

Abstract
Congenital hearing loss affects one in 500 newborns. Sequence variations in OTOF, which encodes the calcium-binding protein otoferlin, are responsible for 1–8% of congenital, nonsyndromic hearing loss and are the leading cause of auditory neuropathy spectrum disorders. The natural history of otoferlin-related hearing loss, the relationship between OTOF genotype and hearing loss phenotype, and the outcomes of clinical practices in patients with this genetic disorder are incompletely understood because most analyses have reported on small numbers of cases with homogeneous OTOF genotypes. Here, we present the first systematic, quantitative literature review of otoferlin-related hearing loss, which analyzes patient-specific data from 422 individuals across 61 publications. While most patients display a typical phenotype of severe-to-profound hearing loss with prelingual onset, 10–15% of patients display atypical phenotypes, including mild-to-moderate, progressive, and temperature-sensitive hearing loss. Patients’ phenotypic presentations appear to depend on their specific genotypes. For example, non-truncating variants located in and immediately downstream of the C2E calcium-binding domain are more likely to produce atypical phenotypes. Additionally, the prevalence of certain sequence variants and their associated phenotypes varies between populations due to evolutionary founder effects. Our analyses also suggest otoacoustic emissions are less common in older patients and those with two truncating OTOF variants. Critically, our review has implications for the application and limitations of clinical practices, including newborn hearing screenings, hearing aid trials, cochlear implants, and upcoming gene therapy clinical trials. We conclude by discussing the limitations of available research and recommendations for future studies on this genetic cause of hearing loss.
Introduction
Hearing loss is the fourth leading cause of disability worldwide, affecting an estimated 6.8% of the global population (Neumann et al. 2019). One out of every 500 children is born with hearing loss, and approximately half of these cases have severe-to-profound bilateral hearing loss (Lieu et al. 2020). The majority of congenital hearing loss cases are caused by genetic variations (Shearer et al. 1999; Lieu et al. 2020). Sequence variations in the OTOF gene, which encodes the membrane-associated protein otoferlin, are responsible for 1–8% of congenital, nonsyndromic hearing loss cases and are the leading cause of auditory neuropathy spectrum disorders (ANSD; Azaiez et al. 1993). Otoferlin is a calcium-sensing protein involved in inner hair cell (IHC) vesicular transport and exocytosis, which are critical processes for IHC signaling to auditory nerve fibers (Michalski et al. 2017). IHCs and spiral ganglion neurons form specialized ribbon synapses for transmitting acoustic signals with high temporal fidelity. This fidelity is critical for certain aspects of auditory perception, including sound localization and speech comprehension (Nouvian et al. 2006). OTOF sequence variants can result in deficient or non-functional otoferlin protein, thus disrupting synaptic transmission and causing otoferlin-related hearing loss.
Also known as DFNB9, otoferlin-related hearing loss follows an autosomal recessive inheritance pattern and typically presents as severe-to-profound sensorineural hearing loss (SNHL) with congenital or prelingual onset (Azaiez et al. 1993). However, the literature is replete with reports of atypical hearing phenotypes, including mild-to-moderate (Yildirim-Baylan et al. 2014; Fedick et al. 2016; Wang et al. 2018; Santarelli et al. 2021; Iwasa et al. 2022), progressive (Chiu et al. 2010; Matsunaga et al. 2012; Fedick et al. 2016), and even temperature-sensitive (Varga et al. 2006; Marlin et al. 2010; Zhang et al. 2016b; Zhu et al. 2021) hearing loss. The latter occurs when an individual’s auditory threshold varies with fluctuations in body temperature. Importantly, the tone-detection thresholds used to determine the severity of hearing loss do not always correlate with functional deficits. As in other forms of ANSD, speech comprehension in otoferlin-related SNHL can be poorer than expected, based on hearing thresholds in less severe phenotypes (Santarelli et al. 2013; Vona et al. 2020).
The phenotypic spectrum of otoferlin-related SNHL is likely a consequence of the diversity of OTOF sequence variants. Over 200 pathogenic and likely pathogenic OTOF variants have been reported, yet the relationship between phenotype and OTOF genotype is poorly understood (Vona et al. 2020). Consisting of 48 exons spanning 90 kb of DNA, OTOF yields five isoforms in humans (Yasunaga et al. 2000). The canonical otoferlin isoform in human cochlea contains six C2 domains, which likely bind calcium or interact with proteins, one putative C2 domain, at least one Fer domain hypothesized to interact with phospholipid membranes, and a transmembrane domain (Yasunaga et al. 2000; Pangršič et al. 2012; Harsini et al. 2018; Vona et al. 2020; Dominguez et al. 2022; Liu et al. 2023). These domains subserve otoferlin’s functions in vesicular tethering, exocytosis, and replenishment at the presynaptic axon terminals of IHCs (Johnson and Chapman 2010; Helfmann et al. 2011; Padmanarayana et al. 2014; Meese et al. 2017; Michalski et al. 2017; Vona et al. 2020; Liu et al. 2023). However, the precise structure of otoferlin is still being elucidated—recent studies identified a previously unannotated exon (Ranum et al. 2019) and a novel truncated isoform in mouse IHCs (Liu et al. 2023), and a recent in silico analysis suggested the existence of a novel “C2-FerA” domain (Dominguez et al. 2022).
Although OTOF variants can disrupt the physiology of synaptic transmission at the ribbon synapse (Roux et al. 2006; Santarelli et al. 2009, 2011, 2015; Santarelli 2010), their impact on cochlear anatomy may be minimal. There is evidence that the IHCs, outer hair cells (OHCs), and auditory nerve develop normally, at least initially. In mouse models of otoferlin deficiency, early postnatal development of IHCs and ribbon synapses appears normal, although the quantity and morphology of ribbon synapses are later altered (Roux et al. 2006; Al-Moyed et al. 2019; Stalmann et al. 2021). Many patients with otoferlin-related SNHL initially have preserved otoacoustic emissions (OAEs), which indicate that OHCs are present and functional (Varga et al. 2003; Iwasa et al. 2022). However, multiple reports indicate that OAEs appear to diminish in amplitude and eventually become absent over time, indicating that OHCs may gradually die or become non-functional as a result of OTOF sequence variants (Rodríguez-Ballesteros et al. 2003; Loundon et al. 2005; Rouillon et al. 2006; Chiu et al. 2010; Kitao et al. 2019; Thorpe et al. 2022).
Due to the initial preservation of OAEs, children with otoferlin-related hearing loss commonly pass newborn hearing screening (NBHS) based only on OAE detection (Varga et al. 2003; Iwasa et al. 2022). Upon identification of SNHL, hearing aids are recommended as first-line intervention (Azaiez et al. 1993). However, with the sound-amplifying OHCs initially intact, additional sound amplification from hearing aids may not compensate for dysregulated exocytosis downstream at the ribbon synapse (Rouillon et al. 2006; Chiu et al. 2010; Marlin et al. 2010; Santarelli et al. 2021). Hearing aids have also been hypothesized to accelerate the death of OHCs and the loss of OAEs in these patients through acoustic trauma mechanisms (Rouillon et al. 2006; Kitao et al. 2019; Vona et al. 2020). In contrast, cochlear implants are expected to provide clinical benefit in most patients with otoferlin-related SNHL because they bypass the dysfunctional ribbon synapse to stimulate the auditory nerve directly (Rouillon et al. 2006; Zheng and Liu 2020; Iwasa et al. 2022). Conversely, patients with an ANSD phenotype [preserved OAEs and abnormal auditory brainstem response (ABR)] arising from postsynaptic dysfunction are less likely to benefit from cochlear implantation (Shearer et al. 2017; De Siati et al. 2020). For this reason, in the absence of an identified genetic etiology that indicates presynaptic dysfunction, cochlear implantation may be delayed in patients with ANSD. In the future, genetic therapies that restore functional otoferlin to IHCs may offer patients with OTOF-related SNHL more natural hearing than cochlear implants can provide (Akil et al. 2019; Al-Moyed et al. 2019; Vona et al. 2020). Regardless of the clinical interventions under consideration, optimal patient care and translational research require thoroughly understanding the relationship between OTOF variants, phenotypic manifestations, and clinical outcomes.
Despite numerous case reports and small-scale studies of OTOF-related hearing loss, large-scale studies are scarce, and there is an absence of comprehensive meta-analyses or quantitative literature reviews. Vona et al. (2020) provide an excellent qualitative description of the genetic and phenotypic heterogeneity of otoferlin-related SNHL, and Thorpe et al. (2022) analyzed some aspects of the genotype–phenotype relationship by combining original data with data from the literature. However, the natural history, genotype–phenotype relationships, and clinical prognoses of this disorder remain incompletely defined due to a lack of quantitative analyses in large cohorts containing the full genetic spectrum of OTOF-related SNHL. Here, we attempt to elucidate these aspects of otoferlin-related SNHL by conducting a systematic, quantitative literature review. The resulting analyses provide a thorough representation of phenotypes, associated genotypes, and clinical outcomes reported in the literature.
Methods
Search strategy
Original article search was completed using the search term “OTOF” in the PubMed online database (https://pubmed.ncbi.nlm.nih.gov) on February 14, 2022. An abstract scan requiring human data yielded 109 unique publications. After the original query date, two additional papers (Iwasa et al. 2022; Lee et al. 2022) were published and identified via a Pubmed database alert for new articles, yielding a total of 111 publications.
Case and publication selection
The 111 identified publications were reviewed for data on individuals with otoferlin-related hearing loss. Inclusion criteria included: non-syndromic SNHL; biallelic, pathogenic or likely pathogenic OTOF sequence variations confirmed via sequencing; individual data on genotype, phenotype, OAEs, or clinical outcomes. Exclusion criteria included: syndromic or conductive hearing loss; severe prematurity or perinatal hypoxia; treatment with ototoxic agents; perinatal jaundice or hyperbilirubinemia; traumatic brain injury; noise exposure; anatomical abnormalities of the auditory or vestibular systems. One patient with cystic fibrosis, one with osteogenesis imperfecta, and one with type I diabetes were included as these conditions were not expected to have influenced their hearing phenotypes. Cases duplicated between publications were combined into a single entry, though due to unclear language in some publications, we cannot exclude the possibility that a small number of duplicated cases were included in our review. In total, 422 individual patients from 61 publications were included (Fig. 1; Table 1). One additional publication (Lee et al. 2022) with group data, but no individual data, showing speech perception outcomes after cochlear implantation was included in our cochlear implant analysis. All analyses were performed and visualized using GraphPad Prism (version 9.5.0, GraphPad Software, San Diego, CA, USA).
Fig. 1.

Data collection flow diagram. From the 111 publications identified in our initial database search, we analyzed patient-specific data on 422 cases with biallelic OTOF variants across 61 publications, in addition to group data from one publication. OAE otoacoustic emissions, NBHS newborn hearing screening
Table 1.
