

Abstract
Propionic acidemia (PA) is rare autosomal recessive metabolic disorder caused by defects in the mitochondrially localized enzyme propionyl-coenzyme A (CoA) carboxylase. Patients with PA can suffer from lethal metabolic decompensation and cardiomyopathy despite current medical management, which has led to the pursuit of gene therapy as a new treatment option for patients. Here we assess the therapeutic efficacy of a recently described adeno-associated virus (AAV) capsid, AAV44.9, to deliver a therapeutic PCCA transgene in a new mouse model of propionyl-CoA carboxylase α (PCCA) deficiency generated by genome editing. Pcca−/− mice recapitulate the severe neonatal presentation of PA and manifest uniform neonatal lethality, absent PCCA expression, and increased 2-methylcitrate. A single injection of the AAV44.9 PCCA vector in the immediate newborn period, systemically delivered at a dose of 1e11 vector genome (vg)/pup but not 1e10 vg/pup, increased survival, reduced plasma methylcitrate, and resulted in high levels of transgene expression in the liver and heart in treated Pcca−/− mice. Our studies not only establish a versatile and accurate new mouse model of PA but further demonstrate that the AAV44.9 vectors may be suitable for treatment of many metabolic disorders where hepato-cardiac transduction following systemic delivery is desired, such as PA, and, by extension, fatty acid oxidation defects and glycogen storage disorders.
Keywords: AAV44.9, propionic acidemia, systemic gene therapy, mouse model, genome editing, organic acidemia, neonatal gene therapy, cardiac tropism, PCCA, propionylcoa carboxylase deficiency
Graphical abstract
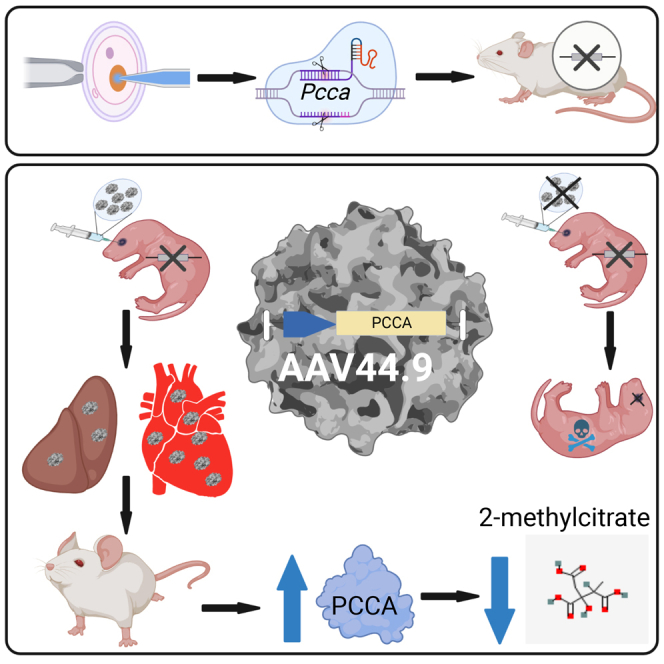
Venditti and colleagues use the AAV44.9 vector to treat a severe murine model of propionic acidemia, a lethal metabolic disorder. The AAV44.9 capsid effectively delivered a PCCA transgene to the liver and heart. AAV44.9 could be applicable to other diseases where hepatic and cardiac gene delivery are required.
Introduction
Propionic acidemia (PA) is a well-recognized autosomal recessive disorder of organic acid metabolism, occurring with an estimated incidence of 1:250,000–1:750,000 births.1,2 It is caused by a deficiency of propionyl-coenzyme A (CoA) carboxylase (PCC), a ubiquitously expressed, mitochondrial enzyme involved primarily in the catabolism of propiogenic amino acids, particularly isoleucine, valine, methionine, and threonine, as well as odd-chain fatty acids.3,4 The enzyme is composed of six α and six β subunits encoded by their respective genes, PCCA and PCCB.5 PA is caused by variants in the PCCA or PCCB genes at equal frequencies.2,6,7,8,9 PCC catalyzes the first step in conversion of propionyl-CoA to D-methylmalonyl-CoA in the pathway of propionyl-CoA oxidation, depicted in (Figure 1). PCC deficiency leads to an increase in the levels of propionyl-CoA-derived metabolites, including 2-methylcitrate,10 propionylcarnitine,11 and 3-hydroxypropionate,12 which have been used as metabolic biomarkers of PA to identify patients on newborn screening, for clinical monitoring, and in genomic therapy studies.13,14,15
Figure 1.

Mitochondrial catabolism of propionyl-CoA to succinyl-CoA
Propionic acidemia (PA) is caused by mutations in the PCCA or PCCB gene, which encode for the respective subunits of propionyl-CoA carboxylase (PCC). Enzymes downstream of PCC in the metabolism of propionyl-CoA to succinyl-CoA, a Krebs cycle intermediate. Other enzymes include D-methylmalonyl-CoA epimerase (MCEE) and methylmalonyl-CoA mutase (MMUT). The figure was created with BioRender.
Most frequently, patients with PA present in the neonatal period with a life-threatening metabolic crisis, characterized by vomiting, poor feeding, hypotonia, ketoacidosis, and hyperammonemia.16 Those who survive suffer from recurrent metabolic instability and can develop multisystemic complications, which can include cardiomyopathy.17,18 Over the decades, it has been recurrently documented that PA patients with an early and severe clinical course experience increased mortality and disease-associated morbidity.2,19,20 The recalcitrant nature of the disorder to conventional medical management, including dietary restriction of amino acid precursors, L-carnitine supplementation, and administration of metronidazole to reduce generation of propionic acid by intestinal bacteria, has led to implementation of elective liver transplantation (LT) as an experimental surgical treatment for PA.21,22,23 While not curative of all aspects of this disorder, successful LT in the setting of PA provides restoration of metabolic stability and protection from early death and therefore represents a clinical benchmark for gene replacement or addition approaches that might increase hepatic PCC expression and activity.22 Whether an LT is protective against development of cardiomyopathy in PA is unknown but seems unlikely, given the recent reports describing progression of cardiac disease after successful LT, leading to death in one reported patient and the need for a heart transplant in another,24,25 highlighting the need for therapies that target extrahepatic organs.
