

Abstract
High systemic doses of adeno-associated viruses (AAVs) have been associated with immune-related serious adverse events (SAEs). Although AAV was well tolerated in preclinical models, SAEs were observed in clinical trials, indicating the need for improved preclinical models to understand AAV-induced immune responses. Here, we show that mice dual-dosed with AAV9 at 4-week intervals better recapitulate aspects of human immunity to AAV. In the model, anti-AAV9 immunoglobulin G (IgGs) increased in a linear fashion between the first and second AAV administrations. Complement activation was only observed in the presence of high levels of both AAV and anti-AAV IgG. Myeloid-derived pro-inflammatory cytokines were significantly induced in the same pattern as complement activation, suggesting that myeloid cell activation to AAV may rely on the presence of both AAV and anti-AAV IgG complexes. Single-cell RNA sequencing of peripheral blood mononuclear cells confirmed that activated monocytes were a primary source of pro-inflammatory cytokines and chemokines, which were significantly increased after a second AAV9 exposure. The same activated monocyte clusters expressed both Fcγ and complement receptors, suggesting that anti-AAV-mediated activation of myeloid cells through Fcγ receptors and/or complement receptors is one mechanism by which anti-AAV antigen complexes may prime antigen-presenting cells and amplify downstream immunity.
Keywords: adeno-associated virus, innate immunity, adaptive immunity, muscular dystrophy, single-cell RNA sequencing, Luminex
Graphical abstract
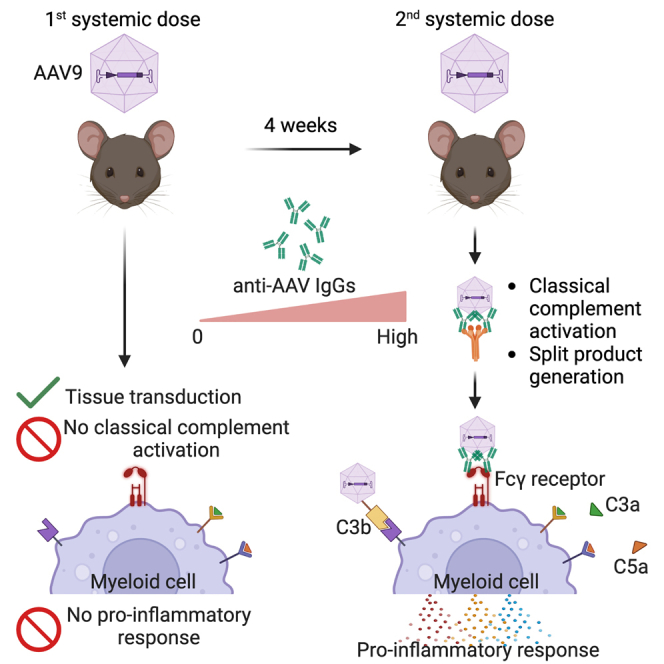
Emami and colleagues employed dual-AAV dosing to model immune responses in mice. This approach revealed activation of the classical complement pathway and identified myeloid-derived chemokines and cytokines dependent on adaptive immunity. The use of dual-AAV dosing holds promise for enhancing future investigations into AAV-based immune responses.
Introduction
Adeno-associated viruses (AAVs) are widely used vectors for delivering nucleic acids for gene therapy approaches. The first systemically administered AAV-based gene therapy was approved by the Food and Drug Administration (FDA) for spinal muscular atrophy (SMA) (Zolgensma). Currently, AAV-based gene delivery of a modified DMD gene, termed micro-dystrophin (μDYS), is being evaluated in clinical trials for Duchenne muscular dystrophy (DMD).1,2,3 However, serious adverse events (SAEs) attributed to innate immune responses against the AAV capsid have arisen in individuals receiving high systemically delivered doses of AAV. The SAEs (grade 3 and above) reported in the DMD clinical trials sponsored by Pfizer and Solid Biosciences occurred in patients receiving AAV doses greater than 2.0 × 1014 vg/kg, in which several patients experienced thrombotic microangiopathy (TMA) and renal failure attributed to complement activation (https://www.solidbio.com/about/media/press-releases/solid-biosciences-announces-fda-removes-clinical-hold-on-sgt-001 and https://www.pfizer.com/news/press-release/press-release-detail/pfizers-new-phase-1b-results-gene-therapy-ambulatory-boys).4 Similarly, several cases of TMA were also reported in pediatric patients dosed with Zolgensma,5,6,7 and in adults in clinical trials for Danon disease (https://ir.rocketpharma.com/news-releases/news-release-details/rocket-pharmaceuticals-announces-positive-updates-phase-1/), Fabry disease (https://ir.4dmoleculartherapeutics.com/news-releases/news-release-details/4d-molecular-therapeutics-presents-interim-data-4d-310-inglaxa), and methylmalonic acidemia (https://www.prnewswire.com/news-releases/logicbio-therapeutics-reports-second-quarter-2022-financial-results-and-provides-corporate-update-301605458.html). While the deleterious consequences of TMA can be ameliorated with the C5 complement inhibitor, eculizumab, it should be noted that inhibiting C5 does not impede the generation of complement split products. These split products may amplify downstream pathways of both innate and adaptive immunity. Thus, although TMA can be pharmacologically resolved, the downstream consequences of complement activation and split product formation persist.
Complement activation occurs through three different mechanisms: the lectin, the alternative, or the classical pathways. All three pathways converge on the formation of C3 convertase, generating split products that mediate protection against pathogens via recruitment of inflammatory cells, pathogen opsonization, and pathogen destruction.8 While systemic administration of AAV-based therapies was well tolerated in small and large animal models, subsequent clinical trials revealed AAV-mediated complement activation.9,10,11 The pathway underlying AAV-mediated complement activation in patients remains unclear, as the clinical evidence is limited to the pathological findings consistent with complement activation and detection of complement split products after AAV dosing.4,5,6,7,12,13 Four separate in vitro studies detected complement activation only when AAV was incubated with human serum containing anti-AAV immunoglobulin G (IgG), strongly implicating the classical pathway, which is initiated by antigen-antibody complexes.12,13,14,15 However, complement split products from the alternative pathway were detected in non-human primates following systemic administration of AAV,16,17 and in some human trials (M. Corti, “Immune modulation as an adjunctive therapy to AAV systemic dosing to improve safety, increase expression and allow for repeated AAV dosing.” FDA + ASGCT Immune Responses to AAV Vectors workshop, 2023). The alternative pathway is in homeostatic balance between initiation and inhibition, but it can be stimulated via a feedforward amplification loop following activation of other pathways.18
Preclinical studies have traditionally relied on a single systemic administration of each therapeutic AAV vector, and have primarily focused on evaluating their therapeutic efficacy. However, the lack of exposure to natural AAV infections may prevent mice from responding strongly to a single AAV dose. In contrast, approximately 30%–60% of the general population is estimated to have pre-existing immunity to AAV, through antigen-reactive memory T cells and/or neutralizing antibodies (NAbs) and/or binding (BAbs) antibodies.19,20 Screening for NAbs and/or interferon (IFN)γ-producing antigen-reactive memory T cells via the enzyme-linked immunospot (ELISPOT) assay is often used to determine pre-existing immunity.21,22,23 However, these assays may not be sensitive enough to detect memory B, effector memory T (TEM), or central memory T (TCM) cells, which are present at low frequencies in circulation and could interfere with therapeutic efficacy.24,25,26
Preclinical studies of AAV-based therapies did not predict the complement activation that has been observed in humans. One explanation is that human immune responses are markedly more sensitive to viral and bacterial stimulation compared with lesser mammals27; however, an alternative explanation is that sterilely housed murine models are not exposed to the same large array of pathogens to which humans are exposed, and this may further contribute to the inability of preclinical studies to predict human immune responses associated with a single administration of AAV. This observation is not only important for mitigating SAEs, but also for developing protocols that enable repeat AAV dosing. Since complement split products amplify both innate and adaptive immune arms, new insights are needed to elucidate how complement split products modulate downstream immune effectors. Thus, there is a need for improved preclinical models to understand the earliest immunomodulatory effectors, including complement, and their roles in stimulating downstream immunity. In order to improve the mouse as a model of human immune responses, we carried out dual-AAV administration, in which the first dose effectively immunizes the mouse, and showed that a dual-dosing approach reveals classical complement pathway,myeloid cell activation and induction of specific chemokines and cytokines such as IP-10 and MCP-1. In summary, our AAV dual-dosing strategy recapitulates aspects of human AAV-immune responses and provides opportunities to model aspects of human immune responses.
