

Abstract
Host cells sense and respond to pathogens by dynamically regulating cell signaling. The rapid modulation of signaling pathways is achieved by post-translational modifications (PTMs) that can alter protein structure, function, and/or binding interactions. By using chemical probes to broadly profile changes in enzyme function or side-chain reactivity, activity-based protein profiling (ABPP) can reveal PTMs that regulate host–microbe interactions. While ABPP has been widely utilized to uncover microbial mechanisms of pathogenesis, in this review, we focus on more recent applications of this technique to the discovery of host PTMs and enzymes that modulate signaling within infected cells. Collectively, these advances underscore the importance of ABPP as a tool for interrogating the host response to infection and identifying potential targets for host-directed therapies.
Keywords: Enzymes, proteomics, protein modifications, host–microbe interactions, infection
1. Introduction
The host response to infection is a complex process that requires the concerted activity of diverse cell types to thwart invading pathogens. Epithelial cells and resident macrophages must rapidly sense and mount a local defense against harmful microbes while producing cytokines that recruit immune cells to the infection site[1]. Activation of such signaling pathways requires dynamic changes in gene transcription, protein synthesis, and enzyme activity[2]. To achieve tight temporal control of these processes, cells often rely on post-translational modifications (PTMs) that can quickly and reversibly modulate protein structure, function, and/or binding interactions[3–5] (Figure 1). In turn, certain pathogens can modify PTMs on host proteins to subvert immune defenses[6–7]. Identifying PTMs that influence cell signaling is therefore essential for understanding the host response to infection.
Figure 1:

Representative classes of PTMs.
Over the past two decades, activity-based protein profiling (ABPP) has emerged as a versatile tool for assessing post-translational changes in protein function within diverse biological systems. Pioneered by Cravatt and coworkers, ABPP uses small-molecule, activity-based probes comprised of an electrophilic warhead, flexible linker, and a reporter tag (e.g., biotin, a fluorophore, or a “clickable” reporter such as an azide) to covalently label and detect specific classes of reactive amino acids within a proteome[8]. Probe-labeled proteins can be visualized using gel-based analyses or enriched for identification by mass spectrometry. Importantly, probe-based enrichment facilitates the detection of low-abundance proteins that are typically obscured by more abundant molecules in conventional proteomic analyses[9]. By quantifying changes in the probe-based enrichment of a given protein, ABPP can uncover PTMs or shifts in enzyme activity associated with a specific biological condition or disease state.
While ABPP has been widely applied to increase understanding of microbial infections, most studies to date have centered on microbial mechanisms of pathogenesis. Using probes that can covalently modify an enzyme’s active site, a number of studies have identified pathogen enzymes active during infection[10–12], facilitating the functional characterization of novel virulence factors[10, 13–14]. In addition, studies comparing the probe-based enrichment of enzymes in cells treated with or without a given drug (i.e., competitive ABPP) have enabled the identification and validation of putative drug targets[15–18]. Several recent review articles provide extensive coverage of these topics[19–22]. Here, we focus on the emerging role of ABPP as a tool for querying the host response to infection (Figure 2). We discuss how ABPP has been used to identify PTMs that regulate the signaling pathways of infected cells (Section 2), to profile functional changes in the host proteome during infection (Section 3), and to generate new leads for host-directed therapies (Section 4). Given recent advances in genome editing, and the continuous development of activity-based probes offering expanded coverage of proteomic space, the time is ripe for the ABPP-driven discovery of host pathways that could provide alternative therapeutic targets for microbial infections in an era of increasing antibiotic resistance.
Figure 2:

Applications of ABPP for interrogating host biology during infection. (A) ABPP can be used to discover post-translationally modified sites within the proteome of infected cells. (B) ABPP can identify host enzymes that are active during infection. Enzymes containing active-site PTMs or autoinhibitory propeptides (represented as small triangles within the active site), or lacking a probe-reactive nucleophile (represented as enzymes with rounded active sites) will not be captured. (C) Competitive ABPP can uncover host enzymes targeted by anti-infective agents. For example, the proteomes of virus-infected cells treated with an antiviral drug (gray rounded rectangle) or vehicle control can be labeled with an activity-based probe. Enzymes that exhibit decreased probe-based enrichment in the presence of drug versus vehicle represent potential drug targets (red enzyme).
2. Post-translational modification of host proteins during infection
PTMs can finely tune protein activity on a rapid timescale, enabling cells to respond quickly to external stimuli[5]. ABPP is particularly well-suited for dissecting dynamic changes in protein function[8, 23]. In the classic model, an activity-based probe selective for a PTM-responsive amino acid is used to assess side-chain reactivity in the presence or absence of a defined stimulus, thereby providing an indirect readout of PTMs within a given proteome[24–25]. Alternatively, a custom probe can be designed to either covalently modify a specified PTM or serve as a chemical reporter of the modification itself, affording direct, probe-based enrichment of modified sites[23]. Activity-based probes have been developed to study a wide range of PTMs, including oxidation, phosphorylation, and glycosylation, using both indirect and direct labeling strategies[15, 26].
In recent years, such probes have found broad application to mapping PTMs at the host–microbe interface (Table 1). PTMs are used by bacterial and host cells alike to adapt to infection conditions. For example, phosphorylation of mitogen-activated protein kinases can induce the expression of pro-inflammatory cytokines[27]; similarly, bacteria rely on two-component phosphorelay pathways to rapidly alter their transcriptional programs[28]. Notably, pathogens can also modulate PTMs on host proteins to promote successful colonization of the host[7]. Certain pathogen-secreted virulence factors directly dephosphorylate or deubiquitinate host proteins to corrupt cell signaling[29], whereas infection-associated conditions like oxidative stress can indirectly modify host proteins[30]. In this section, we discuss representative examples of how ABPP has been applied to interrogate post-translational transactions at the host–microbe interface that shape immune responses and host signaling pathways.
Table 1.
Representative activity-based probes and chemical reporters used to profile host proteins during infection.
| Probe | Chemical structure | Enzyme class/amino acid |
|---|---|---|
| FP-based probes[69, 71–72, 88, 115] |
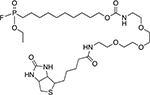
|
Serine hydrolases |
| IA-alkyne[30] |
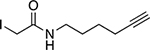
|
Reactive cysteine residues |
| 1-OH-Az[38] |
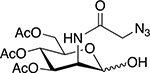
|
Reactive cysteine residues |
| ISG15-propargylamide[45–46] |
|
ISG15-based probe for cysteine proteases |
| DCG-04 linked to biotin[71] |
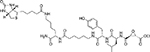
|
Papain-like cysteine proteases |
| AMS101[72] |
|
Vacuolar processing enzymes |
| ABP1[109] |
|
Rupintrivir-based probe for cysteine proteases |
| Desthiobiotin-FP[119] |
|
Serine hydrolases |
| TReNDs[51] |
|
Proteins AMPylated by Fic enzymes |
2.1. Itaconate modification
Macrophages are phagocytic immune cells that are among the “first responders” to an infection[31]. Upon sensing a pathogen, macrophages undergo sweeping changes in gene expression[32] and metabolism[33]. These metabolic changes, collectively known as metabolic reprogramming[34], result in the production of “immunometabolites”—compounds generated via glycolysis or as byproducts of the TCA cycle that regulate immune cell activation[35–36]. Recent studies have shown that certain immunometabolites, such as itaconate, a TCA cycle-derived metabolite, can directly modulate immune signaling through the covalent modification of cellular proteins[37]. For example, cysteine residues of KEAP1 react with itaconate through a Michael addition-based mechanism, resulting in the activation of the transcription factor Nrf2 and a corresponding increase in the expression of genes with antioxidant functions[37].
