

Abstract
Human brain is characterized by extremely sparse extracellular matrix (ECM). Despite its low abundance, the significance of brain ECM in both physiological and pathological conditions should not be underestimated. Brain metastasis is a serious complication of cancer, and recent findings highlighted the contribution of ECM in brain metastasis development. In this review, we provide a comprehensive outlook on how ECM proteins promote brain metastasis seeding. In particular, we discuss (1) disruption of the blood–brain barrier in brain metastasis; (2) role of ECM in modulating brain metastasis dormancy; (3) regulation of brain metastasis seeding by ECM-activated integrin signaling; (4) functions of brain-specific ECM protein reelin in brain metastasis. Lastly, we consider the possibility of targeting ECM for brain metastasis management.
Keywords: Brain metastasis, Extracellular matrix, Blood, Brain barrier, Integrin, Reelin
Introduction
Brain metastasis (BrM) is a devastating complication of several cancer types and a major cause of morbidity and mortality worldwide. Despite advances in cancer treatment, the incidence of BrM continues to rise, affecting about 10% of cancer patients during the course of their disease [1]. BrM is particularly common in patients with melanoma and lung and breast cancers. BrM is manifested by headaches, seizures, cognitive impairment, fatigue, and focal deficits; overall, these symptoms dramatically reduce patients’ quality of life [2]. Unfortunately, the prognosis for patients with symptomatic BrMs remains poor, with a median survival of only 13 months [3].
Current treatment options for patients with BrMs include surgery, chemotherapy, and radiation therapy. All these treatments are mostly palliative and extend patients’ survival by merely a few months. The progress in targeted therapy for BrM is hampered by the lack of BrM-specific, actionable targets. More innovative treatment approaches, such as immunotherapy, are being actively investigated, but the current effectiveness of immune checkpoint blockade monotherapy is very limited in patients with symptomatic BrMs [4–6].
The extracellular matrix (ECM) is a complex network of proteins, glycoproteins, and other molecules that surround and support cells in various tissues throughout the body [7]. The ECM plays a critical role in maintaining tissue structure and function, regulating cell behavior, and modulating cellular responses to environmental cues (Table 1). Further, the ECM acts as a signaling hub, transmitting information from the outside to the inside of cells, and modulating various cellular processes, including proliferation, differentiation, migration, and apoptosis [7]. In addition to its functions in these physiological processes, the ECM plays a key role in the maintenance of tissue homeostasis and repair as well as in pathological processes such as inflammation, fibrosis, and cancer [8]. Fibroblasts are responsible for depositing significant amounts of ECM; as a result, organs with high numbers of fibroblasts, such as the skin, breast, and liver, tend to display an elevated stromal component. It has been proposed that the ECM in these organs may be involved in establishing a pre-metastatic niche [9–11].
Table 1.
A brief summary of ECM molecules (as proposed by Naba et al. [12])
| Domain | Category | Function | Notable examples |
|---|---|---|---|
| Core matrisome | Collagens | Structural | Collagen I, Collagen IV |
| Glycoproteins | Structural, functional, signaling | Laminins, Emilins, Tenascins | |
| Proteoglycans | Functional, signaling | Versican, Perlecan, Biglycan | |
| Matrisome-associated | ECM regulators | Regulatory (ECM-remodeling enzymes, crosslinkers, proteases, regulators etc.) | Lysyl oxidases, Serpins, Cathepsins |
| Secreted factors | Known or suspected to bind core ECM proteins | CXCLs, Angiopoietins, S100A |
To successfully develop BrMs, cancer cells must adapt and survive in the hostile, nutrient-deficient microenvironment of the brain (Fig. 1). Examples of BrM-specific adaptations include activation of oncogenic kinases [13], induction of PTEN-loss [14], establishing an immunosuppressive microenvironment [15], increasing oxidative phosphorylation [16], competition for limited resources such as iron [17], synthesis of fatty acids [18], and induction of vascular co-option [19]. In recent years, substantial evidence indicates the involvement of ECM of distant organs in metastasis [20–22]. The ECM has been implicated in regulating attachment of metastasizing cancer cells [11, 23], controlling metastasis dormancy [24], and mediating the immune escape of metastasis [25]. However, knowledge about the role of the ECM in BrMs is scarce and has not been systemically assessed. In this article, we provide a comprehensive overview of studies investigating the involvement of the ECM in the incidence and pathogenesis of BrM.
Fig. 1.

Schematic explaining the process of BrM establishment. Adapted from “Breast Cancer To Brain Metastasis”, by BioRender.com (2023). Retrieved from https://app.biorender.com/biorender-templates
Brief overview of brain ECM
Securely contained in the cranium, the brain is one of the softest organs in the human body because of minimal concentrations of fibrous ECM, the main contributor of tensile strength in an organ [26]. Approximately 60% of adult human brain is fat, whereas the remaining 40% is water, protein, carbohydrates, and salts. Only ~ 20% of adult brain accounts for the extracellular space, and ECM proteins constitute only a fraction of that amount [27]. Whereas stiffer organs contain an abundance of collagen and elastin, the brain ECM backbone is primarily composed of a disaccharide polymer, hyaluronan (HA), which is a much softer matter [28]. The evolution of the brain has dramatically altered the function and structure of its ECM, which can be categorized into three major compartments: the blood–brain barrier (BBB), perineuronal net (PNN), and neural interstitial matrix [29]. In addition to these categories, some researchers also define perinodal ECM, a separate class of brain matrix that surrounds nodes of Ranvier within axons and whose protein composition resembles that of the PNN [30].
The BBB is formed by endothelial cells that compose the walls of the capillaries, which are ensheathed by basement membrane primarily consisting of type IV and XVIII collagens, laminins, nidogens, and heparin sulfate proteoglycans [31]. Capillary basement membrane is a prime physical barrier protecting the neural tissue from infiltrating cells and chemical compounds, and its breakdown is fraught with BBB dysfunction, which ultimately may lead to instability of the central nervous system and the onset of neurological diseases [32]. However, it is important to note that capillary basement membrane is not the sole type of barrier comprising the BBB, which also includes tight junctions between adjacent endothelial cells; a transport barrier, that is, specific transport mechanisms mediating the flux of ions; and a metabolic barrier, consisting of enzymes that metabolize molecules in transit [33].
The PNN is a scaffold primarily consisting of HA, connecting surrounding neurons and proximal dendrites into a net-like fibrous structure. Besides HA, the PNN consists of tenascins C and R as well as link proteins and chondroitin sulfate proteoglycans, all of which stabilize HA. The exact function of the PNN is incompletely studied; however, it has been strongly implicated in maintaining synaptic plasticity [34, 35] and the formation of long-term memories [36]. In addition, PNNs were reported to protect brain cells from oxidative stress [37] and neurotoxins such as amyloid beta-protein [38]. The perinodal ECM has a very similar structure to the PNN: its core component is HA, which is stabilized by hyaluronan and proteoglycan link protein 2 (HAPLN2) as well as brevican, versican, and neurocan [30]. The primary function of the perinodal ECM is to facilitate the formation of nodes of Ranvier and modulating axon conduction by regulating extracellular electrical resistance [30].
The neural interstitial matrix is an ensemble of all the remaining ECM constituents loosely dispersed in the brain parenchyma. Major components of the neural interstitial matrix are proteoglycans, HA, tenascins, link proteins, collagen XV, elastin, laminin 511, and fibronectin [39]. The function of the neural interstitial matrix is understudied; although it does not provide major structural support to the brain due to its scarcity, it may represent residual structures formerly in control of brain development. Matrix molecules, especially fibronectin, play a dominant role in development of the brain and constitute up to 40% of its mass in the perinatal period [29, 40]. In the postnatal period, the ECM proportion in the brain reduces by twofold. It is possible that individual constituents of the neural interstitial matrix exert special signaling functions, facilitating neuronal function. For example, the ECM glycoprotein reelin directly controls the migration of neurons in the central nervous system and regulates dendritic morphogenesis and neurotransmission [41].