Included publications
| Publication | Cases | Country of origin |
|---|---|---|
| Ahmed et al. (2021) | 7 | Pakistan |
| Alkowari et al. (2017) | 2 | Qatar |
| Almontashiri et al. (2018) | 7 | Saudi Arabia |
| Ammar-Khodja et al. (2015) | 7 | Algeria |
| Bai et al. (2019) | 1 | China |
| Bitarafan et al. (2020) | 1 | Iran |
| Safka Brozkova et al. (2020) | 2 | Czech Republic |
| Chiu et al. (2010) | 6 | Taiwan |
| Choi et al. (2009) | 55 | Pakistan |
| Churbanov et al. (2016) | 2 | Russia |
| Dallol et al. (2016) | 2 | Saudi Arabia |
| de Oliveira Costa et al. (2012) | 2 | Brazil |
| Fareed et al. (2021) | 2 | India |
| Fedick et al. (2016) | 4 | USA |
| Gallo-Terán et al. (2004) | 4 | Spain |
| Gallo-Terán et al. (2006) | 1 | Spain |
| Houseman et al. (2001) | 5 | United Arab Emirates |
| Hutchin et al. (2005) | 2 | United Kingdom |
| Iwasa et al. (2013) | 7 | Japan |
| Iwasa et al. (2019) | 39 | Japan |
| Iwasa et al. (2022) | 64 | Japan |
| Jin et al. (2014) | 3 | Korea |
| Kim et al. (2018) | 13 | Korea |
| Lee et al. (2020) | 2 | Korea |
| Li et al. (2021) | 4 | China |
| Likar et al. (2018) | 1 | Slovenia |
| Loundon et al. (2005) | 1 | France |
| Marlin et al. (2010) | 3 | France |
| Matsunaga et al. (2012) | 13 | Japan |
| Naz et al. (2017) | 7 | Pakistan |
| Noman et al. (2019) | 4 | Pakistan |
| Pandey et al. (2017) | 1 | India |
| Qiu et al. (2019) | 2 | China |
| Rodríguez-Ballesteros et al. (2003) | 21 | Spain |
| Rodríguez-Ballesteros et al. (2008) | 36 | Argentina, Colombia, Spain |
| Romanos et al. (2009) | 3 | Brazil |
| Rouillon et al. (2006) | 2 | France |
| Runge et al. (2013) | 2 | USA |
| Santarelli et al. (2009) | 1 | Italy, Spain |
| Santarelli et al. (2011) | 1 | Italy, Spain |
| Santarelli et al. (2015) | 7 | Italy, Spain |
| Santarelli et al. (2021) | 5 | Italy, Spain |
| Shahin et al. (2010) | 3 | Pakistan |
| Souissi et al. (2021) | 1 | Tunisia |
| Tabatabaiefar et al. (2018) | 1 | Iran |
| Tang et al. (2017) | 2 | China |
| Tekin et al. (2005) | 3 | Turkey |
| Varga et al. (2003) | 3 | USA |
| Varga et al. (2006) | 4 | USA |
| Wang et al. (2010) | 1 | China |
| Wang et al. (2018) | 4 | China |
| Wu et al. (2018) | 10 | China |
| Wu et al. (2020) | 1 | China |
| Xia et al. (2018) | 2 | China |
| Xiang et al. (2020) | 1 | China |
| Yildirim-Baylan et al. (2014) | 9 | Turkey |
| Zadro et al. (2010) | 4 | Italy |
| Zhang et al. (2013) | 1 | China |
| Zhang et al. (2016a) | 3 | China |
| Zhang et al. (2016b) | 11 | China |
| Zhu et al. (2021) | 4 | China |
All 61 publications contained patient-specific data on individuals with confirmed pathogenic or likely-pathogenic, biallelic OTOF sequence variations. In total, data were collected on 422 unique individuals from these publications
Phenotype analyses
To delineate patient phenotypes, we either (1) calculated the air-conduction, pure-tone average hearing thresholds determined by behavioral or electrophysiological audiometry at 0.5, 1, 2, and 4 kHz between ears, or (2) used the average hearing threshold reported by the authors. When a range of hearing thresholds was reported, we used the mean of that range. If a range was unbounded at the upper end (e.g., ≥ 80 dB), we used the lower limit (e.g., 80 dB). We then assigned a hearing phenotype to each individual using a simplified version of the classifications established by Clark (1981): 0–25 dB, normal; 26–40 dB, mild; 41–70 dB, moderate; 71–90 dB, severe; > 90 dB, profound. If a numeric hearing threshold was not reported, we accepted the severity classification assigned by the authors. Patients reported as “severe-profound” (n = 59) were classified as severe, those with thresholds below 25 dB but with complaints of hearing deficits were classified as mild (n = 5), and those with fluctuant hearing thresholds that were neither temperature-sensitive nor unidirectionally progressive were classified as unstable (n = 2). ABRs were classified as absent if no response was detected at the maximum stimulation level, and they were classified as abnormal if an aberrant response was detected in at least one ear. No ABRs were described as normal. The results of ABR-based NBHS were only included in NBHS analyses, not in other ABR analyses.
Genotype analyses
Deletions larger than one residue, nonsense, frameshift, and splice-site variants were considered truncating (T) unless specifically described as non-truncating (n = 1). Single-residue, in-frame deletions and missense variants were classified as non-truncating (NT). In cases of multiple homozygous variants, the most severe variant was used to classify subjects. We defined the boundaries of functional regions within otoferlin according to Vona et al. (2020) and, for the putative C2 domain, C2de, according to Pangršič et al. (2012). One NT splice-site variant was not included in the regional analysis. Assessments of pathogenicity were multi-factorial and included other reports of the variant, sequence conservation, co-segregation of the variant with SNHL, results of mutation prediction software reported by authors, and exclusion of other possible genetic and environmental causes of hearing loss.
Otoacoustic emissions analyses
Distortion product otoacoustic emissions (DPOAEs) and transient evoked otoacoustic emissions (TEOAEs) were all categorized as “OAEs.” OAE results were classified as “present,” “absent,” “reduced,” or, in cases where OAEs were lost over time, “present, then absent.” “Present” was used when (1) qualitatively reported by the authors or, if raw data were provided, (2) OAEs were present (greater than 5 dB signal-to-noise ratio) at 50% or more of the frequencies tested in both ears. “Reduced” was used when (1) both ears had present OAEs in less than 50% of frequencies or (2) only one ear had present OAEs in at least 50% of frequencies. “Absent” was used when (1) described qualitatively by the authors or (2) if raw data indicated absent (less than 5 dB signal-to-noise ratio) OAE responses at all tested frequencies. The results of OAE-based NBHS were only included in NBHS analyses, not in other OAE analyses.
Analyses of clinical outcomes
We compared the results of NBHS that utilized ABRs, OAEs, and unspecified test methodology. Hearing aid outcomes were reported, predominantly in subjective terms, for 32 patients from 12 publications, which we summarized in Table 3. Methodological variability in cochlear implant outcome assessments limited our ability to conduct quantitative analyses, so we summarized the reported outcomes of 100 patients from 17 publications in Table 4.
Table 3.
Hearing aid outcomes
| Publication | Cases | Outcomes |
|---|---|---|
| Chiu et al. (2010) | 6 | “All the 6 homozygotes had limited benefits from hearing aids” |
| Costa et al. (2012) | 2 |
“Could not tell whether the hearing aid was working or not” “The child obtained no benefits from the device” |
| Loundon et al. (2005) | 1 | “The mean threshold in free field… was 75 dB with hearing aids” |
| Marlin et al. (2010) | 3 | “Pure-tone hearing levels improved by 10… and 15 dB… with hearing aid amplification, but did not dramatically improve vocal audiometry performance” |
| Rouillon et al. (2006) | 2 | “No improvement was seen with the bilateral powerful hearing aid” |
| Runge et al. (2013) | 2 | “Aided benefit was reportedly limited to inconsistent increase in sound awareness” |
| Santarelli et al. (2015) | 8 | “Aided thresholds showed a marked improvement in pure tone sensitivity… however, they proved to be above the intensity range of conversational speech” |
| Santarelli et al. (2021) | 2 | “Both adult subjects had tried hearing aids without benefit” |
| Tang et al. (2017) | 1 | “He had worn hearing aids in both ears since… birth, but his ability to communicate was not improved” |
| Varga et al. (2003) | 3 | “All the children reported receiving no benefit from hearing aids” |
| Zadro et al. (2010) | 1 | “The child showed poor perceptive skills, only detection of sounds and words using hearing aids” |
| Zhang et al. (2013) | 1 | “6-month trial of conventional amplification failed to show benefit” |
The outcomes of hearing aids reported in 32 individuals across 12 publications are summarized here. Reports were typically subjective, and no individual received from hearing aids benefit that improved conversational speech perception
Table 4.
Cochlear implant outcomes
| Publication | Cases | Outcomes |
|---|---|---|
| Gallo-Terán et al. (2006) | 1 | Appropriate language development at 26 months of age; does not require lip-reading |
| Iwasa et al. (2022) | 36 | ≥ 2 years post-CI, for 6% CAP = 7, 79% CAP = 6, 9% CAP = 5, 3% CAP = 4, 3% CAP = 3; 47% had 20–29 dB hearing threshold, 42% had 30–39 dB threshold, 11% had 40–49 dB threshold |
| Kim et al. (2018) | 10 | Mean pre-CI CAP = 0.2; 3 months post-CI CAP = 2.2 (n = 10); 12 months post-CI CAP = 4.6 (n = 7); 24 months post-CI CAP = 5.4 (n = 5); 36 months post-CI CAP = 7 (n = 2) |
| Lee et al. (2022) | 10 | At mean of 24.1 months post-CI, mean CAP = 6.2 |
| Loundon et al. (2005) | 1 | Pre-CI 75 dB hearing threshold, 4/40 MAIS; 12 months post-CI, 45 dB hearing threshold, 31/40 MAIS |
| Rodríguez-Ballesteros et al. (2003) | 7 | All “considered successful in terms of sound detection and communication skills” |
| Runge et al. (2013) | 2 | 3 years post-CI on LNT Easy spoken word score, one patient had 80%, one patient had 44% |
| Rouillon et al. (2006) | 2 | One patient 18 months post-CI had 45 dB hearing threshold and 31/40 MAIS; one patient 3 years post-CI had 37 dB hearing threshold and 40/40 MAIS |
| Santarelli et al. (2015) | 6 | 1–1.5 years post-CI, mean hearing threshold was 25–30 dB with 90–100% disyllable recognition |
| Santarelli et al. (2021) | 5 | Authors report improvements in speech perception for all 5 patients |
| Tang et al. (2017) | 2 | Both patients report improvement in speech perception and pronunciation |
| Varga et al. (2006) | 2 | Both patients report benefit |
| Wu et al. (2018) | 10 | Pre-op, median CAP = 1.5; 1 year post-CI CAP = 5; 5 year post-CI CAP = 6 |
| Wu et al. (2020) | 1 | Authors describe outcome as successful; 3 years post-CI, enrolled in regular school |
| Zadro et al. (2010) | 3 | Authors report improvement for all patients |
| Zhang et al. (2013) | 1 | 3 years post-CI, 25 dB hearing thresholds with excellent speech and language recognition, and is enrolled in regular school |
| Zhang et al. (2016a) | 1 | Temperature-sensitive case; 2 years post-CI, 93% open-set word recognition and 98% open-set sentence recognition |
The outcomes of cochlear implants reported in 100 individuals across 17 publications are summarized here. One publication, Lee et al. (2022), did not contain patient-specific data. Nearly all reported cochlear implantations were considered successful
CI cochlear implant, CAP Categories of Auditory Performance, MAIS Meaningful Auditory Integration Scale, LNT Lexical Neighborhood Test
Analyses involving age
Age data were used only if a specific age was indicated for the case. Age ranges were not used unless the entire range fit within a single bin. If hearing thresholds were measured at multiple ages, we used the youngest age. If OAEs were measured and present at multiple ages, we used the oldest age.
Results
Phenotypes
Audiometry results were available for 398 of the 422 individuals with biallelic, pathogenic or likely pathogenic OTOF variants (Fig. 2A). Of those cases, 227 (57%) displayed profound SNHL and 120 (30%) displayed severe SNHL, which are both typical phenotypes. The remaining 51 (13%) patients displayed atypical phenotypes—SNHL was moderate in 14 (3.5%), mild in 13 (3.3%), temperature-sensitive in 14 (3.5%), unstable in two (0.5%), and progressive in eight (2%). These proportions changed minimally when only patients assessed with behavioral audiometry (e.g., pure tone audiometry, visual reinforcement audiometry) were analyzed (n = 321; Fig. 2B) and patients assessed with electrophysiological tests (e.g., ABR audiometry, auditory steady-state response audiometry; n = 17; Fig. 2C) and unspecified methodology (n = 60; Fig. 2D) were excluded. Atypical phenotypes were present in 30 of 154 (19%) cases with absent ABRs at the maximum stimulation tested (Fig. 2E) and in nine of 26 (35%) cases with abnormal ABRs (Fig. 2F). Severe or profound SNHL was present in 70–100% of cases in all age groups (Fig. 3).
Fig. 2.

