

Keywords: epithelial cells, fibrosis, progenitor cells, SASP, senescence
Abstract
Pulmonary fibrosis comprises a range of chronic interstitial lung diseases (ILDs) that impose a significant burden on patients and public health. Among these, idiopathic pulmonary fibrosis (IPF), a disease of aging, is the most common and most severe form of ILD and is treated largely by lung transplantation. The lack of effective treatments to stop or reverse lung fibrosis—in fact, fibrosis in most organs—has sparked the need to understand causative mechanisms with the goal of identifying critical points for potential therapeutic intervention. Findings from many groups have indicated that repeated injury to the alveolar epithelium—where gas exchange occurs—leads to stem cell exhaustion and impaired alveolar repair that, in turn, triggers the onset and progression of fibrosis. Cellular senescence of alveolar epithelial progenitors is a critical cause of stemness failure. Hence, senescence impairs repair and thus contributes significantly to fibrosis. In this review, we discuss recent evidence indicating that senescence of epithelial progenitor cells impairs alveolar homeostasis and repair creating a profibrotic environment. Moreover, we discuss the impact of senescent alveolar epithelial progenitors, alveolar type 2 (AT2) cells, and AT2-derived transitional epithelial cells in fibrosis. Emerging evidence indicates that transitional epithelial cells are prone to senescence and, hence, are a new player involved in senescence-associated lung fibrosis. Understanding the complex interplay of cell types and cellular regulatory factors contributing to alveolar epithelial progenitor senescence will be crucial to developing targeted therapies to mitigate their downstream profibrotic sequelae and to promote normal alveolar repair.
NEW & NOTEWORTHY With an aging population, lung fibrotic diseases are becoming a global health burden. Dysfunctional repair of the alveolar epithelium is a key causative process that initiates lung fibrosis. Normal alveolar regeneration relies on functional progenitor cells; however, the senescence of these cells, which increases with age, hinders their ability to contribute to repair. Here, we discuss studies on the control and consequence of progenitor cell senescence in fibrosis and opportunities for research.
INTRODUCTION
Interstitial lung diseases (ILDs) are a group of respiratory conditions characterized by an excessive deposition of extracellular matrix (ECM), mostly fibrillar collagens (type I, III, others), within interstitial spaces. As for most organs, fibrosis within the lung impairs tissue function, which involves gas exchange within the alveolar compartment (respiratory unit). Among ILDs, idiopathic pulmonary fibrosis (IPF) is the most common. IPF is a devastating, progressive chronic respiratory disease of aging with poor clinical outcomes (1, 2) and is increasing in incidence throughout the world (1, 3). Recently, lung fibrosis associated with postacute SARS-CoV-2 infection (PASC) has gained substantial attention (4). Although the long-term clinical outcomes of PASC lung fibrosis are still being uncovered, this condition shares some key pathological features with IPF (5–8). Hence, understanding the fundamental mechanisms of fibrosis will aid in the identification of processes that can be targeted therapeutically to improve outcomes in this spectrum of lung diseases.
In lung and other tissues, such as liver and kidney, repeated epithelial injury, along with aging, genetic, and environmental factors, contributes to defective repair creating a profibrotic environment. Through mechanisms that are still being uncovered, persistent and impaired epithelial repair stimulates the differentiation of interstitial fibroblasts (and possibly other resident cells) into myofibroblasts that produce and deposit an excess of ECM, which is the hallmark of any form of fibrosis. Thus, a defect in the regenerative activity of epithelial progenitor cells likely contributes to the onset of fibrosis.
Epithelial progenitor cells maintain alveolar homeostasis through their capacity for self-renewal and repopulation of alveolar type I (AT1) cells (9, 10). Due to their thin squamous morphology and intimate association with the alveolar capillaries, AT1 cells are specialized for efficient gas exchange. AT1 cells are derived from alveolar type 2 (AT2) cells via an intermediate population (see Transitional Cells and Fig. 1). AT2 cells are abundant, large cuboidal cells that produce pulmonary surfactants and are the critical progenitor population within the alveolus. Failure or exhaustion of their ability to self-renew and to generate AT1 cells leads to impaired alveolar regeneration and repair, defects that are critical to the onset of lung fibrosis (11, 12).
Figure 1.

Alveolar progenitor cells. Various epithelial cells in the distal airways of lungs can generate alveolar type 2 (AT2) cells. These bipotent progenitor cells include club cells, ciliated cells, tuft cells, pulmonary neuroendocrine cells (PNECs), basal cells, and respiratory airway secretory cell (RAS)/alveolar type 0 (AT0) cells. Except for basal cells, which are found in human and not mouse airways, these distal airway progenitor epithelial cells are found in human and mouse lungs, and some can self-renew. AT2 cells in the alveolar compartment can self-renew and differentiate into AT1 cells via a transitional cell type. (Created with BioRender.com with permission).
Senescence is a common feature of progenitor stem cell exhaustion and is characterized by the permanent loss of a cell’s proliferative capability. Cellular senescence is linked to aging, and age is the main risk factor for developing IPF (13). Supporting a functional link between senescence and fibrosis, senescent alveolar progenitor epithelial cells are abundant in both PASC and IPF lungs (14–16). In this review, we summarize the evidence supporting the idea that the senescence of alveolar epithelial progenitor cells contributes to lung fibrosis.
The alveolar environment comprises epithelial cells, endothelial cells, fibroblasts, and immune cells, as well as the fused basement membranes of the epithelium and endothelium and an elastin-rich interstitium (17, 18). Maintaining alveolar homeostasis requires communication among these cell types through cell-cell and paracrine signaling pathways, as well as through biophysical forces via ligation with the ECM (19–21).
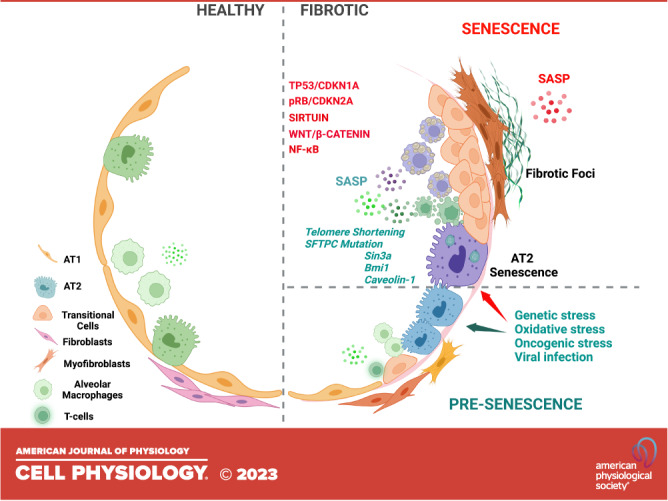
As we discuss here, emerging data indicate that the senescence of lung progenitor epithelial cells can initiate lung fibrosis and promote its progression (10, 22, 23). Senescent cells mediate disease pathogenesis by two broad mechanisms: 1) a failure to regenerate alveolar epithelial cells and restore alveolar structure and function creates a profibrotic environment and 2) the paracrine effects of senescence-associated secretory phenotype (SASP) cause an accumulation of senescent cells in the lung microenvironment that furthers impaired repair processes and promotes fibrosis. Interestingly, and as we discuss Senescence of AT2, whereas senescence of alveolar epithelial cells is detrimental, some studies have reported that senescence of nonepithelial cells, such as fibroblasts, appears to be beneficial for effective tissue regeneration (24, 25). The cell type-specific outcomes of senescence on fibrosis create a clear challenge to targeting this process as a therapeutic means to mitigate disease. However, this challenge also provides the opportunity to uncover pro- and antifibrotic mechanisms of distinct senescent cell types, knowledge that could be used to design effective approaches to lessen or reverse fibrosis.
ALVEOLAR EPITHELIAL PROGENITOR CELLS
In recent years, much progress has been made in the identification of lung epithelial stem and progenitor cells (26, 27). Alveolar progenitor cells can be divided into two groups: 1) AT2 cells in the alveolar niche that are capable of self-renewing and differentiating into AT1 cells and 2) bipotent progenitor cells in distal airways that can generate AT2 cells (Fig. 1). In addition, as they differentiate into AT1 cells, AT2 cells assume a transient “prealveolar type-1 transitional” cell state or PATS. As we discuss, compelling evidence from a variety of mouse models and human specimens has linked senescence of AT2 cells to lung fibrosis and IPF (28, 29). In contrast, evidence has not yet emerged to indicate that senescence of the distal airway progenitor cells contributes to fibrosis. Hence, in this article, we focus on the senescence of AT2 cells.
AT2 Cells
In healthy, mature lungs, AT2 serves critical roles necessary for alveolar function and stability. They produce and recycle surfactants and can self-renew and differentiate into AT1 cells (21, 30, 31). A subset of Wnt-responsive AT2 cells marked by Axin2 has been claimed to serve as major facultative progenitor cells in the mouse distal lung (32, 33). In normal human and mouse lungs, AT2 cells are the primary source of AT1 cells (9, 34). Hence, AT2 cells are critical both to maintaining alveolar structure and function during homeostasis and to the regeneration of damaged alveoli following injury and infection. Most studies linking alveolar progenitor cell senescence to fibrosis have focused on AT2 cells.
Transitional Cells
Recent transcriptomic studies have defined epithelial cells with a molecular phenotype suggestive of a transition state between AT2 and AT1 cells, referred to as “transitional cells,” “damage-associated transitional progenitor,” (DATP) or “prealveolar type-1 transitional cells” (PATS) (29, 35–41). In these studies, transitional cells have been defined as either Krt8+ (29), Trp53+ Krt17+ (35), damage-associated transient progenitors (Krt8+, Cldn4+, and Cdkn1a+) (41), double Krt7+ and Krt8+ cells (42), or transitional Krt5− and Krt17+ cells (37). An accumulation of these transitional cells following infection and injury suggests a connection between senescence and abnormal AT2 differentiation (43), leading to impaired lung repair and fibrosis (35). However, more evidence is required to support this notion.
Distal Airway Progenitor Cells
Epithelial progenitors in terminal respiratory bronchioles play essential roles in alveolar repair and regeneration. In lung repair, the expansion of keratin 5-positive (KRT5+) cells in severe H1N1 influenza virus and likely COVID-19 infection is a crucial example such that the origin of these KRT5+ cells has been traced to distal airway progenitor cells by multiple groups: rare Sox2+ airway progenitor cells (44), Trp63+ and Krt5+ stem cells (44, 45), embryonic p63+ progenitor (46) and Trp63+ cells (47, 48), club cell variant (Scgb1a1+, Upk3a+) (49, 50), lineage-negative epithelial progenitors (LNEPTRP63−, Itgb4+) (51), and H2-K1high (Itgb4+, H2-K1+) (52). Specific distal progenitors may be involved in alveolar regeneration: SCGB3A2+ cells in the human distal airway [also called respiratory airway secretory (RAS) (50), AT0 cells (53)] can give rise to AT2 cells in vitro three-dimensional (3-D) organoid culture and the Sftpc and Scgb1a1 double-positive cells in mouse bronchioalveolar region [bronchioalveolar stem cells, BASC cells (54)] that can differentiate into AT2 and subsequently into AT1 cells. The dual lineage tracing approach of Sftpc-DreER and Scgb1a1-CreER demonstrated that BASCs become activated and exhibit distinct responses to the specific types of lung injury. Notably, BASCs can differentiate into club cells and ciliated cells in naphthalene exposure and AT2 and AT1 cells in bleomycin injury (55). With additional human studies to validate their roles, the distal airway progenitors play critical functions in maintaining terminal bronchiole and alveolar homeostasis following various insults. The decline of progenitor proliferation during aging indirectly implies the involvement of senescence (56). We also demonstrate the role of p53 in regulating distal airway progenitor cell fates (57).
