

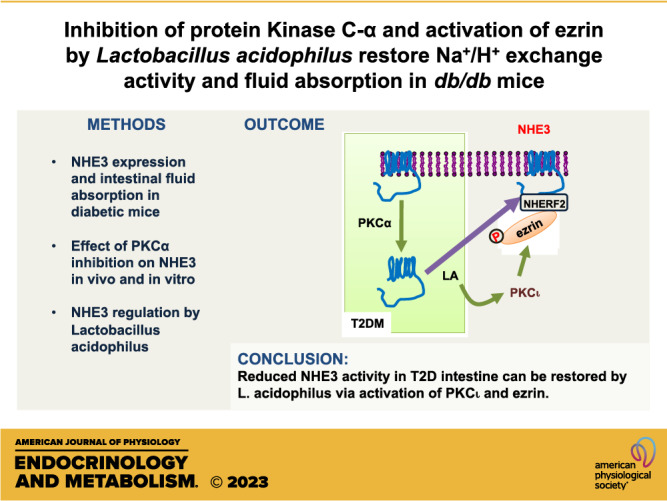
Keywords: diabetes, ezrin, NHE3, protein kinase C
Abstract
Gastrointestinal (GI) complications, including diarrhea, constipation, and gastroparesis, are common in patients with diabetes. Dysregulation of the Na+/H+ exchanger NHE3 in the intestine is linked to diarrhea and constipation, and recent studies showed that NHE3 expression is reduced in type 1 diabetes and metformin causes diarrhea in the db/db mouse model of type 2 diabetes (T2D) via inhibition of NHE3. In this study, we investigated whether NHE3 expression is altered in type 2 diabetic intestine and the underlying mechanism that dysregulates NHE3. NHE3 expression in the brush border membrane (BBM) of the intestine of diabetic mice and humans was decreased. Protein kinase C (PKC) activation is associated with pathologies of diabetes, and immunofluorescence (IF) analysis revealed increased BBM PKCα abundance. Inhibition of PKCα increased NHE3 BBM abundance and NHE3-mediated intestinal fluid absorption in db/db mice. Previous studies have shown that Lactobacillus acidophilus (LA) stimulates intestinal ion transporters. LA increased NHE3 BBM expression and mitigated metformin-mediated inhibition of NHE3 in vitro and in vivo. To understand the underlying mechanism of LA-mediated stimulation of NHE3, we used Caco-2bbe cells overexpressing PKCα that mimic the elevated state of PKCα in T2D. LA diminished PKCα BBM expression, increased phosphorylation of ezrin, and the interaction of NHE3 with NHE regulatory factor 2 (NHERF2). In addition, inhibition of PKCι blocked phosphorylation of ezrin and activation of NHE3 by LA. These findings demonstrate that NHE3 is downregulated in T2D, and LA restores NHE3 expression via regulation of PKCα, PKCι, and ezrin.
NEW & NOTEWORTHY We used mouse models of type 2 diabetes (T2D) and human patient-derived samples to show that Na+/H+ exchanger 3 (NHE3) expression is decreased in T2D. We show that protein kinase C-α (PKCα) is activated in diabetes and inhibition of PKCα increased NHE3 expression and mitigates diarrhea. We show that Lactobacillus acidophilus (LA) stimulates NHE3 via inhibition of PKCα and phosphorylation of ezrin.
INTRODUCTION
Various gastrointestinal (GI) symptoms commonly occur in people with diabetes. The most common lower GI symptoms are constipation, diarrhea, abdominal pain, and distention. The incidence of diarrhea occurring in patients with diabetes has been reported to vary from 10% to as high as 20% (1, 2). Diarrhea is more frequently associated with insulin-dependent or type 1 diabetes (T1D), whereas medications often cause diarrhea in patients with insulin-independent or type 2 diabetes (T2D) (2–4). Constipation is also commonly reported in both T1D and T2D with up to 31% of patients reporting an incidence of chronic constipation (5). The causes of the GI symptoms associated with diabetes are not clear, but studies in animal models and humans have suggested that neural dysfunction and dysbiosis play key fundamental roles in diabetic GI complications (6).
The GI tract is lined with a single layer of epithelium whose main function is the absorption of nutrients and ions. The movement of ions and small organic molecules, including Na+, K+, Cl−, HCO3−, H+, mono- and disaccharides, amino acids, and small lipid molecules, across the epithelium is mediated by numerous transporters and channels. The conventional view is that the movement of water across the epithelium results principally from the osmotic gradient created by the movement of electrolytes and nutrients (7). Yet, the contribution of the transporters and channels in the intestinal epithelium to the adverse effects in the GI tract associated with diabetes is not well studied.
Na+/H+ exchanger 3 (NHE3, Slc9a3) is the major Na+/H+ exchanger at the apical membrane of epithelial cells in the intestine and kidney, where it has a major role in Na+, HCO3−, and fluid absorption (8). Defective NHE3 expression and activity in the intestine are often a culprit for diarrhea (9, 10). On the other hand, increased NHE3 activity depletes water from the intestinal lumen so that hard stools become difficult to pass. A recent work has shown that inhibition of NHE3 is a potential treatment for patients with constipation-predominant irritable bowel syndrome (11). We have shown that NHE3 expression is downregulated in patients with T1D and a rodent model of T1D where NHE3 activity and intestinal fluid absorption are decreased (12). In addition, we found that metformin, a highly effective drug to treat T2D, inhibits NHE3 activity and fluid absorption in the intestine of db/db mice (13).
Persistent hyperglycemia in diabetic conditions contributes to excessive production of diacyglycerol (DAG), which directly or indirectly activates protein kinase C (PKC) (14). The PKC family consists of multiple isoforms of PKC that plays a key role in many cellular functions (15). The conventional PKC isoforms (PKC-α, -β1, -β2, and -γ) are activated by Ca2+ and DAG, whereas novel PKCs (PKC-δ, -ε, -θ, and -η) are activated by DAG, but not by Ca2+. The atypical PKCs (PKC-ζ and -ι/λ) are not activated by Ca2+ or DAG (15). Hyperglycemia-induced diabetic nephropathy involves PKC signal that activates the mitogen-activated protein kinases and PKC isoform-selective inhibitors, including those targeting PKCβ, have shown optimistic outcomes (16). Elevated PKCα signaling in a mouse model of T1D stimulates intestinal iron absorption, contributing to elevation of iron content in serum and liver (17). In addition, activation of PKC leads to the promotion of oxidative stress and inflammation on myocardial and vascular tissues that eventually cause irreversible damage to the cardiovascular system in patients with diabetes (18).
In this study, we determined NHE3 expression in mouse models of T2D. We found that NHE3 expression is downregulated and internalized in diabetic mice compared with control mice. Inhibition of PKCα enhanced NHE3 expression in the brush border membrane (BBM) of intestinal epithelial cells and NHE3-dependent intestinal fluid absorption. In addition, our data show that probiotics can be used to modulate NHE3 activity and to mitigate the inhibitory effects of metformin on NHE3.
MATERIALS AND METHODS
Cell Line, Reagents, and Antibodies
Caco-2bbe cells stably transfected with human NHE3 with a C-terminal vesicular stomatitis virus glycoprotein (VSVG) epitope were cultured as previously described (19). Cells were grown on Transwell (Corning, Tewksbury, MA) for 7 days postconfluence. Gӧ6976 was purchased from Tocris (Minneapolis, MN) and PKCι inhibitor HY-126146 was from MedCheExpress (Monmouth Junction, NJ). All other chemicals were purchased from Sigma Aldrich (St. Louis, MO). Rabbit polyclonal anti-NHE3 (EM450, 1:7,000 dilution), mouse monoclonal anti-VSVG (P5D4, 1:100 dilution), rabbit polyclonal anti-NHERF1 (Ab5199, 1:5,000 dilution), and rabbit polyclonal anti-NHERF2 (Ab2570, 1:2,000 dilution) antibodies were previously described and validated (12, 20–23). Commercial antibodies used in this study are listed in Table 1. Alexa Fluor 594 Phalloidin (A12381; 1:200) was from Thermo Fisher Scientific (Waltham, MA). pHACE harboring PKCα was a gift from Bernard Weinstein (Addgene, Plasmid No. 21232).
Table 1.
