

Abstract
CD8+ cytotoxic T lymphocytes play a crucial role in targeting virus-infected and cancer cells. Although other cytotoxic lymphocytes such as CD4+ T and NK cells, as well as CAR-T cells, can also identify and destroy aberrant cells, they seem to be significantly less potent based on available experimental data. Here, I contemplate the molecular mechanisms controlling the sensitivity and kinetics of granule-mediated CD8+ T cell cytolytic responses. I posit that the clustering of MHC-I molecules and TCRs on the cell surface, as well as the contribution of the CD8 co-receptor, are major factors driving exceptionally potent cytolytic responses. I also contend that CD8+ T cells with known specificity and engineered TCR-T cells might be among the most efficient cytolytic effectors for treating patients suffering from viral infections or cancer.
A model for the cytotoxic potency of CD8+ T lymphocytes
Cytotoxic lymphocytes contain cytolytic granules, i.e., “poison pills”, that can be released to kill target cells. Granule delivery is controlled by establishing a unique contact between the two cellular membranes called the immunological synapse or synaptic interface. The interface structure and the dynamics of its formation and stability play an essential role in the efficiency of granule delivery. The latter is essential for the host in winning the race against the spread of aberrant or infectious cells. In this opinion, I hypothesize that for CD8+ T cells, the clustering of MHC-I and TCR molecules and the binding of CD8, significantly contribute to the potency of CTL cytotoxicity, based on the sensitivity and kinetics of these interactions for granule delivery. I argue that antigen-specific CD8+ T cells and engineered TCR-T cells might be among the most efficient cytotoxic cells. Below, I compare several factors contributing to the regulation of CD8+ and CD4+ cytotoxic lymphocyte effector functions.
CD8+ versus CD4+ cytotoxic T lymphocytes
CD8+ T cells evolved to destroy virus infected and cancer cells by delivering cytotoxic granules [1, 2]. Although other lymphocytes such as CD4+ T cells and NK cells can also exercise cytolytic activity, CD8+ T cells appear to be much more sensitive and efficient killers [3, 4]. Indeed, our group previously showed that even a very few cognate peptide-MHC ligands presented on target cells would be sufficient for CD8+ cytotoxic T lymphocytes (CTL) to recognize and destroy such target cells [5]. Although the recognition parameters of the peptide-MHC-I by the TCR on CD8+ CTLs are thought to be important for mediating the induction of efficient T-cell responses [6, 7], the contribution of the CD8 co-receptor that binds to the non-polymorphic domain of MHC class-I is known to play a key role in enabling recognition and enhancing cytotoxic responses [8, 9]. In fact, CD8 knockout mice (Cd8−/−) harbor very few Cd8-deficient T cells with a potential for CD8+ lineage differentiation and in which the Cd4 locus is shut down [10]. Indeed, there is a limited number of such T cells that are detectable in mouse peripheral lymph nodes. Moreover, the CD8- T cells from Cd8−/− mice can respond to allogeneic but not syngeneic target cells, suggesting that missed CD8 co-receptor contribution diminishes the recognition of syngeneic ligands; this has been attributed to low avidity of the TCR due to the absence of CD8, while higher TCR avidity permits recognition of allogeneic target cells [10]; indeed, very few Cd8−/− T cells were recovered from lymph nodes and spleen [10]. These data indicated that the CD8 co-receptor plays an essential role in contributing to CTL function. By contrast, CD4 knockout mice (Cd4−/−) harbor a relatively high number of CD4-deficient T cells that have been differentiated into the CD4 lineage and can be found in peripheral lymph nodes; these show diminished helper activity for B cell antibody production compared with Cd4+/+ T cells [11]. These T cells, however, have been shown to respond to syngeneic stimulation, and the absence of CD4 in this mouse model did not alter the ability of such T cells to mount antiviral cytolytic responses and produce IL-2, suggesting that absence of the CD4 co-receptor is less critical for Cd4−/− T cell effector function [11]. Even though both CD8 and CD4 co-receptors can deliver the lymphocyte specific tyrosine protein kinase p56lck in close proximity to TCRs, the coreceptors are engaged by either peptide-MHC class I (pMHC-I) or pMHC-II ligands [12–14] and the CD8-MHC-I binding is much stronger than that of CD4-MHC-II; this suggests that the CD4 co-receptor may be more dispensable [15, 16]. Moreover, despite the similar functions of CD4 and CD8 co-receptors in T cells, I argue that the contribution of CD8 to the CTL cytolytic response, seems to be more important than that of CD4; however, this remains conjectural. Of note, CD8 has been reported to significantly accelerate CD8+ CTL-mediated cytotoxicity in contrast to CD4+ CTL (for illustrative purposes only see Figure 1) and increase the sensitivity of the response that is essential for rapid elimination of aberrant cells, thus winning the race against the proliferation of the former [17, 18].
Figure 1. CD8+ T cells can exhibit greater potency to fight aberrant cells than CD4+ T cells.