Brain matrisome proteins come from many different sources. One of the largest producers of ECM in the brain are astrocytes (glial cells), which deposit HA, proteoglycans, and tenascins into the extracellular space [42, 43]. In addition, astrocytes produce versican V2 and brevican for the formation of nodes of Ranvier [30]. Microglia, in contrast, clear the redundant ECM by either secreting proteolytic enzymes or directly engulfing and phagocytosing unwanted ECM components [43]. Neurons have been reported to produce tenascin-C, tenascin-R, and the chondroitin sulfate proteoglycans brevican and neurocan, all of which were shown to be required for establishment of the PNN and synapse formation [44]. Neurons also synthesize reelin, a major ECM chemoattractant and signaling molecule [45]. Various matrix proteins comprising the inner and outer vascular basement membranes of the BBB are produced by cerebral endothelial cells and pericytes and, to a lesser extent, by astrocytes and brain adventitial fibroblasts [33, 46]. It should also be noted that when needed, virtually all brain cell populations have an ability to remodel the surrounding ECM by producing matrix-specific proteolytic enzymes (e.g., MMPs, cathepsins, ADAMs) and their inhibitors (e.g., TIMPs) [43]. Thus, all the brain cell types are involved in production and maintenance of the ECM (Fig. 2).
Fig. 2.

Overview of major ECM constituents of an adult human brain as well as cell types producing them. Abbreviations: HSPGs heparan sulphate proteoglycans, CSPGs chondroitin sulfate proteoglycans, HAPLN hyaluronan and proteoglycan link protein, MMPS matrix metalloproteinases, SPARC secreted protein acidic and rich in cysteine, ECM extracellular matrix. Created with BioRender.com
Disruption of the BBB in BrMs
Early BrMs are located in the brain perivascular niche, where they arrest exclusively in brain capillaries and/or postcapillary venules [47]. Successful dissemination to the brain parenchyma would require cancer cells to partially disintegrate the BBB. Early studies on this topic, led by Josh Fidler, showed that the BBB’s permeability was compromised in several experimental BrM models in vivo [48]. Later, it was demonstrated that brain-seeking 4T1 mammary tumor cells disintegrate BBB through disruption of ZO-1 and claudin-5 tight junctions in vivo [46]. In this study, the frequency of BBB-penetrating cancer cells coincided with increased permeability of the BBB, which indicates that BrM seeding is an active, not passive, process directly linked to tight junction breakdown [49]. Several studies indicated that BBB disruption is a modus operandi for metastasizing cells from triple-negative breast cancer (TNBC) and basal-type breast cancers but not those from HER2/neu-positive breast cancer [50, 51], but the reason for this difference is currently unknown. Our group has found that breast cancer cells harboring activated Src tyrosine kinase can induce severe BBB disruption [13]. Metastatic cells have also been reported to disrupt the BBB through secreting a protease cathepsin [51]. Genetic knockout of the CTSS gene encoding cathepsin S in brain-seeking MDA-MB-231 cells led to a dramatic reduction in metastatic seeding in the mouse brain upon their intracardiac injection [51]. It was further shown that tumor cell-derived cathepsin S actively cleaves tight junction proteins that regulate BBB integrity [51]. In addition to directly secreting ECM-remodeling factors, cancer cells are able to produce exosomes loaded with lnc-MMP2-2 RNA, which promotes BBB disruption in vivo by modulating human brain microvascular endothelial cells [52]. The same study showed that lnc-MMP2-2 functions as a molecular sponge for miR-1207-5p, which is critical for production of tight junction proteins in human brain microvascular endothelial cells, by upregulating EPB41L5. Thus, metastasizing cancer cells can “educate” major BBB-producing cells (i.e., endotheliocytes, astrocytes, etc.) to break down tight junctions and enable further dissemination of BrMs.
These findings from in vivo studies are further supported by human pathology research. Remodeling of the ECM by BrMs was evident from study examining 40 patients with lung and breast cancer-derived BrMs [53]. At both mRNA and protein levels, more than 80% of lung cancer-derived BrMs and 50% of breast cancer-derived BrMs expressed MMP14 [54], a known indicator of poor prognosis in cancer [55–57]. Of note, the authors observed a negligible expression of MMP7 in the BrM lesions [53]. Whether MMP14 plays a direct role in BBB breach remains an open question requiring further investigation.
Multiple studies have demonstrated the contribution of BBB-associated heparan sulfate proteoglycans (HSPGs) in the onset of BrMs. HSPGs are glycoproteins containing covalently attached heparan sulfate chains, a type of glycosaminoglycan (GAG) [58]. Their major functions include: (i) cooperating with integrins to facilitate cell–cell and cell–matrix communications, (ii) storing growth factors and soluble ECM-affiliated molecules which will be released upon HSPG degradation, (iii) defining the basement membrane structure (as a part of BBB), and (iv) acting as receptors and co-receptors for different tyrosine kinase-type growth factor receptors. In the BrM setting, tumor astrocytes were shown to produce large amounts of heparanase, an endoglycosidase which cleaves HSPGs [59]. Furthermore, treatment of BrM cancer cells with astrocyte conditioned media containing heparanase dramatically promoted their migration [60]. Intriguingly, highly brain-tropic human melanoma cells degraded purified ECM HSPG and HSPG cell-surface subpopulations faster than sublines of lower metastatic potential [61], indicating that HSPGs indeed pose a major barrier to metastasizing tumor cells. Accordingly, inhibition of cancer cell invasion by heparanase blockade was further confirmed in pediatric brain tumor cells in vitro [62].
Metabolism of HA can help establishing the brain metastatic niche through controlling the integrity of the BBB. Hyaluronidase 1 (HYAL1), an enzyme that cleaves HA into low molecular weight fragments, was shown to be produced by BrM-initiating cells, and it could mediate the adhesion of BrM cells to primary human brain endothelial cells in vitro [63]. Furthermore, it was found that the HYAL1 gene knockout in BrM cells led to a reduced BrM burden after intracardiac injection, compared to vector control cells [63]. Thus, pericellular HA-coat may facilitate the tumor cell interaction with the brain endothelium and lead to successful BrM seeding of disseminated cancer cells.
Finally, it was reported that the BBB can be selectively disrupted in BrMs [64] at least in part due to inhibition of major facilitator superfamily domain 2a (Mfsd2a), the endothelial cell-expressed docosahexaenoic acid transporter that is critical for the formation and function of the BBB [65]. Compared with normal brain, the expression of the Mfsd2a gene was markedly decreased in BrMs derived from breast cancer, lung cancer, or neuroendocrine prostate cancer. Importantly, BrM-associated vasculature demonstrated a similar dramatic reduction in the expression of the Mfsd2a gene compared to the normal brain vasculature [64]. The same study further demonstrated that perivascular astrocytes promote Mfsd2a expression in endothelial cells through secreting TGFβ and bFGF, while metastasizing tumor cells can disrupt this signaling pathway to downregulate Mfsd2a and thus facilitate BBB degradation [64].
Taken together, these studies indicate that metastasizing tumor cells employ a plethora of means to break down the BBB to extravasate and seed in the brain. Notably, brain-tropic cancer cells can degrade the BBB on their own or through “educating” cells in the brain microenvironment, such as astrocytes.