SENESCENCE AND SASP
Senescence
Cellular senescence increases with age and represents a state of irreversible growth arrest (58, 59). Senescence is characterized by a combination of metabolic changes, acquisition of a distinctive secretome that is commonly referred to as the senescence-associated secretory phenotype (SASP), elevated β-galactosidase activity, oxidative stress, resistance to apoptosis, and morphological changes, such as an enlarged and flattened cell shape, multinucleation, and vacuolization (58, 60). Canonical features of senescent cells include the upregulation of key cell cycle inhibitory proteins, such as p16 and/or p21, cell cycle arrest, and evidence of DNA damage by phosphorylation of H2A histone family member X (γH2AX). A challenge in studying senescence is that the compilation of protein and mRNA markers used to identify senescent cells differs among cell types and is not individually specific to senescent cells (60, 61). Thus, to assure rigor in defining senescent cell types, a range of endpoints must be used.
SASP
The secretome of senescent cells (62) includes proinflammatory cytokines, chemokines, growth factors, proteinases and inhibitors, reactive oxygen species, and other macromolecules (Table 1). Although no individual SASP component can be considered a selective marker of senescence, the combined release of multiple SASP components coupled with the presence of other cell-intrinsic senescence markers/characteristics, especially expression of p16 and/or p21, β-galactosidase activity, and permanent growth arrest, would be confirmatory of a senescent state.
Table 1.
Senescence-associated secretory phenotypes components involved in AT2 cell senescence and lung fibrosis
| Classification | Factors | References |
|---|---|---|
| Inflammatory factors | IL-1α, IL-1β | (63) |
| IL-4 | (64) | |
| IL-6, IL-11 | (65, 66, 67–69) | |
| IL-8 | (63) | |
| IL-13 | (70, 71, 72) | |
| TNFα | (66, 68, 69) | |
| Chemokines | CCL2, CCL5, CCL17 | (73, 74) |
| Cytokines | TGFβ1 | (65, 70, 75, 68) |
| PDGF | (66, 70) | |
| IGFBP3, IGFBP4 | (76) | |
| Proteinases and inhibitors | MMP-2, MMP-9 | (77, 69) |
| MMP-9 | (63, 69) | |
| MMP-12 | (76) | |
| PAI-1 | (78, 79, 70) | |
| Nonprotein | mitoROS | (80) |
AT2, alveolar type 2; CCL, chemokine (CC motif) ligand; IGFBP, insulin-like growth factor binding protein 2; mitoROS, mitochondrial reactive oxygen species; MMP, matrix metalloproteinase; PAI-1, plasma activator inhibitor-1; PDGF, platelet-derived growth factor; TGFβ1, transforming growth factor β1; TNFa, tumor necrotic factor.
SENESCENCE OF AT2 CELLS IN FIBROSIS
Senescence is observed in various cell types throughout fibrotic lungs, and these different cells can independently or collaboratively promote lung fibrosis. Stemness failure associated with alveolar epithelial progenitor senescence not only inhibits proper alveolar repair and regeneration but can also induce the senescence of healthy progenitors and nonprogenitor in the alveolar niches through the paracrine effects of SASP further deteriorating the fibroproliferative processes (15, 21, 26, 75, 81–84). The loss of alveolar epithelial progenitor cells stemness (i.e., exhaustion) through senescence contributes significantly to the progression of lung fibrosis pathogenesis (75, 85–89). Although the mechanisms are not yet fully understood, impaired alveolar epithelial repair and/or sustained alveolar injury activates pathways, such as TGFβ (75) and Wnt signaling, (90) that promote the differentiation and proliferation of fibroblasts into invasive myofibroblasts, which deposit excessive ECM, and facilitate immune cell dysregulation that can further impair alveolar repair (15, 22, 75, 81, 87, 91).
Numerous external stimuli and internal cellular stress contribute to both age-related and premature senescence of AT2 cells. The accumulation of AT2 senescence in IPF and PASC lung fibrosis, as demonstrated by transcriptomic and immune-staining analyses (16, 65, 75, 80, 92)—as discussed in senescence regulatory pathways—provides compelling evidence that senescence of AT2 cells is a critical effector in these progressive conditions. Indeed, transcriptomic quantitation of lung epithelial senescence by a computational scoring approach for top common senescence genes (http://csgene.bioinfo-minzhao.org) highlights the potential significance of this process in AT2 cells in disease pathogenesis (75, 93). In our work, the spontaneous development of lung fibrosis in mice due to AT2 senescence following the deletion of Sin3a in AT2 cells implicates further the pathogenic role of senescence of AT2 cells in lung fibrosis (75).
The plasticity of AT2 cells in maintaining alveolar homeostasis is partially explained by the presence and accumulation of transitional cells following lung injury in mice and in the progression of IPF in humans (28, 29, 41, 93–95) and COVID-19 lung fibrosis (96). The upregulation of senescent signature genes in PATS and their accumulation in fibrotic regions suggests they have roles in disease pathogenesis. For example, Krt5+ transitional cells in IPF express p16 suggesting they are a senescent population in the diseased lung (95). Although the role of senescence in hindering the differentiation of transitional cells into AT1 cells is limited (35–37), transitional cells do show upregulation of canonical senescence markers, indicating some contribution to cellular senescence (27). Both our and public scRNAseq profiling data (28, 29, 35) demonstrate that transitional cells in both control and IPF lungs display a higher senescence score than that of AT2 cells in the same specimens (Fig. 2). Although the impact of senescence on the cellular function of transitional cells remains to be determined, it is reasonable to propose that senescence of these cells hinders their ability to differentiate into AT1 cells, thereby impairing alveolar repair and promoting fibrosis. Indeed, upregulation of senescence markers (p16, CCND1, and GDF15) on transitional cells in IPF and PASC lung fibrosis implicate that senescence of alveolar progenitor differentiation processes is involved in that pathogenesis of these conditions (43, 94, 97).
Figure 2.

Transcriptomic senescence score of alveolar epithelial progenitor cells in idiopathic pulmonary fibrosis (IPF). A: uniform manifold approximation and projection (UMAP) visualization of alveolar progenitor epithelial cells (AT2 and transitional cells). B: average expression of alveolar type 2 (AT2) and transitional cell common markers and transcriptomic senescence score of each cell type in control and IPF. Data are described in two metrics: gene expression and the percentage of expressing cells.
The contribution of SASP from senescent cells to lung fibrosis is established (23, 88). The proinflammatory factors in SASP (Table 1), as discussed in this and impact of at2 cell senescence of other cell types in fibrosis, can activate profibrotic signaling, either directly or indirectly. In addition, SASP can promote the senescence of nearby cells by the paracrine effects of several of the factors released. Thus, in a disease setting like IPF, SASP could function in a deleterious feed-forward manner that promotes senescence, thereby leading to impaired progenitor cell function and interstitial fibrosis.
The regulation of SASP is intertwined with other senescence regulatory pathways (98). Alveolar epithelial progenitors can produce SASP in a senescent microenvironment, thereby augmenting the impact of senescence in hindering proper alveolar repair. Indeed, in the bleomycin model, senescent p21-positive AT2 cells contribute to fibrosis through their SASP-secreted effector proteins (66). Furthermore, the “cytokine storm” concept as a causative event in COVID-19 lung fibrosis makes SASPs relevant as stimulators and effectors of alveolar progenitor senescence (7, 99). Indeed, widespread AT2 senescence and upregulation of SASP genes are observed in lung tissues of patients with fatal COVID-19 and COVID-19 lung fibrosis (100–102). In addition, transcriptomic data, validated with 3-D organoid cultures, provides compelling findings indicating that SASP secreted from senescent AT2 cells and other inflammatory cells are critical culprits in cellular failure in acute and chronic states associated with fibrosis (100–102).
SENESCENCE REGULATORY PATHWAYS
Pathways that regulate cellular senescence are complex and interconnected. Senescence is regulated by various transcriptional factors and cell cycle-associated genes (103). For example, animals with either AT2-specific loss of function (LOF) of Sin3-a (75), a transcriptional repressor, or global deficiency of B cell–specific Moloney murine leukemia virus insertion region 1 (Bmi-1) (65) developed spontaneous AT2 senescence and lung fibrosis. Furthermore, in the Sin-3a LOF model, administration of a “senolytic cocktail,” which stimulated apoptosis in senescent cells, thereby reducing their numbers, alleviated lung fibrosis, substantiating a causative role of AT2 senescence in promoting fibrosis (76). The “senolytic cocktail” used in this study was a combination of an antioxidant (quercetin) and a multikinase inhibitor (dasatinib). The most studied signaling mechanisms controlling senescence of alveolar progenitor cells are the TP53 and retinoblastoma protein (RB) pathways, which, broadly speaking, function to promote or limit senescence, respectively.
TP53 and p21
The transcription factor p53 (TP53) inhibits cell cycle progression in response to a range of stimuli, such as DNA damage, telomere dysfunction, cell-cell fusion, and SASP (104). As such, TP53 has effects on progenitor cell activity, such as promoting differentiation and hindering self-renewal (105), and via these functions, TP53 promotes senescence. An essential effector molecule of TP53-mediated growth arrest is p21/p21WAF1/CIP1/CDKN1A (78, 79), which mediates AT2 senescence-induced lung fibrosis (103, 106). In our work, we found that conditional silencing of Sin3a in AT2 cells leads to spontaneous lung fibrosis by activating p53 and p21 that, in turn, promoted AT2 cell senescence (75). Recently, Kobayashi et al. (35) reported that TP53 signaling hinders the differentiation of transitional cells into AT1 cells.
RB and p16
In contrast to TP53, Rb protein (pRb) is a tumor suppressor whose normal activity regulates cell cycling (107). Unphosphorylated pRb binds E2F1, a transcriptional factor that promotes cell division. When bound to pRb, E2F1 is held in the cytosol leading to G1 arrest (108, 109). Phosphorylation of pRb by cyclin-dependent kinases, CDK1 and CDK4, allows dissociation of E2F1 that then enters the nucleus and drives the transition of cells from G1 to S phase (107). An important player in pRb signaling is p16/CDKN2A, which inhibits CDK1 and CDK4. Thus, by preventing phosphorylation of pRb, p16 promotes senescence by causing cell cycle arrest. Consistent with this idea, compared with wild-type mice, p16-null mice have greater AT2 cell expansion and renewal during adulthood and better alveolar regeneration in response to injury (25), phenotypes that imply reduced senescence. Similar findings were demonstrated in elastase-induced alveolar damage in emphysema (110). These observations suggest that the pRb/p16 pathway functions to moderate senescence and to promote AT2 cell self-renewal and differentiation into transitional cells and transitional cells into AT1 cells.