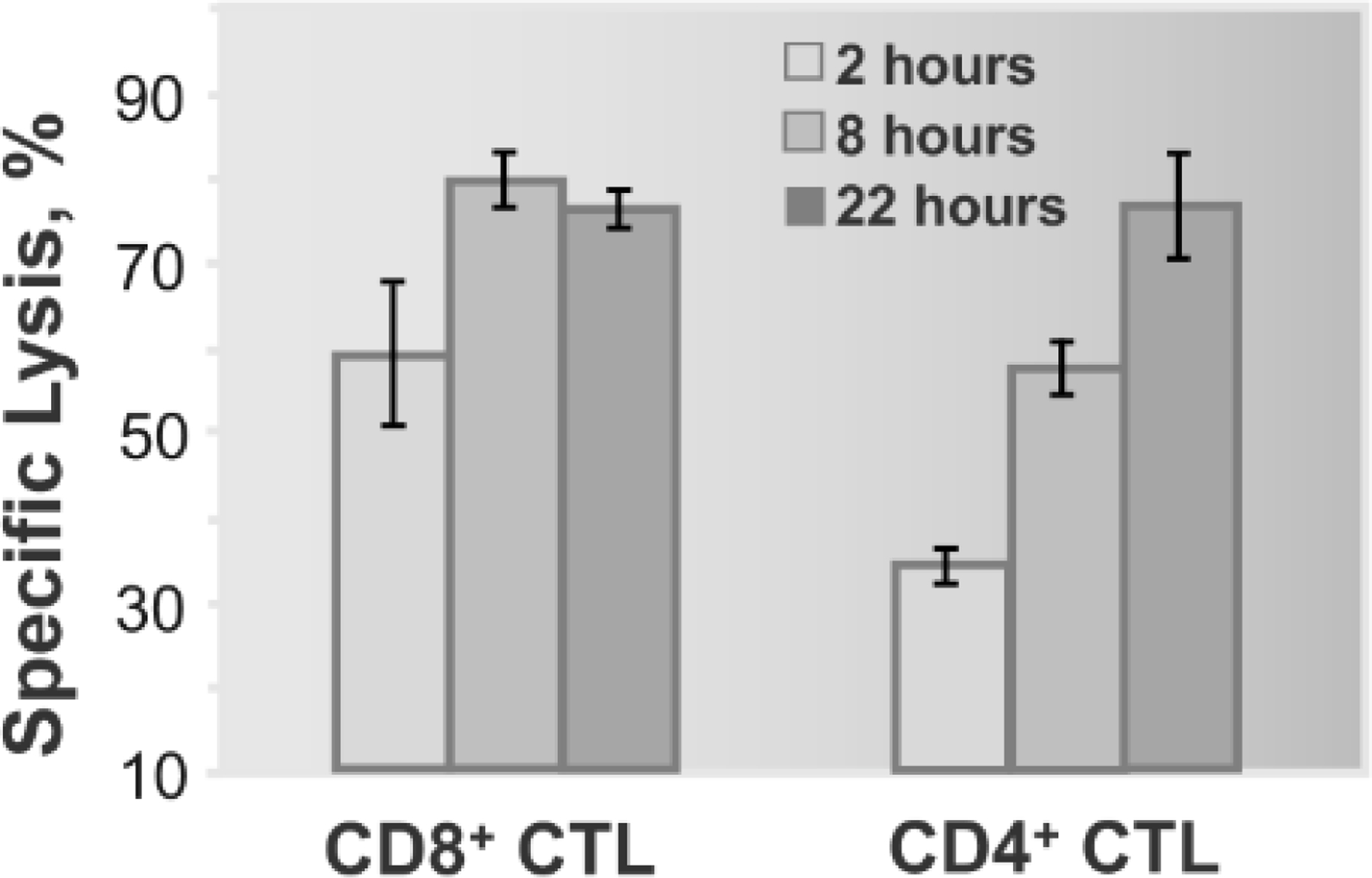
For illustrative purposes only, a previously published experiment is shown [17]. Lymphoblastoid T2 cells displaying both HLA-A2 and HLA-DR1 were sensitized with two different stimulatory peptides at concentrations required for half-maximal lysis. Each target cell was exposed to either CD8+ or CD4+ human cytotoxic T cells (CTL), respectively. This study concluded that target cells were destroyed much fasted by CD8+ CTL than by CD4+ cytolytic effector cells [17].
Noteworthy, engagement of the CD8 co-receptor also limits the affinity of the TCR for pMHC-I ligands, called “affinity ceiling”, indicating that the contribution of CD8 binding to the complex, eliminates a requirement for stronger TCR-pMHC-I interactions to ensue for activation [19]. One research group produced a TCR that recognized cognate pMHC ligands on tumor cells with very high affinity, and which was above the “affinity ceiling” [20]. Specifically, genes encoding a specific TCR were transduced into polyclonal T cells and the engineered TCR-T cells, both CD8+ and CD4+, were injected into mice to target a tumor. Analysis of these T cells derived from lymphoid organs and those infiltrating the tumor showed complete absence of CD8+ T cells, while CD4+ T cells expressing high-affinity TCR were still present in both lymphoid organs and the tumor, consistent with a significantly weaker binding of CD4 to MHC-II as opposed to a CD8-MHC-I interaction which would induce T cell death [20]. The authors determined that the TCR with enhanced affinity bound to the same cognate pMHC ligand with 1000-fold higher affinity compared to the natural TCR. Under these conditions, TCR-mediated signaling was too strong because of high affinity for the TCR, and which was further enhanced by the CD8 co-receptor which bound to the same cognate pMHC ligand on the cell surface, ultimately leading to T cell death [20].