Commercial antibodies used
| Antibody | Source | Catalog Number | Dilution |
|---|---|---|---|
| Anti-NHE3 antibody (NHE31-A) | Alpha Diagnostics, San Antonio, TX, USA | NHE31-A | 1:1,000 |
| Anti-phospho-PKCα | Abcam, Waltham, MA, USA | ab180848 | 1:1,000 |
| Anti-PKCα | Cell Signaling Technologies, Danvers, MA, USA | 2056 | 1:2,000 |
| Anti-villin | Cell Signaling Technologies | 2369 | 1:1,000 |
| Anti-phospho-Ezrin | Cell Signaling Technologies | 3141 | 1:2,000 |
| Anti-ezrin | Cell Signaling Technologies | 3145 | 1:2,000 |
| Anti-HA | Cell Signaling Technologies | 3724 | 1:1,000 |
| Anti-β-actin | Cell Signaling Technologies | 4967 | 1:2,000 |
| Alexa Fluor 488-conjugated goat anti-rabbit IgG | Thermo Fisher Scientific, Waltham, MA, USA | A-11008 | 1:500 |
HA, hemagglutinin; NHE3, Na+/H+ exchanger 3; PKCα, protein kinase C-alpha.
Animals and Treatments
Experiments with animals were conducted under approval by the Institutional Animal Care and Use Committees of the Atlanta Veterans Administration Medical Center and Emory University and in accordance with the National Institutes of Health (NIH)’s Guide for the Care and Use of Laboratory Animals. Six- to seven-week-old male Leprdb/db (db/db), Leprdb/+ (db/+), KK.Cg-Ay/J (KKAy), and a/a mice were obtained from the Jackson Laboratory (Bar Harbor, ME). Mice were acclimated and given standard chow and water ad libitum for 2 wk before the start of the experiments. All measurements and treatments were performed without fasting. Mice were orally administered 150 μL of Gӧ6976 (1 mg/kg) or DMSO once a day for 3 days (days 1, 2, and 3) and were euthanized 24 h after the last treatment. Blood samples were collected, and glucose levels were measured on the day before treatment and on the day of euthanasia with a OneTouch Verio glucose meter (LifeScan, Malvern, PA). At the same time, body weights were measured. Metformin at the concentration of 500 mg/kg was administered once a day for 3 days by gavage as previously described (13).
Human Biopsies
All samples used in this study were obtained with institutional review board approval. Preoperative medical records of patients with a history of T2D and healthy controls were previously described (17).
Lactobacillus acidophilus Culture
Lactobacillus acidophilus (LA) was obtained from American Type Culture Collection (ATCC) (4357), and bacteria were cultured as previously described (24). Briefly, bacteria were grown overnight in MRS (Mann-Rogosa-Sharpe, Difco) broth for 24 h at 37°C without shaking. Next day, bacteria were spun down by centrifuging at 3,000 rpm for 10 min. Culture supernatant was separated from bacterial pellet, filtered through a 0.22 μm filter, and diluted in the cell culture media for further use. For in vivo studies, 3 × 109 colony-forming unit (CFU) of live bacteria suspended in 200 μL of sterile PBS per was gavaged three times every other day per animal. Control mice received 200 μL of sterile PBS. Animals were used for intestinal water flux measurement 24 h later or euthanized for tissue collection.
Intestinal Water Flux Measurement
Intestinal water flux was measured as previously reported (19). Briefly, a mouse anesthetized with sodium pentobarbital was placed on a 37°C heating block, and a small incision was made in the abdomen. A 5 cm loop of the ileum (between 5 and 10 cm upstream of cecum) cannulated at the proximal and distal ends was flushed with saline for 10 min. This was followed by perfusion of prewarmed perfusion solution (mM: 118.4 NaCl, 4.7 KCl, 2.52 CaCl2, 1.18 MgSO4, 25 Na gluconate, 1.18 KH2PO4, pH 7.4) at 1 mL/min for 2 h. The CFTR inhibitor CFTRinh-172 (250 μg/kg) was administered by intraperitoneal injection to each mouse 1 h before the start of perfusion. Intestinal water flux was recorded by calculating a change in the volume of perfusion buffer in a reservoir every 10 min over the course of a 90–120 min perfusion period.
Na+-Dependent Intracellular pH Recovery
The Na+-dependent changes in intracellular pH (pHi) by NHE3 were determined using the ratio-fluorometric, pH-sensitive dye 2′,7′-bis-(2-carboxyethyl)-5-carboxyfluorescein acetoxymethyl ester as previously described (12). Caco-2bbe cells grown for 10 days postconfluence were treated with 1 mM metformin for 30 min during dye loading. In some experiments, cells were pretreated with LA-conditioned media (LCM) for 6 h before metformin treatment. Coverslips were mounted on a perfusion chamber, placed on an inverted microscope, superfused with NH4+ buffer, followed by sequential perfusion with tetramethylammonium (TMA) buffer (130 mM TMA-Cl, 20 mM HEPES, 5 mM KCl, 1 mM TMA-PO4, 2 mM CaCl2, 1 mM MgSO4, and 25 mM glucose) and Na+ buffer that drives Na+-dependent pH recovery. Na+ buffer was supplemented with 30 μM HOE694 or 2 μM dimethyl amiloride to inhibit NHE1 and NHE2 activities. The microfluorometry was performed using the Metafluor software (Molecular Devices, Sunnyvale, CA) and five traces of Na+-dependent pH recovery were captured from each coverslip, each trace originating from an independent group of cells. For each experiment, a minimum of four coverslips per group were studied. Na+/H+ exchange rate was described by the initial rate of pHi recovery, which was calculated by determining slopes along the pHi recovery by linear least-squares analysis over a minimum of 7 s.
Western Blot
Caco-2bbe/NHE3 cells were lysed in cold lysis buffer (No. FNN0011, Invitrogen, Waltham, MA) containing 10 mM Tris, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM NaF, 20 mM Na4P2O7, 2 mM Na3VO4, 1% Triton X-100, 10% glycerol, 0.1% SDS, 0.5% deoxycholate supplemented with protease inhibitors cocktail tablets (Roche, Indianapolis, IN), and 10 µM PMSF before use. To make mouse intestinal mucosa lysates, 6- to 7-wk-old male mice were euthanized and the abdomens were opened, and the small intestine was rapidly removed. Epithelial cells were collected by scrapping the segment 5–12 cm upstream of cecum with a glass coverslip and lysed in the cold lysis buffer as described earlier. The crude lysate from cells or mouse intestine was sonicated twice for 15 s and spun at 14,000 g for 15 min. Protein concentration was determined by the bicinchoninic acid assay (Sigma). Western immunoblotting was performed as previously described (13) and visualization was performed using the LI-COR Odyssey Imaging System (LI-COR Bioscience, Lincoln, NE), and densitometric analysis was performed using Image Studio Ver 5.2 (LI-COR Bioscience).
Surface Biotinylation
Surface biotinylation of NHE3 was performed as previously described (25). Briefly, Caco-2bbe cells were rinsed twice in PBS and incubated in borate buffer (154 mM NaCl, 7.2 mM KCl, 1.8 mM CaCl2, and 10 mM H3BO3, pH 9.0) for 10 min. Cells were then incubated for 40 min with 0.5 mg/mL sulfo-NHS-LC-biotin (Pierce, Rockford, IL) in borate buffer. Unbound sulfo-NHS-LC-biotin was quenched with Tris buffer (20 mM Tris, 120 mM NaCl, pH 7.4). Cells were lysed in lysis buffer (150 mM NaCl, 1 mM β-glycerophosphate, 2.5 mM sodium pyrophosphate, 1 mM Na2EDTA, 1 mM EGTA, 1 mM Na3VO4, 1 μg/mL leupeptin, 1% Triton X-100, and protease inhibitors cocktail tablets) and sonicated for 2 × 15 s. Lysate was agitated for 30 min and spun at 14,000 g for 30 min at 4°C to remove the insoluble cell debris. An aliquot was retained as the total fraction representingthe total cellular NHE3. Protein concentration was determined, and 1 mg of lysate was then incubated with streptavidin-agarose beads (Pierce) for 2 h. The streptavidin-agarose beads were washed three times in lysis buffer and twice in PBS. All the aforementioned procedures were performed at 4°C or on ice. Biotinylated surface proteins were then eluted by boiling the beads at 95°C for 10 min. Dilutions of the total and surface NHE3 were resolved by SDS-PAGE and immunoblotted with an anti-VSVG antibody as described earlier.