Sirtuin Pathways
Impaired autophagy and mitochondria dysfunction can lead to cellular senescence, and these processes are affected by the Sirtuin pathway. For example, SIRT1 activity is downregulated by cigarette smoke and is associated with impaired autophagy, greater mitochondria dysfunction, and senescence of AT2 cells than in room-air controls (80). Pharmacological stimulation of SIRT1 activity with SRT1720 or minimization of mitochondria dysfunction with mitoROS both reversed these disease responses (80). Similarly, stimulating Sirtuin 1 by H2S inhalation reduces AT2 senescence (111). In addition, H2S attenuates bleomycin-induced lung fibrosis in rats (112), although the mechanism of its protective effect is not known. Furthermore, extracellular vesicles released by lung fibroblast contain miR-23b-3p and miR-494-3p that can downregulate SIRT3 (113), leading to mitochondria dysfunction-induced AT2 senescence and enhanced lung fibrosis.
Recent work from our center uncovered a novel mechanism linking the Sirtuin pathway to AT2 cell senescence and fibrosis (114). They found that downregulation of the zinc transporter ZIP8/SLC39A8 (ZIP8) in both IPF lungs and lungs of old mice is associated with AT2 cell senescence and enhanced lung fibrosis and that these phenotypes are dependent on SIRT1 (114). Replenishment with exogenous zinc and SIRT1 activation promoted self-renewal and differentiation of AT2 cells from lung tissues of patients with IPF and old mice. Therapeutic strategies to restore zinc metabolism and appropriate SIRT1 signaling could improve the progenitor function of AT2 cells, thereby moderating ongoing fibrogenesis. Overall, several studies suggest that the Sirtuin pathway functions to mitigate cell senescence and, in turn, fibrosis.
Other Pathways
Caveolin-1, a membrane-associated scaffolding protein of caveolae, mediates cell cycle arrest in stromal cells through TP53/p21 (115). In AT2 cells, caveolin-1 mediates senescence by impairing autophagy via a p53/p21-dependent mechanism (116). Furthermore, Cav1−/− mice have less senescent AT2 cells and reduced fibrosis in response to bleomycin injury compared with wild-type mice, which was linked to enhanced pRB/p16 levels (77). These findings indicate that caveolin-1 promotes fibrosis by being permissive for the onset of senescence.
Other signaling pathways that may regulate senescence include WNT/β-catenin, NF-κB, insulin-like growth factor (IGF), and other tyrosine kinases. Although Wnt signaling is important for maintaining AT2 stemness, chronic stimulation of WNT/β-catenin signaling through WNT3a promotes AT2 senescence in lung fibrosis (117). The loss of PTEN can activate IκB, IKK, and NF-κB (63) and Akt leading to AT2 senescence (81). Moreover, inhibiting NF-κB by gene silencing or drugs hinders the onset of senescence of AT2.
Studies on the IGF pathway have shown that different signaling components can have distinct effects on cellular senescence. For example, whereas IGF1/PI3K/AKT signaling promotes senescence of AT2 cells in IPF (118), IGF binding protein 2 (IGFBP2) hinders AT2 cell senescence in bleomycin-injured mice (119). The TGF-β1/IL-11/MEK/ERK (TIME) pathway has been reported to promote AT2 senescence and lung fibrosis (65). In this model, the TIME pathway was stimulated by deletion of the gene for BMI-1, a polycomb repressor complex 1 protein essential for stem cell self-renewal (65). Hence, this study supports the general concept that impaired progenitor cell function leads to senescence and creates a profibrotic environment.
PROCESSES THAT INITIATE SENESCENCE IN AT2 CELLS
Senescence is initiated by various processes, such as genetic and epigenetic dysregulation, stress- or oncogenic-induced, infection-associated diseases, etc. These factors contribute to alveolar progenitor senescence via both paracrine-affected and autocrine-affected means.
Telomere Dysfunction
In familial pulmonary fibrosis and IPF, shortened telomere length in AT2 cells arises due to genetic abnormalities and/or to age-related vulnerability of the DNA damage response (120, 121). In these and other lung fibrotic diseases, senescent AT2 cells are proximal to fibrotic lesions and, overall, are more abundant than other senescent cell types (120, 121). Mouse models of AT2 cell-conditional telomere dysfunction demonstrated that shortened telomere length leads to senescence of AT2 cells and spontaneous fibrosis (121–124). In contrast, telomere dysfunction in fibroblasts did not lead to lung fibrosis (121–124)—possibly by impairing the differentiation into myofibroblasts—and a recent preclinical study confirmed this conclusion (125). Furthermore, telomere uncapping associated with F-box and WD40 repeat domain-containing 7 binding to telomere protection protein 1 accelerates AT2 senescence and promotes lung fibrosis (126). Moreover, downregulating of telomerase reverse transcriptase (TERT) by overexpression of Krüpple-like-factors 4 reduces AT2 senescence and minimizes bleomycin-induced lung fibrosis (127). Together, these studies indicate that telomere pathology is a significant risk factor for AT2 senescence and fibrosis. To date, it remains unclear if telomere attrition contributes to the accumulation and/or senescence of PATS cells.
ER Stress
Some studies have indicated a causal link between ER stress and AT2 senescence leading to spontaneous lung fibrosis (128, 129). For example, activation of the unfolded protein response (UPR) in AT2 cells due to overexpression of mutant surfactant protein C increases ER stress and induces senescence markers (43, 87, 130). Stimulating ER stress in AT2 cells by reducing or loss of glucose-regulated protein 78 (GRP78), an ER stress chaperone protein, causes senescence and spontaneous lung fibrosis (131). A recent study demonstrated that activation of the UPR via IRE1-α signaling following bleomycin injury promotes AT2 cell senescence, an accumulation of PATS cells, and fibrosis (42). Blocking IRE1-α signaling reduced senescence and promoted the differentiation of PATS cells to AT1 cells resulting in less bleomycin-induced lung fibrosis (42). These data provide compelling evidence for a mechanistic association among ER stress, senescence of AT2 cells, and the onset of lung fibrosis. In addition, these findings suggest that senescence of AT2 cells does not hinder their ability to transition into PATS cells but does impair the ability to fully differentiate into AT1 cells. Additional research is needed to determine how AT2 cell senescence affects PATS cell function.
Autophagy and Mitochondria Dysfunction
Autophagy can both activate and inhibit cellular senescence—and, in turn, fibrosis—depending on the setting and on the presence and selectivity of autophagy regulatory molecules (132). For example, both inhalation of cigarette smoke and single-walled carbon nanotubes impairs autophagy and induces AT2 senescence and lung fibrosis (80, 133). Although these and other studies suggest that impaired autophagy promotes cellular senescence and fibrosis, the senescence-suppressor protein SIRT1 can be degraded through the autophagy-lysosome pathway indicating functional autophagy can limit senescence (134).
Mitochondria dysfunction is tightly correlated with cellular senescence (135). For example, dysfunctional and dysmorphic mitochondria, as evident by the accumulation of mitochondrial DNA (mtDNA), result from the downregulation of macroautophagy and are associated with AT2 senescence in IPF (136, 137). Using aged mice, Bueno et al. (137) demonstrated that PINK1 (PTEN-induced putative kinase 1) deficiency in AT2 cells induces mitochondria dysfunction and AT2 senescence and enhances susceptibility to virus-mediated fibrosis. As reported by Summer et al. (138), another characteristic of mitochondrial dysfunction in senescent cells is activation of the mTOR/PGC-1α/β pathway (mammalian target of rapamycin/peroxisome proliferator-activated receptor gamma, coactivator 1α/β), which promotes mitochondria synthesis, increased oxidative phosphorylation, and mtROS production, which can all further facilitate AT2 senescence. Of interest, Summer et al. (138) found that PINK1 activity was elevated in senescent cells—which contradicts the findings of Bueno et al. (137)—and independent mTOR activity. They found that although rapamycin-mediated inhibition of mTOR activity blocked senescence, it did not reduce elevated PINK1 levels (137). Thus, additional research is needed to determine if and how PINK1 functions in senescence. Furthermore, as seen in both IPF and bleomycin-injured mouse lungs, upregulation of CD38, a nicotinamide adenine dinucleotide (NAD) hydrolase, in AT2 cells leads to NAD deficiency and mitochondria dysfunction promoting AT2 senescence and fibrosis (139).
Metabolic reprogramming of alveolar epithelial progenitor cells through the autophagy-mitochondria axis contributes greatly to progenitor senescence-induced lung fibrosis. This notion is supported by studies showing that AT2 senescence is mitigated in vivo and in vitro by human mesenchymal stromal cells (MSCs) through nicotinamide phosphoribosyl transferase (NAMPT)-mediated NAD metabolism (140); specifically, MSCs upregulated NAMPT expression, increasing NAD+ level in AT2 cells. Thus, targeting metabolic pathways could prevent mitochondria dysfunction in and senescence of AT2 cells and, in turn, fibrosis.
IMPACT OF AT2 CELL SENESCENCE OF OTHER CELL TYPES IN FIBROSIS
Although eliminating senescent epithelial cells with senolytic agents or blocking senescence regulatory pathways alleviates lung fibrosis and improves survival in mouse models (15, 75, 76, 141–145), not all senescent cell types are profibrotic. In addition, not all senescent cells release SASP factors, and the onset of cellular senescence does not require SASP from other senescent cells (146). Another important confounding issue is that some senescent cell types and some SASP products—none of which are specific to senescent cells—can promote repair. For example, p16+ fibroblasts promote airway epithelial repair after naphthalene injuries via macrophage-derived IL-1β (24), and PDGF-AA secreted from senescent fibroblasts and endothelial cells promotes cutaneous wound healing (147), and other studies have concluded that senescent mesenchymal cells can contribute to tissue repair in injured lung, heart, and muscle (24, 148, 149). Along with the many studies demonstrating the profibrotic effects of senescent AT2 cells, as discussed in the preceding section, these studies indicate opposing consequences mediated by the senescence of different cell types in different tissue compartments. For example, senescent interstitial cells, like fibroblasts, could be beneficial in tissue regeneration, possibly by limiting their ability to be fibrotic, senescence of epithelial cells, particularly progenitor cells, would be detrimental by hindering reepithelialization and restoration of tissue function. Thus, understanding the unique mechanisms controlling senescent in specific cell types would be needed to design effective therapeutic approaches.