It is well established that naïve CD4+ T cells can differentiate into various subsets with distinct functions [21]. This includes Th1 and Th2 T cells as well as Th9 and Th17 T cells. CD4+ T cells can also develop into regulatory T cells (Treg) either during selection in the thymus or in the periphery. Other CD4+ T cells include follicular T helper cells, (Tfh), that play a pivotal role in B cell activation, affinity maturation of antibodies, and germinal center formation. In contrast to MHC-I-restricted CD8+ T cells that are present on all nucleated cells, MHC class II-restricted CD4+ T cells are expressed only on specialized antigen-presenting cells (APC), such as monocytes and dendritic cells (DC). However, MHC-II expression on these cells, particularly activated APC, is significantly higher than MHC-I expression on target cells; this is necessary to engage a large number of TCRs on CD4+ T cells to be sufficient for T cell activation [22, 23]. Indeed, upon DC activation, the expression of long-lived pMHC-II is significantly increased at the cell surface to trigger the activation of naïve CD4+ T cells [24]. However, even at a very high level of presentation, naïve CD4+ T cells must make many encounters with APC to induce the activation of the former [25]. This appears to be true for the activation of both MHC-I and MHC-II restricted T cells [26]. However, in contrast to CD8+ T cells, CD4+ T cells typically respond to target cells that present a higher number of pMHC-II ligands [27]. This has been generally attributed to a low affinity of TCR-pMHC-II interactions [15]. Thus, higher amounts of pMHC-II on target cells that are recognizable by CD4+ T cells ensure the engagement of a sufficient number of TCRs to initiate strong enough signaling to achieve a productive CD4+ T cell response. Accordingly, very weak interactions of CD4 with MHC-II have been reported to barely contribute to productive TCR engagement on live CD4+ T cells by pMHC-II [28]. Consistent with this, cytotoxic CD4+ T cells have been found to be less efficient killers and to require a longer time to destroy the same target cells compared with CD8+ CTL [17] (and Figure 1 which is shown for illustrative purposes). Taken together, it seems that an efficient response from a CD4+ T cell relies on the engagement of a larger number of TCRs compared to that required for a CD8+ T cell.
On the mechanism of granule release
Our laboratory group showed that efficient killing by cytotoxic lymphocytes is linked to a short pathway of cytolytic granule delivery to the T cell/target cell interface [3]. The latter is regulated by the rapid kinetics of Ca2+ mobilization upon T cell activation; intracellular Ca2+ mediates the activity of dynein motors that move granules (i.e., specialized lysosomes containing a “poison pill”) along microtubules towards the microtubule organizing center (MTOC) [29, 30]; this is followed by MTOC polarization to the T cell membrane contact interface with a target cell [31, 32] (Figure 2). Subsequently, this leads to lysosome membrane fusion with the T-cell membrane allowing the release of cytolytic granules [33, 34] Analysis of the patterns of Ca2+ mobilization initiated by different strengths of stimulation in CTLs shows that a strong stimulatory signal results in rapid and sustained increase of intracellular Ca2+, while weak stimulation initiates delayed Ca2+ mobilization, leading to an inefficient T cell response [3]. Thus, variations in the signaling strength regulating the kinetics of Ca2+ mobilization determine the dynamics of granule movements along microtubules and ultimately, the pattern of granule release at the T cell membrane contact with the target cell [3]. Granules can be released in the middle of the synaptic interface; specifically, inside the peripheral ring junction of the immunological synapse that does not contain highly polymerized actin, membrane fusion is facilitated and granule release ensues, as evidenced from previous findings [3]. This seems to be the most efficient pathway of granule delivery to target cells that has been observed for highly potent CD8+ T cells. However, for cytotoxic CD4+ T cells, granule release within the peripheral ring junctions is delayed because of a ‘long path’ to granule delivery; specifically, the latter is mediated by slow kinetics of Ca2+ mobilization induced by the initiation of weak TCR-mediated signaling in CD4+ T cells [3]. This in turn, can delay granule movement along microtubules by dynein motors toward MTOC, while recruitment of the MTOC to the contact membrane might still remain rapid [35, 36]. In this scenario, granules can move to the periphery of the synaptic interface where microtubules are attached to the polymerized actin [37] and then cross to the peripheral ring junction to reach the middle of the interface to be released. Such a longer pathway of granule delivery may account for less efficient CD4+ T cells cytolytic responses compared with CD8+ T cells [3, 34–37]. The synaptic interface formed by NK cells is usually less well organized compared to the interface formed by T cells because complete segregation of polymerized actin is often not achieved, and cytolytic granules are usually delivered and released in small areas across the interface that is devoid of highly polymerized actin [38]. Such patterns of granule release by NK cells have been associated with incomplete actin segregation and perhaps might be linked to less potent cytolytic responses compared with T cells.
Figure 2. Mechanism of granule delivery to the T cell synaptic interface.