Phenotypes. The spectrum of reported phenotypes in otoferlin-related sensorineural hearing loss (A) was subdivided into phenotypes classified with behavioral audiometry (B), electrophysiological audiometry (C), and audiometry with unspecified methodology (D). The phenotypes of cases with absent auditory brainstem responses (ABRs) at the maximum stimulation tested are shown in (E) while the phenotypes of cases with abnormal ABRs are shown in (F). Phenotype classification was assigned based on hearing thresholds as follows: 0–25 dB, normal; 26–40 dB, mild; 41–70 dB, moderate; 71–90 dB, severe; > 90 dB, profound. NT non-truncating, T truncating
Fig. 3.

Phenotypes by age. The distribution of hearing phenotypes reported in patients of different ages is shown. The majority of cases in all age groups have severe-to-profound sensorineural hearing loss. The relative paucity of temperature-sensitive, mild, and moderate cases at younger ages is likely a consequence of delayed diagnoses rather than a physiological process. Unstable and progressive cases were omitted due to their variability over time. Year of Life 1 = birth to 1 year old; Year of Life 2 = 1.01 years old to 2 years old; etc.
Genotypes
Of the 398 cases with biallelic, pathogenic or likely pathogenic OTOF variants and phenotypic data, 238 (60%) were homozygotes and 160 (40%) were compound heterozygotes. One hundred and sixty-five cases (41%) had two NT variants (NT/NT; Fig. 4A), 81 (20%) had one NT and one T variant (NT/T; Fig. 4B), and 152 (38%) had two T variants (T/T; Fig. 4C). Typical profound or severe phenotypes were reported in 129 (78%) of 165 NT/NT cases, while the remaining 36 (22%) displayed atypical phenotypes, including 11 (6.7%) with temperature-sensitive SNHL and 8 (4.8%) with progressive SNHL (Fig. 4A). In cases with NT/T genotypes (n = 81), 70 (86%) had profound or severe SNHL, while 11 (14%) displayed atypical phenotypes (Fig. 4B). Among T/T cases (n = 152), 148 (97%) displayed severe or profound SNHL, while only four (2.7%) had atypical phenotypes (Fig. 4C).
Fig. 4.

Phenotypes by genotype. The spectrum of phenotypes in cases with biallelic OTOF variants was subdivided according to genotype. Twenty-two percent of cases with two non-truncating alleles (NT/NT) had atypical (mild, moderate, temperature-sensitive, progressive, or unstable) phenotypes (A). Atypical phenotypes were present in 14% of cases with one non-truncating and one truncating allele (NT/T; B), and in 2.7% of cases with two truncating alleles (T/T; C). Hearing thresholds: 0–25 dB, normal; 26–40 dB, mild; 41–70 dB, moderate; 71–90 dB, severe; > 90 dB, profound
We next investigated whether certain NT variants, or NT variants in certain regions of the gene, might be associated with particular phenotypes (Fig. 5). A schematic of otoferlin protein is shown in Fig. 5A with the locations of functional domains demarcated and common NT variants indicated. Figure 5B shows the number of times NT alleles were reported within each gene region, and the phenotype associated with each allele (e.g., the phenotype of each NT/NT case is represented twice, once for each allele, while NT/T cases are represented once). The majority of NT alleles (total n = 410) were localized to either a C2 domain (n = 201; 49%) or the regions flanking the final C2 domain, C2F (n = 177; 43%). Twenty percent (n = 83) of all NT alleles (n = 410) were associated with atypical phenotypes, but in the C2E domain and the region immediately downstream (C2E-C2F), 57% (n = 35 of 61) of NT alleles were associated with atypical phenotypes. Figure 5C shows the most common NT alleles in our dataset, which were reported in at least five individuals and two unrelated families, and the frequency of phenotypes associated with each allele (each allele is represented once, so the phenotypes of homozygous cases are represented twice). Notably, 78% of p.I1573T alleles (n = 9) were associated with mild or moderate phenotypes, and 41% and 14% of p.E1700Q alleles (n = 29) were associated with progressive or temperature-sensitive phenotypes, respectively. Ten variants in total were associated with temperature-sensitive phenotypes: p.G541S (Matsunaga et al. 2012; Zhang et al. 2016b), p.G614E (Romanos et al. 2009), p.R698T (Zhang et al. 2016a), p.R1080P (Romanos et al. 2009), p.R1157Q (Marlin et al. 2010), p.R1607W (Wang et al. 2010; Zhang et al. 2016b), p.P1628T (Zhu et al. 2021), p.E1661K (Zhang et al. 2016a), p.E1700Q (Zhu et al. 2021), and p.E1804del (Marlin et al. 2010). With the exception of p.E1700Q, these variants were exclusively identified in temperature-sensitive cases within our dataset.
Fig. 5.

Phenotypes by non-truncating variant. A Shows a schematic of otoferlin with specific domains demarcated and the locations of common non-truncating (NT) alleles. B Shows each reported instance of a NT allele within each gene region, as well as the phenotype of the patient, such that the phenotypes of cases with two NT alleles are represented twice (once for each allele) and the phenotypes of cases with one NT allele and one truncating allele are represented once. Pathogenic NT variants were frequently reported in some regions (e.g., C2F, C2F-TM) and not reported in others (e.g., C2A, FerB), and NT variants were more commonly associated with atypical phenotypes in some regions (e.g., C2E) than in others (e.g., C2F-TM). C Shows the most common NT alleles in our dataset and their associated phenotypes (each allele is represented once, so the phenotypes of homozygous cases are represented twice). Variant p.I1573T is notable for its frequent association with mild-to-moderate sensorineural hearing loss, whereas p.E1700Q is commonly associated with progressive or temperature-sensitive sensorineural hearing loss. C2A, C2B, C2C, C2D, C2E, and C2F C2 domains, C2de putative C2 domain, FerA FerB ferlin domains, TM transmembrane domain
As common allelic variants are not evenly distributed across different populations and geographic regions, we tabulated the number and locations of NT and T alleles reported in at least five individuals and two families from our dataset (Table 2).
Table 2.
Geographic distribution of common variants
| Variant | Alleles | Country |
|---|---|---|
| Non-truncating | ||
| p.E841K | 8 | Korea, China |
| p.L1011P | 9 | Turkey, Korea |
| p.R1172Q | 50 | Japan |
| p.I1573T | 9 | Turkey, Japan |
| p.R1583H | 7 | China, Japan |
| p.E1700Q | 29 | China |
| p.E1733K | 21 | Pakistan, China |
| p.R1792H/C | 11 | Saudi Arabia, Japan |
| p.R1856Q/W | 18 | Pakistan, Japan, Czech Republic, Korea |
| p.R1939Q/W | 142 | Japan, Korea, Pakistan |
| Truncating | ||
| p.R237X | 16 | United Arab Emirates, Algeria |
| p.R425X | 13 | Pakistan, China |
| p.Y474X | 16 | Japan |
| p.R708X | 31 | Pakistan, Algeria, India, Spain, Colombia |
| p.E747X | 12 | Qatar, Saudi Arabia, Libya, Italy |
| p.Q829X | 94 | Spain, Colombia, Argentina, France, USA |
| p.Y1064X | 7 | Japan, Korea, Czech Republic |
| p.Q1072X | 5 | Japan |
| c.3289-1G>T | 20 | Pakistan |
The geographic locations of the most common sequence variations in our dataset are listed. All variants were reported in at least five individuals and two unrelated families
Otoacoustic emissions (OAEs)
Of the 231 patients with OAE data, OAEs were present in 166 (72%) and absent in 45 (19%; Fig. 6A). In 15 (6.5%) patients, OAEs were present but significantly reduced or only detected unilaterally. In five (2.2%) patients, OAEs were present at one time and later found to be absent; these patients were reported twice in analyses involving age—once at the oldest age with present OAEs and once at the youngest age with absent OAEs (Fig. 6B, n = 5; Fig. 7D, n = 4; Fig. 7E, n = 1; Fig. 7F, n = 1; Fig. 7G, n = 3). It should be noted, however, that the majority of publications only tested OAEs at a single time point and classified them as either “present” or “absent” without a “reduced” classification. Only patients assessed with behavioral audiometry were included in analyses involving OAEs and phenotype (Figs. 6C, 7A–D). While OAEs were absent in less than 25% of cases up to 10 years of age, OAEs were absent in 33% of patients aged 11–20, absent in 45% and reduced in 27% of patients aged 21–40, and absent in 60% and reduced in 20% of patients aged more than 40 years (Fig. 6B). Regarding phenotypes, OAEs were detected in all mild and temperature-sensitive cases and in approximately 75% of profound, severe, moderate, and progressive cases (Fig. 6C). Similarly, OAEs were present in approximately 80% of NT/NT and NT/T cases, but only in 58% of T/T cases (Fig. 6D).
Fig. 6.

Otoacoustic emissions. Otoacoustic emissions (OAEs) were present, present but reduced, or initially present and lost over time in over 80% of cases (A). The proportion of cases with present OAEs decreases in older age groups (B; n = 5 cases with OAEs that were initially present and lost over time are represented twice, once at their oldest age with present OAEs and once at their youngest age with absent OAEs). Patients with mild or temperature-sensitive sensorineural hearing loss were more likely to have present OAEs than patients with other phenotypes (C), and patients with at least one non-truncating (NT) allele were more likely to have present OAEs than patients with two truncating (T) alleles (D). Only hearing loss phenotypes classified using behavioral audiometry were included in C. Year of Life 1 = birth to 1 year old; Year of Life 2 = 1.01 years old to 2 years old; etc.
Fig. 7.

Otoacoustic emissions by age and phenotype or genotype. The age in years and results of otoacoustic emission (OAE) testing are shown for individuals with mild (A), moderate (B), severe (C), and profound (D) sensorineural hearing loss, and for individuals with two non-truncating (NT) alleles (E), one NT and one truncating (T) allele (F), and two T alleles (G). Individuals with reduced OAEs are represented as gray dots in the “Present” column, and individuals with present OAEs at one time and absent OAEs at a later time are represented twice, once in the “Present” and once in the “Absent” column, with blue dots. Red lines indicate the median age. Only hearing loss phenotypes classified using behavioral audiometry were included in A–D
Next, we examined the distribution of present vs. absent OAEs across ages in patients with specific phenotypes (Fig. 7A–D) and genotypes (Fig. 7E–G). To facilitate data visualization, cases with “reduced” OAEs are shown as gray dots within the “present” column, cases represented twice—once with present and once with absent OAEs—are shown as blue dots, and ages greater than 40 years are presented as 40. Sample sizes were limited for patients with OAE data, age data, and either mild (n = 10 present, n = 0 absent; Fig. 7A), moderate (n = 5 present, n = 3 absent; Fig. 7B), or severe phenotypes (n = 12 present, n = 2 absent; Fig. 7C), or NT/NT (n = 61 present, n = 9 absent) or NT/T (n = 39 present, n = 7 absent) genotypes. However, patients with profound SNHL and present OAEs (n = 79) tended to be younger (median age four years) than patients with profound SNHL and absent OAEs (n = 23; median age seven years; Fig. 7D), and patients with T/T genotypes and present OAEs (n = 43) tended to be younger (median age three years) than those with absent OAEs (n = 17; median age 13 years; Fig. 7G).
Clinical outcomes
NBHS relies on either OAE or ABR testing with pass/fail criteria. In our dataset, NBHS results were available for 73 patients. Of the 35 NBHS that utilized ABRs, 32 (91%) were failed and three (8.6%) were passed (Fig. 8). In contrast, only one of 24 (4.2%) OAE-based NBHS were failed while 23 of 24 (96%) were passed. In 16 cases, the NBHS method was not reported, and of those, 15 (94%) failed and one (6.3%) passed. Two patients are represented twice in Fig. 8 as they received both OAE-based and ABR-based NBHS, and both passed the former and failed the latter.
Fig. 8.