Immunoprecipitation of Surface NHE3
Apical membrane of Caco-2bbe cells were biotinylated as described earlier. Protein concentration was determined, and 1 mg of lysate was then incubated with Monomeric Avidin agarose beads (Pierce) for 1 h. After washing three times with ice-cold lysis buffer, Avidin agarose beads bound proteins were eluted with excessive free biotin and then used for the subsequent immunoprecipitation of NHE3 using anti-VSVG antibody at 4°C for 16 h. The lysates were incubated with Protein G Sepharose beads for 2 h and then washed three times with ice-cold lysis buffer. Proteins were eluted from beads by boiling in reducing sample buffer. Entire eluted proteins were resolved by SDS-PAGE and immunoblotted as described earlier.
Confocal Immunofluorescence Microscopy
Mouse ileal sections were harvested, rinsed in Hank’s buffered saline solution, opened lengthwise, fixed with 4% paraformaldehyde for 30 min at room temperature (RT), embedded with optical cutting temperature (OCT) compound, and sectioned into 6-μm thick sections. Human biopsies were formalin-fixed, paraffin-embedded, and cut into 5 μm sections. Sections were deparaffinized, rehydrated, and antigen unmasking was performed through pressure cooker treatment in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0). Caco-2bbe/NHE3 cells grown on Transwells (Corning, Lowell, MA) were fixed using 100% methanol at −20°C for 20 min. The fluorescence staining procedures were described before (12). Briefly, specimens were blocked with 5% normal goat serum for 1 h at RT, incubated with primary antibody for 12 h at 4°C, washed with PBS three times, and incubated with Alexa Fluor-conjugated secondary antibodies for 1 h at RT. After three 10 min washes with PBS, specimens were mounted with ProLong Glass Antifade Reagent (Thermo Fisher Scientific, Waltham, MA) and observed under a Nikon A1R HD confocal microscope (Nikon Instruments Inc., NY) coupled to a Plan Apo λ ×60 Oli lens and images were analyzed using NIS-Elements Software Ver. 4.50.
Statistical Analysis
Statistical analysis was performed using independent samples two-tailed unpaired Student’s t test or ANOVA, followed by the Tukey post hoc analysis. Results are presented as means ± SE. Statistical analysis was performed using GraphPad Prism software version 9 (La Jolla, CA). A value of P < 0.05 was considered significant.
RESULTS
NHE3 Abundance in the Brush Border Membrane of Diabetic Mouse Intestine Is Reduced
To assess whether NHE3 expression is altered in T2D, we determined NHE3 expression in two mouse models, db/db and KKAy (26). The anti-NHE3 antibody EM450 used here has been validated previously against Nhe3−/− mouse intestinal lysates (23). In both T2D models, Western blot analyses showed that NHE3 protein expression levels in diabetic mouse ileum were reduced compared with their respective controls, when NHE3 expression was normalized to the intestinal epithelial marker villin (Fig. 1, A and B). Given that the changes in NHE3 protein expression were relatively small, we performed confocal immunofluorescence (IF) analysis of mouse intestine to determine whether NHE3 cellular expression was altered. We found that NHE3 signal on the BBM of db/db mouse intestine was diminished, whereas diffused NHE3 IF signal in the cytosol was more prominent compared with control db/+ mouse intestine (Fig. 1C). This pattern of NHE3 protein was not unique to db/db mice and increased levels of cytosolic NHE3 protein were also observed in KKAy mouse intestine relative to control aa mouse intestine (Fig. 1D). To determine whether the expression of NHE3 is also altered in humans, we examined colonic biopsies from patients with T2D and healthy individuals that we have reported previously (17). The biopsies were collected in the distal colons due to the difficulty of getting ileal biopsies from the patients. Representative IF staining shown in Fig. 1E corroborated decreased luminal abundance of NHE3 in human diabetic colonic tissues compared with healthy controls. Na+ absorption by NHE3 in the intestinal epithelial cells facilitates water absorption across the intestinal epithelial barrier. Hence, NHE3 inhibition leads to decreased Na+ absorption, which causes luminal water accumulation, a proxy for diarrhea. We next compared the rate of NHE3-dependent fluid absorption in db/db and db/+ mice in the presence of CFTRinh-172, which blocks the activities of CFTR and ClC-2 chloride channel (27). In db/+ mouse ileal segments, the net rate of fluid absorption was 54.5 ± 6.3 μL/cm/h, which was similar to our previous data using C56BL/6 mice (Fig. 1F) (19, 23). In comparison, db/db mice exhibited a reduced rate of fluid absorption at 32.2 ± 1.8 μL/cm/h, a 41% decrease. These data indicated that the activity of NHE3 in the small intestine is reduced in T2D.
Figure 1.

Decreased Na+/H+ exchanger 3 (NHE3) expression in the intestine of diabetic mice and humans. A: NHE3 protein expression in the ileum of db/db and db/+ mice was determined. Villin was used for a loading control. Quantification of NHE3 protein relative to villin is shown on the bottom. B: NHE3 and villin expression in the ileum of KKAy and a/a control mice are shown. Bottom: NHE3 expression relative to villin expression. *P < 0.05. C and D: representative immunofluorescence (IF) images of NHE3 (green), F-actin (red, phalloidin), and nuclei (blue, DAPI) of mouse ileal epithelial cells. Lower panels show enlarged views of the boxed area. Scale bar, 20 μm. E: representative confocal IF images for NHE3 (green) and bright field in colonic biopsies from patients with type 2 diabetes (T2D) and healthy controls (Con). Arrows indicate NHE3. Scale bar, 10 μm. Right: average fluorescence intensity on each field of view determined using ImageJ. *P < 0.05. F: fluid absorption was measured for 2 h in the ileal section of db/+ and db/db. **P < 0.01. All results are representative of two independent experiments.
Inhibition of Protein Kinase C Increases NHE3 Membrane Abundance and Intestinal Fluid Absorption in db/db Mice
We next questioned what could cause reduced NHE3 expression and activity in diabetic mice. Activation of PKC has been linked to the development of diabetic complications that include retinopathy, nephropathy, and endothelial dysfunction (16, 28). Acute, reversible inhibition of NHE3 by PKC-dependent signaling has been shown by multiple studies and a study using PS120 fibroblasts has implicated the role of PKCα in NHE3 inhibition (29–31). We therefore assessed whether inhibiting PKCα alters NHE3 expression by treating db/db mice with Gӧ6976, which preferentially inhibits PKCα and PKCβ, for 3 days. The blood glucose levels measured on day 0 without fasting before the inhibitor treatment and on day 4, a day after the last treatment, did not reveal a significant difference (Fig. 2A). Similarly, NHE3 protein expression levels were not different between the control and inhibitor-treated groups (Fig. 2B).
Figure 2.

Inhibition of protein kinase C (PKC) increases Na+/H+ exchanger 3 (NHE3) membrane expression in db/db mice. A: blood glucose levels in db/db mice treated with PKC inhibitor Gӧ6976 or the same volume of DMSO were determined. Glucose levels were determined prior to treatment (D0) and on the last day (D4). ns, not significant. B: NHE3 expression in the ileum was determined. NHE3 expression was quantified relative to villin expression. C: representative immunofluorescence (IF) images of PKCα (green), F-actin (red), and nuclei (DAPI, blue) are shown. Bottom panels show enlarged images of the boxed area. Scale bar, 20 μm. D: representative IF images of NHE3 (green), F-actin (red), and nuclei (DAPI, blue) are shown. Scale bar, 20 μm. E: fluid absorption was measured for 2 h in the ileal sections of the mice. **P < 0.01. All results are representative of two independent experiments.