Over the past decade, adenovirus- and adeno-associated virus (AAV)-based gene therapies have been studied in mouse models of PCCA deficiency, with variable success.13,14,19,26 The mouse models used in these studies were made either by creating a large genomic deletion in the Pcca gene or through overexpression of a hypomorphic PCCA allele as a germline transgene under control of a strong heterologous enhancer/promoter.14,27 Here, we used genome editing to create a small deletion in exon 5 of Pcca, resulting in a frameshift (c.398_401delAAGC p.(Gln133Leufs∗41)), similar to the type of variants that occur in some PA patients.28 Homozygous Pccac.398_401delAAGC mice, hereafter designated Pcca−/−, recapitulate the severe neonatal presentation of PA and manifest uniform neonatal lethality, absent PCCA expression, and increased 2-methylcitrate. We next explored the utility of an AAV44.9 pseudoserotyped vector to treat Pcca−/− mice.29,30 This capsid was selected because it has recently been recognized to result in pronounced hepato-cardiac transduction after systemic delivery and to treat mice with the related disorder methylmalonic acidemia (MMA).31 Here we show that a single dose of an AAV44.9-PCCA vector, delivered by retro-orbital injection at a dose of 1e11 vg/pup, increased survival and PCCA mRNA and PCCA protein levels and reduced plasma 2-methylcitrate levels in treated PA mice. Of note, there was enhanced transduction and robust transgene expression in the hearts of the treated Pcca−/− mice, which suggests that AAV44.9 vectors, in addition to restoring hepatic PCC activity, might be useful to treat cardiomyopathy, a common and often lethal complication of PA.
Results
Creation and characterization of a new Pcca allele
CRISPR-Cas9 genome editing was used to engineer a frameshift-stop mutation in the Pcca gene. A CRISPR guide RNA was designed to target the biotin carboxylation domain of the PCCA subunit, and co-injected with Cas9 mRNA were into zygotes, resulting in a 4-bp deletion in exon 5 of the Pcca gene after cleavage and non-homologous end joining (Figure 2). The corresponding variant, Pccac.398_401delAAGC, is predicted to result in the truncated and non-functional protein PCCA p.Gln133Leufs∗41 (Figure S1).
Figure 2.

Schematic of the Pccac.398_401delAAGC allele
Exon 5 of the Pcca was targeted with a single-guide RNA (sgRNA) resulting in a 4-bp deletion after Cas9 cleavage and repair by non-homologous end joining (NEHJ). Shown are Sanger sequencing results of genomic DNA from a mouse with a WT Pcca genotype compared with a mouse homozygous for the Pccac.398_401delAAGC mutation (bottom). The figure was created with BioRender.
Pcca−/− mice display a severe neonatal-lethal phenotype (Figure 3A). At birth, these mice are indistinguishable from their phenotypically wild-type littermates (Pcca+/+ and Pcca+/−) but have significantly decreased survival (p < 0.001, log rank [Mantel-Cox] test), uniformly perishing within the first 2 days of life. Pcca+/+, Pcca+/−, and Pcca−/− mice were culled on day of life 1 to examine metabolic changes and Pcca expression. Pcca−/− newborn pups had significantly elevated plasma 2-methylcitrate (Figure 3B) in comparison with their wild-type newborn littermates (Pcca−/− = 37.89 ± 1.35 μM versus wild type [WT] = 0.62 ± 0.02 μM, Mann-Whitney test, p < 0.001). Total RNA was extracted from the liver of the pups, and qRT-PCR was performed to access the relative transcriptional level to the endogenous murine Pcca mRNA with normalization to β-actin (Actb) mRNA. Pcca+/− and Pcca−/− mice had significantly lower levels Pcca mRNA expression than Pcca+/+ mice (Figure 3C), with mean levels of Pcca mRNA of 56.3% ± 3.8% (p < 0.001, one-way ANOVA) and 11.3% ± 0.3% (p < 0.001, one-way ANOVA), respectively. Pcca−/− mice also had significantly lower levels of Pcca mRNA expression than Pcca+/− mice (p < 0.01, one-way ANOVA). Immunoblotting of liver tissue lysates from Pcca−/− newborn pups with an anti-PCCA antibody failed to detect PCCA protein relative to their phenotypically normal littermates (Figure 3D). While the levels of endogenous PCCA in the livers of Pcca+/− mice appeared to be lower than the PCCA levels in Pcca+/+ mice, this difference was not significant (Figure S2). Hepatic lipidosis was the only histopathology finding in newborn Pcca−/− pups (Figure S3A). Notably, the heart was grossly and microscopically without abnormalities (Figure S3B).
Figure 3.

Clinical and biochemical characterization of Pcca−/− mice
(A) Survival of mice with Pcca+/+,Pcca+/−, and a Pcca−/− genotypes. (B) Plasma 2-methylcitrate levels from Pcca+/+ and Pcca+/− newborn pups (n = 13) in comparison with Pcca−/− newborn pups (n = 6). (C) Pcca mRNA levels in the liver of Pcca+/+ (n = 3), Pcca+/− (n = 3), and Pcca−/− (n = 3) mice, shown as a percentage of WT Pcca mRNA expression normalized to Actb. (D) Immunoblot of 50 μg hepatic protein from Pcca+/+, Pcca+/−, and Pcca−/− newborn pups for the PCCA protein using the mitochondrial protein ubiquinol-cytochrome c reductase core protein 2 (UQCRC2) as a loading control. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Treatment with an AAV44.9 vector increases the survival of Pcca−/− mice and decreases a disease-related biomarker
A previously described AAV PCCA transgene, pAAV-CBA-PCCA, was pseudotyped with an AAV44.9 capsid to create AAV44.9-CBA-PCCA (Figure 4A).13 Pcca−/− mice were injected via the retro-orbital route on the first day of life with either 1e10 vg or 1e11 vg of AAV44.9-CBA-PCCA. The mice treated with a dose of 1e11 vg had a significant increase in survival (Mantel-Cox test, p = 0.003), with a few treated mice surviving longer than 150 days (Figure 4B). By comparison, Pcca−/− mice that were treated with a dose of 1e10 vg or left untreated did not survive more than 2 days. The AAV44.9-CBA-PCCA-treated Pcca−/− mice that survived were smaller than their WT littermates (Figure S4) and appeared otherwise healthy but slowly declined, with ruffled coats, hunching, and progressive lethargy. The cause(s) of demise in the mice was/were undetermined, with most animals found dead and unsuitable for further study. Gross examination of the deceased mice that were studied before autolysis was unremarkable, with no obvious changes in the size or appearance of the internal organs. 4 days (n = 3) and 1–3 months (n = 14) post treatment, the AAV44.9-CBA-PCCA-treated Pcca−/− mice had significantly reduced mean plasma 2-methylcitrate levels (p = 0.002 and 0.02, respectively; one-way ANOVA) of 32.7 ± 5.1 μM in comparison with untreated newborn Pcca−/− pups (n = 13), which had a mean of level of 45.1 ± 2.1 μM (Figure 4C). However, 2-methylcitrate levels in treated Pcca−/− mice remained elevated in comparison with Pcca+/+ mice (n = 5) at 1 month of age, which had a mean level of 0.56 ± 0.21 μM.