To facilitate the identification of itaconate modifications on a proteome-wide scale, Qin et al. developed the monosaccharide-based probe 3,4,5-O-Ac3ManNAz (1-OH-Az), which can facilitate the indirect mapping of itaconate-modified sites[38]. Using isotopic tandem orthogonal proteolysis-ABPP (isoTOP-ABPP), a chemical proteomic workflow for the quantitative analysis of cysteine reactivity[39], the authors identified 260 itaconate-modified cysteines in Raw264.7 macrophages treated with itaconate. Notably, three key glycolytic enzymes—GAPDH, LDHA, and ALDOA—were found to contain itaconate modifications. Through stable-isotope tracing experiments, the authors determined that itaconate treatment decreases the abundance of glycolytic metabolites produced downstream of ALDOA and LDHA. In addition, the authors showed that mutating itaconate-sensitive cysteines in ALDOA and LDHA is sufficient to restore glycolytic flux in the presence of itaconate. Together, these findings suggest itaconate inhibits glycolysis by post-translationally modifying cysteine residues that mediate LDHA and ALDOA activity, illuminating a novel mechanism of immunometabolite signaling. Since itaconate has also been shown to modify bacterial enzymes that regulate pathogen metabolism during infection[40], 1-OH-Az should be a valuable tool for elucidating the role of itaconate modifications in host–pathogen interactions.
2.2. ISGylation
Another immunomodulatory PTM that influences host cell signaling during infection is the conjugation of proteins to interferon-stimulated gene 15 (ISG15), a ubiquitin-like modifier. ISG15 expression is induced by type I interferons and pathogenic stimuli[41]. The 17-kDa protein plays dual roles as both a covalent modifier of intracellular proteins and as an extracellular cytokine[42]. ISG15-conjugation (ISGylation) is mediated by ubiquitin-associated enzymes that attach and remove ISG15 from target proteins[42]. Notably, certain viruses encode de-ISGylating enzymes that can cleave protein-linked ISG15, increasing levels of free ISG15[43]. For example, the papain-like protease (PLpro) of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) decreases protein ISGylation in HeLa cells treated with interferon-α[44]. Using a set of fluorogenic activity-based probes containing an ISG15 or a di-ubiquitin recognition element, Shin et al. found that SARS-CoV-2 PLpro preferentially interacts with ISG15[45]. By contrast, the closely related SARS-CoV PLpro exhibits greater reactivity towards ubiquitin, suggesting SARS-CoV and SARS-CoV-2 may exploit distinct host PTMs to tune innate immune signaling.
Using an ISG15-GlyGly peptidomics pipeline, in which ISGylated proteins are digested with trypsin and ISG15-modified peptides are subsequently enriched by immunoprecipitation of the terminal GlyGly motif, Munnur et al. found that SARS-CoV-2 PLpro substantially decreases the ISGylation of glycolytic enzymes in interferon-stimulated HeLa cells[46]. ISGylation of glycolytic enzymes has previously been implicated in downregulating the expression of pro-inflammatory cytokines[47], suggesting deISGylation by PLpro may contribute to the “cytokine storms” associated with SARS-CoV-2 infection. Along these lines, the authors detected enhanced secretion of pro-inflammatory cytokines by SARS-CoV-2-infected macrophages following siRNA-mediated knockdown of ISGylating enzymes[46]. Together, these findings imply that reduced protein ISGylation and increased free ISG15 signaling activate a pro-inflammatory response in SARS-CoV-2-infected macrophages. Applying similar approaches to study other viral infections could reveal additional proteases that moonlight as deISGylating enzymes and provide further insight into altered ISGylation as a mechanism of viral pathogenesis.
2.3. AMPylation
Several pathogenic bacteria induce the AMPylation (aka adenylylation) of host proteins to disrupt cell signaling in a manner that promotes infection[7, 48]. Protein AMPylation involves the covalent modification of a threonine, serine, or tyrosine side chain with the adenosine 5′-monophosphate (AMP) moiety of adenosine triphosphate (ATP)[49]. AMPylation of host Rho GTPases, for instance, has been shown to inhibit GTPase binding to downstream effectors, thereby blunting major responses to infection such as actin remodeling and pro-inflammatory signaling by NF-κB[50].
One class of bacterial enzymes that catalyzes host cell AMPylation is the filamentation induced by cyclic-AMP (Fic) enzymes, which are typically secreted into host cells during infection[49]. To facilitate the identification of Fic-modified proteins, Gulen et al. developed an approach termed “co-substrate-mediated covalent capture”, in which a recombinant Fic enzyme is tethered to a nucleotide derivative that binds to target proteins[51]. In this manner, the Fic enzyme acts as a macromolecular activity-based probe that can enrich AMPylated proteins for identification by mass spectrometry. This approach was used to identify dozens of previously uncharacterized substrates of Fic enzymes from the bacterial pathogens Histophilus somni and Bartonella henselae, including host proteins that regulate iron homeostasis and integrin signaling, respectively. Given that Fic enzymes are present in all domains of life[52], co-substrate-mediated covalent capture promises to expand the identification of target proteins in a variety of biological contexts.
A complementary strategy for the identification of host proteins that are AMPylated in situ was developed by Rauh et al[53]. The authors designed a cell-permeable, alkyne-containing pronucleotide probe (pro-N6pA) that is used to AMPylate proteins within intact cells. By applying pro-N6pA to HeLa cells infected with the enteric pathogen Vibrio parahaemolyticus, the authors identified new Rho GTPase targets of the AMPylating effector protein VopS. In addition to providing new insights into the role of AMPylation in infection biology, pro-N6pA could also be applied to profile proteins that are intrinsically AMPylated by host enzymes, such as the recently discovered pseudokinase SelO[54].
2.4. Cysteine oxidation
Another mechanism by which host cell signaling can be modulated during infection is cysteine oxidation. Cysteine thiols are one of the primary amino-acid side chains targeted by reactive oxygen species (ROS)[24, 55]. ROS exposure induces oxidative PTMs that decrease cysteine reactivity[24], thereby regulating protein function and binding interactions that modulate cell signaling[56–57]. Several microbial infections induce inflammation in host tissues that promotes ROS accumulation[58]. For example, the stomach pathogen Helicobacter pylori stimulates the production of ROS in gastric tissues, which facilitates the onset of peptic ulcers and gastric cancer in certain hosts[59]. While ROS accumulation is known to induce oxidative DNA damage during H. pylori infection[60–61], very little is known about the host proteins that are modified by these oxidants in infected cells. To address this question, our lab used isoTOP-ABPP to map changes in the cysteine reactivity of human gastric cancer cells following H. pylori infection[30]. We identified 35 cysteines in host proteins with reduced reactivity in H. pylori-infected cells, including Cys219 of the lysosomal protease legumain. Legumain expression and activity have previously been implicated in tumorigenesis[62], suggesting a possible connection between legumain oxidation and cancer signaling. Using genetic and biochemical approaches, we confirmed that legumain Cys219 is oxidized in H. pylori-infected cells and showed that the loss of Cys219 reactivity inhibits the intracellular activation of legumain[30]. Furthermore, mutating Cys219 enhanced the growth of mouse xenograft tumors, suggesting oxidation of this residue during H. pylori infection may promote tumorigenic signaling. Similar applications of ABPP to survey the oxidation of host proteins during chronic infection with pathogens that induce ROS production in host tissues could provide additional insights into how oxidative stress shapes disease development at the molecular level.