Activation of both NHE3 and PKC involves the translocation of proteins to the plasma membrane (8, 32). We therefore determined whether Gӧ6976 alters the cellular localization of NHE3 and PKCα. Confocal IF microscopy analysis of ileal sections of db/db mice using anti-PKCα antibodies showed PKCα IF signal on the brush border and lateral membrane (Fig. 2C, left). Of note, the specificity of the anti-PKCα antibody was previously validated using PKCα−/− mouse (17). In comparison, PKCα IF labeling on the membrane was markedly decreased, whereas the cytoplasmic PKCα labeling was more diffused in Gӧ6976-treated mouse intestine (Fig. 2C, right). The IF staining pattern of NHE3 was opposite to that of PKCα. Gӧ6976 resulted in a decrease in the cytoplasmic presence of NHE3 and the increased appearance of the green and yellow pixels above the red indicated increased expression of NHE3 in the BBM (Fig. 2D, bottom, Gӧ6976). Consistent with the increased NHE3 expression in the BBM, the rate of fluid absorption was significantly increased from 46.4 ± 6.2 μL/cm/h in DMSO-treated to 74.1 ± 4.6 μL/cm/h in Gӧ6976-treated intestine (Fig. 2E).
Lactobacillus acidophilus Mitigates Metformin-Induced Inhibition of NHE3 in Caco-2bb2 Cells
Recently, we have shown that metformin-associated diarrhea in T2D is in part caused by decreased NHE3 activity by metformin (13). Based on previous studies that probiotics stimulate intestinal transporters, including NHE3, we questioned whether probiotics can be used to mitigate the effects of metformin on NHE3, thereby preventing metformin-associated diarrhea (24, 33). Singh et al. (24) reported that out of several Lactobacillus strains they tested, L. acidophilus (LA) was the only strain that increased NHE3 activity. Therefore, Caco-2bbe cells with stable expression of NHE3 were treated with LA-derived conditioned media (LCM) for 4 h according to the previous study (24). NHE3 activity was significantly elevated in cells treated with LCM compared with controls treated with MRS media (Fig. 3A). Metformin reduced NHE3 activity, but the effect of metformin was completely blocked by LCM and NHE3 activity in the presence of metformin + LCM was raised above the basal level (Fig. 3A, Met + LCM vs. Con). These changes in NHE3 activity were confirmed by determining NHE3 expression in the apical membrane of Caco-2bbe cells by surface biotinylation. LCM increased the surface abundance of NHE3 in control cells but did not alter total cellular expression level of NHE3 (Fig. 3B, LCM vs. Con). The lack of effect on NHE3 total expression was not surprising since NHE3 expression in these cells was under the control of the cytomegalovirus (CMV) promoter. LCM treatment completely blocked the inhibitory effect of metformin and led to more than a twofold increase in the surface abundance of NHE3 compared with metformin-treated cells (Fig. 3B, M + LCM vs. M). Confocal IF microscopy analysis further corroborated the stimulatory effects of LCM, resulting in a significant increase of NHE3 IF signal at the apical membrane (Fig. 3C). These results demonstrated that LA activates NHE3 in part by mobilizing NHE3 proteins into the apical membrane.
Figure 3.

Lactobacillus acidophilus (LA) mitigates metformin-induced inhibition of Na+/H+ exchanger 3 (NHE3) in Caco-2bbe cells. A: Caco-2bbe/NHE3 cells were treated with metformin (Met; M), LA conditioned media (LCM), or Met + LCM, and NHE3 activity was determined. B: NHE3 expression in the apical membrane was determined by surface biotinylation. Representative Western blots for surface (S) and total (T) NHE3 expression are shown. S-NHE3/T-NHE3 ratios are shown on the right. **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant. Statistical analysis was performed by two-way ANOVA with Tukey’s post hoc test. Data (means ± SE) are combined from three independent experiments using cells of different passages. C: confocal immunofluorescence (IF) images for NHE3 (green), F-actin (red), and DAPI (blue) are shown. Top: focal plane xy views. Middle: vertical sectional xz view of NHE3. Bottom: vertical sectional xz view of NHE3, F-actin, and DAPI. Scale bar, 10 μm. Con, control.
Lactobacillus acidophilus Stimulates Fluid Absorption in db/db Mice
Having demonstrated that LA stimulates NHE3 activity in Caco-2bbe cells, we next tested whether LA can be used to prevent metformin-induced diarrhea in db/db mice. Mice were treated with LA on days 1, 3, and 5, and metformin was administered on days 4, 5, and 6 (Fig. 4A). Control mice received aliquots of PBS. Neither LA nor metformin altered body weight and mice looked indistinguishable from control animals (Fig. 4B). On the other hand, metformin, but not LA, lowered blood glucose levels, and LA did not interfere with the hypoglycemic effect of metformin (Fig. 4C).
Figure 4.

Lactobacillus acidophilus (LA) does not interfere with metformin-induced effect on blood glucose. A: treatment scheme of LA and metformin (Met). Changes in body weight (B) and blood glucose levels (C) in db/db mice treated with PBS (Con), LA, metformin (M), or metformin + LA (M + LA) are shown. **P < 0.01; ****P < 0.0001; ns, not significant.
We next determined NHE3 expression in mouse ileal epithelial lysates. LA increased NHE3 protein expression consistent with previous studies (24, 34). On the contrary, NHE3 expression was not significantly altered by metformin, as we have shown previously (13). Not surprisingly, increased NHE3 expression was observed in mice treated with both metformin and LA (Fig. 5A). Consistent with the increased NHE3 expression levels, LA markedly increased the rate of intestinal fluid absorption when given alone or together with metformin (Fig. 5B), indicating that LA mitigates metformin-associated diarrhea. Confocal IF analysis of ileal tissues showed that NHE3 IF signal in the BBM was enhanced by LA, suggesting that LA triggers the translocation of NHE3 into the BBM (Fig. 5C, Control vs. LA). Consistent with decreased fluid absorption by metformin, metformin induced retrieval of NHE3 from the BBM, which was completely reversed by LA (Fig. 5C, Met vs. LA + Met).
Figure 5.

Lactobacillus acidophilus (LA) increases Na+/H+ exchanger 3 (NHE3) membrane abundance and intestinal fluid absorption in metformin-treated db/db mice. A: NHE3 protein expression in ileal epithelial lysates was determined. Right: quantification of NHE3 normalized to villin expression. *P < 0.05; **P < 0.01; ns, not significant. B: fluid absorption was measured for 2 h in the ileal sections of the mice. *P < 0.05; **P < 0.01; ****P < 0.0001. C: representative confocal immunofluorescence (IF) images for NHE3 (green), F-actin (red), and DAPI (blue) are shown. Scale bar, 20 μm. Bottom: enlarged images of the boxed area. Con, control; M, Met, metformin.
Lactobacillus acidophilus Retracts PKCα from the BBM
We showed in Fig. 2 that the elevated levels of PKCα activation caused internalization of NHE3 and hence decreased NHE3-mediated fluid absorption in db/db mouse intestine. Therefore, we postulated that LA might restore NHE3 activity through an inhibition of PKCα. We first determined whether PKCα expression or phosphorylation was altered by LA. Western blots of mouse ileal epithelial lysates showed that neither LA nor metformin significantly altered the total PKCα expression level or phosphorylation at S657 of PKCα (Fig. 6A). Since an increase in S657 phosphorylation is not a stringent determinant of PKCα activation (35), we determined whether LA affected the translocation of PKCα to the plasma membrane. As shown earlier, the presence of PKCα in the plasma membrane was evident in control db/db mouse intestine and metformin did not significantly alter the expression pattern of PKCα (Fig. 6B, Con vs. Met). In comparison, plasma membrane-associated PKCα level was reduced in db/db mouse intestines treated with LA alone or together with metformin (Fig. 6B, LA or LA + Met), indicating that LA inhibits PKCα that leads to NHE3 activation.
Figure 6.

Lactobacillus acidophilus (LA) inhibits protein kinase C (PKC)α. A: representative Western blots for phospho-PKCα (p-PKCα) and total PKCα (T-PKCα) in the ileal epithelial cells are shown. Right: relative ratio of p-PKCα to T-PKCα. M, metformin; ns, not significant. B: representative confocal immunofluorescence (IF) images for PKCα (green), F-actin (red), and DAPI (blue) are shown. Scale bar, 20 μm. Bottom: enlarged images of the boxed area. Con, control; Met, metformin.