Figure 4.

AAV44.9 gene delivery of PCCA rescues Pcca−/− mice from neonatal lethality
(A) Schematic of the recombinant AAV vector genome (pAAV-CBA-PCCA) packaged with the AAV44.9 capsid (CMV, cytomegalovirus; CBA, chicken β-actin). Pcca−/− pups received a retro-orbital injection of 1e10 vg or 1e11 vg of AAV44.9-CBA-PCCA on day of life 1 (DOL 1). (B) Survival curve comparing the viability of Pcca−/− mice treated with AAV44.9 at doses of 1e10 vg or 1e11 vg with untreated Pcca−/− mice. (C) Plasma 2-methylcitrate in untreated neonatal Pcca−/− mice and Pcca−/− mice 4 days and 1–3 months after treatment with AAV44.9 at 1e11 vg. (D) Vector biodistribution in liver, heart, brain skeletal muscle, and kidneys from Pcca−/− treated mice (n = 3) 4 days and 1 month post treatment, as determined by ddPCR. (E) PCCA and Pcca mRNA expression in the liver and heart (WT, n = 3, 3; PA, n = 2, 1; AAV44.9, n = 3, 3), shown as a percentage of WT Pcca mRNA expression normalized to Actb (liver) or Gapdh (heart). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; ns, not significant; ND, not detected.
Vector biodistribution, PCCA mRNA expression, and PCCA protein transgene expression
Three AAV44.9 treated Pcca−/− mice were sacrificed at 4 days and 1 month post treatment to determine the biodistribution of the vector and the expression of mRNA and protein from the AAV transgene. Vector copy number in the liver, heart, skeletal muscle, brain, and kidneys was determined using digital droplet PCR (ddPCR) to detect the PCCA transgene and the murine Gapdh gene and is reported as a ratio of the copy number of PCCA to Gapdh (Figure 4D). Pcca−/− mice on day 4 post treatment had mean vector copies in the heart (29.04 ± 7.65, n = 3) and liver (28.53 ± 8.22, n = 3). The ratio of PCCA/Gapdh 30 days post treatment was highest in the heart (3.7 ± 0.6, n = 3) in comparison with the liver (2.2 ± 0.7, n = 3), and both ratios were significantly higher than in skeletal muscle, brain, and kidneys, which all had mean values of less than 0.5 PCCA/Gapdh. Transgene mRNA expression in AAV44.9-treated Pcca−/− mice 30 days post treatment and untreated controls was determined using probes specific to PCCA, Actb (liver), Gapdh (heart), and endogenous Pcca. PCCA mRNA was detectable by qRT-PCR 1 month post treatment in the livers and hearts of AAV44.9-treated Pcca−/− mice at levels of approximately 4% and 160% that of WT endogenous Pcca RNA expression (Figure 4E). Low levels of a Pcca transcript were detectable in the livers and hearts of untreated Pcca−/− mice at approximately 10%–15% of the levels seen in WT mice, and no PCCA transgene mRNA signal was detected in untreated control mice. Immunoblotting with an antibody capable of detecting the endogenous murine and the human PCCA protein was used to determine PCCA expression at 4 days (Figure 5A) and 1 month post treatment with a retro-orbital sinus injection on day of life 1 with 1e11 vg of AAV44.9. While no PCCA expression was detected in untreated Pcca−/− mice, AAV44.9-treated Pcca−/− mice 4 days post treatment were determined to have 68.5% ± 24.4% (n = 3) and 352.4% ± 92.6.0% (n = 2) of WT (Pcca+/+) PCCA protein levels in the liver and heart, respectively (Figures 5A and 5C). Interestingly, PCCA protein expression in the liver of a single treated AAV44.9 Pcca+/− mouse 4 days after treatment was higher than the PCCA protein expression in the liver of an untreated Pcca+/− mouse (Figure 5A) but less than that seen in the treated Pcca−/− mice. By 30 days post treatment, AAV44.9-treated Pcca−/− mice had decreased levels of relative PCCA protein of 49.7% ± 4.4% and 214% ± 19.0% in the liver (n = 3) and heart (n = 3), respectively (Figures 5D and 5C).
Figure 5.

Expression of PCCA protein in the liver and heart
(A) Immunoblot of hepatic and cardiac PCCA protein from untreated Pcca+/+ (n = 1), Pcca−/− (n = 1), and Pcca+/− (n = 1) and AAV44.9-treated Pcca+/− (n = 1) and Pcca−/− (n = 3) mice 4 days post treatment. (B) Immunoblot of hepatic and cardiac PCCA protein from untreated Pcca+/+ (n = 2) and Pcca−/− (n = 2) and AAV44.9-treated Pcca−/− mice (n = 3) 30 days post treatment. (C) Quantification of PCCA protein expression relative to untreated Pcca+/+ mice from the blots depicted in (A) and (B). ∗p < 0.05, ∗∗p < 0.003, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
RNA in situ hybridization to detect PCCA in the liver, heart, skeletal muscle, brain, and kidneys
A hybridization probe was designed specifically to detect the human PCCA mRNA expressed by the AAV vector transgene and not to cross-react with endogenous murine transcripts. PCCA mRNA was detected (punctate red staining) in the liver, heart, and skeletal muscle and variably in the brain but not in the kidneys 1 month post treatment (Figure 6). The greatest amount of PCCA mRNA was observed in the heart. Smaller numbers of positively stained cells were also seen in the liver and skeletal muscle. The distribution of PCCA RNA-positive cells was uniform in the heart, liver, and skeletal muscle. While two of the three AAV44.9-treated mice had minimal positive staining in the brain, a single mouse had a localized area with many positively stained cells (Figure 6C).
Figure 6.

RNA in situ hybridization in tissues 30 days after AAV44.9 gene delivery to detect PCCA expression
PCCA transgene mRNA (punctate red staining) in situ hybridization (magnification, 10× with 20× inset) of liver, heart, skeletal muscle, brain, and kidney tissue from three Pcca−/− AAV44.9-CBA-PCCA-treated mice, labeled A, B, and C.