Certain commensal strains of lactobacilli induce low levels of ROS production in the colonic mucosa that can influence intestinal homeostasis[63–64]. Matthews et al. used thiol-reactive probes to identify proteome-wide changes in the cysteine reactivity of intestinal epithelial isolates from germ-free mice colonized with Lactobacillus rhamnosis GG (LGG) or Escherichia coli K-12[65]. Cys178 of Rac1, a protein that regulates the activity of the ROS-generating enzyme Nox1[66], exhibited reduced reactivity in the colonic mucosa of LGG-colonized mice[65], suggesting a potential role for this residue in regulating redox homeostasis following epithelial contact by LGG. Because several other microbes can stimulate ROS production by epithelial barriers[67–68], these findings motivate future studies investigating the role of microbe-induced redox signaling in the intestine.
3. Profiling changes in host enzyme activity during infection
Enzymes are critical mediators of host–microbe interactions. As such, changes in enzyme activity during infection can point to new mechanisms of microbial pathogenesis or nominate candidate proteins as biomarkers of disease progression. By profiling host enzymes active during infection with wild-type versus mutant strains of a given pathogen, ABPP can provide unique insights into the functional interplay of host and pathogen enzymes in vivo[69–70]. Furthermore, activity-based comparisons of disease-susceptible and disease-tolerant hosts can uncover enzymes that promote resistance to infection[71]. Finally, ABPP followed by the genetic knockdown of targeted enzymes can identify host factors exploited by a pathogen for survival[72–73]. In this section, we draw on examples of such diverse applications to collectively underscore the breadth of ABPP as a tool for probing host biology during infection (Table 1).
3.1. Gastrointestinal diseases
Vibrio cholerae is a Gram-negative, extracellular pathogen that causes the severe diarrheal disease cholera[74]. V. cholerae relies on secreted factors to colonize the intestinal epithelium and escape from the host[75]. To identify enzymes that shape pathogen interactions with the intestine, Hatzios et al. used fluorophosphonate (FP)-containing activity-based probes to globally profile pathogen- and host-secreted serine hydrolases active in V. cholerae-infected infant rabbits and human choleric stool[69]. FP-enriched proteins were digested with trypsin and analyzed by Multidimensional Protein Identification Technology (MudPIT)[76] using label-free quantitation. The authors identified 233 host and 14 bacterial proteins from the cecal fluid of V. cholerae-infected rabbits that exhibited activity-based enrichment[69]. The V. cholerae serine protease IvaP was found to be active in both rabbit cecal fluid and human choleric stool; together with three other secreted V. cholerae enzymes, IvaP was shown to decrease binding of an intestinal lectin to V. cholerae in the gut[69, 77]. Furthermore, activity-based proteomic analyses of cecal fluid from rabbits infected with wild-type V. cholerae or a mutant strain expressing catalytically inactive IvaP identified two rabbit enzymes, kallikrein 1 and cholesterin esterase, with reduced activity during wild-type V. cholerae infection[69]. These findings suggest secreted V. cholerae enzymes may regulate host enzyme activity in the gut and that comparative ABPP analyses using defined bacterial mutants enable quantitative profiling of such interactions during infection.
Recurrent intestinal inflammation associated with inflammatory bowel disease (IBD) is believed to stem in part from microbial imbalances in the gut (dysbiosis)[78]. Identifying microbial strains and/or microbe-derived factors that contribute to IBD progression could generate new therapeutic leads to improve patient care. However, the enormous strain diversity of the gut microbiota makes it challenging to comprehensively profile and deconvolute the proteomes of indigenous microbes in the gut. Liquid chromatography-tandem mass spectrometry (LC-MS/MS)-based metaproteomic techniques use protein reference sequences translated from shotgun metagenomics data to address this problem. Thuy-Boun et al. applied LC-MS/MS-based metaproteomics to identify proteins with altered abundance in the fecal samples of patients with ulcerative colitis (UC; a form of IBD) and healthy volunteers[70]. Differential expression analysis revealed 176 protein groups of host, microbial, and/or dietary origin that were significantly altered between healthy and diseased samples. Of those protein groups that were enriched in UC fecal samples, 29 were host-derived and linked to immune-related secretory events. Enzymes with serine-type endopeptidase activity were also enriched in this cohort, prompting the authors to profile active serine hydrolases in UC fecal samples using a biotinylated FP-based probe and LC-MS/MS analysis. Of the 27 probe-enriched host proteins identified by ABPP, 14 were enzymes with known serine hydrolase activity, including chymotrypsin-like elastases and dipeptidyl peptidase 4. Notably, robust serine hydrolase activity was detected in the UC fecal samples despite the presence of protease-inactivating serpins in the metaproteomic dataset, consistent with prior reports of elevated protease activity in IBD[79]. Altogether, these studies underscore the utility of ABPP in identifying host enzymes with altered activity in the diseased gut. The transfer of defined microbial communities into germ-free mouse models of IBD should allow for more focused studies of microbial and host enzymes that may contribute to UC and associated pathologies.
3.2. Plant diseases
Ralstonia solanacearum is a soil-borne pathogen of significant economic importance that causes bacterial wilt in various plant species including tomato[80]. It infects plants through wounds in the roots and travels through the apoplast (intercellular space) to the xylem vessels, which facilitate pathogen dissemination to other parts of the plant[81]. Because proteases in the apoplast play important immunological roles during bacterial infection[82], Planas-Marquès et al. applied ABPP to monitor changes in the enzyme activity of apoplastic fluid from R. solanacearum-infected tomato plants[71]. Using fluorescent activity-based probes specific for papain-like cysteine proteases (PLCPs) and serine hydrolases, the authors detected elevated PLCP and serine hydrolase activity following R. solanacearum infection. Notably, this phenotype was more pronounced in a tolerant tomato cultivar that survives R. solanacearum-induced tissue necrosis. From a subsequent activity-based proteomic analysis of apoplastic fluid, the PLCPs Pip1 and Rcr3 were found to be highly active in infected plants, along with serine hydrolases from the P69 family. Genetic inactivation of the targeted protease genes should help resolve which enzymes contribute to the resistance phenotype of tolerant cultivars.
The plant nematode Heterodera schachtii is a parasite that invades the root tip and induces the formation of a syncytium, which serves as the primary source of nutrients for the nematode[83]. To assess changes in the functional proteome of root tissues during syncytium formation, Hütten et al. applied ABPP to analyze Arabidopsis thaliana roots infected with H. schachtii[72]. Using a fluorescent activity-based probe for vacuolar processing enzymes (VPEs), a group of cysteine proteases known to influence plant–pathogen interactions[84], the authors detected altered VPE activity in syncytia versus uninfected controls by in-gel fluorescence analysis[72]. A corresponding transcriptional analysis revealed that the expression of certain VPEs is downregulated during syncytium formation. However, deletion of all four VPEs had no effect on the rate of H. schachtii infection or of nematode development, indicating these plant enzymes do not directly influence syncytium formation. Additional studies using probes with expanded coverage of the functional proteome may provide further insights into enzymes that mediate parasite invasion and survival in syncytia.