LA Stimulates Ezrin Phosphorylation
NHE3 function and expression at the plasma membrane are regulated by the interaction of NHE3 with several regulatory proteins, including Na+/H+ exchange regulator factor 1 (NHERF1), NHERF2, and inositol 1,4,5-triphosphate receptor-binding protein (IRBIT) (36, 37). In addition, ezrin plays a critical role in trafficking of NHE3 to the BBM (38, 39). To determine whether LA regulates the expression of these regulatory proteins, we overexpressed PKCα in Caco-2bbe/NHE3 cells to mimic the conditions of diabetes. NHE3 expression was not altered by PKCα overexpression (Fig. 7A). On the other hand, PKCα overexpression reduced NHE3 expression in the apical membrane, whereas LCM led to a significant increase (Fig. 7B). The expression levels of NHERF1 or IRBIT were not altered by PKCα or LCM, but NHERF2 expression was increased by LCM in both control-transfected and PKCα-overexpressing cells (Fig. 7, A and C). In addition, ezrin phosphorylation at T567 was elevated by LCM without altering the expression level of ezrin (Fig. 7, A and C). The effect on ezrin phosphorylation by LA was confirmed in vivo. Comparing ileal epithelial lysates of db/db mice corroborated that LA induced ezrin phosphorylation (Fig. 7D). Because NHERF2 can bind to both NHE3 and ezrin, there is the possibility that LCM regulates the interaction between NHE3 and NHERF2. However, NHERF2 and NHE3 are present in the cytosol as well as on the plasma membrane. To assess the NHE3-NHERF2 interaction at the plasma membrane, we immunoprecipitated NHE3 from the surface biotinylated fraction and assessed the amount of NHERF2 coimmunoprecipitated with NHE3. Interestingly, the NHE3-NHERF2 interaction was reduced by PKCα overexpression (Fig. 7E). On the other hand, the amount of NHERF2 coimmunoprecipitated with NHE3 was elevated by LCM in both control cells and PKCα overexpressing cells. These results indicated that LA stimulates NHE3 expression in the apical membrane of intestinal epithelial cells through the activation of ezrin and enhancing the interaction of NHE3 with NHERF2.
Figure 7.

Lactobacillus acidophilus (LA) increases phosphorylation of ezrin and stimulates Na+/H+ exchanger 3 (NHE3)-NHE regulatory factor 2 (NHERF2) interaction. A: protein expression was determined in Caco-2bbe cells treated with or without LA conditioned media (LCM) for 6 h. Representative blots of three independent experiments are shown. B: surface membrane abundance of NHE3 in Caco-2bbe cells overexpressing protein kinase C (PKC)α was determined. Relative surface to total NHE3 ratios are shown in the lower panel. Data (means ± SE) are combined from four independent experiments using cells of different passages. C: quantification of NHERF2 expression (left) and ezrin phosphorylation (right) in control transfected (clear bars) and PKCα overexpressing cells (shaded bars). Data are combined from two independent experiments. D: expression levels of phospho-ezrin and total ezrin were determined. E: NHE3 was immunoprecipitated from biotinylated membrane pools and coimmunoprecipitated NHERF2 was determined. Clear bars, controls; shade bars, PKCα overexpressing cells. NHERF2 to NHE3 ratios are shown below. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
PKCι Stimulates Ezrin Phosphorylation
Although the data aforementioned indicated that LA enhances ezrin phosphorylation, how the phosphorylation of ezrin is regulated is not clear. PKCα has been implicated in phosphorylation of ezrin but given that the inhibition of PKCα by Gӧ6976 stimulated NHE3 activity, it is unlikely that LA-mediated ezrin phosphorylation is dependent on PKCα (40). A previous study has shown that atypical PKC iota (PKCι) phosphorylates ezrin in Caco-2 cells (41). Therefore, we contemplated whether LA-mediated phosphorylation of ezrin is PKCι dependent. To determine the role of PKCι, cells were treated with LCM in the presence or absence of PKCι inhibitor HY-126146 (42). HY-126146 did not alter basal ezrin phosphorylation, but blocked ezrin phosphorylation by LCM (Fig. 8A). The importance of PKCι was further assessed by determining the effect of HY-126146 on LCM-induced activation of NHE3. Similar to the lack of effect on ezrin phosphorylation under basal conditions, NHE3 activity of Caco-2bbe/NHE3 cells was not significantly altered by the inhibitor. However, HY-126146 completely blocked the activation of NHE3 activity by LCM (Fig. 8B). These results demonstrate the role of PKCι in LA-mediated activation of ezrin and NHE3.
Figure 8.

Lactobacillus acidophilus (LA) phosphorylates ezrin via protein kinase C (PKC)ι. Caco-2bbe cells were treated with LA in the presence or absence of PKCι inhibitor. A: representative Western blots of ezrin phosphorylation are shown. Data (means ± SE) are combined from three independent experiments using cells of different passages. B: NHE3 activity was determined in cells treated with LA conditioned media (LCM; L) or LCM + HY-126146 (HY). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant.
DISCUSSION
We report here that NHE3 expression is reduced in the intestinal BBM of type 2 diabetic mice and human patients with diabetes, and inhibition of PKCα restores NHE3 expression and NHE3-mediated fluid absorption in diabetic mouse intestine. Another finding of the current study is that LA is an effective strain of probiotics in reversing diarrhea often associated with metformin in T2D. LA prevents the inhibition of NHE3 and intestinal fluid absorption by metformin without affecting the antiglycemic effect of metformin. LA stimulates phosphorylation of ezrin and mobilization of NHE3 to the BBM, restoring NHE3 activity and intestinal fluid absorption.
In the current study, we found that NHE3 expression in the ileum of both db/db and KKAy diabetic mice was lower than control mice. The total amount of NHE3 protein was reduced in db/db mice but the most striking difference was seen in the intracellular localization of NHE3 in diabetic mouse ileum, which resulted in reduced BBM expression of NHE3. Reduced NHE3 membrane abundance was also observed in diabetic human colons. In a recent study, NHE3 was found to be upregulated in db/db mouse jejunum, which contrasts our current findings (43). We do not know the reason for the discrepancy between these two studies. The small intestine is the primary site for uptake of dietary glucose by the Na+-dependent glucose transporter 1 (SGLT1) and coordinated activation of SGLT1 and NHE3 has been demonstrated (38, 44). However, the SGLT1 membrane expression in db/db mouse intestine is reduced compared with control mice, making the SGLT1-dependent upregulation of NHE3 unlikely (45). On the other hand, inflammation is considered a sequel to increasing insulin resistance and disturbed glucose metabolism in diabetes and NHE3 is a transcriptional target of inflammatory cytokines (46, 47). Therefore, we speculate that inflammatory cytokines in diabetes contribute to decreased NHE3 expression.
The role of PKCα in the regulation of NHE3 has been demonstrated using cultured cells (29, 30). PKCα mediates Ca2+-dependent inhibition of NHE3 in fibroblasts and gastrin-induced inhibition of NHE3 in a renal proximal tubule cell line was blocked by Gӧ6976 (31, 48). In the current study, the activation state of PKCα in db/db mice correlated with decreased NHE3 activity and expression. Upon activation that involves phosphorylation, PKCs translocate from the cytosol to the plasma membrane where PKCs phosphorylate their substrates (49). We found that inhibition of PKCα by Gӧ6976 decreased the abundance of NHE3 at the BBM without altering total PKCα protein expression levels. Gӧ6976 is a potent inhibitor of both PKCα and PKCβI, although it is also shown to inhibit JAK2 in vitro (50, 51). Although we cannot exclude a contribution by PKCβ, we found previously that PKCβ expression was decreased in the BBM of the streptozotocin-induced diabetic model, whereas BBM expression of PKCα was increased in the same mice (12).
Recent studies have shown that probiotics have the potential to alleviate chronic diarrhea (52, 53). We found here that LA increased fluid absorption in db/db mouse intestine and effectively prevented diarrhea associated with metformin. On the other hand, LA itself had no effect on the blood glucose level or body weight in db/db mice, indicating that it did not alter the metabolic status of diabetes. In contrast to our findings, others have reported hypoglycemic or hypolipidomic effects of probiotics in diabetic humans and animals, but a much longer ingestion period of probiotics was needed for the hypoglycemic effects in these studies (54, 55).
