

Summary
Bioorthogonal labeling and click chemistry techniques allow the detailed examination of cellular physiology through tagging and visualizing newly synthesized proteins. Here, we describe three methods applying bioorthogonal non-canonical amino acid tagging and fluorescent non-canonical amino acid tagging to quantify protein synthesis in microglia. We describe steps for cell seeding and labeling. We then detail microscopy, flow cytometry, and Western blotting techniques. These methods can be easily adapted for other cell types to explore cellular physiology in health and disease.
For complete details on the use and execution of this protocol, please refer to Evans et al. (2021).1
Subject areas: Cell Culture, Flow Cytometry/Mass Cytometry, Cell-based Assays, Microscopy, Neuroscience, Protein Biochemistry, Protein Expression and Purification
Graphical abstract
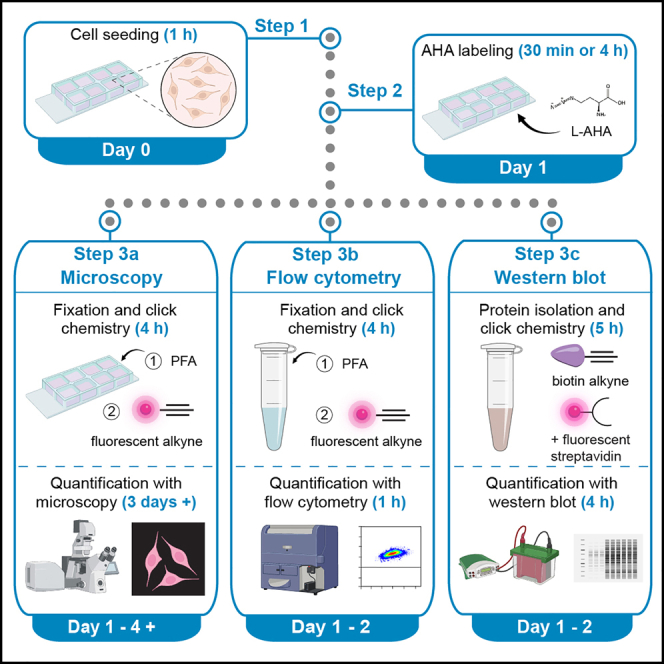
Highlights
-
•
In vitro bioorthogonal labeling of newly synthesized proteins with L-azidohomoalanine
-
•
Detection of newly synthesized proteins via copper (I)-catalyzed click chemistry
-
•
FUNCAT quantification of newly synthesized proteins using three complementary methods
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Bioorthogonal labeling and click chemistry techniques allow the detailed examination of cellular physiology through tagging and visualizing newly synthesized proteins. Here, we describe three methods applying bioorthogonal non-canonical amino acid tagging and fluorescent non-canonical amino acid tagging to quantify protein synthesis in microglia. We describe steps for cell seeding and labeling. We then detail microscopy, flow cytometry, and Western blotting techniques. These methods can be easily adapted for other cell types to explore cellular physiology in health and disease.
Before you begin
In the following protocol, we use BV2 murine microglial cells in combination with bioorthogonal non-canonical amino acid tagging (BONCAT) to illustrate the specific steps for detecting and quantifying de novo protein synthesis in vitro. The BV2 line has been generated by infecting murine primary microglial cell cultures with a v-raf/v-myc oncogene for immortalization2 and since then, BV2 cells were used extensively as an in vitro model to study the physiology of the microglia. However, the protocol presented here can be easily applied to other in vitro cell culture models by adjusting labeling times, as required.
To tag specifically the nascent proteins in cells, we use the non-canonical methionine analog L-azidohomoalanine (AHA), which is efficiently incorporated into proteins by the endogenous cellular translation machinery (Figure 1A).3,4,5,6 Next, fluorescent non-canonical amino acid tagging (FUNCAT) is achieved through a copper (I)-catalyzed click chemistry reaction, in which the azide tag (N3) present on AHA is bonded covalently through a click chemistry reaction to alkynes containing either fluorescent or biotin residues (Figure 1B). The signal from these AHA-labeled fluorescent proteins can then be visualized and quantified using microscopy or flow cytometry. Alternatively, the biotinylated newly synthesized proteins can be visualized on Western blot membranes incubated with fluorescently labeled streptavidin.
Figure 1.

BONCAT bioorthogonal labeling of newly synthesized proteins with L-azidohomoalanine (AHA) and detection via FUNCAT copper (I)-catalyzed click chemistry
(A) The principle of BONCAT (bioorthogonal non-canonical amino acid tagging) methodology: the non-canonical amino acid AHA is charged to methionyl-tRNA (tRNA(Met)) by methionine tRNA synthetase (MetRS) and transported to the ribosome where it is incorporated into proteins during translation.
(B) For FUNCAT (fluorescent non-canonical amino acid tagging)-based detection, a copper (I)-catalyzed click chemistry reaction is performed to bond the azides (N3) on the AHA-labeled newly synthesized proteins to either fluorophore- or biotinyl-tagged alkynes, allowing the subsequent visualization of newly synthesized proteins.
Before starting, prepare the necessary buffers and stock solutions.
BV2 cell culture media preparation
Timing: 1 h
CRITICAL: On the day prior to preparing cell culture media, incubate frozen FBS at room temperature (20°C–25°C) or 4°C to thaw for 16–24 h.
-
1.Heat-inactivate Fetal Bovine Serum (FBS).
-
a.Heat water bath to 56°C.
-
b.Place thawed FBS bottle into the 56°C water and gently swirl to mix FBS every 10 min for a total time of 30 min. Separate into 25 mL aliquots and store at −20°C until use.
-
a.
-
2.Prepare 250 mL of cell culture medium.
-
a.Mix cell culture medium reagents and sterilize the medium through a 0.22 μm pore size filter unit. Store at 4°C.
-
a.
BV2 cell culture maintenance
-
3.Thaw and maintain BV2 cells in a 37°C incubator, with 5% CO2.
-
a.Once thawed, BV2 cells will be ready to use (80% confluency) in 1–3 days.
-
b.BV2 cells require passaging every 3–5 days, depending on when they reach 80% confluency.
-
a.
Note: To avoid changes in cellular phenotype, BV2 should not be used for more than 30 passages.
Preparation of PDL/borate buffer coating solution
-
4.Prepare 20 mg/mL Poly-D-lysine (PDL) stock.
-
a.Mix 100 mg PDL into 5 mL Milli-Q water. Sterilize through a 0.22 μM filter unit. Make 250 μL aliquots and store at −20°C.
-
a.
-
5.Prepare borate buffer.
-
a.Mix boric acid and sodium tetraborate (Borax) in 800 mL Milli-Q water.
-
b.Adjust pH to 8.4 using sodium hydroxide (NaOH 1 M) and/or hydrochloric acid (HCl 1 M) as needed and added dropwise under constant agitation to ensure thorough mixing or the solution during pH measurement.
-
c.Add Milli-Q water up to 1 L.
-
d.Sterilize through a 0.22 μM filter unit and store at 4°C.
-
a.
Preparation of labeling stock solutions
-
6.Prepare 0.4 M stock solution of AHA.
-
a.Reconstitute powdered AHA in Milli-Q water to 0.4 M (72.236 mg/mL) and vortex until dissolved. Sterilize through a 0.22 μM filter, aliquot and store at −20°C for up to 3 months.
-
a.
-
7.Prepare 0.4 M stock solution of methionine.
