

Key Points
-
•
Presence and degree of CRS, alongside high disease burden (≥ 25% marrow blasts), influence risk of ≥Gr3 AEs.
-
•
Nonresponders who developed CRS experienced the highest median number of NC AEs.
Visual Abstract
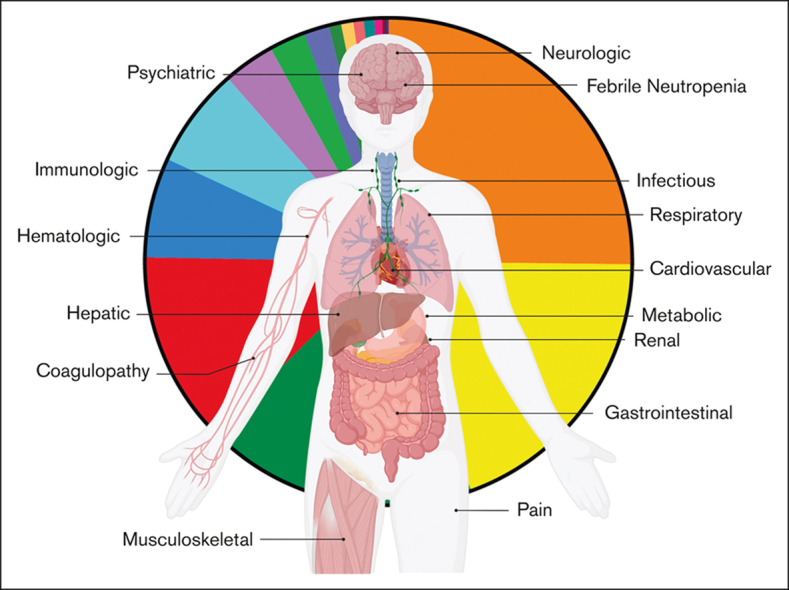
Abstract
The tremendous success of chimeric antigen receptor (CAR) T cells in children and young adults (CAYAs) with relapsed/refractory B-cell acute lymphoblastic leukemia is tempered by toxicities such as cytokine release syndrome (CRS). Despite expansive information about CRS, profiling of specific end-organ toxicities secondary to CAR T-cell therapy in CAYAs is limited. This retrospective, single-center study sought to characterize end-organ specific adverse events (AEs) experienced by CAYAs during the first 30 days after CAR T-cell infusion. AEs graded using Common Terminology Criteria for Adverse Events were retrospectively analyzed for 134 patients enrolled in 1 of 3 phase 1 CAR T-cell trials (NCT01593696, NCT02315612, and NCT03448393), targeting CD19 and/or CD22. A total of 133 patients (99.3%) experienced at least 1 grade ≥3 (≥Gr3) AE across 17 organ systems, of which 75 (4.4%) were considered dose- or treatment-limiting toxicities. Excluding cytopenias, 109 patients (81.3%) experienced a median of 3 ≥Gr3 noncytopenia (NC) AEs. The incidence of ≥Gr3 NC AEs was associated with the development and severity of CRS as well as preinfusion disease burden (≥ 25% marrow blasts). Although those with complete remission trended toward experiencing more ≥Gr3 NC AEs than nonresponders (median, 4 vs 3), nonresponders experiencing CRS (n = 17; 37.8%) had the highest degree of NC AEs across all patients (median, 7 vs 4 in responders experiencing CRS). Greater understanding of these toxicities and the ability to predict which patients may experience more toxicities is critical as the array of CAR T-cell therapies expand. This retrospective study was registered at www.clinicaltrials.gov as NCT03827343.
Introduction
Chimeric antigen receptor (CAR) T cells have revolutionized the field of cancer therapy, especially for B-cell malignancies. With the remarkable ability to overcome chemotherapeutic resistance in patients with relapsed refractory (r/r) disease, this potent immunotherapy bestows cytotoxic T cells with major histocompatibility complex–independent recognition of target antigens. Because of the extraordinary early success, a growing number of CAR T-cell products are approved by the US Food and Drug Administration, with CD19-targeted tisagenlecleucel available for children and adolescent young adults (CAYAs) and brexucabtagene autoleucel for those aged ≥18 years after the promising outcomes of the ELIANA and ZUMA-3 trials respectively.1,2 Numerous other CAR T-cell constructs for pediatric patients with hematologic and solid malignancies are currently in clinical trials.
As novel immunotherapeutic agents, CAR T cells can lead to disease eradication in the most challenging of cases but are marred by considerable toxicities. Most notably, cytokine release syndrome (CRS), characterized by high fevers, hemodynamic instability, and pulmonary compromise and immune effector cell–associated neurotoxicity syndrome (ICANS), which can include symptoms of aphasia, altered mental status, weakness, and seizures, have been well described.3, 4, 5 The incidence of CRS varies based on the CAR construct and target antigen, but in pediatric B-cell acute lymphoblastic leukemia (B-ALL), it has generally ranged from 70% to 90%; and the incidence of ICANS also varies.6, 7, 8, 9, 10 The advent of universal grading systems for both CRS and ICANS has allowed for systematic recognition and reporting of these syndromes.3,5
Although these guidelines, with enriched detection and understanding of CRS and ICANS, have improved outcomes and allowed for preemptive mitigation efforts, the extent of specific end-organ toxicities, such as hepatic, renal, or gastrointestinal toxicities that patients may experience with CAR T-cell therapy remains unclear. Although there are increasing reports describing cardiac, neurologic, and hematologic (coagulopathies and cytopenias) toxicities and infectious events, a comprehensive profile of single end-organ toxicities after CAR T-cell therapy, particularly for CAYAs in early-phase trials is needed.8,11, 12, 13, 14, 15, 16, 17
As CAR T-cell trials expand in CAYAs, understanding the spectrum of toxicities our patients face, determining who is at highest risk, and identifying how to better predict and mitigate toxicities remain critical priorities. We outline our findings based on the experience in CAYAs with r/r B-ALL across 3 phase 1 CAR T-cell trials.
Methods
Study population
This was a retrospective analysis (NCT03827343) evaluating the incidence of grade ≥3 (≥Gr3) AEs in the first 30 days after CAR T-cell infusion in CAYA patients with r/r B-ALL. Eligible patients were treated in 1 of 3 phase 1 CAR T-cell trials (NCT01593696 targeting CD19, NCT02315612 targeting CD22, and NCT03448393 targeting CD19/CD22) at the Pediatric Oncology Branch of the National Cancer Institute from 2012 to 2020 (supplemental Figure 1A). Data cutoff date was 31 December 2020. The clinical trial design and outcome data across these 3 studies have been previously reported.7,8,18, 19, 20
Toxicity assessment
AEs ≥Gr3 occurring between day 0 and day 30 characterized by attribution to therapy and/or underlying disease were prospectively captured by 2 consistent data managers and graded using Common Terminology Criteria for Adverse Events (CTCAE) version 4 (NCT01593696 and NCT02315612) and version 5 (NCT03448393; supplemental Table 1) at baseline (before lymphodepletion [LD]) and from initiation of LD chemotherapy to at least 30 days after CAR T-cell infusion (see supplemental Methods for complete details of AE analysis). Attributions were assigned by a consistent group of providers in real time during the conduct of the clinical trials. This formed the data set that was used for this retrospective analysis and accounted for interrater variability.