Senescent AT2 cells can recruit and activate immune cells, which can promote fibrosis and disease progression. Senescent AT2 cells in mice expressing a mutant surfactant protein C due to activation of UPR and ER stress and stimulate the recruitment of granulocytes (43). Similarly, deficiency of leucine-rich repeat kinase 2 in AT2 cells promotes bleomycin-induced senescence of these cells, which secrete CCL2 to recruit profibrotic macrophages (73). SASP components, such as IL-4 and IL-13, released by injury-induced senescent AT2 cells, can promote the transition of alveolar macrophages to a profibrotic phenotype (70, 150, 151). Findings from a preclinical model of SARS-CoV-2 pneumonia suggest that the virus caused senescence of AT2 cells that stimulated the recruitment of granulocytes and macrophages, which then caused lung injury (14). These findings indicate that the interplay between senescent AT2 cells and immune cells, mainly through SASP components, plays a crucial role in developing and progressing pulmonary fibrosis.
Senescent AT2 cells can modulate phenotypic changes in interstitial cells, such as fibroblasts, and vice versa. For example, SASP components released from senescent AT2 stimulate collagen production from fibroblasts in vitro (63). In addition, single-walled carbon nanotubes-induced AT2 senescence facilitated lung fibrosis by promoting the differentiation of lung fibroblasts to become myofibroblasts (133). Similarly, bleomycin-induced AT2 senescence activated lung fibroblasts through Nanog-WNT/β-catenin signaling pathway to promote lung fibrosis (152). Moreover, the loss of PTEN from AT2 cells promoted their senescence via NF-κB activation (63).
CONCLUSIONS
Senescence of alveolar epithelial progenitors, largely AT2 cells, plays an essential role in promoting progressive lung fibrosis. Current data suggest that alveolar epithelial progenitors are vulnerable to becoming senescent and orchestrating pathological processes in the lung microenvironment. Persistent alveolar injury and/or dysfunctional alveolar repair, such as due to progenitor cell exhaustion, are key initiating mechanisms in fibrotic lung diseases. Cellular senescence is likely the key causative process that impairs stemness capacity of alveolar epithelial progenitor cells, thereby negatively impacting effective lung repairing processes.
In contrast, the beneficial function of senescence during development and the role of senescent (p16+) interstitial cells in promoting repair and/or limiting fibrosis in lung, skeletal muscle, and liver (149) indicates that senescence is not universally bad. The somewhat opposing outcomes of senescence of different cell types in different tissue compartments (i.e., epithelium vs. interstitium) illustrate the complexity of targeting senescence regulatory networks as therapeutic interventions. In addition, these observations underscore the need for specific markers that can distinguish pathological senescent cells from beneficial or benign ones.
Nonmodifiable processes, such as aging and genetic mutation, and modifiable risk factors, including oxidative stress and mitochondria dysregulation, contribute to alveolar progenitor stemness exhaustion in IPF. Controllable risk factors are potential targets for effective prevention or therapeutic intervention, and metabolic reprogramming is emerging as an intervenable senescence regulatory mechanism (153). For example, the ability of zinc replenishment to reverse impaired self-renewal ability of AT2 cells from IPF lungs, which lack a zinc transporter (114), suggests that even approved supplements could improve AT2 progenitor function and, in turn, mitigate ongoing fibrogenesis. Alternative approaches include eradicating senescent cells or inhibiting the downstream effects of senescence. Since some senescent cells convey beneficial roles in lung regeneration, more targeted senescence elimination, either cell-specific or function-specific, would be more effective.
A challenge in preventing alveolar progenitor exhaustion is a lack of cell type-specific modifiable predisposing factors and targets in the early stages of senescence. Developing more appropriate animal models to recapitulate human pathology and using human biospecimens that represent better early cellular and molecular events are essential steps. In addition, the management of alveolar epithelial progenitor senescence should prioritize prevention or containment rather than the total elimination of senescent progenitors. Moreover, efforts should be made to explore more specific approaches to eliminate senescent cells. A first step toward selective eradication would be the identification of specific markers to differentiate pathological senescent cells from nonpathological senescent cells. Because such markers are currently not known, identification of specific features of disease-causing senescent cells would provide a significant advance in our understanding of and treatment of progressive fibrotic conditions.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants K08 HL141590 (to T.P.), R01 HL159953 and R01 HL155759 (to P.C.), P01 HL108793 (to P.W.N. and B.R.S.), R01 HL159953 (to W.C.P.), R01 HL151160 (to B.R.S.), and R35 HL150829 (to P.W.N.); National Institute on Aging Grant R01 AG078655 (to J.L. and P.W.N.); Foundation for the National Institute on Health Grant UCLA CTSI KL2 TR001882 (to T.P.); Parker B. Francis Foundation (to C.Y.); UCLA | Clinical and Translational Science Institute, University of California, Los Angeles (CTSI) Grant KL2-NCATSKL2TR001882 (to C.Y.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.P. and C.Y. prepared figures; T.P. and C.Y. drafted manuscript; T.P., P.C., B.R.S., J.L., D.J., P.W.N., W.C.P., and C.Y. edited and revised manuscript; T.P., P.C., B.R.S., J.L., D.J., P.W.N., W.C.P., and C.Y. approved final version of manuscript.
ACKNOWLEDGMENTS
The graphical abstract was created with BioRender.com.
REFERENCES
- 1. Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med 378: 1811–1823, 2018. doi: 10.1056/NEJMra1705751. [DOI] [PubMed] [Google Scholar]
- 2. Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, , et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med 205: e18–e47, 2022. doi: 10.1164/rccm.202202-0399ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Raghu G, Chen S-Y, Yeh W-S, Maroni B, Li Q, Lee Y-C, Collard HR. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med 2: 566–572, 2014. [Erratum in Lancet Respir Med 2: e12, 2014]. doi: 10.1016/S2213-2600(14)70101-8. [DOI] [PubMed] [Google Scholar]
- 4. Hama Amin BJ, Kakamad FH, Ahmed GS, Ahmed SF, Abdulla BA, Mohammed SH, Mikael TM, Salih RQ, Ali RK, Salh AM, Hussein DA. Post COVID-19 pulmonary fibrosis; a meta-analysis study. Ann Med Surg (Lond) 77: 103590, 2022. doi: 10.1016/j.amsu.2022.103590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bharat A, Querrey M, Markov NS, Kim S, Kurihara C, Garza-Castillon R, Manerikar A, Shilatifard A, Tomic R, Politanska Y, Abdala-Valencia H, Yeldandi AV, Lomasney JW, Misharin AV, Budinger GRS. Lung transplantation for patients with severe COVID-19. Sci Transl Med 12: eabe4282, 2020. doi: 10.1126/scitranslmed.abe4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roach A, Chikwe J, Catarino P, Rampolla R, Noble PW, Megna D, Chen Q, Emerson D, Egorova N, Keshavjee S, Kirklin JK. Lung transplantation for Covid-19-related respiratory failure in the United States. N Engl J Med 386: 1187–1188, 2022. doi: 10.1056/NEJMc2117024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wendisch D, Dietrich O, Mari T, von Stillfried S, Ibarra IL, Mittermaier M, , et al. SARS-CoV-2 infection triggers profibrotic macrophage responses and lung fibrosis. Cell 184: 6243–6261.e27, 2021. doi: 10.1016/j.cell.2021.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Myall KJ, Mukherjee B, Castanheira AM, Lam JL, Benedetti G, Mak SM, Preston R, Thillai M, Dewar A, Molyneaux PL, West AG. Persistent post-COVID-19 interstitial lung disease. An observational study of corticosteroid treatment. Ann Am Thorac Soc 18: 799–806, 2021. doi: 10.1513/AnnalsATS.202008-1002OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Basil MC, Katzen J, Engler AE, Guo M, Herriges MJ, Kathiriya JJ, Windmueller R, Ysasi AB, Zacharias WJ, Chapman HA, Kotton DN, Rock JR, Snoeck H-W, Vunjak-Novakovic G, Whitsett JA, Morrisey EE. The cellular and physiological basis for lung repair and regeneration: past, present, and future. Cell Stem Cell 26: 482–502, 2020. doi: 10.1016/j.stem.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Katzen J, Beers MF. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J Clin Invest 130: 5088–5099, 2020. doi: 10.1172/JCI139519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moss BJ, Ryter SW, Rosas IO. Pathogenic mechanisms underlying idiopathic pulmonary fibrosis. Annu Rev Pathol 17: 515–546, 2022. doi: 10.1146/annurev-pathol-042320-030240. [DOI] [PubMed] [Google Scholar]
- 12. Kropski JA, Blackwell TS. Progress in understanding and treating idiopathic pulmonary fibrosis. Annu Rev Med 70: 211–224, 2019. doi: 10.1146/annurev-med-041317-102715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moore C, Blumhagen RZ, Yang IV, Walts A, Powers J, Walker T, , et al. Resequencing study confirms that host defense and cell senescence gene variants contribute to the risk of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 200: 199–208, 2019. doi: 10.1164/rccm.201810-1891OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee S, Yu Y, Trimpert J, Benthani F, Mairhofer M, Richter-Pechanska P, , et al. Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature 599: 283–289, 2021. doi: 10.1038/s41586-021-03995-1. [DOI] [PubMed] [Google Scholar]