-
a.Reconstitute powdered methionine in Milli-Q water to 0.4 M (59.648 mg/mL) and vortex until dissolved. Sterilize through a 0.22 μM filter, aliquot and store at −20°C for up to 3 months.
-
a.
-
8.Prepare 10 mM stock solution of anisomycin.
-
a.Reconstitute anisomycin in Milli-Q water to 10 mM (2.658 mg/mL). Sterilize through a 0.22 μM filter, aliquot and store at −20°C for up to 3 months.
-
a.
Note: Methionine and/or AHA supplemented with anisomycin (protein synthesis inhibitor) are used as controls to ensure successful bioorthogonal labeling and click chemistry. Other protein synthesis inhibitors such as cycloheximide can also be used.
Preparation of click chemistry reagents
-
9.For microscopy and flow cytometry experiments:
-
a.Alexa Fluor 647 alkyne: reconstitute Alexa Fluor 647 alkyne in DMSO to 1 mM (0.8 mg/mL). Aliquot and store at −20°C.Note: We used Alexa Fluor 647 alkyne, however, other fluorescent alkynes can also be used.
-
b.Prepare the Invitrogen Click-iT® Cell Reaction Buffer Kit reagents as per kit instructions.
-
i.Component A: Reaction Buffer. Dilute 1:10 in Milli-Q water. Store at 4°C.
-
ii.Component C: The entire Reaction Additive. Reconstitute with 4 mL Milli-Q water. Aliquot and store at −20°C until use (stable for up to 1 year).
-
i.
-
a.
-
10.For Western blotting experiments:
-
a.Biotin alkyne: reconstitute biotin alkyne in DMSO to 4 mM (2.115 mg/mL). Vortex until dissolved. Aliquot and store at −20°C until use (stable for up to 1 year).
-
b.Prepare the Invitrogen Click-iT® Protein Reaction Buffer Kit reagents as per kit instructions.
-
i.Component A: Reaction buffer. Dilute 1:2 in Milli-Q water. Store at 4°C.
-
ii.Component C: Reaction buffer additive 1. Mix with 500 μL Milli-Q water until fully dissolved. Aliquot and store at −20°C until use.
-
iii.Component D: Reaction buffer additive 2. Mix with 540 μL Milli-Q water until fully dissolved. Store at 4°C until use.
-
i.
-
a.
Preparation of Western blot reagents
-
11.Prepare Western blot running buffer and transfer buffer.
-
a.Running buffer: mix together 100 mL 10 × Tris/Glycine/SDS buffer in 900 mL Milli-Q water.
-
b.Transfer buffer: mix together 200 mL methanol (100%), 200 mL 5 × Trans-Blot Turbo Buffer, and 600 mL Milli-Q water.
-
a.
-
12.Prepare REVERT wash and REVERT reverse solutions.
-
a.REVERT wash: mix together 0.67 mL Glacial Acetic Acid, 3 mL methanol (100%) and 63.3 mL Milli-Q water.
-
b.REVERT reverse: mix together 0.2 mL 5 M sodium hydroxide, 3 mL methanol (100%), and 6.8 mL Milli-Q water.
-
a.
Preparation of immunocytochemistry reagents
-
13.
Prepare 1 × Tris-buffered saline (TBS) (pH 7.6) solution by diluting 10× TBS solution in Milli-Q water.
-
14.
Prepare 0.2% Triton X-100 in TBS.
-
15.
Prepare 0.05% Tween20 in TBS.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| L-Azidohomoalanine (AHA) | Click chemistry Tools | Cat # 1066-100, CAS 942518-29-8; https://clickchemistrytools.com/product/l-azidohomoalanine-aha/ |
| L-Methionine | Sigma-Aldrich | Cat #M5308, CAS 63-68-3 |
| Anisomycin | Tocris Bioscience | Cat # 1290, CAS 22862-76-6 |
| Biotin alkyne (PEG4 carboxamide-Propargyl biotin) | Thermo Fisher Scientific | Cat #B10185; https://www.thermofisher.com/order/catalog/product/B10185 |
| Alexa fluor 647 alkyne, triethylammonium salt | Thermo Fisher Scientific | Cat # A10278; https://www.thermofisher.com/order/catalog/product/A10278 |
| IRDye 800CW Streptadivin | LI-COR | Cat # 926-32230 |
| Alexa fluor 488 phalloidin | Cell Signaling Technology | Cat # 8878S |
| DAPI (4′, 6-diamidino-2-phenylindole, dihydrochloride) | Thermo Fisher Scientific | Cat #D1306 |
| DAKO fluorescence mounting medium | Agilent DAKO | Cat #S3023 |
| GlutaMAX™ supplement | Gibco | Cat # 35050061 |
| Fetal bovine serum (FBS), heat inactivated | Gibco | Cat # 10100147; https://www.thermofisher.com/order/catalog/product/10100147 |
| ‘BovoStar’ Bovine Serum Albumin (BSA) | Bovogen | Cat # BSAS 0.1 |
| Normal goat serum (NGS) | Cell Signaling Technology | Cat # 5425S |
| Penicillin / Streptomycin | Gibco | Cat # 15140-122 |
| 0.25% Trypsin-EDTA (1×), 100 mL | Gibco | Cat # 25200-056 100 |
| Dulbecco’s modified Eagle’s medium (DMEM) high glucose 1× | Gibco | Cat # 11965-092 |
| Poly-D-lysine hydrobromide (PDL) | Sigma-Aldrich | Cat #P0899, |
| 4× Laemmli sample buffer | Bio-Rad | Cat #1610747 |
| Revert 700 total protein stain | LI-COR | Cat # 926-1102; https://www.licor.com/bio/reagents/revert-700-total-protein-stain |
| 10× TBS buffer (Tris-buffered-saline) | Astral Scientific | Cat # BIOA0027-4L |
| Triton X-100 | Calbiochem, Millipore | Cat # 9410-OP, CAS 9002-93-1 |
| Paraformaldehyde | Sigma-Aldrich | Cat #P6148-500G |
| Tween 20 | Sigma-Aldrich | Cat #P1379-1L |
| 10× Tris/Glycine/SDS buffer | Bio-Rad | Cat #1610772 |
| Trans-blot Turbo 5× Transfer buffer | Bio-Rad | Cat # 10026938 |
| cOmplete mini, EDTA-free Protease inhibitor cocktail Tablets | Roche | Cat # 11836170001 |
| PhosSTOP phosphatase inhibitor cocktail Tablets | Roche | Cat # 04 906 837 001 |
| EveryBlot blocking buffer | Bio-Rad | Cat # 12010020 |
| RIPA buffer (10×) | Cell Signaling Technology | Cat # 9806 |
| Chloroform | Sigma-Aldrich | Cat #C2432-500ML |
| Glacial acetic acid | Merk | Cat # 5438080250 |
| Methanol | Supelco | Cat #106009, CAS 67-56-1 |
| Sodium dodecyl sulfate (SDS) | Sigma-Aldrich | Cat #L4390-1KG |
| Sodium hydroxide (NaOH) solution | Sigma-Aldrich | Cat # 72068, CAS 1310-73-2 |
| Sodium tetraborate (Borax) | Sigma-Aldrich | Cat # 221732 |
| Critical commercial assays | ||
| Click-iT® protein reaction buffer kit | Thermo Fisher Scientific | Cat #C10276; https://www.thermofisher.com/order/catalog/product/C10276 Kit manual: www.thermofisher.com/document-connect/document-connect.html?url= https://assets.thermofisher.com/TFS-Assets%2FLSG%2Fmanuals%2Fmp10276.pdf |
| Click-iT® cell reaction buffer kit | Thermo Fisher Scientific | Cat #C10269; https://www.thermofisher.com/order/catalog/product/C10269 Kit manual: www.thermofisher.com/document-connect/document-connect.html?url = https://assets.thermofisher.com/TFS-Assets%2FLSG%2Fmanuals%2Fmp10269.pdf |
| Pierce BCA protein assay kit | Thermo Fisher Scientific | Cat # 23225 |
| Experimental models: Cell line | ||
| BV2 murine microglia cell line | Blasi et al. 19902 | CVCL_0182 |
| Software and algorithms | ||
| FlowJo™ v10.8.1 | BD Biosciences | https://www.flowjo.com/solutions/flowjo |
| Imaris x64 v9.9.1 | Oxford Instruments IMARIS | https://imaris.oxinst.com/ |
| Image Studio v1.0.19 | LI-COR | https://www.licor.com/bio/image-studio/ |
| Other | ||
| 8-well chamber slides | Sarstedt | Cat # 94.6104.802 |
| Tissue culture plate, 6 well (6-well plate) | Falcon | Cat # 353046 |
| 75 cm2 (T75) Rectangular culture flask | Corning | Cat # 430641U |
| 25 cm2 (T25) Rectangular culture flask | Corning | Cat # 430639 |
| 96-Well clear flat Bottom TC-treated culture MicroPlate | Falcon, corning | Cat # 353072 |
| 4%–15% pre-cast gel | Bio-Rad | Cat # 4561083 or Cat # 4561086 |
| Trans-blot Turbo system | Bio-Rad | Cat # 1704150 |
| Confocal microscope LSM 710- Inverted | Zeiss | N/A |
| BD® LSR II Analyzer flow cytometer | BD Biosciences | N/A |
Materials and equipment
Cell culture solutions
Cell culture medium
| Reagent | Final concentration | Amount |
|---|---|---|
| Dulbecco’s Modified Eagle Medium (DMEM) High Glucose (1×) | 1× | 220 mL |
| Penicillin Streptomycin | 1% | 2.5 mL |
| GlutaMAX | 1% | 2.5 mL |
| Heat inactivated FBS | 10% | 25 mL |
| Total | N/A | 250 mL |
Store at 4°C for up to one month.