The primary cumulative analysis sought to capture the overall toxicity incidence, incorporating recurrent events (eg, thrombocytopenia or hypokalemia) toward the total number of events per patient. Select AE terms (eg, multiorgan failure) capturing duplicative information were excluded. The secondary focused analysis aimed to capture only the highest noncytopenia (NC) AE term per event per patient over the 30-day period to characterize general toxicities experienced. AEs designated as dose- or treatment-limiting toxicities (DLTs/TLTs), as defined in the individual protocols, were identified as such. CRS was graded using American Society for Transplantation and Cellular Therapy guidelines (retrospectively applied to those treated before implementation of consensus guidelines); however, AEs that comprised CRS, including fever, hypotension and hypoxia, were accounted for individually to capture the symptoms experienced.3 Similarly, neurotoxicities were captured based on CTCAE terms because ICANS grading was not established from the start of all 3 trials and cannot be retrospectively applied. Disease response was determined on day 28 (± 4 days) using standard morphology and minimal residual disease flow–based analysis of bone marrow and cerebrospinal fluid. Extramedullary sites and response were evaluated when indicated.
Statistical analysis
With the main aim of analyzing the cumulative set of AEs experienced by patients, the primary analysis sought to describe the median number of ≥Gr3 AEs per patient overall and per trial. After categorization of individual ≥Gr3 AEs based on CTCAE and/or clinically relevant categories (supplemental Table 2), the number of ≥Gr3 AEs were analyzed overall and by protocol to determine the fraction of AEs attributed to each organ system, with a focus on NC events (excluding neutropenia, anemia, and thrombocytopenia), given a degree of expected cytopenias due to underlying disease and effects of LD chemotherapy. After initial descriptive statistics, the median ≥Gr3 NC AEs were characterized across preinfusion characteristics (eg, disease burden, prior therapies, and demographics) and postinfusion outcome measures (eg, CRS development, CRS severity, and response). The secondary focused analysis was performed similarly, with additional details described in the supplemental Methods.
AE data, patient demographics, and disease information were loaded from Excel files into Python 3.9 for preprocessing and statistical analysis. The “pandas” library was used to read and load data, and “numpy” library was used for subsequent numerical and statistical analyses (median, IQR [interquartile range], range, etc). Additional statistical analyses were performed using SAS version 9.4 and GraphPad Prism 9.0.
A variety of statistical tests were used based on outcome or characteristic of interest and presented without correction for multiple comparisons because these results are intended to be primarily descriptive; χ2 test and Mehta modification to Fisher exact test were used for comparisons of baseline demographics, disease status, and treatment history across trials, and Kruskal-Wallis and Mann-Whitney-Wilcoxon tests were used for comparisons of continuous outcomes between groups. The number of ≥Gr3 NC AEs across ordered categories (age and number of prior regimens) were evaluated for statistical association, using Jonckheere-Terpstra test for trends. Those factors found to be at least potentially associated with ≥Gr3 NC AEs on univariate analysis (approximate P ≤ .1) were included in a multivariable linear regression analysis completed separately per trial. Baseline bone marrow disease (based on flow cytometric analysis) was tested as both a continuous and categorical variable in the multivariable regression. Backward elimination was used to arrive at a final model in which the remaining parameters had P < .05 when considered jointly, and adjusted r2 was used for interpretation of goodness of fit.
Results
Demographics
Demographics from 134 patients with r/r ALL were analyzed (Table 1). The median age was 15.2 years (IQR, 9.4-21.2 years). Patients were heavily pretreated, with a median of 5 prior lines of therapy (IQR, 3-6), with patients on the CD22 CAR trial having received more lines of prior therapy (median, 6; overall P = .0002). Preceding immunotherapy exposure was high, including hematopoietic stem cell transplantation (56.7%), blinatumomab (32.8%) or inotuzumab (17.2%), and CAR T cells (40.3%), and were more frequently used in patients before enrolling in CD22 or CD19/CD22 CAR T-cell trials than the CD19 trial. The majority had significant disease burden, with more than two-thirds (67.9%) having ≥5% disease burden (≥M2) before CAR infusion. Of 134 patients, 133 were evaluable for response; 88 patients (66.2%) achieved a CR, of whom 77 (87.5%) were negative for minimal residual disease. Response rates were comparable across trials.
Table 1.
Demographics across trials
| All Patients | CD19 | CD22 | CD19/CD22 | P | |
|---|---|---|---|---|---|
| All patients | n = 134 | n = 50 | n = 68 | n = 16 | n/a |
| Demographics | |||||
| Median age∗, (IQR) | 15.2 (9.4-21.2) | 13.5 (8.6-18.8) | 15.8 (9.5-21.1) | 20.5 (16.2-28.3) | .02 |
| Sex, male, n (%) | 97 (72.3%) | 41 (82%) | 44 (64.7%) | 12 (75%) | .11 |
| Ethnicity, non-Hispanic, n (%) | 94 (70.1%) | 31 (62%) | 52 (76.5%) | 11 (68.8%) | .23 |
| Race, White†, n (%) | 96 (80.7%) | 36 (80%) | 52 (82.5%) | 8 (72.7%) | .74 |
| Prior therapy | |||||
| Median number of prior cycles of therapy not including HSCT (IQR) | 5 (3-6) | 4 (2-5) | 6 (4-7) | 4.5 (3-7) | .0002 |
| Prior HSCT, n (%) | 76 (56.7%) | 22 (44%) | 44 (64.7%) | 10 (62.5%) | .071 |
| Prior CAR, n (%) | 54 (40.3%) | 2 (4%) | 45 (66.2%) | 7 (43.8%) | <.0001 |
| Prior blinatumomab, n (%) | 44 (32.8%) | 4 (8%) | 28 (41.2%) | 12 (75%) | <.0001 |
| Prior inotuzumab, n (%) | 23 (17.2%) | 0 | 18 (26.5%) | 5 (31.3%) | .0002 |
| Disease status‡ | |||||
| No. of pts with ≥M2 marrow, % | 91 (67.9%) | 31 (62%) | 52 (76.5%) | 8 (50%) | .066 |
| No. of pts with CNS2+, % | 7 (5.2%) | 5 (10%) | 2 (2.9%) | 0 | .15 |
| No. of pts with EM§ disease, % | 24 (17.9%) | 4 (8%) | 12 (17.6%) | 8 (50%) | .0007 |
| Disease response‖ | |||||
| No. of pts with CR, % | 88 (66.2%) | 31 (62%) | 49 (73.1%) | 8 (50%) | .16 |
| No. of pts with MRD-neg CR, % | 77 (58.3%) | 27 (54%) | 42 (63.6%) | 8 (50%) | .45 |
CNS2+, CNS involvement with some degree of blasts on cytospin; EM, extramedullary; HSCT, hematopoietic stem cell transplant; MRD, minimal residual disease; pt, patient.