ECM molecules in control of BrM dormancy
The vast majority of cancer patients (> 90%) have resting-state (G0) tumor cells in the brain [66]; these dormant cancer cells may take years or even decades to awake and outgrow as clinically detectable BrMs [67]. Both entering and exiting a dormant state require a permissive milieu, and identification of mechanisms responsible for these events poses a great challenge for scientists. The biology and mechanisms of cellular dormancy in BrM remain incompletely understood, yet it has been shown that patients with BrMs derived from dormant disseminated cells have a longer median survival [68].
Recent studies have demonstrated a key role of the astrocyte-derived ECM in regulating the dormant state of disseminated tumor cells in the brain [69]. During the early steps of BrM progression, astrocytes are stripped from the vasculature and segregated to the border of BrMs. Such eviction of astrocytes coincides with removal of their end-feet as well. Upon contact with disseminated tumor cells, astrocytes then induce their dormancy via deposition of laminin-211 into the parenchymal basement membrane, which transduces pro-dormancy signaling via receptor dystroglycan [69]. It is interesting that integrins, classically recognized laminin receptors, are not involved in this pro-dormancy signaling [69]. Further mechanistic studies revealed that laminin-211–activated dystroglycan promotes BrM cell dormancy by tethering Yes-associated protein (YAP) to the cell membrane [69]. YAP is a major Hippo pathway effector [70] previously linked with lung cancer cell dormancy through repressing the pro-apoptotic protein Bmf via Slug. In the BrM setting, cancer cells with constitutively active YAP, which shuttled freely to the nucleus and back, can escape from dormancy and accelerate brain colonization in vivo [69]. Activation of dystroglycan by astrocyte-derived laminin-211 on the surface of cancer cells resulted in sequestering YAP to the cancer cells’ membrane and promoting the quiescent phenotype in experimental models of mouse BrM [69]. These findings suggest that BrM-associated astrocytes may limit the growth of BrM during early steps of metastatic dissemination, and that escape from BrM dormancy can be triggered by factors that would enable the drift/relocation of brain astrocytes from dormant cells, such as neuroinflammation or trauma. However, it remains unclear whether these findings are clinically relevant, as astrocytes have been widely reported to secrete multiple pro-tumor factors that promote BrM growth, stemness, invasiveness, and chemoresistance [71].
The crosstalk between dormant cancer cells and surrounding ECM can be investigated using hydrogels, representing an engineered native-like ECM structure [72]. In one such study, brain-tropic MDA-MB-231 and BT474 cells, grown as 3D spheroids in HA-based hydrogels, demonstrated a reduction in proliferation marker Ki67 and a higher percentage of cells positive for dormancy marker phospho-p38, compared to spheroids grown in an ECM-free environment [73]. Importantly, transferring dormant spheroids into a scaffold-free culture system reinstated spheroid growth and aborted the dormancy program [73]. These results indicate that the ECM can, in principle, induce dormancy of brain-tropic cells. A similar study investigated the composition of ECM-like hydrogels on the behavior of brain-tropic MDA-MB-231 subline BrM2a-831 [74]. The authors formulated hydrogels of varying degradability and adhesiveness and identified the ECM parameters with the highest impact on cancer cell dormancy in vitro. In particular, they demonstrated that while hydrogels with high adhesivity and degradability promoted cancer cell proliferation, those with no adhesivity but high degradability had a potential to induce dormancy and activate survival programs in BrM2a-831 cells as well as in bone- and lung-tropic MDA-MB-231 sublines [74]. Importantly, the authors could reactivate dormant cells by increasing hydrogel adhesivity in vitro, which enabled the cells to exit dormancy [74]. These results are consistent with a study showing that soft HA-based hydrogels can induce a much stronger dormancy phenotype (as measured by Ki67+ cells) in brain-tropic MDA-MB-231 cells than can stiff hydrogels [75]. Thus, ECM degradability/softness plays a key role in inducing dormancy, whereas its adhesiveness/stiffness may help dormant cells to re-enter the cell cycle (Fig. 3). Further studies to validate these exciting findings in vivo and in human disease are warranted.
Fig. 3.
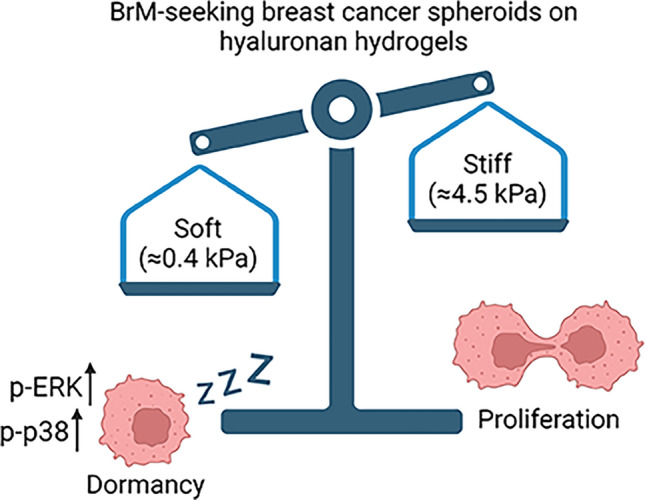
ECM softness controls dormancy state in BrM-seeking cells. Created with BioRender.com
ECM-activated Integrin signaling facilitates BrM seeding
Integrins are a class of molecules primarily responsible for “outside-in” signaling in control of communication between the ECM and surrounding cells [76]. Integrins are composed of two subunits, α and β, which form a heterodimeric structure [77]. There are 18 α and 8 β subunits, which can form 24 distinct integrin receptors, each with unique binding properties and functions [78]. Integrins have multiple roles in the cell; however, they are not constitutively active and require “outside-in” signals from ECM proteins to activate them. Integrins exist in low-affinity “bent” conformation when inactive. Upon contact with an ECM ligand, integrin’s ectodomain turns into “upright” conformation, leading to activation of a cytoplasmic tail of the β subunit, which binds several intracellular anchor proteins, including talin, kindling, etc [78, 79]. If stronger activation is needed, extended integrins can further cluster at cell-ECM contacts to create the foundation for adhesion complexes [80]. Thus, integrins act as signaling receptors, transmitting signals bidirectionally across the cell membrane to control multiple cellular processes (Fig. 4). Dysregulation of integrin function has been implicated in cancer [81], autoimmune disorders [82], and cardiovascular diseases [83].
Fig. 4.

Schematic illustration of integrin structure and activation. Signals both inside and outside the cell can trigger integrin activation, which induces multiple signaling pathways to regulate cytoskeleton assembly, adhesion, migration, proliferation, etc. Reprinted from “Integrin Structure and Activation”, by BioRender.com (2023). Retrieved from https://app.biorender.com/biorender-templates
Certain ECM proteins in the brain perivascular niche could facilitate the adaptation of disseminating cancer cells in the brain. Profiling of the BrM-initiating capability of different cancer cell lines in vivo revealed that human lung cancer line EBC-1 had the highest probability to induce BrM (~ 100%) when injected via the intracardiac route [84]. Follow-up analysis of integrin chains reveled that a BrM-derived EBC-1 subline expressed significantly higher levels of integrin α3 than did parental or bone–homing EBC-1 counterparts [84]. In vitro, this BrM-derived EBC-1 subline exhibited a more pronounced attachment to type I collagen, fibronectin, and laminin, all of which are binding partners of α3β1. Interestingly, the BrM capacity of brain-seeking EBC-1 subclones was impaired upon in vivo treatment with an integrin α3–blocking antibody [84]. While this study implicated integrin α3 in control of BrM formation, another study showed that 4T1 mouse mammary tumor cells with knockdown or pharmacological inhibition of integrin α3 displayed a dramatic reduction of lung metastasis in a tail vein injection model [85], indicating that integrin α3 is also critical for metastasis to other organs. Since cancer cells injected into the tail vein rarely metastasize to the brain, this study is limited in assessing the capability of 4T1 cells to metastasize to the lungs only. Nonetheless, in the BrM setting, the function of integrin α3β1 has been explained by its interaction with αB-crystallin, a molecular chaperone responsible for skeletal myopathies [86] and cardiomyopathies [87]. It was shown that αB-crystallin increases adhesion of TNBC cells to human brain microvascular endothelial cells through an integrin α3β1-dependent mechanism and also promotes adhesion to type I collagen, fibronectin, and laminin [88].