3.3. Viral infections
Several viruses rely on host enzymes to cleave viral proteins required for invasion and replication in host cells[85]. For example, influenza A virus (IAV) uses host serine proteases to process hemagglutinin[86], a surface protein necessary for viral entry into host cells, and serine protease inhibitors can reduce IAV replication in mouse models[87]. To identify other host enzymes that may contribute to IAV pathogenesis, Shahiduzzaman et al. used FP-based probes to profile serine hydrolase activity in IAV-infected lung cells and uninfected controls[88]. Cells labeled with isotopically ‘light’ or ‘heavy’ amino acids were lysed following infection and treated with desthiobiotin-FP to facilitate the enrichment of active serine hydrolases for identification by mass spectrometry. The proteasomal subunit PSMA2 was among the top probe-enriched proteins identified in IAV-infected cells, in line with prior studies linking the proteasome to viral infection. While siRNA-mediated knockdown of PSMA2 attenuated viral replication, proteasomal inhibitors such as MG132 were unable to decrease viral invasion without impairing host cell viability. Nevertheless, such activity-based screens provide insights into host enzymes and cellular processes that play important roles in viral replication and survival and hold promise for uncovering candidate proteins that could be targeted by host-directed antivirals.
Hepatitis C virus (HCV) replication requires specialized lipid membranes that contain cholesterol and unsaturated fatty acids[89]. miRNA-185 is a microRNA that inhibits HCV replication by regulating genes involved in cholesterol metabolism and lipid biosynthesis[90–91]; however, the targets of miRNA-185 are not well defined. Because serine hydrolases play important metabolic roles in the liver[92], Filip et al. used FP-biotin probes to characterize serine hydrolase activity in human hepatoma cells transfected with miRNA-185[73]. Mass spectrometry analysis identified 16 probe-labeled serine hydrolases with roles in lipid or endocannabinoid metabolism that exhibited decreased activity in the presence of miRNA-185, including monoglyceride lipase (MGLL), an enzyme that catalyzes endocannabinoid hydrolysis[93]. siRNA-mediated knockdown of MGLL, as well as administration of an MGLL inhibitor, decreased HCV levels in infected cells[73]. Overall, these findings point to miRNA-185-mediated changes in enzyme activity that may contribute to the antiviral effects of miRNA-185. Given that other miRNAs have been implicated in hepatic host–pathogen interactions[94], similar applications of ABPP could uncover new miRNA targets that regulate the host response to viral infection.
4. Host-directed therapies
While most anti-infectives target microbial proteins, host-directed therapies (HDTs) offer an alternative approach to the treatment of infectious diseases. HDTs function by interfering with host factors necessary for pathogen survival or by augmenting the host immune response[95]. There are several advantages of using HDTs in combination with conventional antimicrobials. First, it is considerably more challenging for pathogens to evolve resistance to HDTs[96–97]. To do so, a pathogen would need to exploit an entirely different host pathway to survive and replicate within the host, which would require extensive mutagenesis. Second, since some pathogens use the same host pathways for survival, HDTs can be designed to treat a broad range of infections[95, 98]. Here we discuss how ABPP can accelerate the identification of candidate enzymes for HDT development, as well as the discovery of host enzymes required for the activation of antimicrobial prodrugs that inhibit pathogen replication through more traditional means.
4.1. Discovery of host drug targets
Malaria is a mosquito-borne disease that is caused by parasites of the Plasmodium genus, primarily Plasmodium falciparum[99]. Asexual replication of P. falciparum occurs in the blood and consists of four stages—ring, trophozoite, schizont, and merozoite—that are collectively termed the erythrocytic cycle[100]. Davison et al. employed ABPP to identify serine hydrolases that are active during asexual replication and could serve as potential antimalarial targets[101]. Chemical proteomic analysis of P. falciparum lysates using an FP-based probe revealed 25 parasitic and eight host serine hydrolases with variable activity across the erythrocytic cycle, suggesting stage-specific roles for these enzymes. To evaluate the importance of human serine hydrolases to parasite replication, P. falciparum was treated with commercial inhibitors targeting each of the eight identified host enzymes. When administered to ring-stage parasites, five of the inhibitors decreased parasite replication. The two most effective inhibitors, targeting the host enzymes APEH and LYPLA1, were found to delay parasite progression to schizogony or arrest parasite development at earlier stages, respectively. While further analysis is needed to rule out possible off-target effects, this study identified two host serine hydrolases that could potentially be targeted for the development of anti-malarial drugs.
Chronic infection with HCV is a leading cause of hepatocellular carcinoma and liver transplantation[102]. While existing therapies focus on targeting viral proteases[103], host cells also encode factors that are crucial for the replication and survival of the virus[104]. Yoo et al. applied thiol-reactive probes to identify host factors with altered cysteine reactivity following HCV infection[105]. Using a fluorescein-iodoacetamide (Flu-IA) probe and in-gel fluorescence analysis, the authors observed marked changes in the thiol reactivity of human hepatoma cells following expression of the HCV genotype 2a viral replicon. They applied a competitive isoTOP-ABPP strategy[106] to identify proteins targeted by the Flu-IA probe and identified 26 candidate proteins, including several host factors previously implicated in HCV replication[105]. Plastin-3 (aka T-plastin), a protein involved in actin crosslinking[107] that exhibited significant Flu-IA labeling in gel-based assays, was selected for further study. siRNA-mediated knockdown of T-plastin decreased HCV replication efficiency[105]; together with prior reports implicating actin polymerization in HCV replication[108], these data support further investigation of T-plastin as a potential antiviral target.
In a related example, Yang et al. screened a library of roughly 200 biaryl-substituted quinolones for compounds with antiviral activity towards HCV[109]. RYL-634, the most potent compound to emerge following hit optimization, inhibited the replication of several RNA viruses, including dengue virus, Zika virus, and human immunodeficiency virus, at low-nanomolar concentrations. To identify proteins targeted by RYL-634, the authors installed a terminal alkyne and diazirine crosslinker on the quinolone scaffold of RYL-634 and identified putative binding partners via irradiation with ultraviolet (UV) light and LC-MS/MS analysis. Through subsequent structural analyses, dihydroorotate dehydrogenase (DHODH) was identified as a potential target of RYL-634. DHODH is a mitochondrial enzyme involved in the biosynthesis of pyrimidine, which is required for viral replication[110]. While previous studies have validated DHODH as an antiviral target[110], RYL-634 exhibits more potent antiviral activity than existing DHODH inhibitors and holds promise as a starting point for further optimization.
