

Abstract
Introduction:
Sirtuins are highly conserved nicotinamide adenine dinucleotide (NAD+) sensors that are considered the guardians of homeostasis. Acute inflammation, which is intended to ward off pathogen invasion, is nature’s highly conserved stress-associated and molecular-based survival mechanism for most life forms. Acute inflammatory responses deviate cells from the homeostasis to enable survival. It is not surprising perhaps, that these two must interact in the most dramatic way to preserve homeostasis and preserve life.
Areas covered:
In this review we present an overview of sirtuin responses in acute life-threatening inflammatory conditions. We examine how the seven sirtuins (sirtuins 1–7) are responsible for modulating the acute inflammatory response in a context-dependent manner, thus presenting novel therapeutic targets. The database search includes Medline (since 1966) and PubMed (since 1996).
Expert opinion
Sirtuins fine-tune inflammatory response to acute infectious and non-infectious inflammatory stimuli. Modulating sirtuin activity leads to profound changes in inflammatory response. Sirtuin-activating and inhibiting agents are emerging as therapeutic agents to resolve inflammation and promote homeostasis in chronic inflammation. The use of sirtuin modulation in acute life-threatening inflammatory conditions has great potential.
Keywords: Acute Inflammation, Acute Lung Injury, Hemorrhagic shock, Sepsis, septic shock, Sirtuin, Sirtuins
1. INTRODUCTION
Sirtuins, first described in yeast as nicotinamide adenine dinucleotide (NAD+) sensors and regulators of metabolism, are a highly conserved family of genes with potent effects on inflammation, innate and adaptive immunity, intermediary metabolism, redox balance, and bioenergetics (1–5). They evolved as stress response genes with an ability to protect homeostasis from life threatening changes in environment. There are seven mammalian homologues of sirtuins: sirtuin1–7. Sirtuin 1, the primordial and well established as a longevity gene, is by far the most extensively studied sirtuin (6, 7). Sirtuin1–7 are dispersed through various cell compartment, but can translocate to other sites. Sirtuins 1, 6 and 7 primarily reside in the nucleus, sirtuins 3, 4 and 5 are mitochondrial, and sirtuin 2 is mainly localized in cytosol; however, under acute cellular stress, sirtuin 2 translocates into the nucleus(1).
The sirtuins depend on NAD+ for their enzyme activation(2). Notably, sirtuin activation requires adequate concentrations of NAD+ in the compartment in which they reside, or to which they transfer (8). Sirtuins are a highly conserved family of proteins, with a core catalytic domain of 275 amino acids. This core is flanked by N- and C- terminal extensions which determine sirtuin activity and sub-cellular localization (9). The catalytic core of sirtuin contains two sub-domains. The larger sub-domain consists of Rossman fold for NAD+ binding and a smaller domain, inserted in the Rossman fold, is formed by a zinc-binding motif and α-helical regions. The acetyl-lysine containing polypeptide and NAD+ substrates bind to the interface of these two domains (9–11). These form a “cofactor binding loop” where the catalytic events occur. The catalytic events comprise of covalent 1’ O-alkylamide intermediate between two substrates with nicotinamide release. This intermediate is further hydrolyzed to form deacetylated substrate polypeptide and 2’-O-acetyl-ADP-ribose(9). Evidence suggests significant conformational changes to the sirtuin molecule upon deacetylation (9).
While nicotinamide is removed during deacetylation reaction, evidence suggests that nicotinamide can promote the reverse reaction where the substrates are formed and sirtuin is inhibited, thus acting as a sirtuin inhibitor (12). Isonicotinamide, with competitive inhibition of nicotinamide, acts as a sirtuin activator (13). sirtuin inhibitors mainly consist of molecules that bind to the NAD+ binding site partially or completely (9). Resveratrol, is a known sirtuin activator. While controversy regarding direct sirtuin1 activation by resveratrol, evidence suggests that resveratrol increases NAD+ levels via activation of AMP kinase with concomitant increase in sirtuin1 deacetylase activity (14).
As a result of diverse location and function, the sirtuin family is able to broadly guard immuno-metabolic and bioenergy homeostasis of many cells and organs (15–17). At this time, we know of no other gene family of homeostasis regulators. Not surprisingly, sirtuins as key protectors and determinants of homeostasis fitness, deal with molecular mechanisms of death and survival.
In this review, we primarily focus on the role of sirtuins in reprogramming severe life threatening acute inflammatory response syndromes of two types: 1) Sepsis inflammation: life threatening stress from uncontrolled and disseminated infection (sepsis) and 2) Non-sepsis inflammation: uncontrolled non-infectious life threatening stress from severe trauma/ hemorrhagic shock or acute lung injury. Together, these two groups can be referred to as inflammatory “shock” syndromes. While there is overlap clinically, for the purpose of this review, they are discussed separately. We introduce the emerging field of sirtuins as “druggable” targets for these life threatening acute inflammatory ‘shock’ syndromes, and highlight gaps in moving the field forward.
2. ACUTE INFLAMMATION
Inflammation is nature’s ancient and highly conserved stress-associated and molecular based survival mechanism for most life forms. It operates at the cell, organ and organism level. It is designed to “defend, adapt, and mend” as a sequential pathway for guarding and restoring homeostasis. As such, acute inflammation is a key component of evolution’s universal principal of survival of the fittest formula: resist or tolerate danger (18). Our contemporary understanding of innate and adaptive immunity as essential components of inflammation grew from historic studies of horseshoe crabs and starfish larvae (19). These ancient creatures and their ancestors have inhabited earth for over 400 million years (20) (21). Striking similarities between mammalian and horseshoe crab-innate immune detection and response systems indicate that the inflammatory response is highly conserved in its core ability to resist or tolerate danger from environmental threats, stemming from an effort to survive (20). The starfish larvae taught us phagocytosis(19, 22); the horseshoe crab about sensing invading microbes by “toxic” constituents like endotoxin(21). The two properties play a crucial role in acute inflammation and innate immunity from pathogen invasion.
While both the acute and chronic inflammatory responses deviate from the homeostasis, the extent and duration of this departure significantly differ in acute versus chronic inflammatory response. For example, a chronic inflammatory response of obesity and ageing are persistent low grade prolonged pro-inflammation (23–25). In contrast, the acute inflammatory response is of shorter duration and greater severity of pro-inflammation and farther departure from homeostasis(26). Acute inflammatory response may or may not transition to anti-inflammatory/ mixed pro- and anti-inflammatory response as described in details in the following sections. We stress acute inflammation in this article, but allude to differences between acute vs chronic inflammation based on sirtuin functions. Expert reviews in chronic inflammation provide more in depth understanding of sirtuins and NAD contributions to chronic inflammatory diseases such as diabetes, metabolic syndrome, obesity and aging (3, 5, 27–29).