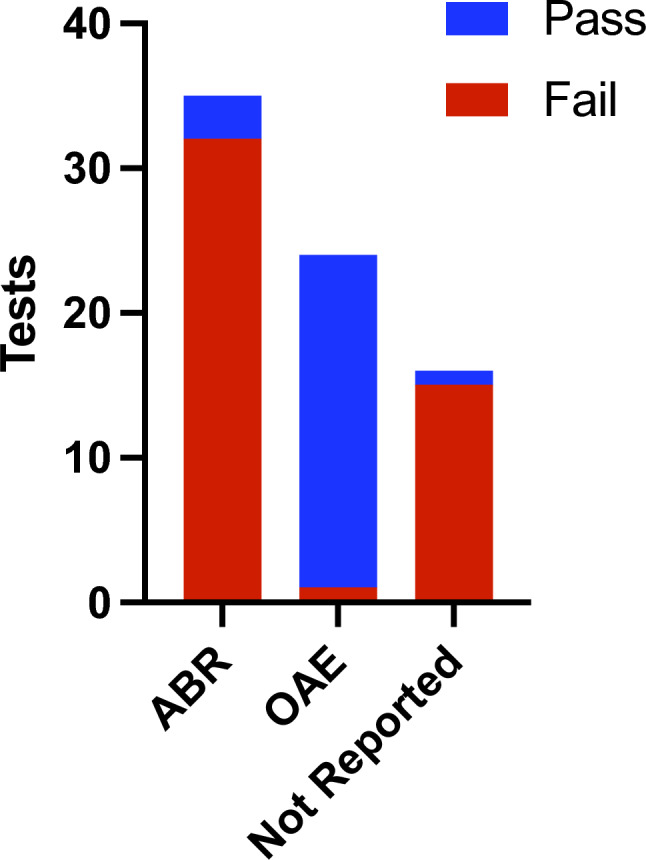
Newborn hearing screening outcomes. The results of auditory brainstem response (ABR)-based and otoacoustic emission (OAE)-based newborn hearing screenings (NBHS), as well as those with unspecified methodology, are shown. Twenty-three of 24 (96%) OAE-based NBHS were inappropriately passed compared to three of 35 (8.6%) ABR-based tests. Two patients underwent both OAE-based and ABR-based screening and are represented once in both the OAE and ABR columns; both patients passed the OAE test and failed the ABR test
Thirty-two cases from 12 publications reported, typically in subjective terms, the efficacy of hearing aids (Table 3). None of the patients who used hearing aids received clinically meaningful benefit that improved conversational speech perception. Additionally, 17 publications provided information on the efficacy of cochlear implants in a total of 100 patients. Although the potential for quantitative analyses was limited due to the heterogeneity of the reported data, the outcomes suggest that cochlear implants reliably improved speech perception (Table 4).
Discussion
Two recent publications have made significant contributions to our understanding of otoferlin-related SNHL. Vona et al. (2020) reviewed both transgenic mouse models and human clinical literature to describe in detail the function and expression patterns of otoferlin isoforms, the spectrum of OTOF sequence variants and their impact on otoferlin function and phenotypes, and the clinical implications of this knowledge. Thorpe et al. (2022) combined audiometric data from original cases and cases reported in the literature to analyze the correlation between genotypic and phenotypic severity in 233 patients with biallelic, pathogenic OTOF variants. Here, we analyzed patient-specific data from 422 unique cases to characterize the genotype–phenotype relationship at the level of individual variants, the relationship between OAEs and multiple case characteristics, and the clinical outcomes of NBHS, hearing aids, and cochlear implants.
Genotype–phenotype relationships
Our comprehensive literature review indicates that 85–90% of cases with biallelic, pathogenic or likely pathogenic OTOF variants are “typical” in that they display prelingual, severe-to-profound SNHL (Fig. 2). However, 10–15% of cases are “atypical” with mild-to-moderate, temperature-sensitive, unstable, or progressive phenotypes. These proportions deviate from those reported in studies of cohorts from geographically or ethnically homogeneous populations. Such studies typically describe relatively homogeneous OTOF genotypes and phenotypes likely due to evolutionary founder effects, though the nature of this homogeny differs between studies. For example, Rodríguez-Ballesteros and colleagues published two reports with a total of 57 cases, all of which had profound SNHL (Rodríguez-Ballesteros et al. 2003, 2008). The majority of these cases were of Spanish descent, 30 (53%) were homozygous for p.Q829X, and 19 (33%) were compound heterozygous for p.Q829X and another pathogenic variant. Two publications by Iwasa and colleagues described slightly more heterogeneous cases in Japan. The first contained 32 cases with phenotypic data, of which 31 (97%) had severe or profound hearing loss and 27 (84%) had at least one p.R1172Q allele (Iwasa et al. 2019). The second publication contained 63 cases with phenotypic data, of which 58 (92%) had severe or profound hearing loss and 58 (92%) had at least one p.R1939Q/W allele (Iwasa et al. 2022). In contrast to the Spanish and Japanese studies, a report of 22 Taiwanese cases described two thirds of their cohort as having moderate SNHL and only one third as having severe-to-profound SNHL (Wu et al. 2019). Twenty (91%) of these patients had at least one p.E1700Q allele, and of the 12 patients with longitudinal data, eight (67%) displayed progressive or fluctuant phenotypes. None of the cases from Wu et al. (2019) were included in our dataset because the publication did not contain any case-specific data. However, of the 29 p.E1700Q alleles in our dataset, 12 (41%) were associated with progressive phenotypes and four (14%) with temperature-sensitive phenotypes (Fig. 5C). Other studies have documented divergent rates of OTOF sequence variations or founder variants in individuals of Pakistani (Choi et al. 2009), Chinese (Zhang et al. 2016a), Turkish (Duman et al. 2011), French (Baux et al. 2017), and Ashkenazi Jewish descent (Fedick et al. 2016). In conjunction with our analyses, and in concurrence with Vona et al. (2020) and Thorpe et al. (2022), these studies demonstrate that OTOF-related phenotypes depend largely on genotypes, and that the prevalence of common genotypes can depend on the ethnicity or geography of a given population (Table 2).
Typical cases likely arise from two non-functional OTOF alleles, which produce proteins that are either truncated and rapidly degraded or carry variations that impair the protein’s ability to bind calcium or phospholipid membranes (Thorpe et al. 2022). Conversely, atypical cases likely occur when at least one allele produces a partially functional protein, which might be sufficient for facilitating calcium-dependent exocytosis but insufficient for replenishing and trafficking vesicles, resulting in rapid vesicle depletion and synaptic fatigue (Pangršič et al. 2010, 2012; Wynne et al. 2013; Santarelli et al. 2015; Vogl et al. 2015; Strenzke et al. 2016; Liu et al. 2023). This may explain why atypical phenotypes almost exclusively arise in cases with at least one NT allele—in our dataset, 22% of NT/NT cases and 14% of NT/T cases were phenotypically atypical (Fig. 4A, B), while just 2.7% of T/T cases were atypical (Fig. 4C). These proportions are remarkably similar to the 22% of missense/missense, 14% of missense/loss-of-function, and 2.3% of loss-of-function/loss-of-function cases Thorpe et al. (2022) described as “less than severe,” though both their analysis and ours drew data from many of the same publications.
The locations of NT sequence variations and the frequencies with which they are reported and associated with particular phenotypes may offer insight into the functional importance of specific protein domains (Fig. 5). Presynaptic calcium plays a central role in exocytosis by regulating vesicle priming, fusion, and replenishment via interactions with multiple proteins, including otoferlin (Beutner et al. 2001; Vona et al. 2020). In our dataset, nearly 50% of reported NT alleles occurred in C2 domains, suggesting the importance of calcium-binding in otoferlin function (Pangršič et al. 2012; Fig. 5B). However, the functionality of each otoferlin C2 domain is debated. C2A does not bind calcium, but it facilitates endocytic membrane retrieval and binding with synaptic proteins (Helfmann et al. 2011; Liu et al. 2023). Liu et al. (2023) recently identified a short otoferlin isoform lacking C2A in mouse IHCs. They demonstrated that transgenic mice expressing only the short isoform and not the canonical isoform had intact hearing thresholds despite abnormal ABRs and reduced sustained exocytosis at the ribbon synapse. If expression of this short isoform is confirmed in humans, it may help explain why we identified no pathogenic NT variants in C2A (Fig. 5B; Liu et al. 2023). The calcium-binding abilities of C2B and C2C, which contain five (1.2%) and 24 (5.9%) NT alleles, respectively, in our dataset, are debated (Johnson and Chapman 2010; Pangršič et al. 2012; Padmanarayana et al. 2014; Meese et al. 2017; Vona et al. 2020). However, there is consensus that C2D, C2E, and C2F, which contain 26 (6.3%), 27 (6.6%), and 74 (18%) NT alleles, respectively, in our dataset, bind calcium and are functionally important (Vona et al. 2020). Sequence variants in C2E, including p.I1573T, are notable for being associated predominately with atypical phenotypes (n = 17 of 27 C2E alleles, or 63%; Fig. 5B, C). Interestingly, the putative C2 domain, C2de, was the third-most common location for NT variants to be reported with 45 (11%) alleles in our dataset, lending support for its hypothesized functionality (Pangršič et al. 2012; Vona et al. 2020; Fig. 5B). Additionally, the number of times NT variants occurring between C2F and the transmembrane domain were reported (n = 143), and the proportion of those alleles associated with typical severe-to-profound phenotypes (n = 135; 94%), suggest that conservation of this region is critical for otoferlin functionality (Fig. 5B).
In addition to the ten alleles associated with temperature-sensitive phenotypes described in the results section, one additional variant, p.I515T, was identified in two siblings with temperature-sensitive DFNB9 phenotypes (Varga et al. 2006). However, these cases were not included in our analyses because biallelic sequence variations could not be identified. In other reports, p.I515T has been associated with a typical phenotype (Leal et al. 1998; Mirghomizadeh et al. 2002). Varga et al. (2006) also reported identifying the p.R1157Q variant in their cohort, though they classified it as a benign polymorphism and did not specify in which subject(s) it was found. Later, Marlin et al. (2010) identified three siblings with temperature-sensitive phenotypes homozygous for both p.R1157Q and p.E1804del. The p.R1157Q variant has been identified in one other patient, though its pathogenicity remains uncertain (Dallol et al. 2016). Consequently, the link between temperature-sensitive phenotypes and the p.I515T, p.R1157Q, and p.E1804del variants is not completely clear. However, the association between temperature-sensitive phenotypes and the eight other variants (p.G541S, p.G614E, p.R698T, p.R1080P, p.R1607W, p.P1628T, p.E1661K, p.E1700Q) described in the results is more certain.
Otoacoustic emissions
Over 80% of cases with OAE data had present, reduced, or present-then-absent OAEs (Fig. 6A), suggesting that a portion of OHCs are functional in a majority of patients with otoferlin-related SNHL, at least in early childhood. When stratified by age, our dataset suggests that OAEs, and thus OHC functionality, are less common in older individuals and may be lost with age. OAEs were absent in only 13% (n = 17 of 136) of children 10 years of age or younger, but they were absent in 40% (n = 16 of 40) of individuals over 10 (Fig. 6B). Although the proportion of cases with OAEs was fairly constant among age bins under 10 years in our dataset, two longitudinal studies have reported OAE deterioration in young children. Kitao et al. (2019) described 10 patients (not included in our dataset due to a lack of case-specific data) with biallelic OTOF variants and showed that OAEs rapidly deteriorated in all patients during the first few years of life. Similarly, Thorpe et al. (2022) conducted serial OAE testing in four patients between one and five years of age and found that OAEs deteriorated in five of eight ears during that time.
OAEs were more likely to be absent in cases with typical phenotypes (n = 35 of 154, or 23%) than in cases with atypical phenotypes (n = 4 of 39, or 10%), and none of the mild (n = 10) or temperature-sensitive (n = 13) cases had absent OAEs (Fig. 6C). Additionally, T/T cases were more than 2.5 times more likely to have absent OAEs than NT/NT or NT/T cases (Fig. 6D). Our analyses stratifying OAE data by age and either phenotype (Fig. 7A–D) or genotype (Fig. 7E–G) must be interpreted cautiously due to small sample sizes in many categories and methodological variability in how sample populations were selected between publications. Nevertheless, the data suggest that NT/NT cases with OAEs tend to be older (median age five years) than NT/T or T/T cases with OAEs (median age three years for both), that T/T cases with OAEs tend to be younger (median age three years) than those without OAEs (median age 13 years), and that profound cases with OAEs tend to be younger (median age four years) than those without OAEs (median age seven years). All these inferences support the hypotheses that OAEs may be lost over time, and that OAEs are more likely to be lost in cases with more severe genotypes and phenotypes. However, a wide range of ages, often spanning from less than one to more than forty years old, were represented among patients both with and without OAEs in most genotype and phenotype categories. This suggests that other variables, including environmental factors like noise exposure, may contribute to OAE loss in otoferlin deficiency. Elucidating the precise mechanisms driving OAE loss is a crucial objective for future research. Prospective studies with serial OAE testing would be particularly informative.