Human hand, foot, and mouth disease is a common pediatric illness characterized by a localized rash that results from infection with enterovirus 71 (EV71)[111]. Following viral entry into host cells, the RNA genome of EV71 is translated into a polyprotein that is hydrolyzed by the viral protease 3Cpro[112]. Although 3Cpro is the primary enzyme responsible for EV71 maturation[113], host enzymes are also believed to facilitate this process[114]. To identify host proteases involved in polyprotein hydrolysis, Sun et al. designed a biotinylated activity-based probe, ABP1, using the EV71 3Cpro inhibitor rupintrivir as a reference compound[114]. By examining host cysteine proteases labeled by ABP1 in infected cells, the authors identified an enzyme associated with autophagy-related antiviral immunity, ATG4B, that can cleave an EV71 3Cpro-specific peptide substrate. shRNA-mediated knockdown of ATG4B increased host cell resistance to EV71 infection and decreased EV71 RNA levels in infected cells. The results of this study support a role for ATG4B in EV71 polyprotein hydrolysis and replication, nominating the enzyme as a potential target for host-directed antiviral therapy.
4.2. Identifying host enzymes that activate antimicrobial prodrugs
The discovery and development of new prodrugs is often hampered by poorly characterized prodrug activation pathways. Identifying the enzyme(s) responsible for activating a given prodrug can help predict pharmacogenomic variability and facilitate the selection of suitable animal models for preclinical testing. Shenoy et al. used competitive ABPP to identify serine hydrolases that activate the antiviral ester prodrug valacyclovir (VACV)[115] (Figure 3A). A novel, FP-based probe was competed against saturating concentrations of VACV in fractions of enterocyte-like Caco-2 cells to identify enzymes with reduced probe labeling in the presence of the prodrug (Figure 3B). One of the candidate enzymes revealed by this screen, RBBP9, was shown to contribute to VACV activation in Caco-2 cells, and similar results were obtained using mouse intestinal perfusions of VACV in the presence or absence of a selective RBBP9 inhibitor. Compared to BPHL, a previously identified VACV-activating enzyme in the liver[116], RBBP9 exhibited much greater activity in the intestine[115], suggesting the biogeography of these enzymes may shape their clinical utility.
Figure 3:

Identification of prodrug-activating enzymes using ABPP. (A) Nucleoside/nucleotide analogs like the antiviral drug acyclovir are often converted into inactive carboxyester prodrugs (e.g., valacyclovir) to enhance cell permeability and intracellular activation. Host esterases hydrolyze the prodrugs to yield the pharmacologically active compounds. (B) Competitive ABPP using FP-based probes to detect serine hydrolases that are active in the presence or absence of prodrug can identify host enzymes involved in ester hydrolysis. Cells grown in medium containing isotopically “light” or “heavy” amino acids are treated with vehicle or prodrug, respectively, prior to probe labeling. Probe-based enrichment, trypsin digestion, and LC-MS/MS analysis of the combined, isotopically labeled proteomes enables quantitative comparisons of peptide abundance. Serine hydrolases that exhibit decreased probe-based enrichment in the presence of prodrug versus vehicle represent potential prodrug-activating enzymes (orange enzyme).
Nucleoside/nucleotide analogs are commonly used as antivirals since they inhibit viral replication[117]. The conversion of these analogs to ester prodrugs is a useful strategy for improving cell permeability and metabolic activation[118]. Indeed, several nucleoside prodrugs are currently in use to treat viral infections: tenofovir alafenamide (TAF), sofosbuvir (SBV), and remdesivir (RDV)[117]. However, the enzymes that facilitate prodrug activation in the host are largely uncharacterized. To identify potential TAF- and SBV-activating enzymes, Li et al. used a desthiobiotin-FP probe to profile active serine hydrolases in human lung tissue homogenates[119]. Three serine hydrolases were enriched within one minute of probe labeling: LYPLA1, CatA, and CSE1. CatA and CES1 exhibited significant hydrolytic activity towards TAF and SBV, respectively. In addition, the CatA inhibitor telaprevir decreased TAF activation, whereas the CES1 inhibitor BNPP had only a nominal effect. Together, these results validate CatA as a TAF-activating enzyme. Given its expression in the lung, CatA could potentially be harnessed for the activation of novel prodrugs targeting diverse respiratory viruses.
5. Summary and outlook
In this review, we have highlighted recent applications of ABPP that demonstrate its versatility in deconstructing host responses to infection. Activity-based probes have been successfully deployed to identify host PTMs that regulate cell signaling during infection[30, 38, 45–46, 51, 53, 65], host enzymes that shape pathological processes[69–73, 88], and host targets for antimicrobial therapy[101, 105, 109, 114]. Future advances in probe design will afford expanded coverage of functionally diverse PTMs that impact host–pathogen interactions. Probes with novel electrophilic warheads and/or recognition elements will add to the growing list of PTMs and enzymes that can be globally profiled using chemical proteomic techniques. Likewise, cell-permeable probes with caged electrophiles that are activated by UV light[120] or infection-associated environmental cues[121] will facilitate the study of transient modifications within infected cells. Finally, combining ABPP with CRISPR screening[122] will provide a systematic way to connect active enzymes or functional sites within a proteome to infection phenotypes. Through continued innovation and broader application to diverse infection models, ABPP promises to uncover new host–microbe interactions that underlie human health and disease while revealing new targets for anti-infective therapies.
Acknowledgments
We thank Prof. Benjamin Cravatt and coworkers for developing activity-based tools that have enriched the work of our lab and countless others. We gratefully acknowledge support from NIH R35 GM137952 (SKH), an Arnold and Mabel Beckman Young Investigator Award (SKH), a Conquer Cancer Now Award from the Concern Foundation (SKH), a Gruber Science Fellowship (RR), and NIH Chemical Biology Training Grant T32 GM067543 (RR).
Biographies
Biographical sketches

Renuka Ramanathan is a Ph.D. candidate in Dr. Stavroula Hatzios’ group in the Department of Molecular, Cellular, and Developmental Biology at Yale University. She received her B.S. in Biochemistry from the University of California, Los Angeles, where she worked with Dr. Robert Prins to investigate the cancer-testis antigen NY-ESO-1 as an immunotherapeutic target for the treatment of glioblastomas. In the Hatzios lab, Renuka is studying redox regulation of host cell signaling during Helicobacter pylori infection.

Stavroula Hatzios is an Assistant Professor of Molecular, Cellular, and Developmental Biology and of Chemistry in the Microbial Sciences Institute at Yale University. She received her B.S. in Chemistry from the Massachusetts Institute of Technology and conducted her graduate work in chemical biology with Dr. Carolyn Bertozzi at the University of California, Berkeley. After earning her Ph.D., Stavroula completed postdoctoral training in microbial pathogenesis with Dr. Matthew Waldor at Brigham and Women’s Hospital/Harvard Medical School. Her lab studies host–microbe interactions in the gastrointestinal tract through the activity-guided discovery of proteins, post-translational modifications, and metabolites that shape cell signaling at the molecular level.
Footnotes
This review is dedicated to Professor Benjamin Cravatt, a recipient of the 2022 Wolf Prize in Chemistry.