Acute inflammatory responses begin within seconds to minutes of sensing an environmental danger signal, and often become clinically evident after a few hours. The five cardinal signs of acute inflammation: calor (increased heat), rubor (redness), dolor (pain) and tumor (swelling) were described by Celcus between 30 BC–38 AD, while the fifth sign, functio laesa (loss of function), was described by Virchow in 19th century. Another important but less famous sign is anorexia (30). After environmental sensing of danger (alarmins) and signaling from external membrane receptors or internal protein sensors inform cytosol and nuclear signaling pathways, the acute inflammatory response, upregulates several thousand genes that reprogram pro-inflammatory resistance responses; in monocytes or macrophages, just as many genes are repressed. Importantly, organ specific cells expressing toll like receptors (TLR) for sensing microbes also reprogram many genes associated with anabolism and catabolism. Adaptive immune cells also increase expression of many overlapping and cell specific cytokines and metabolic pathways (31). Successful acute inflammation returns to homeostasis, but restored homeostasis epigenetically differs from that which inflammation departed (32).
Thus, the fate of cells, organs, and organisms during acute inflammation depends on whether homeostasis can be restored. To complete the homeostasis survival route, acute inflammation must sequentially depart homeostasis enter tolerance/adaptation, and then reprogram inflammation resolution. As such, inflammation deals in life and death by defending (immune resistance), and mending (immune tolerance). Sirtuins play a key role in determining when and whether acute inflammation resolves.
The sirtuin family of NAD+ sensors and signaling mediators plays a critical role in the entire process of acute inflammation and its resolution. To do this, sirtuins lie downstream of alarmin sensors, which initiate a sirtuin-dependent reprogramming network that directs immunity, metabolism of nutrients, mitochondrial bioenergetics and redox balance. A feature of sirtuins that lends itself to molecular targeting is that NAD+ and sirtuin control highly conserved signaling nodes within the complex homeostasis network, which integrates immune and organ cell physiology or pathophysiology. The complex reprogramming by sirtuins informs epigenetic, transcriptional, post-transcriptional, and post-translational cell function and fate.
A critical component of sirtuin sensing and responding is NAD+ availability. NAD+ levels are rate limiting step for sirtuin activation and tightly controlled by the recycling NAD+ via salvage pathway. Recent reports also emphasize an unrecognized role for de novo synthesis of NAD+ dependent on tryptophan conversion to quinolinic acid (33) shows regulation of NAD+ levels as an essential cofactor for the de-acylation activities of most sirtuin family members. In the following sections, we discuss sepsis-related and non-sepsis related life threatening systemic inflammatory processes and sirtuin control of them in details.
2.1. SIRTUINS AND SEPSIS-INDUCED LIFE THREATENING INFLAMMATION:
Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host i infection. Septic shock is a subset of sepsis in which underlying circulatory and cellular/metabolic abnormalities are profound enough to substantially increase mortality (34). Sirtuins play a major role in various aspects of inflammatory response in sepsis.
2.1.1. Immuno-metabolic response:
The acute systemic inflammatory response to the uncontrolled pathogen infection of sepsis reprograms from early/hyper-inflammatory resistance/defend response to a late/hypo-inflammatory and immunosuppressive phenotype (35, 36) as shown in Figure 1. With improved life support technology, a minority of sepsis-related deaths occur during the hyper-inflammation, while over 60% of the sepsis-mortality occurs during hypo-inflammatory phase and up to 1 year after the sepsis episode (37).
Figure 1.
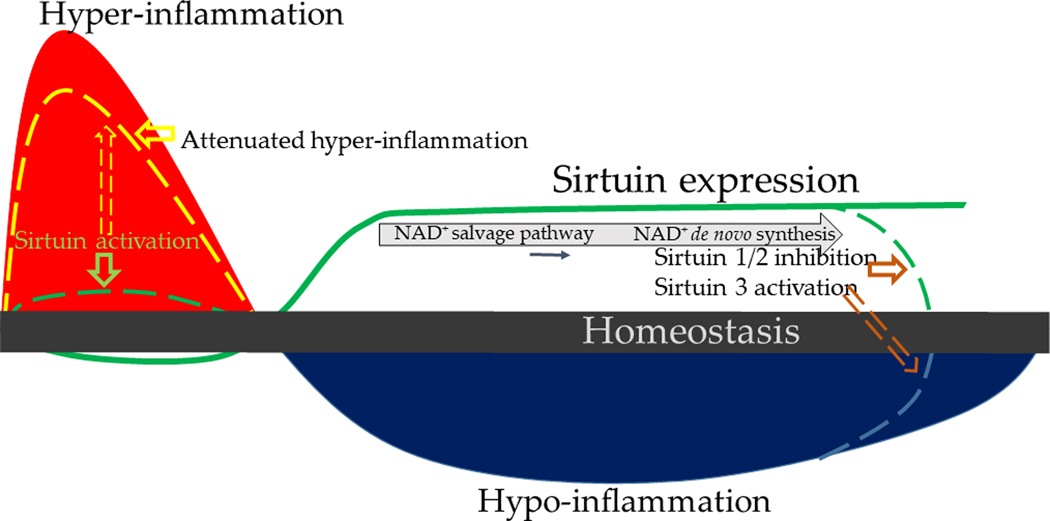
Immune response in sepsis: Immune response to sepsis transitions from early/hyper-inflammatory and endotoxin sensitive to a late/hypo-inflammatory and endotoxin tolerant phase. Hyper-inflammation is associated with sirtuin deficiency posing a therapeutic potential for sirtuin activation during hyper-inflammation. Sirtuins are crucial for the transition from hyper- to a hypo-inflammatory phase; sustained sirtuin expression delays return to homeostasis. Immune cells use NAD+ salvage pathway during early hypo-inflammation switching to de novo synthesis during late hypo-inflammation; NAD+ is required for sustained sirtuin expression. Sirtuin inhibition during late sepsis can achieve return to homeostasis quickly.
Many cell types, including immune and organ specific cells that express microbial sensors such as Toll like receptors (TLR), respond quickly during the hyper-inflammatory phase to kill the invading pathogens. The hyper-inflammatory response first decreases sirtuin as well as NAMPT and AMP kinase levels (38). The sirtuin levels and/or activity increase concomitant with the shift in oxidative state from a pro-oxidant to a reductive state by increased expression and function of antioxidants like GSH and superoxide dismutases (SOD) in cytosol and mitochondria during hypo-inflammation discussed below. The cell migration, phagocytosis and pathogenic killing are energy consuming processes. During hyper-inflammation, the innate immune monocytes or macrophages predominantly derive their early ATP energy “burst” from aerobic glycolysis, similar to that seen in cancer cells. Preferential aerobic glycolysis was first described by Otto Warburg in 1927 in cancer cells(39). With aerobic glycolysis, immune cells not only generate much needed ATP molecules in the cytosol from glycolysis, but also increase reactive oxygen species (ROS) needed for pathogen killing from multiple sources, including the NOX family, xanthine oxidase, and the mitochondria electron transport chain (ETC) (40, 41). Evidence suggests that the hyper-inflammation is at least partly provoked by decreases in sirtuins phenotype (42). Specifically, sirtuins 1, 2, 3 and 6 levels rapidly and transiently decrease during acute hyper-inflammatory phase (33, 43–45). Sirtuin 3 overexpression decreases microvascular dysfunction (44) and attenuates organ dysfunction of early sepsis (43, 45, 46).