In addition to their role in BrM seeding, integrins can also serve as biomarkers of, or therapeutic targets for established BrMs. Berghoff et al. [89] classified invasion patterns in established human BrMs and identified three subtypes characterized by well-demarcated borders, vascular co-option, or diffuse infiltration. Profiling of integrin expression in these subtypes revealed that integrin αvβ6 was significantly overexpressed in the well-demarcated group compared with the vascular co-option and diffuse infiltration groups [89]. Theoretically, well-demarcated BrMs without loci of aggressive infiltration may take a significantly longer time to grow and become symptomatic. In support of this hypothesis, the expression of integrin αvβ6 correlated with lower cell proliferation index in BrMs [90]. Lastly, the expression of integrin αvβ6 in BrMs was associated with a longer overall patient survival after BrM diagnosis [90]. Thus, integrin αvβ6 is a feature of well-demarcated BrMs characterized by slower growth, which could be used in prognostication for BrM patients. Importantly, targeting of integrin αvβ6 by an antipsychotic drug, penfluridol, showed promising results in treating TNBC BrMs in vivo [91], suggesting a strong clinical relevance of this BrM-associated integrin. Inhibition of other αv integrin chains has also been examined in preclinical trials. Wu et al. [92] treated rat BrMs from brain-tropic MDA-MB-231. HER2 subline with intetumumab, an antibody binding αv integrins. The authors observed that intetumumab prevented metastasis formation in 30% of tested rats while the remaining animals displayed a reduction in BrM burden compared to controls [92]. Thus, inhibition of integrins, particularly αv chains, may represent an attractive strategy to treat BrMs.
Pivotal role of Reelin in BrM and central nervous system metastasis
Reelin is a major brain-specific ECM glycoprotein involved in cortical development and synaptic function maintenance [93]. Deposited in the extracellular space, reelin binds to its receptor apolipoprotein E receptor 2 (ApoER2) expressed on the cell membrane of neurons. This leads to activation of a downstream signaling cascade that eventually promotes Ca2+ influx and subsequent release of neurotransmitters. Notably, mutations in the reelin-encoding gene, RELN, are linked to schizophrenia in women [94], autism [95], and Alzheimer disease [96], highlighting the importance of this glycoprotein in normal brain physiology. Even though reelin’s expression is restricted to brain cells, recent studies showed that BrM cancer cells can acquire a neuronal-like metaprogram and express brain-specific proteins to adapt to the microenvironment of the brain [97]. Therefore, it is possible that neuron-like BrM cancer cells can also express reelin. Indeed, it was found that brain-tropic breast cancer cells produce more reelin compared to their parental counterparts [98]. Furthermore, dormant brain-tropic breast cancer sublines of MDA-MB-231 and BT474 overexpress reelin after co-culture with neurons [88, 99]. The knockdown of the RELN gene resulted in downregulation of key synaptic mediators NTRK2, NGFR, NRXN1, and NLGN4X, suggesting that neuronal exposure enhances reelin expression in BrM cells to activate a neuron-like phenotype [98].
Human epidermal growth factor receptor 2 (HER2) overexpression (HER2+) is frequently (15–20%) detected in breast cancer [100], and patients with HER2+ breast cancer have an increased risk of developing BrMs [101]. A recent study showed that reelin is highly expressed in HER2+ BrMs compared to TNBC BrMs, and that reelin co-localizes with HER2 in BrM tissue Sects. [99]. Furthermore, co-immunoprecipitation studies revealed that reelin can form a complex with HER2 [102], implicating a functional link between these two proteins. It was further shown that reelin knockdown inhibited the spheroid-forming ability of BrM-capable HER2+ cells but not TNBC cells [99]. Interestingly, the authors found the reelin-expressing BrM cells are associated with astrocytes and they also showed that conditioned media from astrocytes can induce reelin in HER2+ BrM cells [99], suggesting that not only neurons but also glial cells are able to promote reelin expression in BrM. The functional role of rellin-HER2 complex in BrMs warrants further investigations.
Reelin has recently been implicated in promoting metastasis in medulloblastoma, a rare pediatric tumor of the cerebellum, that metastasizes almost exclusively to the spine or intracranial leptomeninges [103]. The authors of that study identified that overexpression of SMARCD3 in medulloblastoma activates reelin–DAB1–Src signaling-mediated cancer cell migration, which eventually results in metastatic dissemination [104]. In this pathway, the role of reelin is pivotal, as reelin was found to be directly regulated by a key epigenetic modulator, SMARCD3, to launch a cascade of factors that trigger cancer cell metastasis [104]. Thus, it appears that reelin may be a key ECM protein involved in central nervous system metastasis.
Discussion and concluding remarks
BrM remains an unmet clinical need, with devastating incidence rates of 2.8–14.3 per 100,000 people of general population [105]. The advent of single-cell sequencing and spatial profiling in recent years enabled a deeper understanding of BrM mechanisms and pathophysiology, even though this has not yet led to qualitative improvements in treatment. The importance of ECM in progression of primary tumors and metastasis has long been underestimated for several reasons, including the fact that ECM is difficult to extract and investigate owing to crosslinking and insolubility and also because ECM research is at the intersection of cell/molecular biology and biophysics that requires multidisciplinary approaches. In this review, we summarized knowledge about BrM-associated ECM to highlight its significance in the development of disease.
The brain is notable for poor ECM content, mainly due to the lack of fibroblasts, except for brain vascular adventitial fibroblasts. The matrisome of the normal brain is limited to only 69 proteins, among which are only nine proteoglycans and 18 collagens [106]. Nonetheless, the brain ECM plays a major role during development [29] and maintains BBB integrity [31], highlighting the ECM’s unique function in this organ. Importantly, cerebrovascular matrisome may be completely different to the ensemble of ECM proteins produced by metastasizing cancer cells; therefore, when studying the BrM-associated ECM, it is particularly important to identify the source of studied ECM proteins, whether they derive from the brain cells or the tumor cells. In our studies, we observed that in experimental mouse models of BrM, some mouse mammary tumor cell lines produce virtually no ECM (such as EO771 or 4T1), whereas the others display extensive ECM deposits (EMT3). In agreement, a comparative analysis of metastatic matrisome from different organs revealed that BrMs had at least fourfold lower abundance of matrisome proteins compared to metastases to other organs [107]. The exact function of BrM-derived ECM proteins remains to be determined in future studies (Fig. 5).
Fig. 5.