- 15. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, Mazula DL, Brooks RW, Fuhrmann-Stroissnigg H, Pirtskhalava T, Prakash YS, Tchkonia T, Robbins PD, Aubry MC, Passos JF, Kirkland JL, Tschumperlin DJ, Kita H, LeBrasseur NK. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 8: 14532, 2017. doi: 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. D'Agnillo F, Walters K-A, Xiao Y, Sheng Z-M, Scherler K, Park J, Gygli S, Rosas LA, Sadtler K, Kalish H, Blatti CA, Zhu R, Gatzke L, Bushell C, Memoli MJ, O'Day SJ, Fischer TD, Hammond TC, Lee RC, Cash JC, Powers ME, O'Keefe GE, Butnor KJ, Rapkiewicz AV, Travis WD, Layne SP, Kash JC, Taubenberger JK. Lung epithelial and endothelial damage, loss of tissue repair, inhibition of fibrinolysis, and cellular senescence in fatal COVID-19. Sci Transl Med 13: eabj7790, 2021. doi: 10.1126/scitranslmed.abj7790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Planer JD, Morrisey EE. After the storm: regeneration, repair, and reestablishment of homeostasis between the alveolar epithelium and innate immune system following viral lung injury. Annu Rev Pathol 18: 337–359, 2023. doi: 10.1146/annurev-pathmechdis-031621-024344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gillich A, Zhang F, Farmer CG, Travaglini KJ, Tan SY, Gu M, Zhou B, Feinstein JA, Krasnow MA, Metzger RJ. Capillary cell-type specialization in the alveolus. Nature 586: 785–789, 2020. doi: 10.1038/s41586-020-2822-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shiraishi K, Shah PP, Morley MP, Loebel C, Santini GT, Katzen J, Basil MC, Lin SM, Planer JD, Cantu E, Jones DL, Nottingham AN, Li S, Cardenas-Diaz FL, Zhou S, Burdick JA, Jain R, Morrisey EE. Biophysical forces mediated by respiration maintain lung alveolar epithelial cell fate. Cell 186: 1478–1492.e15, 2023. doi: 10.1016/j.cell.2023.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kotton DN, Morrisey EE. Lung regeneration: mechanisms, applications and emerging stem cell populations. Nat Med 20: 822–832, 2014. doi: 10.1038/nm.3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Olajuyin AM, Zhang X, Ji H-L. Alveolar type 2 progenitor cells for lung injury repair. Cell Death Discov 5: 63, 2019. doi: 10.1038/s41420-019-0147-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barnes PJ, Baker J, Donnelly LE. Cellular senescence as a mechanism and target in chronic lung diseases. Am J Respir Crit Care Med 200: 556–564, 2019. doi: 10.1164/rccm.201810-1975TR. [DOI] [PubMed] [Google Scholar]
- 23. Parimon T, Hohmann MS, Yao C. Cellular senescence: pathogenic mechanisms in lung fibrosis. Int J Mol Sci 22: 6214, 2021. doi: 10.3390/ijms22126214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reyes NS, Krasilnikov M, Allen NC, Lee JY, Hyams B, Zhou M, Ravishankar S, Cassandras M, Wang C, Khan I, Matatia P, Johmura Y, Molofsky A, Matthay M, Nakanishi M, Sheppard D, Campisi J, Peng T. Sentinel p16INK4a+ cells in the basement membrane form a reparative niche in the lung. Science 378: 192–201, 2022. doi: 10.1126/science.abf3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zysman M, Baptista BR, Essari L-A, Taghizadeh S, Ménonville C, Giffard C, Issa A, Franco-Montoya M-L, Breau M, Souktani R, Aissat A, Caeymaex L, Lizé M, Nhieu JTV, Jung C, Rottier R, Cruzeiro MD, Adnot S, Epaud R, Chabot F, Lanone S, Boczkowski J, Boyer L. Targeting p16INK4a promotes lipofibroblasts and alveolar regeneration after early-life injury. Am J Respir Crit Care Med 202: 1088–1104, 2020. doi: 10.1164/rccm.201908-1573OC. [DOI] [PubMed] [Google Scholar]
- 26. Alysandratos K-D, Herriges MJ, Kotton DN. Epithelial stem and progenitor cells in lung repair and regeneration. Annu Rev Physiol 83: 529–550, 2021. doi: 10.1146/annurev-physiol-041520-092904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xie T, Lynn H, Parks WC, Stripp B, Chen P, Jiang D, Noble PW. Abnormal respiratory progenitors in fibrotic lung injury. Stem Cell Res Ther 13: 64, 2022. doi: 10.1186/s13287-022-02737-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. DePianto DJ, Heiden JAV, Morshead KB, Sun KH, Modrusan Z, Teng G, Wolters PJ, Arron JR. Molecular mapping of interstitial lung disease reveals a phenotypically distinct senescent basal epithelial cell population. JCI Insight 6: e143626, 2021. doi: 10.1172/jci.insight.143626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Strunz M, Simon LM, Ansari M, Kathiriya JJ, Angelidis I, Mayr CH, Tsidiridis G, Lange M, Mattner LF, Yee M, Ogar P, Sengupta A, Kukhtevich I, Schneider R, Zhao Z, Voss C, Stoeger T, Neumann JHL, Hilgendorff A, Behr J, O'Reilly M, Lehmann M, Burgstaller G, Königshoff M, Chapman HA, Theis FJ, Schiller HB. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat Commun 11: 3559, 2020. doi: 10.1038/s41467-020-17358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW, Hogan BLM. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest 123: 3025–3036, 2013. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liang J, Zhang Y, Xie T, Liu N, Chen H, Geng Y, Kurkciyan A, Mena JM, Stripp BR, Jiang D, Noble PW. Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nat Med 22: 1285–1293, 2016. doi: 10.1038/nm.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, Desai TJ. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 359: 1118–1123, 2018. doi: 10.1126/science.aam6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zacharias WJ, Frank DB, Zepp JA, Morley MP, Alkhaleel FA, Kong J, Zhou S, Cantu E, Morrisey EE. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 555: 251–255, 2018. doi: 10.1038/nature25786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hogan Brigid LM, Barkauskas Christina E, Chapman Harold A, Epstein Jonathan A, Jain R, Hsia Connie CW, Niklason L, Calle E, Le A, Randell Scott H, Rock J, Snitow M, Krummel M, Stripp Barry R, Vu T, White Eric S, Whitsett Jeffrey A, Morrisey Edward E. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell 15: 123–138, 2014. doi: 10.1016/j.stem.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kobayashi Y, Tata A, Konkimalla A, Katsura H, Lee RF, Ou J, Banovich NE, Kropski JA, Tata PR. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat Cell Biol 22: 934–946, 2020. doi: 10.1038/s41556-020-0542-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Heinzelmann K, Hu Q, Hu Y, Dobrinskikh E, Ansari M, Melo-Narváez MC, Ulke HM, Leavitt C, Mirita C, Trudeau T, Saal ML, Rice P, Gao B, Janssen WJ, Yang IV, Schiller HB, Vladar EK, Lehmann M, Königshoff M. Single-cell RNA sequencing identifies G-protein coupled receptor 87 as a basal cell marker expressed in distal honeycomb cysts in idiopathic pulmonary fibrosis. Eur Respir J 59: 2102373, 2022. doi: 10.1183/13993003.02373-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, Peter L, Chung M-I, Taylor CJ, Jetter C, Raju L, Roberson J, Ding G, Wood L, Sucre JMS, Richmond BW, Serezani AP, McDonnell WJ, Mallal SB, Bacchetta MJ, Loyd JE, Shaver CM, Ware LB, Bremner R, Walia R, Blackwell TS, Banovich NE, Kropski JA. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv 6: eaba1972, 2020. doi: 10.1126/sciadv.aba1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jiang P, Gil de Rubio R, Hrycaj SM, Gurczynski SJ, Riemondy KA, Moore BB, Omary MB, Ridge KM, Zemans RL. Ineffectual type 2–to–type 1 alveolar epithelial cell differentiation in idiopathic pulmonary fibrosis: persistence of the KRT8hi transitional state. Am J Respir Crit Care Med 201: 1443–1447, 2020. doi: 10.1164/rccm.201909-1726LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Joshi N, Watanabe S, Verma R, Jablonski RP, Chen CI, Cheresh P, Markov NS, Reyfman PA, McQuattie-Pimentel AC, Sichizya L, Lu Z, Piseaux-Aillon R, Kirchenbuechler D, Flozak AS, Gottardi CJ, Cuda CM, Perlman H, Jain M, Kamp DW, Budinger GRS, Misharin AV. A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M-CSF/M-CSFR signalling in monocyte-derived alveolar macrophages. Eur Respir J 55: 1900646, 2020. doi: 10.1183/13993003.00646-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Riemondy KA, Jansing NL, Jiang P, Redente EF, Gillen AE, Fu R, Miller AJ, Spence JR, Gerber AN, Hesselberth JR, Zemans RL. Single cell RNA sequencing identifies TGFβ as a key regenerative cue following LPS-induced lung injury. JCI Insight 5: e123637, 2019. doi: 10.1172/jci.insight.123637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Choi J, Park J-E, Tsagkogeorga G, Yanagita M, Koo B-K, Han N, Lee J-H. Inflammatory signals induce AT2 cell-derived damage-associated transient progenitors that mediate alveolar regeneration. Cell Stem Cell 27: 366–382.e7, 2020. doi: 10.1016/j.stem.2020.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Auyeung VC, Downey MS, Thamsen M, Wenger TA, Backes BJ, Sheppard D, Papa FR. IRE1α drives lung epithelial progenitor dysfunction to establish a niche for pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 322: L564–L580, 2022. doi: 10.1152/ajplung.00408.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Katzen J, Rodriguez L, Tomer Y, Babu A, Zhao M, Murthy A, Carson P, Barrett M, Basil MC, Carl J, Leach JP, Morley M, McGraw MD, Mulugeta S, Pelura T, Rosen G, Morrisey EE, Beers MF. Disruption of proteostasis causes IRE1 mediated reprogramming of alveolar epithelial cells. Proc Natl Acad Sci USA 119: e2123187119, 2022. doi: 10.1073/pnas.2123187119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ray S, Chiba N, Yao C, Guan X, McConnell AM, Brockway B, Que L, McQualter JL, Stripp BR. Rare SOX2+ airway progenitor cells generate KRT5+ cells that repopulate damaged alveolar parenchyma following influenza virus infection. Stem Cell Reports 7: 817–825, 2016. doi: 10.1016/j.stemcr.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zuo W, Zhang T, Wu DZA, Guan SP, Liew A-A, Yamamoto Y, Wang X, Lim SJ, Vincent M, Lessard M, Crum CP, Xian W, McKeon F. p63+Krt5+ distal airway stem cells are essential for lung regeneration. Nature 517: 616–620, 2015. doi: 10.1038/nature13903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yang Y, Riccio P, Schotsaert M, Mori M, Lu J, Lee D-K, García-Sastre A, Xu J, Cardoso WV. Spatial-temporal lineage restrictions of embryonic p63+ progenitors establish distinct stem cell pools in adult airways. Dev Cell 44: 752–761.e4, 2018. doi: 10.1016/j.devcel.