Poly-D-Lysine (PDL) coating solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Poly-D-Lysine 20 mg/mL stock | 0.5 mg/mL | 250 μL |
| Borate Buffer | N/A | 9.75 mL |
| Total | N/A | 10 mL |
Store at −20°C up to 1 year.
Borate Buffer pH 8.4
| Reagent | Final concentration | Amount |
|---|---|---|
| Boric Acid | 40 mM | 2.48 g |
| Sodium tetraborate (Borax) | 10 mM | 3.8 g |
| Milli-Q | N/A | Up to 1 L |
| Total | N/A | 1 L |
Store at 4°C for up to 1 year.
Buffer and detergent solutions
1× Tris Buffered Saline (TBS)
| Reagent | Final concentration | Amount |
|---|---|---|
| 10× Tris Buffered Saline (TBS) | 1× | 100 mL |
| Milli-Q water | N/A | 900 mL |
| Total | N/A | 1 L |
Store at room temperature (20°C–25°C) for up to 1 year.
0.2% Triton X-100 in TBS
| Reagent | Final concentration | Amount |
|---|---|---|
| Triton X-100 | 0.2% | 2 mL |
| 1× TBS | 1× | 998 mL |
| Total | N/A | 1 L |
Store at room temperature (20°C–25°C) for up to 1 year.
0.05% Tween-20 in TBS
| Reagent | Final concentration | Amount |
|---|---|---|
| Tween-20 | 0.05% | 0.5 mL |
| 1× TBS | 1× | 999.5 mL |
| Total | N/A | 1 L |
Store at room temperature (20°C–25°C) for up to 1 year.
Solutions for click chemistry, microscopy, and flow cytometry
Microscopy FUNCAT blocking buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Bovine Serum Albumin (BSA) | 2% (w/v) | 0.2 g |
| Normal Goat Serum (NGS) | 5% (v/v) | 0.5 mL |
| Triton X-100 | 0.2% (v/v) | 20 μL |
| TBS | 1× | 9.48 mL |
| Total | N/A | 10 mL |
Store at 4°C for up to 1 week.
Flow cytometry FUNCAT blocking buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Bovine Serum Albumin (BSA) | 2% | 0.2 g |
| Normal Goat Serum (NGS) | 5% | 0.5 mL |
| Tween20 | 0.2% | 20 μL |
| TBS | 1× | 9.48 mL |
| Total | N/A | 10 mL |
Store at 4°C for up to 1 week.
Solutions for protein isolation and Western blot
RIPA-based solution master mix
| Reagent | Amount per mL |
|---|---|
| 10× RIPA buffer | 100 μL |
| 10× Complete protease inhibitor | 100 μL |
| 10× PhosStop phosphatase inhibitor | 100 μL |
| 10% SDS | 100 μL |
| Milli-Q | 600 μL |
| Total | 1 mL |
Prepare fresh for every use.
Note: Adding phosphatase inhibitors is not crucial for the analysis of FUNCAT signal only, however, is critical if analysis of any protein phosphorylation will also be performed.
Western blot running buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 10× Tris/glycine/SDS buffer | 1× | 100 mL |
| Milli-Q water | N/A | 900 mL |
| Total | N/A | 1 L |
Store at 4°C for up to 1 year.
Western blot transfer buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Trans-Blot Turbo Buffer 5× | 1× | 200 mL |
| Methanol (100%) | 20% (v/v) | 200 mL |
| Milli-Q water | N/A | 600 mL |
| Total | N/A | 1 L |
Store at 4°C for up to 3 months.
REVERT wash solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Glacial acetic acid | 6.7% (v/v) | 0.67 mL |
| Methanol (100%) | 30% (v/v) | 3 mL |
| Milli-Q water | N/A | 6.33 mL |
| Total | N/A | 10 mL |
Store at room temperature (20°C–25°C) for up to 1 month.
REVERT reverse solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Sodium hydroxide (NaOH) | 0.1 M | 0.2 mL of 5 M |
| Methanol (100%) | 30% (v/v) | 3 mL |
| Milli-Q water | N/A | 6.8 mL |
| Total | N/A | 10 mL |
Store at room temperature (20°C–25°C) for up to 1 month.
Step-by-step method details
Cell seeding – Day 0
The steps below describe the seeding of BV2 cells in preparation for de novo protein synthesis detection using bioorthogonal labeling and click chemistry the following day. For quantification by microscopy, seed BV2 cells into 8-well chamber slides. For flow cytometry and Western blotting, seed them into 6-well plates.
Note: The total number of cells required determines the flask size needed to grow your stock culture. A T25 flask is sufficient for experiments using up to 10 × 8-well chamber slides, and a T75 is sufficient for experiments using 8 × 6 well plates. While handling these numbers is possible, we do not recommend using more than 4 × 8-well chamber slides or 4 × 6-well plates at a time, as this will increase timing and thus variability in sample processing.
-
1.For microscopy only: prepare 8-well chamber slides by coating with PDL for 1 h prior to seeding cells:
-
a.Add 200 μL 0.5 mg/mL PDL to each well and place in a 37°C incubator for at least 1 h.
-
b.Following the incubation period, remove PDL and wash 3 × 3 min each with PBS. Remove PBS after the last wash, and place back in 37°C incubator until use.
-
a.
-
2.