Discussion
PA was first recognized as a human inborn error of metabolism in the 1960s, and the clinical presentation of severe patients has remained unchanged for many decades, with a neonatal metabolic crisis still typical as the first manifestation of illness, even in an era of newborn screening.32 While early recognition of PA and related organic acidemias during infancy has undoubtably led to improved diagnosis and a more rapid implementation of supportive therapies, such as hemodialysis, the outcomes for patients remain guarded.2 The difficult clinical course many patients experience has led to implementation of elective LT as a surgical treatment for PA, providing a strong rationale for development of alternative therapies.33,34,35,36
As part of an effort to develop AAV gene therapy for PA, we critically assessed the current animal models to inform our approach. The first mouse model of PA was generated by targeted disruption of the Pcca gene via homologous recombination in embryonic stem cells.27 The mutant allele, Pccadel, contains a several-kilobase-pair deletion in cis with a neomycin cassette and replaces exons 1–4 of the Pcca gene. Pccadel/del mice perish within the first 48 hours after birth and lack PCCA expression. The earliest gene therapy studies using these mice yielded disappointing results; treatment with an adenoviral vector designed to express the human PCCA cDNA produced an increase in life expectancy of only several hours, with AAV serotype 8 (AAV8)-mediated gene transfer of PCCA equally ineffective.26 However, a later study of the very same Pccadel/del mice, informed by a series of successful experiments with mouse models of MMA, demonstrated that systemic delivery of a reconfigured AAV8 vector that expressed the human PCCA gene under control of a strong heterologous promoter provided effective rescue from neonatal lethality, restoration of hepatic PCC expression, and a substantial reduction in the plasma level of the associated biomarker, 2-methylcitrate.13,37,38,39 Despite successful rescue with an improved AAV8 vector, the fragility of the Pccadel/del mice led Guenzel et al.14,19,40 to develop transgenic mice that overexpress a human mutation, PCCAp.A138T, under control of the hybrid cytomegalovirus early enhancer/chicken β-actin (CAG) promoter. Breeding to create Pccadel/del TgCAG-PCCA(p.A138T)IRES-GFP mice yielded a hypomorphic mouse model of PA. Pccadel/del TgCAG-PCCA(p.A138T)IRES-GFP mice are rescued from severe PA by expression of PCCAp.A138T and are mildly affected, displaying preserved survival into the adult period and only modest elevations of biochemical markers of the disease.14 Even though this model does not recapitulate the most salient clinical features of PA, specifically early lethality, Pccadel/del TgCAG-PCCA(p.A138T)IRES-GFP mice have been used to test adenoviral and AAV8, AAV1, and AAVrh10 PCCA vectors, all of which variably reduce the levels of metabolites after gene delivery.14,19,40 An additional limitation of this model is that the rescue transgene does not uniformly express PCCAp.A138T in different tissues and between sexes. Indeed, Subramanian et al.41 noted that there was highly disparate PCCAp.A138T expression in Pccadel/del TgCAG-PCCA(p.A138T)IRES-GFPmice that did not correlate with the endogenous levels of PCCA, indicating that the rescue transgene is susceptible to epigenic effects or that overexpression of the mutant protein may have other untoward effects, such as interference with formation of a stable PCC heterododecamer. The gaps between the PA models and clinically relevant patient phenotypes led us to create a new Pcca allele, which was then used in a proof-of-concept study with a novel AAV capsid.
Because a majority of the PA patients have small deletions, insertions, and missense mutations in the PCCA gene, and the existing Pccadel/del TgCAG-PCCA(p.A138T)IRES-GFP model does not recapitulate the severe clinical phenotype of PA seen in many patients, especially those who might benefit from AAV gene therapy, we used genome editing to engineer a 4-bp deletion, causing a frameshift mutation (Pcca c.398_401delAAGC).6 Pcca−/− (c.398_401delAAGC) mice have a severe neonatal lethal phenotype and elevated plasma 2-methylcitrate and lack PCCA protein. Interestingly, Pcca+/− (c.398_401delAAGC) mice express less PCCA mRNA than Pcca+/+ mice but have similar levels of PCCA protein. As with carriers of PCCA variants in humans, heterozygous mice do not have elevated plasma 2-methylcitrate or display any phenotypes associated with PA.
The multisystemic presentation of PA and ubiquitous expression of the PCC enzyme led to the design of a vector transgene with a constitutive promoter to test AAV gene therapy and the in vivo potency of the AAV44.9 capsid. A previous study that examined systemic gene delivery in a mouse model of the related disorder MMA reported that AAV44.9 was highly efficient at transducing the liver and heart,31 targets for transgene expression in patients with PA. Indeed, AAV44.9-treated Pcca−/− (c.398_401delAAGC) mice had significantly improved survival and reduced 2-methylcitrate, a disease biomarker. Transduction and transgene expression were detected in multiple tissue types that included the liver, heart, skeletal muscle, and brain. The heart had the highest level transgene expression, followed by the liver. Unfortunately, most of the rescued PA mice died between 1 and 3 months of age, likely because of loss of the episomal AAV vector, a well recognized outcome that follows AAV delivery to neonatal animals.42,43 To date, only AAV44.9 and AAV8 gene delivery have been potent enough to rescue neonatal lethal models of PA, which is important because patients with early and severe presentation of PA have the greatest need for new therapies, and these AAV capsids likely have distinct seroreactivity in humans.31
The AAV44.9 capsid enabled widespread transduction and transgene expression in PA mice. In situ hybridization detected cells expressing PCCA mRNA in the liver, heart, skeletal muscle, and brain at levels consistent with the vector transgene in the respective tissues. The distribution of cells expressing the transgene appeared to be homogeneous except for the brain, where positively stained cells were localized in the forebrain. While two of the AAV44.9-treated mice studied only had rare cells that expressed PCCA, a single mouse had a larger area of positively stained cells. That this was a consequence of an aberrant retro-orbital injection seems likely, but the overall results suggest that AAV44.9 can transduce neurons. More targeted investigations to examine the specific neurotropism of AAV44.9, which was not an objective of the current study, will be necessary but seem promising.
Histological examination of the heart of untreated Pcca−/− (c.398_401delAAGC) neonatal mice in this study did not reveal any cardiac abnormalities (Figure S3). AAV44.9 treatment resulted in significant amounts of cardiac transduction and PCCA protein expression, which could be vital for treatment of the potentially lethal cardiomyopathy associated with PA. While AAV44.9-treated Pcca−/− mice had normal histology, without aged matched untreated controls it is not possible to assess the effects of gene therapy on the heart (Figure S5). Therefore, to truly measure the effects of gene therapy on the cardiomyopathy of PA, the creation of Pcca knockin, conditional, or tissue-specific transgenic rescue mice may be required to generate a model with a consistent cardiac phenotype that replicates the human condition.