Results
Characterization of humoral responses in mdx mice double-dosed with AAV9
We sought to develop a model that emulates features of human immune responses to AAV vectors in mice. Unlike their human counterparts, research mice housed in a sterile vivarium are not exposed to pathogens including wild-type AAV. Therefore, we delivered a sensitizing AAV dose to trigger immunity, followed by a second dose 4 weeks later in a dystrophic mouse model, the hDMD del45 mdx mouse.28 The hDMD del45 mdx mouse model contains an out-of-frame exon 45 deletion in the human DMD gene and a point mutation in exon 23 of the mouse Dmd gene, thereby completely lacking dystrophin protein (hereafter referred to as mdx). The mdx mice were systemically dosed with two administrations of AAV9 at ∼1.16 × 1014 vg/kg carrying three different vectors. AAV9 was packaged with either micro-dystrophin (μDYS), Cas9, or Cas9 with a frameshift mutation (Cas9-FS) that disrupts Cas9 protein production while maintaining a similar genetic payload (Figures S1A–S1C). Dosing with Cas9 and Cas9-FS vectors offered an opportunity to distinguish between AAV9 capsid-specific and transgene-specific immune responses throughout the assays used in the study. SDS-PAGE of packaged vectors confirmed that the capsid proteins (VP1, VP2, and VP3) were visible and were present in the correct stoichiometric ratio 1:1:10 (Figure S1D).
Previous studies have shown that animal models and humans exposed to AAV develop anti-AAV antibodies,29,30,31,32 and we sought to assess the timing of these responses in our model. We evaluated humoral responses using a high-content protein microarray (HCPM) (Table S1), which contains proteins of interest (e.g., AAV capsid) printed directly onto nitrocellulose microarrays.33,34,35 The HCPM assay revealed a significant increase in anti-AAV9 IgG antibodies after a single dose of AAV9, increasing linearly from a single injection and peaking at the Post-2 (2 weeks) time point (Figure 1A and Table S2). A statistically significant increase in anti-AAV9 IgM was observed at the Post-1 (2 weeks) time point relative to baseline (Pre) (Figure 1B). In addition, we detected AAV serotype cross-reactivity against other serotypes such as AAV2, AAV8, and AAVMYO,36 which is a muscle tropic AAV capsid variant identified via high throughput library screening. Similar to the anti-AAV9 IgG levels, anti-AAV2 IgG, anti-AAV8 IgG, and anti-AAVMYO IgG antibodies increased significantly post-dosing, peaking at the Post-2 (2 weeks) time point (Figures 1C, 1E, and S2). A significant increase in anti-AAV8 IgM and anti-AAVMYO IgM antibodies were also observed at the Post-1 (2 weeks) time point relative to baseline (Pre), comparable to anti-AAV9 IgM responses (Figures 1D and 1F). Last, anti-transgene responses were probed using the HCPM assay; however, we did not observe statistical differences in either IgM or IgG levels against Cas9 or μDYS transgenes across all time points (Figures 1G–1J).
Figure 1.

Assessment of anti-AAV and anti-transgene antibody responses using a high-content protein microarray (HCPM)
(A and B) Anti-AAV9 IgG and IgM responses were measured via HCPM from male (n = 9) and female (n = 8) mdx mice at indicated time points. The key indicates the vector that was dosed and the black line represents AAV9-treated groups combined by time point, which was used for multiple comparison statistical analysis. Error bars for all HCPM graphs represent standard deviation. Symbols above time points are used to represent statistical significance, in which p < 0.05, for time points compared with: (#) Pre, (&) Post-1 (2 weeks), and ($) Post-1 (4 weeks). The data and levels of statistical significance between time point comparisons are available in Tables S1 and S2. (C–F) Plasma collected from mice double-dosed with AAV9 was assessed by HCPM for the ability to bind to other AAV serotypes. Capsids of AAV8 and AAVMYO were printed on the chip and plasma from male (n = 9) and female (n = 8) mdx mice was assessed. (G–J) anti-Cas9 and anti-μDYS IgG and IgM responses were assayed via HCPM from male (n = 9) and female (n = 8) mdx mice at indicated time points.
Evaluation of complement responses after dual AAV9 dosing in mdx mice
Ongoing clinical trials for DMD have recently reported evidence of AAV-mediated complement activation following systemic administration of high-dose AAV.4 It has been speculated that the main complement cascade being activated is the classical pathway ,12,13,14,15,37 although there is also evidence for activation of the alternative pathway.16,17 While complement activation has been reported ex vivo and in human clinical trials, there are no published studies to support AAV-mediated complement activation in small animal models, such as the mouse. To examine complement activation in dual-AAV-dosed mice, we probed for complement components C3, C4, and C5b9 for the time points in our study (Tables S3 and S4). These assessments revealed no statistically significant depletion of complement components C3 and C4 at 5 h or 2 weeks after the first dose relative to baseline (Pre), but statistically significant C3 and C4 complement depletion at the Post-2 (5 h) time point relative to Pre (Figures 2A, 2B and S3A), which is suggestive of classical pathway activation since C3 and C4 consumption are only detected when high levels of antibodies and AAV are present in the blood (after the second administration of AAV9) (Figure 1A). Deviations in C5b9 levels were not detected (Figure 2C), which may be attributed to the time points studied in our experiment. In DMD clinical trials, C5b9 levels peaked 3-5 days after AAV dosing,13 whereas we sampled 5 h and 2 weeks post AAV dosing. These results showing statistically significant C3 and C4 consumption 5 h from AAV administration provide a model to study complement activation and downstream consequences after AAV dosing in mice.
Figure 2.

Consumption of complement components, C3 and C4, and concomitant induction of pro-inflammatory chemokines and cytokines after the second dose of AAV
(A–C) Plasma from mdx mice was evaluated by ELISA for levels of complement C3 (n = 9 females), C4 (n = 9 females), and C5b9 (n = 9 females). C3 levels for males (n = 9) and females (n = 9) are shown in Figure S3A. The key indicates the vector that was dosed and the black line represents AAV9-treated groups combined by time point, which was used for multiple comparison statistical analysis. Error bars for all graphs represent standard deviation. Symbols above time points are used to represent statistical significance, in which p < 0.05, for time points compared with (†) Post-2 (5 h). The data and levels of statistical significance between time point comparisons are available in Tables S3 and S4. (D) Levels of immunomodulatory analytes: IP-10 (CXCL10), MIP-1β (CCL4), MCP-1 (CCL2), and TNF-α as measured by Luminex ProcartaPlex, n = 9 males and n = 9 females. Symbols above time points are used to represent statistical significance, in which p < 0.05, for time points compared with (#) Pre, (☨) Post-1 (5 h), and (†) Post-2 (5 h). The data and levels of statistical significance between time point comparisons are available in Tables S5 and S6.
Characterization of AAV-induced cytokine and chemokine responses in dual-dosed mice
We next sought to assess cytokine and chemokine responses that arise in plasma after dual-AAV dosing. Prior studies have attributed AAV-induced innate immune responses to activation of toll-like receptor 9 (TLR9), a pattern recognition receptor (PRR) that senses unmethylated cytosine-phosphate-guanine (CpG) DNA motifs present in AAV genomes.38 TLR9 activation leads to nuclear factor κB (NF-κB) pro-inflammatory cytokine, chemokine and interferon regulatory factor 7 (IRF7) type I interferon responses.38,39 Given the potential of TLR9 to detect the AAV vector genomes used in this study, we calculated the risk potential for TLR9 activation (KTLR9) for each vector, using several equations that consider both stimulatory and inhibitory configurations of CpG DNA motifs as described.40 As expected, AAV9-Cas9 (KTLR9 = 20), AAV9-Cas9-FS (KTLR9 = 20), and AAV9-μDYS (KTLR9 = 13) contain a high-risk potential for TLR9 activation (Table 1). Although the μDYS transgene is CpG depleted and contains a lower KTLR9 compared with the Cas9 vectors, the packaged μDYS vector is not devoid of CpGs and, thus, poses a risk for TLR9 activation.
Table 1.