Place culture medium and 0.25% Trypsin-EDTA in 37°C water bath 10–15 min prior to beginning work.
-
3.Detach cells from the T25 or T75 culture flask using the following steps:
-
a.Remove medium and wash briefly with 5 mL (T25) or 10 mL (T75) PBS at room temperature (20°C–25°C).
-
b.Remove PBS and add pre-warmed 0.25% Trypsin-EDTA depending on the flask size:
-
i.1 mL for T25 stock culture flasks.
-
ii.3 mL For T75 stock culture flasks.
-
i.
-
c.Incubate cells in a 37°C incubator for 3 min.
-
d.Following 3 min incubation, tap sides of flask and check under microscope to ensure all cells have detached.
-
a.
Note: If cells are not all detached, continue tapping the flask edges, or incubate for 2 or more minutes. Do not allow cells to sit in 0.25% Trypsin-EDTA for longer than 10 min. Alternatively, detachment can be forced by more vigorous aspiration and release (i.e., by pipetting) of the Trypsin-EDTA over the cells.
-
4.
Add 1 mL (for T25) or 3 mL (for T75) pre-warmed culture medium to inhibit trypsin.
-
5.
Collect cells from flask into a 15 mL tube by pipetting.
-
6.
Centrifuge the tube at 200 × g for 3 min.
-
7.
Remove medium without disturbing the cell pellet and resuspend in 1 mL fresh culture medium.
-
8.
Perform cell counts to determine cell numbers / mL via preferred cell counting method (i.e., a cell counting chamber or an automated cell counting machine).
-
9.Seed cells as follows:
-
a.For microscopy experiments using 8-well chamber slides, seed 25,000 cells/well.
-
b.For flow cytometry and Western blot experiments using 6-well plates, seed 350,000 cells/well.
-
a.
-
10.
Place seeded cells in a 37°C incubator and leave them to grow overnight (16–20 h).
Figure 2.

Seeding of BV2 cells on Day 0 and imaged on Day 1
(A) To achieve appropriate cell densities on Day 1, cells need to be seeded the day before (Day 0) at 25,000 cells per well in 8-well chamber slides, or 350,000 cells/well in 6-well plates.
(B) Bright field microscopy image showing the density of BV2 cells on Day 1.
AHA labeling – Day 1
and/or
To investigate the process of protein synthesis in BV2 cells, we have used either a 30 min or a 4 h AHA-labeling period. The shorter 30 min time point is useful for studying processes in microglial cells that are fast, for example phagocytosis. The longer time 4 h point allows for observation of protein synthesis and the derived effects later than the 30 min time point. Longer AHA-labeled proteins could help also increase the contrast between background and FUNCAT signal, thus increasing the sensitivity of the method if needed.
As controls, we use methionine and/or AHA supplemented with anisomycin (a protein synthesis inhibitor). These controls are used to ensure successful bioorthogonal labeling and click chemistry. Other protein synthesis inhibitors such as cycloheximide can also be used.
The following steps provide the details of how to perform AHA-labeling of proteins synthesized within a 30 min or 4 h time period (Figure 3).
-
11.
Warm culture medium in a 37°C water bath 10 min prior to beginning the experiment.
-
12.
Thaw 0.4 M AHA, 0.4 M methionine, and 10 mM anisomycin (if required).
-
13.Prepare experimental medium as follows:
-
a.For 8-well chamber slides; 300 μL medium per well:
-
i.For AHA labeling, add 3 μL 0.4 M AHA per 300 μL medium to obtain a working concentration of 4 mM.
-
ii.For methionine control, add 3 μL of 0.4 M methionine per 300 μL medium for a working concentration of 4 mM.
-
iii.For AHA and anisomycin controls, add 3 μL of 0.4 M AHA to obtain a working concentration of 4 mM AHA and 1.2 μL of 10 mM anisomycin per 300 μL medium for a working concentration of 40 μM anisomycin.
-
i.
-
b.For 6-well plates; 1 mL medium per well:
-
i.For AHA labeling, add 10 μL 0.4 M AHA per 1 mL medium to reach a working concentration of 4 mM.
-
ii.For methionine control, add 10 μL of 0.4 M methionine per 1 mL medium for a working concentration of 4 mM.
-
iii.For AHA with anisomycin controls, add 10 μL of 0.4 M AHA plus 4 μL of 10 mM anisomycin per 1 mL medium for a working concentration of 40 μM anisomycin.
-
i.
-
a.
Note: To minimize pipetting errors during the treatment of multiple wells, always prepare a master mix solution (10% in excess) from which appropriate volumes are added to wells.
Note: We have found that 4 h anisomycin treatment is toxic to BV2 cells, but can be used for 30 min.
-
14.
Remove medium from wells and replace with experimental medium containing either methionine, AHA with anisomycin, or AHA (prepared in step 13).
-
15.
Incubate in 37°C incubator for either 30 min OR 4 h depending on desired labeling period.
Note: For labeling of other cell types or different treatment conditions, these labeling periods may require further optimization (see limitations).
Figure 3.

Experimental timeline for 30 min or 4 h AHA-labeling
Seed cells on Day 0 at the required density. The following day, label newly synthesized proteins via AHA incorporation for either 30 min or 4 h, using appropriate controls (i.e., methionine, and/or AHA with the protein synthesis inhibitor anisomycin). Fix cells or collect proteins at the end of the AHA-labeling period.
IMPORTANT: Depending on the experimental design, proceed to either A) Microscopy-based quantification of FUNCAT signal OR B) Flow cytometry-based quantification of FUNCAT signal OR C) Western blotting-based quantification of FUNCAT signal.
Microscopy-based quantification of FUNCAT signal
The following steps describe how to perform cell fixation and copper (I)-catalyzed click chemistry to visualize newly synthesized proteins using FUNCAT and microscopy after AHA-labeling. Microscopy data is quantified as mean FUNCAT signal per cell.
-
16.
Warm medium in 37°C water bath.
-
17.
Remove AHA/methionine/anisomycin-containing medium from wells and add 300 μL pre-warmed fresh medium. For AHA with anisomycin treated cells, add to a concentration of 4 mM AHA and 40 μM anisomycin to the fresh medium to ensure a sustained protein synthesis inhibition.
-
18.
Place back in a 37°C incubator for 10 min.
Note: This 10 min post-treatment incubation with fresh medium is performed to decrease intracellular free AHA and therefore increase signal to noise ratio.
-
19.
Remove medium and wash briefly once with PBS.
-
20.
Remove PBS and add 4% paraformaldehyde (PFA) prepared in PBS); incubate at room temperature (20°C–25°C) for 15 min.
-
21.
During 15 min incubation, prepare Microscopy FUNCAT blocking buffer.
-
22.
Remove PFA and wash twice with 1× TBS for 5 min each at room temperature (20°C–25°C).
Note: From this point, we use TBS rather than PBS, however, continued use of PBS could be also compatible.
Pause point: If needed, leave at 4°C overnight (16–20 h) in TBS.
-
23.
Permeabilize and block with Microscopy FUNCAT blocking buffer for 15 min at room temperature (20°C–25°C).
-
24.
Wash 3 times with 1× TBS for 5 min each at room temperature (20°C–25°C).
-
25.
During washes, prepare click chemistry reaction solution using Invitrogen Click-iT® Cell Reaction Buffer kit and Alexa Fluor 647 alkyne according to the following table, in the exact order of reagents presented.