In conclusion, a new murine model of severe PA caused by PCCA deficiency has been created and characterized. The mutation is similar to those seen in patients and might be amenable to treatments using other forms of genomic therapy, such as prime editing.44 Use of an AAV44.9 vector resulted in striking clinical and biochemical improvements in these severely affected PA mice, with robust transduction of the heart, an important target organ for treatment of PA because severe cardiomyopathy can develop in PA patients, even after LT.24,25 Pseudoserotyped AAV44.9 vectors might therefore be especially well suited for treatment of other metabolic disorders like PA, where hepatic and cardiac targeting is needed, such as glycogen storage disorders and fatty acid oxidation defects.
Materials and methods
Animal studies
Animal work was approved by and performed in accordance with the guidelines for animal care at the National Institutes of Health and the Guide for the Care and Use of Laboratory Animals. All mice were housed in micro-isolators maintained under a 12-h light/12-h dark cycle. Mice were maintained on standard mouse chow (PicoLab Mouse Diet 20, LabDiet, St. Louis, MO, USA) and water, which was available ad libitum. Heterozygous carriers of the Pcca mutation were bred to generate Pcca+/− and Pcca−/− mice.
Generation of a murine model of PA caused by PCCA deficiency using CRISPR-Cas9 genome editing
DESKGEN software was used to identify protospacer-adjacent motif (PAM) sequences (NGG) from Streptococcus pyogenes (SpyCas9) targeting exon 5 of the Pcca gene for mutagenesis. A synthetic single-guide RNA (sgRNA) was custom synthesized by Horizon Discovery (Edit-R predesigned synthetic sgRNA). Zygotes were co-injected with the Cas9 mRNA and sgRNA and cultured at 37°C under 5% CO2 until the blastocyst stage and then transferred into the uterus of pseudo-pregnant females.
The Pcca exon5 sgRNA sequence used was : 5′-GTTCGCTCAGACTCACAGCT-3′ PAM-TGG.
Founder and colony genotyping
DNA was extracted from tail clips using the DNeasy Blood & Tissue Kit (QIAGEN, Hilden, Germany) for PCR amplification. DNA samples from founders were screened using flourescent PCR (fPCR) for gene knockout using primers (Pcca E5_fPCRFor, Pcca E5_fPCRRev) that spanned exon 5 of the Pcca gene and crossed with WT FvBN mice to generate heterozygous carriers of the mutation, which were further bred to generate Pcca−/− mice. The sequence of the mutation was determined by targeted PCR of genomic DNA and Sanger sequencing using primers (Pcca E5for, Pcca E5rev) that flanked exon 5 of the Pcca gene. After confirmation of the genetic mutation, genotyping of tail snip lysate by qPCR was performed by Transnetyx. The qPCR primers and probes were as follows:
Pcca E5_fPCRfor, 5′-TGTAAAACGACGGCCAGTGAGCATCTGTTAATACAGGG-3′
Pcca E5_fPCRrev, 5′-GTGTCTTAACTCCAGCTTTCTGCTC-3′
Pcca E5for, 5′-AGTCACGGGTGATGCTGACC-3′
Pcca E5rev, 5′-CCTGAACTGAAGCTGCTC-3′
qPCR Pcca for, 5′-CTACCTCAACATGGATGCCATCAT-3′
qPCR Pcca rev, 5′-CATATGAAACAGCIGCAGTAGATAGGT-3′
qPCR reporter 1, 5′-TCACAGCTTGGGCCCT-3′
qPCR reporter 2, 5′-AGACTCACAGGGCCCT-3′
Metabolic measurements
Blood samples were collected using retro-orbital sinus plexus sampling with sterile heparinized glass capillary tubes. Samples were immediately centrifuged, and plasma was collected, diluted in water, and frozen at −80°C until measurements were performed. Plasma 2-methylcitrate levels were quantified by gas chromatography-mass spectrometry with stable isotopic internal calibration as described previously.45,46
rAAV44.9 construction, production, and delivery
The viral construct pAAV2.CB7.CI.RBG was graciously provided by the University of Pennsylvania Vector Core (Philadelphia, PA, USA), and a full-length human PCCA cDNA was cloned into this vector as described previously.13 Production and purification of recombinant AAV44.9 were performed using a four-plasmid (AAV44.9) procedure as described previously.30,31 Retro-orbital injections were performed on non-anesthetized neonatal mice, typically within several hours after birth. In treated litters, all pups were injected without genotyping. Viral particles were diluted to a total volume of 40 μL with phosphate-buffered saline immediately before injection.
Titration of AAVs
AAV44.9-CBA-PCCA titer was determined by quantitative real-time PCR analysis with TaqMan gene expression assays (human PCC [Hs01120571_m1]; Applied Biosystems, Foster City, CA, USA).
Vector genome quantification by ddPCR
Genomic DNA from tissue samples was extracted using the DNeasy Blood & Tissue Kit (catalog number 69506; QIAGEN). ddPCR was performed according to the manufacturer’s recommendations using a Bio-Rad (Hercules, CA, USA) QX200 AutoDG ddPCR system with the following probes: Bio-Rad ddPCR copy number variation (CNV) assay Gapdh (assay ID dMumCNS300520369) and PCCA (assay ID dHsaCPE5056812).
Relative PCCA mRNA expression
Total RNA was extracted using RNeasy Mini with DNase digestion using the RNase-Free DNase Set (catalog numbers 74804 and 79254, QIAGEN). A cDNA library was prepared using the High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (catalog number 4374966, Applied Biosystems). TaqMan gene expression assays for human PCCA, murine Pcca, and murine Actb and Gapdh (assay IDs Hs00165407_m1, Mm00454899_m1, and Mm00607939_s1 for liver and Mm99999915_g1 for heart, respectively; Applied Biosystems) were performed on an CFX96 Real-Time System (Bio-Rad). The gene expression assays use primers specific to the mouse Pcca and human PCCA mRNA. Quantification of relative gene expression, presented as percentage of the relevant baseline (WT), was calculated using the 2ˆ-[delta][delta]Cq (quantification cycle) method.
PCCA mRNA in situ hybridization
Liver, heart, brain, kidney, and skeletal muscle tissues were fixed in 4% paraformaldehyde and embedded in paraffin blocks. Five-micrometer sections were cut and stained with the RNAScope 2.5 HD Assay-Brown (322300; ACDBio, Newark, CA, USA), following the manufacturer’s instructions. Slide images were captured using the Carl Zeiss AxioScan Z1 slide scanner and analyzed using Image Pro Premier 3D v.9.3 (Media Cybernetics).