References
- [1].Janeway CA Jr, Travers P, Walport M, Shlomchik MJ, in Immunobiology: The Immune System in Health and Disease. 5th edition, Garland Science, 2001. [Google Scholar]
- [2].Argüello RJ, Rodrigues CR, Gatti E, Pierre P, Current Opinion in Immunology 2015, 32, 28–35. [DOI] [PubMed] [Google Scholar]
- [3].Liu X, Wang Q, Chen W, Wang C, Cytokine & Growth Factor Reviews 2013, 24, 559–570. [DOI] [PubMed] [Google Scholar]
- [4].Maurais AJ, Weerapana E, Current Opinion in Chemical Biology 2019, 50, 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Deribe YL, Pawson T, Dikic I, Nature Structural & Molecular Biology 2010, 17, 666–672. [DOI] [PubMed] [Google Scholar]
- [6].Denzer L, Schroten H, Schwerk C, International Journal of Molecular Sciences 2020, 21, 3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ribet D, Cossart P, FEBS Letters 2010, 584, 2748–2758. [DOI] [PubMed] [Google Scholar]
- [8].Cravatt BF, Wright AT, Kozarich JW, Annual Review of Biochemistry. 2008, 77, 383–414. [DOI] [PubMed] [Google Scholar]
- [9].Mayers MD, Moon C, Stupp GS, Su AI, Wolan DW, Journal of Proteome Research 2017, 16, 1014–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Shindo T, Kaschani F, Yang F, Kovács J, Tian F, Kourelis J, Hong TN, Colby T, Shabab M, Chawla R, Kumari S, PLoS Pathogens 2016, 12, e1005874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Puri AW, Broz P, Shen A, Monack DM, Bogyo M, Nature Chemical Biology 2012, 8, 745–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sanman LE, Bogyo M, Annual Review of Biochemistry 2014, 83, 73. [DOI] [PubMed] [Google Scholar]
- [13].Lentz CS, Sheldon JR, Crawford LA, Cooper R, Garland M, Amieva MR, Weerapana E, Skaar EP, Bogyo M, Nature Chemical Biology 2018, 14, 609–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Song J, Win J, Tian M, Schornack S, Kaschani F, Ilyas M, van der Hoorn RA, Kamoun S, Proceedings of the National Academy of Sciences 2009, 106, 1654–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Benns HJ, Wincott CJ, Tate EW, Child MA, Current Opinion in Chemical Biology 2021, 60, 20–29. [DOI] [PubMed] [Google Scholar]
- [16].Greenbaum DC, Baruch A, Grainger M, Bozdech Z, Medzihradszky KF, Engel J, DeRisi J, Holder AA, Bogyo M, Science 2002, 298, 2002–2006. [DOI] [PubMed] [Google Scholar]
- [17].Larson ET, Parussini F, Huynh M-H, Giebel JD, Kelley AM, Zhang L, Bogyo M, Merritt EA, Carruthers VB, Journal of Biological Chemistry 2009, 284, 26839–26850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Li M, Patel HV, Cognetta III AB, Smith II TC, Mallick I, Cavalier J-F, Previti ML, Canaan S, Aldridge BB, Cravatt BF, Cell Chemical Biology 2022, 29, 883–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kovalyova Y, Hatzios SK, Current Topics in Microbiology and Immunology 2019, 420, 73–91. [DOI] [PubMed] [Google Scholar]
- [20].Wright MH, Proteomics 2018, 18, 1700333. [DOI] [PubMed] [Google Scholar]
- [21].Keller LJ, Babin BM, Lakemeyer M, Bogyo M, Current Opinion in Chemical Biology 2020, 54, 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Desrochers GF, Pezacki JP, Activity-Based Protein Profiling 2018, 420, 131–154. [Google Scholar]
- [23].Shi Y, Carroll KS, Accounts of Chemical Research 2019, 53, 20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Paulsen CE, Carroll KS, Chemical Reviews 2013, 113, 4633–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gordon EM, Hatzios SK, PLoS Pathogens 2020, 16, e1009070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tate EW, Journal of Chemical Biology 2008, 1, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cargnello M, Roux PP, Microbiology and Molecular Biology Reviews 2011, 75, 50–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Capra EJ, Laub MT, Annual Review of Microbiology 2012, 66, 325–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ashida H, Kim M, Sasakawa C, Nature Reviews Microbiology 2014, 12, 399–413. [DOI] [PubMed] [Google Scholar]
- [30].Kovalyova Y, Bak DW, Gordon EM, Fung C, Shuman JH, Cover TL, Amieva MR, Weerapana E, Hatzios SK, Nature Chemical Biology 2022, 18, 698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Riera Romo M, Pérez‐Martínez D, Castillo Ferrer C, Immunology 2016, 148, 125–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mosser DM, Hamidzadeh K, Goncalves R, Cellular & Molecular Immunology 2021, 18, 579–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].He W, Heinz A, Jahn D, Hiller K, Current Opinion in Biotechnology 2021, 68, 231–239. [DOI] [PubMed] [Google Scholar]
- [34].Diskin C, Pålsson-McDermott EM, Frontiers in Immunology 2018, 9, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].O’Neill LA, Kishton RJ, Rathmell J, Nature Reviews Immunology 2016, 16, 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Williams NC, O’Neill LA, Frontiers in Immunology 2018, 9, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, Jedrychowski MP, Costa AS, Higgins M, Hams E, Szpyt J, Runtsch MC, King MS, McGouran JF, Fischer R, Kessler BM, McGettrick AF, Hughes MM, Carroll RG, Booty LM, Knatko EV, Meakin PJ, Ashford MLJ, Modis LK, Brunori G, Sévin DC, Fallon PG, Caldwell ST, Kunji ERS, Chouchani ET, Frezza C, Dinkova-Kostova AT, Hartley RC, Murphy MP, O’Neill LA, Nature 2018, 556, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Qin W, Qin K, Zhang Y, Jia W, Chen Y, Cheng B, Peng L, Chen N, Liu Y, Zhou W, l Wang Y, Chen X, Wang C, Nature Chemical Biology 2019, 15, 983–991. [DOI] [PubMed] [Google Scholar]
- [39].Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, Cravatt BF, Nature 2010, 468, 790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, Binz T, Wegner A, Tallam A, Rausell A, Buttini M, Linster CL, Medina E, Balling R, Hiller K, Proceedings of the National Academy of Sciences 2013, 110, 7820–7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Perng Y-C, Lenschow DJ, Nature Reviews Microbiology 2018, 16, 423–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Durfee LA, Lyon N, Seo K, Huibregtse JM, Molecular Cell 2010, 38, 722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Swaim CD, Canadeo LA, Monte KJ, Khanna S, Lenschow DJ, Huibregtse JM, Cell Reports 2020, 31, 107772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lindner HA, Fotouhi-Ardakani N, Lytvyn V, Lachance P, Sulea T, Ménard R, Journal of Virology 2005, 79, 15199–15208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shin D, Mukherjee R, Grewe D, Bojkova D, Baek K, Bhattacharya A, Schulz L, Widera M, Mehdipour AR, Tascher G, Geurink PP, Wilhelm A, van der Heden van Noort GJ, Ovaa H, Müller S, Knobeloch K.-p, Rajalingam K, Schulman BA, Cinatl J, Hummer G, Ciesek S & Dikic I, Nature 2020, 587, 657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Munnur D, Teo Q, Eggermont D, Lee HH, Thery F, Ho J, van Leur SW, Ng WW, Siu LY, Beling A, Ploegh H, Pinto-Fernandez A, Damianou A, Kessler B, Impens F, Ka Pun Mok C & Sanyal S, Nature Immunology 2021, 22, 1416–1427. [DOI] [PubMed] [Google Scholar]