Clinical implications
Given the genotype–phenotype associations described above, an individual’s specific genotype can inform clinical guidance. Even if a patient’s sequence variations are novel, some insight may be gleaned from both the severity (NT vs. T) of the variant and the region of the protein it affects. For instance, a child born with mild SNHL and NT variants in the C2E domain might have a stable phenotype, whereas mild SNHL in a child with NT variants between C2E and C2F may be more likely to progress (Fig. 5B).
It is also important to recognize that different clinical techniques can yield divergent results. For instance, 30 of 154 (19%) cases with absent ABR at maximum stimulation displayed atypical phenotypes as measured by audiometric hearing thresholds, including seven (4.5%) with mild, eight (5.2%) with moderate, and eight (5.2%) with temperature-sensitive hearing loss (Fig. 2E). This discrepancy between ABR and tone detection thresholds likely occurs because OTOF variants can disrupt temporal dynamics of vesicular exocytosis necessary for synchronous postsynaptic neural activity (measured by ABR) while still allowing enough dysregulated exocytosis for sound detection (measured by audiometric hearing thresholds; Pangršič et al. 2010; Santarelli et al. 2015, 2021; Strenzke et al. 2016; Vona et al. 2020; Liu et al. 2023). Neural synchrony is necessary for encoding auditory signals with high temporal fidelity. Consequently, patients with mild or moderate tone detection thresholds and absent ABRs may have more severe functional deficits (e.g., speech perception) than suggested by behavioral audiometry (Santarelli et al. 2013).
Similarly, NBHS relies on either OAE testing or more time-intensive ABR testing. Unfortunately, the majority of babies born with OTOF-related SNHL or other forms of ANSD pass OAE-based NBHS, leading to delayed diagnoses and interventions (Fig. 8; Kim et al. 2018; Iwasa et al. 2022). To reliably detect ANSD, NBHS must include ABR testing (Fig. 8; Rouillon et al. 2006; Vona et al. 2020). Early detection of hearing loss enables early intervention (e.g., before two years of age) with medical devices during critical periods of brain plasticity and language acquisition, which improves outcomes (e.g., language skills development) for children with congenital SNHL (Nicholas and Geers 2007; Ching et al. 2017; Kral et al. 2019; Azaiez et al. 1993). Consequently, leaders in the field have called for inclusion of ABR in newborn screening and early genetic testing of all children with SNHL (Shearer and Smith 2015; Shearer et al. 2019; Thorpe and Smith 2020; Vona et al. 2020).
Hearing aids are the first-line intervention for children diagnosed with OTOF-related SNHL (Azaiez et al. 1993). However, the 32 cases with hearing aid outcomes reported in the literature suggest that patients with otoferlin-related SNHL do not receive meaningful benefit from so-called “hearing aid trials,” which are currently required for cochlear implant eligibility in all types of congenital SNHL, regardless of etiologic diagnosis (Table 3). In one report, although eight patients had improvements in pure-tone hearing thresholds, their aided thresholds remained “above the intensity range of conversational speech” and all eight went on to receive cochlear implants (Santarelli et al. 2015). Notably, it has been hypothesized that OTOF variants might render OHCs more susceptible to noise-induced damage, and that hearing aids may thereby accelerate the loss of OAEs through constant exposure to high intensity stimuli in an inner ear with normal cochlear amplification mechanisms (Rouillon et al. 2006; Kitao et al. 2019; Vona et al. 2020).
Unlike hearing aids, cochlear implants provided speech perception benefit in nearly all 100 cases reported in our dataset (Table 4). This is consistent with the conclusions of a recent review of cochlear implant outcomes in patients with biallelic OTOF variants (Zheng and Liu 2020). Given the reliable benefit of cochlear implants in these patients, early genetic screening may facilitate prompt cochlear implantation and better long-term outcomes (Shearer and Hansen 2019; Azaiez et al. 1993; Carlson et al. 2023). Furthermore, the stark contrast between hearing aid and cochlear implant outcomes warrants further investigation with prospective studies and a re-evaluation of the existing evidence to support hearing aid trials in children with genetically confirmed otoferlin deficiency.
Due to intact cochlear and neural anatomy in most patients with otoferlin-related SNHL, experimental gene therapies that restore functional otoferlin to IHCs might provide an opportunity for instating the use of natural cochlear processes, physiologic hearing, and potentially superior outcomes compared to management with medical devices (Vona et al. 2020). This approach has proved promising in preclinical studies utilizing animal models of otoferlin deficiency (e.g., otoferlin-knockout mice; Akil et al. 2019; Al-Moyed et al. 2019). Like interventions with medical devices, future gene therapy interventions are also likely to be most effective if initiated at an early age, but not only to coincide with critical periods of brain plasticity and language development. The progressive deterioration of OHC responses (i.e., OAEs) is a key feature of the natural history of otoferlin deficiency (Kitao et al. 2019; Thorpe et al. 2022), and it remains unknown the degree to which OHC and OAE preservation will affect the efficacy of gene therapy. Similarly, it is unclear whether cochlear damage from the surgical implantation of electrodes will preclude the use of gene therapy in ears that previously underwent cochlear implantation (Carlson et al. 2011; Bas et al. 2012). These questions will be important to investigate in the event that gene therapies are shown to be effective treatments for congenital SNHL.
Limitations and recommendations for future research
Our analyses, and the literature on which they are based, have several important limitations, which raise considerations for future research. The majority of the literature on otoferlin-related SNHL is comprised of case reports or cross-sectional studies with small sample sizes limited by the frequency of OTOF variants in a given population. Although challenging to conduct, additional prospective studies with longitudinal data would be valuable for further elucidating the natural history of this disorder. Additionally, most studies were conducted in specific geographic regions (e.g., Japan, Spain), which may differ in clinical practices and OTOF allele frequencies. Consequently, foremost among the potential confounds of our analyses is the disproportionate influence of a small number of papers with large sample sizes derived from populations that may not be representative of the global population. For example, 55 of the 422 cases in our dataset came from 13 consanguineous Pakistani families described in Choi et al. (2009). Additionally, cases with atypical phenotypes—particularly temperature-sensitive and progressive cases—may be more likely to be reported in the literature than cases with typical phenotypes due to their novelty, which could result in these phenotypes being overrepresented in our dataset.
Our analyses were complicated by a large amount of methodological variability between studies and, in some instances, unclear descriptions of methodological details. Studies differed in their clinical techniques (e.g., free field vs. ear-specific air and bone conduction), phenotypic bins (e.g., hearing threshold cutoffs), diagnostic criteria (e.g., definitions of present vs. reduced OAEs), inclusion/exclusion criteria, and genetic sequencing methodologies. For instance, some cases were described as having hearing thresholds “greater than” a certain value (e.g., ≥ 80 dB); as we used the lower limit (e.g., 80 dB) to assign a phenotype to these cases, our analysis may overestimate the number of severe cases and underestimate the number of profound cases. Methodological variability is inevitable, but detailed reporting of methodological techniques and case-specific data (rather than group data only) ensures transparency, facilitates accurate interpretation, and enables subsequent quantitative reviews and meta-analyses.
Moving forward, one open question is how, if at all, OTOF sequence variants affect the vestibular system. Otoferlin is expressed in vestibular hair cells and there are limited reports of patients with both OTOF variants and vestibular dysfunction, but no causal relationship has been established (Varga 2003; Varga et al. 2006; Iwasa et al. 2019; Gao et al. 2021). It is also notable that we documented 42 patients heterozygous for a single OTOF variant and with phenotypes compatible with otoferlin-related SNHL (Rodríguez-Ballesteros et al. 2003; Varga et al. 2003, 2006; Romanos et al. 2009; Chiu et al. 2010; Wang et al. 2010, 2011; Matsunaga et al. 2012; Iwasa et al. 2013), in addition to 14 heterozygous patients with late-onset, unilateral hearing loss that the authors suspected may have been related to otoferlin deficiency (Gao et al. 2021). This observation raises the possibility that some molecular detection methods, such as those commonly employed in older publications, may not reliably detect sequence variants in OTOF. Therefore, it may be worth re-sequencing these ostensibly heterozygous patients using more sensitive next-generation sequencing techniques. Alternatively, there may be more unidentified exons (Ranum et al. 2019) or isoforms (Liu et al. 2023), or heterozygosity might cause a subclinical reduction in otoferlin levels (Pangršič et al. 2010) that leaves individuals more susceptible to other environmental or genetic insults. Additionally, the complexity of otoferlin and the rarity of cases with biallelic OTOF variants make it difficult to discern the pathogenicity of certain variants, particularly those with only one documented occurrence. Given the rarity, genetic heterogeneity, and phenotypic spectrum of otoferlin-related SNHL, every case described in the literature and each variant (regardless of pathogenicity) reported to online databases is valuable. Through the collective effort of researchers and clinicians around the world, OTOF-related hearing loss will continue to be better understood and more effectively treated in the future.
Acknowledgements
The authors would like to thank Dr. Lars Becker and Paul Sommer for their constructive feedback on this manuscript.
Funding
This work was supported by Decibel Therapeutics.
Data availability
All data relevant to the results and conclusions of this study were derived from published literature as referenced within the article or described in the methods.