Age at consent.
Race total, n = 119; race was reported as unknown in 15 patients; non-White patients: African American, Asian, Hawaiian/Pacific Islander, and multirace.
Disease status before lymphodepleting chemotherapy.
EM disease refers to sites of EMD outside the CNS.
One patient was not evaluable for minimal residual disease (MRD) analysis because of inability to obtain aspirate sample, and another was not evaluable for CR or MRD owing to early death; CR (n = 133) and MRD (n = 132); both in the CD22 trial.
Cumulative incidence of ≥Gr3 AEs
Across 134 patients, 133 (99.3%) experienced at least 1 ≥Gr3 AE during the first 30 days after CAR infusion. With a total of 1719 individual ≥Gr3 AEs, each patient experienced a median of 10 (IQR, 4-19) ≥Gr3 AEs across 17 unique organ systems (Figure 1A,B). Cytopenias (neutropenia, thrombocytopenia, and anemia) comprised the majority of the total ≥Gr3 AEs (n = 982; 57.1%), inclusive of recurrent events/patients.
Figure 1.

Incidence of ≥Gr3 AEs, overall and across trials. (A) Flow diagram of median and IQR of all ≥Gr3 overall and separated based on the trial, as well as ≥Gr3 NC AEs based on outcomes. ∗One patient was not evaluable for response owing to early death. (B) Pie graph of all ≥Gr3 AEs based on the system. (C) Pie graph of ≥Gr3 NC AEs based on the system. (D) Pie graph of highest ≥Gr3 NC AEs per patient per secondary analysis that excluded duplicate and overlapping terms. (E,F) Dot plot of the number of ≥Gr3 AEs (and ≥Gr3 NC AEs) per patient based on the trial, with horizontal line representing the median per patient. Pair-wise rank comparisons using Mann-Whitney are represented as P values at the top of graphs, with Kruskal-Wallis comparisons across all 3 trials to the right. GI, gastrointestinal.
Excluding cytopenias, 109 patients (81.3%) experienced 737 ≥Gr3 NC AEs, with a median of 3 ≥Gr3 NC AEs per patient (IQR, 1-8; Figure 1A,C; supplemental Table 3). Restricted to NC AEs, metabolic AEs made up 42.3% (n = 312) of AEs; followed by hepatic toxicities accounting for 15.5% (n = 114); febrile neutropenia, 15.5% (n = 114); and cardiovascular toxicities, 8.0% (n = 59). When limited to reporting the maximum grade NC AE per event per patient (secondary analysis described later in the article), 108 patients (80.6%) experienced 457 ≥Gr3 NC AEs (Figure 1D).
Of all ≥Gr3 AEs, 75 (4.4%) were considered DLTs or TLTs. A total of 5 patients experienced a DLT, including 2 receiving CD19 (CRS), 2 receiving CD22 (diarrhea and hypoxia), and 1 receiving CD1922 (encephalopathy with Gr3 ICANS).18, 19, 20 One patient death occurred due to Gr5 adult respiratory distress syndrome.8 Additional TLTs after determination of the maximum tolerated dose are listed in supplemental Table 4.
AEs across CAR T-cell trials
When stratified based on the trial (CD19 vs CD22 vs CD19/22), the median number of ≥Gr3 AEs experienced per patient differed substantially (7 vs 13.5 vs 5.5, respectively; P = .0007; Figure 1E); patients in the CD22 CAR trial experienced a higher number of overall and NC ≥Gr3 AEs (Figure 1F; supplemental Figure 1B).
AEs based on outcomes: CRS vs no CRS
Across 134 patients, 104 (77.6%) developed CRS. In those experiencing CRS, there was a median of 4.5 (IQR, 1.25-9) ≥Gr3 NC AEs, compared with a median of 1 (IQR, 0-3) in the 30 patients without CRS (P < .0001; Figure 2A). Notably, more than half (56.7%) of the patients without CRS had at least 1 NC ≥Gr3 AE. When stratified based on CRS severity (CRS Gr 1-2 vs Gr 3-4), patients with CRS Gr 3 or 4 had a higher median number of ≥Gr3 NC AEs (medians, 3 vs 7, respectively; P = .0002; Figure 2B). Relative proportions of ≥Gr3 NC AEs in those with and without CRS are shown in Figures 2C,D. In patients who developed CRS (n = 104), 93.1% of NC ≥Gr3 AEs occurred after CRS onset (Figure 2E).
Figure 2.

Incidence of ≥Gr3 NC AEs based on the characteristics of CRS. (A) Dot plot of ≥Gr3 NC AEs in patients based on the presence of CRS. (B) Dot plot of ≥Gr3 NC AEs in patients based on severity of CRS maximum grade 1 or 2 vs 3 or 4 as graded per American Society for Transplantation and Cellular Therapy guidelines. (C) Pie graph of ≥Gr3 NC AEs in patients with CRS based on the system. (D) Pie graph of ≥Gr3 NC AEs in patients without CRS based on the system. (E) Timing of ≥Gr3 NC AEs in patients with CRS separated based on the percentage, with onset before (before CRS, red) or after (after CRS, blue) the onset of CRS.
AEs based on outcomes: CR vs non-CR
Of 133 patients who underwent disease response evaluation, 88 (66.2%) achieved a CR (1 patient was nonevaluable for response because of early death). Patients with CR trended toward having more ≥Gr3 NC AEs per patient (4 [IQR, 1-8]) than nonresponders (3 [IQR, 0-5.5]; P = .10; Figure 3A). Of patients without CR, 32 (71.1%) had at least 1 ≥Gr 3 NC AE. A total of 16 patients without CR developed progressive disease, of whom 13 required initiation of alternative chemotherapy before day +30, beginning at a median of day +17. Among these 13, only 4 patients experienced any ≥Gr3 NC AEs (5 AEs in total) between initiation of alternative therapy and day +30, of whom 3 had CAR T-cell expansion, raising the possibility that these AEs could have been CAR T-cell mediated.
Figure 3.