Features of ECM in a healthy normal brain versus brain metastasis. Normal brain is characterized by sparsely distributed ECM proteins as well as high integrity of the BBB. In brain metastasis, the BBB is disrupted through Mfsd2a downregulation in epithelial cells. Deposition of Laminin-211 by astrocytes promotes cancer cell dormancy via YAP signaling. Cancer cells produce large amounts of integrins (α3, αv) on the cell surface. Metastasis-associated ECM has high concentrations of MMPs, Heparanase, and Reelin. Adapted from “Extracellular Matrix in Brain Tumorigenesis”, by BioRender.com (2023). Retrieved from https://app.biorender.com/biorender-templates
This review has several limitations. Even though this article does not cover the role of immune cells in BrMs, recent studies have demonstrated a complex interaction between tumor-associated ECM and tumor-infiltrating leukocytes, monocytes/macrophages, and granulocytes [108]. In the context of BrMs, collagen deposits were co-localized with CD3+ T cells and CD68+ macrophages [109], so it would be interesting to explore whether BrM-associated ECM may influence immune cell trafficking, antigen recognition, presentation, or cancer cell killing. In addition, this review predominantly covered biochemical cues derived from brain ECM in cancer metastasis; however, biophysical signals are also an important factor to regulate cancer cell dormancy and colonization. Due to the dearth of published studies, this aspect remains poorly understood and warrants further investigation.
Certain components of the tumor microenvironment may be successfully targeted to treat solid tumors. Immunotherapies or anti-angiogenic therapies using monoclonal antibodies to inhibit key proteins in the pro-tumor microenvironment (e.g., PD-L1, VEGF) have achieved dramatic improvement in survival of patients with cancer [110, 111]. Studies discussed in this review raise the possibility of targeting specific brain ECM components for BrM treatment (Tables 2 and 3); this is further supported by our finding that upregulation of ECM genes is generally associated with poor tumor outcomes across multiple cancer types [112], suggesting the potential benefit of normalizing the ECM. However, even though numerous clinical trials were conducted during the past decade to inhibit ECM synthesis or enhance its turnover, these attempts did not bear fruit. Blockade of collagen-crosslinking enzyme lysyl oxidase did not show any survival benefit in pancreatic cancer [113] or colorectal adenocarcinoma [114], yet initial preclinical trials were very optimistic [115]. Furthermore, targeting integrins through FAK inhibition did not improve the outcome of patients with mesothelioma [116]. Along similar lines, breakdown of HA by delivering hyaluronidase in patients with pancreatic cancer did not prove to be effective [117]. Attempts to facilitate the remodeling of tumor-associated ECM by MMP inhibitors failed because of limited effectiveness [118]. The lack of success in these trials of targeting the ECM may be explained by the complexity of ECM and our poor understanding of how/when individual ECM components are promoting cancer progression. In the context of BrM, the situation is additionally complicated by the BBB, which restricts the delivery of many therapeutics to the brain. It has been postulated that permeabilizing the BBB in patients with symptomatic BrMs could facilitate the delivery of drugs and enhance therapeutic efficacy [119]. On the other hand, novel therapies targeting the ECM such as gene therapies that strengthen the BBB’s integrity, may be effective in preventing BrMs in high-risk patients. For example, adeno-associated viruses designed to selectively target cells of the brain endothelium would carry transgenes such as Mfsd2a and be used to increase BBB integrity to build up protection from circulating tumor cells’ entering the brain. Additionally, combining ECM-targeting therapies with other BrM-inhibiting and brain microenvironment-normalizing therapies may lead to a better control of BrM progression. Clearly, disruptive research on BrM-associated ECM is needed to bring about substantially improved treatment for BrM patients in dire need.
Table 2.
A brief summary of ECM molecules (as proposed by Naba et al. [12])
| Domain | Category | Function | Notable examples |
|---|---|---|---|
| Core matrisome | Collagens | Structural | Collagen I, Collagen IV |
| Glycoproteins | Structural, functional, signaling | Laminins, Emilins, Tenascins | |
| Proteoglycans | Functional, signaling | Versican, Perlecan, Biglycan | |
| Matrisome-associated | ECM regulators | Regulatory (ECM-remodeling enzymes, crosslinkers, proteases, regulators etc.) | Lysyl oxidases, Serpins, Cathepsins |
| Secreted factors | Known or suspected to bind core ECM proteins | CXCLs, Angiopoietins, S100A |
Table 3.
Potential ECM targets for BrM therapy and prevention
| ECM component | Targeted therapy potential |
|---|---|
| Heparanase | To prevent BrM seeding [59, 60, 62] |
| Hyaluronidase 1 | To prevent BrM seeding [63] |
| Cathepsin S | To prevent BrM seeding [51] |
| Laminin-211 | To prevent BrM seeding [69] |
| Dystroglycan | To prevent BrM seeding [69] |
| Integrin α3 | To prevent BrM seeding [84] |
| Integrin α3β1 | To prevent BrM seeding [88] |
| Integrin αvβ6 | To reduce BrM growth [91] |
| αv integrins | To prevent BrM seeding [92] |
| Reelin | To prevent early BrM adaptation by acquiring a neuron-like phenotype [98, 99] |
| To limit growth of HER2+ BrMs [99] | |
| To prevent metastasis from cerebellum to other parts of the brain [104] |
Acknowledgements
Editorial support was provided by Bryan Tutt, Scientific Editor, Research Medical Library, MD Anderson. The artwork was created with BioRender.com
Author contributions
A.Y. wrote the manuscript and procured funding, D.Y. edited the manuscript and procured funding.
Funding
This work was supported by National Institutes of Health (NIH) grants R01CA266099 (D.Y.), R01CA208213 (D.Y.), R01CA231149 (D.Y.), METAvivor research grants #56675 and #58284 (D.Y.), DoD Breakthrough Award W81XWH-22-1-0064 (A.Y.). A.Y. is supported by the Odyssey Fellowship Program of MD Anderson. D.Y. is the Hubert L. & Olive Stringer Distinguished Chair in Basic Science at MD Anderson.
Availability of data and material
Not applicable.
Declarations
Competing interests
None declared.