Note: Calculate your required master mix volume for 100 μL per well plus 10% excess (i.e., for 12 samples, prepare a total of 1.32 mL master mix):
Click chemistry reaction master mix
| Reagent | Amount per 100 μL |
|---|---|
| Component A: Reaction Buffer (prepared in before you begin (Prepare click chemistry reagents)) | 88 μL |
| Component C: Reaction Additive (prepared in before you begin (Prepare click chemistry reagents)) | 10 μL |
| 1 mM Alexa Fluor 647 Alkyne (prepared in before you begin (Prepare click chemistry reagents)) | 0.2 μL (2 μM working conc.) |
| Component B: Copper (II) sulfate (CuSO4) | 2 μL |
| Total | 100 μL |
-
26.
Remove the wash solution and add 100 μL click chemistry reaction master mix solution per well; shield from light and incubate at room temperature (20°C–25°C) on a gentle rocker (20 rpm) for 1 h.
Optional: If performing immunostaining after the click reaction, wash three times with 1× TBS, permeabilize and block for 30 min, incubate with primary antibody overnight (16–20 h) at 4°C and 1 h at room temperature (20°C–25°C) with the secondary antibody, as required, before proceeding to step 27. This will add an extra day to the protocol.
-
27.
Wash 3 times with 1× TBS for 5 min each at room temperature (20°C–25°C). Following the last wash, add 1:20 Alexa fluor 488 phalloidin to visualize the cell outline and incubate for 30 min at room temperature (20°C–25°C).
-
28.
Wash 3 times with 1× TBS for 5 min each; in the second wash, add 1:10,000 DAPI (1 μg/mL stock) to label cell nuclei.
-
29.
Remove frame of the well-chambers and gently tap to remove excess liquid; add 3 drops of DAKO mounting medium. Coverslip and seal.
Note: The number of days/h required for imaging and analysis will vary depending on the experimental set up.
-
30.
Image DAPI (405), Phalloidin (488), and FUNCAT (647) channels at an appropriate magnification.
Note: To visualize FUNCAT in BV2 cells, we use 63× magnification and a confocal microscope (but other microscope types and a lower magnification can also be used). Imaging within one week is also recommended. Make sure to record and keep all microscopy parameters consistent between images and microscopy sessions. Ensure even sampling within wells (Figure 4).
Note: Depending on the specifications of the microscope and objectives used, imaging at higher magnification will increase the signal-to-background ratio. However, it is important to note that under identical laser settings, higher magnification objectives can lead to photobleaching. As such, we use a low laser intensity setting. We suggest always to adjust the acquisition setting accordingly on their microscope.
-
31.Quantify FUNCAT fluorescence for each individual cell using the IMARIS software (Figure 5) by using the Phalloidin fluorescence to create a surface of the area of the cells. IMARIS calculates the average fluorescence intensity of the FUNCAT signal (the measure of newly synthesized proteins), within each Phalloidin positive cell surface.
-
a.Convert raw image files to IMARIS format (.ims) using IMARIS Convertor software.
-
b.In IMARIS, use the phalloidin channel to create a surface of the cells in the image (Figure 5A) with the Surface function, using appropriate surface detail and threshold values.
-
i.Use the default ‘Number of Voxels Img = 1’ function and adjust the threshold to remove unwanted small background surfaces.
-
ii.If cells are touching (and therefore surfaces are joined), select the point to be cut using ‘SHIFT + click’, and then separate cells using the ‘cut surface’ IMARIS function (Figures 5B and 5C).
-
i.
-
c.Export data to an excel file using ‘average values’ in the Details tab (Figure 5D). Fluorescence intensity in the FUNCAT channel per cell can then be extracted from this table and analyzed.
-
a.
Note: We suggest only quantifying cells with one nucleus (visualized using DAPI). Phalloidin and FUNCAT fluorescence without DAPI are likely cells that have burst or been lysed during the click chemistry and washing process (these should be very minimal). Cells with multiple nuclei may represent a certain stage in the cell cycle, with potentially altered protein synthesis.
Note: The excel file will contain quantifications for many parameters. Intensity quantification is provided as mean, max, and sum for each channel.
Alternatives: This protocol uses IMARIS to quantify FUNCAT fluorescence, however any program capable of quantifying fluorescence intensity (e.g., FIJI/ImageJ software) can be used with appropriate adjustments.
Figure 4.

A guide for imaging BV2 cells using a confocal microscope at 63× magnification
(A) To decrease sampling bias, we recommend taking multiple images (≥ 9) from different areas of each well, such as in this grid example – red squares depict imaging acquisition areas. This is to ensure a representative capture of the whole well.
(B) Representative image of BV2 cells at 63× magnification with FUNCAT fluorescence in magenta.
Figure 5.

Quantifying FUNCAT fluorescence in BV2 cells using IMARIS
(A) Create cell surface using the channel for the cell outline marker, phalloidin (in this case, green). Apply appropriate cell surface and threshold values to capture the whole cell.
(B) If cells are touching, joining the surfaces of two cells, separate them using the ‘Cut Surface’ function.
(C) Ensure that all cells are properly separated.
(D) Export data as excel file.
Flow cytometry-based quantification of FUNCAT signal
The following steps describe how to perform fixation and copper (I)-catalyzed click chemistry to visualize newly synthesized proteins using FUNCAT and flow cytometry after AHA-labeling. This protocol is specific for the use of 2 × 6-well plates (i.e., 12 wells in total).
-
32.
Warm medium in a 37°C water bath.
-
33.
Remove AHA/methionine/anisomycin-containing medium and add pre-warmed fresh medium. For AHA with anisomycin-treated cells, add anisomycin into fresh medium to ensure continued protein synthesis inhibition.
-
34.
Place back in a 37°C incubator for 10 min.
Note: This 10 min chase with fresh medium is performed to decrease intracellular free AHA and therefore increase signal to noise ratio.
-
35.
Remove medium and wash briefly once with PBS.
-
36.
Remove PBS and add 500 μL 0.25% Trypsin-EDTA to each well; incubate at 37°C for 3 min.
-
37.
Add 500 μL fresh medium to each well; collect total 1 mL cell suspension into respective 1.5 mL tube. Keep on ice.
-
38.
Centrifuge tubes at 300 × g for 5 min at 4°C. Carefully remove supernatant; resuspend pellet in 1 mL ice cold PBS to wash.
-
39.
Centrifuge tubes at 300 × g for 5 min at 4°C. Carefully remove supernatant; resuspend pellet in 500 μL 4% PFA. Incubate at room temperature (20°C–25°C) for 15 min.
-
40.
During this 15 min incubation, prepare Flow cytometry FUNCAT blocking buffer.
-
41.
Centrifuge tubes at 1,000 × g for 5 min. Carefully remove the supernatant; resuspend the pellet in 1 mL TBS.
Note: TBS is used rather than PBS from this point as it is recommended when performing copper click chemistry.
-
42.
Centrifuge tubes at 1,000 × g for 5 min. Carefully remove supernatant and resuspend pellet in 200 μL Flow cytometry FUNCAT blocking buffer and incubate for 15 min at room temperature (20°C–25°C) with rotation to avoid cell settling.
-
43.
Centrifuge tubes at 1,000 × g for 5 min. Carefully remove supernatant; resuspend pellet in 1 mL 1× TBS.
-
44.