Activation potential for toll-like receptor 9 (TLR9)40
| AAV DNA vector | TLR9 activation potential (KTLR9) | Activation (+/−) |
|---|---|---|
| CK8e-Cas9-FS | 19.7 | ++ |
| CK8e-Cas9 | 19.7 | ++ |
| CK8e-μDYS | 12.8 | + |
| AAV2 WT genome | 26.0 | +++ |
| Human genome | 1.0 | – |
We harvested plasma from dual-dosed mice in order to assess AAV-induced innate immune responses downstream of TLR9 stimulation by performing multiplex analyte analysis via Luminex (Figures 2D, S3B, Tables S5, and S6). A total of 27 analytes were probed; however, 10 analytes were omitted from additional analysis as they were above or below the limits of quantification (e.g., IFN-α). We observed that many of the significantly elevated pro-inflammatory chemokines and cytokines peaked after the second dose at the Post-2 (5 h) time point compared with baseline (Pre) (Figure 2D). These elevated myeloid-derived chemokines and cytokines include IP-10 (CXCL10), MIP-1β (CCL4), MCP-1 (CCL2), and tumor necrosis factor (TNF)-α.These findings correlate with published pro-inflammatory responses observed following AAV and CpG agonist oligonucleotide administration studies in mice, non-human primates, and humans.38,41,42,43,44,45 The absence of increases in IP-10 (CXCL10), MIP-1β (CCL4), MCP-1 (CCL2), and TNF-α between Pre and the Post-1 (5 h) time point (Figure 2D), suggests that activation of these chemokines and cytokines may require an adaptive immune response, similar to our observations for complement activation. Previous studies have demonstrated that AAV ineffectively transduces TLR9-expressing antigen-presenting cells, which includes monocytes, dendritic cells, and B cells.15,46 Therefore, the induction of complement and myeloid-derived chemokines and cytokines after the second exposure suggests AAV capsid antibodies may be needed to mediate AAV uptake to prime myeloid cells either through Fcγ receptors and/or complement receptors.
Single-cell RNA sequencing reveals monocyte cell activation after the second dose
We characterized peripheral blood mononuclear cells (PBMCs) from AAV-dosed mice using single-cell RNA sequencing (scRNA-seq). PBMCs were collected at baseline and 2 weeks after the first and second administrations of AAV9. scRNA-seq uniform manifold approximation and projection (UMAP) plots revealed shifts in transcriptional phenotypes between Pre and Post time points across broad immune cell types including T cells, B cells, natural killer (NK) cells, and monocytes (Figures S4A–S4C). In addition, large changes in cell proportions were observed between Post-1 (2 weeks) and Post-2 (2 weeks) time points among CD8T cells and monocytes (Figure S4D).
The activation of complement and the elevated myeloid-derived chemokine and cytokine responses were only observed when blood levels of both anti-AAV IgGs and AAV were high (Figures 1A and 2D), suggesting an important link between humoral immunity, induction of complement, and myeloid cell activation. The data suggest that AAV must complex with anti-AAV IgGs to activate cells of the myeloid lineage. To obtain more granular information about how transcriptional profiles of monocyte sub-populations change with AAV dosing, we performed monocyte subcluster analysis and identified nine unique monocyte clusters with transcriptional shifts that arise after dosing and re-dosing (Figures 3A and 3B). Several monocyte sub-populations contributed to over 40% of the cells in the Post-2 (2 weeks) time point including non-classical (Spn+ Ly6c2−), classical (Spn− Ly6c2+), and other monocyte clusters defined by markers Lilr4b+, Ly6d+, Clec4d+Clec4e+, and Ceacam1+ (Figures 3C and 3D).
Figure 3.

Unbiased characterization via scRNA-seq analysis to identify cell types responding to AAV in peripheral blood mononuclear cells (PBMCs)
(A) Monocyte subcluster UMAP shows heterogeneous sub-populations present in AAV9 double-dosed mice. (B) Monocyte subcluster UMAP, color coded by time point before and after AAV dosing. Pre (blue), Post-1 (pink) and Post-2 (green). (C) Dot plot of top differentially expressed genes among monocyte sub-populations. Color is scaled by average expression and the dot size is proportional to the percent of cells expressing the respective gene. (D) Bar graph shows the percentage of monocyte sub-populations by time point. Pre (blue), Post-1 (pink) and Post-2 (green).
Myeloid-derived monocytes recognize antigen-antibody complexes via Fc receptors and/or complement split products via complement receptors, suggesting that these receptors might mediate the observed chemokine and cytokine responses in monocytes. To further explore this question, we examined transcriptional profiles of monocyte subclusters expressing Fc and complement receptors. Gene expression analysis confirmed that the non-classical (Spn+ Ly6c2−), classical (Spn− Ly6c2+) and Clec4d+Clec4e+ monocyte sub-populations expressed activating FcγRs including Fcgr1 (CD64), Fcgr3 (CD16), Fcgr4 (CD16-2), and Fcer1g, which all bind IgG immune complexes (Figures 4A and S5A). We also observed expression of complement receptors including Cr1l (receptor that binds C3b/C4b), C5ar1 (receptor that binds anaphylatoxin, C5a) and Cd93 (receptor that binds C1q, which initiates the classical complement pathway) (Figures 4A and S5A). Importantly, the expression of activating FcγRs and complement receptors by Post-2 (2 weeks) correlated with an increased pro-inflammatory transcriptional profile in the same monocyte subclusters. Differentially expressed genes (DEGs) in non-classical (Spn+ Ly6c2−), classical (Spn− Ly6c2+), and Clec4d+Clec4e+ monocytes showed several transcripts that were upregulated in Post-2 (2 weeks) relative to baseline (Pre) compared with DEGs upregulated in Post-1 (2 weeks) relative to Pre (Figures 4B–4D). Notably, we observed a significant induction of pro-inflammatory cytokine and chemokine gene expression only after Post-2 (2 weeks) compared with Pre including Tnf (TNF-α), Cxcl2 (MIP-2α), Ccl3 (MIP-1α), Ccl4 (MIP-1β), Ccl5 (RANTES), Cxcl10 (IP-10), Il1a (interleukin [IL]-1α), and Il1b (IL-1β) (Figures 4B and 4C). However, we failed to detect any significant upregulation of cytokine and chemokine responses in Post-1 (2 weeks) relative to Pre (Figure 4D). Similarly, we observed significant upregulation of cytokine and chemokine transcripts at Post-2 (2 weeks) relative to the Post-1 (2 weeks) time point (Figure S5B). Since upregulation of the cytokine and chemokine genes observed in the Clec4d+Clec4e+ monocytes at the Post-2 (2 weeks) time point overlapped with the analytes detected by Luminex at the Post-2 (5 h) time point, we conducted gene set enrichment analysis (GSEA) to investigate the biological implications of DEGs in this sub-population. When comparing Post-2 (2 weeks) with Pre, we found that the top significantly enriched pathways included TNF signaling, TLR signaling, and nuclear factor (NF)-κB signaling (Figure 5A). In addition, Post-2 (2 weeks) compared with Post-1 (2 weeks) time points similarly demonstrated that the top enriched pathways included NF-κB signaling and TNF signaling (Figure 5B). The top enriched pathways upregulated Post-2 (2 weeks) consisted of genes associated with inflammatory responses and were largely driven by Tnf (TNF-α), Cxcl2 (MIP-2α), Cxcl3 (MIP-2β), Ccl3 (MIP-1α), Ccl4 (MIP-1β), Il1a (IL-1α), and Il1b (IL-1β). Conversely, the only down-regulated pathway when comparing Post-2 (2 weeks) with the Pre time point consisted of MHC genes known to be associated with antigen processing and presentation (Figure 5A).
Figure 4.

Monocyte sub-populations demonstrate activation and induction of pro-inflammatory chemokine and cytokine genes after the second, not the first, systemic administration of AAV9
(A) Stacked violin plot shows expression of FcγRs and complement receptor genes present in activated non-classical, classical, and Clec4d+Clec4e+ monocyte populations. (B) Stacked violin plot shows the expression of significantly differentially expressed pro-inflammatory cytokine and chemokine genes upregulated Post-2 (2 weeks). (C and D) Volcano plots of classical, non-classical, and Clec4d+Clec4e+ monocytes showing differentially expressed genes between Post-2 (2 weeks) and Pre (C) and Post-1 (2 weeks) and Pre (D). Significantly up- and down-regulated genes contain a Log2(Fold Change) > 0.5 or Log2(Fold Change) < 0.5 and -Log10(p value) > 2.0.
Figure 5.