Immunoblot analysis
Previously snap-frozen livers and hearts were homogenized in of Tissue Protein Extraction Reagent (catalog number 78510; Thermo Scientific, Waltham, MA, USA) with cOmplete Tablets, Mini EDTA-free (catalog number 04693159001, Roche, Indianapolis, IN, USA). Protein concentrations were determined using a Protein Assay Dye Reagent Concentrate (catalog number 5000006; Bio-Rad) according to the manufacturer’s protocol. A total of 50 μg of total protein was separate by gel electrophoresis using the Mini-PROTEAN Tetra Vertical Electrophoresis Cell with Mini-PROTEAN TGX Gels (catalog numbers 1658004 and 4561084, Bio-Rad, respectively). Protein transfer was accomplished using a Trans-Blot Turbo Transfer Pack (catalog number 1704156, Bio-Rad). EveryBlot Blocking Buffer (catalog number 12010020, Bio-Rad) was used for blocking and antibody hybridization. The following antibodies were used for detection of PCCA (Abcam, ab187686) at a dilution of 1:1,000; UQCRC2 (Abcam, ab14745) at a dilution of 1:2,000; GAPDH (Proteintech, catalog number 60004-1-Ig) at a dilution of 1:10,000; and β-actin (Proteintech, 66009-1-Ig) at a dilution of 1:10,000 in conjunction with the following secondary antibodies at a dilution of 1:20,000: (LI-COR Biosciences, 926-32213) and (LI-COR Biosciences, 926-68072). Blots were imaged using the LI-COR Biosciences Odyssey DLx imaging system and LI-COR Biosciences acquisition software.
Statistical analyses
Prism 9 v.9.2.0 (GraphPad) was used to analyze all data. Results are expressed as mean ± SD. Values of p < 0.05 were considered statistically significant. Depending on the experimental design, Mann-Whitney test, a one-way ANOVA, or log rank (Mantel-Cox) test were used to test for statistical significance, and the test used is specified under Results.
Acknowledgments
We appreciate the assistance of the NHGRI mouse core, especially Gene Elliot and Cecilia Rivas; the NCI Molecular Pathology Lab, especially Andrew Warner; and Darwin Romero for help with treating animals and collecting murine tissues. R.J.C., D.C., E.-Y.C., L.L., S.N.S., J.L.S., and C.P.V. were supported by the Intramural Research Program of the NHGRI through 1ZIAHG200318-16. C.P.V. also acknowledges support from the National Center for Advancing Translational Sciences (NCATS) and the Organic Acid Association. G.D.P. and J.A.C. were supported by the Intramural Research Program of the NIDCR through Z01 DE00695.
Author contributions
R.J.C., conceptualization, methodology, validation, formal analysis, investigation, data curation, writing (first draft, review and editing), visualization, and project administration; C.P.V. and J.A.C., conceptualization, writing (review and editing), funding acquisition, and project administration; G.D.P., D.C., E.-Y.C., S.N.S., J.L.S., V.H., and L.L., formal analysis, investigation, and writing (review and editing).
Declaration of interests
R.J.C., G.D.P., J.A.C., and C.P.V. are inventors on a patent application filed by the NIH on their behalf on use of AAV44.9 as a gene therapy vector to treat methylmalonic acidemia.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2023.06.008.
Supplemental information
Data and code availability
Data supporting the studies presented in this manuscript can be made available by request to the corresponding authors.
References
- 1.Almási T., Guey L.T., Lukacs C., Csetneki K., Vokó Z., Zelei T. Systematic literature review and meta-analysis on the epidemiology of propionic acidemia. Orphanet J. Rare Dis. 2019;14:40. doi: 10.1186/s13023-018-0987-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shchelochkov O.A., Carrillo N., Venditti C. In: GeneReviews((R)) Adam M.P., Everman D.B., Mirzaa G.M., Pagon R.A., Wallace S.E., Bean L.J.H., Gripp K.W., Amemiya A., editors. 1993. Propionic Acidemia. [Google Scholar]
- 3.Gompertz D., Storrs C.N., Bau D.C., Peters T.J., Hughes E.A. Localisation of enzymic defect in propionicacidaemia. Lancet. 1970;1:1140–1143. doi: 10.1016/s0140-6736(70)91216-x. [DOI] [PubMed] [Google Scholar]
- 4.Hsia Y.E., Scully K.J., Rosenberg L.E. Inherited propionyl-Coa carboxylase deficiency in "ketotic hyperglycinemia". J. Clin. Invest. 1971;50:127–130. doi: 10.1172/JCI106466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang C.S., Sadre-Bazzaz K., Shen Y., Deng B., Zhou Z.H., Tong L. Crystal structure of the alpha(6)beta(6) holoenzyme of propionyl-coenzyme A carboxylase. Nature. 2010;466:1001–1005. doi: 10.1038/nature09302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Desviat L.R., Pérez B., Pérez-Cerdá C., Rodríguez-Pombo P., Clavero S., Ugarte M. Propionic acidemia: mutation update and functional and structural effects of the variant alleles. Mol. Genet. Metabol. 2004;83:28–37. doi: 10.1016/j.ymgme.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Ma X., Liu Y., Chen Z.H., Zhang Y., Dong H., Song J.Q., Jin Y., Li M.Q., Kang L.L., He R.X., et al. [Phenotypes and genotypes of 78 patients with propionic acidemia] Zhonghua Yufang Yixue Zazhi. 2022;56:1263–1271. doi: 10.3760/cma.j.cn112150-20220620-00630. [DOI] [PubMed] [Google Scholar]
- 8.Liu Y., Chen Z., Dong H., Ding Y., He R., Kang L., Li D., Shen M., Jin Y., Zhang Y., et al. Analysis of the relationship between phenotypes and genotypes in 60 Chinese patients with propionic acidemia: a fourteen-year experience at a tertiary hospital. Orphanet J. Rare Dis. 2022;17:135. doi: 10.1186/s13023-022-02271-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ugarte M., Perez-Cerda C., Rodriguez-Pombo P., Desviat L.R., Perez B., Richard E., Muro S., Campeau E., Ohura T., Gravel R.A. Overview of mutations in the PCCA and PCCB genes causing propionic acidemia. Hum. Mutat. 1999;14:275–282. doi: 10.1002/(SICI)1098-1004(199910)14:4<275::AID-HUMU1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 10.Ando T., Rasmussen K., Wright J.M., Nyhan W.L. Isolation and identification of methylcitrate, a major metabolic product of propionate in patients with propionic acidemia. J. Biol. Chem. 1972;247:2200–2204. [PubMed] [Google Scholar]