Declarations
Conflict of interest
CLF is a consultant for Decibel Therapeutics. WJR, TQ, OPK, JPW, and VV are employees of, and hold equity in, Decibel Therapeutics.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Ahmed A, Wang M, Khan R, Shah AA, Guo H, Malik S, Xia K, Hu Z. A splice-site variant (c.3289–1G>T) in OTOF underlies profound hearing loss in a Pakistani kindred. BMC Med Genom. 2021;14:2. doi: 10.1186/s12920-020-00859-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akil O, Dyka F, Calvet C, Emptoz A, Lahlou G, Nouaille S, Boutet de Monvel J, Hardelin JP, Hauswirth WW, Avan P, Petit C, Safieddine S, Lustig LR. Dual AAV-mediated gene therapy restores hearing in a DFNB9 mouse model. Proc Natl Acad Sci USA. 2019;116:4496–4501. doi: 10.1073/pnas.1817537116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkowari MK, Vozzi D, Bhagat S, Krishnamoorthy N, Morgan A, Hayder Y, Logendra B, Najjar N, Gandin I, Gasparini P, Badii R, Girotto G, Abdulhadi K. Targeted sequencing identifies novel variants involved in autosomal recessive hereditary hearing loss in Qatari families. Mutat Res. 2017;800–802:29–36. doi: 10.1016/j.mrfmmm.2017.05.001. [DOI] [PubMed] [Google Scholar]
- Almontashiri NAM, Alswaid A, Oza A, Al-Mazrou KA, Elrehim O, Tayoun AA, Rehm HL, Amr SS. Recurrent variants in OTOF are significant contributors to prelingual nonsydromic hearing loss in Saudi patients. Genet Med. 2018;20:536–544. doi: 10.1038/gim.2017.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Moyed H, Cepeda AP, Jung S, Moser T, Kügler S, Reisinger E. A dual-AAV approach restores fast exocytosis and partially rescues auditory function in deaf otoferlin knock-out mice. EMBO Mol Med. 2019 doi: 10.15252/emmm.201809396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammar-Khodja F, Bonnet C, Dahmani M, Ouhab S, Lefèvre GM, Ibrahim H, Hardelin JP, Weil D, Louha M, Petit C. Diversity of the causal genes in hearing impaired Algerian individuals identified by whole exome sequencing. Mol Genet Genom Med. 2015;3:189–196. doi: 10.1002/mgg3.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azaiez H, Thorpe RK, Smith RJH (1993 (updated 2021)) OTOF-related deafness. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A (eds) GeneReviews(®). University of Washington, Seattle [PubMed]
- Bai X, Nian S, Feng L, Ruan Q, Luo X, Wu M, Yan Z. Identification of novel variants in MYO15A, OTOF, and RDX with hearing loss by next-generation sequencing. Mol Genet Genom Med. 2019;7:e808. doi: 10.1002/mgg3.808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bas E, Dinh CT, Garnham C, Polak M, Van de Water TR. Conservation of hearing and protection of hair cells in cochlear implant patients' with residual hearing. Anat Rec. 2012;295:1909–1927. doi: 10.1002/ar.22574. [DOI] [PubMed] [Google Scholar]
- Baux D, Vaché C, Blanchet C, Willems M, Baudoin C, Moclyn M, Faugère V, Touraine R, Isidor B, Dupin-Deguine D, Nizon M, Vincent M, Mercier S, Calais C, García-García G, Azher Z, Lambert L, Perdomo-Trujillo Y, Giuliano F, Claustres M, Koenig M, Mondain M, Roux AF. Combined genetic approaches yield a 48% diagnostic rate in a large cohort of French hearing-impaired patients. Sci Rep. 2017;7:16783. doi: 10.1038/s41598-017-16846-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutner D, Voets T, Neher E, Moser T. Calcium dependence of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse. Neuron. 2001;29:681–690. doi: 10.1016/s0896-6273(01)00243-4. [DOI] [PubMed] [Google Scholar]
- Bitarafan F, Seyedena SY, Mahmoudi M, Garshasbi M. Identification of novel variants in Iranian consanguineous pedigrees with nonsyndromic hearing loss by next-generation sequencing. J Clin Lab Anal. 2020;34:e23544. doi: 10.1002/jcla.23544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson ML, Driscoll CL, Gifford RH, Service GJ, Tombers NM, Hughes-Borst BJ, Neff BA, Beatty CW. Implications of minimizing trauma during conventional cochlear implantation. Otol Neurotol. 2011;32:962–968. doi: 10.1097/MAO.0b013e3182204526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson RJ, Walsh T, Mandell JB, Aburayyan A, Lee MK, Gulsuner S, Horn DL, Ou HC, Sie KCY, Mancl L, Rubinstein J, King M-C. Association of genetic diagnoses of childhood-onset hearing loss with cochlear implant outcomes. JAMA Otolaryngol Head Neck Surg. 2023 doi: 10.1001/jamaoto.2022.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching TYC, Dillon H, Button L, Seeto M, Van Buynder P, Marnane V, Cupples L, Leigh G. Age at intervention for permanent hearing loss and 5-year language outcomes. Pediatrics. 2017 doi: 10.1542/peds.2016-4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YH, Wu CC, Lu YC, Chen PJ, Lee WY, Liu AY, Hsu CJ. Mutations in the OTOF gene in Taiwanese patients with auditory neuropathy. Audiol Neurootol. 2010;15:364–374. doi: 10.1159/000293992. [DOI] [PubMed] [Google Scholar]
- Choi BY, Ahmed ZM, Riazuddin S, Bhinder MA, Shahzad M, Husnain T, Riazuddin S, Griffith AJ, Friedman TB. Identities and frequencies of mutations of the otoferlin gene (OTOF) causing DFNB9 deafness in Pakistan. Clin Genet. 2009;75:237–243. doi: 10.1111/j.1399-0004.2008.01128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churbanov AY, Karafet TM, Morozov IV, Mikhalskaia VY, Zytsar MV, Bondar AA, Posukh OL. Whole exome sequencing reveals homozygous mutations in RAI1, OTOF, and SLC26A4 genes associated with nonsyndromic hearing loss in Altaian families (South Siberia) PLoS One. 2016;11:e0153841. doi: 10.1371/journal.pone.0153841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark JG. Uses and abuses of hearing loss classification. ASHA. 1981;23:493–500. [PubMed] [Google Scholar]
- Costa NT, Martinho-Carvalho AC, Cunha MC, Lewis DR. Auditory and communicative abilities in the auditory neuropathy spectrum disorder and mutation in the Otoferlin gene: clinical cases study. J Soc Bras Fonoaudiol. 2012;24:181–187. doi: 10.1590/s2179-64912012000200016. [DOI] [PubMed] [Google Scholar]
- Dallol A, Daghistani K, Elaimi A, Al-Wazani WA, Bamanie A, Safiah M, Sagaty S, Taha L, Zahed R, Bajouh O, Chaudhary AG, Gari MA, Turki R, Al-Qahtani MH, Abuzenadah AM. Utilization of amplicon-based targeted sequencing panel for the massively parallel sequencing of sporadic hearing impairment patients from Saudi Arabia. BMC Med Genet. 2016;17:67. doi: 10.1186/s12881-016-0329-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Siati RD, Rosenzweig F, Gersdorff G, Gregoire A, Rombaux P, Deggouj N. Auditory neuropathy spectrum disorders: from diagnosis to treatment: literature review and case reports. J Clin Med. 2020 doi: 10.3390/jcm9041074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez MJ, McCord JJ, Sutton RB. Redefining the architecture of ferlin proteins: Insights into multi-domain protein structure and function. PLoS One. 2022;17:e0270188. doi: 10.1371/journal.pone.0270188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman D, Sirmaci A, Cengiz FB, Ozdag H, Tekin M. Screening of 38 genes identifies mutations in 62% of families with nonsyndromic deafness in Turkey. Genet Test Mol Biomark. 2011;15:29–33. doi: 10.1089/gtmb.2010.0120. [DOI] [PubMed] [Google Scholar]
- Fareed M, Sharma V, Singh I, Rehman SU, Singh G, Afzal M. Whole-exome sequencing reveals a rare variant of OTOF gene causing congenital non-syndromic hearing loss among large Muslim families favoring consanguinity. Front Genet. 2021;12:641925. doi: 10.3389/fgene.2021.641925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedick AM, Jalas C, Swaroop A, Smouha EE, Webb BD. Identification of a novel pathogenic OTOF variant causative of nonsyndromic hearing loss with high frequency in the Ashkenazi Jewish population. Appl Clin Genet. 2016;9:141–146. doi: 10.2147/tacg.S113828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo-Terán J, Megía López R, Morales-Angulo C, del Castillo I, Moreno-Pelayo MA, Mazón Gutiérrez A, Moreno Herrero F. Evaluation of a family with sensorineural hearing loss due to the Q829X mutation in the OTOF gene. Acta Otorrinolaringol Esp. 2004;55:120–125. doi: 10.1016/s0001-6519(04)78494-0. [DOI] [PubMed] [Google Scholar]
- Gallo-Terán J, Morales-Angulo C, Sánchez N, Manrique M, Rodríguez-Ballesteros M, Moreno-Pelayo MA, Moreno E, del Castillo I. Auditory neuropathy due to the Q829X mutation in the gene encoding otoferlin (OTOF) in an infant screened for newborn hearing impairment. Acta Otorrinolaringol Esp. 2006;57:333–335. doi: 10.1016/s0001-6519(06)78722-2. [DOI] [PubMed] [Google Scholar]
- Gao Y, Wang HY, Guan J, Lan L, Zhao C, Xie LY, Wang DY, Wang QJ. Genetic susceptibility study of Chinese sudden sensorineural hearing loss patients with vertigo. Curr Med Sci. 2021;41:673–679. doi: 10.1007/s11596-021-2422-2. [DOI] [PubMed] [Google Scholar]
- Harsini FM, Chebrolu S, Fuson KL, White MA, Rice AM, Sutton RB. FerA is a membrane-associating four-helix bundle domain in the ferlin family of membrane-fusion proteins. Sci Rep. 2018;8:10949. doi: 10.1038/s41598-018-29184-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfmann S, Neumann P, Tittmann K, Moser T, Ficner R, Reisinger E. The crystal structure of the C2A domain of otoferlin reveals an unconventional top loop region. J Mol Biol. 2011;406:479–490. doi: 10.1016/j.jmb.2010.12.031. [DOI] [PubMed] [Google Scholar]
- Houseman MJ, Jackson AP, Al-Gazali LI, Badin RA, Roberts E, Mueller RF. A novel mutation in a family with non-syndromic sensorineural hearing loss that disrupts the newly characterised OTOF long isoforms. J Med Genet. 2001;38:E25. doi: 10.1136/jmg.38.8.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchin T, Coy NN, Conlon H, Telford E, Bromelow K, Blaydon D, Taylor G, Coghill E, Brown S, Trembath R, Liu XZ, Bitner-Glindzicz M, Mueller R. Assessment of the genetic causes of recessive childhood non-syndromic deafness in the UK—implications for genetic testing. Clin Genet. 2005;68:506–512. doi: 10.1111/j.1399-0004.2005.00539.x. [DOI] [PubMed] [Google Scholar]
- Iwasa Y, Nishio SY, Yoshimura H, Kanda Y, Kumakawa K, Abe S, Naito Y, Nagai K, Usami S. OTOF mutation screening in Japanese severe to profound recessive hearing loss patients. BMC Med Genet. 2013;14:95. doi: 10.1186/1471-2350-14-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasa YI, Nishio SY, Sugaya A, Kataoka Y, Kanda Y, Taniguchi M, Nagai K, Naito Y, Ikezono T, Horie R, Sakurai Y, Matsuoka R, Takeda H, Abe S, Kihara C, Ishino T, Morita SY, Iwasaki S, Takahashi M, Ito T, Arai Y, Usami SI. OTOF mutation analysis with massively parallel DNA sequencing in 2,265 Japanese sensorineural hearing loss patients. PLoS One. 2019;14:e0215932. doi: 10.1371/journal.pone.0215932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasa YI, Nishio SY, Yoshimura H, Sugaya A, Kataoka Y, Maeda Y, Kanda Y, Nagai K, Naito Y, Yamazaki H, Ikezono T, Matsuda H, Nakai M, Tona R, Sakurai Y, Motegi R, Takeda H, Kobayashi M, Kihara C, Ishino T, Morita SY, Iwasaki S, Takahashi M, Furutate S, Oka SI, Kubota T, Arai Y, Kobayashi Y, Kikuchi D, Shintani T, Ogasawara N, Honkura Y, Izumi S, Hyogo M, Ninoyu Y, Suematsu M, Nakayama J, Tsuchihashi N, Okami M, Sakata H, Yoshihashi H, Kobayashi T, Kumakawa K, Yoshida T, Esaki T, Usami SI. Detailed clinical features and genotype-phenotype correlation in an OTOF-related hearing loss cohort in Japan. Hum Genet. 2022;141:865–875. doi: 10.1007/s00439-021-02351-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin YJ, Park J, Kim AR, Rah YC, Choi BY. Identification of a novel splice site variant of OTOF in the Korean nonsyndromic hearing loss population with low prevalence of the OTOF mutations. Int J Pediatr Otorhinolaryngol. 2014;78:1030–1035. doi: 10.1016/j.ijporl.2014.03.033. [DOI] [PubMed] [Google Scholar]
