sensitizes NF-κB activation in pancreatic acinar cells through effects on protein kinase C-epsilon. Am J Physiol Gastrointest Liver Physiol 291: G432–G438, 2006. doi: 10.1152/ajpgi.00579.2005. [DOI] [PubMed] [Google Scholar]
- 49. Tsukamoto H, French SW, Reidelberger RD, Largman C. Cyclical pattern of blood alcohol levels during continuous intragastric ethanol infusion in rats. Alcohol Clin Exp Res 9: 31–37, 1985. doi: 10.1111/j.1530-0277.1985.tb05046.x. [DOI] [PubMed] [Google Scholar]
- 50. Jones AW, Holmgren A. Age and gender differences in blood-alcohol concentration in apprehended drivers in relation to the amounts of alcohol consumed. Forensic Sci Int 188: 40–45, 2009. doi: 10.1016/j.forsciint.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 51. Mareninova OA, Vegh ET, Shalbueva N, Wightman CJ, Dillon DL, Malla S, Xie Y, Takahashi T, Rakonczay Z Jr, French SW, Gaisano HY, Gorelick FS, Pandol SJ, Bensinger SJ, Davidson NO, Dawson DW, Gukovsky I, Gukovskaya AS. Dysregulation of mannose-6-phosphate-dependent cholesterol homeostasis in acinar cells mediates pancreatitis. J Clin Invest 131: e146870, 2021. doi: 10.1172/JCI146870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 117: 2805–2812, 2004. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- 53. Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, Khare S, Kondo S, Kondo Y, Yu Y, Mills GB, Liao WS, Bast RC Jr. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J Clin Invest 118: 3917–3929, 2008. doi: 10.1172/JCI35512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Park NY, Jo DS, Cho DH. Post-translational modifications of ATG4B in the regulation of autophagy. Cells 11: 1330, 2022. doi: 10.3390/cells11081330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gukovskaya A, Mareninova O, Ding W, Habtezion A, Gukovsky I. Models of pancreatitis caused by genetic blockage of autophagy/lysosomal pathway. Pancreapedia 2021. doi: 10.3998/panc.2021.12. [DOI] [Google Scholar]
- 56. Lugea A, Tischler D, Nguyen J, Gong J, Gukovsky I, French SW, Gorelick FS, Pandol SJ. Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology 140: 987–997, 2011. doi: 10.1053/j.gastro.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available upon reasonable request.