- 11.Chace D.H., DiPerna J.C., Kalas T.A., Johnson R.W., Naylor E.W. Rapid diagnosis of methylmalonic and propionic acidemias: quantitative tandem mass spectrometric analysis of propionylcarnitine in filter-paper blood specimens obtained from newborns. Clin. Chem. 2001;47:2040–2044. [PubMed] [Google Scholar]
- 12.la Marca G., Malvagia S., Pasquini E., Innocenti M., Donati M.A., Zammarchi E. Rapid 2nd-tier test for measurement of 3-OH-propionic and methylmalonic acids on dried blood spots: reducing the false-positive rate for propionylcarnitine during expanded newborn screening by liquid chromatography-tandem mass spectrometry. Clin. Chem. 2007;53:1364–1369. doi: 10.1373/clinchem.2007.087775. [DOI] [PubMed] [Google Scholar]
- 13.Chandler R.J., Chandrasekaran S., Carrillo-Carrasco N., Senac J.S., Hofherr S.E., Barry M.A., Venditti C.P. Adeno-associated virus serotype 8 gene transfer rescues a neonatal lethal murine model of propionic acidemia. Hum. Gene Ther. 2011;22:477–481. doi: 10.1089/hum.2010.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guenzel A.J., Hofherr S.E., Hillestad M., Barry M., Weaver E., Venezia S., Kraus J.P., Matern D., Barry M.A. Generation of a hypomorphic model of propionic acidemia amenable to gene therapy testing. Mol. Ther. 2013;21:1316–1323. doi: 10.1038/mt.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang L., Park J.S., Yin L., Laureano R., Jacquinet E., Yang J., Liang S., Frassetto A., Zhuo J., Yan X., et al. Dual mRNA therapy restores metabolic function in long-term studies in mice with propionic acidemia. Nat. Commun. 2020;11:5339. doi: 10.1038/s41467-020-19156-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pena L., Franks J., Chapman K.A., Gropman A., Ah Mew N., Chakrapani A., Island E., MacLeod E., Matern D., Smith B., et al. Natural history of propionic acidemia. Mol. Genet. Metabol. 2012;105:5–9. doi: 10.1016/j.ymgme.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 17.Baumgartner D., Scholl-Bürgi S., Sass J.O., Sperl W., Schweigmann U., Stein J.I., Karall D. Prolonged QTc intervals and decreased left ventricular contractility in patients with propionic acidemia. J. Pediatr. 2007;150:192–197.e1. doi: 10.1016/j.jpeds.2006.11.043. [DOI] [PubMed] [Google Scholar]
- 18.North K.N., Korson M.S., Gopal Y.R., Rohr F.J., Brazelton T.B., Waisbren S.E., Warman M.L. Neonatal-onset propionic acidemia: neurologic and developmental profiles, and implications for management. J. Pediatr. 1995;126:916–922. doi: 10.1016/s0022-3476(95)70208-3. [DOI] [PubMed] [Google Scholar]
- 19.Guenzel A.J., Collard R., Kraus J.P., Matern D., Barry M.A. Long-term sex-biased correction of circulating propionic acidemia disease markers by adeno-associated virus vectors. Hum. Gene Ther. 2015;26:153–160. doi: 10.1089/hum.2014.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van der Meer S.B., Poggi F., Spada M., Bonnefont J.P., Ogier H., Hubert P., Depondt E., Rapoport D., Rabier D., Charpentier C., et al. Clinical outcome and long-term management of 17 patients with propionic acidaemia. Eur. J. Pediatr. 1996;155:205–210. doi: 10.1007/BF01953939. [DOI] [PubMed] [Google Scholar]
- 21.Charbit-Henrion F., Lacaille F., McKiernan P., Girard M., de Lonlay P., Valayannopoulos V., Ottolenghi C., Chakrapani A., Preece M., Sharif K., et al. Early and late complications after liver transplantation for propionic acidemia in children: a two centers study. Am. J. Transplant. 2015;15:786–791. doi: 10.1111/ajt.13027. [DOI] [PubMed] [Google Scholar]
- 22.Curnock R., Heaton N.D., Vilca-Melendez H., Dhawan A., Hadzic N., Vara R. Liver Transplantation in Children With Propionic Acidemia: Medium-Term Outcomes. Liver Transpl. 2020;26:419–430. doi: 10.1002/lt.25679. [DOI] [PubMed] [Google Scholar]
- 23.Vara R., Turner C., Mundy H., Heaton N.D., Rela M., Mieli-Vergani G., Champion M., Hadzic N. Liver transplantation for propionic acidemia in children. Liver Transpl. 2011;17:661–667. doi: 10.1002/lt.22279. [DOI] [PubMed] [Google Scholar]
- 24.Berry G.T., Blume E.D., Wessel A., Singh T., Hecht L., Marsden D., Sahai I., Elisofon S., Ferguson M., Kim H.B., et al. The re-occurrence of cardiomyopathy in propionic acidemia after liver transplantation. JIMD Rep. 2020;54:3–8. doi: 10.1002/jmd2.12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hejazi Y., Hijazi Z.M., Al-Saloos H., Omran T.B. The re-occurrence of dilated cardiomyopathy in propionic acidemia after liver transplantation requiring heart transplant, first case from Middle East. Cardiol. Young. 2022;33:86–89. doi: 10.1017/S104795112200035X. [DOI] [PubMed] [Google Scholar]
- 26.Hofherr S.E., Senac J.S., Chen C.Y., Palmer D.J., Ng P., Barry M.A. Short-term rescue of neonatal lethality in a mouse model of propionic acidemia by gene therapy. Hum. Gene Ther. 2009;20:169–180. doi: 10.1089/hum.2008.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyazaki T., Ohura T., Kobayashi M., Shigematsu Y., Yamaguchi S., Suzuki Y., Hata I., Aoki Y., Yang X., Minjares C., et al. Fatal propionic acidemia in mice lacking propionyl-CoA carboxylase and its rescue by postnatal, liver-specific supplementation via a transgene. J. Biol. Chem. 2001;276:35995–35999. doi: 10.1074/jbc.M105467200. [DOI] [PubMed] [Google Scholar]
- 28.Kraus J.P., Spector E., Venezia S., Estes P., Chiang P.W., Creadon-Swindell G., Müllerleile S., de Silva L., Barth M., Walter M., et al. Mutation analysis in 54 propionic acidemia patients. J. Inherit. Metab. Dis. 2012;35:51–63. doi: 10.1007/s10545-011-9399-0. [DOI] [PubMed] [Google Scholar]