During hypo-inflammatory phase, we showed sustained increase in sirtuin 1 in lean (35) and sirtuin 2 in obese mice (47) with sepsis. In both these cases, we showed, that sirtuins target NFκB p65 to deacetylate and deactivate it (47, 48). Consequently, NFκB p65-dependent pro-inflammatory genes such as TNF-α, IL-1β, E-selectin and ICAM-1 are suppressed during hypo-inflammation and bacterial clearance impaired (47–49). These changes are at least partially reversed with sirtuin inhibition during hypo-inflammation (35, 40, 47, 50)
2.1.2. Redox regulation:
Hyper-inflammatory phase of sepsis is a pro-oxidant. Evidence suggests that S-glutathionylation decreases sirtuin 1 and 3 activities. Interestingly, while S-glutathionylation does not seem to decrease basal deacetylation activity of sirtuin1, it inhibits stimulation of sirtuin1 activity by sirtuin activator resveratrol (51). Sirtuin 3 deacetylates and activates superoxide dismutase 2 (SOD2); sirtuin 3 inactivation due to S-glutathionylation leads to hyper-acetylation of SOD2 and exacerbates angiotensin II-induced hypertension in sirtuin 3 knockout and SOD2 depleted mice(52). Sirtuin 3 activity is also regulated by mitochondrial function and matrix ph. Reduced membrane potential decreases sirtuin3 activity and changes substrate from carbohydrate oxidation to lactate production. This switch in substrate utilization increases NADH/NAD+ ratio with increased Acetyl Co-A levels which in turn decrease sirtuin 3 activity (53).
Similarly, we showed that the sirtuins 6 and 2 are inactivated by cysteine thiol oxidation (50, 54) during hyper-inflammatory phase of sepsis. Oxidized and inactivated sirtuin 2 shows decreased ability to deacetylate NFκB p65 and exaggerated pro-inflammatory response to LPS stimulation in macrophages (50). Together, these studies suggest that during pro-oxidant stress, such as hyper-inflammation, direct oxidation and inactivation of sirtuins may assist in the pro-inflammatory response that is which is ultimately intended for pathogen clearance. However, in a dysregulated hyper-inflammatory process, an excessive induction of ROS and NOS breaks down mitochondria and nuclear DNA, which within and without cells further activates hyper-inflammatory injury (55). Nuclear sirtuin 6 promotes nuclear DNA repair (56, 57).
The resistance and tolerance molecular reprogramming sequence is time and danger dose dependent. During sepsis, the magnitude of hyper-inflammation may generate rapid cardiovascular collapse and shock. In survivors of the hyper-inflammatory state, reprogramming to tolerance occurs within 2–4 h (58) and may reach the suspended animation low energy state that is immune and metabolic paralysis that cannot regenerate immune and organ cell physiologic competence. This state, referred to as hypo-inflammation here, is characterized by endotoxin tolerance and sustained departure from homeostasis with a low ATP energy and antioxidant state (Figure 1). The presence of endotoxin tolerance of monocytes is a good biomarker of the hypo-inflammatory state of sepsis in mice and humans with its sustained “immuno-metabolic paralysis” and increasing mortality (59) (58). The strength of the danger and the host immuno-metabolic and energy reserve play a major role in outcome and persistent NAD+ generation and sirtuin activation that underlie resistance and tolerance dysregulation (35, 42, 47).
The hypo-inflammatory phase of sepsis is characterized by endotoxin tolerance and decreased pathogen clearance (35), as well as antioxidant activity. The immune cells undergo substantial metabolic changes during the transition from hyper-inflammatory to hypo-inflammatory molecular phenotypes. Specifically, the immune cells switch the from glucose and glutamine nutrient fueling of glycolysis and mitochondrial bioenergetics to lipolysis and fatty acid oxidation. Sirtuins 1 and 6 coordinate the switch to tolerance; sirtuin 6 inhibits glycolysis while sirtuin 1 increases fatty acid oxidation in a coordinated fashion (42). During the nuclear-mitochondrial reprogramming, sirtuin 1 also induces mitochondrial sirtuin 3 via activation of RelB to ultimately increase mitochondrial biogenesis (60). We found in lean mice with sepsis that sirtuin 1 inhibition during hypo-inflammatory phase broadly promotes immune and metabolic homeostasis, and improves cardiac function, concomitant with reducing mortality (61) (35).
Obesity prolongs the hypo-inflammatory state following sirtuin 2 induction; surprisingly that sirtuin 1 does not play a part in this process. Sirtuin 2 inhibition during the hypo-inflammatory phase of obese mice with sepsis shortens hypo-inflammation and improves survival (47). Others have also observed that sirtuin 2 inhibition protects rodent sepsis by improving bacterial clearance (62, 63). As another function of sirtuins, mitochondrial sirtuin 4 is induced during late sepsis in cultured monocyte, which counters sirtuin 1 metabolic regulation of lipid oxidation. The sirtuin 4 causes metabolic reprogramming in a feedforward manner to use glucose as energy-fuel and supports resolution of acute inflammation (64). An important feature of sustained sirtuin 1 activation is that de novo synthesis of NAD+ and not NAMPT expression promotes tolerance (33).Emerging evidence suggests that the NAD+ salvage pathway switches to de novo synthesis of NAD+ to sustain increased NAD+ levels and in turn increased sirtuin expression during hypo-inflammation and endotoxin tolerance (33) (Figure 2). Sirtuin 2 activation and nuclear translocation also promotes immune function in obese septic mice after its nuclear translocation (47, 49). Increases in sirtuin 3 expression and its transfer from the nucleus mitochondrial also coordinate the nutrient shift to mitochondria fatty acid oxidation (60). Sirtuin 1 is an essential determinant of survival in septic mice. Mechanistically this occurs when persistent elevation of sirtuin 1 leads to immuno-metabolic repression. Sirtuin 1 inhibitor EX-527 treatment during hypo-inflammatory phase reversed the immuno-metabolic repression and improved survival in mice (35).
Figure 2.