Incidence of ≥Gr3 NC AEs based on response and disease burden. (A) Dot plot of ≥Gr3 NC AEs in patients with complete response (CR) compared with that in those without CR (no CR). (B) Dot plot of ≥Gr3 NC AEs in patients based on both CRS and response. (C) Heat map of ≥Gr3 NC AEs based on the system in nonresponders (no CR) with CRS and without CRS. Deeper blue corresponds to greater number of events per patient. (D) Dot plot of ≥Gr3 NC AEs in patients based on baseline bone marrow disease as determined based on the percent mononuclear cells via flow cytometry: M1, <5%; M2, from 5% to 25%; M3, >25%. (E) Combined dot plots evaluating number ≥Gr3 NC AEs in patients based on baseline bone marrow disease in each of 4 response cohorts defined in panel B.
AEs based on outcomes: CRS and response rates
Evaluating AEs across CRS and responses (Figure 1A), the highest degree of toxicity was experienced by nonresponders who developed CRS. Nonresponders experiencing CRS (–CR/+CRS) trended toward experiencing more ≥Gr3 NC AEs than those with CRS who achieved a CR (+CR/+CRS) (median, 7 [IQR, 4-11] vs 4 [IQR, 1-8.3]; P = .07; Figure 3B). The degree of CRS in nonresponders was low, with 15 patients (88.2%) having Gr 1 or 2 CRS, similar to responders, in whom 66 (76.7%) had Gr 1 or 2 CRS (P = .29). Only 2 patients had a CR without experiencing CRS, only 1 of whom had a single ≥Gr3 NC AE. Unsurprisingly, among nonresponders, there were more ≥Gr3 NC AEs in patients who had CRS (–CR/+CRS) than those who did not (–CR/–CRS) (median, 7 [IQR, 4-11] vs median, 1 [IQR, 0-3], respectively; P < .0001; Figure 3B). Evaluation of toxicities experienced by nonresponders revealed metabolic toxicities and febrile neutropenia to be the most prevalent NC AEs (Figure 3C).
AEs in relation to preinfusion disease burden
Given the association of high disease burden with both CRS severity and nonresponse,21,22 disease burden was evaluated in relationship to the number of ≥Gr3 NC AEs overall (Figure 3D) and across the 4 response categories (Figure 3E). Although the difference in ≥Gr3 NC AEs was minimal between patients with M1 (< 5% disease burden) and those with M2 (5%-25%) marrows (median, 1 vs 2, respectively; P = .21), patients with an M3 (>25% disease burden) marrow did develop significantly more NC AEs (median, 5.5; P < .0001). Additionally, in patients with +CR/+CRS, disease burden influenced the median number of ≥Gr3 NC AEs (P < .0001). For patients with CRS without response (–CR/+CRS) (n = 17), because only 2 patients had low-disease burden, assessing the impact of disease on toxicity was limited. Lastly, in those without CRS or response (–CR/–CRS) (n = 28), baseline disease burden did not substantially affect the number of ≥Gr3 NC AEs (P = .82). Interestingly, the 2 responding patients without CRS (+CR/–CRS) both had an M1 marrow, and only 1 had a single ≥Gr3 NC AE. Collectively, although disease burden positively correlated with greater ≥Gr3 NC AEs overall and in those with +CR/+CRS, the relationship of disease burden and toxicity in nonresponders warrants further study. The influence of disease burden on ≥Gr3 NC AEs did not extend to the presence of non–central nervous system (CNS) extramedullary disease (EMD) or CNS disease, as neither had a statistical association with development of ≥Gr3 NC AEs (Figures 4A,B).
Figure 4.

Incidence of ≥Gr3 NC AEs in patients based on pre-CAR factors. (A) Dot plot of ≥Gr3 NC AEs in patients based on the presence of EM (extramedullary) disease. (B) Dot plot of ≥Gr3 NC AEs in patients based on the presence of CNS disease. (C) Dot plot of ≥Gr3 NC AEs in patients based on the age. (D) Dot plot of ≥Gr3 NC AEs in patients based on the sex. (E) Dot plot of ≥Gr3 NC AEs in patients based on the ethnicity. (F) Dot plot of ≥Gr3 NC AEs in patients based on the race; because of small numbers, analysis was grouped into White vs non-White, with non-White including African American, Asian, Hawaiian/Pacific Islander, and multirace; 15 patients with unknown race were excluded from this analysis. (G) Dot plot of ≥Gr3 NC AEs in patients based on the number of prior treatment regimens, excluding prior hematopoietic stem cell transplantation (HSCT). (H) Dot plot of ≥Gr3 NC AEs in patients based on prior HSCTs. (I) Dot plot of ≥Gr3 NC AEs in patients based on prior CAR T-cell therapy. (J) Dot plot of ≥Gr3 NC AEs in patients based on the prior receipt of blinatumomab. (K) Dot plot of ≥Gr3 NC AEs in patients based on the prior receipt of inotuzumab. CNS1, 0 blasts on cytospin; CNS2+, some degree of CNS disease, either CNS2 (<5/μL white blood cells, cytospin positive for blasts) or CNS3 (≥ 5/μL white blood cells, cytospin positive for blasts).
AEs in relation to baseline demographics
There were no meaningful associations between patient demographics and toxicity, with ≥Gr3 NC AEs not differing across age groups (<12, 12-17, and ≥18 years). However, there was a slight trend toward greater number of AEs in those aged ≥18 years (median, 5 vs 3 and 2 in the youngest and middle cohorts, respectively; P = .13; Figure 4C). There were also no differences in ≥Gr3 NC AEs based on sex, ethnicity, or race, noting that because of small numbers, race categories had to be combined for purposes of analysis (Figure 4D-F).
AEs in relation to prior therapy
Prior lines of therapy did not associate with the degree of ≥Gr3 NC AEs (P = .23; Figure 4G). Although prior hematopoietic stem cell transplantation, prior CAR T-cell therapy, and blinatumomab administration (Figures 4H-J) demonstrated no independent association with development of toxicities, receipt of prior inotuzumab (n = 23) was associated with a higher median number of ≥Gr3 NC AEs (median, 6 [IQR, 2-14] vs 3 [IQR, 1-6]; P = .012; Figure 4K). Of note, <8% of ≥Gr3 NC AEs in patients who received prior inotuzumab were hepatic toxicities (supplemental Figure 2A).
AEs attributed to underlying leukemia vs CAR T cells
Given the toxicity profile of those experiencing nonresponse, ≥Gr3 AEs were further stratified based on their attribution (disease vs research vs LD, as described in supplemental Methods) to distinguish those attributed to underlying disease and not CAR T cells. Restricted to those AEs attributed to disease, there were 460 ≥Gr3 AEs (26.8%), of which 123 were ≥Gr3 NC AEs (supplemental Figure 2B). Metabolic toxicities were the most common, followed by febrile neutropenia and hepatic and cardiovascular toxicities.