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors give consent to publish this manuscript.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Nayak L, Lee EQ, Wen PY. Epidemiology of brain metastases. Curr Oncol Rep. 2012;14:48–54. doi: 10.1007/s11912-011-0203-y. [DOI] [PubMed] [Google Scholar]
- 2.Achrol AS, et al. Brain metastases. Nat Rev Dis Primers. 2019 doi: 10.1038/s41572-018-0055-y. [DOI] [PubMed] [Google Scholar]
- 3.Sperduto PW, et al. Survival in patients with brain metastases: summary report on the updated diagnosis-specific graded prognostic assessment and definition of the eligibility quotient. J Clin Oncol. 2020;38:3773–3784. doi: 10.1200/JCO.20.01255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldberg SB, et al. Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol. 2016;17:976–983. doi: 10.1016/S1470-2045(16)30053-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parakh S, et al. Efficacy of anti-PD-1 therapy in patients with melanoma brain metastases. Br J Cancer. 2017;116:1558–1563. doi: 10.1038/bjc.2017.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tawbi HA, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med. 2018;379:722–730. doi: 10.1056/NEJMoa1805453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK. Extracellular matrix structure. Adv Drug Deliv Rev. 2016;97:4–27. doi: 10.1016/j.addr.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15:786–801. doi: 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patras L, Paul D, Matei IR. Weaving the nest: extracellular matrix roles in pre-metastatic niche formation. Front Oncol. 2023 doi: 10.3389/fonc.2023.1163786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu YL, et al. Tetrathiomolybdate (TM)-associated copper depletion influences collagen remodeling and immune response in the pre-metastatic niche of breast cancer. NPJ Breast Cancer. 2021 doi: 10.1038/s41523-021-00313-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuzhalin AE, et al. Colorectal cancer liver metastatic growth depends on PAD4-driven citrullination of the extracellular matrix. Nat Commun. 2018;9:4783. doi: 10.1038/s41467-018-07306-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naba A, et al. The matrisome: In Silico definition and In Vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteom. 2012;11:M111.014647. doi: 10.1074/mcp.M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang S, et al. Src family kinases as novel therapeutic targets to treat breast cancer brain metastases. Cancer Res. 2013;73:5764–5774. doi: 10.1158/0008-5472.CAN-12-1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang L, et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature. 2015;527:100–104. doi: 10.1038/nature15376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang L, et al. Blocking immunosuppressive neutrophils deters pY696-EZH2-driven brain metastases. Sci Transl Med. 2020 doi: 10.1126/scitranslmed.aaz5387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer GM, et al. Molecular profiling reveals unique immune and metabolic features of melanoma brain metastases. Cancer Discov. 2019;9:628–645. doi: 10.1158/2159-8290.CD-18-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chi Y, et al. Cancer cells deploy lipocalin-2 to collect limiting iron in leptomeningeal metastasis. Science. 2020;369:276–282. doi: 10.1126/science.aaz2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferraro GB, et al. Fatty acid synthesis is required for breast cancer brain metastasis. Nature Cancer. 2021;2:414–428. doi: 10.1038/s43018-021-00183-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valiente M, et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell. 2014;156:1002–1016. doi: 10.1016/j.cell.2014.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilkes DM, Semenza GL, Wirtz D. Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat Rev Cancer. 2014;14:430–439. doi: 10.1038/nrc3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kai FB, Drain AP, Weaver VM. The extracellular matrix modulates the metastatic journey. Dev Cell. 2019;49:332–346. doi: 10.1016/j.devcel.2019.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eble JA, Niland S. The extracellular matrix in tumor progression and metastasis. Clin Exp Metastasis. 2019;36:171–198. doi: 10.1007/s10585-019-09966-1. [DOI] [PubMed] [Google Scholar]
- 23.Tan X, et al. Transcriptional control of a collagen deposition and adhesion process that promotes lung adenocarcinoma growth and metastasis. JCI Insight. 2022 doi: 10.1172/jci.insight.153948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Di Martino JS, et al. A tumor-derived type III collagen-rich ECM niche regulates tumor cell dormancy. Nat Cancer. 2022;3:90–107. doi: 10.1038/s43018-021-00291-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mariathasan S, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Axpe E, Orive G, Franze K, Appel EA. Towards brain-tissue-like biomaterials. Nat Commun. 2020;2020(11):1–4. doi: 10.1038/s41467-020-17245-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nicholson C, Syková E. Extracellular space structure revealed by diffusion analysis. Trends Neurosci. 1998;21:207–215. doi: 10.1016/S0166-2236(98)01261-2. [DOI] [PubMed] [Google Scholar]
- 28.Deasy SK, Erez N. A glitch in the matrix: organ-specific matrisomes in metastatic niches. Trends Cell Biol. 2022;32:110–123. doi: 10.1016/j.tcb.2021.08.001. [DOI] [PubMed] [Google Scholar]
- 29.Yuzhalin AE. Parallels between the extracellular matrix roles in developmental biology and cancer biology. Semin Cell Dev Biol. 2022;128:90–102. doi: 10.1016/j.semcdb.2021.09.010. [DOI] [PubMed] [Google Scholar]
- 30.Fawcett JW, Oohashi T, Pizzorusso T. The roles of perineuronal nets and the perinodal extracellular matrix in neuronal function. Nature Rev Neurosci. 2019;20:451–465. doi: 10.1038/s41583-019-0196-3. [DOI] [PubMed] [Google Scholar]
- 31.Daneman R, Prat A. The blood-brain barrier. Cold Spring Harbor Perspect. 2015 doi: 10.1101/cshperspect.a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19:1584–1596. doi: 10.1038/nm.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ. Structure and function of the blood–brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 34.McRae PA, Porter BE. The perineuronal net component of the extracellular matrix in plasticity and epilepsy. Neurochem Int. 2012;61:963–972. doi: 10.1016/j.neuint.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carulli D, et al. Animals lacking link protein have attenuated perineuronal nets and persistent plasticity. J Neurol. 2010 doi: 10.1093/brain/awq145. [DOI] [PubMed] [Google Scholar]
- 36.Tsien RY. Very long-term memories may be stored in the pattern of holes in the perineuronal net. Proc Natl Acad Sci USA. 2013;110:12456–12461. doi: 10.1073/pnas.1310158110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cabungcal JH, et al. Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc Natl Acad Sci USA. 2013;110:9130–9135. doi: 10.1073/pnas.1300454110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyata S, Nishimura Y, Nakashima T. Perineuronal nets protect against amyloid beta-protein neurotoxicity in cultured cortical neurons. Brain Res. 2007;1150:200–206. doi: 10.1016/j.brainres.2007.02.066. [DOI] [PubMed] [Google Scholar]
- 39.Jain D, Mattiassi S, Goh E, Yim E. Extracellular matrix and biomimetic engineering microenvironment for neuronal differentiation. Neural Regen Res. 2020;15:573–585. doi: 10.4103/1673-5374.266907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bondareff W, Pysh JJ. Distribution of the extracellular space during postnatal maturation of rat cerebral cortex. Anat Rec. 1968;160:773–780. doi: 10.1002/ar.1091600412. [DOI] [PubMed] [Google Scholar]
- 41.Faini G, Del Bene F, Albadri S. Reelin functions beyond neuronal migration: from synaptogenesis to network activity modulation. Curr Opin Neurobiol. 2021;66:135–143. doi: 10.1016/j.conb.2020.10.009. [DOI] [PubMed] [Google Scholar]
- 42.Pu A, Stephenson EL, Yong VW. The extracellular matrix: focus on oligodendrocyte biology and targeting CSPGs for remyelination therapies. Glia. 2018;66:1809–1825. doi: 10.1002/glia.23333. [DOI] [PubMed] [Google Scholar]