Gene sent enrichment analysis of Clec4d+Clec4e+ monocytes reveal inflammatory response pathways enriched after AAV re-administration
(A) Differential gene expression analysis between Post-2 and Pre time points and gene set enrichment analysis (GSEA) identified inflammatory response pathways including cytokine signaling, TLR signaling, and NF-κB signaling. Table of GSEA pathways and associated genes are listed in Table S7. (B) Differential gene expression analysis between Post-2 and Post-1 time points and GSEA similarly identified inflammatory response pathways enriched after AAV re-administration compared with the first administration. Table of GSEA pathways and associated genes are listed in Table S8.
Activated monocyte sub-populations also expressed PRRs including Tlr9, Tlr2, Cd14, and Myd88, which are needed for downstream TLR signal transduction (Figure S5A). While TLR9 senses hypomethylated AAV DNA leading to activation,38 TLR2 has been shown to associate with CD14 and activate primary liver cells after recognition of the AAV capsid.47 Previous studies have revealed that stimulating FcγRs, TLR9, or complement receptors can induce NF-κB-dependent gene expression in plasmacytoid dendritic cells (pDCs) and monocytes,39,48,49,50 resulting in the production of pro-inflammatory responses similar to those we observed via Luminex and scRNA-seq. These findings lend support for the notion that AAV capsid antibodies in complex with AAV lead to complement activation, which can prime myeloid cells by engaging FcγRs, TLRs, and/or complement receptors to induce downstream NF-κB-dependent gene expression.
Discussion
Innate and adaptive immune responses pose a significant challenge for safe and efficacious clinical application of AAV-based gene therapies. Three components of AAV vectors can trigger immunity: (1) capsid, (2) unmethylated CpGs in the nucleic acid cargo, and (3) the transgene protein.51 These immune responses compromise the safety and efficacy of AAV-based therapies and have resulted in at least 11 patient deaths observed across eight different trials (M. Corti, “Immune modulation as an adjunctive therapy to AAV systemic dosing to improve safety, increase expression and allow for repeated AAV dosing.” FDA + ASGCT Immune Responses to AAV Vectors workshop, 2023). Additionally, two deaths have been reported in pediatric patients with SMA treated with FDA-approved Zolgensma.5,7 The high-dose systemic administration required for treating patients with neuromuscular diseases makes these patients more susceptible to immune-mediated SAEs that can lead to death even in the presence of immunosuppressive corticosteroids.
Complement-related SAEs reported in many different clinical trials with different vectors suggest that pre-dose screening is insufficient to detect all immune responses that contribute to AAV-induced immunotoxicities. While TMA can be resolved with a C5 complement inhibitor, inhibition of C5 is incomplete and the downstream immune consequences of split product generation are likely vast.52 Thus, there is a new appreciation for understanding the role of AAV-complement interactions and the impact of complement split products on downstream immunity. Better preclinical models are needed to elucidate how AAV-complement interactions interface with other arms of immunity.
Our study demonstrates that dual-AAV dosing in mice induces classical complement activation and cytokine/chemokine induction that is not detected in singly dosed mice at 5 h or 2 weeks post dosing. By administering a second AAV dose 4 weeks after the first, anti-AAV IgG and AAV are able to form a complex, leading to classical complement activation and priming of myeloid cells followed by cytokine/chemokine release. The observation that myeloid-derived pro-inflammatory chemokines and cytokines were induced after the second AAV administration suggests that myeloid cell activation depends on adaptive immunity to allow AAV entry through antibody-dependent enhancement (ADE). This observation has been demonstrated previously with human blood or cells exposed to AAV.14,53,54 Here, we show the first demonstration of AAV-mediated ADE in mice.
We confirmed anti-AAV9 IgGs cross-reacted with commonly used serotypes, including AAV2 and AAV8, and confirmed cross-reactivity against a newly discovered muscle tropic variant of AAV9, termed AAVMYO,36 which suggests that even novel variants may be neutralized by pre-existing capsid antibodies. The absence of detectable anti-transgene responses for the time points assessed is likely because we utilized a muscle-specific promoter, CK8e.55 Thus, non-target tissues transduced by AAV, such as antigen-presenting cells, will not express μDYS or Cas9.56 Our anti-AAV findings correlate with studies in which antibodies from individuals with pre-existing immunity showed similar cross-reactivity across many serotypes.19 Therefore, it is unlikely that simply switching AAV serotypes will overcome pre-existing immunity. Induction of anti-AAV9 IgG response and subsequent complement activation correlate with the data reported in the Pfizer DMD clinical trial, in which two participants, with rapidly rising anti-AAV9 levels, experienced complement-related adverse events post-dosing.13 Also, Pfizer and Solid Biosciences reported SAEs associated with complement activation,4,12,13 and demonstrated that complement activation relies on the presence of pre-existing antibodies in vitro. The Byrne and Corti labs have shown that blocking both B and T cells prior to AAV administration can mitigate complement activation (M. Corti, “Immune modulation as an adjunctive therapy to AAV systemic dosing to improve safety, increase expression and allow for repeated AAV dosing.” FDA + ASGCT Immune Responses to AAV Vectors workshop, 2023),57 while Smith et al.14 showed that anti-AAV complexes facilitate AAV entry to myeloid cells and chemokine/cytokine induction. Together, these findings demonstrate the important link between AAV IgG complexes and promotion of both complement activation and myeloid cell priming.
We posit that AAV Ig complexes mediate pro-inflammatory responses through two mechanisms. One is through FcγR-mediated uptake allowing for TLR9 recognition and myeloid activation. Second, AAV IgG complexes can initiate the classical pathway of complement to activate myeloid cells by either complement receptor-mediated uptake for TLR9 recognition and/or by complement C3a and C5a split products that bind anaphylatoxin receptors. Prior evidence demonstrated that AAV associates with complement C3 and split products C3b and iC3b in vitro.15 Our hypothesis is supported by Smith et al.,14 in which serum containing anti-AAV IgGs and complement C3 incubated with AAV resulted in detectable complement activation, AAV uptake in myeloid cells, and induction of inflammatory cytokines in vitro while C3-depleted serum containing anti-AAV IgGs significantly reduced AAV uptake.15 Likewise, an independent study confirmed that the presence of AAV NAbs in whole blood led to complement activation, uptake of AAV in myeloid-derived cells, and subsequent induction of pro-inflammatory responses including IP-10, TNF-α, MCP-1, IL-1β, etc.14 One study revealed that AAV-specific BAbs enhanced AAV uptake in HEK293 and Hepa 1–6 cells in vitro and introducing an anti-Fc antibody partially blocked AAV uptake.53 These data support the model that AAV capsid antibodies present by the second dose mediate monocyte activation by Fcγ receptor stimulation and/or complement receptor activation via split products generated by the classical complement pathway.
Furthermore, scRNA-seq analysis revealed that only the activated monocyte sub-populations express FcγRs and complement receptors. Comparable to Luminex cytokine analysis, the identified monocyte sub-populations demonstrated significant upregulation of NF-κB-dependent genes only after the second dose, including Tnf (TNF-α), Cxcl2 (MIP-2α), Ccl3 (MIP-1α), Ccl4 (MIP-1β), Ccl5 (RANTES), Cxcl10 (IP-10), Il1a (IL-1α), and Il1b (IL-1β). The cytokine responses we observed were also reported in an in vitro study that incubated AAV with human whole blood containing myeloid cells including macrophages, monocytes, and dendritic cells.14 However, the same study also identified neutrophils as one of the most prevalent subsets to internalize AAV and produce cytokines when incubated with serum containing AAV NAbs.14 The lack of neutrophils detected in our scRNA-seq data can be attributed to two main factors. First, neutrophils and other granulocytes sediment with erythrocytes due to their higher densities when isolating PBMCs using the Ficoll-Paque density gradient.58 Second, granulocytes such as neutrophils are relatively short-lived and do not endure the cryopreservation-thawing process.59 Although we fail to detect neutrophils in our scRNA-seq data, there may be neutrophil-specific AAV responses occurring in our pre-existing mouse model that warrant further investigation.
Another important variable to consider for future studies is to examine the effect of empty AAV capsids in our dual-dosing model and delineate immune responses attributed to the capsid antigen and capsids packaged with vector genomes. While we systemically administered a dose of ∼1.16 × 1014 vg/kg, the AAV preparations were purified via iodixanol gradient ultracentrifugation and thus, our total capsid antigen dose is likely much greater since empty capsids are typically the most abundant particle type in cell culture preparations of AAV.60 Lastly, future studies will focus on uncovering how pre-existing anti-AAV antibodies trigger AAV uptake and their role in amplifying downstream innate and adaptive immune responses. Additional work will focus on adapting the dual-dosing strategy by reducing the initial priming dose to more accurately reflect naturally acquired AAV immunity. Our findings offer a novel paradigm for future preclinical studies using a dual-AAV dosing strategy and the mouse immune system to recapitulate human immune responses and thus, offer the potential to identify immunomodulatory targets to develop re-dosing strategies and to thwart adverse responses.