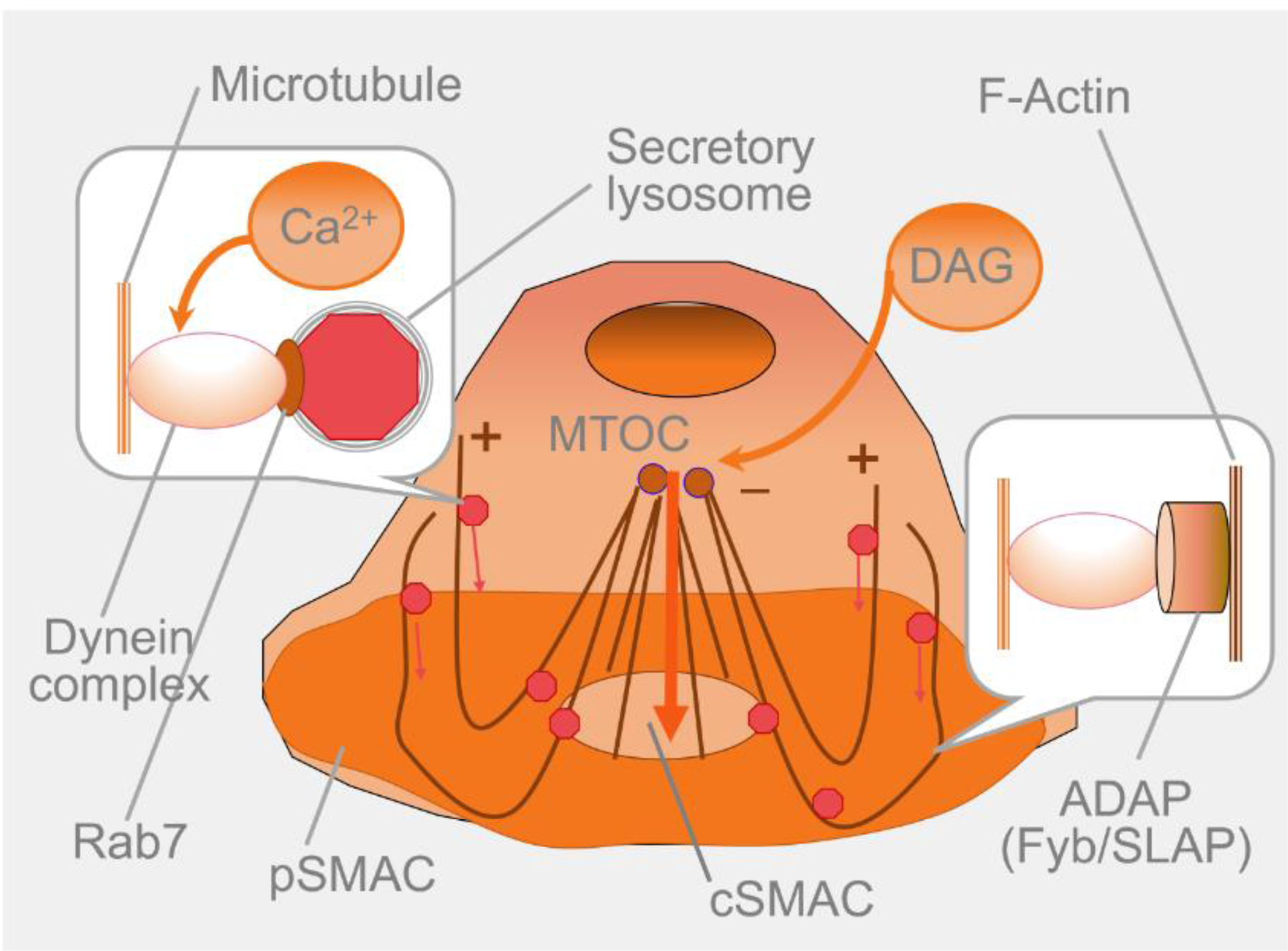
It is well-established that strong T cell stimulation by cognate peptide-MHC on the surface of a target cell leads to Ca2+ influx activating cytoplasmic dynein motors that are linked to microtubules via Rab7, a member of the RAB family of RAS-related GTP-binding proteins. Activated dynein moves secretory lysosomes containing cytolytic granules from the plus end of microtubules towards the microtubule organizing center (MTOC). At the same time, another variant of the dynein motor linked by adhesion- and degranulation-promoting adapter protein (ADAP) to highly polymerized actin at the peripheral supramolecular activating cluster (pSMAC) functions as a fishing rod that reels in microtubules, pooling MTOC toward the center of the T cell/target cell interface, i.e., central supramolecular activating cluster (cSMAC). Diacylglycerol (DAG) that is produced as a result of the hydrolysis of a membrane lipid PIP2 (phosphatidylinositol 4,5-bisphosphate) during T cell activation facilitates MTOC recruitment to the contact interface [31–37]. This seems to be the most efficient pathway of cytolytic granule delivery by CD8+ CTL to target cells.
Another significant factor that controls the efficiency of CD8+ T cell responses is the extent of clustering of TCR molecules on T cells and pMHC-I ligands on target cells. For instance, the extent of TCR clustering depends on the stage of T cell differentiation: Naïve T cells display small size nanoclusters that do not include CD8 co-receptors, while activated T cells present large clusters of TCR molecules that co-cluster with CD8 [39]. In addition, TCR and CD8 co-clustering enhance the recognition of a very small number of cognate pMHC-I, facilitating the initiation of signaling within a cluster where only a few or perhaps even a single TCR are bound to stimulatory pMHC-I ligands [5, 40]. Thus, closely positioned TCR and CD8 molecules [39] mediate signal spread within clusters, resulting in the activation of TCRs that are bound to self pMHC-I ligands, and not necessarily to stimulatory pMHC-I (Figure 3). This has been documented to lead to signal amplification and enhanced sensitivity of the CD8+ T cell response [40]. It is well-established that clustering of pMHC-I on target and APCs further facilitates CD8+ T cells responses [41]. Our group exploited nanoparticles to assemble pMHC-I on the surface of such nanoparticles; these were used to mimic membrane patches presenting pMHC-I at various densities [42, 43]. CD8+ T cells stimulation with the pMHC-I/nanoparticle cluster to model membrane clusters demonstrated the dependence on the density of pMHC-I ligands displayed on nanoparticles was associated with the efficiency of T cell responses, as evidenced from the kinetics and patten of Ca2+ mobilization [42, 43]. Thus, clustering of antigen-specific receptors on T cells and their ligands on antigen-presenting and target cells as well as the contribution of the CD8 co-receptor play essential roles in facilitating the efficiency of T cell responses [40, 43].
Figure 3. On the mechanism of signal spread within TCR clusters.