- 43.Crapser JD, Arreola MA, Tsourmas KI, Green KN. Microglia as hackers of the matrix: sculpting synapses and the extracellular space. Cell Mol Immunol. 2021;18:2472. doi: 10.1038/s41423-021-00751-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Geissler M, et al. Primary hippocampal neurons, which lack four crucial extracellular matrix molecules, display abnormalities of synaptic structure and function and severe deficits in perineuronal net formation. J Neurosci. 2013;33:7742–7755. doi: 10.1523/JNEUROSCI.3275-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jossin Y. Reelin functions, mechanisms of action and signaling pathways during brain development and maturation. Biomolecules. 2020;10:1–31. doi: 10.3390/biom10060964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang HD, Rätsep MT, Chapman A, Boyd R. Adventitial fibroblasts in vascular structure and function: the role of oxidative stress and beyond. This review is one of a selection of papers published in a Special Issue on Oxidative Stress in Health and Disease. Canadian J Physiol Pharmacol. 2010 doi: 10.1139/Y10-01588,177-186. [DOI] [PubMed] [Google Scholar]
- 47.Lorger M, Felding-Habermann B. Capturing changes in the brain microenvironment during initial steps of breast cancer brain metastasis. Am J Pathol. 2010;176:2958–2971. doi: 10.2353/ajpath.2010.090838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang RD, Price JE, Fujimaki T, Bucana CD, Fidler IJ. Differential permeability of the blood-brain barrier in experimental brain metastases produced by human neoplasms implanted into nude mice. Am J Pathol. 1992;141:1115. [PMC free article] [PubMed] [Google Scholar]
- 49.Avraham HK, et al. Angiopoietin-2 mediates blood-brain barrier impairment and colonization of triple-negative breast cancer cells in brain. J Pathol. 2014;232:369–381. doi: 10.1002/path.4304. [DOI] [PubMed] [Google Scholar]
- 50.Yonemori K, et al. Disruption of the blood brain barrier by brain metastases of triple-negative and basal-type breast cancer but not HER2/neu-positive breast cancer. Cancer. 2010;116:302–308. doi: 10.1002/cncr.24735. [DOI] [PubMed] [Google Scholar]
- 51.Sevenich L, et al. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat Cell Biol. 2014;16:876–888. doi: 10.1038/ncb3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu D, et al. TGF-β1-mediated exosomal lnc-MMP2–2 increases blood–brain barrier permeability via the miRNA-1207–5p/EPB41L5 axis to promote non-small cell lung cancer brain metastasis. Cell Death Dis. 2021 doi: 10.1038/s41419-021-04072-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoshida S, Takahashi H. Expression of extracellular matrix molecules in brain metastasis. J Surg Oncol. 2009;100:65–68. doi: 10.1002/jso.21296. [DOI] [PubMed] [Google Scholar]
- 54.Yoshida S, Takahashi H. Expression of extracellular matrix molecules in brain metastasis. J Surg Oncol. 2009;100:65–68. doi: 10.1002/jso.21296. [DOI] [PubMed] [Google Scholar]
- 55.Cui G, Cai F, Ding Z, Gao L. MMP14 predicts a poor prognosis in patients with colorectal cancer. Hum Pathol. 2019;83:36–42. doi: 10.1016/j.humpath.2018.03.030. [DOI] [PubMed] [Google Scholar]
- 56.Jin Y, Liang ZY, Zhou WX, Zhou L. High MMP14 expression is predictive of poor prognosis in resectable hepatocellular carcinoma. Pathology. 2020;52:359–365. doi: 10.1016/j.pathol.2020.01.436. [DOI] [PubMed] [Google Scholar]
- 57.Wang X, Meng Q, Wang Y, Gao Y. Overexpression of MMP14 predicts the poor prognosis in gastric cancer: Meta-analysis and database validation. Medicine. 2021;100:e26545. doi: 10.1097/MD.0000000000026545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol. 2011;3:1–33. doi: 10.1101/cshperspect.a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marchetti D, Nicolson GL. Human heparanase: A molecular determinant of brain metastasis. Adv Enzyme Regul. 2001;41:343–359. doi: 10.1016/S0065-2571(00)00016-9. [DOI] [PubMed] [Google Scholar]
- 60.Marchetti D, Li J, Shen R. Astrocytes contribute to the brain-metastatic specificity of melanoma cells by producing heparanase. Cancer Res. 2000;60:4767–4770. [PubMed] [Google Scholar]
- 61.Denkins Y, et al. Brain metastases in melanoma: Roles of neurotrophins. Neuro Oncol. 2004;6:154–165. doi: 10.1215/S115285170300067X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spyrou A, et al. Inhibition of heparanase in pediatric brain tumor cells attenuates their proliferation, invasive capacity, and in vivo tumor growth. Mol Cancer Ther. 2017;16:1705–1716. doi: 10.1158/1535-7163.MCT-16-0900. [DOI] [PubMed] [Google Scholar]
- 63.Hamester F, et al. Key role of hyaluronan metabolism for the development of brain metastases in triple-negative breast cancer. Cells. 2022;11:3275. doi: 10.3390/cells11203275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tiwary S, et al. Metastatic brain tumors disrupt the blood-brain barrier and alter lipid metabolism by inhibiting expression of the endothelial cell fatty acid transporter Mfsd2a. Sci Rep. 2018 doi: 10.1038/s41598-018-26636-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ben-Zvi A, et al. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature. 2014;509:507–511. doi: 10.1038/nature13324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Noltenius C, Noltenius H. Dormant tumor cells in liver and brain. An autopsy study on metastasizing tumors. Pathol Res Pract. 1985;179:504–511. doi: 10.1016/S0344-0338(85)80191-6. [DOI] [PubMed] [Google Scholar]
- 67.Phan TG, Croucher PI. The dormant cancer cell life cycle. Nat Rev Cancer. 2020;20:398–411. doi: 10.1038/s41568-020-0263-0. [DOI] [PubMed] [Google Scholar]
- 68.Ferlini L, Peluso L, Lolli V, Gaspard N, Lefranc F. Prognosis of patients treated in a single neurosurgical reference centre for brain metastasis caused by dormant disseminated cells. Oncol Lett. 2021 doi: 10.3892/ol.2021.12715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dai J, et al. Astrocytic laminin-211 drives disseminated breast tumor cell dormancy in brain. Nat Cancer. 2022;3:25–42. doi: 10.1038/s43018-021-00297-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kurppa KJ, et al. Treatment-induced tumor dormancy through YAP-mediated transcriptional reprogramming of the apoptotic pathway. Cancer Cell. 2020;37:104–122.e12. doi: 10.1016/j.ccell.2019.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wasilewski D, Priego N, Fustero-Torre C, Valiente M. Reactive astrocytes in brain metastasis. Front Oncol. 2017;7:1. doi: 10.3389/fonc.2017.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Geckil H, Xu F, Zhang X, Moon S, Demirci U. Engineering hydrogels as extracellular matrix mimics. Nanomedicine (Lond) 2010;5:469–484. doi: 10.2217/nnm.10.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kondapaneni RV, Shevde LA, Rao SS. A biomimetic hyaluronic acid hydrogel models mass dormancy in brain metastatic breast cancer spheroids. Adv Biol. 2023 doi: 10.1002/adbi.202200114. [DOI] [PubMed] [Google Scholar]
- 74.Farino Reyes CJ, Pradhan S, Slater JH. The influence of ligand density and degradability on hydrogel induced breast cancer dormancy and reactivation. Adv Healthc Mater. 2021 doi: 10.1002/adhm.202002227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kondapaneni RV, Rao SS. Matrix stiffness and cluster size collectively regulate dormancy versus proliferation in brain metastatic breast cancer cell clusters. Biomater Sci. 2020;8:6637–6646. doi: 10.1039/D0BM00969E. [DOI] [PubMed] [Google Scholar]
- 76.Cooper J, Giancotti FG. Integrin signaling in cancer: mechanotransduction, stemness, epithelial plasticity, and therapeutic resistance. Cancer Cell. 2019;35:347–367. doi: 10.1016/j.ccell.2019.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Campbell ID, Humphries MJ. Integrin structure, activation, and interactions. Cold Spring Harb Perspect Biol. 2011;3:1–14. doi: 10.1101/cshperspect.a004994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang L, et al. EZH2 engages TGFβ signaling to promote breast cancer bone metastasis via integrin β1-FAK activation. Nat Commun. 2022 doi: 10.1038/s41467-022-30105-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Welf ES, Naik UP, Ogunnaike BA. A spatial model for integrin clustering as a result of feedback between integrin activation and integrin binding. Biophys J. 2012;103:1379–1389. doi: 10.1016/j.bpj.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hamidi H, Ivaska J. Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer. 2018;18:533–548. doi: 10.1038/s41568-018-0038-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bednarczyk M, Stege H, Grabbe S, Bros M. β2 Integrins-multi-functional leukocyte receptors in health and disease. Int J Mol Sci. 2020;21:1402. doi: 10.3390/ijms21041402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Clemetson KJ, Clemetson JM. Integrins and cardiovascular disease. Cell Mol Life Sci. 1998;54:502–513. doi: 10.1007/s000180050179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yoshimasu T, et al. Increased expression of integrin alpha3beta1 in highly brain metastatic subclone of a human non-small cell lung cancer cell line. Cancer Sci. 2004;95:142–148. doi: 10.1111/j.1349-7006.2004.tb03195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhou B, et al. Integrin α3β1 can function to promote spontaneous metastasis and lung colonization of invasive breast carcinoma. Mol Cancer Res. 2014;12:143–154. doi: 10.1158/1541-7786.MCR-13-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vicart P, et al. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet. 1998;20:92–95. doi: 10.1038/1765. [DOI] [PubMed] [Google Scholar]