Evaluating predictors of ≥Gr3 NC AEs
After univariate analysis of preinfusion factors associated with ≥Gr3 NC AEs, a multivariable linear regression model was developed per protocol to determine the factors that may jointly predict the development of ≥Gr3 NC AEs. This was performed separately by protocol, given the difference in ≥Gr3 AE incidence across trials identified in the univariate analysis, reflecting inherent differences across CAR T-cell constructs and accounting for referral bias (ie, more heavily pretreated patients with CD19– disease were referred for CD22 CAR therapy).
Across protocols, only 1 consistent factor was predictive of the number of ≥Gr3 NC AEs: the baseline disease burden. Disease burden was considered for inclusion both as a categorical variable (M1 vs M2 vs M3) and a continuous variable based on the percentage of disease burden via flow cytometry. Although the specific models are shown in Table 2, in addition to disease burden, age and receipt of prior CAR showed some signal of influencing ≥Gr3 NC AEs, which warrants further study. The fit of these models for CD19 and CD22 could be interpreted as being at least marginally acceptable, with adjusted r2 values of 0.46 and 0.51, respectively (supplemental Figures 3A,B).
Table 2.
Multivariable linear regression
| Protocol | Primary cumulative analysis | Secondary focused analysis | Interpretation |
|---|---|---|---|
| CD19 | 0.20 × age + 0.034 × actual (0-99) marrow disease level Adjusted r2 = 0.46 |
0.94×(1, 2, or 3 depending on marrow disease) + 2.33 (if EM disease) Adjusted r2 = 0.53 |
Cumulative analysis: increasing age and marrow disease are predictive of higher NC AEs Focused analysis: presence of EM disease, rather than age, and categorical marrow disease were more accurate predictors |
| CD22 | 4.07×(1, 2, or 3 depending on marrow disease) − 3.87 (if prior CAR) Adjusted r2 = 0.51 |
2.465×(1, 2, or 3 depending on marrow disease) − 2.094 (if prior CAR) Adjusted r2 = 0.58 |
Cumulative analysis: higher marrow disease as a categorical factor influences NC AEs, whereas receipt of prior CAR decreases the number of predicted NC AEs Focused analysis: Same factors influence NC AEs but to a lesser degree |
| CD1922 | 4.81 – 1.89×agecat + 1.16×(1, 2, or 3 depending on marrow disease) Agecat = 1 if <12 y Agecat = 2 if 12-17 y Agecat = 3 if ≥18 y Adjusted r2 = 0.60 |
2.1207 – 2.5172 (if age ≥18) + 0.7241× (1, 2, or 3 depending on marrow disease) Adjusted r2 = 0.65 |
Cumulative analysis: NC AEs are predicted based on the categorical marrow disease; increased age decreases the number of predicted NC AEs Focused analysis: Same factors influence NC AEs but to a lesser degree in secondary analysis |
Secondary focused analysis
Across the 457 ≥Gr3 NC AEs experienced by 108 patients (80.6%) (Figure 1D), the median number of ≥Gr3 NC AEs was 3 (IQR, 1-5), with the majority experiencing at least 1 ≥Gr3 metabolic event and 1 ≥Gr3 febrile neutropenia and nearly one-third developing at least 1 ≥Gr3 hepatic event (supplemental Table 3).
As shown in supplemental Table 5 and supplemental Figure 4A,B, overall outcomes were comparable with those of the primary analysis. Incidence varied based on the trial, with more events per patient receiving CD22 CAR T cells than CD19 and CD19/22 (median, 4 vs 2 and 1, respectively; P < .0001). Univariate analysis again showed that CRS incidence and severity as well as ≥25% bone marrow disease were correlated with a higher number of ≥Gr3 NC AEs (supplementary Figure 4C-E). Notably, on secondary analysis, there was a greater degree of difference in the median number of NC AEs in patients with CR vs no CR (median, 3 vs 2; P = .037; supplementary Figure 4F). There were no other significant differences in the univariate analysis between the secondary and primary analyses (supplementary Figures 4G, 5A-K). Multivariable linear regression also demonstrated the consistent importance of baseline disease burden in predicting the number of ≥Gr3 NC AEs (Table 2). The median duration of ≥Gr3 NC AEs per patient was 2 days (IQR, 1-5 days; maximum, 25 days). The median duration did differ based on the trial, with a higher duration of 3 days per NC AE in both CD22 and CD19/22 trials (IQR, 1-6 and 1-5, respectively) compared with 2 days in the CD19 trial (IQR, 1-3; P = .0006).
Discussion
With the advent of CAR T-cell therapies, CAYAs with r/r B-ALL are subject to a new set of inflammatory toxicities not previously associated with standard chemotherapy-based treatments. Particularly counterintuitive to chemotherapy-related toxicities, in which AEs are not generally associated with response, experiencing some inflammatory toxicities after CAR T cells is often desired and frequently associates with efficacy. However, with early-phase studies in which efficacy may be unknown and in patients with multiply r/r disease who may be more prone to complications, delineating the full spectrum of toxicities is imperative to understand what patients may experience, especially as novel CAR T-cell constructs are being developed. Although the capture, reporting, and interpretation of AEs are imperfect and newer more subjective tools such as patient reported outcomes (PROs) are rapidly becoming the standard of care, AEs serve as a useful objective measure of the degree of end-organ toxicity and morbidity experienced in clinical trials.23, 24, 25, 26, 27 Accordingly, our study provides a comprehensive characterization of end-organ toxicities experienced by CAYAs with r/r B-ALL in the first 30 days after CAR T-cell therapy.
Among 134 CAYAs with r/r B-ALL receiving a phase 1 CAR T-cell construct, we found that most patients (n = 109; 81.3%) experienced at least 1 ≥Gr3 NC AE, with a median of 3 ≥Gr3 NC AEs across 14 unique categories of toxicity. In comparison with an adult study of 60 patients with diffuse large B-cell lymphoma treated with CD19 CAR T cells, Wudhikarn et al identified 289 cumulative toxicities ≥Gr3 up to 1 year after infusion experienced by 53 patients (88.3%) patients, which, if equally distributed, would amount to greater than 5 ≥Gr3 AEs per patient, establishing an important foundation to understand the full spectrum of CAR T-cell toxicities.28 Similarly, in the pediatric setting, a safety analysis of 137 patients receiving tisagenlecleucel in 2 phase 2 trials similarly showed that the majority (77%) experienced at least 1 ≥Gr3 AE, inclusive of cytopenias; whereas a study of a dual specificity CAR with humanized anti-CD19 and anti-CD22 CAR in Europe demonstrated that 60% of children (9 of 15) experienced grade 3 or 4 toxicity by day 30.29,30
Understanding how CAR T-cell toxicities compare with standard chemotherapy and/or alternative immunotherapy associated toxicities remains of great interest and an important benchmark for comparison of the toxicities that emerge from novel CAR T-cell trials. Incorporation of AE reporting, primarily based on CTCAE, across all clinical trials helps facilitate intertrial comparisons. For example, in the phase 3 Children’s Oncology Group trial AALL1331 (NCT02101853), which randomly assigned patients with B-ALL to blinatumomab or standard chemotherapy after induction for first relapse, the majority of CAYA patients (>80% receiving blinatumomab and >90% receiving chemotherapy) experienced at least 1 ≥Gr3 AE,31 suggesting a high incidence of high-grade toxicities with standard regimens. Although individual patient data were not provided, the majority of ≥Gr3 AEs were cytopenias, with high rates of febrile neutropenia (58%), transaminitis (alanine aminotransferase elevated; 41%), mucositis (28%), and sepsis (27%) in the chemotherapy arm.