Sources of NAD during sepsis: Immune cells use NAD salvage pathway during early sepsis switching to de novo synthesis of NAD during late sepsis. Increased NAD concentrations then lead to increased expression of NAD sensors, the sirtuins
2.1.3. Epigenetic reprogramming:
A major feature of acute inflammation from sepsis is epigenetic reprogramming of immunity, intermediary metabolism, and mitochondrial bioenergetics (41). Others and we showed that the monocyte reprogramming transition from hyper- to hypo-inflammatory phase requires gene specific switching of signaling responsive euchromatin (hyper-inflammation) to silencing facultative heterochromatin (hypo-inflammation) (65, 66). This switch repositions nucleosomes to expose or cover DNA accessibility of protein complexes required to activate or repress of acute inflammation regulatory. Mechanistically, we found that that the euchromatin to heterochromatin transition requires a switch from NFκB p65 to RelB (26, 67–69) that requires sirtuin 1. Sirtuin 1 supports RelB loading on DNA, followed by reprogramming the histone and DNA epigenetic code to facultative heterochromatin formation (41). We reported that sirtuin 1 induces RelB during the hypo-inflammation to participate in this epigenetic reprogramming (60).
Taken together, many studies support that both shifts in redox and activation of sirtuins by NAD direct immuno-metabolic and bioenergy shifts during sepsis. Excessive induction of ROS and NOS breaks down mitochondria and nuclear DNA, which within and without cells further activates hyper-inflammatory injury (55). Nuclear sirtuin 6 promotes nuclear DNA repair (56).
Summary:
Sirtuins play a critical role in controlling immuno-metabolic and energy reprogramming at multiple levels during sepsis, including epigenetics, transcription, post translational modifications, and shifts in the redox code. Importantly, sirtuin 1 and 2 are emerging as potential druggable targets for promoting homeostasis during the acute inflammatory responses of sepsis in lean and obese mice, respectively.
2.2. SIRTUINS AND NON-SEPSIS INDUCED LIFE THREATENING INFLAMMATION
Acute systemic inflammatory response to non-sepsis related injuries such as trauma, hemorrhagic shock and acute lung injury due to various non-infectious causes also are associated with increased mortality in critically ill patients. While the initial pro-inflammatory response is practically indistinguishable from that of sepsis-related systemic inflammation, the course of the disease is quite different. As depicted in Figure 3, while the departure from homeostasis in the initial phase is not quite as severe in magnitude as sepsis-related inflammation, the pro-inflammatory phase is prolonged and is associated with decreased sirtuin expression. Two specific examples of this type of inflammation are described below:
Figure 3.
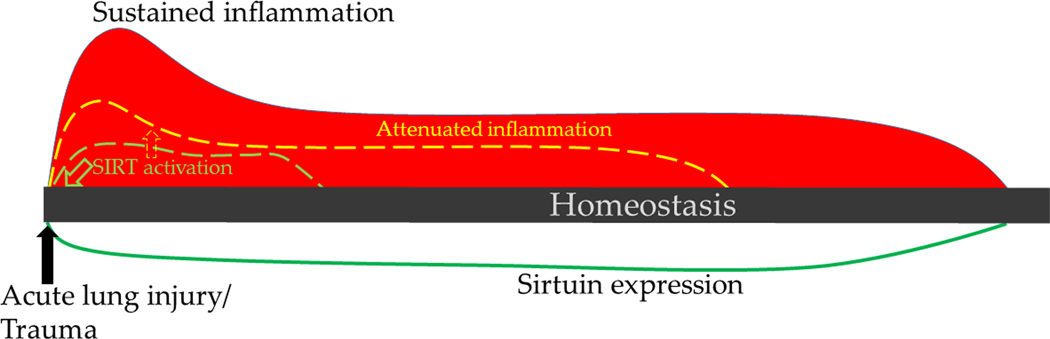
Immune response in acute lung injury and trauma: Immune response to acute lung injury and trauma exhibit pro-inflammatory phenotype. This is associated with decreased sirtuin expression and delayed return to homeostasis. Sirtuin activation early during the injury can attenuate pro-inflammatory response to achieve return to homeostasis quickly
2.2.1. Acute traumatic injury of lung injury:
Like in early sepsis, acute lung trauma generates alarmins that activates the key pro-inflammatory and immune axis that requires the NFκB p65-dependent pro-inflammatory response. Moreover, during direct severe lung injury from trauma, the early response “primes” immune cells for further injury to lung and other organs if a second environmental alarm such as infection occurs. The exaggerated pro-inflammatory response to “second hit” with another inflammatory stress response accentuates the pro-inflammatory early cytokine storm. Sirtuin levels, specifically sirtuin1 mRNA and protein levels decrease in response to traumatic lung injury as a key feature of “priming”. Activating sirtuin1 by resveratrol can attenuate the second hit inflammatory response and decrease the acute lung failure syndrome. Traumatic lung injury by activating NFkB and other pro-inflammatory signaling events increase the production of reactive oxygen species from NADPH oxidases and inducible nitric oxide synthase results in nitrosylation and inactivation of sirtuin1, as a mechanism contributing to priming in response to lung trauma. The importance of ROS and NOS in “priming” is supported by studies showing that anti-oxidant N-acetylcysteine attenuates reductions in sirtuin1 (70, 71). The kinetics of acute lung trauma with sepsis contrasts with the sepsis without trauma. In sepsis, sirtuin1 reductions rapidly shift to increases in NAD and sirtuin1 in the nucleus with transition of hyper-inflammatory dominance to hypo-inflammatory dominance of the tolerant state and pronounced immune suppression. Thus, sirtuin1 and perhaps other sirtuins not studied yet, support context-dependent modulation sirtuin contributions among the acute inflammatory syndromes like environmental physical trauma (35, 47). In this paradigm, sirtuin reductions promote priming; conversely sirtuin1 agonist protect against a second hit dissemination on inflammation and accentuation of the local pulmonary inflammation.
2.2.2. Hemorrhagic shock.
Another example of the role of sirtuins an inflammatory shock syndrome is in the context of hemorrhagic shock in animal models, which prompts post ischemia-reperfusion injuries in cardiac tissue. This also occurs myocardial infarction. The models descriptively identify mitochondrial dysfunction, decreased levels of sirtuin1 mRNA, and increased c-myc growth factor. Increasing sirtuin1 therapeutically in post ischemia perfusion injury and shock, in stark contrast to inhibiting sirtuin1 in sepsis, improves left ventricular dysfunction (72). Trauma-hemorrhagic shock-associated kidney injury was also shown to be associated with mitochondrial dysfunction via decreased sirtuin1 expression; sirtuin1 activators rescued mitochondrial dysfunction but not organ dysfunction (73).
2.2.3. Acute Lung Injury:
Acute lung injury (ALI) with respiratory failure is a distinct life threatening acute inflammatory reaction in the lung from multitude of causes. It is characterized by non-cardiogenic pulmonary edema, increased vascular permeability, cellular infiltration of alveolar space and decreased aerated lung space often requiring ventilatory support. While overall mortality of acute lung failure has substantially decreased over the past two decades, the mortality continues to be unacceptably high (33). A growing body of evidence suggests a substantial role for sirtuins in ALI with respiratory failure syndrome. Decreased sirtuin1 protein expression in alveolar epithelial cells (AEC) is central to the pathogenesis of pro-inflammatory phenotype in ARDS. Sirtuin 1 attenuation, which may follow direct oxidation injury to sirtuin thiols, is associated with increased pro-inflammatory cytokine expression. Mechanistically, sirtuin1 regulates lung inflammation via regulation of autophagy (74) as well as that of MAP kinase and NF kappa b pathways. Moreover, in experimental model of ARDS, increased miR199a was shown to promote inflammatory response via downregulation of sirtuin 1(74).