- [47].Yan S, Kumari M, Xiao H, Jacobs C, Kochumon S, Jedrychowski M, Chouchani E, Ahmad R, Rosen ED, The Journal of Clinical Investigation 2021, 131, e144888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Woolery AR, Luong P, Broberg CA, Orth K, Frontiers in Microbiology 2010, 1, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Itzen A, Blankenfeldt W, Goody RS, Trends in Biochemical Sciences 2011, 36, 221–228. [DOI] [PubMed] [Google Scholar]
- [50].Woolery AR, Yu X, LaBaer J, Orth K, Journal of Biological Chemistry 2014, 289, 32977–32988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gulen B, Rosselin M, Fauser J, Albers MF, Pett C, Krisp C, Pogenberg V, Schlüter H, Hedberg C, Itzen A, Nature Chemistry 2020, 12, 732–739. [DOI] [PubMed] [Google Scholar]
- [52].Stanger FV, Burmann BM, Harms A, Aragão H, Mazur A, Sharpe T, Dehio C, Hiller S, Schirmer T, Proceedings of the National Academy of Sciences 2016, 113, E529–E537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Rauh T, Brameyer S, Kielkowski P, Jung K, Sieber SA, ACS Infectious Diseases 2020, 6, 3277–3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sreelatha A, Yee SS, Lopez VA, Park BC, Kinch LN, Pilch S, Servage KA, Zhang J, Jiou J, Karasiewicz-Urbańska M, Łobocka M, Grishin NV, Orth K, Kucharczyk R, Pawłowski K, Tomchick DR, Tagliabracci VS, Cell 2018, 175, 809–821. e819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Janssen-Heininger YM, Mossman BT, Heintz NH, Forman HJ, Kalyanaraman B, Finkel T, Stamler JS, Rhee SG, van der Vliet A, Free Radical Biology and Medicine 2008, 45, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Schieber M, Chandel NS, Current Biology 2014, 24, R453–R462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang J.-k., Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS, Thomas CJ, Vander Heiden MG, Cantley LC, Science 2011, 334, 1278–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Medzhitov R, Nature 2008, 454, 428–435. [DOI] [PubMed] [Google Scholar]
- [59].Tegtmeyer N, Backert S, Molecular Pathogenesis and Signal Transduction by Helicobacter pylori, Springer, 2017, 400, 1–27. [Google Scholar]
- [60].Chaturvedi R, de Sablet T, Asim M, Piazuelo MB, Barry DP, Verriere TG, Sierra JC, Hardbower DM, Delgado AG, Schneider BG, Israel DA, Romero-Gallo J, Nagy TA, Morgan DR, Murray-Stewart T, Bravo LE, Peek RM Jr., Fox JG, Woster PM, Casero RA Jr., Correa P, Wilson KT, Oncogene 2015, 34, 3429–3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Butcher LD, den Hartog G, Ernst PB, Crowe SE, Cellular and Molecular Gastroenterology and Hepatology 2017, 3, 316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Dall E, Brandstetter H, Biochimie 2016, 122, 126–150. [DOI] [PubMed] [Google Scholar]
- [63].Hertzberger R, Arents J, Dekker HL, Pridmore RD, Gysler C, Kleerebezem M, de Mattos MJT, Applied and Environmental Microbiology 2014, 80, 2229–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Jones RM, Neish AS, Free Radical Biology and Medicine 2017, 105, 41–47. [DOI] [PubMed] [Google Scholar]
- [65].Matthews JD, Reedy AR, Wu H, Hinrichs BH, Darby TM, Addis C, Robinson BS, Go Y-M, Jones DP, Jones RM, Neish AS, Redox Biology 2019, 20, 526–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Cheng G, Diebold BA, Hughes Y, Lambeth JD, Journal of Biological Chemistry 2006, 281, 17718–17726. [DOI] [PubMed] [Google Scholar]
- [67].Kumar A, Wu H, Collier‐Hyams LS, Hansen JM, Li T, Yamoah K, Pan ZQ, Jones DP, Neish AS, The EMBO Journal 2007, 26, 4457–4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ashida H, Ogawa M, Kim M, Mimuro H, Sasakawa C, Nature Chemical Biology 2012, 8, 36–45. [DOI] [PubMed] [Google Scholar]
- [69].Hatzios SK, Abel S, Martell J, Hubbard T, Sasabe J, Munera D, Clark L, Bachovchin DA, Qadri F, Ryan ET, Davis BM, Weerapana E, Waldor MK, Nature Chemical Biology 2016, 12, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Thuy-Boun PS, Wang AY, Crissien-Martinez A, Xu JH, Chatterjee S, Stupp GS, Su AI, Coyle WJ, Wolan DW, Molecular & Cellular Proteomics 2022, 21, 100197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Planas-Marquès M, Bernardo-Faura M, Paulus J, Kaschani F, Kaiser M, Valls M, van der Hoorn RA, Coll NS, Molecular & Cellular Proteomics 2018, 17, 1112–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Hütten M, Geukes M, Misas-Villamil JC, van der Hoorn RA, Grundler FM, Siddique S, Plant Physiology and Biochemistry 2015, 97, 36–43. [DOI] [PubMed] [Google Scholar]
- [73].Filip R, Desrochers GF, Lefebvre DM, Reed A, Singaravelu R, Cravatt BF, Pezacki JP, Cell Chemical Biology 2021, 28, 202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Ritchie JM, Waldor MK, Molecular Mechanisms of Bacterial Infection via the Gut 2009, 337, 37–59. [Google Scholar]
- [75].Sikora AE, PLoS Pathogens 2013, 9, e1003126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Weerapana E, Speers AE, Cravatt BF, Nature Protocols 2007, 2, 1414–1425. [DOI] [PubMed] [Google Scholar]
- [77].Howell M, Dumitrescu DG, Blankenship LR, Herkert D, Hatzios SK, Journal of Biological Chemistry 2019, 294, 9888–9900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Caruso R, Lo BC, Núñez G, Nature Reviews Immunology 2020, 20, 411–426. [DOI] [PubMed] [Google Scholar]
- [79].Jablaoui A, Kriaa A, Mkaouar H, Akermi N, Soussou S, Wysocka M, Wołoszyn D, Amouri A, Gargouri A, Maguin E, Lesner A, Rhimi M, Frontiers in Cellular and Infection Microbiology 2020, 10, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Peeters N, Guidot A, Vailleau F, Valls M, Molecular Plant Pathology 2013, 14, 651–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Vasse J, Frey P, Trigalet A, Molecular Plant-Microbe Interactions 1995, 8, 241–251. [Google Scholar]
- [82].Gupta R, Lee SE, Agrawal GK, Rakwal R, Park S, Wang Y, Kim ST, Frontiers in Plant Science 2015, 6, 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Sobczak M, Golinowski W, Grundler FM, European Journal of Plant Pathology 1997, 103, 113–124. [Google Scholar]