Although most data regarding CAR T-cell toxicities in CAYA have emerged from experiences with CD19 targeting, recognizing that toxicities may differ across constructs will be particularly critical as the field expands. Accordingly, we identified that the degree of toxicity differed based on the trials analyzed in our study, with higher ≥Gr3 AEs and NC AEs in patients who received CD22 CAR T cells. With both higher incidence of CRS in the CD22 CAR T-cell trial than in the 2 CD19-based trials and the predilection of the CD22 CAR–treated population to have been more heavily pretreated, multiple factors likely contributed to this finding.18 Patients receiving CD22 CAR T cells also experienced a greater incidence of hemophagocytic lymphohistiocystosis–like toxicities (recently defined as immune effector cell–associated hemophagocytic lymphohistiocytosis–like syndrome),32 likely accounting for the higher number of hepatic and coagulopathic AEs in this trial.16,33
Not surprisingly, patients with CRS experienced more ≥Gr3 AEs than those without CRS; the majority of ≥Gr3 NC AEs occurred after CRS onset, suggesting that toxicities are highly associated with cytokine driven inflammation. However, separating out which end-organ toxicities occur as a direct sequela of CRS vs an independent coexisting pathologic process remains challenging. Because ≥Gr3 AEs also occurred in all 30 patients without CRS, additional factors such as LD chemotherapy and progression of underlying disease may contribute to the toxicity profile.
Aligned with the association of disease burden and CRS severity,7,34 we found that leukemia involvement also influenced development of ≥Gr3 AEs, with disease burden as the only consistent predictor of ≥Gr3 NC AEs across all 3 trials, upon multivariable analysis performed using both cumulative and focused data sets. Although CNS disease and EMD did not generally associate with an increase in ≥Gr3 NC AEs, we have shown that site-specific involvement of EMD can be associated directly with CAR T-cell toxicity.35 Although the focused multivariable analysis showed some correlation of EMD with toxicity after CD19 CAR, among patients with EMD across trials, the overall degree of CRS was mild (only 3 of 24 had CRS grade ≥3), explaining fewer high-grade NC AEs overall. Thus, individual patient considerations and experiences remain critical, even when not captured as a factor in a larger analysis.
Although the successes of CAR T cells have been widely acknowledged, the experience of nonresponders has been critically limited in CAR T-cell literature. Given the poor overall survival in nonresponders to tisagenlecleucel or alternative CD19 CAR T cells, the degree of toxicities experienced without the benefit of response has yet to be described.21,36,37 Unfortunately, our data indicated that those who developed CRS in the absence of clinical response had the highest degree of ≥Gr3 NC AEs. This finding suggests that early disease assessment in patients with severe CRS may be warranted to evaluate for disease progression as a contributor to toxicity severity.38,39 Notably, the majority (–CR, +CRS) had low grade (1-2) CRS, suggesting that higher toxicity was not driven by CRS alone. Furthermore, when evaluating toxicities in all patients attributable to underlying disease and not CAR T cells, metabolic, febrile neutropenia, hepatic and cardiovascular events comprised the majority of ≥Gr 3 NC AEs identified. This suggests that patients with underlying r/r B-ALL may experience toxicity due to the natural progression of their disease process, independent of CAR T-cell infusion. Moreover, it is important to acknowledge that in these early-phase dose-escalation trials, rates of nonresponse were low because of the remarkable efficacy of CAR T cells in B-cell malignancies; however, such responses may not be assured with alternative CAR T-cell trials for other diseases, highlighting the critical need to capture the experience of nonresponders.
The granularity of this data begins to illustrate the full profile of toxicities that CAYA patients experience after CAR T-cell therapy, regardless of whether they experience CRS or have a clinically meaningful response. Importantly, we show that toxicity profiles may differ across constructs, which may link directly to both incidence of CRS and characteristics of each unique CAR, an important consideration as novel CAR T cells are tested in the clinic. Understanding how CAR-associated toxicities compare with those caused by standard chemotherapy (which has not worked in our patients) is imperative, given the clear association of inflammatory toxicities with response, which is an entirely different approach to chemotherapy-based strategies.5 This is a particularly critical consideration when developing new CAR T-cell trials in which the use of preemptive strategies for toxicity mitigation, which is largely derived from experience with CD19 CAR T cells, may have uncertain implications on the efficacy of novel CAR T-cell constructs.
Because our analysis was limited to those toxicities experienced in the first 30 days after CAR T-cell infusion, this study does not provide insight into protracted findings or late onset toxicities, both of which need to be studied in more detail. Furthermore, as a referral center with variability in documentation of prior patient histories, it was not feasible to analyze prior toxicities in relation to post-CAR toxicities. Additionally, although age showed some correlation in predicting NC AEs upon multivariable analysis, the exclusion of infants and toddlers aged <3 years per the inclusion criteria of our center likely skews this analysis. Future analyses of toxicity based on age as well as cytogenetics are needed. Additionally, although inclusion of recurrent events in the cumulative analysis, which was meant to provide a full picture of ≥Gr3 AEs, might have inflated the degree of toxicity experienced, the focused analysis resulted in the same median number of events per patient, with similar findings upon both univariate and multivariable analyses. Given the limitations of CTCAE reporting (recall bias, chart abstraction errors, and over- and/or under-reporting)40,41 alongside increasing data supporting the incorporation of PRO measures into pediatric oncology trials, the importance of incorporating the patient voice into toxicity assessments cannot be overemphasized.25 Accordingly, our center, along with others, have recently begun to incorporate PROs into novel CAR T-cell trials for CAYAs. Indeed, given the improvements in quality-of-life scores seen across CAYA receiving tisagenlecleucel, these assessments will be critical in further analyzing the risk-benefit ratios of these novel therapies.42
Although our data provide a much-needed benchmark for end-organ specific toxicities experienced by CAYAs in phase 1 CAR T-cell trials for B-ALL, a critical next step is to develop a predictive tool to determine which patients may be at highest risk of severe end-organ toxicities after CAR T-cell therapy. Although our initial multivariable analysis of pre-CAR characteristics suggests that high disease burden predicts ≥Gr3 NC toxicities, incorporating additional biologic correlatives (eg, endothelial markers) warrants further study. Recently developed risk-scores in adults receiving CAR T cells (eg, Modified EASIX and CAR-HEMATOTOX) incorporate biomarker-based models and need to be validated in children.43,44 Understanding the impact of baseline comorbidities on outcomes is similarly imperative, particularly given the extensive prior therapy our patients might have received.45, 46, 47 Considering the limited treatment options for patients seeking CAR T cells and acknowledging that toxicities may be associated with clinical response, it is essential to optimize strategies to reduce CAR T-cell–associated toxicities in patients at high risk to improve overall outcomes in responders and nonresponders alike.