Emerging evidence indicates a role for sirtuin3 contributes to the pathophysiology of ALI in experimental models. Specifically, in ALI, the expression of sirtuin3 levels are decreased in lung tissue and are associated with pro-inflammatory response. sirtuin3 deficient mice exhibit increased pro-inflammatory phenotype and oxidant damage in lungs via activation of NLRP3 inflammasome pathway; sirtuin3 activation reduces lung injury and oxidant damage as well(75). In hyperoxia model of ALI (76), overexpression of sirtuin3 is associated with attenuation of pro-inflammatory phenotype via decreased oxidative stress.
Endothelial barrier dysfunction in ALI leads to accumulation of protein-rich fluid and inflammatory cell accumulation in the alveolar spaces. Sirtuin 3 deficiency is shown to decrease this barrier and increase lung inflammation in ALI model as well (77). Thus, targeting sirtuin3 may become a therapeutic option in near future.
Our view is that, although context specific, these lung dysfunction syndromes with acute life threatening injury are part of a sirtuin response spectrum. Overlaps are likely and sirtuin targeting may require precision medicine, unless a common overlapping sirtuin or NAD generating node is identified. More basic translational research is needed in the fields of non-sepsis, as well as the sepsis syndromes, including more than gene expression analysis. Unbiased metabolomics and proteomics with single cell analyses will enlighten similarities and differences of the entire sirtuin network in inflammatory “shock” syndromes. The importance of mitochondrial bioenergy reprogramming cannot be over emphasized.
3. SIRTUIN THERAPEUTIC TARGETING.
3.1. SEPSIS:
The field of sirtuin based treatment of life threatening acute inflammatory syndromes is embryonic, but promising. Sirtuin family members are likely determinants of outcome of inflammatory shock and its reprogramming of metabolism, immunity, and bioenergetics. The present state of knowledge suggests that sirtuin targeting should be context dependent and assessed at different times of acute inflammation reprogramming. While the response in sepsis and septic shock goes through the early/hyper-inflammatory to late/hypo-inflammatory response before resolution occurs; traumatic injury and ALI-related inflammatory response may not. More kinetic analyses of tolerance, including metabolomics are needed to be sure that tolerance is absent. We doubt it, particularly in ALI.
Sirtuin based therapies in sepsis and septic shock are nearing a promise and should be applied to human clinical trials. As described above, the sirtuins levels and function are biphasic in sepsis. First there is early direct oxidation and enzymatic inactivation of sirtuin shortly after acute inflammation generates ROS and NOS. Second there is increased expression, increased NAD+ generation, and persistent deacetylation activity of sirtuins 1 and 2. This is uncertain for other sirtuins. It is important to point out that the sirtuin family members vary in their time of induction, as exemplified for sirtuin4 promoter of sepsis inflammation resolution in mice. Sirtuin 4 expression occurs after sirtuin1/2, 6, and 3 peak. We deem that activation of sirtuins during hyper-inflammation and sirtuin1 and/or sirtuin2 inhibition during the hypo-inflammatory tolerant state should continue toward human clinical trials.
Sirtuin activation during hyper-inflammation:
sirtuin 1 and 2 levels are decreased during the hyper-inflammatory phase of sepsis, indicating potential therapeutic role for sirtuin activation. Indeed, others and we showed that sirtuin 1 activation during hyper-inflammation using resveratrol increased survival in a rodent model of sepsis (48, 78, 79).
Sirtuin inhibition during hypo-inflammation:
Modulation of sirtuins during hypo-inflammation is more complicated, for at this time it appears time and context dependent. For example, we found that sirtuin1 played a critical role in switch from hyper-to hypo-inflammation in lean sepsis, but did not contribute to this axis in obese mice with sepsis(35, 47). Instead, sirtuin2 treatment during tolerance obese mice promoted survival. This presents a conundrum, since many sepsis victims are obese.
3.2. NON SEPSIS ACUTE INFLAMMATION:
3.2.1. Sirtuin based therapies in acute traumatic injury and hemorrhagic shock:
Acute systemic inflammatory response in traumatic injury differs from that of sepsis and septic shock. Evidence suggests that the pro-inflammatory state in acute traumatic injury may not enter the universal survival tolerance phenotype; more studies however are needed. We deem that further developing sirtuin1 agonist treatment of acute traumatic lung injury is warranted. Resveratrol has been shown to attenuate cardiac and hepatic injury in experimental models of trauma-hemorrhage (72, 80). Interestingly, a specific sirtuin 1 agonist SRT1720 was shown to improve hepatic and immune function as well as attenuate mitochondrial dysfunction via PGC1-α modification in trauma-hemorrhage model (81).
3.2.2. Sirtuin based therapies in ALI:
Acute lung injury, a devastating condition in critically ill patients not only increases mortality but also increases long term disability in the surviving patients. Therapeutic strategies to target faster recovery are essential. Sirtuin 1 based therapies including resveratrol prevent lung damage in rodent models (79, 82, 83). Moreover, miRNA based therapies that act via sirtuin 1 activation are also shown to be beneficial (84). Sirtuin 3 overexpressing mice showed decreased lung injury (76). Sirtuin 3 activation with viniferin also reversed lung inflammation via NLRP inflammasome pathway (75) and honokiol via effect on endothelial barrier function (77) in rodent model of ALI. Thus, emerging data indicates therapeutic potential for sirtuin 1 and sirtuin 3 based therapies in ALI.
4. CONCLUSION
Acute inflammation is a highly conserved process to preserve life; it must deviate from homeostasis to “defend and mend” to ultimately restore homeostasis. Sirtuins, the NAD+ sensors, are the guardians of homeostasis. The interactions between the two are context-dependent and evolving as acute inflammation progresses. These interactions present sirtuins as potential “druggable” therapeutic targets.
5. EXPERT OPINION
Dramatic perturbations in sirtuin pathway occur as acute inflammation progresses to ultimately restore homeostasis. These perturbations pose sirtuins as unique therapeutic targets. Literature suggests that these perturbations occur in a context and time-dependent manner in the disseminated acute inflammatory response of sepsis and non-sepsis. Furthermore, evidence suggests that while sirtuins affect acute inflammatory phenotype, oxidant stress of acute inflammation also modulates sirtuin activity in return.