Prepare click chemistry reaction solution using Invitrogen Click-iT® Cell Reaction Buffer kit and Alexa Fluor 647 alkyne using the following table, in the order of reagents presented. Calculate your required master mix volume for 50 μL per sample plus 10% excess (i.e., for 12 samples, make a total of 660 μL master mix):
Click chemistry reaction master mix
| Reagent | Amount per 100 μL |
|---|---|
| Component A: Reaction Buffer (prepared in before you begin (Prepare click chemistry reagents)) | 88 μL |
| Component C: Reaction Additive (prepared in before you begin (Prepare click chemistry reagents)) | 10 μL |
| 1 mM Alexa Fluor 647 alkyne (prepared in before you begin (Prepare click chemistry reagents)) | 0.2 μL (2 μM working conc.) |
| Component B: Copper (II) sulfate (CuSO4) | 2 μL |
| Total | 100 μL |
-
45.
Centrifuge tubes at 1,000 × g for 5 min. Carefully remove supernatant; resuspend pellet in 50 μL click reaction solution per tube; shield from light and incubate at room temperature (20°C–25°C) with rotation for 1 h.
-
46.
Centrifuge tubes at 1,000 × g for 5 min. Carefully remove supernatant; resuspend pellet in 1 mL 1× TBS. Repeat this wash step.
-
47.
Centrifuge tubes at 1,000 × g for 5 min. Carefully remove supernatant; resuspend pellet in 200 μL TBS + 1% BSA and pipette into flow cytometry tube.
Note: we add 1% BSA to the final cell suspension as we have found this increases the total number of cells and decreases cell count variability between wells.
Note: We use a BD® LSR II Flow Cytometer (BD Biosciences), however, any flow cytometer capable of reading fluorescent intensity in the required FUNCAT channel will work.
-
48.Quantify FUNCAT fluorescence using a flow cytometer with a 633 laser:Note: any laser configuration that captures emission from AlexaFluor647 (e.g. 640 nm) can be used in this step.
-
a.Set up cell debris gate, doublet gate 1, and doublet gate 2 as required by the sample properties (Figures 6A–6C).
-
b.Use the methionine control to set background fluorescence threshold (Figure 6D). Cells above this threshold will be FUNCAT positive.
-
c.Quantify equal numbers of cells for each sample (e.g., 20,000 cells per sample).
-
d.Analyze data using the mean fluorescence intensity and percent cell count above threshold (i.e., percent FUNCAT positive cells).
-
a.
Figure 6.

Gating strategies for quantifying FUNCAT fluorescence using flow cytometry
(A) Exclude debris by creating a gate around cells based on the FSC-A and SSC-A parameters.
(B) Create a second gate using FSC-H and FSC-W parameters to remove cell doublets from the population analyzed.
(C) Create a third gate using SSC-H and SSC-W to further remove doublets from the population.
(D) Use the methionine control sample (FUNCAT negative background) to set background threshold for FUNCAT positive fluorescence.
Western blotting-based quantification of FUNCAT signal
The protocol below describes how to perform protein isolation and copper (I)-catalyzed click chemistry to visualize protein synthesis with FUNCAT and Western blot following AHA-labeling. This protocol is specific for 2 × 6-well plates (i.e., 12 wells). In this protocol, the protein isolation and protein concentration quantification occur on Day 1 (following AHA labeling), and the click chemistry and Western blotting occurs on Day 2.
-
49.
Warm medium in a 37°C water bath and label 1.5 mL tubes (one per well).
-
50.
Set a centrifuge to cool down to 4°C.
-
51.
Prepare Radio-Immunoprecipitation Assay (RIPA)-based lysis solution master mix.
-
52.
Remove AHA/methionine/anisomycin-containing medium and chase with 1 mL pre-warmed fresh medium per well. For AHA with anisomycin treated cells, add anisomycin into fresh medium to ensure continued protein synthesis inhibition.
-
53.
Place back in a 37°C incubator for 10 min.
Note: This 10 min chase with fresh medium is performed to decrease intracellular free AHA. This step is optional for WB.
-
54.
Following a 10 min chase, remove medium and add 1 mL ice-cold PBS. At this point, put plates on ice and keep cells cold until protein isolation is complete.
-
55.
Use a cell scraper to detach cells from wells. Briefly check under microscope to ensure no cells remain attached.
-
56.
Transfer cells into 1.5 mL tubes using a pipette.
-
57.
Centrifuge 5 min at 200 × g at 4°C.
-
58.
Remove supernatant, but be careful not to disturb the cell pellet. Add 100 μL RIPA-based solution per tube.
-
59.
Sonicate to lyse the cells.
Note: We use a Sonic Vibra-Cell sonicator machine with the following settings: 20%, 10 sec, no pulse. Repeat sonication if necessary (i.e., sample is still ‘sticky’). However, alternative cell lysis methods will also work.
-
60.
Centrifuge 15 min at 14,000 × g at 4°C.
-
61.
Carefully collect supernatant and transfer into new 1.5 mL tube, being careful not to disturb the pellet. Throw away the pellet.
-
62.
Denature proteins by incubating at 95°C for 10 min.
-
63.
Separate into aliquots (20–30 μL each), plus one 12 μL aliquot for use with BCA Assay.
-
64.Quantify protein concentration using Pierce BCA Assay:
-
a.Dilute 12 μL aliquot of sample 1:3 in RIPA-based solution.Note: we have found a 1:3 dilution to consistently provide optical density measurements within the standard curve range, however, it is possible that this dilution may need to be adjusted by the end user.
-
b.Prepare 8‒10 standards using the provided 2 μg/μL BSA using a serial dilution in RIPA-based solution. Use RIPA-based solution as blank.
-
c.Mix Pierce BCA reagent A:B at 50:1 ratio for 200 μL per well.
-
d.Add 10 μL sample, standard, or blank to wells in a 96 well plate.
-
e.Add 200 μL Reagent A:B mix to each well.
-
f.Protect from light and incubate at 37°C for 30 min.
-
g.Following incubation, read absorbance at 562 wavelength using a plate reader.
-
a.
-
65.
Create a standard curve and use linear regression to calculate the protein concentration of each sample from average optical densities (OD). Multiply by dilution factor (here: 3).
-
66.Perform the click chemistry reaction using the Click-iT® Protein Reaction Buffer Kit and Biotin Alkyne (prepared in before you begin):
-
a.For each sample: add 10–20 μg protein into a protein-free 1.5 mL tube. Add 1× RIPA solution up to 20 μL.
-
b.Mix Reaction Buffer (Component A; prepared in before you begin (Prepare click chemistry reagents)) with the biotin alkyne to a working concentration of 40 μM biotin alkyne.
-
c.Add 50 μL Reaction Buffer/biotin-alkyne mix to each sample. Vortex for 3 s.
-
d.Add 10 μL Milli-Q water per sample and vortex.
-
e.Add 5 μL copper (II) sulfate (CuSO4) and vortex.
-
f.Add 5 μL Buffer Additive I (Component C; prepared in before you begin (Prepare click chemistry reagents)). Vortex to mix. Wait for 2–3 min, but make sure to not exceed 5 min before step h (incubation step).
-
g.Add 10 μL Additive 2 (Component D; prepared in before you begin (Prepare click chemistry reagents)). Vortex to mix. The solution will turn bright orange.
-
h.Rotate end-over-end for 20 min at room temperature (20°C–25°C).
-
i.Add 300 μL methanol and vortex briefly.
-
j.Add 75 μL chloroform and vortex briefly.
-
k.Add 200 μL Milli-Q water and vortex briefly.
-
l.Centrifuge 5 min 14,000 × g. Carefully discard the supernatant (i.e., the orange upper aqueous phase). The precipitated protein will be situated at the interface between the orange upper layer and the clear lower layer.