Previous studies have shown that LA attenuates PKC expression in the enteric smooth muscle of mice with a traumatic brain injury although which PKC isoform is regulated is not clear (56). Interestingly, LA can also activate a PKC-dependent pathway in regulation of innate immune response in RAW 264.7 macrophages (57). We provide evidence that LA-induced regulation of NHE3 is PKCα dependent that LA diminished PKCα expression in the BBM of db/db mouse intestine, an effect similar to when treated with Gӧ6976.
Another observation in our current study is the regulation of ezrin phosphorylation by LA. The importance of ezrin phosphorylation on NHE3 activation has been demonstrated previously (58). Several kinases, including PKCα, PKCξ, PKCγ, PkCι, Akt2, and Rho-kinase, have been implicated in phosphorylation of ezrin (40, 59–62). We ruled out PKCα based on our data that the retrieval of PKCα from the BBM correlated with increased ezrin phosphorylation. Although Akt2 was shown to phosphorylate ezrin to trigger NHE3 translocation to the apical membrane of Caco-2 cells, LA inhibits the PI3K/Akt/mammalian target of rapamycin (mTOR) signaling cascade, suggesting that LA-mediated phosphorylation of ezrin via Akt is unlikely (61, 63). Instead, we focused on PKCι based on a previous study that PKCι phosphorylates ezrin in the apical domain of Caco-2 cells (41). In addition, the role of PKCι in intestinal epithelial barrier integrity and epithelial cell differentiation has been implicated (41, 64, 65). We found here that the inhibition of PKCι using HY-126146 attenuated LCM-mediated ezrin phosphorylation and NHE3 activation, suggesting that LA regulates NHE3 via phosphorylation of ezrin by PKCι. The caveat of the current study, however, is that we did not confirm the specificity of HY-126146 by silencing of PKCι. Although the specificity of HY-126146 on PKCι was previously confirmed by PKCι knockdown on the growth of HUH-7 cells, HY-126146 has limited selectivity within PKC family (42). Future studies are needed to validate the role of PKCι in LA-mediated regulation of ezrin and NHE3 in diabetic intestine.
NHE3 can interact with ezrin directly or indirectly (39). Indirect interaction is mediated by NHERF1 or NHERF2, which can simultaneously bind NHE3 and ezrin (20, 36). We found that LA stimulated NHERF2 expression but not NHERF1 or IRBIT expression. This differs from our previous findings that insulin increases expression of NHERF1 and IRBIT, but not NHERF2 (12). Although we did not determine whether LA induces ezrin interaction with NHE3, phosphorylation of ezrin is required for NHE3 activation and LA is expected to enhance the NHE3-ezrin interaction (12, 61, 66). However, a question remains whether NHERF2 is required for phosphorylation of ezrin by PKCι.
In summary, we have shown that NHE3 expression is reduced in T2D and inhibition of PKCα enhances NHE3 expression and NHE3-dependent fluid absorption. We also showed that LA effectively prevents metformin-mediated inhibition of NHE3 and fluid absorption in diabetic intestine. LA enhances translocation of NHE3 to the BBM via inactivation of PKCα and phosphorylation of ezrin by PKCι.
DATA AVAILABILITY
The data that support the findings of this study are available within the article. The data are available upon request from the corresponding author.
GRANTS
This work was supported by a grant from the Veterans Administration Merit Award I01BX004459. Confocal microscopic analysis was supported in part by the Emory University Integrated Cellular Imaging Core of the Winship Cancer Institute of Emory University and NIH/National Cancer Institute (NCI) under Award P30CA138292.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.H. and C.C.Y. conceived and designed research; Y.H. performed experiments; Y.H., S.S., and C.C.Y. analyzed data; Y.H. and C.C.Y. interpreted results of experiments; Y.H. and C.C.Y. prepared figures; Y.H. and C.C.Y. drafted manuscript; Y.H., S.S., and C.C.Y. approved final version of manuscript.
REFERENCES
- 1. Feldman M, Schiller LR. Disorders of gastrointestinal motility associated with diabetes mellitus. Ann Intern Med 98: 378–384, 1983. doi: 10.7326/0003-4819-98-3-378. [DOI] [PubMed] [Google Scholar]
- 2. Ogbonnaya KI, Arem R. Diabetic diarrhea. Pathophysiology, diagnosis, and management. Arch Intern Med 150: 262–267, 1990. doi: 10.1001/archinte.150.2.262. [DOI] [PubMed] [Google Scholar]
- 3. Gould M, Sellin JH. Diabetic diarrhea. Curr Gastroenterol Rep 11: 354–359, 2009. doi: 10.1007/s11894-009-0054-y. [DOI] [PubMed] [Google Scholar]
- 4. McGovern A, Tippu Z, Hinton W, Munro N, Whyte M, de Lusignan S. Comparison of medication adherence and persistence in type 2 diabetes: a systematic review and meta-analysis. Diabetes Obes Metab 20: 1040–1043, 2018. doi: 10.1111/dom.13160. [DOI] [PubMed] [Google Scholar]
- 5. Prasad VG, Abraham P. Management of chronic constipation in patients with diabetes mellitus. Indian J Gastroenterol 36: 11–22, 2017. doi: 10.1007/s12664-016-0724-2. [DOI] [PubMed] [Google Scholar]
- 6. Chandrasekharan B, Srinivasan S. Diabetes and the enteric nervous system. Neurogastroenterol Motil 19: 951–960, 2007. doi: 10.1111/j.1365-2982.2007.01023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Surawicz CM. Mechanisms of diarrhea. Curr Gastroenterol Rep 12: 236–241, 2010. doi: 10.1007/s11894-010-0113-4. [DOI] [PubMed] [Google Scholar]
- 8. He P, Yun CC. Mechanisms of the regulation of the intestinal Na+/H+ exchanger NHE3. J Biomed Biotechnol 2010: 238080, 2010. doi: 10.1155/2010/238080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Siddique I, Hasan F, Khan I. Suppression of Na+/H+ exchanger isoform-3 in human inflammatory bowel disease: lack of reversal by 5'-aminosalicylate treatment. Scand J Gastroenterol 44: 56–64, 2009. doi: 10.1080/00365520802321253. [DOI] [PubMed] [Google Scholar]
- 10. Janecke AR, Heinz-Erian P, Yin J, Petersen BS, Franke A, Lechner S, Fuchs I, Melancon S, Uhlig HH, Travis S, Marinier E, Perisic V, Ristic N, Gerner P, Booth IW, Wedenoja S, Baumgartner N, Vodopiutz J, Frechette-Duval MC, De Lafollie J, Persad R, Warner N, Tse CM, Sud K, Zachos NC, Sarker R, Zhu X, Muise AM, Zimmer KP, Witt H, Zoller H, Donowitz M, Müller T. Reduced sodium/proton exchanger NHE3 activity causes congenital sodium diarrhea. Hum Mol Genet 24: 6614–6623, 2015. doi: 10.1093/hmg/ddv367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rosenbaum DP, Yan A, Jacobs JW. Pharmacodynamics, safety, and tolerability of the NHE3 inhibitor tenapanor: two trials in healthy volunteers. Clin Drug Investig 38: 341–351, 2018. doi: 10.1007/s40261-017-0614-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. He P, Zhao L, Zhu L, Weinman EJ, De Giorgio R, Koval M, Srinivasan S, Yun CC. Restoration of Na+/H+ exchanger NHE3-containing macrocomplexes ameliorates diabetes-associated fluid loss. J Clin Invest 125: 3519–3531, 2015. doi: 10.1172/JCI79552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Han Y, Yun CC. Metformin inhibits Na(+)/H(+) exchanger NHE3 resulting in intestinal water loss. Front Physiol 13: 867244, 2022. doi: 10.3389/fphys.2022.867244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science 258: 607–614, 1992. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 15. Newton AC. Protein kinase C: structure, function, and regulation. J Biol Chem 270: 28495–28498, 1995. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- 16. Meier M, Menne J, Haller H. Targeting the protein kinase C family in the diabetic kidney: lessons from analysis of mutant mice. Diabetologia 52: 765–775, 2009. doi: 10.1007/s00125-009-1278-y. [DOI] [PubMed] [Google Scholar]