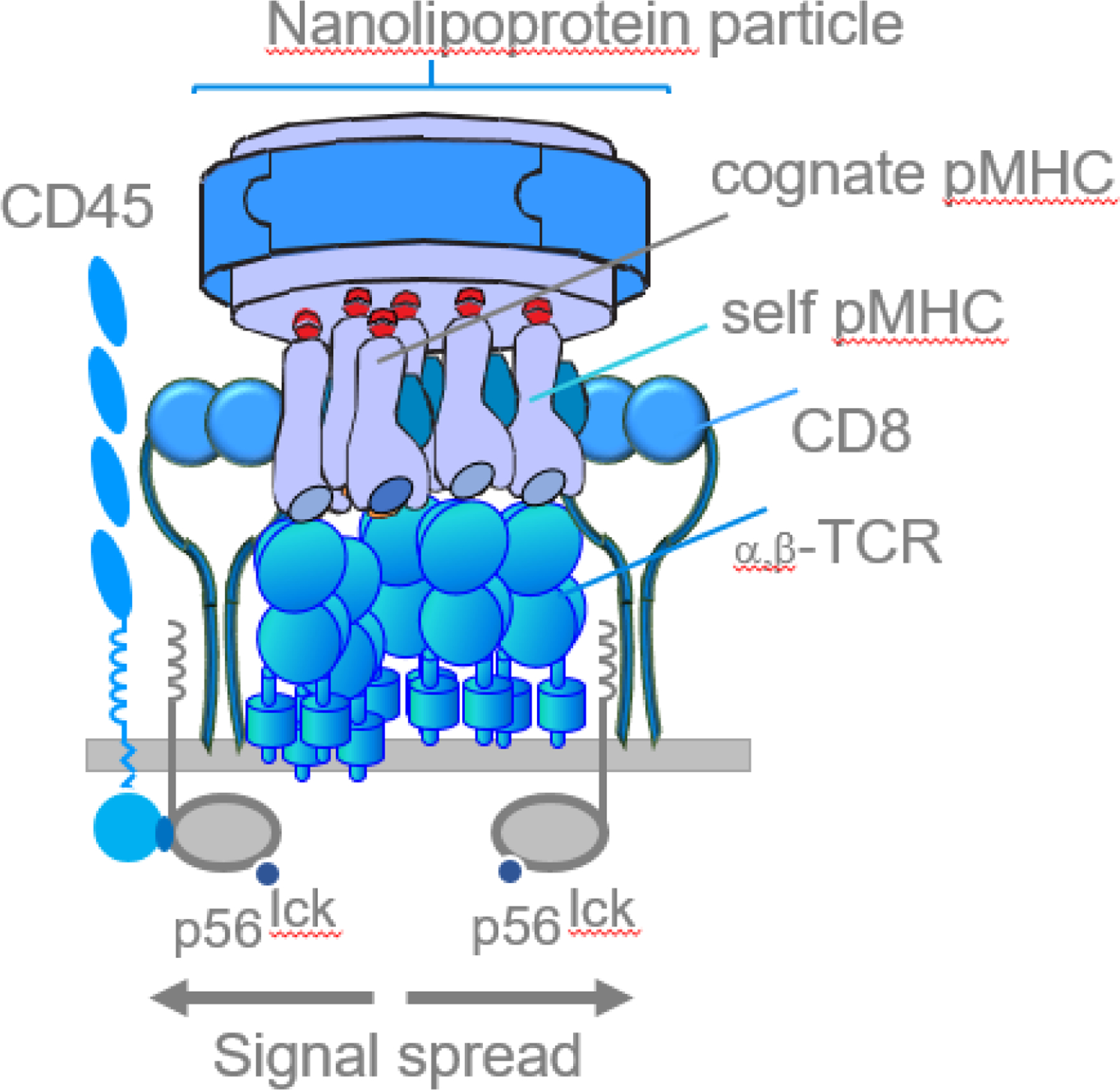
Interaction of clustered TCRs on T cells with peptide-MHC ligand clusters displayed on nanolipoprotein particles has led to the engagement of a single stimulatory peptide-MHC by a TCR [43]. It is well-established that recruitment of activated tyrosine kinases (p56lck) by CD45 protein initiates phosphorylation of the CD3 cytoplasmic tails of the engaged receptor. Binding of CD8 co-receptor to MHC-I further facilitates recruitment of p56lck [12–14]. At the same time, CD3 ITAM motifs of other TCRs bound to self-peptide-MHC ligands that are in a very close proximity can also become phosphorylated by the p56lck that is recruited to activated TCR, spreading the signal within the cluster. Thus, clustering of MHC-I and TCR and CD8 on the T cells leads to productive engagement of the TCRs facilitating potency of CD8+ T cell responses [12]15].
Engineering T cells
The repertoire of available antigen-specific receptors on T cells is unique in every individual. Therefore, certain individuals harbor T cells that are more efficient at controlling viruses and transformed cells compared to others [44, 45]. Indeed, selected individuals from this category bear exceptionally potent T cells recognizing virus-infected cells (e.g. HIV-1), precluding the typical symptoms of infection, termed “elite controllers” [46]. The repertoire of the receptors available on T cells in other individuals may not mediate an efficient response against unwanted cells, resulting in the development of a severe infection. Even though similar considerations might be applied to T cells identifying and destroying tumor cells, other factors such as tumor microenvironment may negatively influence T cell functions [47].