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Huang H, Fang Y, Jiang M, Zhang Y, Biermann J, Melms JC, Danielsson JA, Yang Y, Qiang L, Liu J, Zhou Y, Wang M, Hu Z, Wang TC, Saqi A, Sun J, Matsumoto I, Cardoso WV, Emala CW, Zhu J, Izar B, Mou H, Que J. Contribution of Trp63CreERT2-labeled cells to alveolar regeneration is independent of tuft cells. eLife 11: e78217, 2022. doi: 10.7554/eLife.78217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xi Y, Kim T, Brumwell AN, Driver IH, Wei Y, Tan V, Jackson JR, Xu J, Lee DK, Gotts JE, Matthay MA, Shannon JM, Chapman HA, Vaughan AE. Local lung hypoxia determines epithelial fate decisions during alveolar regeneration. Nat Cell Biol 19: 904–914, 2017. doi: 10.1038/ncb3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zuo WL, Rostami MR, LeBlanc M, Kaner RJ, O'Beirne SL, Mezey JG, Leopold PL, Quast K, Visvanathan S, Fine JS, Thomas MJ, Crystal RG. Dysregulation of club cell biology in idiopathic pulmonary fibrosis. PLoS One 15: e0237529, 2020. doi: 10.1371/journal.pone.0237529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Basil MC, Cardenas-Diaz FL, Kathiriya JJ, Morley MP, Carl J, Brumwell AN, Katzen J, Slovik KJ, Babu A, Zhou S, Kremp MM, McCauley KB, Li S, Planer JD, Hussain SS, Liu X, Windmueller R, Ying Y, Stewart KM, Oyster M, Christie JD, Diamond JM, Engelhardt JF, Cantu E, Rowe SM, Kotton DN, Chapman HA, Morrisey EE. Human distal airways contain a multipotent secretory cell that can regenerate alveoli. Nature 604: 120–126, 2022. doi: 10.1038/s41586-022-04552-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vaughan AE, Brumwell AN, Xi Y, Gotts JE, Brownfield DG, Treutlein B, Tan K, Tan V, Liu FC, Looney MR, Matthay MA, Rock JR, Chapman HA. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature 517: 621–625, 2015. doi: 10.1038/nature14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kathiriya JJ, Brumwell AN, Jackson JR, Tang X, Chapman HA. Distinct airway epithelial stem cells hide among club cells but mobilize to promote alveolar regeneration. Cell Stem Cell 26: 346–358.e44, 2020. doi: 10.1016/j.stem.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kadur Lakshminarasimha Murthy P, Sontake V, Tata A, Kobayashi Y, Macadlo L, Okuda K, Conchola AS, Nakano S, Gregory S, Miller LA, Spence JR, Engelhardt JF, Boucher RC, Rock JR, Randell SH, Tata PR. Human distal lung maps and lineage hierarchies reveal a bipotent progenitor. Nature 604: 111–119, 2022. doi: 10.1038/s41586-022-04541-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee JH, Bhang DH, Beede A, Huang TL, Stripp BR, Bloch KD, Wagers AJ, Tseng YH, Ryeom S, Kim CF. Lung stem cell differentiation in mice directed by endothelial cells via a BMP4-NFATc1-thrombospondin-1 axis. Cell 156: 440–455, 2014. doi: 10.1016/j.cell.2013.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu Q, Liu K, Cui G, Huang X, Yao S, Guo W, Qin Z, Li Y, Yang R, Pu W, Zhang L, He L, Zhao H, Yu W, Tang M, Tian X, Cai D, Nie Y, Hu S, Ren T, Qiao Z, Huang H, Zeng YA, Jing N, Peng G, Ji H, Zhou B. Lung regeneration by multipotent stem cells residing at the bronchioalveolar-duct junction. Nat Genet 51: 728–738, 2019. [Erratum in Nat Genet 2019 ]. doi: 10.1038/s41588-019-0346-6. [DOI] [PubMed] [Google Scholar]
- 56. Watson JK, Sanders P, Dunmore R, Rosignoli G, Julé Y, Rawlins EL, Mustelin T, May R, Clarke D, Finch DK. Distal lung epithelial progenitor cell function declines with age. Sci Rep 10: 10490, 2020. doi: 10.1038/s41598-020-66966-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McConnell A, Yao C, Yeckes A, Wang Y, Selvaggio AS, Tang J, Kirsch D, Stripp B. p53 Regulates progenitor cell quiescence and differentiation in the airway. Cell Rep 17: 2173–2182, 2016. doi: 10.1016/j.celrep.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 58. Di Micco R, Krizhanovsky V, Baker D, d'Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol 22: 75–95, 2021. doi: 10.1038/s41580-020-00314-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huang W, Hickson LJ, Eirin A, Kirkland JL, Lerman LO. Cellular senescence: the good, the bad and the unknown. Nat Rev Nephrol 18: 611–627, 2022. doi: 10.1038/s41581-022-00601-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends Cell Biol 28: 436–453, 2018. doi: 10.1016/j.tcb.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 61. González-Gualda E, Baker AG, Fruk L, Muñoz-Espín D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J 288: 56–80, 2021. doi: 10.1111/febs.15570. [DOI] [PubMed] [Google Scholar]
- 62. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5: 99–118, 2010. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tian Y, Li H, Qiu T, Dai J, Zhang Y, Chen J, Cai H. Loss of PTEN induces lung fibrosis via alveolar epithelial cell senescence depending on NF-κB activation. Aging Cell 18: e12858, 2019. doi: 10.1111/acel.12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chung EJ, Reedy JL, Kwon S, Patil S, Valle L, White AO, Citrin DE. 12-Lipoxygenase is a critical mediator of type II pneumocyte senescence, macrophage polarization and pulmonary fibrosis after irradiation. Radiat Res 192: 367–379, 2019. doi: 10.1667/RR15356.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chen H, Chen H, Liang J, Gu X, Zhou J, Xie C, Lv X, Wang R, Li Q, Mao Z, Sun H, Zuo G, Miao D, Jin J. TGF-β1/IL-11/MEK/ERK signaling mediates senescence-associated pulmonary fibrosis in a stress-induced premature senescence model of Bmi-1 deficiency. Exp Mol Med 52: 130–151, 2020. doi: 10.1038/s12276-019-0371-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yamada Z, Nishio J, Motomura K, Mizutani S, Yamada S, Mikami T, Nanki T. Senescence of alveolar epithelial cells impacts initiation and chronic phases of murine fibrosing interstitial lung disease. Front Immunol 13: 935114, 2022. [Erratum in Front Immunol 14: 1201209, 2023]. doi: 10.3389/fimmu.2022.935114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ng B, Cook SA, Schafer S. Interleukin-11 signaling underlies fibrosis, parenchymal dysfunction, and chronic inflammation of the airway. Exp Mol Med 52: 1871–1878, 2020. doi: 10.1038/s12276-020-00531-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang L, Tong X, Huang J, Wu M, Zhang S, Wang D, Liu S, Fan H. Fisetin alleviated bleomycin-induced pulmonary fibrosis partly by rescuing alveolar epithelial cells from senescence. Front Pharmacol 11: 553690, 2020. doi: 10.3389/fphar.2020.553690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Aoshiba K, Tsuji T, Kameyama S, Itoh M, Semba S, Yamaguchi K, Nakamura H. Senescence-associated secretory phenotype in a mouse model of bleomycin-induced lung injury. Exp Toxicol Pathol 65: 1053–1062, 2013. doi: 10.1016/j.etp.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 70. Rana T, Jiang C, Liu G, Miyata T, Antony V, Thannickal VJ, Liu R-M. PAI-1 regulation of TGF-β1–induced alveolar type II cell senescence, SASP secretion, and SASP-mediated activation of alveolar macrophages. Am J Respir Cell Mol Biol 62: 319–330, 2020. doi: 10.1165/rcmb.2019-0071OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chung SI, Horton JA, Ramalingam TR, White AO, Chung EJ, Hudak KE, Scroggins BT, Arron JR, Wynn TA, Citrin DE. IL-13 is a therapeutic target in radiation lung injury. Sci Rep 6: 39714, 2016. doi: 10.1038/srep39714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Murray LA, Zhang H, Oak SR, Coelho AL, Herath A, Flaherty KR, Lee J, Bell M, Knight DA, Martinez FJ, Sleeman MA, Herzog EL, Hogaboam CM. Targeting interleukin-13 with tralokinumab attenuates lung fibrosis and epithelial damage in a humanized SCID idiopathic pulmonary fibrosis model. Am J Respir Cell Mol Biol 50: 985–994, 2014. doi: 10.1165/rcmb.2013-0342OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tian Y, Lv J, Su Z, Wu T, Li X, Hu X, Zhang J, Wu L. LRRK2 plays essential roles in maintaining lung homeostasis and preventing the development of pulmonary fibrosis. Proc Natl Acad Sci USA 118: e2106685118, 2021. doi: 10.1073/pnas.2106685118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Liu S, Liu C, Wang Q, Liu S, Min J. CC Chemokines in idiopathic pulmonary fibrosis: pathogenic role and therapeutic potential. Biomolecules 13: 333, 2023. doi: 10.3390/biom13020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yao C, Guan X, Carraro G, Parimon T, Liu X, Huang G, Mulay A, Soukiasian HJ, David G, Weigt SS, Belperio JA, Chen P, Jiang D, Noble PW, Stripp BR. Senescence of alveolar type 2 cells drives progressive pulmonary fibrosis. Am J Respir Crit Care Med 203: 707–717, 2021. doi: 10.1164/rccm.202004-1274OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lehmann M, Korfei M, Mutze K, Klee S, Skronska-Wasek W, Alsafadi HN, Ota C, Costa R, Schiller HB, Lindner M, Wagner DE, Günther A, Königshoff M. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur Respir J 50: 1602367, 2017. doi: 10.1183/13993003.02367-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Shivshankar P, Brampton C, Miyasato S, Kasper M, Thannickal VJ, Saux CJL. Caveolin-1 deficiency protects from pulmonary fibrosis by modulating epithelial cell senescence in mice. Am J Respir Cell Mol Biol 47: 28–36, 2012. doi: 10.1165/rcmb.2011-0349OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Jiang C, Liu G, Luckhardt T, Antony V, Zhou Y, Carter AB, Thannickal VJ, Liu R-M. Serpine 1 induces alveolar type II cell senescence through activating p53-p21-Rb pathway in fibrotic lung disease. Aging Cell 16: 1114–1124, 2017. doi: 10.1111/acel.12643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Longhorne FL, Wilkinson HN, Hardman MJ, Hart SP. Dexamethasone induces senescence of lung epithelial cells and augments TGF-β1-mediated production of the fibrosis mediator serpin E1 (plasminogen activator inhibitor-1) (Preprint). bioRxiv, 2011. doi: 10.1101/2021.11.29.470337. [DOI]
- 80. Zhang Y, Huang W, Zheng Z, Wang W, Yuan Y, Hong Q, Lin J, Li X, Meng Y. Cigarette smoke-inactivated SIRT1 promotes autophagy-dependent senescence of alveolar epithelial type 2 cells to induce pulmonary fibrosis. Free Radic Biol Med 166: 116–127, 2021. doi: 10.1016/j.freeradbiomed.2021.02.013. [DOI] [PubMed] [Google Scholar]
- 81. Qiu T, Tian Y, Gao Y, Ma M, Li H, Liu X, Wu H, Zhang Y, Ding H, Cao M, Zhang J, Dai J, Chen J, Cai H. PTEN loss regulates alveolar epithelial cell senescence in pulmonary fibrosis depending on Akt activation. Aging (Albany NY) 11: 7492–7509, 2019. doi: 10.18632/aging.102262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhou S, Zhu J, Zhou P-K, Gu Y. Alveolar type 2 epithelial cell senescence and radiation-induced pulmonary fibrosis. Front Cell Dev Biol 10: 999600, 2022. doi: 10.3389/fcell.2022.999600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hong X, Wang L, Zhang K, Liu J, Liu J-P. Molecular mechanisms of alveolar epithelial stem cell senescence and senescence-associated differentiation disorders in pulmonary fibrosis. Cells 11: 877, 2022. doi: 10.3390/cells11050877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Aoshiba K, Tsuji T, Nagai A. Bleomycin induces cellular senescence in alveolar epithelial cells. Eur Respir J 22: 436–443, 2003. doi: 10.1183/09031936.03.00011903. [DOI] [PubMed] [Google Scholar]