Materials and methods
Mice
All animal care and work were conducted under protocols approved by the UCLA Animal Research Committee in the Office of Animal Research Oversight. hDMD del45 mdx mice were generated and genotyped as described.28
AAV plasmid constructs used in the study
pAAV-CK8e-Cas9 and pAAV-FLAG-CK8e-μDYS vectors were a gift from Dr. Jeffrey Chamberlain and were generated as described.61,62 pAAV-CK8e-Cas9 was modified to introduce an FLAG tag as described below to construct pAAV-FLAG-CK8e-Cas9 and to introduce a frameshift mutation in Cas9 to generate pAAV-FLAG-CK8e-Cas9-FS.
Generation of pAAV-FLAG-CK8e-Cas9 and pAAV-FLAG-CK8e-Cas9-FS vectors: NEBuilder HiFi DNA Assembly (New England BioLabs) was used to generate pAAV-FLAG-CK8e-Cas9 and pAAV-FLAG-CK8e-Cas9-FS. A 386 base pair (bp) gBlock (IDT) containing an FLAG tag at the N terminus and part of the Cas9 coding sequence was synthesized (FLAG-Cas9). A 385-bp gBlock (IDT) identical to FLAG-Cas9, but with a 1-nt deletion to encode for a frameshifting (FS) mutation in Cas9 was synthesized (FLAG-Cas9-FS). The FLAG-Cas9 and FLAG-Cas9-FS gBlocks were cloned in HincII- and BglII-digested pAAV-CK8e-Cas9 using NEBuilder HiFi DNA Assembly according to the manufacturer’s instructions to create pAAV-FLAG-CK8e-Cas9 and pAAV-FLAG-CK8e-Cas9-FS.
Recombinant AAV production
AAV9-FLAG-CK8e-Cas9, AAV9-FLAG-CK8e-Cas9-FS, and AAV9-FLAG-CK8e-μDYS for mdx study: pAAV-FLAG-CK8e-Cas9, pAAV-FLAG-CK8e-Cas9-FS, and pAAV-FLAG-CK8e-μDYS were shipped to Vigene Biosciences Inc. for large-scale AAV9 production. Vigene Biosciences Inc. purified recombinant AAVs using iodixanol gradient ultracentrifugation and quantified viral titer by qPCR using their ITR primers. Purity of capsid viral proteins was determined by SDS-PAGE (9% polyacrylamide gels).
AAV9 double-dosing study in mdx mice in vivo
The double-dosing study was conducted using ∼12-week-old mdx male and female mice. Three AAV serotype 9 vectors carrying either FLAG-CK8e-Cas9, FLAG-CK8e-Cas9-FS, or FLAG-CK8e-μDYS were systemically injected via retro-orbital (r.o.) injection at ∼1.16 × 1014 vg/kg. Each vector was administered in n = 3 males and n = 3 females for a total of nine mice dosed per sex. A second dose (re-dosing) was with the same AAV vector via r.o. injection at ∼1.16 × 1014 vg/kg ∼4 weeks after the first dose. The mice were culled ∼6 weeks after the second dose (i.e., ∼10 weeks after the first dose) and the heart, skeletal muscle tissues, lung, liver, and spleen were harvested and flash frozen in isopentane.
For scRNA-seq, PBMCs were collected from all mice at the following time points: prior to AAV exposure (Pre), 2 weeks after the first dose (Post-1 2 weeks), 4 weeks after the first dose (Post-1 4 weeks), 2 weeks after the second dose (Post-2 2 weeks), and 6 weeks after the second dose (Post-2 6 weeks). Approximately 0.2 mL of whole blood was harvested in EDTA-containing BD Microtainer Capillary Blood Collection Tubes (BD Biosciences). Whole blood was diluted 1:1 in DPBS and carefully layered over Ficoll-Paque Premium (Fisher Scientific) in a 2:1 ratio of diluted blood to Ficoll. Samples were centrifuged at 800 rcf for 25 min with the brakes and acceleration turned off. The PBMC layer (buffy coat) was extracted, washed with DPBS, and resuspended in red blood cell (RBC) lysis buffer (Qiagen) for 1–2 min. PBMCs were then centrifuged using a low-speed spin (150 rcf for 15 min) to remove dead RBCs and platelets and cryopreserved in 90% fetal bovine serum (Thermo Fisher) and 10% dimethyl sulfoxide (DMSO, Millipore Sigma). For scRNA-seq, only two of the three male mice per AAV vector (n = 6 male mice total) were processed for scRNA-seq at the following time points: Pre, Post-1 2 weeks, and Post-2 2 weeks.
For Luminex, enzyme-linked immunosorbent assays (ELISAs), and HCPM analysis, plasma was collected at the following time points: prior to AAV exposure (Pre), ∼5 h after the first dose (Post-1 5 h), 2 weeks after the first dose (Post-1 2 weeks), 4 weeks after the first dose (Post-1 4 weeks), ∼5 h after the second dose (Post-2 5 h), 2 weeks after the second dose (Post-2 2 weeks), and 6 weeks after the second dose (Post-2 6 weeks). Plasma was isolated during PBMC isolation, discussed earlier, in which plasma is the layer above the PBMC buffy coat.
10x library preparation, sequencing, and alignment
scRNA-seq libraries were generated using Chromium Single Cell 3′ v.3 (10x Genomics). Libraries were sequenced using NovaSeq 6000 S4 (Illumina) 2 × 100 bp paired-end reading strategy. Cells were called using Cell Ranger (v.6.1.2, 10x Genomics) with the mouse reference genome (mm10) to generate raw gene expression matrices for each sample.
scRNA-seq data analysis and clustering
The raw gene expression matrices were analyzed by R software (v.4.1.2) with the Seurat package v.4.1.0.63 Low-quality cells were removed when more than 20% of the UMIs were derived from mitochondrial genes, when less than 200 features and more than 1,800 unique features were detected. The data were then normalized using the default NormalizeData function parameters, the FindVariableFeatures function to select 2,000 genes with the highest standardized variance, and the ScaleData function to perform Z score transformation. We then analyzed the integrated samples by using RunUMAP, FindNeighbors, and FindClusters functions for UMAP visualization. The clusters were manually annotated using the top 50 DEGs produced by the FindAllMarkers function. For subcluster analysis of monocytes, we used the subset function in combination with the same methods discussed to analyze the integrated data.
Gene set enrichment analysis
Gene set enrichment analysis was performed with the WEB-based Gene Set Analysis Tool Kit (https://www.webgestalt.org/) using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database in order to identify enriched pathways using DEGs between baseline and AAV administration time points.64
Detection of complement components with ELISAs
Murine plasma samples were used to measure complement C3 (ab157711, Abcam), C4 (NBP2-70040, Novus Biologicals), and C5b9 (OKCD01374, Aviva Systems Biology) levels according to the manufacturer’s instructions. Plasma samples were diluted to 1:50,000, 1:20, and 1:30 to measure C3, C4, and C5b9, respectively. Each sample and time point were run in triplicate.
Multiplex cytokine analysis using Luminex
A custom ProcartaPlex panel (Thermo Fisher) was designed and consisted of 27 analytes: IFN-α, IFN-β, IFN-γ, IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-9, IL-10, IL-12p70, IL-13, IL-15, IL-17A, IL-18, IP-10, MCP-1, MCP-3, MIG, MIP-1α, MIP-1β, MIP-2a, RANTES, and TNF-α. Analytes were measured on a Luminex 200 instrument (Immune Assessment Core, UCLA) using mouse plasma samples according to the manufacturer’s instructions. All male and female plasma samples were assessed at the following time points: Pre, Post-1 (5 h), Post-1 (2 weeks), Post-2 (5 h), and Post-2 (2 weeks). The cloud-based ProcartaPlex Analysis application (Thermo Fisher) was used to confirm the raw data were in the working range, in which the observed concentration was between 70% and 130% of the expected concentration from standard controls using either the four-parameter log-logistic (4PL) or five-parameter log-logistic (5PL) standard curve. Cytokine analytes were expressed in picograms per milliliter (pg/mL). Analytes observed at the lower limits of quantification and upper limits of quantification were omitted from additional analysis (Figure S3B).