Conflict-of-interest disclosure: N.N.S. receives royalties from CARGO Therapeutics. The remaining authors declare no competing financial interests.
Acknowledgments
The authors gratefully acknowledge the study participants and their families, referring medical care teams, the faculty, and staff of the NIH Clinical Center who provided their expertise in the management of the study participants, and the data managers and research nurses and patient care coordinators involved with this work. The authors acknowledge Crystal L. Mackall, Alan S. Wayne, and Terry J. Fry, along with Cindy L. Delbrook, for their leadership in implementing these studies at the National Cancer Institute. Figures were created with BioRender.com and Prism 9.0.
This work was supported, in part, by the Intramural Research Program, Center of Cancer Research, National Cancer Institute, NIH Clinical Center, National Institutes of Health, and the Warren Grant Magnuson Clinical Center. All funding was provided by the NIH Intramural Research Program (ZIA BC 011823; N.N.S.). No nonauthor wrote the first draft or any part of the paper. The content of this publication does not necessarily reflect the views of policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Authorship
Contribution: S.K.S. and N.N.S wrote the first draft of the manuscript; S.K.S, S.M., S.M.S., E.M.H., B.Y., A.S., and N.N.S. performed primary data analysis and evaluated correlative studies; B.Y., H.S., D.W.L., and N.N.S., provided patient care and contributed critically to the manuscript; and all authors contributed to reviewing the final manuscript and have agreed to be coauthors.
Footnotes
All data are available on request to the corresponding authors, Sara Silbert (sara.silbert@nih.gov) and Nirali N. Shah (nirali.shah@nih.gov).
The full-text version of this article contains a data supplement.
Contributor Information
Sara K. Silbert, Email: sara.silbert@nih.gov.
Nirali N. Shah, Email: nirali.shah@nih.gov.
Supplementary Material
References
- 1.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shah BD, Ghobadi A, Oluwole OO, et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet. 2021;398(10299):491–502. doi: 10.1016/S0140-6736(21)01222-8. [DOI] [PubMed] [Google Scholar]
- 3.Lee DW, Santomasso BD, Locke FL, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019;25(4):625–638. doi: 10.1016/j.bbmt.2018.12.758. [DOI] [PubMed] [Google Scholar]
- 4.Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127(26):3321–3330. doi: 10.1182/blood-2016-04-703751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maus MV, Alexander S, Bishop MR, et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immune effector cell-related adverse events. J Immunother Cancer. 2020;8(2) doi: 10.1136/jitc-2020-001511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shah NN, Lee DW, Yates B, et al. Long-term follow-up of CD19-CAR T-cell therapy in children and young adults with B-ALL. J Clin Oncol. 2021;39(15):1650–1659. doi: 10.1200/JCO.20.02262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shah NN, Highfill SL, Shalabi H, et al. CD4/CD8 T-cell selection affects chimeric antigen receptor (CAR) T-cell potency and toxicity: updated results from a phase I anti-CD22 CAR T-cell trial. J Clin Oncol. 2020;38(17):1938–1950. doi: 10.1200/JCO.19.03279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Curran KJ, Margossian SP, Kernan NA, et al. Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL. Blood. 2019;134(26):2361–2368. doi: 10.1182/blood.2019001641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gardner RA, Finney O, Annesley C, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129(25):3322–3331. doi: 10.1182/blood-2017-02-769208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shalabi H, Sachdev V, Kulshreshtha A, et al. Impact of cytokine release syndrome on cardiac function following CD19 CAR-T cell therapy in children and young adults with hematological malignancies. J Immunother Cancer. 2020;8(2) doi: 10.1136/jitc-2020-001159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burstein DS, Maude S, Grupp S, Griffis H, Rossano J, Lin K. Cardiac profile of chimeric antigen receptor T cell therapy in children: a single-institution experience. Biol Blood Marrow Transplant. 2018;24(8):1590–1595. doi: 10.1016/j.bbmt.2018.05.014. [DOI] [PubMed] [Google Scholar]
- 13.Shalabi H, Martin S, Yates B, et al. Neurotoxicity following CD19/CD28ζ CAR T-cells in children and young adults with B-cell malignancies. Neuro Oncol. 2022;24(9):1584–1597. doi: 10.1093/neuonc/noac034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mikkilineni L, Yates B, Steinberg SM, et al. Infectious complications of CAR T-cell therapy across novel antigen targets in the first 30 days. Blood Adv. 2021;5(23):5312–5322. doi: 10.1182/bloodadvances.2021004896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fried S, Avigdor A, Bielorai B, et al. Early and late hematologic toxicity following CD19 CAR-T cells. Bone Marrow Transplant. 2019;54(10):1643–1650. doi: 10.1038/s41409-019-0487-3. [DOI] [PubMed] [Google Scholar]
- 16.Lichtenstein DA, Schischlik F, Shao L, et al. Characterization of HLH-like manifestations as a CRS variant in patients receiving CD22 CAR T cells. Blood. 2021;138(24):2469–2484. doi: 10.1182/blood.2021011898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buechner J, Grupp SA, Hiramatsu H, et al. Practical guidelines for monitoring and management of coagulopathy following tisagenlecleucel CAR T-cell therapy. Blood Adv. 2021;5(2):593–601. doi: 10.1182/bloodadvances.2020002757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shalabi H, Qin H, Su A, et al. CD19/22 CAR T-cells in children and young adults with B-ALL: phase I results and development of a novel Bicistronic CAR. Blood. 2022;140(5):451–463. doi: 10.1182/blood.2022015795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20–28. doi: 10.1038/nm.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schultz LM, Eaton A, Baggott C, et al. Outcomes after nonresponse and relapse post-tisagenlecleucel in children, adolescents, and young adults with B-cell acute lymphoblastic leukemia. J Clin Oncol. 2023;41(2):354–363. doi: 10.1200/JCO.22.01076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Myers RM, Taraseviciute A, Steinberg SM, et al. Blinatumomab nonresponse and high-disease burden are associated with inferior outcomes after CD19-CAR for B-ALL. J Clin Oncol. 2022;40(9):932–944. doi: 10.1200/JCO.21.01405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller T, Getz K, Li Y, et al. Rates of laboratory adverse events by course in paediatric leukaemia ascertained with automated electronic health record extraction: a retrospective cohort study from the Children's Oncology Group. Lancet Haematol. 2022;9:e678–e688. doi: 10.1016/S2352-3026(22)00168-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller TP, Aplenc R. Evolution of hematology clinical trial adverse event reporting to improve care delivery. Curr Hematol Malig Rep. 2021;16(2):126–131. doi: 10.1007/s11899-021-00627-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Freyer DR, Lin L, Mack JW, et al. Lack of concordance in symptomatic adverse event reporting by children, clinicians, and caregivers: implications for cancer clinical trials. J Clin Oncol. 2022;40(15):1623–1634. doi: 10.1200/JCO.21.02669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacobs SS, Withycombe JS, Castellino SM, et al. Longitudinal use of patient reported outcomes in pediatric leukemia and lymphoma reveals clinically relevant symptomatic adverse events. Pediatr Blood Cancer. 2022;69(12) doi: 10.1002/pbc.29986. [DOI] [PubMed] [Google Scholar]