One way to overcome the problem of an inefficient response is to engineer potent T cells with the specificity of interest. Indeed, engineered T cells endowed with either chimeric antigen receptor (CAR-T cells) or T-cell receptor (TCR-T cells) have been produced to target aberrant cells in the human body – a technology that has emerged in recent years to fight cancerous and infected cells. However, the efficiency of these T-cells remains to be further and rigorously studied. The TCR expression on natural T cells can vary from 30,000 to 100,000 per cell depending on stage of differentiation and other factors [48]. However, the expression of CARs on T cells might not always be thoroughly measured. Our research group reported that the transduction of human polyclonal T cells derived from cord blood with second generation CARs that were specific for High Molecular Weight Melanoma Associated protein (HMM-MAA) from melanoma cells, led to the expression of 10,000 CARs per CD8+ T cell and was similar to the expression of transgenic TCR on CD8+ T cells, i.e., 12,000 TCRs per cell [49]. This was significantly lower than the expression of endogenous TCR on T cells but the reasons for lower expression of CAR and transgenic TCR molecules on T cells remain to be dissected. A study comparing the efficiency of CD8+ T cell cytolytic responses with those of CAR-T cells in a standard CTL assay against HLA-A2-positive uveal melanoma cells (CM006) revealed a large difference in the sensitivity of T cells responses mediated by either CAR or endogenous TCR [49]. Specifically, our group compared the extent of lysis of CM006 cells that presented 120,000 HMM-MAA ligands/cell that were recognizable by CAR-T cells, with the killing of the same cells presenting less than 7,000 pHLA-A2 ligands/cell, recognized by specific CD8+ CTL cells at the same effector-to-target ratio (5:1) [49]. Importantly, the intrinsic affinity of the CAR for HMM-MAA (2×107 L/M) was significantly higher than the affinity of the TCR for cognate pMHC (2.4×105 L/M). Less than 20% of the target cells displaying 120,000 l HMM-MAA molecules/cell were killed by CAR-T cells while about 30% the same target cells presenting about 7,000 cognate pHLA-A2 ligands/cell were destroyed by CD8+ CTL. These data indicated that the engagement of a larger number of CARs was necessary to initiate T-cell cytotoxic responses as opposed to a relatively small number of engaged TCRs. This led us to suggest that the sensitivity of CAR T cell responses might be significantly lower than that of CD8+ T cell responses [49], although this warrants further investigation. I hypothesize that this is reminiscent of differences in the sensitivity of cytotoxic responses between CD8+ and CD4+ T cells [43], and argue that the responsiveness of CAR-T cells might be similar to that of CD4+ T cells, although this again, remains conjectural.
In the same study, our group also compared engineered TCR-T cells with ‘natural’ CD8+ T cells responding to the same CM006 melanoma cells [49]. A greater CD8+ CTL sensitivity for cytotoxicity was observed, suggesting that engaging natural TCR could initiate significant signal amplification (see Figure 3), although this remains to be directly demonstrated. Therefore, even though transgenic TCR induced more sensitive CTL responses compared to CAR-T cells, the TCR-T cell response remained less sensitive than that of natural CD8+ T cells [49]. It is currently unclear whether lower transgenic TCR expression is solely responsible for a less sensitive TCR-T cell cytotoxic response compared with that of natural CD8+ T cells. A thorough and robust comparison of the triggering mechanisms initiated by transgenic TCR, CARs, and natural TCRs might help clarify the functional differences between different engineered T cells.
Concluding remarks
Effector cells, particularly CD8+ T cells from some individuals, are potent cytotoxic cells that attack virus-infected or transformed cells, while other individuals fail to control infected or transformed cells with the same potency. One endeavor to improve immune defenses is to engineer efficient cytolytic effector cells capable of killing aberrant target cells. Some of these engineering approaches requires the determination of the best ways to express genes encoding α and β TCR in CD8+ T cells. Random transduction of the TCR of interest into freshly isolated T cells generally results in low transgenic TCR expression compared to endogenous TCR. Moreover, it is well recognized that genes encoding transduced TCR are randomly incorporated into the genome, resulting in heterogeneity of TCR expression and distribution on the T cell surface. In addition, this might lead to mispairing of endogenous and transduced α and β TCR chains, further diminishing the expression of the TCR of interest. I posit that this strategy might be potentially improved by removing endogenous TCR genes to allow increased expression of the TCR of interest, ideally eliminating competition between natural and transgenic TCRs (see outstanding questions). Of note, one of the latest strategies has been to replace endogenous TCR genes with TCR genes of interest [50, 51]. Also, one research group utilized the same strategy to replace genes of a natural TCR with the gene encoding a CAR; CAR expression on peripheral blood CD8+ and CD4+ T cells was more uniform than randomly transduced CAR, and these CAR-T cells demonstrated a greater potency against tumor cells compared to conventional CAR-T cells [52]. Thus, I propose that a thorough analysis of the targeting potency of engineered TCR-T and CAR-T cells vs natural CD8+ (or CD4+) T cells against unwanted cells might pave the way to selection of the most potent cytolytic effectors for testing their ability to control aberrant cells in animal models and clinical trials.
Outstanding Questions.
What specific factors determine the differential cytolytic potencies of cytotoxic lymphocytes?
How can these presumed differences across CD4+, CD8+ and NK cytotoxic lymphocytes be leveraged to better engineer chimeric antigen receptor (CAR)-T cells CAR-NK and specific TCR-T cells for therapeutic benefit?
What are the best approaches to removing endogenous TCR genes to allow increased expression of specific TCRs for engineered T cells?
Can eliminating the competition between natural and transgenic TCRs be realistically achieved in engineered T cells? If so, how?
What are the best experimental methods for modulating the cytotoxic potencies of engineered TCR-T and CAR-T cells vs natural CD8+ or CD4+ T cells?
Highlights.
Ligands on cell surfaces are recognized by specialized receptors on immune cells and serve as an indicator for immunosurveillance by the host.
The expression amounts of ligands and receptors and the extent of their clustering seems to be essential to modulate the strength of T cell stimulation and the quality of response.