- [84].Rojo E, Martın R, Carter C, Zouhar J, Pan S, Plotnikova J, Jin H, Paneque M, Sánchez-Serrano JJ, Baker B, Ausubel FM, Raikhel NV, Current Biology 2004, 14, 1897–1906. [DOI] [PubMed] [Google Scholar]
- [85].Shahiduzzaman M, Coombs KM, Frontiers in Microbiology 2012, 3, 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Bosch FX, Garten W, Klenk H-D, Rott R, Virology 1981, 113, 725–735. [DOI] [PubMed] [Google Scholar]
- [87].Dittmann M, Hoffmann H-H, Scull MA, Gilmore RH, Bell KL, Ciancanelli M, Wilson SJ, Crotta S, Yu Y, Flatley B, Xiao JW, l Casanova J, Wack A, Bieniasz PD, Rice CM, Cell 2015, 160, 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Shahiduzzaman M, Ezatti P, Xin G, Coombs KM, Journal of Proteome Research 2014, 13, 2223–2238. [DOI] [PubMed] [Google Scholar]
- [89].Chukkapalli V, Heaton NS, Randall G, Current Opinion in Microbiology 2012, 15, 512–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Yang M, Liu W, Pellicane C, Sahyoun C, Joseph BK, Gallo-Ebert C, Donigan M, Pandya D, Giordano C, Bata A, Nickels JT, Journal of Lipid Research 2014, 55, 226–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Singaravelu R, O’hara S, Jones DM, Chen R, Taylor NG, Srinivasan P, Quan C, Roy DG, Steenbergen RH, Kumar A, Lyn RK, Özcelik D, Rouleau Y, Nguyen M.-a., Rayner KJ, Hobman TC, Tyrrell DL, Russell RS, Pezacki JP, Nature Chemical Biology 2015, 11, 988–993. [DOI] [PubMed] [Google Scholar]
- [92].Thomas G, Betters JL, Lord CC, Brown AL, Marshall S, Ferguson D, Sawyer J, Davis MA, Melchior JT, Blume LC, Howlett AC, Ivanova PT, Milne D SB. Myers S, Mrak I, Leber V, Heier C, Taschler U, Blankman JL Cravatt BF, Lee RG, Crooke RM, Graham MJ, Zimmermann R, Brown HA, Brown JM, Cell Reports 2013, 5, 508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Scalvini L, Piomelli D, Mor M, Chemistry and Physics of Lipids 2016, 197, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Morishita A, Oura K, Tadokoro T, Fujita K, Tani J, Masaki T, International Journal of Molecular Sciences 2021, 22, 3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Kaufmann SH, Dorhoi A, Hotchkiss RS, Bartenschlager R, Nature Reviews Drug Discovery 2018, 17, 35–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Zumla A, Rao M, Wallis RS, Kaufmann SH, Rustomjee R, Mwaba P, Vilaplana C, Yeboah-Manu D, Chakaya J, Ippolito G, Azhar E, Hoelscher M, Maeurer M, The Lancet Infectious Diseases 2016, 16, e47–e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Hawn TR, Shah JA, Kalman D, Immunological Reviews 2015, 264, 344–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Ni Y, Lempp FA, Mehrle S, Nkongolo S, Kaufman C, Fälth M, Stindt J, Königer C, Nassal M, Kubitz R, Sültmann H, Urban S, Gastroenterology 2014, 146, 1070–1083. [DOI] [PubMed] [Google Scholar]
- [99].Tuteja R, The FEBS Journal 2007, 274, 4670–4679. [DOI] [PubMed] [Google Scholar]
- [100].Venugopal K, Hentzschel F, Valkiūnas G, Marti M, Nature Reviews Microbiology 2020, 18, 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Davison D, Howell S, Snijders AP, Deu E, Iscience 2022, 25, 104996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].McGlynn KA, Petrick JL, El‐Serag HB, Hepatology 2021, 73, 4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Hézode C, Forestier N, Dusheiko G, Ferenci P, Pol S, Goeser T, Bronowicki J-P, Bourlière M, Gharakhanian S, Bengtsson L, McNair L, George S, Kieffer T, Kwong A, Kauffman RS, Alam J, m Pawlotsky J, Zeuzem S, New England Journal of Medicine 2009, 360, 1839–1850. [DOI] [PubMed] [Google Scholar]
- [104].Moradpour D, Penin F, Rice CM, Nature Reviews Microbiology 2007, 5, 453–463. [DOI] [PubMed] [Google Scholar]
- [105].Yoo Y-H, Yun J, Yoon CN, Lee J-S, Scientific Reports 2015, 5, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Wang C, Weerapana E, Blewett MM, Cravatt BF, Nature Methods 2014, 11, 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Wolff L, Strathmann EA, Müller I, Mählich D, Veltman C, Niehoff A, Wirth B, Cellular and Molecular Life Sciences 2021, 78, 5275–5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Bost AG, Venable D, Liu L, Heinz BA, Journal of Virology 2003, 77, 4401–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Yang Y, Cao L, Gao H, Wu Y, Wang Y, Fang F, Lan T, Lou Z, Rao Y, Journal of Medicinal Chemistry 2019, 62, 4056–4073. [DOI] [PubMed] [Google Scholar]
- [110].K Vyas V, Ghate M, Mini Reviews in Medicinal Chemistry 2011, 11, 1039–1055. [DOI] [PubMed] [Google Scholar]
- [111].Chan KP, Goh KT, Chong CY, Teo ES, Lau G, Ling AE, Emerging Infectious Diseases 2003, 9, 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Wang J, Fan T, Yao X, Wu Z, Guo L, Lei X, Wang J, Wang M, Jin Q, Cui S, Journal of Virology 2011, 85, 10021–10030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Shang L, Wang Y, Qing J, Shu B, Cao L, Lou Z, Gong P, Sun Y, Yin Z, Antiviral Research 2014, 112, 47–58. [DOI] [PubMed] [Google Scholar]
- [114].Sun Y, Zheng Q, Wang Y, Pang Z, Liu J, Yin Z, Lou Z, Journal of Virology 2019, 93, e01092–01019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Shenoy VM, Thompson BR, Shi J, Zhu H-J, Smith DE, Amidon GL, Molecular Pharmaceutics 2020, 17, 1706–1714. [DOI] [PubMed] [Google Scholar]
- [116].Kim I, Chu X.-y., Kim S, Provoda CJ, Lee K-D, Amidon GL, Journal of Biological Chemistry 2003, 278, 25348–25356. [DOI] [PubMed] [Google Scholar]
- [117].De Clercq E, Li G, Clinical Microbiology Reviews 2016, 29, 695–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].De Schutter C, Ehteshami M, Hammond ET, Amblard F, Schinazi RF, Current Pharmaceutical Design 2017, 23, 6984–7002. [DOI] [PubMed] [Google Scholar]
- [119].Li J, Liu S, Shi J, Zhu H-J, Pharmaceutics 2021, 13, 1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Abo M, Weerapana E, Journal of the American Chemical Society 2015, 137, 7087–7090. [DOI] [PubMed] [Google Scholar]
- [121].Iwashita H, Castillo E, Messina MS, Swanson RA, Chang CJ, Proceedings of the National Academy of Sciences 2021, 118, e2018513118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Benns HA, Storch M, Falco JA, Fisher FR, Tamaki F, Alves E, Wincott CJ, Milne R, Wiedemar N, Craven G, Baragaña B, Wyllie S, Baum J, Baldwin GS, Weerapana E, Tate EW, Child MA, Nature Microbiology 2022, 7, 1891–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]