- 17. Zhao L, Bartnikas T, Chu X, Klein J, Yun C, Srinivasan S, He P. Hyperglycemia promotes microvillus membrane expression of DMT1 in intestinal epithelial cells in a PKCα-dependent manner. FASEB J 33: 3549–3561, 2019. doi: 10.1096/fj.201801855R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jubaidi FF, Zainalabidin S, Taib IS, Abdul Hamid Z, Mohamad Anuar NN, Jalil J, Mohd Nor NA, Budin SB. The role of PKC-MAPK signalling pathways in the development of hyperglycemia-induced cardiovascular complications. Int J Mol Sci 23: 8582, 2022. doi: 10.3390/ijms23158582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lin S, Yeruva S, He P, Singh AK, Zhang H, Chen M, Lamprecht G, de Jonge HR, Tse M, Donowitz M, Hogema BM, Chun J, Seidler U, Yun CC. Lysophosphatidic acid stimulates the intestinal brush border Na+/H+ exchanger 3 and fluid absorption via LPA5 and NHERF2. Gastroenterology 138: 649–658, 2010. doi: 10.1053/j.gastro.2009.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yun CHC, Lamprecht G, Forster DV, Sidor A. NHE3 kinase A regulatory protein E3KARP binds the epithelial brush border Na+/H+ exchanger NHE3 and the cytoskeletal protein ezrin. J Biol Chem 273: 25856–25863, 1998. doi: 10.1074/jbc.273.40.25856. [DOI] [PubMed] [Google Scholar]
- 21. Murtazina R, Kovbasnjuk O, Chen TE, Zachos NC, Chen Y, Kocinsky HS, Hogema BM, Seidler U, de Jonge HR, Donowitz M. NHERF2 is necessary for basal activity, second messenger inhibition, and LPA stimulation of NHE3 in mouse distal ileum. Am J Physiol Cell Physiol 301: C126–C136, 2011. doi: 10.1152/ajpcell.00311.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yoo BK, Yanda MK, No YR, Yun CC. Human intestinal epithelial cell line SK-CO15 is a new model system to study Na(+)/H(+) exchanger 3. Am J Physiol Gastrointest Liver Physiol 303: G180–G188, 2012. doi: 10.1152/ajpgi.00069.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jenkin KA, Han Y, Lin S, He P, Yun CC. Nedd4-2-dependent ubiquitination potentiates the inhibition of human NHE3 by cholera toxin and enteropathogenic Escherichia coli. Cell Mol Gastroenterol Hepatol 13: 695–716, 2022. doi: 10.1016/j.jcmgh.2021.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Singh V, Raheja G, Borthakur A, Kumar A, Gill RK, Alakkam A, Malakooti J, Dudeja PK. Lactobacillus acidophilus upregulates intestinal NHE3 expression and function. Am J Physiol Gastrointest Liver Physiol 303: G1393–G1401, 2012. doi: 10.1152/ajpgi.00345.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. No YR, He P, Yoo BK, Yun CC. Unique regulation of human Na+/H+ exchanger 3 (NHE3) by Nedd4-2 ligase that differs from non-primate NHE3s. J Biol Chem 289: 18360–18372, 2014. doi: 10.1074/jbc.M113.541706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. King AJ. The use of animal models in diabetes research. Br J Pharmacol 166: 877–894, 2012. doi: 10.1111/j.1476-5381.2012.01911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cuppoletti J, Chakrabarti J, Tewari KP, Malinowska DH. Differentiation between human ClC-2 and CFTR Cl− channels with pharmacological agents. Am J Physiol Cell Physiol 307: C479–C492, 2014. doi: 10.1152/ajpcell.00077.2014. [DOI] [PubMed] [Google Scholar]
- 28. Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res 106: 1319–1331, 2010. doi: 10.1161/CIRCRESAHA.110.217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tse CM, Levine SA, Yun CH, Brant SR, Pouyssegur J, Montrose MH, Donowitz M. Functional characteristics of a cloned epithelial Na+/H+ exchanger (NHE3): resistance to amiloride and inhibition by protein kinase C. Proc Natl Acad Sci USA 90: 9110–9114, 1993. doi: 10.1073/pnas.90.19.9110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wiederkehr MR, Zhao H, Moe OW. Acute regulation of Na/H exchanger NHE3 activity by protein kinase C: role of NHE3 phosphorylation. Am J Physiol Cell Physiol 276: C1205–C1217, 1999. doi: 10.1152/ajpcell.1999.276.5.C1205. [DOI] [PubMed] [Google Scholar]
- 31. Lee-Kwon W, Kim JH, Choi JW, Kawano K, Cha B, Dartt DA, Zoukhri D, Donowitz M. Ca2+-dependent inhibition of NHE3 requires PKC α which binds to E3KARP to decrease surface NHE3 containing plasma membrane complexes. Am J Physiol Cell Physiol 285: C1527–C1536, 2003. doi: 10.1152/ajpcell.00017.2003. [DOI] [PubMed] [Google Scholar]
- 32. Feng X, Becker KP, Stribling SD, Peters KG, Hannun YA. Regulation of receptor-mediated protein kinase C membrane trafficking by autophosphorylation. J Biol Chem 275: 17024–17034, 2000. doi: 10.1074/jbc.275.22.17024. [DOI] [PubMed] [Google Scholar]
- 33. Kumar A, Hecht C, Priyamvada S, Anbazhagan AN, Alakkam A, Borthakur A, Alrefai WA, Gill RK, Dudeja PK. Probiotic Bifidobacterium species stimulate human SLC26A3 gene function and expression in intestinal epithelial cells. Am J Physiol Cell Physiol 307: C1084–C1092, 2014. doi: 10.1152/ajpcell.00194.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kumar A, Anbazhagan AN, Coffing H, Chatterjee I, Priyamvada S, Gujral T, Saksena S, Gill RK, Alrefai WA, Borthakur A, Dudeja PK. Lactobacillus acidophilus counteracts inhibition of NHE3 and DRA expression and alleviates diarrheal phenotype in mice infected with Citrobacter rodentium. Am J Physiol Gastrointest Liver Physiol 311: G817–G826, 2016. doi: 10.1152/ajpgi.00173.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bornancin F, Parker PJ. Phosphorylation of protein kinase C-alpha on serine 657 controls the accumulation of active enzyme and contributes to its phosphatase-resistant state. J Biol Chem 272: 3544–3549, 1997. [Erratum in J Biol Chem 272: 13458, 1997]. doi: 10.1074/jbc.272.6.3544. [DOI] [PubMed] [Google Scholar]
- 36. Yun CH, Oh S, Zizak M, Steplock D, Tsao S, Tse CM, Weinman EJ, Donowitz M. cAMP-mediated inhibition of the epithelial brush border Na+/H+ exchanger, NHE3, requires an associated regulatory protein. Proc Natl Acad Sci USA 94: 3010–3015, 1997. [Erratum in Proc Natl Acad Sci USA 94: 10006, 1997]. doi: 10.1073/pnas.94.7.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. He P, Zhang H, Yun CC. IRBIT, inositol 1,4,5-triphosphate (IP3) receptor-binding protein released with IP3, binds Na+/H+ exchanger NHE3 and activates NHE3 activity in response to calcium. J Biol Chem 283: 33544–33553, 2008. doi: 10.1074/jbc.M805534200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhao H, Shiue H, Palkon S, Wang Y, Cullinan P, Burkhardt JK, Musch MW, Chang EB, Turner JR. Ezrin regulates NHE3 translocation and activation after Na+-glucose cotransport. Proc Natl Acad Sci USA 101: 9485–9490, 2004. doi: 10.1073/pnas.0308400101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cha B, Tse M, Yun C, Kovbasnjuk O, Mohan S, Hubbard A, Arpin M, Donowitz M. The NHE3 juxtamembrane cytoplasmic domain directly binds ezrin: dual role in NHE3 trafficking and mobility in the brush border. Mol Biol Cell 17: 2661–2673, 2006. doi: 10.1091/mbc.e05-09-0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ng T, Parsons M, Hughes WE, Monypenny J, Zicha D, Gautreau A, Arpin M, Gschmeissner S, Verveer PJ, Bastiaens PI, Parker PJ. Ezrin is a downstream effector of trafficking PKC-integrin complexes involved in the control of cell motility. EMBO J 20: 2723–2741, 2001. doi: 10.1093/emboj/20.11.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wald FA, Oriolo AS, Mashukova A, Fregien NL, Langshaw AH, Salas PJ. Atypical protein kinase C (iota) activates ezrin in the apical domain of intestinal epithelial cells. J Cell Sci 121: 644–654, 2008. doi: 10.1242/jcs.016246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kwiatkowski J, Baburajendran N, Poulsen A, Liu B, Tee DHY, Wong YX, Poh ZY, Ong EH, Dinie N, Cherian J, Jansson AE, Hill J, Keller TH, Hung AW. Fragment-based discovery of a small-molecule protein kinase C-iota inhibitor binding post-kinase domain residues. ACS Med Chem Lett 10: 318–323, 2019. doi: 10.1021/acsmedchemlett.8b00546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chan LKY, Wang Y, Ng EKW, Leung PS. Na(+)/H(+) exchanger 3 blockade ameliorates type 2 diabetes mellitus via inhibition of sodium-glucose co-transporter 1-mediated glucose absorption in the small intestine. Diabetes Obes Metab 20: 709–717, 2018. doi: 10.1111/dom.13151. [DOI] [PubMed] [Google Scholar]