- 27.Basch E, Reeve BB, Mitchell SA, et al. Development of the National Cancer Institute's patient-reported outcomes version of the common terminology criteria for adverse events (PRO-CTCAE) J Natl Cancer Inst. 2014;106(9) doi: 10.1093/jnci/dju244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wudhikarn K, Pennisi M, Garcia-Recio M, et al. DLBCL patients treated with CD19 CAR T cells experience a high burden of organ toxicities but low nonrelapse mortality. Blood Adv. 2020;4(13):3024–3033. doi: 10.1182/bloodadvances.2020001972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levine JE, Grupp SA, Pulsipher MA, et al. Pooled safety analysis of tisagenlecleucel in children and young adults with B cell acute lymphoblastic leukemia. J Immunother Cancer. 2021;9(8) doi: 10.1136/jitc-2020-002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cordoba S, Onuoha S, Thomas S, et al. CAR T cells with dual targeting of CD19 and CD22 in pediatric and young adult patients with relapsed or refractory B cell acute lymphoblastic leukemia: a phase 1 trial. Nat Med. 2021;27(10):1797–1805. doi: 10.1038/s41591-021-01497-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown PA, Ji L, Xu X, et al. Effect of postreinduction therapy consolidation with blinatumomab vs chemotherapy on disease-free survival in children, adolescents, and young adults with first relapse of B-cell acute lymphoblastic leukemia: a randomized clinical trial. JAMA. 2021;325(9):833–842. doi: 10.1001/jama.2021.0669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hines MR, Knight TE, McNerney KO, et al. Immune effector cell-associated hemophagocytic lymphohistiocytosis-like syndrome. Transplant Cell Ther. 2023;29(7):438.e1–438.e16. doi: 10.1016/j.jtct.2023.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McNerney KO, Lim SS, Dreyzin A, et al. LBA1--CAR-associated hemophagocytic lymphohistiocytosis (HLH) with use of commercial tisagenlecleucel in the Pediatric Real World CAR Consortium (PRWCC): risk factors and outcomes. Transplantation and Cellular Therapy. 2022;28(suppl 3):S473–S475. [Google Scholar]
- 34.Hay KA, Hanafi LA, Li D, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood. 2017;130(21):2295–2306. doi: 10.1182/blood-2017-06-793141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holland EM, Yates B, Ling A, et al. Characterization of extramedullary disease in B-ALL and response to CAR T-cell therapy. Blood Adv. 2022;6(7):2167–2182. doi: 10.1182/bloodadvances.2021006035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Good Z, Spiegel JY, Sahaf B, et al. Post-infusion CAR TReg cells identify patients resistant to CD19-CAR therapy. Nat Med. 2022;28(9):1860–1871. doi: 10.1038/s41591-022-01960-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haradhvala NJ, Leick MB, Maurer K, et al. Distinct cellular dynamics associated with response to CAR-T therapy for refractory B cell lymphoma. Nat Med. 2022;28(9):1848–1859. doi: 10.1038/s41591-022-01959-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gardner RA, Ceppi F, Rivers J, et al. Preemptive mitigation of CD19 CAR T-cell cytokine release syndrome without attenuation of antileukemic efficacy. Blood. 2019;134(24):2149–2158. doi: 10.1182/blood.2019001463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kadauke S, Myers RM, Li Y, et al. Risk-adapted preemptive tocilizumab to prevent severe cytokine release syndrome after CTL019 for pediatric B-cell acute lymphoblastic leukemia: a prospective clinical trial. J Clin Oncol. 2021;39(8):920–930. doi: 10.1200/JCO.20.02477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller TP, Li Y, Kavcic M, et al. Accuracy of adverse event ascertainment in clinical trials for pediatric acute myeloid leukemia. J Clin Oncol. 2016;34(13):1537–1543. doi: 10.1200/JCO.2015.65.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller TP, Marx MZ, Henchen C, et al. Challenges and barriers to adverse event reporting in clinical trials: a children's oncology group report. J Patient Saf. 2022;18(3):e672–e679. doi: 10.1097/PTS.0000000000000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laetsch TW, Myers GD, Baruchel A, et al. Patient-reported quality of life after tisagenlecleucel infusion in children and young adults with relapsed or refractory B-cell acute lymphoblastic leukaemia: a global, single-arm, phase 2 trial. Lancet Oncol. 2019;20(12):1710–1718. doi: 10.1016/S1470-2045(19)30493-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pennisi M, Sanchez-Escamilla M, Flynn JR, et al. Modified EASIX predicts severe cytokine release syndrome and neurotoxicity after chimeric antigen receptor T cells. Blood Adv. 2021;5(17):3397–3406. doi: 10.1182/bloodadvances.2020003885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rejeski K, Perez A, Sesques P, et al. CAR-HEMATOTOX: a model for CAR T-cell-related hematologic toxicity in relapsed/refractory large B-cell lymphoma. Blood. 2021;138(24):2499–2513. doi: 10.1182/blood.2020010543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sorror ML. How I assess comorbidities before hematopoietic cell transplantation. Blood. 2013;121(15):2854–2863. doi: 10.1182/blood-2012-09-455063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sorror ML, Maris MB, Storb R, et al. Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood. 2005;106(8):2912–2919. doi: 10.1182/blood-2005-05-2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith AR, Majhail NS, MacMillan ML, et al. Hematopoietic cell transplantation comorbidity index predicts transplantation outcomes in pediatric patients. Blood. 2011;117(9):2728–2734. doi: 10.1182/blood-2010-08-303263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.