- Johnson CP, Chapman ER. Otoferlin is a calcium sensor that directly regulates SNARE-mediated membrane fusion. J Cell Biol. 2010;191:187–197. doi: 10.1083/jcb.201002089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BJ, Jang JH, Han JH, Park HR, Oh DY, Lee S, Kim MY, Kim AR, Lee C, Kim NKD, Park WY, Choung YH, Choi BY. Mutational and phenotypic spectrum of OTOF-related auditory neuropathy in Koreans: eliciting reciprocal interaction between bench and clinics. J Transl Med. 2018;16:330. doi: 10.1186/s12967-018-1708-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitao K, Mutai H, Namba K, Morimoto N, Nakano A, Arimoto Y, Sugiuchi T, Masuda S, Okamoto Y, Morita N, Sakamoto H, Shintani T, Fukuda S, Kaga K, Matsunaga T. Deterioration in distortion product otoacoustic emissions in auditory neuropathy patients with distinct clinical and genetic backgrounds. Ear Hear. 2019;40:184–191. doi: 10.1097/aud.0000000000000586. [DOI] [PubMed] [Google Scholar]
- Kral A, Dorman MF, Wilson BS. Neuronal development of hearing and language: cochlear implants and critical periods. Annu Rev Neurosci. 2019;42:47–65. doi: 10.1146/annurev-neuro-080317-061513. [DOI] [PubMed] [Google Scholar]
- Leal SM, Apaydin F, Barnwell C, Iber M, Kandogan T, Pfister M, Braendle U, Cura O, Schwalb M, Zenner HP, Vitale E. A second middle eastern kindred with autosomal recessive non-syndromic hearing loss segregates DFNB9. Eur J Hum Genet. 1998;6:341–344. doi: 10.1038/sj.ejhg.5200201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Oh DY, Han JH, Kim MY, Kim B, Kim BJ, Song JJ, Koo JW, Lee JH, Oh SH, Choi BY. Flexible real-time polymerase chain reaction-based platforms for detecting deafness mutations in Koreans: a proposed guideline for the etiologic diagnosis of auditory neuropathy spectrum disorder. Diagnostics (basel) 2020 doi: 10.3390/diagnostics10090672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CY, Lin PH, Tsai CY, Chiang YT, Chiou HP, Chiang KY, Chen PL, Hsu JS, Liu TC, Wu HP, Wu CC, Hsu CJ. Comprehensive etiologic analyses in pediatric cochlear implantees and the clinical implications. Biomedicines. 2022 doi: 10.3390/biomedicines10081846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Su J, Zhang J, Pei J, Li D, Zhang Y, Li J, Chen M, Zhu B. Targeted next-generation sequencing of deaf patients from Southwestern China. Mol Genet Genom Med. 2021;9:e1660. doi: 10.1002/mgg3.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieu JEC, Kenna M, Anne S, Davidson L. Hearing loss in children: a review. JAMA. 2020;324:2195–2205. doi: 10.1001/jama.2020.17647. [DOI] [PubMed] [Google Scholar]
- Likar T, Hasanhodžić M, Teran N, Maver A, Peterlin B, Writzl K. Diagnostic outcomes of exome sequencing in patients with syndromic or non-syndromic hearing loss. PLoS One. 2018;13:e0188578. doi: 10.1371/journal.pone.0188578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Liu H, Wang L, Song L, Jiang G, Lu Q, Yang T, Peng H, Cai R, Zhao X, Zhao T, Wu H. Cochlear transcript diversity and its role in auditory functions implied by an otoferlin short isoform. Nat Commun. 2023;14:3085. doi: 10.1038/s41467-023-38621-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loundon N, Marcolla A, Roux I, Rouillon I, Denoyelle F, Feldmann D, Marlin S, Garabedian EN. Auditory neuropathy or endocochlear hearing loss? Otol Neurotol. 2005;26:748–754. doi: 10.1097/01.mao.0000169044.63970.4a. [DOI] [PubMed] [Google Scholar]
- Marlin S, Feldmann D, Nguyen Y, Rouillon I, Loundon N, Jonard L, Bonnet C, Couderc R, Garabedian EN, Petit C, Denoyelle F. Temperature-sensitive auditory neuropathy associated with an otoferlin mutation: deafening fever! Biochem Biophys Res Commun. 2010;394:737–742. doi: 10.1016/j.bbrc.2010.03.062. [DOI] [PubMed] [Google Scholar]
- Matsunaga T, Mutai H, Kunishima S, Namba K, Morimoto N, Shinjo Y, Arimoto Y, Kataoka Y, Shintani T, Morita N, Sugiuchi T, Masuda S, Nakano A, Taiji H, Kaga K. A prevalent founder mutation and genotype-phenotype correlations of OTOF in Japanese patients with auditory neuropathy. Clin Genet. 2012;82:425–432. doi: 10.1111/j.1399-0004.2012.01897.x. [DOI] [PubMed] [Google Scholar]
- Meese S, Cepeda AP, Gahlen F, Adams CM, Ficner R, Ricci AJ, Heller S, Reisinger E, Herget M. Activity-dependent phosphorylation by CaMKIIδ alters the Ca2+ affinity of the multi-C2-domain protein otoferlin. Front Synapt Neurosci. 2017 doi: 10.3389/fnsyn.2017.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski N, Goutman JD, Auclair SM, Boutet de Monvel J, Tertrais M, Emptoz A, Parrin A, Nouaille S, Guillon M, Sachse M, Ciric D, Bahloul A, Hardelin JP, Sutton RB, Avan P, Krishnakumar SS, Rothman JE, Dulon D, Safieddine S, Petit C. Otoferlin acts as a Ca(2+) sensor for vesicle fusion and vesicle pool replenishment at auditory hair cell ribbon synapses. Elife. 2017 doi: 10.7554/eLife.31013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirghomizadeh F, Pfister M, Apaydin F, Petit C, Kupka S, Pusch CM, Zenner HP, Blin N. Substitutions in the conserved C2C domain of otoferlin cause DFNB9, a form of nonsyndromic autosomal recessive deafness. Neurobiol Dis. 2002;10:157–164. doi: 10.1006/nbdi.2002.0488. [DOI] [PubMed] [Google Scholar]
- Naz S, Imtiaz A, Mujtaba G, Maqsood A, Bashir R, Bukhari I, Khan MR, Ramzan M, Fatima A, Rehman AU, Iqbal M, Chaudhry T, Lund M, Brewer CC, Morell RJ, Friedman TB. Genetic causes of moderate to severe hearing loss point to modifiers. Clin Genet. 2017;91:589–598. doi: 10.1111/cge.12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann K, Chadha S, Tavartkiladze G, Bu X, White KR. Newborn and infant hearing screening facing globally growing numbers of people suffering from disabling hearing loss. Int J Neonatal Screen. 2019;5:7. doi: 10.3390/ijns5010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas JG, Geers AE. Will they catch up? The role of age at cochlear implantation in the spoken language development of children with severe to profound hearing loss. J Speech Lang Hear Res. 2007;50:1048–1062. doi: 10.1044/1092-4388(2007/073). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noman M, Ishaq R, Bukhari SA, Ahmed ZM, Riazuddin S. Delineation of homozygous variants associated with prelingual sensorineural hearing loss in Pakistani families. Genes (basel) 2019 doi: 10.3390/genes10121031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouvian R, Beutner D, Parsons TD, Moser T. Structure and function of the hair cell ribbon synapse. J Membr Biol. 2006;209:153–165. doi: 10.1007/s00232-005-0854-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanarayana M, Hams N, Speight LC, Petersson EJ, Mehl RA, Johnson CP. Characterization of the lipid binding properties of otoferlin reveals specific interactions between PI(4,5)P2 and the C2C and C2F domains. Biochemistry. 2014;53:5023–5033. doi: 10.1021/bi5004469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey N, Rashid T, Jalvi R, Sharma M, Rangasayee R, Andrabi KI, Anand A. Mutations in OTOF, CLDN14 and SLC26A4 genes as major causes of hearing impairment in Dhadkai village, Jammu and Kashmir, India. Indian J Med Res. 2017;146:489–497. doi: 10.4103/ijmr.IJMR_635_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangršič T, Lasarow L, Reuter K, Takago H, Schwander M, Riedel D, Frank T, Tarantino LM, Bailey JS, Strenzke N, Brose N, Müller U, Reisinger E, Moser T. Hearing requires otoferlin-dependent efficient replenishment of synaptic vesicles in hair cells. Nat Neurosci. 2010;13:869–876. doi: 10.1038/nn.2578. [DOI] [PubMed] [Google Scholar]
- Pangršič T, Reisinger E, Moser T. Otoferlin: a multi-C2 domain protein essential for hearing. Trends Neurosci. 2012;35:671–680. doi: 10.1016/j.tins.2012.08.002. [DOI] [PubMed] [Google Scholar]
- Qiu Y, Chen S, Xie L, Xu K, Lin Y, Bai X, Zhang HM, Liu XZ, Jin Y, Sun Y, Kong WJ. Auditory neuropathy spectrum disorder due to two novel compound heterozygous OTOF mutations in two Chinese families. Neural Plast. 2019;2019:9765276. doi: 10.1155/2019/9765276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranum PT, Goodwin AT, Yoshimura H, Kolbe DL, Walls WD, Koh JY, He DZZ, Smith RJH. Insights into the biology of hearing and deafness revealed by single-cell RNA sequencing. Cell Rep. 2019;26:3160–3171.e3. doi: 10.1016/j.celrep.2019.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Ballesteros M, del Castillo FJ, Martín Y, Moreno-Pelayo MA, Morera C, Prieto F, Marco J, Morant A, Gallo-Terán J, Morales-Angulo C, Navas C, Trinidad G, Tapia MC, Moreno F, del Castillo I. Auditory neuropathy in patients carrying mutations in the otoferlin gene (OTOF) Hum Mutat. 2003;22:451–456. doi: 10.1002/humu.10274. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Ballesteros M, Reynoso R, Olarte M, Villamar M, Morera C, Santarelli R, Arslan E, Medá C, Curet C, Völter C, Sainz-Quevedo M, Castorina P, Ambrosetti U, Berrettini S, Frei K, Tedín S, Smith J, Cruz Tapia M, Cavallé L, Gelvez N, Primignani P, Gómez-Rosas E, Martín M, Moreno-Pelayo MA, Tamayo M, Moreno-Barral J, Moreno F, del Castillo I. A multicenter study on the prevalence and spectrum of mutations in the otoferlin gene (OTOF) in subjects with nonsyndromic hearing impairment and auditory neuropathy. Hum Mutat. 2008;29:823–831. doi: 10.1002/humu.20708. [DOI] [PubMed] [Google Scholar]
- Romanos J, Kimura L, Fávero ML, Izarra FA, de Mello Auricchio MT, Batissoco AC, Lezirovitz K, Abreu-Silva RS, Mingroni-Netto RC. Novel OTOF mutations in Brazilian patients with auditory neuropathy. J Hum Genet. 2009;54:382–385. doi: 10.1038/jhg.2009.45. [DOI] [PubMed] [Google Scholar]
- Rouillon I, Marcolla A, Roux I, Marlin S, Feldmann D, Couderc R, Jonard L, Petit C, Denoyelle F, Garabédian EN, Loundon N. Results of cochlear implantation in two children with mutations in the OTOF gene. Int J Pediatr Otorhinolaryngol. 2006;70:689–696. doi: 10.1016/j.ijporl.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Roux I, Safieddine S, Nouvian R, Grati Mh, Simmler M-C, Bahloul A, Perfettini I, Le Gall M, Rostaing P, Hamard G, Triller A, Avan P, Moser T, Petit C. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell. 2006;127:277–289. doi: 10.1016/j.cell.2006.08.040. [DOI] [PubMed] [Google Scholar]
- Runge CL, Erbe CB, McNally MT, Van Dusen C, Friedland DR, Kwitek AE, Kerschner JE. A novel otoferlin splice-site mutation in siblings with auditory neuropathy spectrum disorder. Audiol Neurootol. 2013;18:374–382. doi: 10.1159/000354978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safka Brozkova D, Poisson Marková S, Mészárosová AU, Jenčík J, Čejnová V, Čada Z, Laštůvková J, Rašková D, Seeman P. Spectrum and frequencies of non GJB2 gene mutations in Czech patients with early non-syndromic hearing loss detected by gene panel NGS and whole-exome sequencing. Clin Genet. 2020;98:548–554. doi: 10.1111/cge.13839. [DOI] [PubMed] [Google Scholar]
- Santarelli R. Information from cochlear potentials and genetic mutations helps localize the lesion site in auditory neuropathy. Genome Med. 2010;2:91. doi: 10.1186/gm212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarelli R, del Castillo I, Rodríguez-Ballesteros M, Scimemi P, Cama E, Arslan E, Starr A. Abnormal cochlear potentials from deaf patients with mutations in the otoferlin gene. J Assoc Res Otolaryngol. 2009;10:545–556. doi: 10.1007/s10162-009-0181-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarelli R, Starr A, del Castillo I, Huang T, Scimemi P, Cama E, Rossi R, Arslan E. Presynaptic and postsynaptic mechanisms underlying auditory neuropathy in patients with mutations in the OTOF or OPA1 gene. Audiol Med. 2011;9:59–66. doi: 10.3109/1651386X.2011.558764. [DOI] [Google Scholar]