- 29.Boye S.L., Choudhury S., Crosson S., Di Pasquale G., Afione S., Mellen R., Makal V., Calabro K.R., Fajardo D., Peterson J., et al. Novel AAV44.9-Based Vectors Display Exceptional Characteristics for Retinal Gene Therapy. Mol. Ther. 2020;28:1464–1478. doi: 10.1016/j.ymthe.2020.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Di Pasquale G., Perez Riveros P., Tora M., Sheikh T., Son A., Teos L., Grewe B., Swaim W.D., Afione S., Zheng C., et al. Transduction of Salivary Gland Acinar Cells with a Novel AAV Vector 44.9. Mol. Ther. Methods Clin. Dev. 2020;19:459–466. doi: 10.1016/j.omtm.2020.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chandler R.J., Di Pasquale G., Sloan J.L., McCoy S., Hubbard B.T., Kilts T.M., Manoli I., Chiorini J.A., Venditti C.P. Systemic gene therapy for methylmalonic acidemia using the novel adeno-associated viral vector 44.9. Mol. Ther. Methods Clin. Dev. 2022;27:61–72. doi: 10.1016/j.omtm.2022.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martín-Rivada Á., Cambra Conejero A., Martín-Hernández E., Moráis López A., Bélanger-Quintana A., Cañedo Villarroya E., Quijada-Fraile P., Bellusci M., Chumillas Calzada S., Bergua Martínez A., et al. Newborn screening for propionic, methylmalonic acidemia and vitamin B12 deficiency. Analysis of 588,793 newborns. J. Pediatr. Endocrinol. Metab. 2022;35:1223–1231. doi: 10.1515/jpem-2022-0340. [DOI] [PubMed] [Google Scholar]
- 33.Barshes N.R., Vanatta J.M., Patel A.J., Carter B.A., O'Mahony C.A., Karpen S.J., Goss J.A. Evaluation and management of patients with propionic acidemia undergoing liver transplantation: a comprehensive review. Pediatr. Transplant. 2006;10:773–781. doi: 10.1111/j.1399-3046.2006.00569.x. [DOI] [PubMed] [Google Scholar]
- 34.Critelli K., McKiernan P., Vockley J., Mazariegos G., Squires R.H., Soltys K., Squires J.E. Liver Transplantation for Propionic Acidemia and Methylmalonic Acidemia: Perioperative Management and Clinical Outcomes. Liver Transpl. 2018;24:1260–1270. doi: 10.1002/lt.25304. [DOI] [PubMed] [Google Scholar]
- 35.Molema F., Martinelli D., Hörster F., Kölker S., Tangeraas T., de Koning B., Dionisi-Vici C., Williams M., additional individual contributors of MetabERN Liver and/or kidney transplantation in amino and organic acid-related inborn errors of metabolism: An overview on European data. J. Inherit. Metab. Dis. 2021;44:593–605. doi: 10.1002/jimd.12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pillai N.R., Stroup B.M., Poliner A., Rossetti L., Rawls B., Shayota B.J., Soler-Alfonso C., Tunuguntala H.P., Goss J., Craigen W., et al. Liver transplantation in propionic and methylmalonic acidemia: A single center study with literature review. Mol. Genet. Metabol. 2019;128:431–443. doi: 10.1016/j.ymgme.2019.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carrillo-Carrasco N., Chandler R.J., Chandrasekaran S., Venditti C.P. Liver-directed recombinant adeno-associated viral gene delivery rescues a lethal mouse model of methylmalonic acidemia and provides long-term phenotypic correction. Hum. Gene Ther. 2010;21:1147–1154. doi: 10.1089/hum.2010.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chandler R.J., Venditti C.P. Long-term rescue of a lethal murine model of methylmalonic acidemia using adeno-associated viral gene therapy. Mol. Ther. 2010;18:11–16. doi: 10.1038/mt.2009.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chandler R.J., Venditti C.P. Pre-clinical efficacy and dosing of an AAV8 vector expressing human methylmalonyl-CoA mutase in a murine model of methylmalonic acidemia (MMA) Mol. Genet. Metabol. 2012;107:617–619. doi: 10.1016/j.ymgme.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guenzel A.J., Hillestad M.L., Matern D., Barry M.A. Effects of adeno-associated virus serotype and tissue-specific expression on circulating biomarkers of propionic acidemia. Hum. Gene Ther. 2014;25:837–843. doi: 10.1089/hum.2014.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Subramanian C., Frank M.W., Tangallapally R., Yun M.K., White S.W., Lee R.E., Rock C.O., Jackowski S. Relief of CoA sequestration and restoration of mitochondrial function in a mouse model of propionic acidemia. J. Inherit. Metab. Dis. 2023;46:28–42. doi: 10.1002/jimd.12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cunningham S.C., Spinoulas A., Carpenter K.H., Wilcken B., Kuchel P.W., Alexander I.E. AAV2/8-mediated correction of OTC deficiency is robust in adult but not neonatal Spf(ash) mice. Mol. Ther. 2009;17:1340–1346. doi: 10.1038/mt.2009.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang L., Bell P., Lin J., Calcedo R., Tarantal A.F., Wilson J.M. AAV8-mediated hepatic gene transfer in infant rhesus monkeys (Macaca mulatta) Mol. Ther. 2011;19:2012–2020. doi: 10.1038/mt.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen P.J., Liu D.R. Prime editing for precise and highly versatile genome manipulation. Nat. Rev. Genet. 2023;24:161–177. doi: 10.1038/s41576-022-00541-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marcell P.D., Stabler S.P., Podell E.R., Allen R.H. Quantitation of methylmalonic acid and other dicarboxylic acids in normal serum and urine using capillary gas chromatography-mass spectrometry. Anal. Biochem. 1985;150:58–66. doi: 10.1016/0003-2697(85)90440-3. [DOI] [PubMed] [Google Scholar]
- 46.Allen R.H., Stabler S.P., Savage D.G., Lindenbaum J. Elevation of 2-methylcitric acid I and II levels in serum, urine, and cerebrospinal fluid of patients with cobalamin deficiency. Metabolism. 1993;42:978–988. doi: 10.1016/0026-0495(93)90010-l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting the studies presented in this manuscript can be made available by request to the corresponding authors.