- 85. Wu H, Yu Y, Huang H, Hu Y, Fu S, Wang Z, Shi M, Zhao X, Yuan J, Li J, Yang X, Bin E, Wei D, Zhang H, Zhang J, Yang C, Cai T, Dai H, Chen J, Tang N. Progressive pulmonary fibrosis is caused by elevated mechanical tension on alveolar stem cells. Cell 180: 107–121.e17, 2020. doi: 10.1016/j.cell.2019.11.027. [DOI] [PubMed] [Google Scholar]
- 86. Juul NH, Stockman CA, Desai TJ. Niche cells and signals that regulate lung alveolar stem cells in vivo. Cold Spring Harb Perspect Biol 12: a035717, 2020. [Erratum in Cold Spring Harb Perspect Biol 12: a040303, 2020]. doi: 10.1101/cshperspect.a035717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Nureki S-I, Tomer Y, Venosa A, Katzen J, Russo SJ, Jamil S, Barrett M, Nguyen V, Kopp M, Mulugeta S, Beers MF. Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J Clin Invest 128: 4008–4024, 2018. doi: 10.1172/JCI99287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hamsanathan S, Alder JK, Sellares J, Rojas M, Gurkar AU, Mora AL. Cellular senescence: the trojan horse in chronic lung diseases. Am J Respir Cell Mol Biol 61: 21–30, 2019. doi: 10.1165/rcmb.2018-0410TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zhang K, Wang L, Hong X, Chen H, Shi Y, Liu Y, Liu J, Liu JP. Pulmonary alveolar stem cell senescence, apoptosis, and differentiation by p53-dependent and -independent mechanisms in telomerase-deficient mice. Cells 10: 2892, 2021. doi: 10.3390/cells10112892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Lv X, Liu C, Liu S, Li Y, Wang W, Li K, Hua F, Cui B, Zhang X, Yu J, Yu J, Hu Z. The cell cycle inhibitor P21 promotes the development of pulmonary fibrosis by suppressing lung alveolar regeneration. Acta Pharm Sin B 12: 735–746, 2022. doi: 10.1016/j.apsb.2021.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Álvarez D, Cárdenes N, Sellarés J, Bueno M, Corey C, Hanumanthu VS, Peng Y, D’Cunha H, Sembrat J, Nouraie M, Shanker S, Caufield C, Shiva S, Armanios M, Mora AL, Rojas M. IPF lung fibroblasts have a senescent phenotype. Am J Physiol Lung Cell Mol Physiol 313: L1164–L1173, 2017. doi: 10.1152/ajplung.00220.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lipskaia L, Maisonnasse P, Fouillade C, Sencio V, Pascal Q, Flaman J-M, Born E, Londono-Vallejo A, Grand RL, Bernard D, Trottein F, Adnot S. Evidence that SARS-CoV-2 induces lung cell senescence: potential impact on COVID-19 lung disease. Am J Respir Cell Mol Biol 66: 107–111, 2022. doi: 10.1165/rcmb.2021-0205LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, , et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med 199: 1517–1536, 2019. doi: 10.1164/rccm.201712-2410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, Chu SG, Raby BA, DeIuliis G, Januszyk M, Duan Q, Arnett HA, Siddiqui A, Washko GR, Homer R, Yan X, Rosas IO, Kaminski N. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv 6: eaba1983, 2020. doi: 10.1126/sciadv.aba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Chilosi M, Carloni A, Rossi A, Poletti V. Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. Transl Res 162: 156–173, 2013. doi: 10.1016/j.trsl.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 96. Parimon T, Espindola M, Marchevsky A, Rampolla R, Chen P, Hogaboam CM. Potential mechanisms for lung fibrosis associated with COVID-19 infection. QJM hcac206, 2022. doi: 10.1093/qjmed/hcac206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kim Y-I, Shin H-W, Chun Y-S, Cho C-H, Koh J, Chung DH, Park J-W. Epithelial cell-derived cytokines CST3 and GDF15 as potential therapeutics for pulmonary fibrosis. Cell Death Dis 9: 506, 2018. doi: 10.1038/s41419-018-0530-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Han X, Lei Q, Xie J, Liu H, Li J, Zhang X, Zhang T, Gou X. Potential regulators of the senescence-associated secretory phenotype during senescence and aging. J Gerontol A Biol Sci Med Sci 77: 2207–2218, 2022. doi: 10.1093/gerona/glac097. [DOI] [PubMed] [Google Scholar]
- 99. Cui L, Fang Z, De Souza CM, Lerbs T, Guan Y, Li I, Charu V, Chen S-Y, Weissman I, Wernig G. Innate immune cell activation causes lung fibrosis in a humanized model of long COVID. Proc Natl Acad Sci USA 120: e2217199120, 2023. doi: 10.1073/pnas.2217199120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Evangelou K, Veroutis D, Paschalaki K, Foukas PG, Lagopati N, Dimitriou M, Papaspyropoulos A, Konda B, Hazapis O, Polyzou A, Havaki S, Kotsinas A, Kittas C, Tzioufas AG, de Leval L, Vassilakos D, Tsiodras S, Stripp BR, Papantonis A, Blandino G, Karakasiliotis I, Barnes PJ, Gorgoulis VG. Pulmonary infection by SARS-CoV-2 induces senescence accompanied by an inflammatory phenotype in severe COVID-19: possible implications for viral mutagenesis. Eur Respir J 60: 2102951, 2022. doi: 10.1183/13993003.02951-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Sinha S, Castillo V, Espinoza CR, Tindle C, Fonseca AG, Dan JM, Katkar GD, Das S, Sahoo D, Ghosh P. COVID-19 lung disease shares driver AT2 cytopathic features with idiopathic pulmonary fibrosis. eBioMedicine 82: 104185, 2022. doi: 10.1016/j.ebiom.2022.104185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Paschalaki K, Evangelou K, Veroutis D, Foukas P, Kittas C, Tzioufas A, De Leval L, Vassilakos D, Barnes P, Gorgoulis V. Alveolar type II cells harbouring SARS-CoV-2 show senescence with a proinflammatory phenotype. Eur Respir J 58: OA4312, 2021. doi: 10.1183/13993003.congress-2021.OA4312. [DOI] [Google Scholar]
- 103. Martínez-Zamudio RI, Robinson L, Roux PF, Bischof O. SnapShot: cellular senescence pathways. Cell 170: 816–816.e1, 2017. doi: 10.1016/j.cell.2017.07.049. [DOI] [PubMed] [Google Scholar]
- 104. Mijit M, Caracciolo V, Melillo A, Amicarelli F, Giordano A. Role of p53 in the regulation of cellular senescence. Biomolecules 10: 420, 2020. doi: 10.3390/biom10030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Spike BT, Wahl GM. p53, Stem cells, and reprogramming: tumor suppression beyond guarding the genome. Genes Cancer 2: 404–419, 2011. doi: 10.1177/1947601911410224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Yosef R, Pilpel N, Papismadov N, Gal H, Ovadya Y, Vadai E, Miller S, Porat Z, Ben-Dor S, Krizhanovsky V. p21 Maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J 36: 2280–2295, 2017. doi: 10.15252/embj.201695553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Safwan-Zaiter H, Wagner N, Wagner K-D. P16INK4A: more than a senescence marker. Life (Basel) 12: 1332, 2022. doi: 10.3390/life12091332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev 9: 1149–1163, 1995. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- 109. Chellappan SP, Hiebert S, Mudryj M, Horowitz JM, Nevins JR. The E2F transcription factor is a cellular target for the RB protein. Cell 65: 1053–1061, 1991. doi: 10.1016/0092-8674(91)90557-f. [DOI] [PubMed] [Google Scholar]
- 110. Ribeiro Baptista B, Justeau G, Toigo M, Zysman M, Thiebaut De Menonville C, Belgacemi R, Chabot F, Lanone S, Derumeaux G, Boczkowski J, Boyer L. Targeting p16 to induce Krt8+ transitional stem cell state and promote alveolar regeneration. Eur Respir J 60: 3173, 2022. doi: 10.1183/13993003.congress-2022.3173. [DOI] [Google Scholar]
- 111. Guan R, Cai Z, Wang J, Ding M, Li Z, Xu J, Li Y, Li J, Yao H, Liu W, Qian J, Deng B, Tang C, Sun D, Lu W. Hydrogen sulfide attenuates mitochondrial dysfunction-induced cellular senescence and apoptosis in alveolar epithelial cells by upregulating sirtuin 1. Aging (Albany NY) 11: 11844–11864, 2019. doi: 10.18632/aging.102454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Fang L, Li H, Tang C, Geng B, Qi Y, Liu X. Hydrogen sulfide attenuates the pathogenesis of pulmonary fibrosis induced by bleomycin in rats. Can J Physiol Pharmacol 87: 531–538, 2009. doi: 10.1139/y09-039. [DOI] [PubMed] [Google Scholar]
- 113. Kadota T, Yoshioka Y, Fujita Y, Araya J, Minagawa S, Hara H, Miyamoto A, Suzuki S, Fujimori S, Kohno T, Fujii T, Kishi K, Kuwano K, Ochiya T. Extracellular vesicles from fibroblasts induce epithelial-cell senescence in pulmonary fibrosis. Am J Respir Cell Mol Biol 63: 623–636, 2020. doi: 10.1165/rcmb.2020-0002OC. [DOI] [PubMed] [Google Scholar]
- 114. Liang J, Huang G, Liu X, Taghavifar F, Liu N, Wang Y, Deng N, Yao C, Xie T, Kulur V, Dai K, Burman A, Rowan SC, Weigt SS, Belperio J, Stripp B, Parks WC, Jiang D, Noble PW. The ZIP8/SIRT1 axis regulates alveolar progenitor cell renewal in aging and idiopathic pulmonary fibrosis. J Clin Invest 132: e157338, 2022. doi: 10.1172/JCI157338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Galbiati F, Volonté D, Liu J, Capozza F, Frank PG, Zhu L, Pestell RG, Lisanti MP. Caveolin-1 expression negatively regulates cell cycle progression by inducing G0/G1 arrest via a p53/p21WAF1/Cip1-dependent mechanism. Mol Biol Cell 12: 2229–2244, 2001. doi: 10.1091/mbc.12.8.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Venkatesan S, Fan L, Tang H, Konduru NV, Shetty S. Caveolin-1 scaffolding domain peptide abrogates autophagy dysregulation in pulmonary fibrosis. Sci Rep 12: 11086, 2022. doi: 10.1038/s41598-022-14832-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Lehmann M, Hu Q, Hu Y, Hafner K, Costa R, van den Berg A, Königshoff M. Chronic WNT/β-catenin signaling induces cellular senescence in lung epithelial cells. Cell Signal 70: 109588, 2020. doi: 10.1016/j.cellsig.2020.109588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Sun W, Jing X, Yang X, Huang H, Luo Q, Xia S, Wang P, Wang N, Zhang Q, Guo J, Xu Z. Regulation of the IGF1 signaling pathway is involved in idiopathic pulmonary fibrosis induced by alveolar epithelial cell senescence and core fucosylation. Aging (Albany NY) 13: 18852–18869, 2021. doi: 10.18632/aging.203335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Chin C, Ravichandran R, Sanborn K, Fleming T, Wheatcroft SB, Kearney MT, Tokman S, Walia R, Smith MA, Flint DJ, Mohanakumar T, Bremner RM, Sureshbabu A. Loss of IGFBP2 mediates alveolar type 2 cell senescence and promotes lung fibrosis. Cell Rep Med 4: 100945, 2023. doi: 10.1016/j.xcrm.2023.100945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Snetselaar R, van Batenburg AA, van Oosterhout MFM, Kazemier KM, Roothaan SM, Peeters T, van der Vis JJ, Goldschmeding R, Grutters JC, van Moorsel CHM. Short telomere length in IPF lung associates with fibrotic lesions and predicts survival. PLoS One 12: e0189467, 2017. doi: 10.1371/journal.pone.0189467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. van Batenburg AA, Kazemier KM, van Oosterhout MFM, van der Vis JJ, Grutters JC, Goldschmeding R, van Moorsel CHM. Telomere shortening and DNA damage in culprit cells of different types of progressive fibrosing interstitial lung disease. ERJ Open Res 7: 00691-2020, 2021. doi: 10.1183/23120541.00691-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Naikawadi RP, Disayabutr S, Mallavia B, Donne ML, Green G, La JL, Rock JR, Looney MR, Wolters PJ. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight 1: e86704, 2016. doi: 10.1172/jci.insight.86704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Povedano JM, Martinez P, Flores JM, Mulero F, Blasco MA. Mice with pulmonary fibrosis driven by telomere dysfunction. Cell Rep 12: 286–299, 2015. doi: 10.1016/j.celrep.2015.06.028. [DOI] [PubMed] [Google Scholar]