These receptor-ligand interactions at the T-cell/target cell interface significantly contribute to the fine-tuning mechanism controlling T cell cytolytic activity.
Lymphocytes that include CD8+ and CD4+ T cells as well as NK cells exploit two major tools to attack aberrant cells. These include the production of cytokines, which regulate the activities of lymphocytes, and delivery of granule ‘poison pills’ to kill target cells.
Available data suggest that the sensitivity and potency of lymphocyte responses cover a very wide range. Sensitivity and potency depend on the efficiency of the response exercised by each kind of lymphocyte.
I posit that there are various factors controlling the potency of different cytotoxic lymphocytes and that antigen-specific CD8+ T cells may be among the most cytotoxic killers.
Significance.
Virus-infected or transformed cells in the body carry significant danger. Generally, infection and transformation of healthy cells cannot be reversed, and these cells must be eliminated to stop their spread. Efficient destruction of unwanted cells depends to a large extent on the potency of cytolytic lymphocytes. Even a relatively small number of highly potent cytolytic effectors might be sufficient to eliminate aberrant cells. Thus, characterizing the differential potencies of cytotoxic lymphocytes such as CD8+ T cells becomes essential for advancing therapeutic endeavors.
Acknowledgments
This work was supported by NIH Grants to Yuri Sykulev (CA217714, R01AI118694, U01AI14811). I am grateful to my longstanding colleague Dr. Nadia Anikeeva for critical reading of the manuscript and useful comments. I also appreciate meaningful discussions with Sergey Panteleev, graduate student in my lab who is working on the development and testing of engineered TCR T cells. This article is prepared without being comprehensive but is rather focused on particular aspects of exceptional CTL potency. I regret that many relevant publications were not included in the reference list, which was assembled to reflect this opinon’s particular focus.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pasternack MS and Eisen HN (1985) A novel serine esterase expressed by cytotoxic T lymphocytes. Nature 314 (6013), 743–5. [DOI] [PubMed] [Google Scholar]
- 2.Pasternack MS et al. (1986) Serine esterase in cytolytic T lymphocytes. Nature 322 (6081), 740–3. [DOI] [PubMed] [Google Scholar]
- 3.Beal AM et al. (2009) Kinetics of early T cell receptor signaling regulate the pathway of lytic granule delivery to the secretory domain. Immunity 31 (4), 632–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenberg J and Huang J (2018) CD8(+) T Cells and NK Cells: Parallel and Complementary Soldiers of Immunotherapy. Curr Opin Chem Eng 19, 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sykulev Y et al. (1996) Evidence that a single peptide-MHC complex on a target cell can elicit a cytolytic T cell response. Immunity 4, 565–571. [DOI] [PubMed] [Google Scholar]
- 6.Sykulev Y et al. (1994a) Kinetics and affinity of reactions between an antigen-specific T-cell receptor and peptide-MHC complexes. Immunity 1, 15–22. [DOI] [PubMed] [Google Scholar]
- 7.Sykulev Y et al. (1994b) High-affinity reactions between antigen-specific T-cell receptors and peptides associated with allogeneic and syngeneic major histocompatibility complex class I proteins. Proc. Natl. Acad. Sci. USA 91, 14487–14491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cole DK et al. (2012) The molecular determinants of CD8 co-receptor function. Immunology 137 (2), 139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clement M et al. (2021) CD8 coreceptor-mediated focusing can reorder the agonist hierarchy of peptide ligands recognized via the T cell receptor. Proc Natl Acad Sci U S A 118(29):e2019639118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fung-Leung WP et al. (1991) CD8 is needed for development of cytotoxic T cells but not helper T cells. Cell 65 (3), 443–9. [DOI] [PubMed] [Google Scholar]
- 11.Rahemtulla A et al. (1991) Normal development and function of CD8+ cells but markedly decreased helper cell activity in mice lacking CD4. Nature 353 (6340), 180–4. [DOI] [PubMed] [Google Scholar]
- 12.Abraham N et al. (1991) Enhancement of T-cell responsiveness by the lymphocyte-specific tyrosine protein kinase p56lck. Nature 350 (6313), 62–6. [DOI] [PubMed] [Google Scholar]
- 13.Artyomov MN et al. (2010) CD4 and CD8 binding to MHC molecules primarily acts to enhance Lck delivery. Proc Natl Acad Sci U S A 107 (39), 16916–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rudd CE (2021) How the Discovery of the CD4/CD8-p56(lck) Complexes Changed Immunology and Immunotherapy. Front Cell Dev Biol 9, 626095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiong Y et al. (2001) T Cell Receptor Binding to a pMHCII Ligand Is Kinetically Distinct from and Independent of CD4. J Biol Chem 276 (8), 5659–67. [DOI] [PubMed] [Google Scholar]
- 16.Rushdi MN et al. (2022) Cooperative binding of T cell receptor and CD4 to peptide-MHC enhances antigen sensitivity. Nat Commun 13 (1), 7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sykulev Y (2010) T cell receptor signaling kinetics takes the stage. Sci Signal 3 (153), pe50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anikeeva N and Sykulev Y (2011) Mechanisms controlling granule-mediated cytolytic activity of cytotoxic T lymphocytes. Immunol Res 51 (2–3), 183–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sykulev Y et al. (1995) The law of mass action governs antigen-stimulated cytolytic activity of CD8+ cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA 92, 11990–11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chervin AS et al. (2013) Design of T-cell receptor libraries with diverse binding properties to examine adoptive T-cell responses. Gene Ther 20 (6), 634–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Golubovskaya V and Wu L (2016) Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers (Basel) 8(3):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gorga JC et al. (1987) Purification and characterization of class II histocompatibility antigens from a homozygous human B cell line. J Biol Chem 262 (33), 16087–94. [PubMed] [Google Scholar]