Acute life threatening inflammation of sepsis and septic shock are the leading causes of death globally. During early/hyper-inflammation of sepsis sirtuin activation is deemed beneficial. However, sepsis is a dynamic disease and sepsis-inflammation transitions from hyper- to hypo-inflammation. Sepsis-related mortality occurs during late/hypo-inflammatory phase while most therapeutic trials have focused on treating early sepsis. There is a dire need for specific therapeutic agents to decrease mortality in sepsis. While the awareness of hypo-inflammatory phase in the field of Critical Care Medicine is increasing, as yet, the lack of reliable biomarkers to distinguish the phases in a patient make therapeutics difficult.
Others and we have shown that sirtuins are critically important for transition to and sustenance of hypo-inflammatory phase of late sepsis. Furthermore, within the sepsis-related injury, sirtuin involvement seems to be in a context-dependent (obese vs. lean) as well. Evidence suggests that sirtuin inhibition during the hypo-inflammatory phase is beneficial. Thus, sirtuin activation during early/hyper-inflammation and sirtuin inhibition during late/hypo-inflammation are needed. This means recognition of the exact phase of sepsis at any given time is critical before we employ sirtuin-based phase-specific therapy. To further complicate the matters, during acute critical illness, the therapeutics need to be quick and precise to make a difference in the care of a patient. In non-sepsis life threatening injuries, the evidence suggests, that this is largely a pro-inflammatory response and phase transition does not occur. The sirtuin involvement is largely that of sirtuin deficiency. Thus, the therapeutic use of sirtuin pathway although promising, seems to be extremely complicated.
Additionally, while we have clearly laid out the sepsis vs. non-sepsis related injuries this review, it is quite difficult to differentiate the two clinically. Thus, biomarkers to not only differentiate sepsis from non-sepsis in addition to the phase of sepsis (hyper- or hypo-inflammatory) at any given time are needed.
To summarize, the challenges to precise treatment in acute systemic inflammatory response syndrome seems to be that of 1) early recognition of the exact etiology (sepsis vs. non-sepsis-related injuries) and 2) within the sepsis related injury state, the phase of sepsis.
On the brighter side, the data implicating sirtuin deficiency and therapeutic efficacy of sirtuin activation in non-sepsis related injuries is convincing. We project that testing for the sirtuin deficiency, targeting for sirtuin activating therapies and ultimately homeostasis promoting concept in non-sepsis related life threatening conditions in humans will occur first and within the next few years. With increasing awareness of phase-shifts in sepsis, within the decade, multiple biomarkers/biomarker panels that will pave the way for early and timely recognition of phase of sepsis and phase-specific sirtuin modulation in sepsis as well.
Article Highlights.
Immune response to acute inflammation must deviate cells from homeostasis to defend and mend; the extent and duration of this deviation is different in acute vs. chronic inflammation.
Evidence suggests that the bioenergy source-selection and metabolism in immune cells dictate immune response to acute inflammation.
Sirtuins, a highly conserved family of nutrient/bioenergy sensors with anti-inflammatory properties are in a unique position to fine-tune the immuno-metabolic response to acute inflammation.
The interaction between acute inflammatory response and sirtuins is bidirectional; acute inflammatory response may decrease sirtuin activity/function via direct oxidation during hyper-inflammatory phase.
The interplay of acute inflammatory response and sirtuin activity/function ultimately decide fate of an immune cell/tissue in an organism making sirtuins unique therapeutic targets during acute inflammation
Funding
The work of the authors is funded by the National Institutes of Health (NIH).
Footnotes
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers
- 1.Guarente L. Sirtuins, aging, and metabolism. Cold Spring Harbor symposia on quantitative biology. 2011;76:81–90. [DOI] [PubMed] [Google Scholar]
- 2.Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends in cell biology. 2014;24(8):464–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annual review of pathology. 2010;5:253–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nogueiras R, Habegger KM, Chaudhary N, Finan B, Banks AS, Dietrich MO, et al. Sirtuin 1 and sirtuin 3: physiological modulators of metabolism. Physiol Rev. 2012;92(3):1479–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130(6):1095–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bai B, Vanhoutte PM, Wang Y. Loss-of-SIRT1 function during vascular ageing: hyperphosphorylation mediated by cyclin-dependent kinase 5. Trends in cardiovascular medicine. 2014;24(2):81–4. [DOI] [PubMed] [Google Scholar]
- 7.Guarente L. Calorie restriction and SIR2 genes--towards a mechanism. Mechanisms of ageing and development. 2005;126(9):923–8. [DOI] [PubMed] [Google Scholar]
- 8.Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Molecular biology of the cell. 2005;16(10):4623–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moniot S, Weyand M, Steegborn C. Structures, substrates, and regulators of Mammalian sirtuins - opportunities and challenges for drug development. Frontiers in pharmacology. 2012;3:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwer B, North BJ, Frye RA, Ott M, Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. The Journal of cell biology. 2002;158(4):647–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao K, Chai X, Clements A, Marmorstein R. Structure and autoregulation of the yeast Hst2 homolog of Sir2. Nature structural biology. 2003;10(10):864–71. [DOI] [PubMed] [Google Scholar]
- 12.Sauve AA. Sirtuin chemical mechanisms. Biochimica et biophysica acta. 2010;1804(8):1591–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sauve AA, Moir RD, Schramm VL, Willis IM. Chemical activation of Sir2-dependent silencing by relief of nicotinamide inhibition. Molecular cell. 2005;17(4):595–601. [DOI] [PubMed] [Google Scholar]
- 14.Villalba JM, Alcain FJ. Sirtuin activators and inhibitors. BioFactors. 2012;38(5):349–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Canto C, Menzies KJ, Auwerx J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015;22(1):31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tao R, Xiong X, DePinho RA, Deng CX, Dong XC. FoxO3 transcription factor and Sirt6 deacetylase regulate low density lipoprotein (LDL)-cholesterol homeostasis via control of the proprotein convertase subtilisin/kexin type 9 (Pcsk9) gene expression. The Journal of biological chemistry. 2013;288(41):29252–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhong L, D’Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010;140(2):280–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science. 2012;335(6071):936–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steven JL. Metchnikoff on the Comparative Pathology of Inflammation. Glasgow Med J. 1892;38(3):195–205. [PMC free article] [PubMed] [Google Scholar]