-
m.Add 450 μL methanol and vortex briefly.
-
n.Centrifuge 5 min 14,000 × g. Carefully discard supernatant.
-
o.Repeat wash step with 450 μL methanol and vortex briefly.Note: This step removes residual reaction components.
-
p.Centrifuge 5 min 14,000 × g. Carefully discard supernatant.
-
q.Cover the open tubes with lint-free tissue and place in fume hood until remaining methanol has evaporated (15 min–1 h). Alternatively, leave covered tubes on benchtop for 1 h to overnight (16–20 h).
-
r.Once methanol has evaporated, close tubes and store at −20°C.Pause point: Store at −20°C until ready to run Western blot.
-
a.
-
67.
Resuspend samples in 10 μL RIPA buffer. Add 2.5 μL 4× Laemmli buffer and denature at 95°C for 5 min.
-
68.Run sample on Western blot:
-
a.Carefully load 2 μL protein ladder (e.g., Bio-Rad Precision Plus protein standard) and samples into 4%–15% pre-cast gel.
-
b.Run at 90 V until samples have entered the gel, then increase the voltage to 120 V.
-
c.Transfer to low fluorescence (LF) PVDF membrane. We perform semidry transfer using the High-MW 10 min setting on the Trans-Blot Turbo Transfer System.
-
a.
Alternatives: Any method of Western blot transfer for mixed to high molecular weight proteins is suitable.
-
69.
Wash the membrane briefly in Milli-Q water.
-
70.Perform a total protein stain (e.g., REVERT 700):
-
a.Incubate membrane in REVERT 700 Total protein stain for 5 min on gentle rocker.
-
b.Wash twice for 30 s each with REVERT wash.
-
c.Briefly wash twice in Milli-Q water.
-
a.
-
71.
Read on Odyssey Li-Cor using 700 wavelength.
-
72.
Remove Milli-Q and add REVERT Reverse for 8 min, rocking gently.
-
73.
Wash briefly with Milli-Q twice. Wash once with TBS to equilibrate the membrane at room temperature (20°C–25°C).
-
74.
Block with BioRad EveryBlot Blocking Buffer for 15 min on a rocker.
-
75.
Add 1:20,000 IRDye 800CW Streptavidin in EveryBlot Blocking Buffer. Incubate at room temperature (20°C–25°C) for 30 min on a rocker.
-
76.
Wash with TBS three times for 10 min each.
-
77.
Visualize in the 800 nm channel (Figure 9A).
-
78.
Quantify the fluorescence intensity using appropriate software and background subtraction.
Note: We use a LI-COR Odyssey Fc with Image Studio™ software to visualize membranes. Alternative systems or methods of quantifying the WB signal can also be used. Alternative detection methods, such as chemiluminescence, could provide increased detection sensitivity. The researcher should use the detection method appropriate to their needs and equipment.
Figure 9.

Expected quantification of protein synthesis in BV2 microglia using Western blotting
(A) Detection of protein synthesis, as shown by FUNCAT signal, following 30 min and 4 h AHA-labeling periods. Newly synthesized proteins are clicked to biotinylated alkynes, run on a Western blot, and visualized using fluorescent streptavidin. The total protein of each sample is detected by using REVERT stain reagent.
(B) Western blot quantification of the FUNCAT to total protein ratio reveals that AHA-labeled proteins are significantly more after 30 min incubation compared to cells treated with AHA in the presence of anisomycin protein synthesis inhibitor. This effect is further increased after 4 h incubation with AHA. Methionine was used as additional time-dependent incubation control and shows no significant difference compared with AHA with anisomycin experimental group. One-way ANOVA with multiple comparisons test; ∗p ≤ 0.05, ∗∗∗p ≤ 0.001; N = 3 wells/experimental group (excepting Met 4 h: N = 2). Data presented as mean ± SEM.
Expected outcomes
Successful bioorthogonal incorporation of AHA into microglial proteins and FUNCAT detection using the copper (I)-catalyzed click chemistry allows for clear visualization and quantification of protein synthesis using three complementary methods: microscopy, flow cytometry, or Western blotting. Examples of physiological protein synthesis in BV2 microglial cells that is occurring within 30 min and 4 h are presented for each technique in Figure 7 (microscopy), Figure 8 (flow cytometry), and Figure 9 (Western blot).
Figure 7.

Expected FUNCAT fluorescence and quantification in BV2 microglia using microscopy
(A and B) (A) Composite images of FUNCAT fluorescence (magenta) and the nucleic marker DAPI (blue) in BV2 cells acquired using microscopy after 30 min (A) or 4 h (B) of AHA-labeling, with methionine and AHA with anisomycin (protein synthesis inhibitor) controls.
(C) Quantification of mean FUNCAT intensity per cell with 30 min AHA-labeling achieves sufficient FUNCAT signal from background controls, and this ratio is further increased by 4 h AHA-labeling (Data presented as mean ± SEM. One-way ANOVA with multiple comparisons, ∗∗∗∗p ≤ 0.0001, n > 70 cells per group).
Figure 8.

Expected quantification of FUNCAT signal in BV2 microglia using flow cytometry
(A and B) Following a 30 min AHA-labeling period, BV2 cells had significantly higher mean FUNCAT fluorescence intensity compared to methionine and AHA with anisomycin controls (one-way ANOVA with multiple comparisons, N = 3 with 30,000 cells per group, ∗∗∗∗p ≤ 0.0001).
(C) Following a 4 h labeling period, the number of BV2 cells above the methionine control threshold is further increased from 30 min labeling.
(D) Quantification of FUNCAT signal shows mean fluorescence intensity of FUNCAT positive AHA-labeled cells is significantly higher than methionine control (Student’s t-test, N = 3 with 9,000 cells per group, ∗∗∗p ≤ 0.001). Data presented as mean ± SEM.
Limitations
AHA-labeling in combination with copper (I)-catalyzed click chemistry is a reliable and non-toxic method for investigating protein synthesis within limitations.
Firstly, AHA, as a surrogate of methionine, competes with methionine present in the culture medium to be incorporated into newly synthesized proteins. We and others7,8 use a concentration of 4 mM AHA, which is much higher than that of methionine in the medium (around 200 μM), and as such, AHA is more readily available for incorporation into new proteins. AHA at a concentration of 4 mM has been found previously to provide robust labeling and is not toxic to cells.9 To further increase the efficiency of AHA-labeling, it is possible to use methionine-free medium, however, as methionine is a key amino acid and the universal initiator of protein synthesis, its depletion prior to AHA labeling (as is suggested in protocols using methionine-free medium) may induce cellular metabolic changes or stress. As such, the suitability of this alternative must be considered depending on the experimental questions being asked. Our protocol demonstrates robust incorporation of AHA in newly synthesized proteins already after 30 min of AHA-labeling without the use of methionine-free medium.
With appropriate adjustments, our protocols can be used with other treatment paradigms and cell types. For the latter, optimization of AHA-labeling time and concentration are recommended. It should be noted that a limitation of using AHA is its lack of cellular specificity, which is a confounding factor when cells are grown in co-culture. Cell-specific labeling is possible using the non-canonical methionine analog L-azidonorleucine (ANL), however, this requires the presence of a mutated form of methionine-tRNA synthetase (MetRS).10 Finally, FUNCAT is a measure of global protein synthesis rates only, and as such this method does not allow for the identification of the newly synthesized proteins. For this, FUNCAT may be combined with a proximal ligation assay (FUNCAT-PLA), or BONCAT with an additional affinity purification of newly synthesized proteins step for mass spectrometry analysis.4,10
Troubleshooting
Problem 1
Uneven cell seeding within and/or between wells (related to Step 9 in cell seeding – Day 0).