Humoral IgM and IgG responses measured by HCPM
HCPM chips printed with AAV capsids, Cas9, or μDYS were used to measure humoral IgM and IgG antibody levels. HCPM chips were generated as previously described.33,34,35 In brief, proteins of interest (i.e., antigens) were printed directly onto nitrocellulose microarrays at concentrations of 0.1 mg/mL, 0.05 mg/mL, 0.025 mg/mL, and 0.0125 mg/mL. The antigens of interest included the following: AAV2 (AAV2-EMPTY, Virovek Inc.), AAV8 (AAV8-EMPTY, Virovek Inc.), AAV9 (AAV9-EMPTY, Virovek Inc.), SpCas9 (CAS9PROT-50UG, Millipore Sigma), AAVMYO (a gift from Dr. Xiangmin Xu, Professor of Anatomy and Neurobiology at the University of California, Irvine), and mini-dystrophin protein was provided by Pfizer. Plasma samples were serially diluted for each microarray to determine optimal dilution and ranged from: 1:200, 1:2,000, 1:20,000, and 1:200,000. Finally, antigen-specific humoral responses were assessed for male and female plasma samples at the following time points: prior to AAV exposure (Pre), 2 weeks after the first dose (Post-1 2 weeks), 4 weeks after the first dose (Post-1 4 weeks), 2 weeks after the second dose (Post-2 2 weeks), and 6 weeks after the second dose (Post-2 6 weeks). Quantitative analyses were performed by the Vaccine R&D Center at the University of California, Irvine, as previously described by their team.33,34,35
Statistical analysis
AAV9-treated groups were combined by time point for multiple comparison statistical analysis. Results presented in Figures 1 and S2 are shown as mean ± standard deviation (SD) and comparison between time points (groups) was evaluated using one-way analysis of variance (ANOVA) mixed effects model with repeated measures followed by Tukey’s post hoc test. Results presented in Figures 2 and S3 are shown as mean ± SD and comparison between time points (groups) was evaluated using one-way ANOVA with repeated measures followed by Tukey’s post hoc test. p < 0.05 (∗), p < 0.01 (∗∗), p < 0.001 (∗∗∗), p < 0.0001 (∗∗∗∗) were considered significant. Statistical analysis and graphs were generated using GraphPad Prism 9 software.
Data availability
All data used to evaluate the conclusions of the article are present in the paper and/or supplementary material. Further inquiries regarding the data can be directed to the corresponding author and will be made available upon request.
Acknowledgments
We would like to thank Jane Wen and Diana Becerra for technical assistance. The authors thank and acknowledge the use of services from the following cores: the Technology Center for Genomics & Bioinformatics (TCGB), the Immune Assessment Core at UCLA, the Institute for Quantitative and Computational Biosciences (QC Bio) at UCLA, and the UCI Vaccine R&D Center. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE-1650604 (M.R.E.). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. Funding was also provided by the Ruth L. Kirschstein National Research Service Award GM007185 (M.R.E.), Ruth L. Kirschstein National Research Service Award (NIH NIAMS) T32 AR065972 (M.R.E.), the Center for DMD at UCLA Azrieli Graduate student Award (M.R.E.), the Muscular Dystrophy Association MDA635862 (C.S.Y. and M.J.S.), and the Whitcome Predoctoral Fellowship in Molecular Biology (M.R.E.). Additional funding was provided by the National Institutes of Health (R01NS117912, M.J.S. and J.S.C. U54 AR052646-07, M.J.S. and RO1 AR064327, A.D.P.) the Department of Defense (MD190060, M.J.S. and S.A.V.), NIH NIAMS sponsored SBIR (1R44AR075469, C.S.Y.), and the California Institute for Regenerative Medicine (CIRM DISC0-13765, M.J.S and A.D.P., and CIRM DISC2-08824, A.D.P.).
Author contributions
Conceptualization, Methodology, Visualization, Project Administration – M.R.E., M.J.S., C.S.Y.; Investigation – M.R.E., C.S.Y., P.K.H., S.A.V.; Software, Data Curation – A.E., F.M.; Formal analysis – M.R.E., A.E., F.M.; Writing – Original Draft – M.R.E.; Writing – Review and Editing – M.R.E., C.S.Y., S.A.V., A.D.P., M.J.S.; Resources – J.S.C., M.P., X.X., P.K.H., S.A.V., P.L.F.; Supervision – M.J.S.; Funding Acquisition – C.S.Y., S.A.V., M.J.S.
Declaration of interests
M.J.S., A.D.P., and C.S.Y. are co-founders of MyoGene Bio, a startup spun out of UCLA developing gene editing therapies for Duchenne muscular dystrophy.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2023.06.002.
Supplemental information
References
- 1.Greer C., Kozyak B., Stedman H. Challenges at the Crossroads: Myopathy Trials in 2020 Hindsight. Mol. Ther. 2021;29:420–421. doi: 10.1016/j.ymthe.2021.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manini A., Abati E., Nuredini A., Corti S., Comi G.P. Adeno-Associated Virus (AAV)-Mediated Gene Therapy for Duchenne Muscular Dystrophy: The Issue of Transgene Persistence. Front. Neurol. 2021;12:814174. doi: 10.3389/fneur.2021.814174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mendell J.R., Al-Zaidy S.A., Rodino-Klapac L.R., Goodspeed K., Gray S.J., Kay C.N., Boye S.L., Boye S.E., George L.A., Salabarria S., et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther. 2021;29:464–488. doi: 10.1016/j.ymthe.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fortunato F., Rossi R., Falzarano M.S., Ferlini A. Innovative Therapeutic Approaches for Duchenne Muscular Dystrophy. J. Clin. Med. 2021;10:820. doi: 10.3390/jcm10040820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chand D.H., Zaidman C., Arya K., Millner R., Farrar M.A., Mackie F.E., Goedeker N.L., Dharnidharka V.R., Dandamudi R., Reyna S.P. Thrombotic Microangiopathy Following Onasemnogene Abeparvovec for Spinal Muscular Atrophy: A Case Series. J. Pediatr. 2021;231:265–268. doi: 10.1016/j.jpeds.2020.11.054. [DOI] [PubMed] [Google Scholar]
- 6.Day J.W., Mendell J.R., Mercuri E., Finkel R.S., Strauss K.A., Kleyn A., Tauscher-Wisniewski S., Tukov F.F., Reyna S.P., Chand D.H. Clinical Trial and Postmarketing Safety of Onasemnogene Abeparvovec Therapy. Drug Saf. 2021;44:1109–1119. doi: 10.1007/s40264-021-01107-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guillou J., De Pellegars A., Porcheret F., Frémeaux-Bacchi V., Allain-Launay E., Debord C., Denis M., Péréon Y., Barnérias C., Desguerre I., et al. Fatal Thrombotic Microangiopathy Case following Adeno-Associated Viral SMN Gene Therapy. Blood Adv. 2022;6:4266–4270. doi: 10.1182/bloodadvances.2021006419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lo M.W., Woodruff T.M. Complement: Bridging the innate and adaptive immune systems in sterile inflammation. J. Leukoc. Biol. 2020;108:339–351. doi: 10.1002/jlb.3mir0220-270r. [DOI] [PubMed] [Google Scholar]
- 9.Ferla R., Alliegro M., Marteau J.B., Dell'Anno M., Nusco E., Pouillot S., Galimberti S., Valsecchi M.G., Zuliani V., Auricchio A. Non-clinical Safety and Efficacy of an AAV2/8 Vector Administered Intravenously for Treatment of Mucopolysaccharidosis Type VI. Molecular therapy. Mol. Ther. Methods Clin. Dev. 2017;6:143–158. doi: 10.1016/j.omtm.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Majowicz A., Salas D., Zabaleta N., Rodríguez-Garcia E., González-Aseguinolaza G., Petry H., Ferreira V. Successful Repeated Hepatic Gene Delivery in Mice and Non-human Primates Achieved by Sequential Administration of AAV5(ch) and AAV1. Mol. Ther. 2017;25:1831–1842. doi: 10.1016/j.ymthe.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelson C.E., Wu Y., Gemberling M.P., Oliver M.L., Waller M.A., Bohning J.D., Robinson-Hamm J.N., Bulaklak K., Castellanos Rivera R.M., Collier J.H., et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019;25:427–432. doi: 10.1038/s41591-019-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez J.P., Brown K.J., Lawlor M.W., Shanks C., S S.J., Morris C.A. SGT-001 Microdystrophin gene therapy for Duchenne muscular dystrophy. Mol. Ther. 2020;28:1–592. doi: 10.1016/j.ymthe.2020.04.019. [DOI] [Google Scholar]