- Santarelli R, del Castillo I, Starr A. Auditory neuropathies and electrocochleography. Hear Balance Commun. 2013;11:130–137. doi: 10.3109/21695717.2013.815446. [DOI] [Google Scholar]
- Santarelli R, del Castillo I, Cama E, Scimemi P, Starr A. Audibility, speech perception and processing of temporal cues in ribbon synaptic disorders due to OTOF mutations. Hear Res. 2015;330:200–212. doi: 10.1016/j.heares.2015.07.007. [DOI] [PubMed] [Google Scholar]
- Santarelli R, Scimemi P, Costantini M, Domínguez-Ruiz M, Rodríguez-Ballesteros M, Del Castillo I. Cochlear synaptopathy due to mutations in OTOF gene may result in stable mild hearing loss and severe impairment of speech perception. Ear Hear. 2021;42:1627–1639. doi: 10.1097/aud.0000000000001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahin H, Walsh T, Rayyan AA, Lee MK, Higgins J, Dickel D, Lewis K, Thompson J, Baker C, Nord AS, Stray S, Gurwitz D, Avraham KB, King MC, Kanaan M. Five novel loci for inherited hearing loss mapped by SNP-based homozygosity profiles in Palestinian families. Eur J Hum Genet. 2010;18:407–413. doi: 10.1038/ejhg.2009.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer AE, Hansen MR. Auditory synaptopathy, auditory neuropathy, and cochlear implantation. Laryngoscope Investig Otolaryngol. 2019;4:429–440. doi: 10.1002/lio2.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer AE, Smith RJH. Massively parallel sequencing for genetic diagnosis of hearing loss: the new standard of care. Otolaryngol Head Neck Surg. 2015;153:175–182. doi: 10.1177/0194599815591156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer AE, Hildebrand MS, Smith RJH (1999 (updated 2017)) Hereditary hearing loss and deafness overview. In: Adam ME, Mirzaa GM et al (eds) GeneReviews [Internet]. University of Washington, Seattle
- Shearer AE, Eppsteiner RW, Frees K, Tejani V, Sloan-Heggen CM, Brown C, Abbas P, Dunn C, Hansen MR, Gantz BJ, Smith RJH. Genetic variants in the peripheral auditory system significantly affect adult cochlear implant performance. Hear Res. 2017;348:138–142. doi: 10.1016/j.heares.2017.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer AE, Shen J, Amr S, Morton CC, Smith RJ. A proposal for comprehensive newborn hearing screening to improve identification of deaf and hard-of-hearing children. Genet Med. 2019;21:2614–2630. doi: 10.1038/s41436-019-0563-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souissi A, Ben Said M, Ben Ayed I, Elloumi I, Bouzid A, Mosrati MA, Hasnaoui M, Belcadhi M, Idriss N, Kamoun H, Gharbi N, Gibriel AA, Tlili A, Masmoudi S. Novel pathogenic mutations and further evidence for clinical relevance of genes and variants causing hearing impairment in Tunisian population. J Adv Res. 2021;31:13–24. doi: 10.1016/j.jare.2021.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalmann U, Franke AJ, Al-Moyed H, Strenzke N, Reisinger E. Otoferlin is required for proper synapse maturation and for maintenance of inner and outer hair cells in mouse models for DFNB9. Front Cell Neurosci. 2021;15:677543. doi: 10.3389/fncel.2021.677543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strenzke N, Chakrabarti R, Al-Moyed H, Müller A, Hoch G, Pangršič T, Yamanbaeva G, Lenz C, Pan KT, Auge E, Geiss-Friedlander R, Urlaub H, Brose N, Wichmann C, Reisinger E. Hair cell synaptic dysfunction, auditory fatigue and thermal sensitivity in otoferlin Ile515Thr mutants. EMBO J. 2016;35:2519–2535. doi: 10.15252/embj.201694564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabatabaiefar MA, Pourreza MR, Tahmasebi P, Saki N, Hashemzadeh Chaleshtori M, Salehi R, Mohammadi-Asl J. A novel pathologic variant in OTOF in an Iranian family segregating hereditary hearing loss. Otolaryngol Head Neck Surg. 2018;158:1084–1092. doi: 10.1177/0194599818759007. [DOI] [PubMed] [Google Scholar]
- Tang F, Ma D, Wang Y, Qiu Y, Liu F, Wang Q, Lu Q, Shi M, Xu L, Liu M, Liang J. Novel compound heterozygous mutations in the OTOF gene identified by whole-exome sequencing in auditory neuropathy spectrum disorder. BMC Med Genet. 2017;18:35. doi: 10.1186/s12881-017-0400-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekin M, Akcayoz D, Incesulu A. A novel missense mutation in a C2 domain of OTOF results in autosomal recessive auditory neuropathy. Am J Med Genet A. 2005;138:6–10. doi: 10.1002/ajmg.a.30907. [DOI] [PubMed] [Google Scholar]
- Thorpe RK, Smith RJH. Future directions for screening and treatment in congenital hearing loss. Precis Clin Med. 2020;3:175–186. doi: 10.1093/pcmedi/pbaa025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorpe RK, Azaiez H, Wu P, Wang Q, Xu L, Dai P, Yang T, Schaefer GB, Peters BR, Chan KH, Schatz KS, Bodurtha J, Robin NH, Hirsch Y, Rahbeeni ZA, Yuan H, Smith RJH. The natural history of OTOF-related auditory neuropathy spectrum disorders: a multicenter study. Hum Genet. 2022;141:853–863. doi: 10.1007/s00439-021-02340-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga R (2003) Clinical and molecular characterization of auditory neuropathy. Dissertation, Creighton University
- Varga R, Kelley PM, Keats BJ, Starr A, Leal SM, Cohn E, Kimberling WJ. Non-syndromic recessive auditory neuropathy is the result of mutations in the otoferlin (OTOF) gene. J Med Genet. 2003;40:45–50. doi: 10.1136/jmg.40.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga R, Avenarius MR, Kelley PM, Keats BJ, Berlin CI, Hood LJ, Morlet TG, Brashears SM, Starr A, Cohn ES, Smith RJ, Kimberling WJ. OTOF mutations revealed by genetic analysis of hearing loss families including a potential temperature sensitive auditory neuropathy allele. J Med Genet. 2006;43:576–581. doi: 10.1136/jmg.2005.038612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogl C, Cooper BH, Neef J, Wojcik SM, Reim K, Reisinger E, Brose N, Rhee JS, Moser T, Wichmann C. Unconventional molecular regulation of synaptic vesicle replenishment in cochlear inner hair cells. J Cell Sci. 2015;128:638–644. doi: 10.1242/jcs.162099. [DOI] [PubMed] [Google Scholar]
- Vona B, Rad A, Reisinger E. The Many faces of DFNB9: relating OTOF variants to hearing impairment. Genes. 2020;11:1411. doi: 10.3390/genes11121411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DY, Wang YC, Weil D, Zhao YL, Rao SQ, Zong L, Ji YB, Liu Q, Li JQ, Yang HM, Shen Y, Benedict-Alderfer C, Zheng QY, Petit C, Wang QJ. Screening mutations of OTOF gene in Chinese patients with auditory neuropathy, including a familial case of temperature-sensitive auditory neuropathy. BMC Med Genet. 2010;11:79. doi: 10.1186/1471-2350-11-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Fan YY, Wang SJ, Liang PF, Wang JL, Qiu JH. Variants of OTOF and PJVK genes in Chinese patients with auditory neuropathy spectrum disorder. PLoS One. 2011;6:e24000. doi: 10.1371/journal.pone.0024000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lu Y, Cheng J, Zhang L, Han D, Yuan H. Novel OTOF gene mutations identified using a massively parallel DNA sequencing technique in DFNB9 deafness. Acta Otolaryngol. 2018;138:865–870. doi: 10.1080/00016489.2018.1476777. [DOI] [PubMed] [Google Scholar]
- Wu CC, Hsu CJ, Huang FL, Lin YH, Lin YH, Liu TC, Wu CM. Timing of cochlear implantation in auditory neuropathy patients with OTOF mutations: our experience with 10 patients. Clin Otolaryngol. 2018;43:352–357. doi: 10.1111/coa.12949. [DOI] [PubMed] [Google Scholar]
- Wu CC, Tsai CY, Lin YH, Chen PY, Lin PH, Cheng YF, Wu CM, Lin YH, Lee CY, Erdenechuluun J, Liu TC, Chen PL, Hsu CJ. Genetic epidemiology and clinical features of hereditary hearing impairment in the Taiwanese population. Genes (basel) 2019 doi: 10.3390/genes10100772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Huang W, Xu Z, Li S, Zhang J, Chen X, Tang Y, Qiu J, Wang Z, Duan X, Zhang L. Clinical and genetic study of 12 Chinese Han families with nonsyndromic deafness. Mol Genet Genom Med. 2020;8:e1177. doi: 10.1002/mgg3.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynne DP, Zeng F-G, Bhatt S, Michalewski HJ, Dimitrijevic A, Starr A. Loudness adaptation accompanying ribbon synapse and auditory nerve disorders. Brain. 2013;136:1626–1638. doi: 10.1093/brain/awt056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H, Huang X, Xu H, Guo Y, Hu P, Deng X, Yang Z, Liu A, Deng H. An OTOF frameshift variant associated with auditory neuropathy spectrum disorder. Curr Genom. 2018;19:370–374. doi: 10.2174/1389202919666171113152951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang YB, Xu CY, Xu YZ, Li HZ, Zhou LL, Xu XQ, Chen ZH, Tang SH. Next-generation sequencing identifies rare pathogenic and novel candidate variants in a cohort of Chinese patients with syndromic or nonsyndromic hearing loss. Mol Genet Genom Med. 2020;8:e1539. doi: 10.1002/mgg3.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasunaga S, Grati MH, Chardenoux S, Smith TN, Friedman TB, Lalwani AK, Wilcox ER, Petit C. OTOF encodes multiple long and short isoforms: genetic evidence that the long ones underlie recessive deafness DFNB9. Am J Hum Genet. 2000;67:591–600. doi: 10.1086/303049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildirim-Baylan M, Bademci G, Duman D, Ozturkmen-Akay H, Tokgoz-Yilmaz S, Tekin M. Evidence for genotype–phenotype correlation for OTOF mutations. Int J Pediatr Otorhinolaryngol. 2014;78:950–953. doi: 10.1016/j.ijporl.2014.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadro C, Ciorba A, Fabris A, Morgutti M, Trevisi P, Gasparini P, Martini A. Five new OTOF gene mutations and auditory neuropathy. Int J Pediatr Otorhinolaryngol. 2010;74:494–498. doi: 10.1016/j.ijporl.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Zhang LP, Chai YC, Yang T, Wu H. Identification of novel OTOF compound heterozygous mutations by targeted next-generation sequencing in a Chinese patient with auditory neuropathy spectrum disorder. Int J Pediatr Otorhinolaryngol. 2013;77:1749–1752. doi: 10.1016/j.ijporl.2013.08.007. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Lan L, Shi W, Yu L, Xie LY, Xiong F, Zhao C, Li N, Yin Z, Zong L, Guan J, Wang D, Sun W, Wang Q. Temperature sensitive auditory neuropathy. Hear Res. 2016;335:53–63. doi: 10.1016/j.heares.2016.01.008. [DOI] [PubMed] [Google Scholar]
- Zhang QJ, Han B, Lan L, Zong L, Shi W, Wang HY, Xie LY, Wang H, Zhao C, Zhang C, Yin ZF, Wang DY, Petit C, Guan J, Wang QJ. High frequency of OTOF mutations in Chinese infants with congenital auditory neuropathy spectrum disorder. Clin Genet. 2016;90:238–246. doi: 10.1111/cge.12744. [DOI] [PubMed] [Google Scholar]
- Zheng D, Liu X. Cochlear implantation outcomes in patients with OTOF mutations. Front Neurosci. 2020;14:447. doi: 10.3389/fnins.2020.00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YM, Li Q, Gao X, Li YF, Liu YL, Dai P, Li XP. Familial temperature-sensitive auditory neuropathy: distinctive clinical courses caused by variants of the OTOF gene. Front Cell Dev Biol. 2021;9:732930. doi: 10.3389/fcell.2021.732930. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data relevant to the results and conclusions of this study were derived from published literature as referenced within the article or described in the methods.