Potential solution
-
•
Create a master cell suspension containing the cells needed to seed all wells with the same amount of medium. Make sure to thoroughly but gently mix the master mix between aliquoting for each well to avoid cell settling.
-
•
After seeding the cells, avoid swirling medium in a circle as this can lead to accumulation of cells at the edges or in the middle of the wells. Rather, we recommend briefly but gently rocking the plate or chamber slide side-to-side and back-and-forth.
Problem 2
Cells do not detach from the culture flask (related to step 36 in flow cytometry-based quantification of FUNCAT signal).
Potential solution
-
•
Ensure Trypsin-EDTA 0.25% is sufficiently pre-warmed to 37°C prior to use.
-
•
Increase Trypsin-EDTA incubation to 5–6 min. Do not allow cells to sit in Trypsin-EDTA for longer than 10 min.
-
•
Continue to tap solidly on the flask edges.
-
•
If none of the above leads to detachment of all the cells, more vigorous aspiration and release by pipetting of the Trypsin-EDTA over the cells can be used to force detachment.
-
•
Alternatively, cell scrapers can be used, however, this will increase the cellular debris in the sample.
Problem 3
Cell loss during centrifugation (related to all centrifugation steps in in flow cytometry-based quantification of FUNCAT signal).
Potential solution
-
•
When removing supernatant, make sure not to disturb the pellet. Tilt the 1.5 mL tube and watch the pellet as you slowly draw up liquid. Leave part of the supernatant in the tube if the pellet begins to move. Using a smaller (e.g., 200 μL) pipette tip rather than larger tips may also help.
-
•
Increase the duration of centrifugation.
-
•
Increase the gravitational force of centrifugation; live cells can be centrifuged up to 700 × g; fixed cells can resist even higher centrifugation force.
-
•
Increase the number of cells seeded into each well. If increasing the seeding number, ensure this is kept consistent across experiments as cell density will affect protein synthesis rates and therefore FUNCAT signal.
-
•
Decrease the concentration of detergent in the blocking solution. In this protocol, we use Tween20 as opposed to the harsher Triton X-100.
Problem 4
High background FUNCAT signal in negative controls (related to the quantification of FUNCAT with microscopy, flow cytometry, or Western blotting).
Potential solution
-
•
Increase the timing or number of washing steps.
-
•
Add 0.1 M glycine to the blocking buffer to decrease any unspecific reaction of alkyne to aldehydes in PFA-fixed cells.9
-
•
If protein synthesis levels are low in the cell type of choice, it may be necessary to increase the duration of AHA-labeling.
-
•
Use methionine-free medium (but see limitations).
-
•
Decrease the concentration of 647 alkyne used.
Problem 5
Low cell count when quantifying with flow cytometer even when pellet is visible in the last centrifugation step (related to steps 47 and 48 in in flow cytometry-based quantification of FUNCAT signal).
Potential solution
-
•
We have found that the addition of 1% BSA to the TBS for the final cell suspension (step 47) consistently increases cell number.
-
•
Don’t filter cells. Filtering cells just prior to flow cytometry in order to remove cell doublets is sometimes recommended, however, we have found this may contribute to cell loss. We have found pipetting up and down gently but firmly in the final 1% TBS solution is enough to create a single cell suspension. Any remaining doublets can be removed using gating during flow cytometry quantification.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Liviu Bodea (l.bodea@uq.edu.au).
Materials availability
This study did not generate new unique materials.
Acknowledgments
A.K.C.’s position is supported by the Australian Government Research Training Program (RTP) Scholarship. J.G.’s position and research are supported by the National Health and Medical Research Council of Australia (GNT1176326, GNT1145580, and GA39196) and the Terry and Maureen Hopkins Foundation. L.-G.B.’s position and research are supported by a Dementia Australia Research Foundation MCR Fellowship. We would like to thank and acknowledge the Queensland Brain Institute Microscopy and Flow Cytometry facilities for their excellent technical support, without which this work would not be possible. The graphical abstract and schematic figures were created using BioRender.com.
Author contributions
A.K.C. optimized the protocol, performed experiments, and conducted data analysis with input from L.-G.B and J.G. The manuscript was written by A.K.C. and L.-G.B. J.G. and L.-G.B. provided the funding and supervised the study. All authors reviewed and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Data and code availability
This study did not generate new datasets or custom code.
References
- 1.Evans H.T., Taylor D., Kneynsberg A., Bodea L.G., Götz J. Altered ribosomal function and protein synthesis caused by tau. Acta Neuropathol. Commun. 2021;9:110. doi: 10.1186/s40478-021-01208-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blasi E., Barluzzi R., Bocchini V., Mazzolla R., Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J. Neuroimmunol. 1990;27:229–237. doi: 10.1016/0165-5728(90)90073-v. [DOI] [PubMed] [Google Scholar]
- 3.Dieterich D.C., Hodas J.J.L., Gouzer G., Shadrin I.Y., Ngo J.T., Triller A., Tirrell D.A., Schuman E.M. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat. Neurosci. 2010;13:897–905. doi: 10.1038/nn.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dieterich D.C., Link A.J., Graumann J., Tirrell D.A., Schuman E.M. Selective identification of newly synthesised proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT) Proc. Natl. Acad. Sci. USA. 2006;103:9482–9487. doi: 10.1073/pnas.0601637103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steward K.F., Eilers B., Tripet B., Fuchs A., Dorle M., Rawle R., Soriano B., Balasubramanian N., Copié V., Bothner B., Hatzenpichler R. Metabolic implications of using BioOrthogonal non-canonical amino acid tagging (BONCAT) for tracking protein synthesis. Front. Microbiol. 2020;11:197. doi: 10.3389/fmicb.2020.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ullrich M., Liang V., Chew Y.L., Banister S., Song X., Zaw T., Lam H., Berber S., Kassiou M., Nicholas H.R., Götz J. Bio-orthogonal labeling as a tool to visualize and identify newly synthesized proteins in Caenorhabditis elegans. Nat. Protoc. 2014;9:2237–2255. doi: 10.1038/nprot.2014.150. [DOI] [PubMed] [Google Scholar]
- 7.Alvarez-Castelao B., Schanzenbächer C.T., Langer J.D., Schuman E.M. Cell-type-specific metabolic labeling, detection and identification of nascent proteomes in vivo. Nat. Protoc. 2019;14:556–575. doi: 10.1038/s41596-018-0106-6. [DOI] [PubMed] [Google Scholar]
- 8.Evans H.T., Benetatos J., van Roijen M., Bodea L.G., Götz J. Decreased synthesis of ribosomal proteins in tauopathy revealed by non-canonical amino acid labelling. EMBO J. 2019;38:e101174. doi: 10.15252/embj.2018101174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tom Dieck S., Muller A., Nehring A., Hinz F.I., Bartnik I., Schuman E.M., Dieterich D.C. Metabolic labeling with noncanonical amino acids and visualization by chemoselective fluorescent tagging. Curr. Protoc. Cell Biol. 2012 doi: 10.1002/0471143030.cb0711s56. Chapter 7, Unit7 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Evans H.T., Blackmore D., Götz J., Bodea L.G. De novo proteomic methods for examining the molecular mechanisms underpinning long-term memory. Brain Res. Bull. 2021;169:94–103. doi: 10.1016/j.brainresbull.2020.12.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate new datasets or custom code.