- 13.Moorehead T., Yong F., Neelakantan S., Beaverson K., Binks M. Safety and tolerability of PF-06939926 in ambulatory boys with Duchenne muscular dystrophy: a phase 1b multicenter, open-label, dose ascending study. Mol. Ther. 2020;28:1–592. doi: 10.1016/j.ymthe.2020.04.019. [DOI] [Google Scholar]
- 14.Smith C.J., Ross N., Kamal A., Kim K.Y., Kropf E., Deschatelets P., Francois C., Quinn W.J., 3rd, Singh I., Majowicz A., et al. Pre-existing humoral immunity and complement pathway contribute to immunogenicity of adeno-associated virus (AAV) vector in human blood. Front. Immunol. 2022;13:999021. doi: 10.3389/fimmu.2022.999021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zaiss A.K., Cotter M.J., White L.R., Clark S.A., Wong N.C.W., Holers V.M., Bartlett J.S., Muruve D.A. Complement is an essential component of the immune response to adeno-associated virus vectors. J. Virol. 2008;82:2727–2740. doi: 10.1128/JVI.01990-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hordeaux J., Song C., Wielechowski E., Ramezani A., Dyer C., Buza E.L., Chichester J., Bell P., Wilson J.M. Characterization of acute toxicity after high-dose systemic adeno-associated virus in nonhuman primates, including impact of vector characteristics. Mol. Ther. 2021;29:1–427. doi: 10.1016/j.ymthe.2021.04.019. [DOI] [Google Scholar]
- 17.Palazzi X., Pardo I.D., Sirivelu M.P., Newman L., Kumpf S.W., Qian J., Franks T., Lopes S., Liu J., Monarski L., et al. Biodistribution and Tolerability of AAV-PHP.B-CBh-SMN1 in Wistar Han Rats and Cynomolgus Macaques Reveal Different Toxicologic Profiles. Hum. Gene Ther. 2022;33:175–187. doi: 10.1089/hum.2021.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lachmann P.J. The amplification loop of the complement pathways. Adv. Immunol. 2009;104:115–149. doi: 10.1016/s0065-2776(08)04004-2. [DOI] [PubMed] [Google Scholar]
- 19.Louis Jeune V., Joergensen J.A., Hajjar R.J., Weber T. Pre-existing anti-adeno-associated virus antibodies as a challenge in AAV gene therapy. Hum. Gene Ther. Methods. 2013;24:59–67. doi: 10.1089/hgtb.2012.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rajavel K., Ayash-Rashkovsky M., Tang Y., Gangadharan B., de la Rosa M., Ewenstein B. Co-Prevalence of Pre-Existing Immunity to Different Serotypes of Adeno-Associated Virus (AAV) in Adults with Hemophilia. Blood. 2019;134:3349. doi: 10.1182/blood-2019-123666. [DOI] [Google Scholar]
- 21.Mendell J.R., Connolly A.M., Lehman K.J., Griffin D.A., Khan S.Z., Dharia S.D., Quintana-Gallardo L., Rodino-Klapac L.R. Testing preexisting antibodies prior to AAV gene transfer therapy: rationale, lessons and future considerations. Molecular therapy. Mol. Ther. Methods Clin. Dev. 2022;25:74–83. doi: 10.1016/j.omtm.2022.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patton K.S., Harrison M.T., Long B.R., Lau K., Holcomb J., Owen R., Kasprzyk T., Janetzki S., Zoog S.J., Vettermann C. Monitoring cell-mediated immune responses in AAV gene therapy clinical trials using a validated IFN-γ ELISpot method. Molecular therapy. Mol. Ther. Methods Clin. Dev. 2021;22:183–195. doi: 10.1016/j.omtm.2021.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith J.G., Liu X., Kaufhold R.M., Clair J., Caulfield M.J. Development and validation of a gamma interferon ELISPOT assay for quantitation of cellular immune responses to varicella-zoster virus. Clin. Diagn. Lab. Immunol. 2001;8:871–879. doi: 10.1128/cdli.8.5.871-879.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masopust D., Vezys V., Marzo A.L., Lefrançois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science (New York, N.Y.) 2001;291:2413–2417. doi: 10.1126/science.1058867. [DOI] [PubMed] [Google Scholar]
- 25.Palm A.K.E., Henry C. Remembrance of Things Past: Long-Term B Cell Memory After Infection and Vaccination. Front. Immunol. 2019;10:1787. doi: 10.3389/fimmu.2019.01787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sallusto F., Lenig D., Förster R., Lipp M., Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 27.Hawash M.B.F., Sanz-Remón J., Grenier J.C., Kohn J., Yotova V., Johnson Z., Lanford R.E., Brinkworth J.F., Barreiro L.B. Primate innate immune responses to bacterial and viral pathogens reveals an evolutionary trade-off between strength and specificity. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2015855118. e2015855118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Young C.S., Mokhonova E., Quinonez M., Pyle A.D., Spencer M.J. Creation of a Novel Humanized Dystrophic Mouse Model of Duchenne Muscular Dystrophy and Application of a CRISPR/Cas9 Gene Editing Therapy. J. Neuromuscul. Dis. 2017;4:139–145. doi: 10.3233/JND-170218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herzog R.W., Yang E.Y., Couto L.B., Hagstrom J.N., Elwell D., Fields P.A., Burton M., Bellinger D.A., Read M.S., Brinkhous K.M., et al. Long-term correction of canine hemophilia B by gene transfer of blood coagulation factor IX mediated by adeno-associated viral vector. Nat. Med. 1999;5:56–63. doi: 10.1038/4743. [DOI] [PubMed] [Google Scholar]
- 30.Kotterman M.A., Yin L., Strazzeri J.M., Flannery J.G., Merigan W.H., Schaffer D.V. Antibody neutralization poses a barrier to intravitreal adeno-associated viral vector gene delivery to non-human primates. Gene Ther. 2015;22:116–126. doi: 10.1038/gt.2014.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verdera H.C., Kuranda K., Mingozzi F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol. Ther. 2020;28:723–746. doi: 10.1016/j.ymthe.2019.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y.C., Powers M., Wasserfall C., Brusko T., Song S., Flotte T., Snyder R.O., Potter M., Scott-Jorgensen M., Campbell-Thompson M., et al. Immunity to adeno-associated virus serotype 2 delivered transgenes imparted by genetic predisposition to autoimmunity. Gene Ther. 2004;11:233–240. doi: 10.1038/sj.gt.3302144. [DOI] [PubMed] [Google Scholar]
- 33.Davies D.H., Liang X., Hernandez J.E., Randall A., Hirst S., Mu Y., Romero K.M., Nguyen T.T., Kalantari-Dehaghi M., Crotty S., et al. Profiling the humoral immune response to infection by using proteome microarrays: high-throughput vaccine and diagnostic antigen discovery. Proc. Natl. Acad. Sci. USA. 2005;102:547–552. doi: 10.1073/pnas.0408782102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davies D.H., Molina D.M., Wrammert J., Miller J., Hirst S., Mu Y., Pablo J., Unal B., Nakajima-Sasaki R., Liang X., et al. Proteome-wide analysis of the serological response to vaccinia and smallpox. Proteomics. 2007;7:1678–1686. doi: 10.1002/pmic.200600926. [DOI] [PubMed] [Google Scholar]