- 20.Opal SM, Esmon CT. Bench-to-bedside review: functional relationships between coagulation and the innate immune response and their respective roles in the pathogenesis of sepsis. Critical care. 2003;7(1):23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iwanaga S. The molecular basis of innate immunity in the horseshoe crab. Current opinion in immunology. 2002;14(1):87–95. [DOI] [PubMed] [Google Scholar]
- 22.Furukawa R, Takahashi Y, Nakajima Y, Dan-Sohkawa M, Kaneko H. Defense system by mesenchyme cells in bipinnaria larvae of the starfish, Asterina pectinifera. Dev Comp Immunol. 2009;33(2):205–15. [DOI] [PubMed] [Google Scholar]
- 23.Nathan C. Epidemic inflammation: pondering obesity. Mol Med. 2008;14(7–8):485–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson AR, Milner JJ, Makowski L. The inflammation highway: metabolism accelerates inflammatory traffic in obesity. Immunol Rev. 2012;249(1):218–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sorgdrager FJH, Naude PJW, Kema IP, Nollen EA, Deyn PP. Tryptophan Metabolism in Inflammaging: From Biomarker to Therapeutic Target. Frontiers in immunology. 2019;10:2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vachharajani V, Liu T, McCall CE. Epigenetic coordination of acute systemic inflammation: potential therapeutic targets. Expert review of clinical immunology. 2014;10(9):1141–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305(5682):390–2. [DOI] [PubMed] [Google Scholar]
- 28.Li X. SIRT1 and energy metabolism. Acta biochimica et biophysica Sinica. 2013;45(1):51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Molecular cell. 2007;28(1):91–106. [DOI] [PubMed] [Google Scholar]
- 30.Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell. 2010;140(6):771–6. [DOI] [PubMed] [Google Scholar]
- 31.Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14(4):528–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carson WF, Cavassani KA, Dou Y, Kunkel SL. Epigenetic regulation of immune cell functions during post-septic immunosuppression. Epigenetics. 2011;6(3):273–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang J, Tao J, Ling Y, Li F, Zhu X, Xu L, et al. Switch of NAD Salvage to de novo Biosynthesis Sustains SIRT1-RelB-Dependent Inflammatory Tolerance. Frontiers in immunology. 2019;10:2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA : the journal of the American Medical Association. 2016;315(8):801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vachharajani VT, Fu Liu T, Brown CM, Wang X, Buechler NL, Wells JD, et al. SIRT1 inhibition during the hypoinflammatory phenotype of sepsis enhances immunity and improves outcome. J Leukoc Biol. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA : the journal of the American Medical Association. 2011;306(23):2594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Otto GP, Sossdorf M, Claus RA, Rodel J, Menge K, Reinhart K, et al. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Critical care. 2011;15(4):R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qin K, Han C, Zhang H, Li T, Li N, Cao X. NAD(+) dependent deacetylase Sirtuin 5 rescues the innate inflammatory response of endotoxin tolerant macrophages by promoting acetylation of p65. Journal of autoimmunity. 2017;81:120–9. [DOI] [PubMed] [Google Scholar]
- 39.Warburg O, Gawehn K, Geissler AW. [Metabolism of leukocytes]. Zeitschrift fur Naturforschung Teil B: Chemie, Biochemie, Biophysik, Biologie. 1958;13B(8):515–6. [PubMed] [Google Scholar]
- 40.Wang X, Buechler NL, Woodruff AG, Long DL, Zabalawi M, Yoza BK, et al. Sirtuins and Immuno-Metabolism of Sepsis. International journal of molecular sciences. 2018;19(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vachharajani V, McCall CE. Epigenetic and metabolic programming of innate immunity in sepsis. Innate Immun. 2019;25(5):267–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu TF, Yoza BK, El Gazzar M, Vachharajani VT, McCall CE. NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. The Journal of biological chemistry. 2011;286(11):9856–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu S, Gao Y, Zhang Q, Wei S, Chen Z, Dai X, et al. SIRT1/3 Activation by Resveratrol Attenuates Acute Kidney Injury in a Septic Rat Model. Oxidative medicine and cellular longevity. 2016;2016:7296092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeng H, He X, Tuo QH, Liao DF, Zhang GQ, Chen JX. LPS causes pericyte loss and microvascular dysfunction via disruption of Sirt3/angiopoietins/Tie-2 and HIF-2alpha/Notch3 pathways. Scientific reports. 2016;6:20931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao WY, Zhang L, Sui MX, Zhu YH, Zeng L. Protective effects of sirtuin 3 in a murine model of sepsis-induced acute kidney injury. Scientific reports. 2016;6:33201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun F, Si Y, Bao H, Xu Y, Pan X, Zeng L, et al. Regulation of Sirtuin 3-Mediated Deacetylation of Cyclophilin D Attenuated Cognitive Dysfunction Induced by Sepsis-Associated Encephalopathy in Mice. Cell Mol Neurobiol. 2017;37(8):1457–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Buechler NL, Martin A, Wells J, Yoza B, McCall CE, et al. Sirtuin-2 Regulates Sepsis Inflammation in ob/ob Mice. PloS one. 2016;11(8):e0160431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang X, Buechler NL, Yoza BK, McCall CE, Vachharajani VT. Resveratrol attenuates microvascular inflammation in sepsis via SIRT-1-Induced modulation of adhesion molecules in ob/ob mice. Obesity. 2015;23(6):1209–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buechler N, Wang X, Yoza BK, McCall CE, Vachharajani V. Sirtuin 2 Regulates Microvascular Inflammation during Sepsis. Journal of immunology research. 2017;2017:2648946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang X, Buechler NL, Long DL, Furdui CM, Yoza BK, McCall CE, et al. Cysteine thiol oxidation on SIRT2 regulates inflammation in obese mice with sepsis. Inflammation. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zee RS, Yoo CB, Pimentel DR, Perlman DH, Burgoyne JR, Hou X, et al. Redox regulation of sirtuin-1 by S-glutathiolation. Antioxidants & redox signaling. 2010;13(7):1023–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dikalova AE, Itani HA, Nazarewicz RR, McMaster WG, Flynn CR, Uzhachenko R, et al. Sirt3 Impairment and SOD2 Hyperacetylation in Vascular Oxidative Stress and Hypertension. Circulation research. 2017;121(5):564–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dikalov SI, Dikalova AE. Crosstalk Between Mitochondrial Hyperacetylation and Oxidative Stress in Vascular Dysfunction and Hypertension. Antioxidants & redox signaling. 2019;31(10):710–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Long D, Wu H, Tsang AW, Poole LB, Yoza BK, Wang X, et al. The Oxidative State of Cysteine Thiol 144 Regulates the SIRT6 Glucose Homeostat. Scientific reports. 2017;7(1):11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yao X, Carlson D, Sun Y, Ma L, Wolf SE, Minei JP, et al. Mitochondrial ROS Induces Cardiac Inflammation via a Pathway through mtDNA Damage in a Pneumonia-Related Sepsis Model. PloS one. 2015;10(10):e0139416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lombard DB, Schwer B, Alt FW, Mostoslavsky R. SIRT6 in DNA repair, metabolism and ageing. Journal of internal medicine. 2008;263(2):128–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, et al. SIRT6 promotes DNA repair under stress by activating PARP1. Science. 2011;332(6036):1443–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cheng SC, Scicluna BP, Arts RJ, Gresnigt MS, Lachmandas E, Giamarellos-Bourboulis EJ, et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nature immunology. 2016;17(4):406–13. [DOI] [PubMed] [Google Scholar]