- 124. Yu TY, Kao YW, Lin JJ. Telomeric transcripts stimulate telomere recombination to suppress senescence in cells lacking telomerase. Proc Natl Acad Sci USA 111: 3377–3382, 2014. doi: 10.1073/pnas.1307415111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Piñeiro-Hermida S, Martínez P, Bosso G, Flores JM, Saraswati S, Connor J, Lemaire R, Blasco MA. Consequences of telomere dysfunction in fibroblasts, club and basal cells for lung fibrosis development. Nat Commun 13: 5656, 2022. doi: 10.1038/s41467-022-32771-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Wang L, Chen R, Li G, Wang Z, Liu J, Liang Y, Liu JP. FBW7 mediates senescence and pulmonary fibrosis through telomere uncapping. Cell Metab 32: 860–877.e9, 2020. doi: 10.1016/j.cmet.2020.10.004. [DOI] [PubMed] [Google Scholar]
- 127. Wang H, Xu H, Lyu W, Xu Q, Fan S, Chen H, Wang D, Chen J, Dai J. KLF4 regulates TERT expression in alveolar epithelial cells in pulmonary fibrosis. Cell Death Dis 13: 435, 2022. doi: 10.1038/s41419-022-04886-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Lawson WE, Cheng D-S, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, Newcomb DC, Jones BR, Roldan J, Lane KB, Morrisey EE, Beers MF, Yull FE, Blackwell TS. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci USA 108: 10562–10567, 2011. doi: 10.1073/pnas.1107559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Katzen J, Wagner BD, Venosa A, Kopp M, Tomer Y, Russo SJ, Headen AC, Basil MC, Stark JM, Mulugeta S, Deterding RR, Beers MF. An SFTPC BRICHOS mutant links epithelial ER stress and spontaneous lung fibrosis. JCI Insight 4: e126125, 2019. doi: 10.1172/jci.insight.126125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Alysandratos KD, Russo SJ, Petcherski A, Taddeo EP, Acín-Pérez R, Villacorta-Martin C, Jean JC, Mulugeta S, Rodriguez LR, Blum BC, Hekman RM, Hix OT, Minakin K, Vedaie M, Kook S, Tilston-Lunel AM, Varelas X, Wambach JA, Cole FS, Hamvas A, Young LR, Liesa M, Emili A, Guttentag SH, Shirihai OS, Beers MF, Kotton DN. Patient-specific iPSCs carrying an SFTPC mutation reveal the intrinsic alveolar epithelial dysfunction at the inception of interstitial lung disease. Cell Rep 36: 109636, 2021. doi: 10.1016/j.celrep.2021.109636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Borok Z, Horie M, Flodby P, Wang H, Liu Y, Ganesh S, Firth AL, Minoo P, Li C, Beers MF, Lee AS, Zhou B. Grp78 Loss in epithelial progenitors reveals an age-linked role for endoplasmic reticulum stress in pulmonary fibrosis. Am J Respir Crit Care Med 201: 198–211, 2020. doi: 10.1164/rccm.201902-0451OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Kang C, Elledge SJ. How autophagy both activates and inhibits cellular senescence. Autophagy 12: 898–899, 2016. doi: 10.1080/15548627.2015.1121361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Zhang X, Hu X, Zhang Y, Liu B, Pan H, Liu Z, Yao Z, Zhu Q, Wu C, Shen T. Impaired autophagy-accelerated senescence of alveolar type II epithelial cells drives pulmonary fibrosis induced by single-walled carbon nanotubes. J Nanobiotechnology 21: 69, 2023. doi: 10.1186/s12951-023-01821-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Xu C, Wang L, Fozouni P, Evjen G, Chandra V, Jiang J, Lu C, Nicastri M, Bretz C, Winkler JD, Amaravadi R, Garcia BA, Adams PD, Ott M, Tong W, Johansen T, Dou Z, Berger SL. SIRT1 is downregulated by autophagy in senescence and ageing. Nat Cell Biol 22: 1170–1179, 2020. doi: 10.1038/s41556-020-00579-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Mora AL, Bueno M, Rojas M. Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J Clin Invest 127: 405–414, 2017. doi: 10.1172/JCI87440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Schuliga M, Kanwal A, Read J, Blokland KEC, Burgess JK, Prêle CM, Mutsaers SE, Grainge C, Thomson C, James A, Bartlett NW, Knight DA. A cGAS-dependent response links DNA damage and senescence in alveolar epithelial cells: a potential drug target in IPF. Am J Physiol Lung Cell Mol Physiol 321: L859–L871, 2021. doi: 10.1152/ajplung.00574.2020. [DOI] [PubMed] [Google Scholar]
- 137. Bueno M, Lai YC, Romero Y, Brands J, St Croix CM, Kamga C, Corey C, Herazo-Maya JD, Sembrat J, Lee JS, Duncan SR, Rojas M, Shiva S, Chu CT, Mora AL. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest 125: 521–538, 2015. doi: 10.1172/JCI74942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Summer R, Shaghaghi H, Schriner D, Roque W, Sales D, Cuevas-Mora K, Desai V, Bhushan A, Ramirez MI, Romero F. Activation of the mTORC1/PGC-1 axis promotes mitochondrial biogenesis and induces cellular senescence in the lung epithelium. Am J Physiol Lung Cell Mol Physiol 316: L1049–L1060, 2019. [Erratum in Am J Physiol Lung Cell Mol Physiol 320: L468–L471, 2021]. doi: 10.1152/ajplung.00244.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Cui H, Xie N, Banerjee S, Dey T, Liu RM, Antony VB, Sanders YY, Adams TS, Gomez JL, Thannickal VJ, Kaminski N, Liu G. CD38 Mediates lung fibrosis by promoting alveolar epithelial cell aging. Am J Respir Crit Care Med 206: 459–475, 2022. doi: 10.1164/rccm.202109-2151OC. [DOI] [PubMed] [Google Scholar]
- 140. Lai X, Huang S, Lin S, Pu L, Wang Y, Lin Y, Huang W, Wang Z. Mesenchymal stromal cells attenuate alveolar type 2 cells senescence through regulating NAMPT-mediated NAD metabolism. Stem Cell Res Ther 13: 172, 2022. doi: 10.1186/s13287-022-02869-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Hohmann MS, Habiel DM, Coelho AL, Verri WA Jr, Hogaboam CM. Quercetin enhances ligand-induced apoptosis in senescent idiopathic pulmonary fibrosis fibroblasts and reduces lung fibrosis in vivo. Am J Respir Cell Mol Biol 60: 28–40, 2019. doi: 10.1165/rcmb.2017-0289OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Nambiar A, Kellogg D 3rd, Justice J, Goros M, Gelfond J, Pascual R, Hashmi S, Masternak M, Prata L, LeBrasseur N, Limper A, Kritchevsky S, Musi N, Tchkonia T, Kirkland J. Senolytics dasatinib and quercetin in idiopathic pulmonary fibrosis: results of a phase I, single-blind, single-center, randomized, placebo-controlled pilot trial on feasibility and tolerability. eBioMedicine 90: 104481, 2023. doi: 10.1016/j.ebiom.2023.104481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Mora AL, Rojas M, Pardo A, Selman M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat Rev Drug Discov 16: 810, 2017. doi: 10.1038/nrd.2017.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Birch J, Gil J. Senescence and the SASP: many therapeutic avenues. Genes Dev 34: 1565–1576, 2020. doi: 10.1101/gad.343129.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Gasek NS, Kuchel GA, Kirkland JL, Xu M. Strategies for targeting senescent cells in human disease. Nat Aging 1: 870–879, 2021. doi: 10.1038/s43587-021-00121-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Coppé JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem 286: 36396–36403, 2011. doi: 10.1074/jbc.M111.257071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Demaria M, Ohtani N, Youssef Sameh A, Rodier F, Toussaint W, Mitchell James R, Laberge R-M, Vijg J, Van Steeg H, Dollé Martijn ET, Hoeijmakers Jan HJ, de Bruin A, Hara E, Campisi J. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 31: 722–733, 2014. doi: 10.1016/j.devcel.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Shi J, Sun J, Liu L, Shan T, Meng H, Yang T, Wang S, Wei T, Chen B, Ma Y, Wang Q, Wang H, Liu J, Wang L. P16ink4a overexpression ameliorates cardiac remodeling of mouse following myocardial infarction via CDK4/pRb pathway. Biochem Biophys Res Commun 595: 62–68, 2022. doi: 10.1016/j.bbrc.2022.01.077. [DOI] [PubMed] [Google Scholar]
- 149. Chikenji TS, Saito Y, Konari N, Nakano M, Mizue Y, Otani M, Fujimiya M. p16INK4A-Expressing mesenchymal stromal cells restore the senescence-clearance-regeneration sequence that is impaired in chronic muscle inflammation. EBioMedicine 44: 86–97, 2019. doi: 10.1016/j.ebiom.2019.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Chung EJ, Kwon S, Reedy JL, White AO, Song JS, Hwang I, Chung JY, Ylaya K, Hewitt SM, Citrin DE. IGF-1 receptor signaling regulates type II pneumocyte senescence and resulting macrophage polarization in lung fibrosis. Int J Radiat Oncol Biol Phys 110: 526–538, 2021. doi: 10.1016/j.ijrobp.2020.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Takenouchi Y, Kitakaze K, Tsuboi K, Okamoto Y. Growth differentiation factor 15 facilitates lung fibrosis by activating macrophages and fibroblasts. Exp Cell Res 391: 112010, 2020. doi: 10.1016/j.yexcr.2020.112010. [DOI] [PubMed] [Google Scholar]
- 152. Chen X, Xu H, Hou J, Wang H, Zheng Y, Li H, Cai H, Han X, Dai J. Epithelial cell senescence induces pulmonary fibrosis through Nanog-mediated fibroblast activation. Aging (Albany NY) 12: 242–259, 2019. doi: 10.18632/aging.102613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Wiley CD, Campisi J. The metabolic roots of senescence: mechanisms and opportunities for intervention. Nat Metab 3: 1290–1301, 2021. doi: 10.1038/s42255-021-00483-8. [DOI] [PMC free article] [PubMed] [Google Scholar]