- 35.Vigil A., Davies D.H., Felgner P.L. Defining the humoral immune response to infectious agents using high-density protein microarrays. Future Microbiol. 2010;5:241–251. doi: 10.2217/fmb.09.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weinmann J., Weis S., Sippel J., Tulalamba W., Remes A., El Andari J., Herrmann A.K., Pham Q.H., Borowski C., Hille S., et al. Identification of a myotropic AAV by massively parallel in vivo evaluation of barcoded capsid variants. Nat. Commun. 2020;11:5432. doi: 10.1038/s41467-020-19230-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hamilton B.A., Wright J.F. Challenges Posed by Immune Responses to AAV Vectors: Addressing Root Causes. Front. Immunol. 2021;12:675897. doi: 10.3389/fimmu.2021.675897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martino A.T., Suzuki M., Markusic D.M., Zolotukhin I., Ryals R.C., Moghimi B., Ertl H.C.J., Muruve D.A., Lee B., Herzog R.W. The genome of self-complementary adeno-associated viral vectors increases Toll-like receptor 9-dependent innate immune responses in the liver. Blood. 2011;117:6459–6468. doi: 10.1182/blood-2010-10-314518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sasai M., Linehan M.M., Iwasaki A. Bifurcation of Toll-like receptor 9 signaling by adaptor protein 3. Science (New York, N.Y.) 2010;329:1530–1534. doi: 10.1126/science.1187029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wright J.F. Quantification of CpG Motifs in rAAV Genomes: Avoiding the Toll. Mol. Ther. 2020;28:1756–1758. doi: 10.1016/j.ymthe.2020.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guillerey C., Mouriès J., Polo G., Doyen N., Law H.K.W., Chan S., Kastner P., Leclerc C., Dadaglio G. Pivotal role of plasmacytoid dendritic cells in inflammation and NK-cell responses after TLR9 triggering in mice. Blood. 2012;120:90–99. doi: 10.1182/blood-2012-02-410936. [DOI] [PubMed] [Google Scholar]
- 42.Krieg A.M., Efler S.M., Wittpoth M., Al Adhami M.J., Davis H.L. Induction of systemic TH1-like innate immunity in normal volunteers following subcutaneous but not intravenous administration of CPG 7909, a synthetic B-class CpG oligodeoxynucleotide TLR9 agonist. J. Immunother. 2004;27:460–471. doi: 10.1097/00002371-200411000-00006. [DOI] [PubMed] [Google Scholar]
- 43.Krogmann A.O., Lüsebrink E., Steinmetz M., Asdonk T., Lahrmann C., Lütjohann D., Nickenig G., Zimmer S. Proinflammatory Stimulation of Toll-Like Receptor 9 with High Dose CpG ODN 1826 Impairs Endothelial Regeneration and Promotes Atherosclerosis in Mice. PLoS One. 2016;11:e0146326. doi: 10.1371/journal.pone.0146326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kwissa M., Nakaya H.I., Oluoch H., Pulendran B. Distinct TLR adjuvants differentially stimulate systemic and local innate immune responses in nonhuman primates. Blood. 2012;119:2044–2055. doi: 10.1182/blood-2011-10-388579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mathes A.L., Rice L., Affandi A.J., DiMarzio M., Rifkin I.R., Stifano G., Christmann R.B., Lafyatis R. CpGB DNA activates dermal macrophages and specifically recruits inflammatory monocytes into the skin. Exp. Dermatol. 2015;24:133–139. doi: 10.1111/exd.12603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ellis B.L., Hirsch M.L., Barker J.C., Connelly J.P., Steininger R.J., 3rd, Porteus M.H. A survey of ex vivo/in vitro transduction efficiency of mammalian primary cells and cell lines with Nine natural adeno-associated virus (AAV1-9) and one engineered adeno-associated virus serotype. Virol. J. 2013;10:74. doi: 10.1186/1743-422x-10-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hösel M., Broxtermann M., Janicki H., Esser K., Arzberger S., Hartmann P., Gillen S., Kleeff J., Stabenow D., Odenthal M., et al. Toll-like receptor 2-mediated innate immune response in human nonparenchymal liver cells toward adeno-associated viral vectors. Hepatology. 2012;55:287–297. doi: 10.1002/hep.24625. [DOI] [PubMed] [Google Scholar]
- 48.Alonso A., Bayón Y., Renedo M., Crespo M.S. Stimulation of Fc gamma R receptors induces monocyte chemoattractant protein-1 in the human monocytic cell line THP-1 by a mechanism involving I kappa B-alpha degradation and formation of p50/p65 NF-kappa B/Rel complexes. Int. Immunol. 2000;12:547–554. doi: 10.1093/intimm/12.4.547. [DOI] [PubMed] [Google Scholar]
- 49.Song W.C. Crosstalk between complement and toll-like receptors. Toxicol. Pathol. 2012;40:174–182. doi: 10.1177/0192623311428478. [DOI] [PubMed] [Google Scholar]
- 50.Thieblemont N., Haeffner-Cavaillon N., Haeffner A., Cholley B., Weiss L., Kazatchkine M.D. Triggering of complement receptors CR1 (CD35) and CR3 (CD11b/CD18) induces nuclear translocation of NF-kappa B (p50/p65) in human monocytes and enhances viral replication in HIV-infected monocytic cells. J. Immunol. 1995;155:4861–4867. [PubMed] [Google Scholar]
- 51.Ronzitti G., Gross D.A., Mingozzi F. Human Immune Responses to Adeno-Associated Virus (AAV) Vectors. Front. Immunol. 2020;11:670. doi: 10.3389/fimmu.2020.00670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harder M.J., Kuhn N., Schrezenmeier H., Höchsmann B., von Zabern I., Weinstock C., Simmet T., Ricklin D., Lambris J.D., Skerra A., et al. Incomplete inhibition by eculizumab: mechanistic evidence for residual C5 activity during strong complement activation. Blood. 2017;129:970–980. doi: 10.1182/blood-2016-08-732800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fitzpatrick Z., Leborgne C., Barbon E., Masat E., Ronzitti G., van Wittenberghe L., Vignaud A., Collaud F., Charles S., Simon Sola M., et al. Influence of Pre-existing Anti-capsid Neutralizing and Binding Antibodies on AAV Vector Transduction. Molecular therapy. Mol. Ther. Methods Clin. Dev. 2018;9:119–129. doi: 10.1016/j.omtm.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mori S., Takeuchi T., Kanda T. Antibody-dependent enhancement of adeno-associated virus infection of human monocytic cell lines. Virology. 2008;375:141–147. doi: 10.1016/j.virol.2008.01.033. [DOI] [PubMed] [Google Scholar]
- 55.Himeda C.L., Chen X., Hauschka S.D. Design and testing of regulatory cassettes for optimal activity in skeletal and cardiac muscles. Methods Mol. Biol. 2011;709:3–19. doi: 10.1007/978-1-61737-982-6_1. [DOI] [PubMed] [Google Scholar]
- 56.Weeratna R.D., Wu T., Efler S.M., Zhang L., Davis H.L. Designing gene therapy vectors: avoiding immune responses by using tissue-specific promoters. Gene Ther. 2001;8:1872–1878. doi: 10.1038/sj.gt.3301602. [DOI] [PubMed] [Google Scholar]
- 57.Salabarria S.M., Nair J., Clement N., Smith B.K., Raben N., Fuller D.D., Byrne B.J., Corti M. Advancements in AAV-mediated Gene Therapy for Pompe Disease. J. Neuromuscul. Dis. 2020;7:15–31. doi: 10.3233/jnd-190426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lan K., Verma S.C., Murakami M., Bajaj B., Robertson E.S. Isolation of human peripheral blood mononuclear cells (PBMCs) Curr Protoc Microbiol. 2007 doi: 10.1002/9780471729259.mca04cs6. Appendix Appendix 4, Appendix 4C, Appendix 4C. [DOI] [PubMed] [Google Scholar]
- 59.Nishimura M., Mitsunaga S., Juji T. Frozen-stored granulocytes can be used for an immunofluorescence test to detect granulocyte antibodies. Transfusion. 2001;41:1268–1272. doi: 10.1046/j.1537-2995.2001.41101268.x. [DOI] [PubMed] [Google Scholar]
- 60.Wright J.F. AAV vector manufacturing process design and scalability - Bending the trajectory to address vector-associated immunotoxicities. Mol. Ther. 2022;30:2119–2121. doi: 10.1016/j.ymthe.2022.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bengtsson N.E., Hall J.K., Odom G.L., Phelps M.P., Andrus C.R., Hawkins R.D., Hauschka S.D., Chamberlain J.R., Chamberlain J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017;8:14454. doi: 10.1038/ncomms14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramos J.N., Hollinger K., Bengtsson N.E., Allen J.M., Hauschka S.D., Chamberlain J.S. Development of Novel Micro-dystrophins with Enhanced Functionality. Mol. Ther. 2019;27:623–635. doi: 10.1016/j.ymthe.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang J., Vasaikar S., Shi Z., Greer M., Zhang B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017;45 doi: 10.1093/nar/gkx356. W130-w137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used to evaluate the conclusions of the article are present in the paper and/or supplementary material. Further inquiries regarding the data can be directed to the corresponding author and will be made available upon request.