- 44. Lin R, Murtazina R, Cha B, Chakraborty M, Sarker R, Chen TE, Lin Z, Hogema BM, de Jonge HR, Seidler U, Turner JR, Li X, Kovbasnjuk O, Donowitz M. d-Glucose acts via sodium/glucose cotransporter 1 to increase NHE3 in mouse jejunal brush border by a Na+/H+ exchange regulatory factor 2-dependent process. Gastroenterology 140: 560–571, 2011. doi: 10.1053/j.gastro.2010.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dominguez Rieg JA, Chirasani VR, Koepsell H, Senapati S, Mahata SK, Rieg T. Regulation of intestinal SGLT1 by catestatin in hyperleptinemic type 2 diabetic mice. Lab Invest 96: 98–111, 2016. doi: 10.1038/labinvest.2015.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol 11: 98–107, 2011. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 47. Amin MR, Malakooti J, Sandoval R, Dudeja PK, Ramaswamy K. IFN-γ and TNF-α regulate human NHE3 gene expression by modulating the Sp family transcription factors in human intestinal epithelial cell line C2BBe1. Am J Physiol Cell Physiol 291: C887–C896, 2006. doi: 10.1152/ajpcell.00630.2005. [DOI] [PubMed] [Google Scholar]
- 48. Liu T, Jose PA. Gastrin induces sodium-hydrogen exchanger 3 phosphorylation and mTOR activation via a phosphoinositide 3-kinase-/protein kinase C-dependent but AKT-independent pathway in renal proximal tubule cells derived from a normotensive male human. Endocrinology 154: 865–875, 2013. doi: 10.1210/en.2012-1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Igumenova TI. Dynamics and membrane interactions of protein kinase C. Biochemistry (Mosc ) 54: 4953–4968, 2015. doi: 10.1021/acs.biochem.5b00565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schachtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J Biol Chem 268: 9194–9197, 1993. [PubMed] [Google Scholar]
- 51. Grandage VL, Everington T, Linch DC, Khwaja A. Go6976 is a potent inhibitor of the JAK 2 and FLT3 tyrosine kinases with significant activity in primary acute myeloid leukaemia cells. Br J Haematol 135: 303–316, 2006. doi: 10.1111/j.1365-2141.2006.06291.x. [DOI] [PubMed] [Google Scholar]
- 52. Guarino A, Guandalini S, Lo Vecchio A. Probiotics for prevention and treatment of diarrhea. J Clin Gastroenterol 49, Suppl 1: S37–S45, 2015. doi: 10.1097/MCG.0000000000000349. [DOI] [PubMed] [Google Scholar]
- 53. Ishaque SM, Khosruzzaman SM, Ahmed DS, Sah MP. A randomized placebo-controlled clinical trial of a multi-strain probiotic formulation (Bio-Kult(R)) in the management of diarrhea-predominant irritable bowel syndrome. BMC Gastroenterol 18: 71, 2018. doi: 10.1186/s12876-018-0788-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Negm El-Dein A, Ezzat A, Aly HF, Awad G, Farid M. Lactobacillus-fermented yogurt exerts hypoglycemic, hypocholesterolemic, and anti-inflammatory activities in STZ-induced diabetic Wistar rats. Nutr Res 108: 22–32, 2022. doi: 10.1016/j.nutres.2022.10.003. [DOI] [PubMed] [Google Scholar]
- 55. Tao YW, Gu YL, Mao XQ, Zhang L, Pei YF. Effects of probiotics on type II diabetes mellitus: a meta-analysis. J Transl Med 18: 30, 2020. [Erratum in J Transl Med 18: 105, 2020]. doi: 10.1186/s12967-020-02213-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sun B, Hu C, Fang H, Zhu L, Gao N, Zhu J. The effects of Lactobacillus acidophilus on the intestinal smooth muscle contraction through PKC/MLCK/MLC signaling pathway in TBI mouse model. PLoS One 10: e0128214, 2015. doi: 10.1371/journal.pone.0128214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cai ZD, Wang PY, Guo YX, Zeng XQ, Wu Z, Pan DD. S-layer protein modulates the stimulatory effects of Lactobacillus acidophilus CICC 6074 by triggering PKC signaling cascade in RAW 264.7 cells. J Funct Foods 67: 103841, 2020. doi: 10.1016/j.jff.2020.103841. [DOI] [Google Scholar]
- 58. Di Sole F, Babich V, Moe OW. The calcineurin homologous protein-1 increases Na(+)/H(+) -exchanger 3 trafficking via ezrin phosphorylation. J Am Soc Nephrol 20: 1776–1786, 2009. doi: 10.1681/ASN.2008121255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Simons PC, Pietromonaco SF, Reczek D, Bretscher A, Elias L. C-terminal threonine phosphorylation activates ERM proteins to link the cell's cortical lipid bilayer to the cytoskeleton. Biochem Biophys Res Commun 253: 561–565, 1998. doi: 10.1006/bbrc.1998.9823. [DOI] [PubMed] [Google Scholar]
- 60. Matsui T, Maeda M, Doi Y, Yonemura S, Amano M, Kaibuchi K, Tsukita S, Tsukita S. Rho-kinase phosphorylates COOH-terminal threonines of ezrin/radixin/moesin (ERM) proteins and regulates their head-to-tail association. J Cell Biol 140: 647–657, 1998. doi: 10.1083/jcb.140.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shiue H, Musch MW, Wang Y, Chang EB, Turner JR. Akt2 phosphorylates ezrin to trigger NHE3 translocation and activation. J Biol Chem 280: 1688–1695, 2005. doi: 10.1074/jbc.M409471200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ren L, Hong SH, Cassavaugh J, Osborne T, Chou AJ, Kim SY, Gorlick R, Hewitt SM, Khanna C. The actin-cytoskeleton linker protein ezrin is regulated during osteosarcoma metastasis by PKC. Oncogene 28: 792–802, 2009. doi: 10.1038/onc.2008.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Saber R, Zadeh M, Pakanati KC, Bere P, Klaenhammer T, Mohamadzadeh M. Lipoteichoic acid-deficient Lactobacillus acidophilus regulates downstream signals. Immunotherapy 3: 337–347, 2011. doi: 10.2217/imt.10.119. [DOI] [PubMed] [Google Scholar]
- 64. Kovac J, Oster H, Leitges M. Expression of the atypical protein kinase C (aPKC) isoforms iota/lambda and zeta during mouse embryogenesis. Gene Expr Patterns 7: 187–196, 2007. doi: 10.1016/j.modgep.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 65. Banan A, Zhang LJ, Farhadi A, Fields JZ, Shaikh M, Forsyth CB, Choudhary S, Keshavarzian A. Critical role of the atypical lambda isoform of protein kinase C (PKC-λ) in oxidant-induced disruption of the microtubule cytoskeleton and barrier function of intestinal epithelium. J Pharmacol Exp Ther 312: 458–471, 2005. doi: 10.1124/jpet.104.074591. [DOI] [PubMed] [Google Scholar]
- 66. Babich V, Di Sole F. The Na+/H+ exchanger-3 (NHE3) activity requires ezrin binding to phosphoinositide and its phosphorylation. PLoS One 10: e0129306, 2015. doi: 10.1371/journal.pone.0129306. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available within the article. The data are available upon request from the corresponding author.