- 87.Brodehl A, et al. The novel αB-crystallin (CRYAB) mutation p.D109G causes restrictive cardiomyopathy. Hum Mutat. 2017;38:947–952. doi: 10.1002/humu.23248. [DOI] [PubMed] [Google Scholar]
- 88.Malin D, et al. αB-crystallin: a novel regulator of breast cancer metastasis to the brain. Clin Cancer Res. 2014;20:56–67. doi: 10.1158/1078-0432.CCR-13-1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Berghoff AS, et al. Invasion patterns in brain metastases of solid cancers. Neuro Oncol. 2013;15:1664–1672. doi: 10.1093/neuonc/not112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Berghoff AS, et al. αvβ3, αvβ5 and αvβ6 integrins in brain metastases of lung cancer. Clin Exp Metastasis. 2014;31:841–851. doi: 10.1007/s10585-014-9675-0. [DOI] [PubMed] [Google Scholar]
- 91.Ranjan A, Gupta P, Srivastava SK. Penfluridol: an antipsychotic agent suppresses metastatic tumor growth in triple-negative breast cancer by inhibiting integrin signaling axis. Cancer Res. 2016;76:877–890. doi: 10.1158/0008-5472.CAN-15-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wu YJ, et al. Targeting αV-integrins decreased metastasis and increased survival in a nude rat breast cancer brain metastasis model. J Neurooncol. 2012;110:27. doi: 10.1007/s11060-012-0942-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ishii K, Kubo KI, Nakajima K. Reelin and neuropsychiatric disorders. Front Cell Neurosci. 2016 doi: 10.3389/fncel.2016.00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shifman S, et al. Genome-wide association identifies a common variant in the reelin gene that increases the risk of schizophrenia only in women. PLoS Genet. 2008;4:e28. doi: 10.1371/journal.pgen.0040028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang Z, et al. Reelin gene variants and risk of autism spectrum disorders: an integrated meta-analysis. Am J Med Genet B Neuropsychiatr Genet. 2014;165B:192–200. doi: 10.1002/ajmg.b.32222. [DOI] [PubMed] [Google Scholar]
- 96.Fehér Á, Juhász A, Pákáski M, Kálmán J, Janka Z. Genetic analysis of the RELN gene: gender specific association with Alzheimer’s disease. Psychiatry Res. 2015;230:716–718. doi: 10.1016/j.psychres.2015.09.021. [DOI] [PubMed] [Google Scholar]
- 97.Biermann J, et al. Dissecting the treatment-naive ecosystem of human melanoma brain metastasis. Cell. 2022;185:2591–2608.e30. doi: 10.1016/j.cell.2022.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Deshpande K, et al. Neuronal exposure induces neurotransmitter signaling and synaptic mediators in tumors early in brain metastasis. Neuro Oncol. 2022;24:914–924. doi: 10.1093/neuonc/noab290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jandial R, Choy C, Levy DM, Chen MY, Ansari KI. Astrocyte-induced Reelin expression drives proliferation of Her2+ breast cancer metastases. Clin Exp Metastasis. 2017;34:185–196. doi: 10.1007/s10585-017-9839-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Krishnamurti U, Silverman JF. HER2 in breast cancer: a review and update. Adv Anat Pathol. 2014;21:100–107. doi: 10.1097/PAP.0000000000000015. [DOI] [PubMed] [Google Scholar]
- 101.Garcia-Alvarez A, Papakonstantinou A, Oliveira M. Brain metastases in HER2-positive breast cancer: current and novel treatment strategies. Cancers (Basel) 2021;13:2927. doi: 10.3390/cancers13122927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Neman J, et al. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc Natl Acad Sci U S A. 2014;111:984–989. doi: 10.1073/pnas.1322098111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Northcott PA, et al. Medulloblastoma. Nat Rev Dis Primers. 2019 doi: 10.1038/s41572-019-0063-6. [DOI] [PubMed] [Google Scholar]
- 104.Zou H, et al. A neurodevelopmental epigenetic programme mediated by SMARCD3-DAB1-Reelin signalling is hijacked to promote medulloblastoma metastasis. Nat Cell Biol. 2023;25:493–507. doi: 10.1038/s41556-023-01093-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yuzhalin AE, Yu D. Brain Metastasis Organotropism. Cold Spring Harb Perspect Med. 2020;10:a037242. doi: 10.1101/cshperspect.a037242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pokhilko A, et al. Global proteomic analysis of extracellular matrix in mouse and human brain highlights relevance to cerebrovascular disease. J Cereb Blood Flow Metab. 2021;41:2423. doi: 10.1177/0271678X211004307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hebert JD, et al. Proteomic profiling of the ECM of xenograft breast cancer metastases in different organs reveals distinct metastatic niches. Cancer Res. 2020;80:1475–1485. doi: 10.1158/0008-5472.CAN-19-2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gordon-Weeks A, Yuzhalin AE. Cancer extracellular matrix proteins regulate tumour immunity. Cancers (Basel) 2020;12:1–25. doi: 10.3390/cancers12113331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Widodo SS, et al. Spatial analysis of the metastatic brain tumor immune and extracellular matrix microenvironment. Adv Cancer Biol Metastasis. 2023;7:100096. doi: 10.1016/j.adcanc.2023.100096. [DOI] [Google Scholar]
- 110.Bejarano L, Jordāo MJC, Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Discov. 2021;11:933–959. doi: 10.1158/2159-8290.CD-20-1808. [DOI] [PubMed] [Google Scholar]
- 111.Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther. 2021;221:107753. doi: 10.1016/j.pharmthera.2020.107753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yuzhalin AE, Urbonas T, Silva MA, Muschel RJ, Gordon-Weeks AN. A core matrisome gene signature predicts cancer outcome. Br J Cancer. 2018;118:435–440. doi: 10.1038/bjc.2017.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Benson AB, et al. A phase II randomized, double-blind, placebo-controlled study of simtuzumab or placebo in combination with gemcitabine for the first-line treatment of pancreatic adenocarcinoma. Oncologist. 2017;22:241–e15. doi: 10.1634/theoncologist.2017-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hecht JR, et al. A phase II, randomized, double-blind, placebo-controlled study of simtuzumab in combination with FOLFIRI for the second-line treatment of metastatic KRAS mutant colorectal adenocarcinoma. Oncologist. 2017;22:243–e23. doi: 10.1634/theoncologist.2016-0479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Johnston KA, Lopez KM. Lysyl oxidase in cancer inhibition and metastasis. Cancer Lett. 2018;417:174–181. doi: 10.1016/j.canlet.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 116.Fennell DA, et al. Maintenance defactinib versus placebo after first-line chemotherapy in patients with merlin-stratified pleural mesothelioma: COMMAND-a double-blind, randomized. Phase II Study J Clin Oncol. 2019;37:790–798. doi: 10.1200/JCO.2018.79.0543. [DOI] [PubMed] [Google Scholar]
- 117.Hakim N, Patel R, Devoe C, Saif MW. Why HALO 301 Failed and implications for treatment of pancreatic cancer. Pancreas (Fairfax) 2019;3:e1–e4. doi: 10.17140/POJ-3-e010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Winer A, Adams S, Mignatti P. Matrix metalloproteinase inhibitors in cancer therapy: turning past failures into future successes. Mol Cancer Ther. 2018;17:1147–1155. doi: 10.1158/1535-7163.MCT-17-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hersh DS, et al. Evolving drug delivery strategies to overcome the blood brain barrier. Curr Pharm Des. 2016;22:1177. doi: 10.2174/1381612822666151221150733. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.