- 59.Allantaz-Frager F, Turrel-Davin F, Venet F, Monnin C, De Saint Jean A, Barbalat V, et al. Identification of biomarkers of response to IFNg during endotoxin tolerance: application to septic shock. PloS one. 2013;8(7):e68218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu TF, Vachharajani V, Millet P, Bharadwaj MS, Molina AJ, McCall CE. Sequential actions of SIRT1-RELB-SIRT3 coordinate nuclear-mitochondrial communication during immunometabolic adaptation to acute inflammation and sepsis. The Journal of biological chemistry. 2015;290(1):396–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith LM, Yoza BK, Hoth JJ, McCall CE, Vachharajani V. SIRT1 Mediates Septic Cardiomyopathy in a Murine Model of Polymicrobial Sepsis. Shock. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ciarlo E, Heinonen T, Theroude C, Herderschee J, Mombelli M, Lugrin J, et al. Sirtuin 2 Deficiency Increases Bacterial Phagocytosis by Macrophages and Protects from Chronic Staphylococcal Infection. Frontiers in immunology. 2017;8:1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao T, Alam HB, Liu B, Bronson RT, Nikolian VC, Wu E, et al. Selective Inhibition of SIRT2 Improves Outcomes in a Lethal Septic Model. Current molecular medicine. 2015;15(7):634–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tao J, Zhang J, Ling Y, McCall CE, Liu TF. Mitochondrial Sirtuin 4 Resolves Immune Tolerance in Monocytes by Rebalancing Glycolysis and Glucose Oxidation Homeostasis. Frontiers in immunology. 2018;9:419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yoza BK, McCall CE. Facultative heterochromatin formation at the IL-1 beta promoter in LPS tolerance and sepsis. Cytokine. 2011;53(2):145–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lyn-Kew K, Rich E, Zeng X, Wen H, Kunkel SL, Newstead MW, et al. IRAK-M regulates chromatin remodeling in lung macrophages during experimental sepsis. PloS one. 2010;5(6):e11145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.El Gazzar M, Yoza BK, Hu JY, Cousart SL, McCall CE. Epigenetic silencing of tumor necrosis factor alpha during endotoxin tolerance. The Journal of biological chemistry. 2007;282(37):26857–64. [DOI] [PubMed] [Google Scholar]
- 68.El Gazzar M, Yoza BK, Chen X, Garcia BA, Young NL, McCall CE. Chromatin-specific remodeling by HMGB1 and linker histone H1 silences proinflammatory genes during endotoxin tolerance. Molecular and cellular biology. 2009;29(7):1959–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McCall CE, Yoza B, Liu T, El Gazzar M. Gene-specific epigenetic regulation in serious infections with systemic inflammation. J Innate Immun. 2010;2(5):395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hoth JJ, Smith LM, Furdui CM, Wells JD, Yoza BK, McCall CE. Antioxidant treatment after injury suppresses second hit immune priming. The journal of trauma and acute care surgery. 2018;85(2):367–74. [DOI] [PubMed] [Google Scholar]
- 71.Smith LM, Wells JD, Vachharajani VT, Yoza BK, McCall CE, Hoth JJ. SIRT1 mediates a primed response to immune challenge after traumatic lung injury. The journal of trauma and acute care surgery. 2015;78(5):1034–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jian B, Yang S, Chaudry IH, Raju R. Resveratrol improves cardiac contractility following trauma-hemorrhage by modulating Sirt1. Mol Med. 2012;18:209–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang H, Guan Y, Karamercan MA, Ye L, Bhatti T, Becker LB, et al. Resveratrol Rescues Kidney Mitochondrial Function Following Hemorrhagic Shock. Shock. 2015;44(2):173–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu P, Huang G, Wei T, Gao J, Huang C, Sun M, et al. Sirtuin 3-induced macrophage autophagy in regulating NLRP3 inflammasome activation. Biochimica et biophysica acta. 2018;1864(3):764–77. [DOI] [PubMed] [Google Scholar]
- 75.Kurundkar D, Kurundkar AR, Bone NB, Becker EJ Jr., Liu W, Chacko B, et al. SIRT3 diminishes inflammation and mitigates endotoxin-induced acute lung injury. JCI insight. 2019;4(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tian YG, Zhang J. Protective effect of SIRT3 on acute lung injury by increasing manganese superoxide dismutase-mediated antioxidation. Molecular medicine reports. 2018;17(4):5557–65. [DOI] [PubMed] [Google Scholar]
- 77.Chen L, Li W, Qi D, Lu L, Zhang Z, Wang D. Honokiol protects pulmonary microvascular endothelial barrier against lipopolysaccharide-induced ARDS partially via the Sirt3/AMPK signaling axis. Life sciences. 2018;210:86–95. [DOI] [PubMed] [Google Scholar]
- 78.An R, Zhao L, Xu J, Xi C, Li H, Shen G, et al. Resveratrol alleviates sepsisinduced myocardial injury in rats by suppressing neutrophil accumulation, the induction of TNFalpha and myocardial apoptosis via activation of Sirt1. Molecular medicine reports. 2016;14(6):5297–303. [DOI] [PubMed] [Google Scholar]
- 79.Li T, Zhang J, Feng J, Li Q, Wu L, Ye Q, et al. Resveratrol reduces acute lung injury in a LPSinduced sepsis mouse model via activation of Sirt1. Molecular medicine reports. 2013;7(6):1889–95. [DOI] [PubMed] [Google Scholar]
- 80.Liu FC, Tsai YF, Tsai HI, Yu HP. Anti-Inflammatory and Organ-Protective Effects of Resveratrol in Trauma-Hemorrhagic Injury. Mediators of inflammation. 2015;2015:643763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Luciano JA, Kautza B, Darwiche S, Martinez S, Stratimirovic S, Waltz P, et al. Sirtuin 1 Agonist Minimizes Injury and Improves the Immune Response Following Traumatic Shock. Shock. 2015;44 Suppl 1:149–55. [DOI] [PubMed] [Google Scholar]
- 82.Akhmedov A, Camici GG, Reiner MF, Bonetti NR, Costantino S, Holy EW, et al. Endothelial LOX-1 activation differentially regulates arterial thrombus formation depending on oxLDL levels: role of the Oct-1/SIRT1 and ERK1/2 pathways. Cardiovascular research. 2017;113(5):498–507. [DOI] [PubMed] [Google Scholar]
- 83.Yang K, Gao B, Wei W, Li Z, Pan L, Zhang J, et al. Changed profile of microRNAs in acute lung injury induced by cardio-pulmonary bypass and its mechanism involved with SIRT1. International journal of clinical and experimental pathology. 2015;8(2):1104–15. [PMC free article] [PubMed] [Google Scholar]
- 84.Liu Y, Guan H, Zhang JL, Zheng Z, Wang HT, Tao K, et al. Acute downregulation of miR-199a attenuates sepsis-induced acute lung injury by targeting SIRT1. American journal of physiology Cell physiology. 2018;314(4):C449–C55. [DOI] [PubMed] [Google Scholar]