- 23.Potolicchio I et al. (2005) Conformational variation of surface class II MHC proteins during myeloid dendritic cell differentiation accompanies structural changes in lysosomal MIIC. J Immunol 175 (8), 4935–47. [DOI] [PubMed] [Google Scholar]
- 24.Cella M et al. (1997) Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells [see comments]. Nature 388 (6644), 782–7. [DOI] [PubMed] [Google Scholar]
- 25.Celli S et al. (2007) Real-time manipulation of T cell-dendritic cell interactions in vivo reveals the importance of prolonged contacts for CD4+ T cell activation. Immunity 27 (4), 625–34. [DOI] [PubMed] [Google Scholar]
- 26.Bousso P and Robey E (2003) Dynamics of CD8+ T cell priming by dendritic cells in intact lymph nodes. Nat Immunol 4 (6), 579–85. [DOI] [PubMed] [Google Scholar]
- 27.Harding CV and Unanue ER (1990) Quantitation of antigen-presenting cell MHC class II/peptide complexes necessary for T-cell stimulation. Nature 346, 574–576. [DOI] [PubMed] [Google Scholar]
- 28.Matsui K et al. (1991) Low affinity interaction of peptide-MHC complexes with T cell receptors. Science 254, 1788–1791. [DOI] [PubMed] [Google Scholar]
- 29.Poenie M et al. (2004) Real-time visualization of the cytoskeleton and effector functions in T cells. Curr Opin Immunol 16 (4), 428–38. [DOI] [PubMed] [Google Scholar]
- 30.Ribeiro M and McNamara JC (2007) Calcium movements during pigment aggregation in freshwater shrimp chromatophores. Pigment Cell Res 20 (1), 70–7. [DOI] [PubMed] [Google Scholar]
- 31.Stinchcombe JC and Griffiths GM (2007) Secretory mechanisms in cell-mediated cytotoxicity. Annu Rev Cell Dev Biol 23, 495–517. [DOI] [PubMed] [Google Scholar]
- 32.Huse M et al. (2008) Shouts, whispers and the kiss of death: directional secretion in T cells. Nat Immunol 9 (10), 1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuhn JR and Poenie M (2002) Dynamic polarization of the microtubule cytoskeleton during CTL-mediated killing. Immunity 16 (1), 111–21. [DOI] [PubMed] [Google Scholar]
- 34.Stinchcombe JC et al. (2006) Centrosome polarization delivers secretory granules to the immunological synapse. Nature 443 (7110), 462–5. [DOI] [PubMed] [Google Scholar]
- 35.Huse M et al. (2007) Spatial and temporal dynamics of T cell receptor signaling with a photoactivatable agonist. Immunity 27 (1), 76–88. [DOI] [PubMed] [Google Scholar]
- 36.Huse M (2012) Microtubule-organizing center polarity and the immunological synapse: protein kinase C and beyond. Front Immunol 3, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Combs J et al. (2006) Recruitment of dynein to the Jurkat immunological synapse. Proc Natl Acad Sci U S A 103 (40), 14883–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rak GD et al. (2011) Natural killer cell lytic granule secretion occurs through a pervasive actin network at the immune synapse. PLoS Biol 9 (9), e1001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhong L et al. (2009) NSOM/QD-based direct visualization of CD3-induced and CD28-enhanced nanospatial coclustering of TCR and coreceptor in nanodomains in T cell activation. PLoS One 4 (6), e5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anikeeva N et al. (2006) Quantum dot/peptide-MHC biosensors reveal strong CD8-dependent cooperation between self and viral antigens that augment the T cell response. Proc Natl Acad Sci U S A 103 (45), 16846–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edidin M (2010) Class I MHC molecules as probes of membrane patchiness: from biophysical measurements to modulation of immune responses. Immunol Res 47 (1–3), 265–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anikeeva N et al. (2019) Extent of MHC Clustering Regulates Selectivity and Effectiveness of T Cell Responses. J Immunol 202 (2), 591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anikeeva N et al. (2012) Evidence that the density of self peptide-MHC ligands regulates T-cell receptor signaling. PLoS One 7 (8), e41466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmidt ME and Varga SM (2018) The CD8 T Cell Response to Respiratory Virus Infections. Front Immunol 9, 678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raskov H et al. (2021) Cytotoxic CD8(+) T cells in cancer and cancer immunotherapy. Br J Cancer 124 (2), 359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walker B (2007) Elite control of HIV Infection: implications for vaccines and treatment. Top HIV Med 15 (4), 134–136. [PubMed] [Google Scholar]
- 47.Anderson KG et al. (2017) Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies. Cancer Cell 31 (3), 311–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schodin BA et al. (1996) Correlation between the number of T cell receptors required for T cell activation and TCR-ligand affinity. Immunity 5, 137–146. [DOI] [PubMed] [Google Scholar]
- 49.Anikeeva N et al. (2021) Efficient killing of tumor cells by CAR-T cells requires greater number of engaged CARs than TCRs. J Biol Chem 297 (3), 101033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schober K et al. (2019) Orthotopic replacement of T-cell receptor alpha- and beta-chains with preservation of near-physiological T-cell function. Nat Biomed Eng 3 (12), 974–984. [DOI] [PubMed] [Google Scholar]
- 51.Foy SP et al. (2023) Non-viral precision T cell receptor replacement for personalized cell therapy. Nature 615 (7953), 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eyquem J et al. (2017) Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543 (7643), 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]